Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results and Discussion
AuNP-DPA specifically enriches...
Conclusions
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(19):5320-5335. doi:10.7150/thno.27165 This issue Cite
Research Paper
Awakening p53 in vivo by D-peptides-functionalized ultra-small nanoparticles: Overcoming biological barriers to D-peptide drug delivery
Zhenyuan Bian1,2*, Jin Yan3,4*, Simeng Wang5*, Yijie Li1, Yi Guo4, Bohan Ma6, Hao Guo7, Zhijie Lei7, Chun Yin7, Yi Zhou8, Min Liu8, Kaishan Tao1 ![]() , Peng Hou5
, Peng Hou5 ![]() , Wangxiao He6,9
, Wangxiao He6,9 ![]()
1. Department of Hepatobiliary Surgery, Xijing Hospital, Fourth Military Medical University, Xi'an, 710032, Shaanxi Province, China.
2. Department of general surgery, General Hospital of Shenyang Military Area Command, Shenyang, 110016, Liaoning Province, China.
3. Department of Biologic and Materials Sciences, Department of Biomedical Engineering, Macromolecular Science and Engineering Center, Department of Materials Science and Engineering, University of Michigan, Ann Arbor, Michigan 48109, USA
4. Frontier Institute of Science and Technology, State Key Laboratory for Mechanical Behavior of Materials, Xi'an Jiaotong University, Xi'an 710049, China
5. Key Laboratory for Tumor Precision Medicine of Shaanxi Province, and Department of Endocrinology, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, China
6. Center for Translational Medicine, Key Laboratory of Biomedical Information Engineering of Ministry of Education, School of Life Science and Technology and Frontier Institute of Science and Technology, Xi'an Jiaotong University, Xi'an 710049, China.
7. Fourth Military Medical University, Xi'an 710032, Shaanxi Province, China.
8. Department of Infectious Diseases, The First Affiliated Hospital of Xi'an Jiaotong University, Xi'an 710061, China
9. Institute of Human Virology and Department of Biochemistry and Molecular Biology, University of Maryland School of Medicine, Baltimore, MD 21201, USA.
*These authors contributed equally.
Received 2018-5-8; Accepted 2018-10-5; Published 2018-10-22
Abstract

Peptides are a rapidly growing class of therapeutics with many advantages over conventional small molecule drugs. Dextrorotary (D)-peptides, with increased enzymatic stability and prolonged plasma half-life in comparison with natural L-peptides, are considered to have great potential as recognition molecules and therapeutic agents. However, the in vivo efficacy of current therapeutic D-peptides is hindered by their inefficient cellular uptake in diseased tissues.
Methods: To overcome physiological and cellular barriers to D-peptides, we designed a gold-based ultra-small nanocarrier coupled with polylysine (PLL) and a receptor-targeted peptide to deliver therapeutic D-peptides. Using a D-peptide p53 activator (DPA) as a proof of concept, we synthesized, functionalized and characterized gold- and DPA-based nanoparticles termed AuNP-DPA.
Results: AuNP-DPA were effectively enriched in tumor sites and subsequently internalized by cancer cells, thereby suppressing tumor growth via reactivating p53 signaling. More importantly, through a series of in vivo experiments, AuNP-DPA showed excellent biosafety without the common side effects that hinder p53 therapies in clinic trials.
Conclusion: The present study not only sheds light on the development of AuNP-DPA as a novel class of antitumor agents for drugging the p53 pathway in vivo, but also supplies a new strategy to use D-peptides as intracellular PPI inhibitors for cancer-targeted therapy.
Keywords: gold nanoparticle, ultra-small nanoparticle, dextrorotary peptide, p53, biosafety
Introduction
Protein-protein interactions (PPIs) mediate a great number of regulatory pathways and play a key role in many pathologies including tumorigenesis, and therefore represent an important and yet largely unexploited class of therapeutic targets [1, 2]. However, compared to the generally well-defined active sites of enzymes, receptors and ion channels, the interaction surfaces of PPIs are always flat or moderately convex, which is much more challenging for traditional drug discovery and development efforts of small molecules [3, 4]. Fortunately, peptides can overcome this obstacle due to their large interacting interface with chemical and structural diversity [5]. Despite their obvious advantages over small molecules for targeting intramolecular PPIs, peptides suffer poor stability, making them unusable as drugs [6]. In recent years, an array of techniques has been developed to improve the stability of peptides through chemical and/or structural strategies, thereby leading to a rapid expansion of the repertoire of peptide drug candidates for clinical evaluation. One of the most effective approaches to increase peptide stabilization is D-enantiomerization [5, 7].
D-peptides, composed entirely of unnatural D-amino acid residues, are resistant to proteolysis because the steric incompatibility of D-peptides and naturally occurring L-proteases affords an exceedingly high free energy barrier to catalysis [8]. As a result, D-peptides are highly stable not only intracellularly but also in circulation [8, 9]. An additional advantage of D-peptide therapeutics is that they are less immunogenic than L-forms because of their poor efficiency in proteasome-processed antigen presentation [10]. Several D-peptide antagonists with antitumor activity have recently been identified using mirror image phage display techniques [11-14], promising a novel class of peptide therapeutics with clinical benefits.
Although the facile access to proteolytically stable D-peptide inhibitors makes it possible to target intracellular PPIs responsible for disease initiation and progression, there are still some major obstacles that need to be solved such as poor membrane permeability and low therapeutic target enrichment of D-peptides [15-17]. Notwithstanding D-peptides can be introduced intracellularly for functional, mechanistic and efficacy studies via different delivery systems such as cell-penetrating peptides (CPPs) [18, 19] and liposomes [20]. However, these delivery vehicles have serious drawbacks and disadvantages, such as rapid elimination by the liver and spleen, non-specific cellular uptake by normal tissues and organs, and toxicity, thereby preventing them from becoming therapeutically viable [21-23]. Thus, there is a compelling need to develop new strategies for efficient intracellular delivery of D-peptides for clinical use.
Hitherto, aurous nanoparticle (AuNP)-based nanomedicine have emerged as an important strategy to deliver various payloads (such as small molecules, peptides and RNAs) into cells and target intracellular molecules [24-26]. Moreover, AuNPs have irreplaceable advantages such as essential chemical inertness, non-toxicity, simple design and economic cost [24, 27]. Thus, AuNP-based therapies have tremendous potential for clinical application, and some of them have entered clinical trials, such as CYT-6091 (complete phase 1 of clinical trials), AuroLase TM (complete early clinical trials), C19-A3 GNP (entered phase 1 of clinical trials) and so on [28, 29]. Considering the specific tumor accumulation by enhanced permeability and retention (EPR) effect and intracellular-redox-triggered drug release by thiol-gold conjugation, AuNP-based drug delivery systems are particularly attractive for the treatment of solid tumors [30-32]. In addition, receptor-ligand modification can promote transportation of nanoparticles carrying therapeutic molecules into cells through receptor-mediated endocytosis [25, 33, 34]. Altogether, we hypothesize that receptor-ligand-modified AuNPs may be used as efficient carriers to deliver D-peptides for cancer therapy.
p53, a tumor suppressor that inhibits growth and induces apoptosis in response to cellular stress, plays a key role in preventing injured cells from becoming cancerous [35, 36]. Dysfunction of p53 occurs in many human cancers, in which wild-type p53 is functionally inhibited by MDM2 [37-39]. Mounting evidence has shown that inhibiting MDM2-p53 interactions is a viable strategy for cancer therapy, and some small molecule antagonists have entered into clinical trials [40, 41]. However, without exception, off-target toxicity poses a big challenge to the application of MDM2 antagonists, for which strict dose constraints has resulted in sluggish development and clinical application [42]. Therefore, there is a compelling need to develop new p53 therapeutics with low nonspecific toxicity.
In this work, to overcome the biological obstacles of D-peptide application and develop new potent and safe p53 therapeutics, we investigated ultra-small AuNPs (<10 nm) as a high-loading drug delivery vehicle conjugated with D-peptide p53 activator DPA (TAWYANFEALLR) [20] for cancer therapy. Meanwhile, an RGD-derived peptide termed RGDDP (CRGDKRGDSP) was used to modify AuNPs in order to enhance their tumor targeting through specific binding to integrins, which have been demonstrated to be highly expressed on cancer cells but not on normal and stem cells [43-45]. Our data indicate that AuNP-DPA may be used as a potential D-peptide-based therapeutic agent for cancer therapy.
Results and Discussion
Synthesis and characterization of AuNP-DPA
The preparation of ultra-small AuNPs-DPA is outlined in Figure 1. The Au core is an ultra-small gold nanoparticle, which was synthesized using the 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) procedure. In detail, 1 mL 10 mM HAuCl4 solution was mixed with 9 mL HEPES buffer (50 mM, pH 7.4) under vigorous stirring. After ~5 min, the solution color changed to red, indicating successful formation of the Au core. Transmission electron microscopy (TEM) (Figure S1) and dynamic light scattering (DLS) measurements (Table S1) of the Au core showed a 4 nm particle diameter and 5 nm hydrated particle size (PDI: 0.02). For Au core conjugation, an extra cysteine was added to the C terminus of DPA (DPA-Cys: TAWYANFEALLRC; Figure S2A), and the amino group at the N terminal was acetylated. Au-DPA was then obtained after 20 min vigorous stirring after adding 2 mL DPA-Cys solution (1 mg/mL in 50% acetonitrile and 50% PBS buffer) into the 10 mL Au core solution, following ultrafiltration (cut off: 10000 Da) and cleaning with the PBS 3 times to remove unconjugated DPA. Next, Fourier transform infrared (FTIR) spectroscopy confirmed conjugation of DPA to the Au core by the characteristic peaks of peptide at 3400 and 1650 cm-1 (Figure S3). Notably, to investigate the effect of particle size on DPA loading, four Au cores with hydrated particle sizes of ~5 nm (PDI: 0.02), 10 nm (PDI: 0.05), 20 nm (PDI: 0.06) and 40 nm (PDI: 0.10) were prepared and used to load DPA. As shown in Table S2, the smallest Au core (ultra-small gold nanocore) had the maximum drug loading, which is mainly because this Au core has the maximum specific surface area.
Synthesis of AuNp-DPA. Schematic depiction for the synthesis of AuNp-DPA and their enrichment in the tumor site.
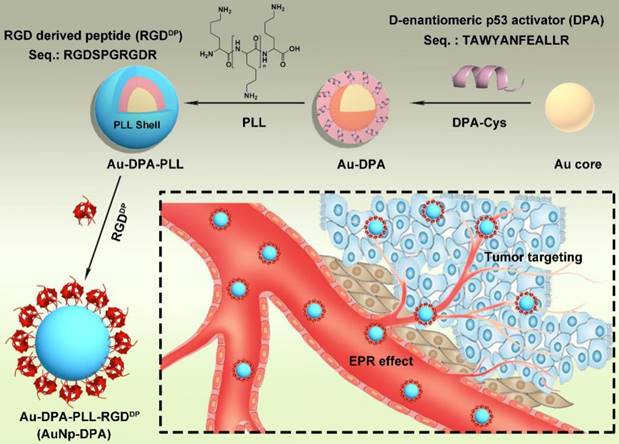
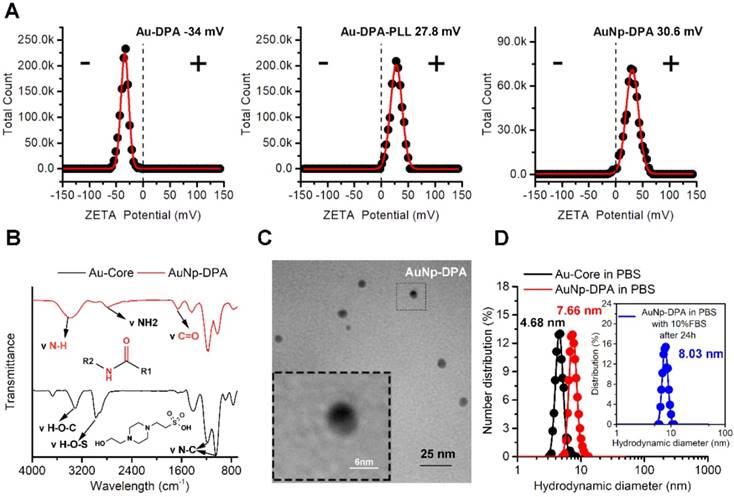
To endow the ultra-small nanoparticles with better hydrophilicity and more functional groups to link with targeting molecules, polylysine (PLL) was used to encapsulate Au-DPA (Figure 1). Considering that that the Au core was formed by the HEPES method and the pKa values for HEPES are pKa,1 = 3 and pKa,2 = 7.5, to guarantee sufficient deprotonation of PLL, pH 8.0 was chosen to promote PLL binding to Au-DPA. In this case, a negatively charged Au-DPA surface is also suitable for PLL electrostatic attachment. After this process, the intergranular electrostatic repulsion from the PLL-coated nanoparticle should prevail over the weak Van der Waals forces toward aggregation, thereby increasing the stability of the gold colloid [46, 47]. The Zeta potential confirmed PLL binding as evidenced by a surface charge increase from -34.0 mV for Au-DPA to 27.8 mV for Au-DPA-PLL (Figure 2A). In addition, compared to the Au core, the size of Au-DPA-PLL increased, as shown by TEM (Figure S4) and DLS (Table S1), further proving the PLL and DPA conjugation.
Physicochemical properties of AuNP-DPA. (A) The surface charge (Zeta potential) of Au-DPA, Au-DPA-PLL and AuNP-DPA measured in PBS buffer (pH 7.4) by DLS at 37 °C. All data points are the average of 20 measurements and were fitted by to a Gaussian distribution. (B) FTIR spectra of AuNP-DPA and Au-core, demonstrating their surface chemical structures before and after peptides as well as PLL conjugation. (C) TEM image of AuNP-DPA at pH 7.4; the embedded image is the magnified image of the outlined region. (D) AuNp-DPA and Au-core were diluted in PBS, and AuNp-DPA was diluted in PBS with 10% FBS for 24 h. Hydrodynamic distributions were then measured by dynamic light scattering. The results show that AuNp-DPA has a narrow size distribution, indicating their stability under physiological conditions.
With an aim of take full advantage of active targeting in vivo, integrin was selectively targeted to achieve strong tumor targeting and enhance delivery of the therapeutic molecules. Towards this end, an RGD-derived peptide termed RGDDP (RGDSPRGDR; Figure S2B) was used to modify Au-DPA-PLL. The large number of amino groups in PLL supplies sufficient sites for peptide conjugation. The reaction between the carboxyl of the C-terminal of RGDDP and the amino groups of PLL was catalyzed by EDC and NHS, both of which helped in the formation of more amide bonds by protecting them from hydrolysis [26, 48]. Next, the final product named AuNP-DPA was collected by centrifugation and lyophilization. Of note, no aggregation was observed during all of these processes.
AuNP-DPA were firstly characterized by Zeta potential measurements (Figure 2A), UV-vis spectroscopy (Figure S5), FTIR spectroscopy (Figure 2B), TEM (Figure 2C) and DLS (Figure 2D). In brief, after RGDDP conjugation, the charge shifted from 27.8 mV (Au-DPA-PLL) to 30.6 mV (AuNP-DPA), indicating successful modification with RGDDP (Figure 2A). The characteristic IR absorbance of peptide (amido bond: ~3400 and 1650 cm-1) and PLL (free amino group: ~3000 cm-1) in FTIR spectroscopy further confirmed the successful synthesis of AuNP-DPA (Figure 2B). In addition, we found that the surface plasmon peak band of AuNP-PDA was indistinct in the UV-vis absorbance measurement, probably due to the small Au core size and PLL coating (Figure S5). TEM analysis revealed that AuNP-DPA was ~5 nm, spherical and had a uniform dispersion (Figure 2C), which was consistent with the DLS results (Figure 2D). Moreover, AuNp-PDA was very easily soluble in water and maintained monodispersity after 24 h incubation in 10% fetal bovine serum (FBS) in PBS buffer at pH 7.4 (Figure 2D), indicating that AuNP-PDA is stable under serum condition. Additionally, to determine the peptide loading, AuNP-PDA was dissolved in a test buffer containing 6 M GuHCl (to eliminate the interaction between DPA and PLL) and 1 M DTT (to break the conjugation between peptide and Au nanoparticle). The amount of released peptide was quantified by HPLC and calculated by the standard curve method, and the loading of DPA-Cys was found to be 0.5 mmol/g (mmol/g = peptide molarity/Au mass) (Figure S6).
AuNP-DPA is efficiently internalized by cancer cells via integrin-dependent micropinocytosis
In general, high cell internalization and stimuli-responsive cargo release are required for delivering D-peptide into cancer cells and to awake the function of the therapeutic peptides. Based on our design, we consider that the positive charge from PLL and RGDDP conjugation can endow AuNP-DPA with good ability to be internalized by cancer cells. Thus, to determine this, we firstly treated an integrin-positive cell line HCT116 with AuNP-DPA (20 μg/mL) and its RGDDP-deleted counterpart (20 μg/mL) for 1 h, 2 h, 4 h and 6 h, and performed laser scanning confocal microscopy (LSCM) to detect their cellular uptake (Figure S7-9). After 6 h incubation, bright green fluorescence from FITC-labeled DPA was found in the AuNP-DPA-treated cells, whereas RGDDP-deleted counterpart-treated cells presented relatively weaker fluorescence at the same exposure time (Figure 3A). Notably, free DPA and AuNP-DPA without PLL and RGDDP showed nearly no cellular uptake (Figure 3A), suggesting that cellular internalization of AuNP-DPA stems from the positive charge and RGDDP conjugation. To further clarify the advantages of the PLL coating and RGDDP modification, we quantified the fluorescence intensity of the LSCM images (Figure 3B-C), and the results showed that the increase in fluorescence intensity from intracellular uptake of AuNP-DPAFITC was significantly faster than that of AuNP-DPAFITC without RGDDP (Figure 3B). In addition, the average fluorescence intensity of AuNP-DPAFITC-treated cells was ~4-fold higher than that of cells treated by the RGDDP-deleted counterpart, and ~8-fold higher than that of cells treated by the RGDDP & PLL-deleted counterpart (Figure 3C). This result was also supported by flow cytometry, in which AuNP-DPAFITC was shown to be taken up by the cells up to 98.5%, whereas the cellular uptakes of RGDDP-deleted counterpart and RGDDP & PLL-deleted counterpart were only 60.1% and 10%, respectively (Figure 3D). Collectively, our data indicate that PLL-coated and RGDDP-modified gold nanoparticles can be taken up by cancer cells.
Cell uptake ability of AuNP-DPA in vitro. (A) CLSM images of HCT116 cells incubated with FITC-labeled DPA, AuNP-DPA W/O RGDDP&PLL, AuNP-DPA W/O RGDDP and AuNP-DPA at a concentration of 200 μg/mL for 6 h. All images were taken in the same condition about exciting light and detector gain (scale bar: 40 μm). (B-C) The average fluorescence intensity of FITC analyzed from green channel images using Image J (n = 3/sample). (D) Cellular uptake measured by flow cytometry analysis via the green fluorescent FITC. HCT116 cells were incubated with FITC-labeled DPA, AuNP-DPA W/O RGDDP&PLL, AuNP-DPA W/O RGDDP and AuNP-DPA at a concentration of 200 μg/mL for 4 h. (E) Typical images acquired of HCT116 cells in control condition (PBS control) and after pretreatment with RGD (50 μM), Amiloride (3 mM) or Cytochalasin D (2 μM) for 12 h before AuNP-DPA incubation. Statistically significant differences are indicated: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
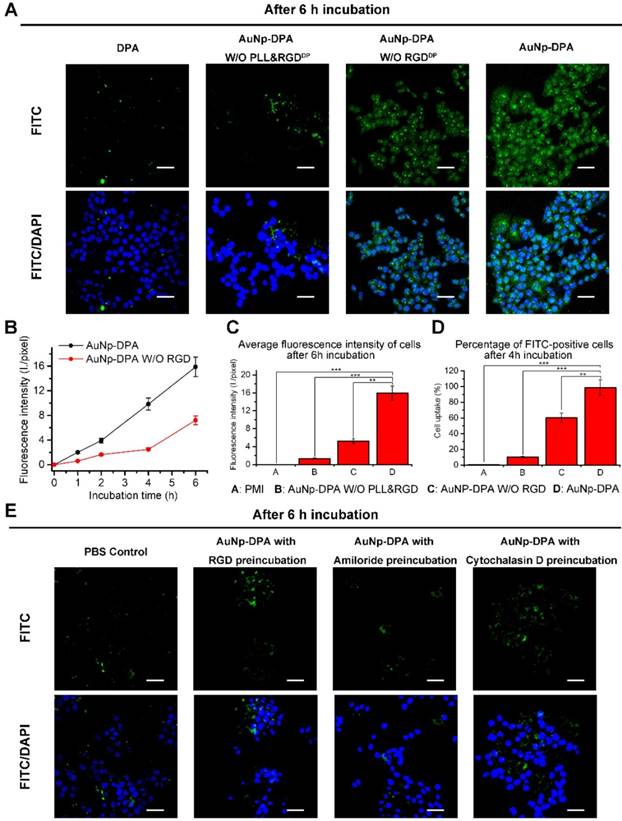
To explore the cellular uptake pathway of AuNP-DPA, free RGDDP and Amiloride (a specific inhibitor of micropinocytosis) were used to block internalization. By this way, we found that the cellular uptake of AuNP-DPA was completely inhibited after RGDDP or amiloride preincubation (Figure 3E), indicating that the cellular uptake was very likely contributed by micropinocytosis. Additionally, as micropinocytosis is actin-dependent, the cellular uptake of AuNP-DPA was also sufficiently inhibited by the actin inhibitor cytochalasin D (Cyto D) at a nontoxic concentration (Figure 3E). Collectively, these findings demonstrate that AuNP-DPA internalizes into cancer cells via integrin-dependent micropinocytosis.
Next, we investigated the intracellular distribution of AuNP-DPA following micropinocytosis. To this end, HCT116 cells were incubated with AuNP-DPA (20 μg/mL) for 6 h and then dyed with known markers for early endosomes (EEA1), late endosomes, and lysosomes (Lysotracker). As shown in Figure S10, the image of red-dye-labeled subcellular organelles and FITC-labeled nanoparticle presented that AuNP-DPA did not colocalize to late endosomes and lysosomes, but there was some overlap with early endosomes. These results demonstrate that AuNP-DPA can escape from early endosomes, which effectively avoids sequestration and degradation of nanoparticle or cargo in lysosomes.
AuNP-DPA releases DPA triggered by the reductive intracellular environment
The therapeutic efficacy of AuNP-DPA depends on efficient release of DPA to the cytosol. This process can be triggered by an intracellular reductant such as GSH to break the gold-thiolate bonds, as shown in Figure 4A. To test this, we incubated AuNP-DPA (~0.5 M, DPA) in PBS without GSH for two weeks and then, after centrifugation, the supernatant was detected by HPLC.
Redox-dependent release of peptides from AuNP-DPA. (A) Schematic depiction of the mechanism underlying the enhanced tumor targeting of AuNP-DPA by RGD conjugation and stimuli-responsive release of its cargo. (B-D) AuNP-DPA was incubated in PBS solution (pH 7.4) without or with GSH, and redox-dependent release of peptides was characterized by HPLC and ESI-MASS. (E) DPA-Cys release from AuNP-DPA in intracellular redox environment mimic solution (10 mM GSH in PBS buffer, pH 7.4). DPA-Cys release was quantified by HPLC.
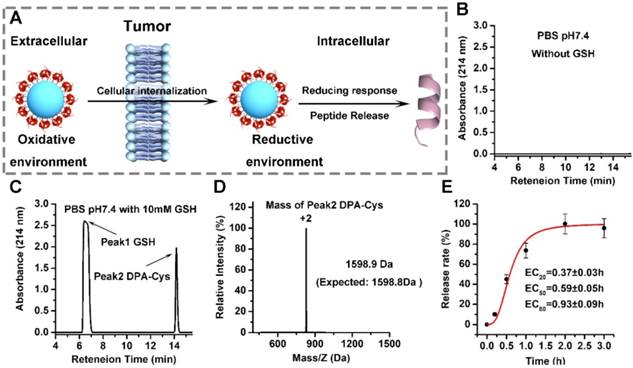
No DPA peptide was found in the supernatant, suggesting that the gold-thiolate bond is highly stable, thereby prolonging the in vivo circulation half-life of AuNP-DPA (Figure 4B). In sharp contrast, DPA-Cys peptides were detected and quantitatively recovered after 4 h incubation with intracellular redox environment mimic solution (10 mM GSH in PBS buffer, pH 7.4) (Figure 4C-D). Next, HPLC was used to monitor the release kinetics of DPA-Cys from AuNP-DPA. Incubation in redox environment mimic solution resulted in ∼50% cumulative release by ~0.6 h (Figure 1G) and ∼100% release by 2 h (Figure 4E), indicating that conjugated DPA-Cys may be efficiently released by intracellular reducing environments.
AuNP-DPA inhibits cancer cell growth in vitro in a p53-dependent manner
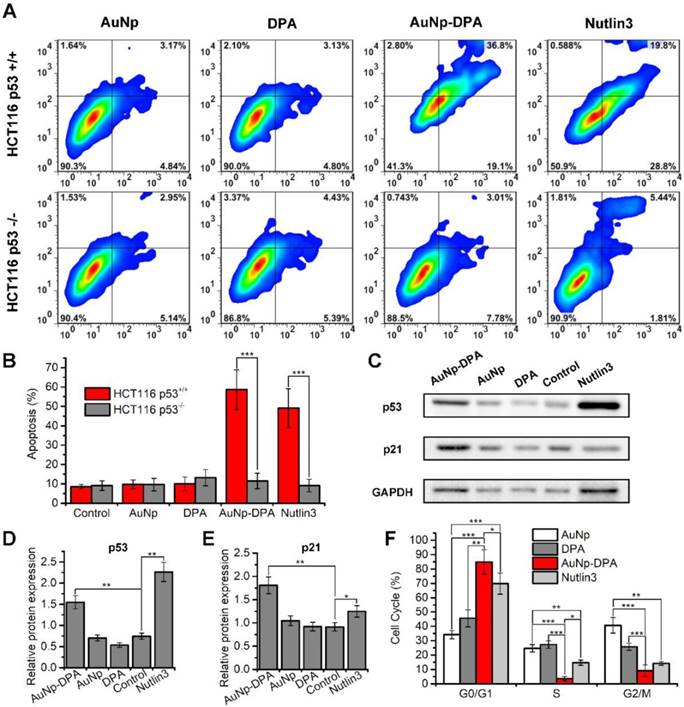
It is well known that approximately one-third of all human cancers suffer from p53 dysfunction [38, 49, 50]. Previous reports found that DPA reactivates p53, which, in turn, induces cell cycle arrest and apoptosis [20]. To verify this, we treated isogenic HCT116 colon cancer cells carrying intact p53 (HCT116 p53+/+) or lacking p53 (HCT116 p53-/-) with 2 μM AuNP-DPA, 2 μM free DPA, 4 μg/mL AuNP and 2 μM Nutlin3. Of these treatment groups, ~4 μg/mL AuNP, equivalent in weight to 2 μM AuNP-DPA, was used as a negative control, and 2 μM Nutlin3, a small -molecular activator of p53 [51], served as positive control. 72 h after treatment, AuNP-DPA and Nutlin3 significantly induced apoptosis of HCT116 p53+/+ cells, whereas they had almost no effect on the apoptosis of HCT116 p53-/- cells (Figure 5A-B and Figure S11). To confirm that the delivered PMI inhibits p53-MDM2/MDMX interaction, we explored the expression of p53 and p21 in HCT116 cells by western blot (Figure 5C). As shown in the quantitative data from three independent experiments in Figure 5D-E, after 24 h treatment, AuNP-DPA and Nutlin3 increased the expression of p53 and its downstream gene p21 [52, 53] significantly. All of these results suggest that DPA inhibited cancer cell proliferation in a p53-dependent manner. In addition, we also evaluated the effect of AuNP-DPA on cell cycle distributions of HCT116 p53+/+ and HCT116 p53-/- cells. The results showed that AuNP-DPA and Nutlin3 significantly induced cycle arrest of HCT116 p53+/+ cells at G0/G1 phase in comparation to PDA and AuNP (Figure 5F), whereas treatment with AuNP-DPA and Nutlin3 almost did not affect the cell cycle distributions of HCT116 p53-/- cells (Figure S12). These findings further support that AuNP-DPA inhibits cancer cell growth in a p53-dependent manner.
In vitro anticancer efficacy of AuNP-DPA. (A-B) HCT116 p53+/+ and HCT116 p53-/- cells were treated with 2 μM AuNP-DPA, 4 μg/mL AuNP, 2 μM DPA and 2 μM Nutlin-3 for 72 h, and cell apoptosis was evaluated by Annexin V-PI staining (abscissa: Annexin V; ordinate: PI) and flow cytometric analysis. The data are presented as mean ± s.d. of values from three independent experiments. (C) HCT116 p53+/+ cells were treated with 1 μM AuNP-DPA, 2 μg/mL AuNP, 1 μM DPA and 1 μM Nutlin-3 for 24 h, and western blot was performed to analyze the expressions of p53 and p21 proteins. β-actin was used as loading control. (D-E) Quantification of the western blot results. The data are presented as mean ± s.d. of values from three independent experiments. (F) HCT116 p53+/+ cells were treated with 1 μM AuNP-DPA, 2 μg/mL AuNP, 1 μM DPA and 1 μM Nutlin-3 for 24 h, and cell cycle distributions were analyzed by flow cytometry. The data are presented as mean ± s.d. of values from three independent experiments. Statistically significant differences are indicated: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
AuNP-DPA specifically enriches in the tumor sites
Based on our design combining the inherent EPR effect with integrin targeting, AuNP-DPA can specifically accumulate in the tumor site. The EPR effect is a phenomenon observed in solid tumors where excessive angiogenic signals result in the formation of defective 'leaky' tumor vasculature, through which nanoparticles (NP) <150 nm in size can extravasate to the tumor microenvironment [54]. Additionally, the increased tumor volume also leads to ineffective lymphatic drainage, further promoting NP retention [55]. In addition to the EPR effect, actively targeted delivery of antitumor drugs/molecules to cancer cells is an optimized and effective strategy to improve the efficacy of anticancer therapies [56]. Nanoparticle selectivity can be improved by specifically binding to proteins specifically upregulated in cancer cells such as integrins [57, 58]. In this context, RGDDP, consisting of two integrin-binding motifs RGDK [43] and RGDSP [44], was linked to the nanoparticles to further improve tumor targeting.
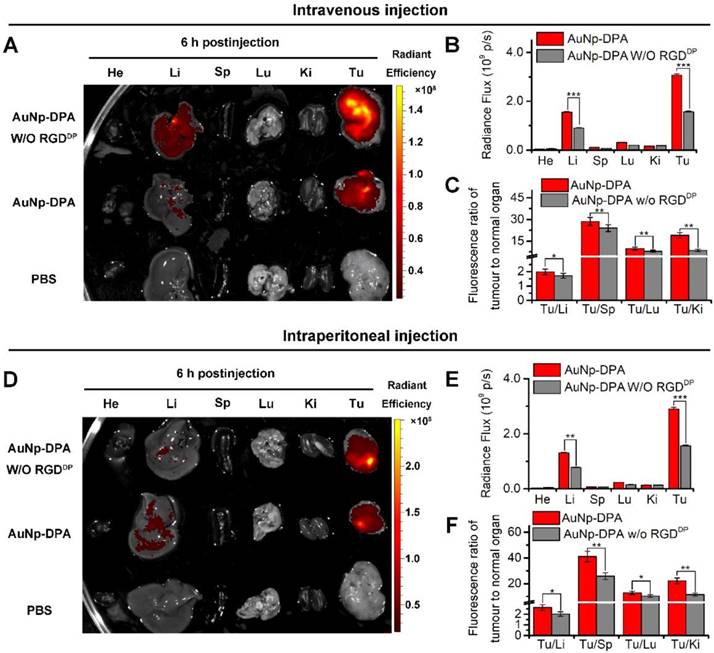
Long blood circulation time is an important requirement for in vivo target-specific drug delivery [59]. We explored the circulation time of AuNP-DPA with an injection dose of 2 mg/kg in mice, and the nanoparticle presented a long half-life of 7.5 h with a first order elimination rate (Figure S13) [60]. To assess tumor targeting of AuNP-DPA, the bio-distributions of AuNP-DPA and its counterpart without RGDDP were examined in normal organs and tumor sites. Toward this end, a red fluorescent dye, sulforhodamine 101 acid chloride, was first conjugated to the amino terminal of DPA. The semi-quantitative results showed that AuNP-DPA exhibited higher accumulation in the tumor sites than its counterpart without RGDDP at 6 h after intravenous injection (iv) (Figure 6A-C) or intraperitoneal injection (ip) (Figure 6D-E). Through further analysis of the fluorescence distribution, either via ip or iv, we found AuNP-DPA expectedly showed 2-fold greater tumor fluorescence than its RGDDP-deleted counterpart (Figure 6B, E), suggesting that RGDDP further increased enrichment of the nanoparticles in the tumor sites. In addition, all tumor-to-organ ratios for AuNP-DPA were significantly higher than those for its RGDDP-deleted counterpart (Figure 6C, E). Taken together, these findings indicate that integrin targeting can improve the tumor selectivity of AuNP-DPA.
Targeted tumor enrichment ability of AuNP-DPA ex vivo. (A) Representative ex vivo fluorescence image of major organs and tumor at 6 h after intravenous injection of AuNP-DPA and its RGDDP-deleted counterpart (AuNP-DPAW/O RGDDP) in mice bearing HCT116 xenograft tumors (dose: 100 μg per mouse). He: heart; Li: liver; Sp: spleen; Lu: lung; Ki: kidneys; Tu: tumor; Br: Brain. (B) Ex vivo semi-quantitative analysis of biodistribution. Fluorescence intensity in each organ was determined using Living Image 3.0. software from IVIS fluorescence data expressed as radiant efficiency [(photons s-1 cm-2 sr-1)/(μW cm-2)]. (C) Tumor-to-background (normal organ or tissue) ratios for AuNP-DPA and AuNP-DPAW/O RGDDP at 6 h post-injection (iv) from the data in (A) (n = 3, mean ± s.d.). (D-F) Ex vivo fluorescence images and biodistribution analysis of AuNP-DPA and AuNP-DPAW/O RGDDP at 6 h following intraperitoneal injection.
Potent antitumor activity of AuNP-DPA in HCT116 p53+/+ xenograft model
To explore the anti-cancer activity of AuNP-DPA in vivo, 25 mice xenografting HCT116 p53+/+ tumors (50-100 mm3) were randomly and equally divided into five groups: AuNP-DPA, AuNP (the DPA-deleted counterpart of AuNP-DPA), free DPA, doxorubicin (positive control) and PBS (control). Given that there exists no difference in the bio-distributions of AuNP-DPA administered iv or ip (Figure S14), user-friendly ip administration was adopted and the mice were treated every other day at a dosage of 2 mg/kg for 13 days. The study design is illustrated in Figure 7A. Of note, there are two irresistible reasons for us to choose doxorubicin (DOX) as a positive control in this experiment: (1) DOX is a first-line chemotherapy and has been used for a long time, whereas Nutlin3 is only at the clinic trial stage; and (2) DOX treatment was more effective than Nutlin3 at the same dose toward this model (Figure S15). After 13 days of treatment, free DPA peptide and AuNP displayed hardly any tumor-inhibitory effect (Figure 7B and Figure S16). In a sharp contrast, AuNP-DPA inhibited tumor growth by 88% on day 13 compared to the control, even better than DOX (inhibition ratio of 64.3% in comparison with the control), also shown in Figure 7B and Figure S16.
In vivo antitumor activity of AuNP-DPA. (A) Schematic diagram of therapeutic treatments. Athymic nude mice (BALB/c) bearing HCT116 p53+/+ xenograft tumors were established and randomly divided into five groups (n = 5/group). Mice were then treated by intraperitoneal injection every other day for 6 times with PBS, 1.5 mg/kg doxorubicin, 2 mg/kg DPA, 2 mg/kg AuNP-DPA or 2 mg/kg AuNP. (B) Tumor growth curves during the 13-day treatment. (mean ± s.e., n =5). (C) Photos of tumors collected after the 13-day treatment. (D) Mice tumor weights after the 13-day treatment. (E&F) Representative tumor sections after the 13-day treatment staining by H&E (E) and TUNEL (F) (scale bar: 50 μm). All statistical analysis in this figure was performed using a t-test. (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
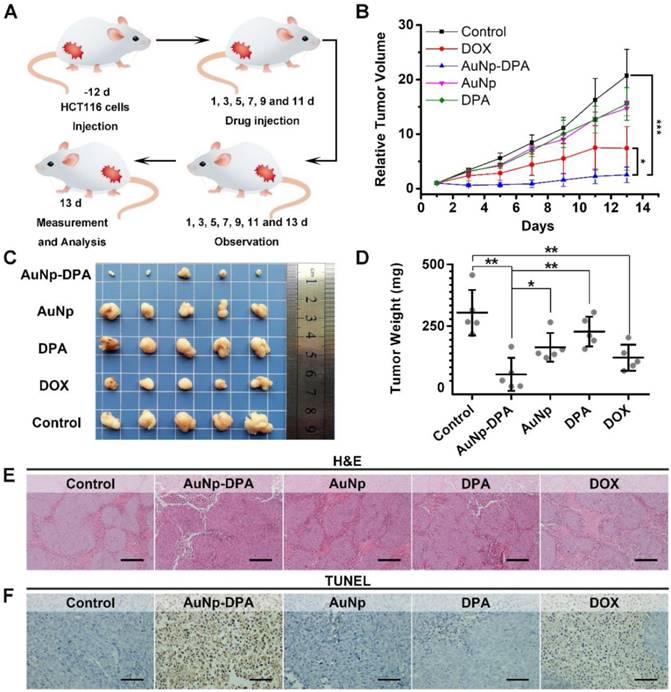
At the end of the experiments, the xenograft tumors were isolated and weighed. As expected, the tumor in the AuNP-DPA-treated mice were much smaller than those in the AuNP-, DPA- or PBS- treated mice (Figure 7C-D). Notably, the average tumor weight of AuNP-treated mice was ~6-fold larger than that of AuNP-DPA-treated mice, suggesting that the main therapeutic effect is due to the anticancer activity of DPA rather than nonspecific toxicity from the nanoparticles. Meanwhile, the geometric mean tumor weight of the AuNP-DPA-treated mice (26.4 mg) was just 12% that of the free DPA-treated mice (221.2 mg, P < 0.01), indicating that this nanosystem awakens the function of DPA.
To further verify the antitumor activity of AuNP-DPA at a pathological level, we first examined the above xenograft tumor tissues with H&E staining. As shown in Figure 7E, morphological integrity was observed in the PBS-, DPA-, and AuNP-treated groups. Conversely, we observed apoptotic or necrotic tumor tissues in the DOX- and AuNP-DPA-treated mice, particularly in the latter. Next, TUNEL assay was performed for the evaluation of the extent of apoptosis. Expectably, a crowd of TUNEL-positive (apoptotic) cells can be found in the DOX- and AuNP-DPA- tumors, whereas scarcely any apoptosis can be observed in the DPA- or AuNP- treated samples (Figure 7F). Collectively, our results provide an abundance of evidence that AuNP-DPA possessed potent in vivo anticancer efficacy even better than the first-line chemotherapy drug DOX.
AuNP-DPA inhibits tumor growth in vivo through reactivating the p53 signaling pathway
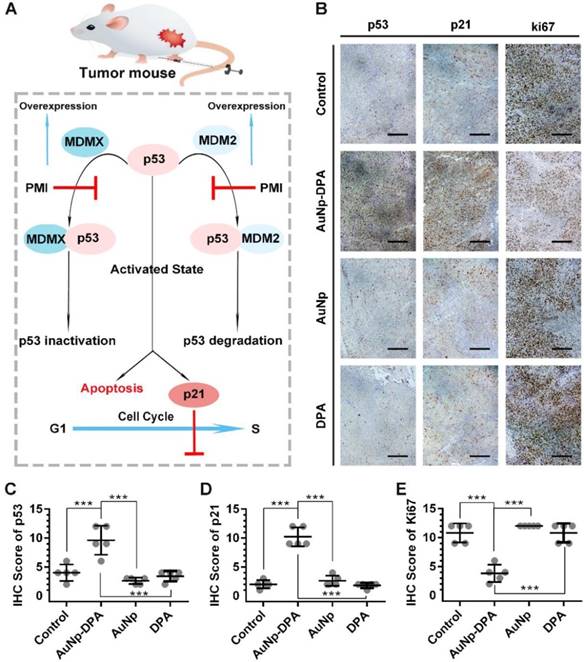
The tumor suppressor protein p53 plays a critical role in maintaining genomic stability as well as inhibiting malignant proliferation, and impairment of p53 signaling is a hallmark of cancers [42, 61]. There is evidence demonstrating MDM2 and/or MDMX antagonism by small molecules or peptides is a clinical strategy for cancer therapy [42]. The DPA used in the present study is a D-enantiomeric peptide with high affinity for both MDM2 and MDMX [20]. Thus, AuNP-DPA should exert its antitumor effect through reactivating the function of p53 via releasing p53 from the MDM2 (MDMX)/p53 complex (Figure 8A). To validate this, immunohistochemistry staining of p53, p21 and Ki67 were observed in all tumors with serial sections. In line with the in vitro WB results, p53 and p21 were apparently up-regulated in the AuNP-DPA-treated tumors compared to PBS-treated tumors, whereas DPA or AuNP treatment almost did not affect the levels of p53 and p21 relative to those of the control (Figure 8B-C). This was consistent with the TUNEL results shown in Figure 7F. In addition, all of the PBS-, DPA- or AuNP- treated tumors showed extensive positive staining of the proliferation marker- Ki67, whereas the number of Ki67-positive cells decreased significantly in the AuNP-DPA-treated tumor (Figure 8E). Collectively, our data indicate that AuNP-DPA inhibits tumor growth in vivo via reactivating p53 signaling.
Tumor-inhibitory effect of AuNP-DPA by reactivating p53 signaling. (A) Schematic diagram for AuNP-DPA inhibition of tumor growth by p53 restoration. (B-E) Representative IHC images and IHC score for p53, p21 and Ki67 (scale bar: 50 μm) in tumor sections. All statistical analysis in this figure was performed using a t-test (***, p < 0.001).
Safety evaluation of AuNP-DPA
As mentioned above, MDM2 and/or MDMX antagonism could be an effective strategy for cancer therapy through reactivating p53 pathway [49, 62]. Thus, in recent years, a number of selective small molecule MDM2/MDMX inhibitors have been widely developed including a class of imidazoline compounds termed nutlins, several of which have entered in clinical trials [4, 40]. Nutlins have been demonstrated to effectively kill B-cell chronic lymphocytic leukemia (B-CLL) cells; however, they also induced some toxicities to normal cells: such as B lymphocytes and hematopoietic stem cell [63]. What's worse, in a clinical trial of nutlin derivative, termed RG7112, almost all patients endured at least one drug-related side effects, including but not limited to thrombocytopenia, leukopenia and neutropenia. [64]. In addition, it has been reported that nutlins can directly cause DNA damage of the normal cells and indirectly promote stem cell differentiation [40-42]. Thus, there is a compelling need to develop new therapeutics with lower nonspecific toxicity.
To overcome the systemic toxicity from off-targeting, a feasible route is to load the anti-cancer drug in a tumor-specific vehicle [65, 66]. To minimize side effects, a two-stage targeting strategy was designed in AuNP-DPA (Figure 9A). Benefited from the EPR effect (first targeting) and RGDDP conjugation (second targeting), AuNp-DPA can accumulate at the tumor sites specifically, resulting in minimized off-target effects and subsequent side effects. Moreover, DPA has strong specificity towards MDM2, which is specifically overexpressed in cancer cells. AuNp-DPA, as expected, had no effect on normal cells and a p53-null cancer cell line SW480, whereas DOX showed potent cytotoxicity. (Figure S17).
Safety evaluation of AuNP-DPA in vivo. (A) Schematic diagram for the biosafety of AuNP-DPA attributed to the targeted accumulation of AuNP-DPA in the tumor region rather than in other organs. (B) Changes in body weight of mice with xenograft tumors upon various treatments. (C-G) The count of RBC, hemoglobin, WBC, thrombocyte and eosinophils. p values were calculated by t-test (unmarked p > 0.05).
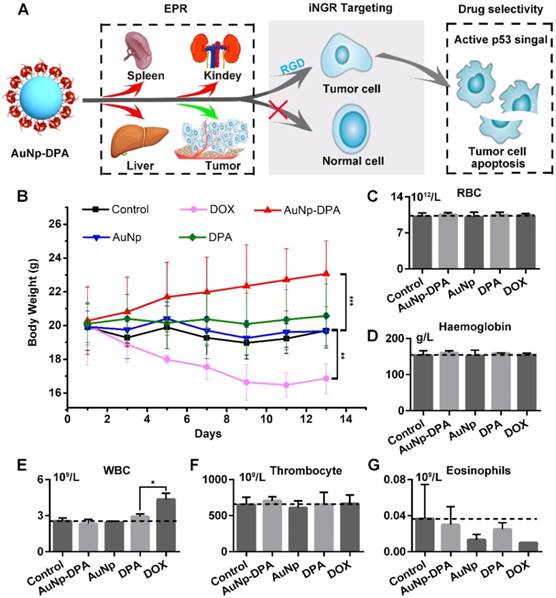
To verify the in vivo biosafety of AuNP-DPA, comprehensive medical examinations were performed during and after anticancer treatment. After 13 days of administration, the body weights of DOX-treated mice significantly decreased, whereas no significant weight loss was found in other groups (Figure 9B). This was supported by several previous studies that DOX always suffered from some undesirable off-target effects [67, 68]. Notably, except for DOX-treated mice, all other mice almost did not (each group respectively compared to control group by t-test, p>0.05) show significant haematological toxicities (Figure 9C-G), suggesting a relatively low toxicity of AuNP-DPA.
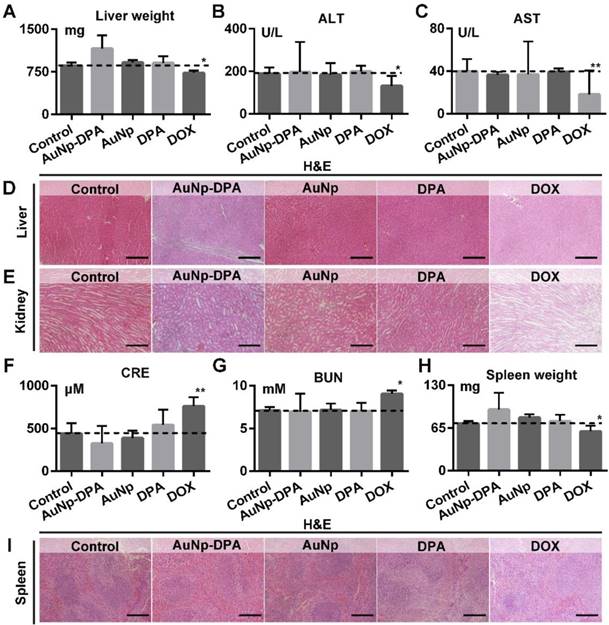
Given that liver and kidney are almost entirely responsible for drug biotransformation and elimination, we comprehensive assessed the health of these two organs. As shown in Figure 10A, AuNp or DPA treatment almost did not affect the liver weight of mice compared to the control (t-test, p>0.05), whereas DOX-treated mice presented a statistically significant loss of liver weight relative to control mice (t-test, p<0.05). In addition, the levels of ALT and AST maintain normality in AuNP-DPA-treated mice (t-test, p>0.05) but increased significantly in DOX-treated mice (t-test, p<0.05), suggesting that DOX leaded to the abnormal hepatic function (Figure 10B-C). This result proved again by H&E staining: DOX-treated mice presented medium, spotty necrosis of the liver (Figure 10D). Next, kidney condition was evaluated by H&E staining (Figure 10E), CRE (Figure 10F) and BUN (Figure 10G). Similarly, mice treated by DOX showed increased levels of CRE and BUN and symptoms of glomerular lesions compared to control mice, whereas the other treatments had no harmful effects on the kidneys (Figure 10E-G). Moreover, mice treated by DOX showed spleen damage compared to control mice, while the spleen in other groups stay healthy (Figure 10H-I). It was observed that all treatments in this work, including DOX, didn't presented any other toxicities, for instance, cardiac trauma (Figure S18) or allergic lung resistance (Figure S19). Overall, AuNP-DPA is safe enough for future clinical use.
The impact of AuNP-DPA on the function of liver, kidney and spleen of mice with different treatments. (A) Mice liver weight after the 13-day treatment. (B-C) The activities of two liver enzymes ALT and AST in mice blood after the 13-day treatment. (D-E) Representative H&E staining of liver and kidney sections from mice after the 13-day treatment (scale bar: 50 µm). (F&G) Measurement of renal indicators (CRE, (F); BUN, (G)) in mice blood after the 13-day treatment. (H) Mice spleen weight after the 13-day treatment. (I) Representative H&E staining of spleen sections (scale bar: 50 µm). p values were calculated by t-test (*, p < 0.05 and **, p < 0.01 in comparison with the control).
Conclusions
Redox-responsive delivery is a robust platform in nanomedicine for the development of peptide-derived anticancer therapeutics. The present study developed a gold-based nanocarrier decorated with a D-enantiomeric peptide coupled with PLL and receptor-targeted peptide (termed AuNP-DPA) for targeted tumor therapy in vivo. Profited by the EPR effect and RGD targeting, AuNP-DPA can successfully deliver DPA into cancer cells and specifically accumulate at tumor sites. Through a series of in vitro and in vivo experiments, we demonstrated that AuNP-DPA has excellent biosafety and potent antitumor activity. In summary, the ultra-small gold nanoparticles functionalized with therapeutic and targeting peptides may be a promising anticancer nanosystem to overcome physiological and cellular barriers for targeted delivery of therapeutic D-peptides and further awaken their antitumor efficacy.
Methods
Materials
HAuCl4·xH2O was purchased from Aladdin Chemicals. All synthetic peptides were obtained from CS Bio in Shanghai, China. We purchased chemicals from Sigma-Aldrich unless otherwise specified. Acetonitrile and water (HPLC grade) were purchased from Fisher Scientific Ltd. All products were used as received without further purification.
Fabrication of Au core, Au-DPA, Au-DPA-PLL and AuNP-DPA
An aqueous solution of tetrachloroauric acid (HAuCl4·xH2O, 1 mL, 10 mM) was mixed with 9 mL HEPES buffer (pH 7.4, 50 mM). After 10 min magnetic stirring, the solution color changed from golden yellow to wine red, which is the Au core. After this, 2 mg DPS-Cys was added into the Au core solution with stirring. Then, the color changed to bluish violet, indicating that DPA-Cys was successfully conjugated to the Au-core to form Au-DPA. Next, 1 mg PLL (MW 1500-2000) was added into the mixture, followed by a 10-min ultrasonic dissolution. The Au-DPA-PLL and its RGDDP-deleted counterpart (termed AuNP-DPA W/O-RGDDP) were collected by 14000 ×g centrifugation and lyophilization. To prepare AuNp-DPA, carboxylates (-COOH) of RGDDP-Cys were firstly reacted to a bifunctional linker N-succinimidyl maleimidoacetate. In detail, 0.5 mg RGDDP-Cys was dissolved in 1 mL DMF containing 0.5 mM N-succinimidyl maleimidoacetate. After 20-min reaction at room temperature, 1 mL DMF mixture was added dropwise into the 10 mL Au-DPA-PLL solution (1 mg/mL in PBS). After 30 min of stirring, the excess reactants were removed by dialysis (cutoff, 10 kDa) and washed three times with PBS buffer. AuNP-DPA were then freeze-dried for subsequent experiments.
Physicochemical properties of AuNP-DPA
TEM images were taken by an HT7700 operated at 100 kV acceleration voltages, to explore the morphology and lattice structure of the nanomaterials. DLS performed by Malvern Zetasizer Nano ZS system, was used to obtain the hydrodynamic size distribution of nanoparticle solutions (1 mg/mL in PBS, 1 mL). For Zeta potential measurement, the nanoparticles were solved in PBS at the concentration of 1 mg/mL, and measured at 37 °C. The FTIR was obtained by Nicolet 6700 via measuring the KBr-included freeze-dried sample powder. The UV-vis absorption spectroscopy was measured by Shimadzu 3000 spectrophotometer, at a concentration of 0.2mg/ml.
Quantification of drug loading and GSH-responsive drug release
To quantify drug loading, we firstly solve 6 M GuHCL and 1 M DTT into PBS buffer (pH 7.4). AuNP-DPA were dissolved in the prepared solvent, and the amount of released peptide was quantified by HPLC. To test GSH-responsive drug release, AuNP-DPA were dissolved in PBS buffer (pH 7.4) containing 10 mM GSH, and the nanoparticles were then removed by 14000 ×g centrifugation. Following this, the supernatants were quantified by HPLC and authenticated by ESI-MASS.
Cellular uptake
Cellular uptake of AuNP-DPA was detected by CLSM (FV1200, Olympus) and flow cytometry (BD Biosciences, NJ). FITC was firstly labeled to the N-terminal of DPA. Similarly, AuNp-DPAFITC and AuNp-DPAFITC W/O-RGDDP were prepared as above. We prepared solutions of DPAFITC, AuNp-DPAFITC and AuNp-DPAFITC W/O-RGDDP in culture medium at a concentration of 200 μg/mL. HCT116 cells were first cultured for 24 h. The medium was then replaced with the medium containing AuNp-DPAFITC or AuNp-DPAFITC W/O-RGDDP at pH 7.4, following 6h incubation at 37 °C. After washing the cells with PBS twice, cell imaging or flow cytometry analysis were carried out. DAPI (Molecular probes) was used to mark cell nucleus. Paraformaldehyde was used to fix the cells before observation. With regard to CLSM, the excitation wavelengths were 405 nm (3.15 mW) and 543 nm (0.7 mW).
Ethics statement
All animal procedures were approved by the Ethics Committee of Xi'an Jiaotong University, and performed following Institution Guidelines of Laboratory Animal Center of Xi'an Jiaotong University.
In vivo bio-distribution
Sulforhodamine 101 acid chloride-labelled DPA-Cys was firstly prepared to synthesize fluorescent AuNp-DPA or AuNp-DPA W/O-RGDDP. In detail, Texas Red™-X, Succinimidyl Ester purchased from Thermo Fisher was incubated with the N-terminal-free DPA-Cys in PBS buffer for 2 h and then purified by HPLC. From this, fluorescent DPA-Cys was obtained, and was used to synthesize fluorescent AuNp-DPA or AuNp-DPA W/O-RGDDP as in the above protocol.
Tumor cells (HCT116 p53+/+, 4×106 cells/site) were injected subcutaneously into BALB/c nude mice with four to five weeks old. After three to four weeks, each tumor-bearing mouse was injected with 200 μL fluorescent AuNp-DPA or AuNp-DPA W/O-RGDDP (2 mg/kg). Mice were humanely sacrificed at each indicated time, and the organ and tumor were immediately collected. IVIS Spectrum System was then used to semi-quantitatively explore the biodistribution of the nanoparticles (λex 590 nm; λem 610 nm).
In vivo circulation time
Pharmacokinetic studies were performed using the athymic BALB/c nude mice as In vivo bio-distribution. 200 μL Sulforhodamine 101 acid chloride-labelled AuNp-DPA (2 mg/kg) were administered by intravenous injection via tail vein. Approximately 100μL blood samples were collected from the retro-orbital sinus of anesthetized mice at indicated time points (n=3 at each time points) and reserved in heparin anticoagulation tube. The blood samples were determined using a microplate reader (λex 586 nm; λem 605 nm). The blood samples from PBS-administered mice as negative control, and these blood samples also were used to dilute the high-concentration AuNp-DPA as standard to quantify the blood samples from administered mice.
In vivo treatment response
HCT116 subcutaneous tumor models were established as described above. When all tumors grew up to 50-100 mm3, the mice were randomly divided into different groups (five mice per group). AuNP-DPA, AuNP and DPA wre administered at dose of 2 mg/kg on days 1, 3, 5, 7, 9, and 11. PBS solution was used as a negative control. Owing to the fact that (1) the amount of DPA in 2 mg/kg AuNP-DPA is approximately equivalent in weight to 1.5 mg/kg DPA, and (2) the dose over.
1.5 mg/kg would result in the drug-related death of BALB/c nude mice, the administration dose of DOX was 1.5 mg/kg every other day. Tumor volume was calculated as follow: volume = length × width2 / 2. Before H&E or immunohistochemical staining, the tumor and organ tissues were fixed and sliced into 5.0 μm sections. In addition, routine blood examination and Clinical biochemical indices were detected by the Clinical Laboratory of the First Affiliated Hospital of Xi'an Jiaotong University according to standard clinical laboratory procedures.
In vivo toxicity
To evaluate potential toxicities of AuNP-DPA, we monitored the body weight of all mice over the course of treatment and measured hematological indices as well as organ function indices after 13 days of treatment. Control mice were only implanted with xenograft tumor and did not receive any treatment. Forty-eight hours after the last dose, mice were anesthetized. We collected the blood to evaluate CBC including WBC, RBC, haemoglobin and platelet counts. Additionally, blood serum was used for ALT, AST, BUN and CRE assessment using ELISA kits. Animals were then euthanized according to the corresponding procedure. Prepared tissue slices were stained with H&E.
Statistical analysis
Statistical analyses were performed using two-sided Student's t-test. P < 0.05 was considered significant. Data are expressed as mean ± s.d. or s.e.
Abbreviations
PLL: polylysine; DPA: D-peptide p53 activator; PPIs: protein-protein interactions; CPP: cell-penetrating peptides; NHS: N-hydroxysuccinimide; EDC: (1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimide); GuHCL: guanidine hydrochloride; DTT: dithiothreitol; HPLC: high performance liquid chromatography; GSH: glutathione; TEM: transmission electron microscope; LC-MS: liquid chromatography-mass spectrometry; EPR: enhanced permeability and retention; CBC: complete blood count; WBC: white blood cell; RBC: red blood cell count; ALT: alanine aminotransferase, AST: aspartate transaminase; BUN: blood urea nitrogen; CRE: creatinine; LSCM: laser scanning confocal microscopy; GSH: glutathione; Cyto D: cytochalasin D; FBS: fetal bovine serum; DLS: Dynamic light scattering; FTIR: Fourier-transform infrared spectroscopy.
Supplementary Material
Supplementary methods about necessary synthetic procedure and cell experimental methods, figures S1-S19 and tables S1 and S2. Supplementary figures and tables.
Acknowledgements
This work was supported by the Clinical Research Award of the First Affiliated Hospital of Xi'an Jiaotong University (No. XJTU1AF-CRF-2017-003 to P. H. and W. H.). The authors gratefully acknowledge financial support from the China Scholarship Council (to W. H. and J. Y.) and thank Prof. Bert Vogelstein (Ludwig Center at John Hopkins, USA) for kindly providing the human colon cancer cell HCT116 p53-/- cell line with both p53 alleles deleted.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001-9
2. Yin H, Hamilton AD. Strategies for targeting protein-protein interactions with synthetic agents. Angew Chem Int Ed Engl. 2005;44:4130-63
3. Mullard A. Protein-protein interaction inhibitors get into the groove. Nat Rev Drug Discov. 2012;11:173-5
4. Zhao Y, Aguilar A, Bernard D, Wang S. Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment: miniperspective. J Med Chem. 2014;58:1038-52
5. Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. 2015;20:122-8
6. Yan J, He W, Yan S, Niu F, Liu T, Ma B. et al. Self-assembled peptide-lanthanide nanoclusters for safe tumor therapy: overcoming and utilizing biological barriers to peptide drug delivery. ACS Nano. 2018;12:2017-26
7. Corbi-Verge C, Garton M, Nim S, Kim PM. Strategies to develop inhibitors of motif-mediated protein-protein interactions as drug leads. Annu Rev Pharmacol Toxicol. 2017;57:39-60
8. Konno R. D-amino acids: A New Frontier in Amino Acids and Protein Research: Practical Methods and Protocols. New York, USA: Nova Science Publishers. 2007
9. Funke SA, Willbold D. Mirror image phage display—a method to generate D-peptide ligands for use in diagnostic or therapeutical applications. Mol Biosyst. 2009;5:783-6
10. Dintzis HM, Symer DE, Dintzis RZ, Zawadzke LE, Berg JM. A comparison of the immunogenicity of a pair of enantiomeric proteins. Proteins. 1993;16:306-8
11. Liu M, Pazgier M, Li C, Yuan W, Li C, Lu W. A left-handed solution to peptide inhibition of the p53-MDM2 interaction. Angew Chem Int Ed Engl. 2010;49:3649-52
12. Eckert DM, Malashkevich VN, Hong LH, Carr PA, Kim PS. Inhibiting HIV-1 entry: discovery of D-peptide inhibitors that target the gp41 coiled-coil pocket. Cell. 1999;99:103-15
13. Schumacher TN, Mayr LM, Minor DL, Milhollen MA, Burgess MW, Kim PS. Identification of D-peptide ligands through mirror-image phage display. Science. 1996;271:1854-7
14. Chang HN, Liu BY, Qi YK, Zhou Y, Chen YP, Pan KM. et al. Blocking of the PD-1/PD-L1 interaction by a D-peptide antagonist for cancer immunotherapy. Angew Chem Int Ed Engl. 2015;54:11760-4
15. Kuang Y, Shi J, Li J, Yuan D, Alberti KA, Xu Q. et al. Pericellular hydrogel/nanonets inhibit cancer cells. Angew Chem Int Ed Engl. 2014;53:8104-7
16. Hayashi T, Sun Y, Tamura T, Kuwata K, Song Z, Takaoka Y. et al. Semisynthetic lectin-4-dimethylaminopyridine conjugates for labeling and profiling glycoproteins on live cell surfaces. J Am Chem Soc. 2013;135:12252-8
17. Zhou J, Du X, Li J, Yamagata N, Xu B. Taurine boosts cellular uptake of small D-peptides for enzyme-instructed intracellular molecular self-assembly. J Am Chem Soc. 2015;137:10040-3
18. Wender PA, Galliher WC, Goun EA, Jones LR, Pillow TH. The design of guanidinium-rich transporters and their internalization mechanisms. Adv Drug Deliv Rev. 2008;60:452-72
19. Rothbard JB, Jessop TC, Lewis RS, Murray BA, Wender PA. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J Am Chem Soc. 2004;126:9506-7
20. Liu M, Li C, Pazgier M, Li C, Mao Y, Lv Y. et al. D-peptide inhibitors of the p53-MDM2 interaction for targeted molecular therapy of malignant neoplasms. Proc Natl Acad Sci U S A. 2010;107:14321-6
21. Sakai N, Matile S. Anion-mediated transfer of polyarginine across liquid and bilayer membranes. J Am Chem Soc. 2003;125:14348-56
22. Allen TM, Cullis PR. Liposomal drug delivery systems: from concept to clinical applications. Adv Drug Deliv Rev. 2013;65:36-48
23. Ulbrich K, Hola K, Subr V, Bakandritsos A, Tucek J, Zboril R. Targeted drug delivery with polymers and magnetic nanoparticles: covalent and noncovalent approaches, release control, and clinical studies. Chem Rev. 2016;116:5338-431
24. Paciotti GF, Kingston DG, Tamarkin L. Colloidal gold nanoparticles: a novel nanoparticle platform for developing multifunctional tumor-targeted drug delivery vectors. Drug Dev Res. 2006;67:47-54
25. Ghosh P, Han G, De M, Kim CK, Rotello VM. Gold nanoparticles in delivery applications. Adv Drug Deliv Rev. 2008;60:1307-15
26. Kumar A, Ma H, Zhang X, Huang K, Jin S, Liu J. et al. Gold nanoparticles functionalized with therapeutic and targeted peptides for cancer treatment. Biomaterials. 2012;33:1180-9
27. He W, Huang H, Yan J, Zhu J. Photocatalytic and antibacterial properties of Au-TiO2 nanocomposite on monolayer graphene: From experiment to theory. J Appl Phys. 2013;114:204701
28. NCT00356980, NCT00848042 and NCT02837094. Accessed October 2018. http://clinicaltrials.gov
29. Libutti S K, Paciotti G F, Myer L. et al. Results of a completed phase I clinical trial of CYT-6091: A pegylated colloidal gold-TNF nanomedicine. J Clin Oncol. 2009;27:3586
30. Pissuwan D, Niidome T, Cortie MB. The forthcoming applications of gold nanoparticles in drug and gene delivery systems. J Control Release. 2011;149:65-71
31. Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65:71-9
32. Torchilin V. Tumor delivery of macromolecular drugs based on the EPR effect. Adv Drug Deliv Rev. 2011;63:131-5
33. Yan J, He W, Li N, Yu M, Du Y, Lei B. et al. Simultaneously targeted imaging cytoplasm and nucleus in living cell by biomolecules capped ultra-small GdOF nanocrystals. Biomaterials. 2015;59:21-9
34. Zhang J, Yan J, Yang Q, Yan Y, Li S, Wang L. et al. Arginine-modified dual emission photoluminescent nanocrystals for bioimaging at subcellular resolution. J Biomater Appl. 2017;32:533-42
35. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307-10
36. Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991;351:453
37. Hu K, Yin F, Yu M, Sun C, Li J, Liang Y. et al. In-tether chiral center induced helical peptide modulators target p53-MDM2/MDMX and inhibit tumor growth in stem-like cancer cell. Theranostics. 2017;7:4566-76
38. Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299-303
39. Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25-7
40. Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E. Clinical overview of MDM2/X-targeted therapies. Front Oncol. 2016;6:7
41. Prives C. Signaling to p53: breaking the MDM2-p53 circuit. Cell. 1998;95:5-8
42. Khoo KH, Verma CS, Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nat Rev Drug Discov. 2014;13:217-36
43. Barui S, Saha S, Mondal G, Haseena S, Chaudhuri A. Simultaneous delivery of doxorubicin and curcumin encapsulated in liposomes of pegylated RGDK-lipopeptide to tumor vasculature. Biomaterials. 2014;35:1643-56
44. Schuler M, Owen GR, Hamilton DW, de Wild M, Textor M, Brunette DM. et al. Biomimetic modification of titanium dental implant model surfaces using the RGDSP-peptide sequence: a cell morphology study. Biomaterials. 2006;27:4003-15
45. Park S-H, Zheng JH, Nguyen VH, Jiang S-N, Kim D-Y, Szardenings M. et al. RGD Peptide cell-surface display enhances the targeting and therapeutic efficacy of attenuated salmonella-mediated cancer therapy. Theranostics. 2016;6:1672
46. Guo Y, Ma Y, Xu L, Li J, Yang W. Conformational change induced reversible assembly/disassembly of poly-L-lysine-functionalized gold nanoparticles. J Phys Chem C. 2007;111:9172-6
47. Stobiecka M, Hepel M. Double-shell gold nanoparticle-based DNA-carriers with poly-L-lysine binding surface. Biomaterials. 2011;32:3312-21
48. de la Fuente JM, Berry CC. Tat peptide as an efficient molecule to translocate gold nanoparticles into the cell nucleus. Bioconjug Chem. 2005;16:1176-80
49. Wade M, Li Y-C, Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer. 2013;13:83-96
50. Zhang Q, Zeng SX, Lu H. Targeting p53-MDM2-MDMX loop for cancer therapy. Subcell Biochem. 2014;85:281-319
51. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z. et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844-8
52. Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. nature. 1993;366:701-4
53. Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown J. et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497-501
54. Acharya S, Sahoo SK. PLGA nanoparticles containing various anticancer agents and tumour delivery by EPR effect. Adv Drug Deliv Rev. 2011;63:170-83
55. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653-64
56. Kamaly N, Xiao Z, Valencia PM, Radovic-Moreno AF, Farokhzad OC. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev. 2012;41:2971-3010
57. Graf N, Bielenberg DR, Kolishetti N, Muus C, Banyard J, Farokhzad OC. et al. αVβ3 integrin-targeted PLGA-PEG nanoparticles for enhanced anti-tumor efficacy of a Pt (IV) prodrug. ACS Nano. 2012;6:4530-9
58. Tucker GC. Integrins: molecular targets in cancer therapy. Curr Oncol Rep. 2006;8:96-103
59. Park JH, von Maltzahn G, Zhang L, Schwartz MP, Ruoslahti E, Bhatia SN. et al. Magnetic iron oxide nanoworms for tumor targeting and imaging. Adv Mater. 2008;20:1630-5
60. Wunderbaldinger P, Josephson L, Weissleder R. Tat peptide directs enhanced clearance and hepatic permeability of magnetic nanoparticles. Bioconjug Chem. 2002;13:264-8
61. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307-10
62. Tan BX, Liew HP, Chua JS, Ghadessy FJ, Tan YS, Lane DP. et al. Anatomy of Mdm2 and Mdm4 in evolution. J Mol Cell Biol. 2017;9:3-15
63. Secchiero P, Barbarotto E, Tiribelli M, Zerbinati C, di Iasio MG, Gonelli A. et al. Functional integrity of the p53-mediated apoptotic pathway induced by the nongenotoxic agent nutlin-3 in B-cell chronic lymphocytic leukemia (B-CLL). Blood. 2006;107:4122-9
64. Ray-Coquard I, Blay J-Y, Italiano A, Le Cesne A, Penel N, Zhi J. et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol. 2012;13:1133-40
65. Wang H, Yu J, Lu X, He X. Nanoparticle systems reduce systemic toxicity in cancer treatment. Nanomedicine (Lond). 2016;11:103-6
66. Wang H, Agarwal P, Zhao S, Yu J, Lu X, He X. A biomimetic hybrid nanoplatform for encapsulation and precisely controlled delivery of theranostic agents. Nat Commun. 2015;6:10081
67. Coates A, Abraham S, Kaye SB, Sowerbutts T, Frewin C, Fox R. et al. On the receiving end—patient perception of the side-effects of cancer chemotherapy. Eur J Cancer Clin Oncol. 1983;19:203-8
68. de Boer-Dennert M, De Wit R, Schmitz P, Djontono J, v Beurden V, Stoter G. et al. Patient perceptions of the side-effects of chemotherapy: the influence of 5HT3 antagonists. Br J Cancer. 1997;76:1055-61
Author contact
![]() Corresponding authors: Email (W. He): WHeumaryland.edu; Email (P. Hou): phouedu.cn; Email (K. Tao): taokaishan0686com.
Corresponding authors: Email (W. He): WHeumaryland.edu; Email (P. Hou): phouedu.cn; Email (K. Tao): taokaishan0686com.