Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Sources of CAFs
Activation of CAFs
Hallmarks of pancreatic cancer
CAFs affect the proliferation...
CAFs regulate the invasion and...
CAFs and angiogenesis in PDAC
CAFs regulate tumor immunity in...
CAFs and tumor metabolism in PDAC
CAFs and chemoresistance in PDAC
Stromal CAFs: friend or foe?
Antistroma therapies
Conclusions
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(18):5072-5087. doi:10.7150/thno.26546 This issue Cite
Review
The impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer
Qiqing Sun1,2,3,4#, Bo Zhang1,2,3,4#, Qiangsheng Hu1,2,3,4, Yi Qin1,2,3,4, Wenyan Xu1,2,3,4, Wensheng Liu1,2,3,4, Xianjun Yu1,2,3,4 ![]() , Jin Xu1,2,3,4
, Jin Xu1,2,3,4 ![]()
1. Department of Pancreatic Surgery, Fudan University Shanghai Cancer Center, Shanghai 200032, China.
2. Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, China.
3. Pancreatic Cancer Institute, Fudan University, Shanghai 200032, China.
4. Shanghai Pancreatic Cancer Institute, Shanghai 200032, China.
# These authors contributed equally to this article.
Received 2018-4-8; Accepted 2018-9-4; Published 2018-10-6
Abstract

Pancreatic ductal adenocarcinoma (PDAC) constitutes one of the most challenging lethal tumors and has a very poor prognosis. In addition to cancer cells, the tumor microenvironment created by a repertoire of resident and recruited cells and the extracellular matrix also contribute to the acquisition of hallmarks of cancer. Among these factors, cancer-associated fibroblasts (CAFs) are critical components of the tumor microenvironment. CAFs originate from the activation of resident fibroblasts and pancreatic stellate cells, the differentiation of bone marrow-derived mesenchymal stem cells and epithelial-to-mesenchymal transition. CAFs acquire an activated phenotype via various cytokines and promote tumor proliferation and growth, accelerate invasion and metastasis, induce angiogenesis, promote inflammation and immune destruction, regulate tumor metabolism, and induce chemoresistance; these factors contribute to the acquisition of major hallmarks of PDAC. Therefore, an improved understanding of the impact of CAFs on the major hallmarks of PDAC will highlight the diagnostic and therapeutic values of these targeted cells.
Keywords: pancreatic cancer, tumor microenvironment, hallmarks of cancer, cancer-associated fibroblast
Introduction
Pancreatic ductal adenocarcinoma (PDAC), which is among the most lethal tumors and has a very poor prognosis, constitutes the fourth most common cause of cancer-related death in the United States. The American Cancer Society estimates that there will be approximately 55,440 new cases of pancreatic cancer and approximately 44, 330 new deaths in 2018 [1]. Patients with precancerous lesions and early PDAC typically exhibit no obvious symptoms [2]. Accordingly, owing to a lack of appropriate methods of early diagnosis, only 20% of patients are diagnosed at an operative stage, contributing to the poor survival rate of only 8% over five years, as reported by the most recent SEER database from 2007-2013 [3].
The tumor microenvironment (TME) is considered to play a crucial role in tumor proliferation, progression, invasion, metastasis, angiogenesis, metabolism, immunosuppression and chemoresistance. The TME comprises the extracellular matrix (ECM) as well as cellular components, including cancer-associated fibroblasts (CAFs), cancer-associated macrophages, immune inflammatory cells, and local or bone marrow-derived mesenchymal stem cells (MSCs), all of which communicate with cancer cells at different levels, affecting tumor prognosis and offering novel therapeutic targets (Figure 1A). PDAC is characterized by the existence of an ample desmoplastic stroma, which constitutes up to 90% of the tumor volume [2, 4, 5]. It is generally accepted that CAFs are crucial for the formation of the desmoplastic stroma; these cells promote tumor progression and metastasis, and might also be implicated in chemoresistance and immunosuppression [4, 6]. In this review, we discuss some novel discoveries related to CAFs in PDAC to illustrate the impact of CAFs on the major hallmarks of PDAC (Table 1), and highlight promising directions for future research.
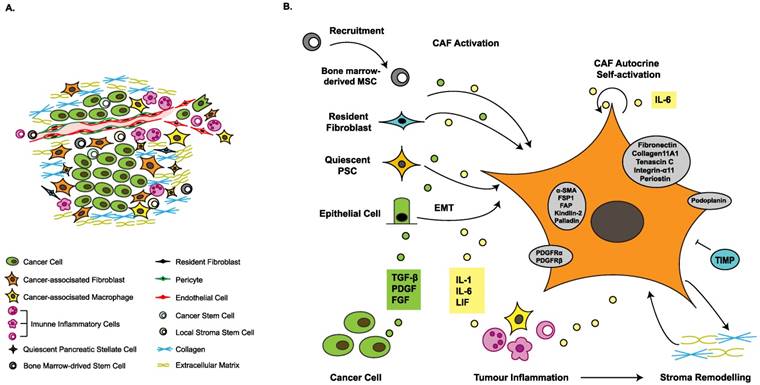
Heterogeneous origins and activation of cancer-associated fibroblasts. (A) The tumor microenvironment is composed of heterogeneous cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), immune inflammatory cells, cancer cells, various progenitor cells, blood vessels and extracellular matrix. (B) The sources of CAFs include recruited bone marrow-derived mesenchymal stem cells (MSCs), resident fibroblasts, quiescent pancreatic stellate cells (PSCs) and epithelial-to-mesenchymal transition (EMT). Growth factors stimulate CAF activation. The inflammatory environment facilitates acquisition of contractile and secretory phenotypes. Stromal modification contributes to phenotype maintenance. Activated CAFs express several marker proteins, such as alpha-smooth muscle actin (α-SMA), fibroblast-specific protein 1 (FSP1) and fibroblast activation protein (FAP), indicating a switch from the quiescent state to the activated state. α-SMA: alpha-smooth muscle actin; DNA: deoxyribonucleic acid; PDGF: platelet-derived growth factor; PDGFR: platelet-derived growth factor receptor; TGF-β: transforming growth factor beta; TIMP: tissue inhibitor of metalloproteinases.
Strengths and weakness of preclinical models of pancreatic cancer-associated fibroblasts/tumor interaction.
| Model | Application | Strengths | Weaknesses | References |
|---|---|---|---|---|
| In vitro | ||||
| 2D cell line culture | Analysis of signaling pathways, e.g., cytokines, exosomes and autophagy Drug development | Low cost and time effort Rapid biomarker and drug screening | Lack of heterogeneity Lack of tumor microenvironment Aggressive clone selection Genetic drift | Takikawa et al. [51] Endo et al. [53] Wang et al. [60] Doi et al. [135] |
| 2D cell coculture | Bidirectional interaction between cancer-associated fibroblasts and pancreatic cancer cells Analysis of signaling pathways | Low cost and time effort Flexible culture conditions (e.g., hypoxia) | Lack of heterogeneity Lack of tumor microenvironment Aggressive clone selection Genetic drift | Yoshida et al. [14] Tjomsland et al. [42] Kikuta et al. [58] |
| 3D organoids | Bidirectional interaction between cancer-associated fibroblasts and other cells Early events in tumor progression, cell invasion and migration | A perfect intermediate between 2D cultures and genetically engineered mouse models Optional and quantitative model components | Insufficient primary stellate cells for replicates Lack of models of terminated stages Technical difficulty | Kadaba et al. [54] Erdogan et al. [62] Koikawa et al. [63] Kuen et al. [84] |
| In vivo | ||||
| Cell line- derived coinjection xenografts | Fibrosis, tumor growth, metastasis and angiogenesis Drug development | Moderate cost and time effort | Immunodeficiency Lack of other stroma components Lack of tumor heterogeneity | Xu et al. [38] Hwang et al. [45] Endo et al. [53] |
| Patient-derived xenografts | Drug screening | Tumor heterogeneity Consistent biological properties Personalized therapy predictions | Aggressive cancer clone selection Selected patients Low engraftment rate | Ernsting et al. [127] |
| Genetically engineered mouse models | Genetic analyses of cancer-associated fibroblasts on tumor progression Tumor microenvironment and immune surveillance Drug screening | Intact immune system Close imitation of the human cancer | Lack of genetic complexity High cost and time effort Dependent on limited cancer-related genes | Lo et al. [48] Rhim et al. [76] Özdemir et al. [77] Ene-Obong et al. [89] Mace et al. [136] |
Sources of CAFs
CAFs are characterized by their diverse origins, including the activation of resident fibroblasts and pancreatic stellate cells (PSCs), the recruitment and differentiation of bone marrow-derived MSCs and epithelial-to-mesenchymal transition (EMT) (Figure 1B). In particular, EMT involves processes related to cellular plasticity, including the loss of epithelial traits and the acquisition of mesenchymal characteristics and cancer stem cell traits [7]. Primary and metastatic tumors actively attract MSCs from the bone marrow and other sites where they become CAFs and contribute to the TME [8]. The most prominent source of CAFs in PDAC is PSCs, the resident mesenchymal cells of the pancreas, which were found to be the main source of collagen in the tumor stroma [9]. In the quiescent state of PSCs, they display ample cellular vitamin A-containing lipid droplets. Once activated, the cells exhibit a loss of vitamin A reserves, switch to a contractile and secretory phenotype, and secrete large amounts of structural matrix components, such as collagens, fibronectin and tenascin C [10].
According to their different functions, fibroblasts can be divided into two stages—the quiescent state and the activated state. Quiescent fibroblasts express vimentin, whereas activated CAFs exhibit myofibroblast-like characteristics and stain for alpha-smooth muscle actin (α-SMA), also known as α actin 2 (ACTA2), a cytoskeletal protein that identifies smooth muscle cells [11]. In addition to α-SMA, CAFs express a variety of proteins, including fibroblast-specific protein 1 (FSP1), platelet-derived growth factor (PDGF) receptor-α (PDGFRα), PDGFRβ, tenascin C, fibroblast activation protein (FAP), kindlin-2, palladin, fibronectin, integrin-α11, and podoplanin [4, 12-15]. These proteins are considered markers of activated fibroblasts; however, the expression levels of these proteins vary in different CAF populations, and none are unique to activated fibroblasts. A recent study identified two CAF subpopulations, inflammatory fibroblasts (iCAFs) and myofibroblasts (myCAFs), based on different levels of α-SMA [16]. Characterized by significantly higher α-SMA expression levels, myCAFs are located immediately adjacent to neoplastic cells. Located more distantly from neoplastic cells in the tumor stroma, iCAFs are identified by their high expression of tumor-promoting cytokines and chemokines [16]. The strategy to distinguish different CAF subpopulations based on diverse expression levels of biomarkers may assist in identifying the multiple functions of CAFs.
Activation of CAFs
To acquire activated phenotypes, quiescent fibroblasts undergo activation via diverse mechanisms (Figure 1B). Growth factors, such as transforming growth factor beta (TGF-β), PDGF and fibroblast growth factor (FGF), could stimulate the recruitment and activation of CAFs [2, 17-19]. TGF-β, which can be secreted by both cancer cells and stromal cells [19], can activate fibroblasts into myofibroblasts (increased α-SMA expression), as shown in TGF-β treatment in vitro [20]. TGF-β treatment can also modulate the cell shape, stiffness and invasion of pancreatic CAFs; however, TGF-β alone is not sufficient to endow fibroblasts with all the features of activated CAFs in vitro [20]. PDGF constitutes another important growth factor that is enriched in the TME and can be secreted by both cancer cells and stromal cells [21, 22]. In contrast to TGF-β, the primary functions of PDGF are recruiting fibroblasts and enhancing their growth and proliferation [23, 24]. Upon activation, the expression profile of microRNAs (miRNAs) in CAFs changes substantially, which suggests that miRNAs are involved in the process of activation [25]. DNA methylation has also been found to be involved in the activation process, as the DNA methylation of SOCS1 in CAF activated the phosphorylation of STAT3 and increased the expression of insulin-like growth factor 1 (IGF-1) to promote pancreatic cancer cell (PCC) proliferation [26].
Altered ECM composition constitutes another stimulus for the process of activation. PCCs could produce different inflammatory cytokines, such as tumor necrosis factor α (TNF-α), interleukin 6 (IL-6), interleukin 8 (IL-8), microphage inhibitory factor (MIF) and interleukin 1β (IL-1β), to interact with other cell types, participating in forming an inflammatory microenvironment [27]. Inflammation plays a crucial role in expediting the development of the TME and accelerates the CAF tumor-promoting phenotypic switch. For example, leukemia inhibitory factor (LIF), an IL-6 class proinflammatory cytokine, has recently been considered a desmoplasia promoter that mediates the proinvasive activation of fibroblasts independent of α-SMA expression [28]. LIF production stimulated by a TGF-β pulse mediates STAT3 phosphorylation, which induces the emergence of proinvasive characteristics of fibroblasts via the actomyosin contractility of CAFs and ECM remodeling in vitro and in vivo. The underlying mechanism is dependent on crosstalk between the JAK1/STAT3 and RhoA/ROCK/MLC2 signaling pathways and remains to be fully clarified [28]. Galectin 3 (GAL3), a member of the lectin family in the cytoplasm that can be secreted by cancer cells, has been found to trigger the proliferation and invasion of CAFs when cocultured with PCCs [29]. Enhanced expression of proinflammatory cytokines such as interleukin 8 (IL-8) and chemokine (C-C motif) ligand 2 (CCL-2) in CAFs is observed with GAL3 treatment. Mechanistically, the production of IL-8 is induced by GAL3 at the transcriptional level via ITGB1/ILK/NF-κB signaling in CAFs [29]. Zhang et al. [30] found that PCC-secreted IL-1β could activate interleukin-1 receptor-associated kinase 4 (IRAK4) in CAFs, which drives NF-κB activity in CAFs in vitro. Furthermore, it has been discovered that PCCs can promote CAF phenotype switching via activation of signaling downstream of sonic hedgehog (SHH), which is overexpressed in PCCs [31]. As a member of a family of nonreceptor serine/threonine kinases expressed in CAFs, p21-activated kinase 1 (PAK1) has also been identified as a regulator of CAF activation [32].
Following activation, CAFs can secrete abundant structural components of ECM, such as collagens, fibronectin, tenascin C, metalloproteinases (MMPs) and their inhibitors, which contribute to dynamic matrix remodeling in PDAC [5, 6]. Matrix remodeling is essential for maintenance of the activated phenotype in CAFs and thus for sustained function. MMPs and their inhibitors, the tissue inhibitor of metalloproteinase (TIMP) family, participate in stromal homeostasis under physiological conditions, and matrix remodeling is to some extent regulated by the levels of both MMPs and TIMPs [33]. Timp loss has been considered sufficient for the acquisition of the CAF phenotype because disintegrin and metalloproteinase domain-containing protein 10 (ADAM10)-rich exosomes secreted by TIMP knockout fibroblasts facilitate tumor proliferation and metastasis [34]. Activated CAFs can promote matrix remodeling via a ROCK-dependent pathway and actomyosin function. A study showed that CAFs could promote ECM remodeling and stiffness through the activation of the Yes-associated protein (YAP) transcription factor, which could regulate the expression of actomyosin [35]. In turn, ECM stiffness may further reinforce YAP activation, thus developing a self-reinforcing loop that stabilizes the activated phenotype of CAFs [35].
Hallmarks of pancreatic cancer
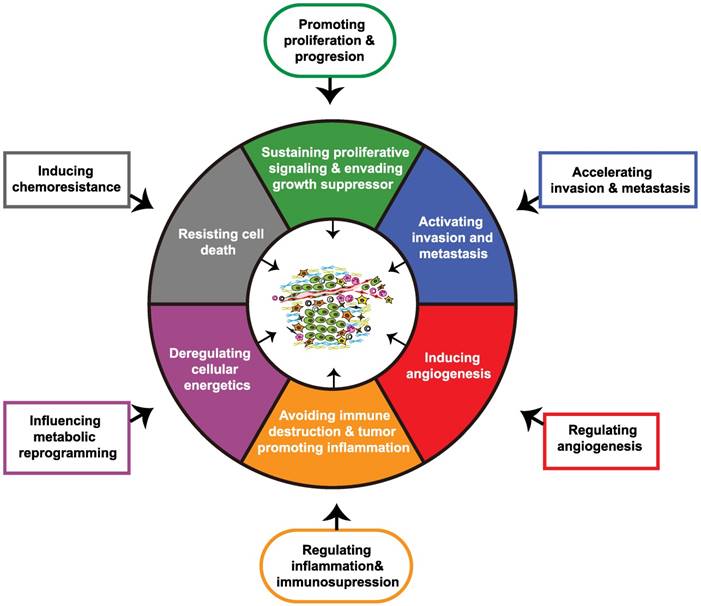
The hallmarks of cancer constitute six biological characteristics that are acquired during the different stages of tumor evolution, including sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis. Genome instability and mutation, along with tumor-promoting inflammation are two underlying hallmarks that precede the emergence of the mentioned tumor biological capabilities. Subsequent to these changes, the reprogramming of energy metabolism and the evasion of immune destruction are also included in cancer hallmarks. Additionally, generation of the TME by a repertoire of recruited and resident cells and extracellular matrix also contributes to the acquisition of hallmarks of cancer [36]. As critical players in the TME, CAFs in PDAC have been demonstrated by previous studies to promote tumor proliferation and growth, accelerate invasion and metastasis, induce angiogenesis, promote inflammation and immune destruction, regulate tumor metabolism, and induce chemoresistance, and these effects contribute to the acquisition of the major hallmarks of pancreatic cancer (Figure 2) [37-44].
Impact of cancer-associated fibroblasts on major hallmarks of pancreatic cancer. Through crosstalk with the tumor microenvironment, cancer-associated fibroblasts (CAFs) acquire physiological functions that ultimately regulate major hallmarks of pancreatic cancer.
CAFs affect the proliferation and growth of PDAC
Studies have demonstrated that CAFs facilitate the proliferation and growth of cancer cells [6, 45]. The primary tumor size was greater after the injection of CAFs in an orthotopic model of PDAC, which indicates that CAFs promote local tumor growth [45, 46]. Activated CAFs produce high levels of growth factors and inflammatory factors that maintain their activated phenotype, with inflammation functioning as a promoter of tumorigenesis [6]. Tjomsland et al. [42] showed that the coculture of PDAC and CAF cell lines increased the production of inflammatory factors including IL-1α, IL-6, chemokine (C-X-C motif) ligand 8 (CXCL8), vascular endothelial growth factor-A (VEGF-A), cyclooxygenase 2 (COX-2), and CCL20, among which, IL-1α was considered the initiator of the enhanced inflammatory response through binding to interleukin-1 receptor 1 (IL-1R1) expressed predominantly by CAFs. Thus, CAFs contribute to the establishment and sustenance of the inflammatory environment, which benefits tumor survival through the IL-1α signaling cascade [42].
Several proteins expressed in CAFs are involved in the promotion of tumor growth. In particular, kindlin-2, a focal adhesion protein that regulates the activation of β-integrins, plays a role in the growth and progression of PDAC [13]. In addition, in a subcutaneous tumor model in nude mice, the coinjection of PSCs and PCCs enhanced tumor size, and this effect was abolished by kindlin-2 knockdown in PSCs [13]. FAP is another key protein involved in tumor-stroma crosstalk: overexpression of FAP in the stroma is associated with tumor progression and poor prognosis [47-49]. Kawase et al. [48] demonstrated that the cell cycle shift of PCCs is dramatically activated during coculture with FAP+ fibroblasts compared to coculture with FAP- fibroblasts. Lo et al. [49] reported that genetic depletion of FAP in a mouse model promoted tumor progression and decreased murine survival. However, apart from stromal cells, FAP was also detected in CD90+ mesenchymal-like PCCs (accounting for 20% of total intratumoral FAP+ cells), which indicated that FAP may also regulate tumor progression in a cell-autonomous manner [49].
In addition to classical autocrine and paracrine interactions between CAFs and PCCs, exosome-mediated CAF-PCC cross talk has recently been considered to play a crucial role in intercellular communication by regulating gene expression and cell functions in receiving cells [50]. Takikawa et al. [51] demonstrated that human PSCs release exosomes containing a variety of microRNAs including miR-21-5p, which stimulate PCC proliferation and migration along with chemokine mRNA expression in PCCs. In turn, PCC-derived exosomes could help maintain the activated CAF phenotype. For example, Masamune et al. [52] demonstrated that PCC-derived exosomes stimulated PSC proliferation and migration, along with Erk and Akt activation, α-SMA and fibrosis-related gene mRNA expression, and procollagen type I C-peptide production in PSCs. Autophagy is also involved in the interaction between CAFs and PCCs. Endo et al. [53] found that autophagic PSCs produce ECM molecules and IL-6 and are associated with poorer survival and disease recurrence in patients with PDAC.
CAFs regulate the invasion and metastasis of PDAC
In an orthotopic model of PDAC, coinjection of PSCs increased the proportion of mice with distant metastasis [45, 46]. CAFs regulate PDAC invasion and metastasis by mediating multiple pathways, and modulate a range of cellular processes in tumor cells in a dose-dependent manner [54]. Recently, several unique features have emerged to describe tumor invasion and metastasis. Whatcott et al. [55] provided clinical evidence that both primary and metastatic lesions of PDAC exhibit high levels of desmoplasia. Some metastasis models have suggested that the CAFs in distant metastases originate from the primary site, while desmoplasia in liver metastatic tumors is additionally considered to be a local physiological response [38, 55, 56]. Endo et al. [53] found that inhibitors of PSC autophagy decreased the tumor size in nude mice, as well as distant metastasis. In addition, although autophagy inhibition caused by Atg7 knockdown in PSCs did not affect PCC proliferation, it decreased cancer cell invasion, suggesting that the decreased metastasis was not only due to the reduced number of primary PCCs, but also due to the inhibition of PCC invasiveness [53].
EMT has been demonstrated to play a vital role in PCC invasiveness under the regulation of several matrix proteins, such as N-cadherin and E-cadherin [57]. PSCs have been demonstrated to promote EMT in PCCs in vitro, with decreased expression of the epithelial markers E-cadherin and cytokeratin, and increased expression of mesenchymal markers such as vimentin [58]. The underlying mechanisms of CAF-induced EMT in PCCs need to be clarified, but some groundwork has provided potential directions. Wu et al. [59] provided in vitro evidence that PSC-secreted IL-6 enhanced EMT in PCCs. The IL-6 secreted by PSCs could stimulate the activation of STAT3 in PCCs, which leads to the induction of Nrf2 activities, thus enhancing EMT in PCCs with altered EMT-related gene expression [59]. High expression of asporin, an EMT protein, has been found in CAFs, and high expression of this protein is associated with poor prognosis in patients with PDAC [60]. Wang et al. [60] showed that exogenous asporin activated NF-κB/p65 in PCCs to induce EMT through interaction with CD44 expressed on cancer cells. In addition, miRNAs may also be involved in PSC-induced EMT in PCCs, and upregulated miR-210 expression in PCCs was noted when PCCs were cocultured with CAFs [61]. Inhibition of miR-210 reduced cell migration and CAF-induced EMT in PCCs, which suggests that miR-210 may play a role in CAF-induced EMT [61].
CAFs have been considered to have the ability to physically modify the tumor stroma in PDAC, with the modified stroma subsequently facilitating PCC invasion. Erdogan et al. [62] showed that CAFs could produce and organize fibronectin-rich ECM with an anisotropic fiber orientation, guiding directional cancer cell migration in a 3D cell-derived matrices (CDMs) model. Koikawa et al. [63] found that CAFs invaded together with PCCs and assisted in PCC invasion by remodeling the ECM by changing the alignment of collagen fibers in a 3D matrix remodeling assay. This assistance could be attenuated by knockdown of Endo180 (urokinase-type plasminogen activator receptor-associated protein) in CAFs, as shown by decreased invasiveness of both cocultured CAFs and PCCs and altered orientation of collagen fibers in coinjection mouse models; this finding suggests that CAFs could remodel the stroma to support tumor invasion, possibly through interaction with Endo180 to rebuild the actin cytoskeleton [63]. CAFs can also create tunnels through the matrix using invadopodia to degrade the matrix [12, 64]. Palladin, an actin-binding protein that is highly expressed in cancer cells and CAFs, has been found to enhance PDAC invasion both in vitro and in vivo [64]. Upon stimulation with phorbol 12-myristate 13-acetate (PMA), palladin could accumulate in invadopodia in CAFs to increase the assembly of matrix-degrading invodapodia by regulating the activity of the small GTPase Cdc42 in vitro [64]. Proteolytic enzymes in invadopodia, such as MMPs and cathepsin, can degrade the ECM to form tunnels, thus assisting in tumor cell invasion [12].
Moreover, it has recently been reported that hepatocyte growth factor (HGF) is involved in tumor-CAF crosstalk. HGF is secreted mainly by stromal cells and promotes cell mitosis and motility by binding to the specific receptor, c-Met, which is expressed on the surface of PCCs [65]. Yang et al. [66] recently demonstrated that PSCs could promote PCC migration via the HGF/c-Met pathway, mediating increased survivin expression, the level of which is closely correlated with metastasis. Rucki et al. [67] discovered that both the IGF1 and HGF pathways independently upregulate AnxA2 phosphorylation, that the expression level of IGF-1 is regulated by SHH signaling, and only inhibition of all these pathways could effectively suppress phosphorylation of the downstream molecule AnxA2, along with PCC development and metastasis. Thus, this discovery may to some extent explain the ineffectiveness of single inhibition of the SHH or Met pathways, and provided a strategy of dual inhibition of these stromal signaling pathways [68, 69].
CAFs and angiogenesis in PDAC
Evidence has suggested that CAFs serve the function of regulating tumor angiogenesis. It has been reported that CAFs constitutively produce VEGF, the production of which is increased by hypoxia [39, 70]. In addition to VEGF, CAFs also express several regulatory molecules of angiogenesis such as VEGF receptors (VEGFR), angiopoietin-1 and its receptor Tie-2, and vasohibin-1. There is also histological evidence that CAF-conditioned media induces tumor angiogenesis, as shown by increased tube formation on Matrigel in vitro and by directed vessel formation in nude mice in vivo [39]. Xu et al. [38] observed induced angiogenesis when PCCs were injected together with PSCs in an orthotopic model of PDAC. Additionally, PSCs have been revealed to be able to intravasate/extravasate through blood vessels and assist in PCC metastasis [38]. Mechanically, it has been proposed that the HGF/c-MET pathway plays a main role in PSC-induced tube formation in the human microvasculature in vitro [71]. The CXCL12/CXCR4 axis, which plays a pivotal role in embryonic hematopoiesis and angiogenesis [72, 73], has also been demonstrated to promote tumor neovascularization in a VEGF-dependent manner in PDAC [40]. Matsuo et al. [74] found that CAF-derived CXCL12 increased the growth and invasiveness of human umbilical vein endothelial cells (HUVECs) in vitro. In addition, CAF-derived CXCL12 could greatly enhance PCC-derived CXCL8 production, which indirectly facilitated HUVEC tube formation [74].
Notably, although CAFs have been considered to facilitate tumor angiogenesis, depletion of the tumor-promoting function of CAFs may evoke a strong angiogenic response instead of an attenuated one. Mathew et al. [75] discovered that despite initial expectations that inhibition of the SHH pathway would result in an impaired ability of CAFs to promote tumor growth, these cells instead exhibited significantly greater potential to support tumor growth compared to that of wild-type fibroblasts, as the reduced levels of SHH signaling increased angiogenesis mediated by the tumor stroma. In addition, Rhim et al. [76] demonstrated that genetic depletion of SHH resulted in increased vascularity in a mouse model of PDAC. Conversely, Özdemir et al. [77] showed that, in contrast to the inhibition of certain signaling pathway, the depletion of myofibroblasts resulted in suppressed neovascularization in a transgenic mouse model of PDAC. These results indicate the existence of multiple functions of CAFs in angiogenesis, which requires further research for full clarification.
CAFs regulate tumor immunity in PDAC
The extremely strong immunosuppression in the TME is a hallmark of PDAC, with ineffective T lymphocyte infiltration, immunosuppressive cytokine production, and increased immunosuppressor cells, such as regulatory T cells (Treg), tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs) [78]. The dense desmoplastic stroma of PDAC may correlate with tumor immune evasion. A recent study identified focal adhesion kinase (FAK) signaling as a central driver of desmoplasia and immunosuppression, with FAK inhibition reversing unresponsiveness to immunotherapy in a KPC mouse model [79]. This study has led to a phase I trial of defactinib, a FAK inhibitor combined with checkpoint immunotherapy in patients with advanced PDAC (NCT02546531).
CAFs interact closely with tumor immunity because they produce large amounts of cytokines and chemokines, such as IL-6, IL-1β, CXCL1, CXCL2, CXCL12, CXCL13, CCL-5 and M-CSF [43, 80, 81]. TGF-β secreted by CAFs suppresses the activation of natural killer (NK) cells and cytotoxic T lymphocytes (CTLs) and triggers the differentiation of Treg cells [82]. FAP+ CAFs are the main tumoral source of CXCL12, which was involved in disadvantageous T cell formation in the TME of a mouse model of PDAC [83]. 3D coculture of monocytes with PCCs and fibroblasts was shown to increase the production of immunosuppressive cytokines, such as IL-6, IL-8, IL-10, GM-CSF and M-CSF, which are known to promote the polarization of M2-like macrophages and MDSCs [84]. Another study found that activated CAFs could induce peripheral blood mononuclear cells to differentiate into MDSCs via a STAT3-dependent mechanism [43]. Zhang et al. [85] revealed that pancreatic CAFs were able to induce the M2 polarization of macrophages, the phenotype of which could promote EMT and PCC invasiveness [86], partly through enhanced cellular reactive oxygen species (ROS) production in monocytes triggered by CAF-secreted M-CSF. In addition, immune cells can also facilitate CAF activation and tumor-promoting functions; for example, quiescent PSCs could be activated and exhibited increased cytokine production when cocultured with macrophage cell lines [81]. TAMs could promote the activation of resident hepatic stellate cells (hStCs) into myofibroblasts and stimulate their production of periostin via granulin secretion, thus inducing liver fibrosis to support PDAC liver metastasis [87].
It has been demonstrated that more than 90% of tumor-infiltrating T cells are either disadvantaged or do not reach the tumor cells because they were trapped in peritumoral tissue [88]. Ene-Obong et al. [89] showed that activated CAFs produce immune inflammatory factor cytokines, as well as adhesion molecules that influence the migration of T lymphocytes to reach effector sites. In particular, they found that CD8+ CTLs separated from human pancreatic cancer tumor tissues exhibited increased tropism toward activated PSCs through the CXCL12/CXCR4 axis, in comparison to quiescent PSCs or PCCs, leading to the reduced CD8+ CTL migration to juxtatumoral stromal compartments, where T cells could interact with PCCs [89].
CAFs might play a role in promoting immunosuppression via increasing T helper type 2 (Th2)-mediated inflammation [90, 91]. De Monte et al. [90] found that thymic stromal lymphopoietin (TSLP) secreted by activated CAFs in PDAC could induce the activation and maturation of tumor antigen-loaded resident dendritic cells (DCs), which could then migrate to the draining lymph nodes, where they activated tumor antigen-specific CD4+ Th2 cells and induced tumor Th2 immune deviation. Therefore, the balance between Th2 and Th1 lymphocytes is broken, resulting in altered immune cytokines in the TME, which may in turn enhance fibrotic desmoplasia [90]. Galectin 1 secreted by PSCs impaired the viability of both CD4+ and CD8+ T cells, and overexpression of galectin 1 in PSCs induced a drastic increase of Th2 cytokine secretion and decreased Th1 cytokines, which indicate that galectin 1 might regulate the PSC-dependent immune privilege in PDAC [91]. These data suggest that CAFs can not only directly regulate the biological behavior of cancer cells, but also contribute to the formation of an immunosuppressive TME in PDAC to assist in tumor evasion.
CAFs and tumor metabolism in PDAC
PDAC exhibits a disorganized, hypovascular, and dense stroma, with mechanically poor perfusion that creates the highly hypoxic, nutrient-poor microenvironment within PDAC, which may further influence tumor metabolic reprogramming, another emerging hallmark of cancer [80, 92, 93]. Emerging evidence indicates that pancreatic CAFs could promote tumor progression in a nonvascular manner by regulating cancer cell metabolic reprogramming, although the detailed mechanism remains to be investigated [94]. A recent study reported that cytokines derived from Kras mutations in PDAC mediate reciprocal CAF-PCC metabolic interactions, which induce the reprogramming of energy metabolism [95]. Notably, tumor cells could highjack CAFs to provide them with energy and nutrients. CAFs surrounding PCCs undergo metabolic transition, similar to the phenotype correlated with the Warburg effect, and activated CAFs take up more glucose and produce more lactate than quiescent fibroblasts, which could be actively taken up by PCCs to support their growth [96]. Meanwhile, tumor cells stabilize HIF-1α in CAFs by increasing ROS production to increase glycolysis, leading to the formation of a “pseudohypoxic” environment for CAFs [97]. In addition, Hirakawa et al. [98] demonstrated that the motility of PCCs cocultured with CAFs was greater under hypoxic conditions than under normoxic conditions, partly via paracrine IGF1/IGF1R signaling.
Furthermore, CAFs have recently been considered to regulate tumor cell metabolic activities through autophagic alanine secretion [99]. Previous studies found that alanine, which is secreted by CAFs through CAF autophagy stimulated by cancer cells, could outcompete glucose and glutamine to support the tricarboxylic acid (TCA) cycle, and thus nonessential amino acid (NEAA) and lipid biosynthesis in PDAC. Therefore, CAF-derived alanine serves as an alternative carbon source, suggesting a novel metabolic interaction between CAFs and PCCs [99]. Furthermore, CAF-derived exosomes (CDEs) provide another source of CAF-derived alanine to fuel cancer cells. Notably, in contrast to the macropinosome, during the medium of macrocinobytosis, which has been exclusively reported in PCCs with mutant Kras, CDE-mediated metabolic remodeling was described to be independent of Kras mutation [100]. Collectively, these CAF-PCC metabolic crosstalk pathways further illustrate the impact of CAFs in providing heterodox methods to help PCCs thrive in a hostile environment.
CAFs and chemoresistance in PDAC
CAFs have been shown to promote chemoresistance via physical barrier methods, biophysical drug scavenging and CAF-PCC cross talk. The PDAC stroma exhibits several characteristics that can be considered physical barriers to effective drug delivery, including a disorganized, hypovascular, and dense stroma and decreased cellular transporters [5, 101-103]. A three-dimensional organ culture model of angiogenesis showed that a dense stroma is correlated with decreased neovascularization and network formation [104]. Similar to other drugs, chemotherapeutic agents must cross the vessel wall, traversing the extracellular compartment and entering the cytoplasm of PCCs to ultimately reach their target sites [105]. However, the dense stroma of PDAC was shown to form a high interstitial fluid pressure (IFP) that prevented movement of the chemotherapeutic agent gemcitabine from the vasculature to the extracellular compartment [106]. In comparison, Rice et al. [107] reported that matrix stiffness induced chemoresistance to paclitaxel, but not to gemcitabine in vitro, suggesting that environmental rigidity underlies an aspect of chemoresistance. CAFs could also interact with PCCs to hinder cellular uptake of gemcitabine. A recent study showed that CAFs could serve as a major source of cysteine-rich angiogenic inducer 61 (CYR61) when activated by TGF-β-ALK5-Smad2/3 signaling, and thus induce chemoresistance by downregulating the nucleoside transporters hENT1 and hCNT3 that mediate cellular uptake of gemcitabine in coculture models [103].
CAFs can also act as a biophysical barrier to drug delivery. A recent study showed that fibroblast drug scavenging accumulated intratumoral gemcitabine and made it unavailable to PCCs through entrapping active gemcitabine within CAFs [108]. Both in vitro and in vivo, in comparison to tumor cells, CAFs exhibit lower expression levels of key gemcitabine-inactivating enzymes, thus increasing the intracellular concentration of gemcitabine [108].
Apart from tumor-promoting effects, various tumor-CAF crosstalk networks are also involved in mediating chemoresistance. Ireland et al. [109] found that CAFs support PCC chemoresistance by secreting insulin-like growth factors (IGF) 1 and 2, which bind to the IGF receptors on the surface of PCCs, whereas pharmacologic inhibition of IGF resensitized PDAC to gemcitabine in vivo. Long et al. [110] showed that STAT3 activation in PCCs induced by CAF-derived IL-6 could mediate chemoresistance in PDAC; thus, disrupting IL-6 signaling using anti-IL6R antibodies holds promise for improving chemotherapeutic efficacy in PDAC. Additionally, Duluc et al. [111] demonstrated the role of the mTOR/4E-BP1 pathway in promoting chemoresistance via the autocrine and paracrine IL-6 loop. Moreover, exosomes have recently been found to participate in the tumor-CAF crosstalk associated with chemoresistance. A recent study found that CDEs could increase the chemoresistance-inducing factor Snail in recipient tumor cells and promote tumor proliferation and drug resistance [112]. Treatment of gemcitabine-exposed CAFs with an inhibitor of exosome release, GW4869, significantly increased cancer cell apoptosis in coculture models [112]. Together, these results suggest that further original research is required to elucidate the role of CAFs in the chemoresistance of pancreatic cancer and determine whether to deplete or utilize this important interstitial component.
Stromal CAFs: friend or foe?
It has been over five decades since the pioneering study of stroma-tumor interactions revealed that normal fibroblasts from Syrian hamsters suppress the survival of transformed kidney cells [113]. The desmoplastic response is considered to be a defense mechanism resembling the wound healing process; thus, cancer is considered the 'wound that never heals' [114]. However, many experimental studies have reported that CAFs promote tumor growth and progression and thus invasion and metastasis. Notably, however, the majority of preclinical studies have failed in clinical trials, which raises doubts about the efficacy of stroma-targeting therapy. Several recent experimental results have provided possible explanations for the inefficacy of these stroma-targeting therapies. Özdemir et al. [77] showed that the depletion of myofibroblasts in a mouse model of PDAC correlated with poor survival with increased cell aggressiveness, undifferentiated histology and suppressed immune response. In addition, Rhim et al. [76] demonstrated that PDAC tumors exhibited enhanced malignant biological behaviors and increased vascularity through genetic abortion of SHH in mouse models, indicating that stromal components may to some extent restrain the development of PDAC. In accordance with these experimental results, a recent study conducted on 155 surgically resected PDACs and 48 normal/benign pancreas samples, showed that the presence of more FAP+ CAFs (>100/high-power field) in the TME correlated with prolonged survival, rather than being a marker of poor prognosis as indicated in most previous studies [115]. Discussion has thus been initiated regarding whether the PDAC stroma represents a 'friend or foe'.
One reason for this phenomenon may be the heterogeneous characteristics of CAFs, which are based on their diverse sources, as well as their multiple functions owing to different expressed markers and secreted proteins. Fujiwara et al. [116] found that stromal CD271 expression is significantly associated with prolonged survival in patients with PDAC, with CD271+ PSCs located on the edge of solid tumors [116]. In contrast, Ikenaga et al. [117] defined another group of CAFs based on CD10 and showed that CD10+ PSCs enhance the progression of PCCs. In addition, periostin, which is secreted by CAFs, has been found to have a biphasic effect on PDAC development that correlates with EMT in human PCCs. While slight overexpression of periostin leads to reduced EMT and metastasis, high levels of periostin lead to the opposite effect [118].
As observed for the differentiation of T lymphocytes, CAFs may possess the ability to differentiate into diverse subpopulations with exclusive functions. As discussed in the context of the sources of CAFs, Öhlund et al. [16] identified two CAF subpopulations based on different levels of αSMA. Notably, defined by high αSMA expression, myCAFs lack expression of tumor-promoting inflammatory cytokines [16]. These findings may partly explain the decreased survival in mouse models that result from depleting CAFs based on their αSMA expression [77]; this strategy might eliminate myCAFs while preserving other subpopulations of CAFs. The stroma types of PDAC may also differ among different patients, resulting in distinct prognose. Moffitt et al. [119] identified two distinct stroma types based on gene expression levels, “normal” and “activated”, with patients with an activated stroma exhibiting a poorer prognosis than those with a normal stroma (median survival, 15 months vs. 25 months; 1-year survival, 60% vs. 82%). Additionally, the investigators found that “normal” stroma expressed relatively high levels of CAF markers, such as α-SMA, vimentin, and desmin, while “activated” stroma tended to express more genes related to macrophages [119]. This difference may lead to a hypothesis that different stroma types may play opposing roles; the “normal” stroma may function as a “good” stroma, while the “activated” stroma may be accountable for the inflammatory stromal response that promotes cancer progression as illustrated in some previous studies [9, 119]. Therefore, the traditional view of the tumor stroma as a foe must be reconsidered, as certain CAF subtypes might have pro-tumorigenic properties, whereas others might exhibit antitumorigenic features.
In addition, the function of CAFs is likely dynamic during cancer progression, and the role of CAFs may depend on the stroma-tumor balance in the TME. Possible modifications of the stroma during tumor development or anticancer treatments could induce changes in CAF function [120]. Fujiwara et al. [116] showed that although the mRNA expression of CD271 in PSCs increased during coculture with PCCs, the expression level decreased after a transient increase when PSCs moved toward PCCs through Matrigel, which indicates a defensive effect of stromal cells in tumorigenesis that might change dynamically with tumor evolution. Leca et al. [121] revealed that in a hostile environment in vitro, CAFs increased PCC invasiveness via a CAF-specific complex composed of ANXA6, LRP1, and TSP1, with extracellular vesicle (EV)-mediated support, highlighting the phenomenon that a hostile environment may evoke significant modifications of CAF activity. This observation suggests that in the stage of hyperplasia, similar to the classic wound healing response, CAFs may play a role as inhibitors. With tumor progression, CAFs thus promote the proliferation and migration of tumor cells and induce angiogenesis through various mechanisms [4, 9, 11].
Antistroma therapies
Many of the studies in this review concerning tumor progression also illustrate potential targets for PDAC, some of which have already been tested both preclinically and clinically (Table 2). One of the most encouraging stroma-targeting treatments in PDAC has been the degradation of hyaluronan (HA). It has been demonstrated that HA contributes to the increased IFP in the TME, giving rise to compressed vascularization and hypoperfusion [101, 122]. The depletion of HA is considered to correlate with elevated drug delivery to the tumor compartment, according to preclinical results [101, 122]. A Phase Ib study reported that when PEGylated recombinant hyaluronidase (PEGPH20) was applied in combination with gemcitabine, the median progression-free survival and overall survival rates were 7.2 and 13.0 months, respectively, in patients with high tissue HA levels, whereas these values were 3.5 and 5.7 months in patients with low tissue HA levels [123]. In a recent Phase II study comparing PEGPH20 plus nab-paclitaxel/gemcitabine (PAG) and nab-paclitaxel/gemcitabine (AG) in patients with metastatic PDAC. Progression-free survival significantly increased (PAG v AG, hazard ratio [HR], P = .049) and in patients with high tissue HA (HR, 0.51; P = .048) [124]. Thus, these studies may indicate that gemcitabine plus PEGPH20 exhibits promising therapeutic benefits in patients with advanced PDAC, especially in the subgroup with high HA content. In addition, PAG versus AG in HA-high PDAC patients is currently being studied in a Phase III clinical trial (NCT02715804).
Clinical trials targeting stroma/cancer-associated fibroblasts in pancreatic ductal adenocarcinoma.
| NCT number | Targeted group | Regiments | Treatment arms | Phase | Primary objectives | Accrual status | Clinical results |
|---|---|---|---|---|---|---|---|
| NCT00844649 | Metastatic adenocarcinoma of the pancreas | Albumin-bound paclitaxel | Experimental: Albumin-bound paclitaxel +Gemcitabine (AG) Active Comparator: Gemcitabine | III | Overall survival (OS) | Completed | OS was 8.5 and 6.7 months AG vs. Gem (hazard ratio [HR], 0.72, p <0.0001) |
| NCT01839487 | Stage IV untreated pancreatic cancer | PEGylated recombinant human hyaluronidase (PEGPH20) | Experimental: PEGPH20 + Nab-paclitaxel + Gemcitabine (PAG) Active Comparator: Placebo + Nab-paclitaxel + Gemcitabine (AG) | II | Progression-free survival (PFS) & thromboembolic events in PAG | Completed | PFS HR 0.73 PAG vs. AG, P = .049; for patients with hyaluronan-high tumors HR, 0.51; P = .048 |
| NCT02715804 | Hyaluronan-high stage IV previously untreated pancreatic ductal adenocarcinoma | PEGylated recombinant human hyaluronidase (PEGPH20) | Experimental: PEGPH20 + nab-paclitaxel + Gemcitabine Active Comparator: Nab-paclitaxel + Gemcitabine | III | PFS & OS comparison | Recruiting | NA |
| NCT01130142 | Metastatic pancreatic cancer | IPI 926 (an inhibitor of the Hedgehog pathway) | Experimental (Phase 2): IPI-926 + Gemcitabine Active Comparator (Phase 2): Placebo + Gemcitabine | Ib/II | Evaluation of safety profile & OS comparison | Completed | NA |
| NCT01064622 | Recurrent or metastatic pancreatic cancer | Vismodegib (an inhibitor of the Hedgehog pathway) | Active Comparator: Gemcitabine + Placebo (GP) Experimental: Gemcitabine + Vismodegib (GV) | Ib/II | PFS comparison | Completed | Median PFS was 2.5 and 4.0 months GP vs. GV (adjusted hazard ratio, 0.81; P= .30) |
| NCT02119663& NCT02117479 | Advanced or metastatic pancreatic ductal adenocarcinoma who have failed or were intolerant to first-line chemotherapy | Ruxolitinib (a JAK2 inhibitor) | Experimental: Ruxolitinib + Capecitabine Active Comparator: Placebo + Capecitabine | III | OS | Terminated | OS HR 0.969, ruxolitinib vs. placebo |
| NCT00892242 | Resectable pancreatic cancer | Zoledronic acid | Experimental: Neoadjuvant Zoledronic Acid | I | Safety and feasibility & OS or PFS | Terminated | NA |
| NCT03117920 | Refractory pancreatic cancer (MinPAC) | Minnelide | Experimental: Minnelide | II | disease control rate | Recruiting | NA |
| NCT02546531 | Advanced cancer | Defactinib (a FAK inhibitor) | Experimental: Dose escalation Experimental: Dose expansion | I | Recommended phase II dose | Recruiting | NA |
| NCT03307148 | Pancreatic cancer | All-trans retinoic acid (ATRA) | Experimental: ATRA + Gemcitabine + Nab-Paclitaxel | Ib | Dose-limiting toxicities & optimum biological dose | Recruiting | NA |
| NCT03331562 | Stage IV pancreatic cancer who have been placed in best possible response | Paricalcitol (a vitamin D receptor agonist) | Experimental: pembrolizumab & paricalcitol Placebo Comparator: pembrolizumab & placebo | II | Percent of progression (by RECIST 1.1) at 6 months | Recruiting | NA |
AG: albumin-bound paclitaxel +gemcitabine; ATRA: all-trans retinoic acid; FAK: focal adhesion kinase; Gem: gemcitabine; GP: gemcitabine + placebo; GV: gemcitabine + vismodegib; HR: hazard ratio; JAK: Janus kinase; PAG: PEGPH20 + nab-paclitaxel + gemcitabine; PEGPH20: PEGylated recombinant human hyaluronidase; PFS: progression-free survival; OS: overall survival.
Several novel experimental results also appear promising. It has been reported that compared to gemcitabine alone, nab-paclitaxel plus gemcitabine significantly improves the prognosis of metastatic PDAC [125]. Nab-paclitaxel is considered to deplete the dense stroma and accumulate chemotherapy drugs in the tumor compartment [126]. A recent study targeted CAFs with a carboxymethylcellulose-docetaxel nanoparticle (CellaxTM-DTX), demonstrating that over 90% of CellaxTM-DTX particles concentrate in SMA+ CAFs and result in long-term degradation of this stromal component, an effect that was not observed with nab-paclitaxel [127]. In addition, compared with the current therapy of gemcitabine or nab-paclitaxel, CellaxTM-DTX treatment increased tumor perfusion and prolonged murine survival in a metastatic model of PDAC [127]. Thus, CellaxTM-DTX may provide a novel strategy in therapeutic reduction of desmoplasia to possibly confine the primary tumor, but whether it will benefit human PDAC therapy remains to be clarified.
As CAFs interact closely with cancer cells to promote tumor progression upon activation, one therapeutic strategy would be to inactivate CAFs. Minnelide, the water-soluble prodrug of triptolide, a diterpene triepoxide from the Chinese plant Tripterygium wilfordii, has been found to effectively revert activated CAFs to an inactive state via downregulation of the TGF-β signaling pathway in vitro, as shown by increased vitamin A-containing lipid droplets, decreased expression of α-SMA, and reduced ECM secretion [128]. Therefore, treatments that inactivate CAF may provide a novel strategy for the treatment of this lethal disease, and a Phase II trial of Minnelide in patients with refractory pancreatic cancer is underway (NCT03117920). Another experimental attempt to support the strategy to induce CAF quiescence is all-trans retinoic acid (ATRA) [129], and ATRA plus gemcitabine and nab-Paclitaxel is currently under a phase Ib clinical trial in patients with pancreatic cancer (NCT03307148).
Some promising results, such as those obtained with PEGPH20 [124], affirm the feasibility of the strategy of depleting stromal components. However, although IPI-926, an SHH pathway inhibitor, achieved promising results in a preclinical study [130], it showed no clinical benefit in clinical trials (NCT01130142 and NCT01383538). Another inhibitor of the SHH pathway, vismodegib, also exhibited no significant effect as part of a combination therapy with gemcitabine in metastatic PDAC in a Phase Ib/II clinical trial [69]. One explanation for these results may be the heterogeneity between human disease and mouse models (Table 1). There is also the possibility that simple depletion of the tumor stroma may result in increased cancer cell spreading. Mathew et al. [75] showed that SHH dosage is a key consideration in antitumor therapy, as reduced levels of SHH signaling evoked a potent angiogenic response. Thus, an angiogenic response owing to a dosage-specific SHH response might partly contribute to the failure of clinical trials (NCT01130142 and NCT01383538) [69, 75]. In addition, simple depletion of the stroma may block the beneficial effects of stromal compartments. Djurec et al. [131] have shown that although high expression of serum amyloid A 1 (SAA1) in the stroma is associated with poorer survival, the ablation of germline Saa3 in PCCs did not inhibit tumor progression of PDAC in mice. They observed that Saa3-competent CAFs stimulated the growth of tumor cells, whereas Saa3-null CAFs inhibited tumor growth in an orthotopic mode. Although depletion of Saa3 in PCCs avoids the tumor-promoting effects of Saa3-competent CAFs, it also blocks the inhibitory impact of Saa3-null CAFs [131]. These findings support the selective blocking concept concerning the heterogeneous functions of CAFs mentioned above.
Under the assumption that some stromal constituents may play a role in restraining tumor progression, stroma remodeling might be a strategy to improve the efficiency of anticancer agents without causing the adverse impacts of stroma depletion. Activation of vitamin D receptors, which are expressed in PSCs, has been revealed as a critical transcriptional regulator of stroma modification, and vitamin D ligand (calcipotriol) plus gemcitabine analog increased intratumoral drug concentration, which resulted in prolonged survival of KPC mice compared to mice treated with gemcitabine alone [132]. Paricalcitol, a vitamin D receptor agonist, has recently been applied along with pembrolizumab, an immune checkpoint inhibitor, to patients with terminal stage PDAC in a phase II clinical trial (NCT03331562). Another study showed that blockage of the STAT3 pathway, in combination with gemcitabine, could enhance the efficacy of drug delivery in a mouse model of pancreatic cancer, thus improving the treatment effect in human patients with pancreatic cancer through stroma modification and downregulation of cytidine deaminase instead of depletion of the tumor stromal compartment [133]. In a Phase II study, ruxolitinib, a JAK2 inhibitor, combined with capecitabine in metastatic pancreatic patients exhibited no overall survival benefit (HR, 0.79; P = .025) but demonstrated improved overall survival (HR, 0.47; P .011) in a subgroup of patients with inflammation (serum C-reactive protein levels higher than the study population median, i.e., 13 mg/L) [134]. However, the subsequent Phase III trial was terminated due to a lack of therapeutic benefit (NCT02119663 and NCT02117479). One reason for the failure of the trial might be the inappropriate therapeutic combination. Altered immune cell infiltration correlated with STAT3 activation in both murine and human PDAC was observed in a recent study [135], and another study reported that a JAK2 inhibitor could decrease the expression of programmed death-ligand 1 (PD-L1) on cancer cells in vitro, which is enhanced by anticancer agents in a dose-dependent manner [136]. A very recent study in murine models confirmed this possible strategy, as preclinical results demonstrated that the inhibition of the JAK activator IL-6 might increase the therapeutic response to anti-PD-L1 treatment in PDAC [137]. Thus, the possible role of these stroma modification effects in immune escape might lead to a potential combination therapy strategy to address the dilemma of immune checkpoint inhibitors [138]. These findings may suggest that further research regarding the design of antistroma treatments in combination with chemotherapy and immunotherapy may be worthwhile.
Conclusions
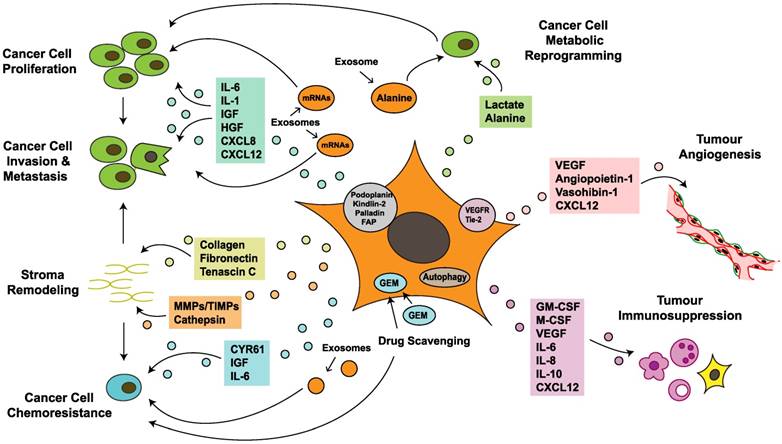
Research into CAFs in PDAC has the potential to improve the prognosis of patients with this lethal tumor. CAFs have a considerable impact on the acquisition of major hallmarks of pancreatic cancer by regulating tumorigenesis, progression, invasion, metastasis, angiogenesis, immunosuppression and chemoresistance (Figure 3). CAFs exhibit several features in tumor-stroma interactions, including creation of an inflammatory environment, modification of the TME and reprogramming of tumor cell metabolism, which further regulate the PCC phenotype and biological behavior. CAFs interact at different levels with PCCs and other TME components via different signaling pathways, paracrine and autocrine mechanisms, direct contact, exosome secretion and autophagy; these processes form a molecular network, making it difficult to elucidate certain biological functions (Figure 3). With recent discussion about whether the stroma is a friend or foe, a reasonable classification strategy for CAF subsets based on distinct biomarkers and functions is needed. A recent study in breast cancer identified four CAF subsets with distinct functions and levels of activation and defined the CAF-S1 fibroblast subset as a pivotal regulator of tumor immunosuppression [139]. This result highlights the selective inhibition concept and the strategy of modifying rather than simply depleting the tumor stroma, which provides a novel direction for studies of pancreatic cancer. Another recent study on breast cancer indicated that the TME is capable of regulating breast cancer subtypes via a PDGF-CC-dependent pathway, which provides promising target ideas for PDAC to reexamine the role of the microenvironment [140]. Although there have been numerous studies on CAFs, the absence of large-scale randomized clinical trials to support these experimental data is apparent. The nonspecific targets of anti-CAF treatments may partly explain the inefficacy of stroma-targeting therapies when applied to routine clinical application. Further original research is needed to elucidate the therapeutic values of these targeted cells and their effects on this lethal disease.
Cancer-associated fibroblasts crosstalk with the tumor microenvironment in pancreatic cancer. Cancer-associated fibroblasts (CAFs) promote cancer cell growth, survival, invasiveness and chemoresistance via paracrine, exosome-mediated or autophagic mechanisms. Some proteins expressed on CAFs also correlate with tumor progression and poor prognosis. In addition, CAFs mediate stromal remodeling, which affects cancer cell migratory tracking and drug delivery. Metabolic reprogramming of CAFs fuels proliferation and renders invasive advantages to pancreatic cancer cells. Furthermore, CAFs also regulate tumor angiogenesis and immunosuppression, which indirectly influence tumor growth and migration. CXCL: chemokine (C-X-C motif) ligand; CYR: cysteine-rich angiogenic inducer; Endo180: urokinase-type plasminogen activator receptor-associated protein; FAP: fibroblast activation protein; GEM: gemcitabine; GM-CSF: granulocyte-macrophage colony-stimulating factor; HGF: hepatocyte growth factor; IL: interleukin; IGF: insulin-like growth factor; miR-210: microRNA-210; M-CSF: macrophage colony-stimulating factor; MMP: matrix metalloproteinases; PDGF: platelet-derived growth factor; TIMP: tissue inhibitor of metalloproteinases; VEGF: vascular endothelial growth factor; VEGFR: vascular endothelial growth factor receptor.
Abbreviations
ACTA2: alpha actin 2; ADAM10: metalloproteinase domain-containing protein 10; α-SMA: alpha-smooth muscle actin; CAF: cancer-associated fibroblast; CCL: chemokine (C-C motif) ligand; CDEs: CAF-derived exosomes; CDMs: cell-derived matrices; CellaxTM-DTX: carboxymethylcellulose-docetaxel nanoparticle; COL11A1: collagen11A1; COX-2: cyclooxygenase 2; CTLs: cytotoxic T lymphocytes; CXCL8: chemokine (C-X-C motif) ligand 8; CYR61: cysteine-rich angiogenic inducer 61; DCs: dendritic cells; EMT: epithelial-to-mesenchymal transition; EV: extracellular vesicle; FAK: focal adhesion kinase; FAP: fibroblast activation protein; FSP1: fibroblast specific protein 1; GAL3: galectin 3; HA: hyaluronan; HGF: hepatocyte growth factor; HR: hazard ratio; hStCs: hepatic stellate cells; HUVECs: human umbilical vein endothelial cells; iCAF: inflammatory cancer associated fibroblasts; IFP: interstitial fluid pressure; IGF-1: insulin-like growth factor 1; IL-6: interleukin 6; IL-8: interleukin 8; IL-1β: interleukin 1β; IL-1R1: interleukin-1 receptor 1; IRAK4: interleukin-1 receptor associated kinase 4; LIF: leukemia inhibitory factor; MDSC: myeloid-derived suppressor cell; MIF: microphage inhibitory factor; miRNAs: microRNAs; MMP: matrix metalloproteinase; MSC: mesenchymal stem cell; NEAA: nonessential amino acid; NK: natural killer; PAK: p21-activated kinase; PCC: pancreatic cancer cell; PDAC: pancreatic ductal adenocarcinoma; PDGF: platelet-derived growth factor; PDGFR: platelet derived growth factor receptor; PD-L1: programmed death-ligand 1; PEGPH20: PEGylated recombinant hyaluronidase; PMA: phorbol 12-myristate 13-acetate; PSC: pancreatic stellate cell; ROS: reactive oxygen species; SAA: serum amyloid A; SHH: sonic hedgehog; TAM: tumor-associated macrophage; TβRII: transforming growth factor beta type II receptor; TCA: tricarboxylic acid; TF: transcription factor; TGF-β: transforming growth factor beta; Th2: T helper type 2; Timp: tissue inhibitor of metalloproteinases; TME: tumor microenvironment; Treg: regulatory T cell; TSLP: thymic stromal lymphopoietin; VEGF: vascular endothelial growth factor; uPA: urokinase plasminogen activator; YAP: Yes-associated protein.
Acknowledgements
This work was supported by the National Natural Science Foundation (81502031, 81772555 and 81602085); the National Science Fund for Distinguished Young Scholars (81625016); and the Shanghai Sailing Program (16YF1401800).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7-30
2. Hidalgo M. Pancreatic cancer. N Engl J Med. 2010;362:1605-17
3. ACS: Atlanta, the United States. Key statistics for pancreatic cancer. Revised 4 January 2018. https://www.cancer.org/cancer/pancreatic-cancer/about/key-statistics.html
4. Erkan M, Michalski CW, Rieder S, Reiser-Erkan C, Abiatari I, Kolb A. et al. The activated stroma index is a novel and independent prognostic marker in pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol. 2008;6:1155-61
5. Neesse A, Algul H, Tuveson DA, Gress TM. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut. 2015;64:1476-84
6. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582-98
7. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871-90
8. Bergfeld SA, DeClerck YA. Bone marrow-derived mesenchymal stem cells and the tumor microenvironment. Cancer Metast Rev. 2010;29:249-61
9. Vonlaufen A, Joshi S, Qu C, Phillips PA, Xu Z, Parker NR. et al. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 2008;68:2085-93
10. McCarroll JA, Phillips PA, Santucci N, Pirola RC, Wilson JS, Apte MV. Vitamin A inhibits pancreatic stellate cell activation: implications for treatment of pancreatic fibrosis. Gut. 2006;55:79-89
11. Micallef L, Vedrenne N, Billet F, Coulomb B, Darby IA, Desmouliere A. The myofibroblast, multiple origins for major roles in normal and pathological tissue repair. Fibrogenesis Tissue Repair. 2012;5:S5
12. Brentnall TA, Lai LA, Coleman J, Bronner MP, Pan S, Chen R. Arousal of cancer-associated stroma: overexpression of palladin activates fibroblasts to promote tumor invasion. PLoS One. 2012;7:e30219
13. Yoshida N, Masamune A, Hamada S, Kikuta K, Takikawa T, Motoi F. et al. Kindlin-2 in pancreatic stellate cells promotes the progression of pancreatic cancer. Cancer Lett. 2017;390:103-14
14. Shindo K, Aishima S, Ohuchida K, Fujiwara K, Fujino M, Mizuuchi Y. et al. Podoplanin expression in cancer-associated fibroblasts enhances tumor progression of invasive ductal carcinoma of the pancreas. Mol Cancer. 2013;12:168
15. Kuzet S-E, Gaggioli C. Fibroblast activation in cancer: when seed fertilizes soil. Cell Tissue Res. 2016;365:607-19
16. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M. et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med. 2017;214:579-96
17. Masamune A, Shimosegawa T. Signal transduction in pancreatic stellate cells. J Gastroenterol. 2009;44:249-60
18. Apte MV, Haber PS, Darby SJ, Rodgers SC, McCaughan GW, Korsten MA. et al. Pancreatic stellate cells are activated by proinflammatory cytokines: implications for pancreatic fibrogenesis. Gut. 1999;44:534-41
19. Massagué J. TGFbeta in Cancer. Cell. 2008;134:215-30
20. Stylianou A, Gkretsi V, Stylianopoulos T. Transforming growth factor-beta modulates pancreatic cancer associated fibroblasts cell shape, stiffness and invasion. Biochim Biophys Acta. 2018;1862:1537-1546
21. Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283-316
22. Elenbaas B, Weinberg RA. Heterotypic signaling between epithelial tumor cells and fibroblasts in carcinoma formation. Exp Cell Res. 2001;264:169-84
23. Dong J, Grunstein J, Tejada M, Peale F, Frantz G, Liang WC. et al. VEGF-null cells require PDGFR alpha signaling-mediated stromal fibroblast recruitment for tumorigenesis. EMBO J. 2004;23:2800-10
24. Cadamuro M, Nardo G, Indraccolo S, Dall'olmo L, Sambado L, Moserle L. et al. Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology. 2013;58:1042-53
25. Masamune A, Nakano E, Hamada S, Takikawa T, Yoshida N, Shimosegawa T. Alteration of the microRNA expression profile during the activation of pancreatic stellate cells. Scand J Gastroenterol. 2014;49:323-31
26. Xiao Q, Zhou D, Rucki AA, Williams J, Zhou J, Mo G. et al. Cancer-associated fibroblasts in pancreatic cancer are reprogrammed by tumor-induced alterations in genomic DNA methylation. Cancer Res. 2016;76:5395-404
27. Roshani R, McCarthy F, Hagemann T. Inflammatory cytokines in human pancreatic cancer. Cancer Lett. 2014;345:157-63
28. Albrengues J, Bourget I, Pons C, Butet V, Hofman P, Tartare-Deckert S. et al. LIF mediates proinvasive activation of stromal fibroblasts in cancer. Cell Rep. 2014;7:1664-78
29. Zhao W, Ajani JA, Sushovan G, Ochi N, Hwang R, Hafley M. et al. Galectin-3 mediates tumor cell-stroma interactions by activating pancreatic stellate cells to produce cytokines via integrin signaling. Gastroenterology. 2018;154:1524-1537
30. Zhang D, Li L, Jiang H, Li Q, Wang-Gillam A, Yu J. et al. Tumor-stroma IL-1beta-IRAK4 feedforward circuitry drives tumor fibrosis, chemoresistance, and poor prognosis in pancreatic cancer. Cancer Res. 2018;78:1700-1712
31. Thayer SP, di Magliano MP, Heiser PW, Nielsen CM, Roberts DJ, Lauwers GY. et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425:851-6
32. Yeo D, Phillips P, Baldwin GS, He H, Nikfarjam M. Inhibition of group 1 p21-activated kinases suppresses pancreatic stellate cell activation and increases survival of mice with pancreatic cancer. Int J Cancer. 2017;140:2101-11
33. Khokha R, Murthy A, Weiss A. Metalloproteinases and their natural inhibitors in inflammation and immunity. Nat Rev Immunol. 2013;13:649-65
34. Shimoda M, Principe S, Jackson HW, Luga V, Fang H, Molyneux SD. et al. Loss of the Timp gene family is sufficient for the acquisition of the CAF-like cell state. Nat Cell Biol. 2014;16:889-901
35. Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI. et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat Cell Biol. 2013;15:637-46
36. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
37. Hartel M, Di Mola FF, Gardini A, Zimmermann A, Di Sebastiano P, Guweidhi A. et al. Desmoplastic reaction influences pancreatic cancer growth behavior. World J Surg. 2004;28:818-25
38. Xu Z, Vonlaufen A, Phillips PA, Fiala-Beer E, Zhang X, Yang L. et al. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am J Pathol. 2010;177:2585-96
39. Masamune A, Kikuta K, Watanabe T, Satoh K, Hirota M, Shimosegawa T. Hypoxia stimulates pancreatic stellate cells to induce fibrosis and angiogenesis in pancreatic cancer. Am J Physiol Gastrointest Liver Physiol. 2008;295:G709-17
40. Guleng B, Tateishi K, Ohta M, Kanai F, Jazag A, Ijichi H. et al. Blockade of the stromal cell-derived factor-1/CXCR4 axis attenuates in vivo tumor growth by inhibiting angiogenesis in a vascular endothelial growth factor-independent manner. Cancer Res. 2005;65:5864-71
41. Liang C, Qin Y, Zhang B, Ji S, Shi S, Xu W. et al. Metabolic plasticity in heterogeneous pancreatic ductal adenocarcinoma. Biochim Biophys Acta. 2016;1866:177-88
42. Tjomsland V, Spångeus A, Välil, Auml J, Sandström P, Borch K. et al. Interleukin 1α sustains the expression of inflammatory factors in human pancreatic cancer microenvironment by targeting cancer-associated fibroblasts. Neoplasia. 2011;13:664-75
43. Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS. et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013;73:3007-18
44. Muerkoster SS, Werbing V, Koch D, Sipos B, Ammerpohl O, Kalthoff H. et al. Role of myofibroblasts in innate chemoresistance of pancreatic carcinoma—epigenetic downregulation of caspases. Int J Cancer. 2008;123:1751-60
45. Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A. et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918-26
46. Vonlaufen A, Phillips PA, Xu Z, Goldstein D, Pirola RC, Wilson JS. et al. Pancreatic stellate cells and pancreatic cancer cells: an unholy alliance. Cancer Res. 2008;68:7707-10
47. Shi M, Yu DH, Chen Y, Zhao CY, Zhang J, Liu QH. et al. Expression of fibroblast activation protein in human pancreatic adenocarcinoma and its clinicopathological significance. World J Gastroenterol. 2012;18:840-6
48. Kawase T, Yasui Y, Nishina S, Hara Y, Yanatori I, Tomiyama Y. et al. Fibroblast activation protein-alpha-expressing fibroblasts promote the progression of pancreatic ductal adenocarcinoma. BMC Gastroenterol. 2015;15:109
49. Lo A, Li CP, Buza EL, Blomberg R, Govindaraju P, Avery D. et al. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight. 2017;2:e92232
50. Mathivanan S, Ji H, Simpson RJ. Exosomes: extracellular organelles important in intercellular communication. J Proteomics. 2010;73:1907-20
51. Takikawa T, Masamune A, Yoshida N, Hamada S, Kogure T, Shimosegawa T. Exosomes derived from pancreatic stellate cells: microRNA signature and effects on pancreatic cancer cells. Pancreas. 2017;46:19-27
52. Masamune A, Yoshida N, Hamada S, Takikawa T, Nabeshima T, Shimosegawa T. Exosomes derived from pancreatic cancer cells induce activation and profibrogenic activities in pancreatic stellate cells. Biochem Biophys Res Commun. 2018;495:71-7
53. Endo S, Nakata K, Ohuchida K, Takesue S, Nakayama H, Abe T. et al. Autophagy is required for activation of pancreatic stellate cells, associated with pancreatic cancer progression and promotes growth of pancreatic tumors in mice. Gastroenterology. 2017;152:1492-506.e24
54. Kadaba R, Birke H, Wang J, Hooper S, Andl CD, Di Maggio F. et al. Imbalance of desmoplastic stromal cell numbers drives aggressive cancer processes. J Pathol. 2013;230:107-17
55. Whatcott CJ, Diep CH, Jiang P, Watanabe A, LoBello J, Sima C. et al. Desmoplasia in primary tumors and metastatic lesions of pancreatic cancer. Clin Cancer Res. 2015;21:3561-8
56. Duda DG, Duyverman AM, Kohno M, Snuderl M, Steller EJ, Fukumura D. et al. Malignant cells facilitate lung metastasis by bringing their own soil. Proc Natl Acad Sci U S A. 2010;107:21677-82
57. Huang RY, Guilford P, Thiery JP. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. J Cell Sci. 2012;125:4417-22
58. Kikuta K, Masamune A, Watanabe T, Ariga H, Itoh H, Hamada S. et al. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem Biophys Res Commun. 2010;403:380-4
59. Wu YS, Chung I, Wong WF, Masamune A, Sim MS, Looi CY. Paracrine IL-6 signaling mediates the effects of pancreatic stellate cells on epithelial-mesenchymal transition via Stat3/Nrf2 pathway in pancreatic cancer cells. Biochim Biophys Acta. 2017;1861:296-306
60. Wang L, Wu H, Wang L, Zhang H, Lu J, Liang Z. et al. Asporin promotes pancreatic cancer cell invasion and migration by regulating the epithelial-to-mesenchymal transition (EMT) through both autocrine and paracrine mechanisms. Cancer Lett. 2017;398:24-36
61. Takikawa T, Masamune A, Hamada S, Nakano E, Yoshida N, Shimosegawa T. miR-210 regulates the interaction between pancreatic cancer cells and stellate cells. Biochem Biophys Res Commun. 2013;437:433-9
62. Erdogan B, Ao M, White LM, Means AL, Brewer BM, Yang L. et al. Cancer-associated fibroblasts promote directional cancer cell migration by aligning fibronectin. J Cell Biol. 2017;216:3799-816
63. Koikawa K, Ohuchida K, Takesue S, Ando Y, Kibe S, Nakayama H. et al. Pancreatic stellate cells reorganize matrix components and lead pancreatic cancer invasion via the function of Endo180. Cancer Lett. 2018;412:143-54
64. Goicoechea SM, Garcia-Mata R, Staub J, Valdivia A, Sharek L, McCulloch CG. et al. Palladin promotes invasion of pancreatic cancer cells by enhancing invadopodia formation in cancer-associated fibroblasts. Oncogene. 2014;33:1265-73
65. Pothula SP, Xu Z, Goldstein D, Merrett N, Pirola RC, Wilson JS. et al. Targeting the HGF/c-MET pathway: stromal remodelling in pancreatic cancer. Oncotarget. 2017;8:76722-39
66. Yang XP, Liu SL, Xu JF, Cao SG, Li Y, Zhou YB. Pancreatic stellate cells increase pancreatic cancer cells invasion through the hepatocyte growth factor /c-Met/survivin regulated by P53/P21. Exp Cell Res. 2017;357:79-87
67. Rucki AA, Foley K, Zhang P, Xiao Q, Kleponis J, Wu AA. et al. Heterogeneous stromal signaling within the tumor microenvironment controls the metastasis of pancreatic cancer. Cancer Res. 2017;77:41-52
68. Kundranda M, Kachaamy T. Promising new therapies in advanced pancreatic adenocarcinomas. Future Oncol. 2014;10:2629-41
69. Catenacci DVT, Junttila MR, Karrison T, Bahary N, Horiba MN, Nattam SR. et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J Clin Oncol. 2015;33:4284-92
70. Beckermann BM, Kallifatidis G, Groth A, Frommhold D, Apel A, Mattern J. et al. VEGF expression by mesenchymal stem cells contributes to angiogenesis in pancreatic carcinoma. Br J Cancer. 2008;99:622-31
71. Patel MB, Pothula SP, Xu Z, Lee AK, Goldstein D, Pirola RC. et al. The role of the hepatocyte growth factor/c-MET pathway in pancreatic stellate cell-endothelial cell interactions: antiangiogenic implications in pancreatic cancer. Carcinogenesis. 2014;35:1891-900
72. Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998;393:595-9
73. Tachibana K, Hirota S, Iizasa H, Yoshida H, Kawabata K, Kataoka Y. et al. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature. 1998;393:591-4
74. Matsuo Y, Ochi N, Sawai H, Yasuda A, Takahashi H, Funahashi H. et al. CXCL8/IL-8 and CXCL12/SDF-1alpha co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int J Cancer. 2009;124:853-61
75. Mathew E, Zhang Y, Holtz AM, Kane KT, Song JY, Allen BL. et al. Dosage-dependent regulation of pancreatic cancer growth and angiogenesis by hedgehog signaling. Cell Rep. 2014;9:484-94
76. Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA. et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735-47
77. Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR. et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719-34
78. Martinez-Bosch N, Vinaixa J, Navarro P. Immune evasion in pancreatic cancer: from mechanisms to therapy. Cancers. 2018;10:6
79. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA. et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22:851-60
80. Shalapour S, Karin M. Immunity, inflammation, and cancer: an eternal fight between good and evil. J Clin Invest. 2015;125:3347-55
81. Shi C, Washington MK, Chaturvedi R, Drosos Y, Revetta FL, Weaver CJ. et al. Fibrogenesis in pancreatic cancer is a dynamic process regulated by macrophage-stellate cell interaction. Lab Invest. 2014;94:409-21
82. Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010;31:220-7
83. Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS. et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110:20212-7
84. Kuen J, Darowski D, Kluge T, Majety M. Pancreatic cancer cell/fibroblast co-culture induces M2 like macrophages that influence therapeutic response in a 3D model. PLoS One. 2017;12:e0182039
85. Zhang A, Qian Y, Ye Z, Chen H, Xie H, Zhou L. et al. Cancer-associated fibroblasts promote M2 polarization of macrophages in pancreatic ductal adenocarcinoma. Cancer Med. 2017;6:463-70
86. Liu CY, Xu JY, Shi XY, Huang W, Ruan TY, Xie P. et al. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells, partially through TLR4/IL-10 signaling pathway. Lab Inves. 2013;93:844-54
87. Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A. et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol. 2016;18:549-60
88. von Bernstorff W, Voss M, Freichel S, Schmid A, Vogel I, Johnk C. et al. Systemic and local immunosuppression in pancreatic cancer patients. Clin Cancer Res. 2001;7:925s-32s
89. Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC. et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145:1121-32
90. De Monte L, Reni M, Tassi E, Clavenna D, Papa I, Recalde H. et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. J Exp Med. 2011;208:469-78
91. Tang D, Yuan Z, Xue X, Lu Z, Zhang Y, Wang H. et al. High expression of Galectin-1 in pancreatic stellate cells plays a role in the development and maintenance of an immunosuppressive microenvironment in pancreatic cancer. Int J Cancer. 2012;130:2337-48
92. Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E. et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015;75:544-53
93. Chauhan VP, Boucher Y, Ferrone CR, Roberge S, Martin JD, Stylianopoulos T. et al. Compression of pancreatic tumor blood vessels by hyaluronan is caused by solid stress and not interstitial fluid pressure. Cancer Cell. 2014;26:14-5
94. Shan T, Chen S, Chen X, Lin WR, Li W, Ma J. et al. Cancer-associated fibroblasts enhance pancreatic cancer cell invasion by remodeling the metabolic conversion mechanism. Oncol Reps. 2017;37:1971-9
95. Tape CJ, Ling S, Dimitriadi M, McMahon KM, Worboys JD, Leong HS. et al. Oncogenic KRAS regulates tumor cell signaling via stromal reciprocation. Cell. 2016;165:1818
96. Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I. et al. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Sign. 2012;16:1264-84
97. Yoshida GJ. Metabolic reprogramming: the emerging concept and associated therapeutic strategies. J Exp Clin Cancer Res. 2015;34:111
98. Hirakawa T, Yashiro M, Doi Y, Kinoshita H, Morisaki T, Fukuoka T. et al. Pancreatic fibroblasts stimulate the motility of pancreatic cancer cells through IGF1/IGF1R signaling under hypoxia. PLoS One. 2016;11:e0159912
99. Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L. et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536:479-83
100. Zhao H, Yang L, Baddour J, Achreja A, Bernard V, Moss T. et al. Tumor microenvironment derived exosomes pleiotropically modulate cancer cell metabolism. eLife. 2016;5:e10250
101. Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21:418-29
102. Pries AR, Hopfner M, le Noble F, Dewhirst MW, Secomb TW. The shunt problem: control of functional shunting in normal and tumour vasculature. Nat Rev Cancer. 2010;10:587-93
103. Hesler RA, Huang JJ, Starr MD, Treboschi VM, Bernanke AG, Nixon AB. et al. TGF-beta-induced stromal CYR61 promotes resistance to gemcitabine in pancreatic ductal adenocarcinoma through downregulation of the nucleoside transporters hENT1 and hCNT3. Carcinogenesis. 2016;37:1041-51
104. Edgar LT, Underwood CJ, Guilkey JE, Hoying JB, Weiss JA. Extracellular matrix density regulates the rate of neovessel growth and branching in sprouting angiogenesis. PLoS One. 2014;9:e85178
105. Koay EJ, Truty MJ, Cristini V, Thomas RM, Chen R, Chatterjee D. et al. Transport properties of pancreatic cancer describe gemcitabine delivery and response. J Clin Invest. 2014;124:1525-36
106. DuFort CC, DelGiorno KE, Carlson MA, Osgood RJ, Zhao C, Huang Z. et al. Interstitial pressure in pancreatic ductal adenocarcinoma is dominated by a gel-fluid phase. Biophys J. 2016;110:2106-19
107. Rice AJ, Cortes E, Lachowski D, Cheung BCH, Karim SA, Morton JP. et al. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis. 2017;6:e352
108. Hessmann E, Patzak MS, Klein L, Chen N, Kari V, Ramu I. et al. Fibroblast drug scavenging increases intratumoural gemcitabine accumulation in murine pancreas cancer. Gut. 2018;67:497-507
109. Ireland L, Santos A, Ahmed MS, Rainer C, Nielsen SR, Quaranta V. et al. Chemoresistance in pancreatic cancer is driven by stroma-derived insulin-like growth factors. Cancer Res. 2016;76:6851-63
110. Long KB, Tooker G, Tooker E, Luque SL, Lee JW, Pan X. et al. IL6 receptor blockade enhances chemotherapy efficacy in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2017;16:1898-908
111. Duluc C, Moatassim-Billah S, Chalabi-Dchar M, Perraud A, Samain R, Breibach F. et al. Pharmacological targeting of the protein synthesis mTOR/4E-BP1 pathway in cancer-associated fibroblasts abrogates pancreatic tumour chemoresistance. EMBO Mol Med. 2015;7:735-53
112. Richards KE, Zeleniak AE, Fishel ML, Wu J, Littlepage LE, Hill R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene. 2017;36:1770-8
113. Stoker M, Macpherson I. Syrian hamster fibroblast cell line BHK21 and its derivatives. Nature. 1964;203:1355-7
114. Dvorak HF. Tumors: wounds that do not heal. N Engl J Med. 1986;315:1650-9
115. Park H, Lee Y, Lee H, Kim JW, Hwang JH, Kim J. et al. The prognostic significance of cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Tumour Biol. 2017;39:1010428317718403
116. Fujiwara K, Ohuchida K, Mizumoto K, Shindo K, Eguchi D, Kozono S. et al. CD271(+) subpopulation of pancreatic stellate cells correlates with prognosis of pancreatic cancer and is regulated by interaction with cancer cells. PLoS One. 2012;7:e52682
117. Ikenaga N, Ohuchida K, Mizumoto K, Cui L, Kayashima T, Morimatsu K. et al. CD10+ pancreatic stellate cells enhance the progression of pancreatic cancer. Gastroenterology. 2010;139:1041-51 51.e1-8
118. Kanno A, Satoh K, Masamune A, Hirota M, Kimura K, Umino J. et al. Periostin, secreted from stromal cells, has biphasic effect on cell migration and correlates with the epithelial to mesenchymal transition of human pancreatic cancer cells. Int J Cancer. 2008;122:2707-18
119. Moffitt RA, Marayati R, Flate EL, Volmar KE, Loeza SG, Hoadley KA. et al. Virtual microdissection identifies distinct tumor-and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015;47:1168-78
120. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328-37
121. Leca J, Martinez S, Lac S, Nigri J, Secq V, Rubis M. et al. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J Clin Invest. 2016;126:4140-56
122. Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK. et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62:112-20
123. Hingorani SR, Harris WP, Beck JT, Berdov BA, Wagner SA, Pshevlotsky EM. et al. Phase Ib study of PEGylated recombinant human hyaluronidase and gemcitabine in patients with advanced pancreatic cancer. Clin Cancer Res. 2016;22:2848-54
124. Hingorani SR, Zheng L, Bullock AJ, Seery TE, Harris WP, Sigal DS. et al. HALO 202: randomized phase II study of PEGPH20 plus nab-paclitaxel/gemcitabine versus nab-paclitaxel/gemcitabine in patients with untreated, metastatic pancreatic ductal adenocarcinoma. J Clin Oncol. 2018;36:359-66
125. Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M. et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369:1691-703
126. Alvarez R, Musteanu M, Garcia-Garcia E, Lopez-Casas PP, Megias D, Guerra C. et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Br J Cancer. 2013;109:926-33
127. Ernsting MJ, Hoang B, Lohse I, Undzys E, Cao P, Do T. et al. Targeting of metastasis-promoting tumor-associated fibroblasts and modulation of pancreatic tumor-associated stroma with a carboxymethylcellulose-docetaxel nanoparticle. J Control Release. 2015;206:122-30
128. Dauer P, Zhao X, Gupta VK, Sharma NS, Kesh K, Gnamlin P. et al. Inactivation of cancer-associated-fibroblasts disrupts oncogenic signaling in pancreatic cancer cells and promotes its regression. Cancer Res. 2018;78:1321-1333
129. Froeling FE, Feig C, Chelala C, Dobson R, Mein CE, Tuveson DA. et al. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology. 2011;141:1486-97 97.e1-14
130. Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D. et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457-61
131. Djurec M, Grana O, Lee A, Troule K, Espinet E, Cabras L. et al. Saa3 is a key mediator of the protumorigenic properties of cancer-associated fibroblasts in pancreatic tumors. Proc Natl Acad Sci U S A. 2018;115:E1147-e56
132. Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H. et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80-93
133. Nagathihalli NS, Castellanos JA, Shi C, Beesetty Y, Reyzer ML, Caprioli R. et al. Signal transducer and activator of transcription 3, mediated remodeling of the tumor microenvironment results in enhanced tumor drug delivery in a mouse model of pancreatic cancer. Gastroenterology. 2015;149:1932-43.e9
134. Hurwitz HI, Uppal N, Wagner SA, Bendell JC, Beck JT, Wade SM 3rd. et al. Randomized, double-blind, phase II study of ruxolitinib or placebo in combination with capecitabine in patients with metastatic pancreatic cancer for whom therapy with gemcitabine has failed. J Clin Oncol. 2015;33:4039-47
135. Wormann SM, Song L, Ai J, Diakopoulos KN, Kurkowski MU, Gorgulu K. et al. Loss of p53 function activates JAK2-STAT3 signaling to promote pancreatic tumor growth, stroma modification, and gemcitabine resistance in mice and is associated with patient survival. Gastroenterology. 2016;151:180-93.e12
136. Doi T, Ishikawa T, Okayama T, Oka K, Mizushima K, Yasuda T. et al. The JAK/STAT pathway is involved in the upregulation of PD-L1 expression in pancreatic cancer cell lines. Oncol Rep. 2017;37:1545-54
137. Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S. et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut. 2018;67:320-32
138. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P. et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366:2455-65
139. Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M. et al. Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell. 2018;33:463-79.e10
140. Roswall P, Bocci M, Bartoschek M, Li H, Kristiansen G, Jansson S. et al. Microenvironmental control of breast cancer subtype elicited through paracrine platelet-derived growth factor-CC signaling. Nat Med. 2018;24:463-473
Author contact
![]() Corresponding authors: Xianjun Yu, MD, PhD, Department of Pancreatic Surgery, Shanghai Cancer Center, Fudan University, No. 270 DongAn Road, Shanghai, P. R. China, Postal code: 200032. Tel: +86-021-64175590 ext 1307, Fax: +86-021-64031446. E-mail: yuxianjunorg Jin Xu, Department of Pancreatic Surgery, Fudan University Shanghai Cancer Center, Shanghai 200032, China; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, China; Pancreatic Cancer Institute, Fudan University, Shanghai 200032, China; Shanghai Pancreatic Cancer Institute, Shanghai 200032, China. Tel: +86-21-64175590, Fax: +86-21-64031446. E-mail: xujinorg.
Corresponding authors: Xianjun Yu, MD, PhD, Department of Pancreatic Surgery, Shanghai Cancer Center, Fudan University, No. 270 DongAn Road, Shanghai, P. R. China, Postal code: 200032. Tel: +86-021-64175590 ext 1307, Fax: +86-021-64031446. E-mail: yuxianjunorg Jin Xu, Department of Pancreatic Surgery, Fudan University Shanghai Cancer Center, Shanghai 200032, China; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, China; Pancreatic Cancer Institute, Fudan University, Shanghai 200032, China; Shanghai Pancreatic Cancer Institute, Shanghai 200032, China. Tel: +86-21-64175590, Fax: +86-21-64031446. E-mail: xujinorg.