Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(18):4995-5011. doi:10.7150/thno.26627 This issue Cite
Research Paper
Novel hybrid molecule overcomes the limited response of solid tumours to HDAC inhibitors via suppressing JAK1-STAT3-BCL2 signalling
Zhi Huang1#, Wei Zhou1#, Yongtao Li1, Mei Cao 1, Tianqi Wang 1, Yakun Ma 1, Qingxiang Guo 1, Xin Wang1, Chao Zhang1, Chenglan Zhang1, Wenzhi Shen6, Yanhua Liu2, Yanan Chen2, Jianyu Zheng5, Shengyong Yang4, Yan Fan1,2,3 ![]() , Rong Xiang1,2,3
, Rong Xiang1,2,3 ![]()
1. Department of Medicinal Chemistry, School of Medicine, Nankai University, Tianjin 300071, China
2. State Key Laboratory of Medicinal Chemical Biology, Nankai University, Tianjin 300071, China
3. 2011 Project Collaborative Innovation Center for Biotherapy of Ministry of Education, Tianjin 300071, China
4. State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Collaborative Innovation Center for Biotherapy and Sichuan University, Chengdu 610041, China
5. State Key Laboratory and Institute of Elemento-Organic Chemistry, Collaborative Innovation Center of Chemical Science and Engineering, Nankai University, Tianjin 300071, China
6. Department of Pathology and Institute of Precision Medicine, Jining Medical University, Jining 272067, China.
# Zhi Huang and Wei Zhou contributed equally to this work.
Received 2018-4-11; Accepted 2018-9-8; Published 2018-10-5
Abstract

Despite initial progress in preclinical models, most known histone deacetylase inhibitors (HDACis) used as a single agent have failed to show clinical benefits in nearly all types of solid tumours. Hence, the efficacy of HDACis in solid tumours remains uncertain. Herein, we developed a hybrid HDAC inhibitor that sensitized solid tumours to HDAC-targeted treatment.
Methods: A hybrid molecule, Roxyl-zhc-84 was designed and synthesized with novel architecture. The pharmacokinetics and toxicity of Roxyl-zhc-84 were analysed. The antitumour effects of Roxyl-zhc-84 on solid tumours were investigated by assessing cell growth, apoptosis and cell cycle in vitro and in three in vivo mouse models and compared to those of corresponding control inhibitors alone or in combination. Gene set enrichment analysis was performed, and relevant JAK1-STAT3-BCL2 signalling was identified in vitro and in vivo in mechanistic studies.
Results: Roxyl-zhc-84 showed excellent pharmacokinetics and low toxicity. The novel hybrid inhibitor Roxyl-zhc-84 induced cell apoptosis and G1-phase arrest in breast cancer and ovarian cancer cell lines. In three mouse models, oral administration of Roxyl-zhc-84 led to significant tumour regression without obvious toxicity. Moreover, Roxyl-zhc-84 dramatically improved the limited response of traditional HDAC inhibitors in solid tumours via overcoming JAK1-STAT3-BCL2-mediated drug resistance. Roxyl-zhc-84 treatment exhibited vastly superior efficacy than the combination of HDAC and JAK1 inhibitors both in vitro and in vivo.
Conclusion: Concurrent inhibition of HDAC and CDK using Roxyl-zhc-84 with additional JAK1 targeting resolved the limited response of traditional HDAC inhibitors in solid tumours via overcoming JAK1-STAT3-BCL2-mediated drug resistance, providing a rational multi-target treatment to sensitize solid tumours to HDACi therapy.
Keywords: HDAC1, CDK4/6, JAK1, inhibitor, solid tumour
Introduction
Cancer is a complex multigenic disease involving complicated signalling networks and multiple cellular pathways [1]. This characteristic of cancer offers an opportunity for drug combination treatments that target two or more pathways. Hybrid molecules, which are made using two different molecular units from independently acting drugs with different pharmacological effects, have recently risen in significance [2, 3]. Strategies using hybrid molecules avoid the use of expensive and time-consuming optimization trials in determine the best drug combinations. These hybrid molecules display enhanced pharmacological properties, are of novel architecture and have been applied as a standard treatment for cancer to overcome drug resistance, reduce side effects and so on [4].
Histone deacetylases (HDACs) regulate chromatin remodelling, gene transcription, and the activity of partner proteins, controlling cell growth, cell cycle regulation, DNA mismatch repair, proliferation and apoptosis [5, 6]. HDAC overexpression has been identified in a variety of human cancers, including haematological neoplasms and solid tumours. Association of HDACs with oncogenic DNA-binding fusion proteins and other repressive transcription factors constitutively restrains the expression of specific tumour suppressor genes. Therefore, HDACs represent a rational target for cancer treatment. As a result, the use of HDACis in cancer therapy is an area of active investigation.
HDACis selectively alter gene transcription and represent the first successful epigenetic-based cancer therapy [7, 8]. Several HDACis have been approved for cancer treatment by the FDA and have been shown to have clinical benefits in multi-type haematological malignancies [9-11]. Vorinostat is approved for the treatment of cutaneous T-cell lymphoma (CTCL). Romidepsin is approved for the treatment of CTCL and peripheral T-cell lymphoma (PTCL) [12]. Belinostat is approved for the treatment of relapsed or refractory PTCL, and panobinostat is approved for the treatment of multiple myeloma (MM) [13]. Despite some progress in clinical trials, most known HDACis have failed to show clinical benefits in nearly all types of solid tumours, such as breast cancer, renal cancer and prostate cancer [11, 14-16]. Hence, the efficacy of HDACis in solid tumours remains unsatisfying [17, 18], such as for the approved pan-HDAC inhibitor vorinostat. Meanwhile, a lack of solid tumour uptake and extensive tissue distribution has resulted in clinical ineffectiveness and off-target effects, such as myelosuppression, fatigue, and cardiac toxicity [19, 20]. The synergistic effects of HDAC inhibitors with other antitumour agents is an ideal strategy to use multifunctional inhibitors to simultaneously interact with multiple targets with high potency and acceptable toxicity. Recent progress has shown success in combining the HDAC pharmacophore with other enzyme inhibitor activities, such as kinases [21-24].
To overcome the weakness of HDACis in solid tumours, we proposed a hybrid molecule strategy [25]. Activity recognition is a key property for hybrid molecules [26, 27], so to identify synergistic combinations [28] with HDACis, we examined our selective inhibitors. From these experiments, we determined that vorinostat [29], a pan-HDAC inhibitor, was significantly synergistic with abemaciclib [30], a CDK4/6 inhibitor [31] (Figure S1). Cyclin-dependent kinases (CDKs) are protein kinases involved in important cellular processes, such as cell cycle or transcription regulation. The first gap phase, G1, of the cell cycle comprises important preliminary steps for the initiation of DNA replication and subsequent cell division in human embryogenesis and homeostasis, as well as in cancer development. In particular, CDK4/6 functions as a critical activator of G1 phase progression. The aim of therapeutic CDK4/6 targeting in cancer cells is to stop cell proliferation and restore cell cycle arrest in G1 phase, while minimizing or avoiding adverse side effects for cancer patients.
In this context, we designed and synthesized a series of novel hybrid compounds, including two essential pharmacophoric elements for HDAC and CDK, to identify combination solutions and augment response of HDACis in solid tumours [32]. As indicated by the suppression of HDAC and CDK, the hybrid molecule Roxyl-zhc-84 exhibits remarkable antitumour activity both in vitro and in vivo (Figure S1). Insight into the mechanism of this novel hybrid inhibitor solution revealed potent induction of apoptosis via overcoming JAK1-STAT3-BCL2-mediated drug resistance.
Methods
Cell lines and culture conditions
Murine and human cancer cell lines (4T1, MDA-MB-231, MDA-MB-468, SK-OV-3, OVCAR-5 and H460) were obtained from the American Type Culture Collection (ATCC). All cell lines were recently authenticated by cellular morphology and short tandem repeat analysis at Microread Inc. (Beijing, China) according to guidelines from the ATCC. MDA-MB-231 and MDA-MB-468 cells were cultured in Leibovitz's L-15 medium (Biological Industries, Israel) at 37 °C in a 0.2% CO2 incubator. 4T1, SK-OV-3, OVCAR-5 and H460 cells were cultured in RPMI 1640 medium at 37 °C in a 5% CO2 incubator. All medium was supplemented with 10% fetal bovine serum (FBS), 100 unit/mL penicillin and 100 μg/mL streptomycin.
Immunoblotting analysis
Immunoblotting analysis was conducted as previously described [33]. Compound-treated cells were resuspended in RIPA lysis buffer. After centrifugation at 16,200 g for 10 min at 4 °C, supernatants were collected in a pre-cooled microfuge tube. Protein concentration was determined by the BCA Protein Assay. Equal amounts of protein were loaded into 10% SDS-PAGE, and proteins were further transferred onto PVDF membranes after electrophoresis. Membranes were blocked with 5% FBS in TBST for 1 h at room temperature followed by incubation with primary antibodies, including antibodies against CDK4, CDK6, β-actin (Santa Cruz Biotechnology, sc-23896, sc-7961, sc-8432), BCL-2 (BD Biosciences, 560637), cyclin D1, p-Rb, Rb, Ac-Histone H3, caspase-3, p-JAK1, JAK1, p-STAT3 and STAT3 (Cell Signaling Technology, CST# 2978, 8516, 9309, 8173, 9662, 3331, 3332, 9145, 4904) overnight at 4 °C. Membranes were washed with TBST three times followed by incubation in HRP-labelled secondary antibodies for 1 h at room temperature. Signal was detected using Millipore Immobilon ECL reagent.
Protein signal density was quantified using ImageJ software (Version 1.60, National Institutes of Health, USA) and subsequently labelled under the bands. All lanes were normalized by dividing the density by the density of β-actin.
Cell cycle assay
Cells were cultured in 6-wells plates with compounds at different concentrations for 48 h. Cells were then digested, harvested and fixed in 75% ice-cold ethanol, and stored at 4 °C for 24 h. After fixation, cells were resuspended in 1 mL of PBS containing 50 μg/mL of propidium iodide (PI) and 50 μg/mL of RNase A. PI-stained cells were analysed using BD Biosciences FACS Calibur (BD, San Jose, CA, USA), and data analysis for the population of cells in G1, S and G2/M phase of the cell cycle was performed using FlowJo 7.6.1 software.
Apoptosis analysis
Cells were treated with compounds for 48 h before staining. Cells were then harvested and stained with PI staining buffer (50 μg/mL PI in PBS) to determine apoptotic stages. To evaluate early stages of apoptosis, treated cells were stained with FITC labelled Annexin V in binding buffer for 15 min at room temperature, avoiding light exposure. Data were analysed with a FACS Calibur flow cytometer.
Drug treatment in vivo
Animal studies were conducted under the approval of the Experimental Animal Management Committee of Nankai University. Tumour bearing mice were randomized into groups, and dosing began when the average tumour volume reached 50-100 mm3. Roxyl-zhc-84 (15-60 mg/kg), vorinostat (60 mg/kg), abemaciclib (60 mg/kg), and filgotinib (60 mg/kg) were given orally daily for the number of days indicated. For combination treatment, two drugs were given concurrently. Tumour growth was monitored by measurement of tumour size using callipers twice per week using the formula (length×width2)/2. Mice were euthanized after the last drug dose, and tumour tissues were collected and prepared for H&E or immunohistochemistry staining.
Immunohistochemistry staining
Histological analysis was conducted as previously described [34]. Briefly, tumours were harvested, freed from soft tissue and fixed in 4% PFA. After the tissue was embedded, paraffin blocks were sectioned into 5 μm-thick slices. Tissue sections were incubated with primary antibodies against Ki67 (1:100, Abcam ab15580), BCL-2 (1:100, BD Biosciences, 560637), p-STAT3 (1:200, CST 9145) or STAT3 (1:1000, CST 4904) diluted in 5% goat serum in 0.1% PBST at 4 °C overnight. Antibody detection was achieved using a biotinylated secondary antibody followed by addition of avidin-peroxidase and developed with 3, 3'-diaminobenzidine hydrochloride (DAB; ZSGB-Bio ZLI-9017, China). Secondary antibodies were purchased from ZSGB-Bio (Peroxidase-conjugated goat anti-rabbit IgG, ZB-2301). For microscopic evaluation, an Olympus microscope was used.
Synergistic effect analysis
Combination index (CI) was calculated as follows:
CI = ( (cA + cB) - cA × cB ) / cAB
where cA represents the inhibitory rate of compound A, cB represents the inhibitory rate of compound B, and cAB represents the inhibitory rate of combination treatment of compound A and B. Synergy is defined as CI lower than 1.0, and antagonism is defined as CI significantly greater than 1.0.
Assessment of pharmacokinetic properties
Pharmacokinetic analysis of Roxyl-zhc-84 was conducted in male Sprague-Dawley rats (Chinese Academy of Medical Science, Beijing, China). Briefly, catheters were surgically placed into the jugular veins of rats to collect serial blood samples. Roxyl-zhc-84 was dissolved in saline with 5% (v/v) DMSO. Animals were administered a single dose of 10 mg/kg Roxyl-zhc-84 by intravenous injection or oral injection after fasting overnight. Blood was collected and centrifuged immediately to isolate plasma, and plasma concentrations were determined using high-performance liquid chromatography (HPLC) analysis on a Shimadzu Prominence-i LC-2030C 3D system and were identified by tandem mass spectrometric detection (3200 QTRAP system, Applied Biosystems). Noncompartmental pharmacokinetic parameters were fitted using DAS software (Enterprise, version 2.0, Mathematical Pharmacology Professional Committee of China).
Docking studies
Since there is no 3D structure of the CDK4-ligand complex reported at present, the structure of CDK6 in complex with an inhibitor (PDB entry: 4EZ5) was used as a template for homology modelling. MODELLER [35] in the Discovery Studio (Accelrys Inc., San Diego, CA, USA) (DS) was employed for homology modelling. The Roxyl-zhc-84 compound was docked to CDK4, HDAC1 (PDB code: 4BKX) and JAK1 (PDB code: 4EHZ) by GOLD (version 5.0). Hydrogen atoms were added to proteins using Discovery Studio 3.1. GoldScore was selected as the scoring function, and other parameters were set as default. Images were created using PyMOL [36].
Bioinformatics analysis
Gene set enrichment analysis (GSEA) was performed according to the authors' guidelines published on the Broad Institute web pages (http://www.broadinstitute.org/gsea/index.jsp) using KEGG_JAK_STAT gene set using Signal2Noise ratios. Data (GSE53603, GSE27226) was acquired on GEO database.
Statistical analysis
Data are represented as the mean ± s.d. All statistical analysis was performed using GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA). Unpaired Student's t-test was used for comparisons between two different groups. p<0.05 was considered as significant. * represents p<0.05; ** represents p<0.005; *** represents p<0.001.
Results
Roxyl-zhc-84 is the most potent antitumour compound among the derivatives
Based on the synergistic experimental results with mean CI < 1 across relevant concentrations of both vorinostat and abemaciclib in vitro and in vivo (Figure S1), in our hybrid strategy, we merged two different pharmacophores to link two inhibitors together with neither a long chain nor high molecular weight. Vorinostat has been reported to bind strongly to the zinc cation or monodentate group, which plays a critical role in chelation to zinc ions for HDAC inhibitors. Analysing the X-ray co-crystal structure of CDK4/6 and abemaciclib, we found that the 2-aminopyrimidine group interacted with the hinge region, as its core CDK4/6-binding scaffold provided a suitable moiety. Thus, we rationally designed and synthesized a series of merged molecules (Scheme 1-3 and synthesis of compounds in Supplementary Material) [37, 38] including these two essential pharmacophoric elements.
A general full description of the synthesis of the final compounds. Reagents and conditions: (a) EDCI, DMAP, DIEA, rt, 12 h; (b) Pd(AcO)2, BINAP, Cs2CO3, 100 °C, 12 h; (c) CH3OH, reflux, 18 h.
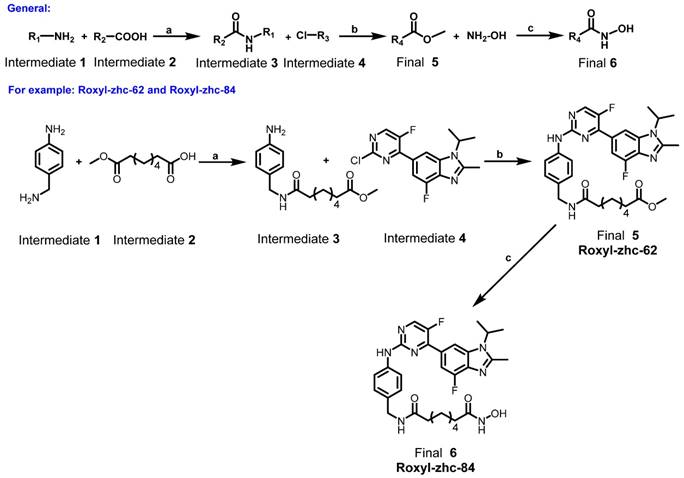
Synthesis of the intermediate aniline reagents 24-50. Reagents and conditions: (a) EDCI, DMAP, DIEA, rt, 12 h, 53% yield; (b) con HCl, THF, 66% yield; (c) SOCl2, pyridine, THF, 80 °C, 6 h, 10% yield; (d) Pd/C, ammonium formate, EtOH, 80 °C, 12 h, 88% yield; (e) EDCI, HOBt, DIEA, rt, 12 h, 39-87% yield.
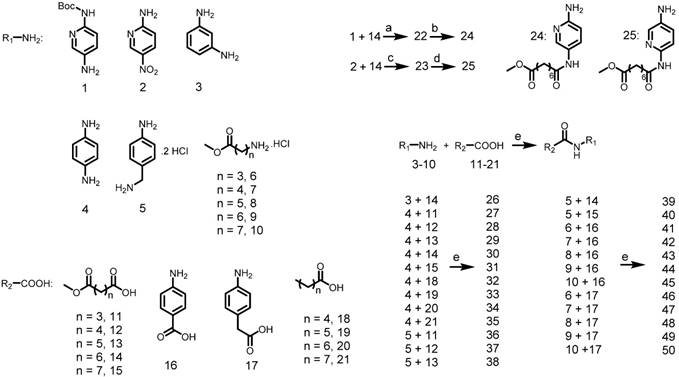
Preparation of final compounds Roxyl-zhc-52-101. Reagents and conditions: (a) Pd(AcO)2, BINAP, Cs2CO3, 100 °C, 12 h, 46-89% yield; (b) NH2OH, CH3OH, reflux, 18 h, 32-76% yield; (c) NaOH, THF/CH3OH/H2O, rt, 12 h, 93% yield.
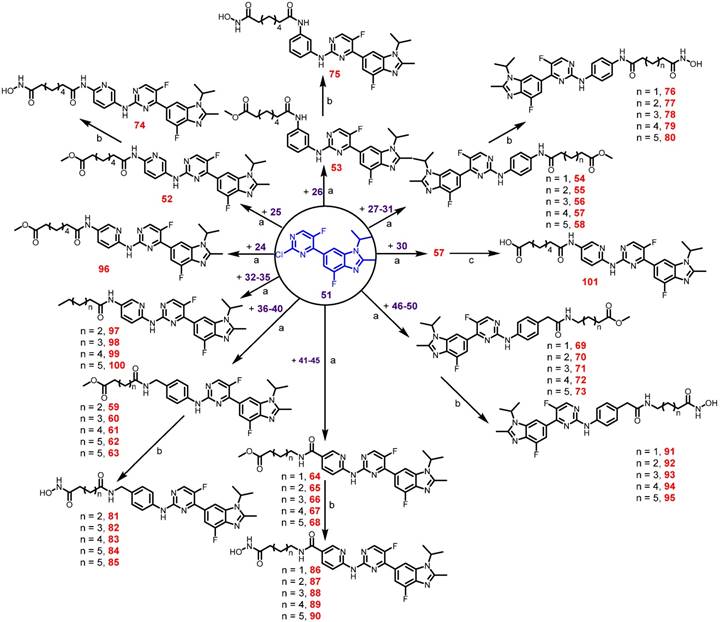
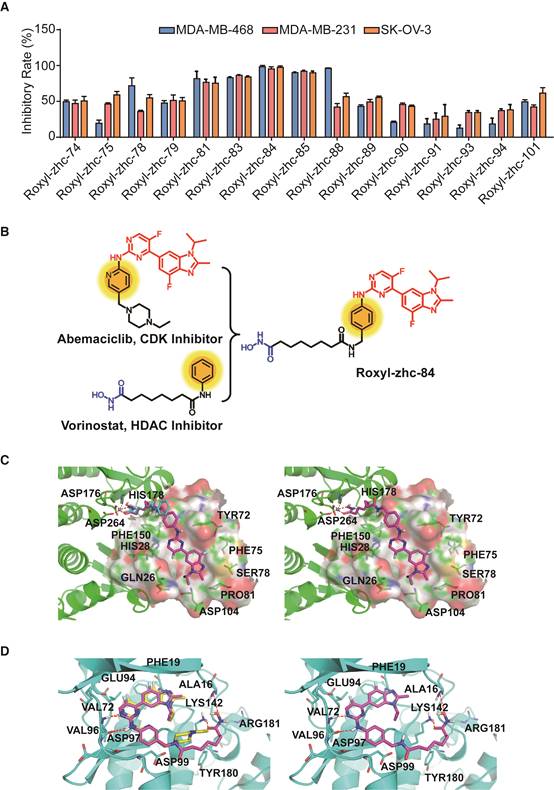
First, we used in vitro enzymatic assays to examine the inhibition of compounds on HDAC1 and CDK4 activity. Fifteen compounds (74, 75, 78, 79, 81, 83-85, 88-91, 93, 94 and 101) inhibited both HDAC1 and CDK4 activity with IC50 values in the nanomolar range, performing better than or comparable to those of positive inhibitors (Table S1). Structure-activity relationship (SAR) studies are summarized in Supplementary Material. In addition to screening for enzyme inhibition activity, we preliminarily screened the ability of these dual-acting compounds to inhibit proliferation in a panel of cancer cell lines using standard CCK8 assay. Of the 15 derivatives, Roxyl-zhc-81, 83, 84, 85 and 88 exhibited potent cytotoxicity in three cancer cell lines (Figure 1A). We further tested the inhibitory rates of the selected candidate compounds on six cancer cell lines (Table S2). Compound Roxyl-zhc-84 displayed superior activity on all the solid cancer cell lines. Because the main purpose of our research was to identify dual inhibitors for solid tumour therapy, we focused our attention on identifying target compounds with potent activities against solid tumour cell lines. Therefore, we performed all subsequent in vitro and in vivo biological studies using Roxyl-zhc-84 (Figure 1B). To obtain evidence for HDAC isoform selectivity, the inhibitory activity of compound Roxyl-zhc-84 was examined against recombinant HDAC1-HDAC11 (Table S3 and Figure S2). Roxyl-zhc-84 displayed much better selectivity than vorinostat among the human HDAC superfamily. The results indicated that the structural design of our compound improved HDAC isoform selectivity. We further tested Roxyl-zhc-84 in a kinase panel composed of 170 kinases from across the kinome (Table S4). Except for CDK, JAKs were the most strongly affected off-target family. Therefore, Roxyl-zhc-84 is a multi-kinase inhibitor acting against HDAC and CDK, with the additional benefit of targeting JAKs. Of note, Roxyl-zhc-84 did not show obvious activity against other kinases, indicating good kinase selectivity for this compound. Molecular docking studies illustrated predicted binding modes and detailed protein-inhibitor interactions of Roxyl-zhc-84 with HDAC1 (Figure 1C) and CDK4 (Figure 1D). Collectively, Roxyl-zhc-84 was identified as the most potent antitumour inhibitor among all derivatives tested.
Roxyl-zhc-84 is the most potent antitumour compound among the derivatives. (A) Roxyl-zhc-84 exhibits the greatest cytotoxicity among all tested compounds. MDA-MB-468, MDA-MB-231 and SK-OV-3 cells were treated with each compound (300 nM) for 48 h, and CCK-8 assay was performed. DMSO was used as a negative control. (B) Schematic showing design of the novel hybrid inhibitor Roxyl-zhc-84. (C-D) Closer view of binding of Roxyl-zhc-84 to HDAC1 and CDK4. (C) Predicted binding mode of Roxyl-zhc-84 (purple) and vorinostat (blue) with HDAC1. The crystal structure of HDAC1 was taken from the RCSB Protein Data Bank (PDB entry: 4BKX). (D) Representation of the predicted binding modes of Roxyl-zhc-84 (purple) and abemaciclib (yellow) in the ATP pocket of CDK4, which employed CDK6 (PDB entry 4EZ5) as the template for homology modelling. Dash lines indicate H-bond interactions between compounds and enzymes.
Roxyl-zhc-84 effectively enhances antitumour activity on cell viability
Anti-viability activities of Roxyl-zhc-84 against various cell lines, including those for 3 aggressive breast cancer cell lines, 2 high grade serous ovarian cancer cell lines and a non-small cell lung cancer cell line, were examined. The IC50 values ranged from 10 nM to 40 nM (Table 1 and Figure S3), exhibiting a highly suppressive effect on cell proliferation. In contrast to Roxyl-zhc-84, the reference drugs vorinostat and abemaciclib only slightly influenced cell viability at a nanomolar potency. The IC50 values of vorinostat and abemaciclib in these cell lines were higher than 1 μM. In cellar assays and a mouse model of breast cancer, in spite of the synergistic effect between vorinostat and abemaciclib, Roxyl-zhc-84 dramatically suppressed tumour growth more effectively than the combination of vorinostat and abemaciclib both in vitro and in vivo (Figure S1A-B). Hence, these results suggest that the hybrid strategy of Roxyl-zhc-84 displays dramatic improvement compared to positive compounds.
IC50 values of Roxyl-zhc-84 against six cancer cell lines. The cytotoxic effects of Roxyl-zhc-84, vorinostat and abemaciclib in six solid tumour cell lines were assayed using CCK-8 assay.
| IC50 Value (nM) | |||
|---|---|---|---|
| Roxyl-zhc-84 | Vorinostat | Abemaciclib | |
| 4T1 | 18.3 | >>2000 | >>2000 |
| MDA-MB-468 | 32.3 | >>2000 | 710.8 |
| MDA-MB-231 | 23.9 | 1718 | 1667 |
| SK-OV-3 | 44 | 1275 | 1621 |
| OVCAR-5 | 15.8 | 1318 | 2504.5 |
| H460 | 32.77 | >>2000 | 961.8 |
Roxyl-zhc-84 presents favourable pharmacokinetic properties [39]
Due to the elevated enzymatic activity and antitumour effects of Roxyl-zhc-84 in vitro, pharmacokinetic evaluation in rats following intravenous and oral administration of this compound were subsequently evaluated (Table 2 and Figure S4). Roxyl-zhc-84 showed an ideal oral bioavailability of 77.39% with an AUC(0-∞) = 9.806 mg/L·h. The oral maximum plasma concentration (Cmax) was 0.803 mg/L, and Tmax was 4 h. The oral half-life (T1/2) was 8.361 h, and mean residence time (MRT0-t) was 9.098 h. In addition, Roxyl-zhc-84 showed an oral apparent distribution volume (Vss) of 12.142 L/kg and a clearance of 1.027 L/h/kg. Its half-life (t1/2 = 8.361 h) and clearance rate (CL = 1.027 L/h/kg) in pharmacokinetic studies indicated that Roxyl-zhc-84 displays good stability in vivo. Together, these results indicate that Roxyl-zhc-84 presents favourable pharmacokinetic properties and is a promising candidate for tumour treatment. Furthermore, Roxyl-zhc-84 exhibited low toxicity, as evidenced by hemogram results in mouse experiments. Primary organs, including heart, liver, spleen, lung, kidney and brain, were barely affected, indicating that the Roxyl-zhc-84 exhibits only slight toxicity to the circulatory system and normal tissues (Table 3 and Figure S5).
Analysis of the main pharmacokinetic parameters of Roxyl-zhc-84 after oral and intravenous administration (n = 5 for each group).
| Parameters | iv | po |
|---|---|---|
| AUC(0-t) (mg/L·h) | 10.923 | 8.453 |
| AUC(0-∞) | 12.474 | 9.806 |
| AUMC(0-t) | 51.04 | 76.923 |
| MRT(0-t) (h) | 4.675 | 9.098 |
| VRT(0-t) (h2) | 18.451 | 46.698 |
| t1/2 (h) | 4.557 | 8.361 |
| Tmax (h) | 0.05 | 4 |
| V (L/kg) | 5.256 | 12.142 |
| CL (L/h/kg) | 0.804 | 1.027 |
| Cmax (mg/L) | 9.013 | 0.803 |
| F a (%) | 77.39 |
AUC: area under the curve; AUMC: the area under the first moment of the plasma concentration-time curve; CL: clearance; Cmax: maximum plasma concentration; Fa (oral bioavailability) = AUC0-t(po)/AUC0-t(iv) × 100%; MRT: mean residence time; t1/2: half-life; Tmax: the time to peak; V: distribution volume; VRT: variance of residence time.
Effect of Roxyl-zhc-84 on haemogram results in Balb/C mice.
| Parameters | DMSO | Roxyl-zhc-84 (60 mg/kg) | Reference Value |
|---|---|---|---|
| WBC | 4.85±1.25 | 5.65±0.55 | 4-9 |
| Neutrophil Granulocyte (%) | 17.5±4.3 | 14.45±1.15 | 42-85 |
| Lymphocyte (%) | 81.2±3 | 82.6±0.9 | 11-49 |
| Monocyte (%) | 0.15±0.15 | 0.75±0.15 | 0-9 |
| Eosinophilic Granulocyte (%) | 1.15±1.15 | 2.2±0.1 | 0-3 |
| Basophilic Granulocyte (%) | 0 | 0 | 0-2 |
| RBC | 9.17±0.27 | 9.45±0.24 | 3.8-5.3 |
| Haemoglobin | 127.5±2.5 | 130±5.5 | 120-180 |
| Haematocrit | 32.2±0.6 | 32.3±0.9 | 36-56 |
| MCV | 35.1±0.4 | 34.2±0.1 | 80-100 |
| MCH | 13.9±0.1 | 13.8±0.2 | 27-32 |
| MCHC | 396±13 | 404±6 | 320-360 |
| RDW | 13.5±0.2 | 13.3±0.1 | 10-16.5 |
| PLT | 280±252 | 368.5±11.5 | 120-380 |
| PDW | 13.65±0.45 | 12.75±0.05 | 12-18 |
| MPV | 5.55±0.75 | 4.65±0.15 | 5-10 |
| Plateletcrit | 0.14±0.12 | 0.17±0.01 | 0.1-1 |
All values are expressed as the mean ± s.d. (n = 3 in each group). MCH: mean corpuscular haemoglobin; MCHC: mean corpuscular haemoglobin concentration; MCV: mean corpuscular volume; MPV: mean platelet volume; PDW: platelet distribution width; PLT: blood platelet; RDW: red blood cell distribution width; WBC: white blood cell; RBC: red blood cell.
Roxyl-zhc-84 induces apoptosis in both breast and ovarian cancer cell lines
Inhibition of HDAC induces programmed cell death. To understand the mechanism by which Roxyl-zhc-84 treatment inhibits cellular proliferation, we first tested its effect on apoptosis by examining the apoptosis level and changes in expression of apoptotic associated proteins. We chose two breast cancer cell lines (MDA-MB-231/MDA-MB-468) and two ovarian cancer cell lines (OVCAR-5/SK-OV-3) with malignant phenotypes and rapid proliferation ability. Cells were treated with Roxyl-zhc-84, reference drugs vorinostat and abemaciclib or vehicle (DMSO) for 48 h, followed by Annexin V/PI staining. Roxyl-zhc-84 caused a dose-dependent induction of apoptosis in all cell lines. The percentage of apoptotic cells in response to Roxyl-zhc-84 were significantly higher than in response to the positive controls at the same concentrations (Figure 2A-D). The positive compounds did not exhibit pro-apoptosis effects at low concentrations.
Roxyl-zhc-84 induces cell apoptosis in both breast and ovarian cancer cell lines. (A-D) Apoptosis analysis in response to Roxyl-zhc-84 performed in MDA-MB-468, MDA-MB-231, SK-OV-3 and OVCAR-5 cells using one-quarter IC50 value, half of IC50 value or approximate IC50 value concentrations. (E-H) Immunoblot of Histone H3 acetylation, BCL-2, p21 and caspase-3 expression in cell lysates. Data are shown as the mean ± s.d. *p<0.05, **p<0.01, ***p<0.001.
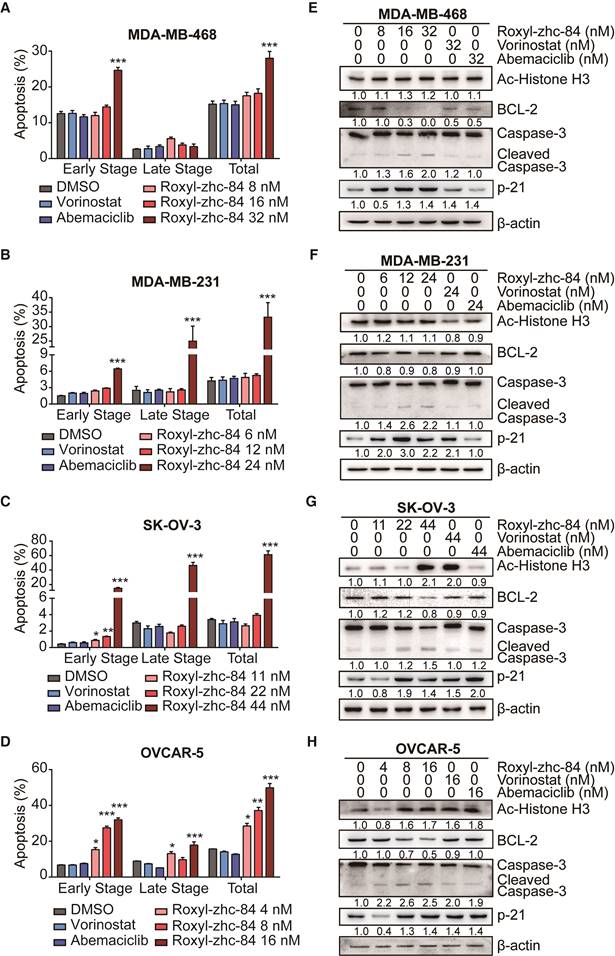
To further confirm apoptosis induction by Roxyl-zhc-84, we next examined expression of apoptosis proteins in these cancer cell lines. Parallel to HDAC inhibition, Roxyl-zhc-84 treatment significantly increased the levels of histone H3 in SK-OV-3 and OVCAR-5 cell lines as a histone deacetylase inhibitor. At a similar concentration, Roxyl-zhc-84 resulted in a slight increase in acetylated histone H3 in MDA-MB-468 and MDA-MB-231 cells. In addition, the antiapoptotic protein BCL-2 was downregulated via Roxyl-zhc-84 treatment. Roxyl-zhc-84 also led to significant induction of caspase-3 cleavage (Figure 2E-H). The observed modulations were primarily attributed to retained HDAC activity in response to Roxyl-zhc-84 treatment. Furthermore, as a target of HDAC, p21 is a cyclin-dependent kinase inhibitor and thus regulates apoptosis. Expression of p21 was increased in Roxyl-zhc-84-treated groups of all cell lines. These data indicate that Roxyl-zhc-84 induces apoptosis in both breast and ovarian cancer cell lines.
Roxyl-zhc-84 induces G1-phase cell cycle arrest in both breast and ovarian cancer cell lines
Given the CDK activity of Roxyl-zhc-84, we next assessed the influence of this compound on cell cycle regulation in solid cancer cells. We treated cells with Roxyl-zhc-84 at concentrations according to IC50 values (a quarter of IC50, half of IC50 and IC50, respectively) for 48 h. Cell cycle analysis indicated that Roxyl-zhc-84 increased the percentage of G1-phase and decreased the percentage of S-phase cells compared to DMSO and reference compounds in all cell lines (Figure 3A-D).
Roxyl-zhc-84 induces G1-phase cell cycle arrest in both breast and ovarian cancer cell lines. (A-D) 48 h cell cycle arrest analysis of Roxyl-zhc-84 was performed in MDA-MB-468, MDA-MB-231, SK-OV-3 and OVCAR-5 cells at one quarter of IC50 value, half of IC50 value or approximate IC50 value concentrations. Effects of vorinostat and abemaciclib are shown as positive controls. (E-H) Immunoblot of CDK4, CDK6, cyclin D1 expression and Rb phosphorylation in cell lysates. Cells were treated with Roxyl-zhc-84, vorinostat or abemaciclib. Data are shown as the mean ± s.d. *p<0.05, **p<0.01, ***p<0.001.
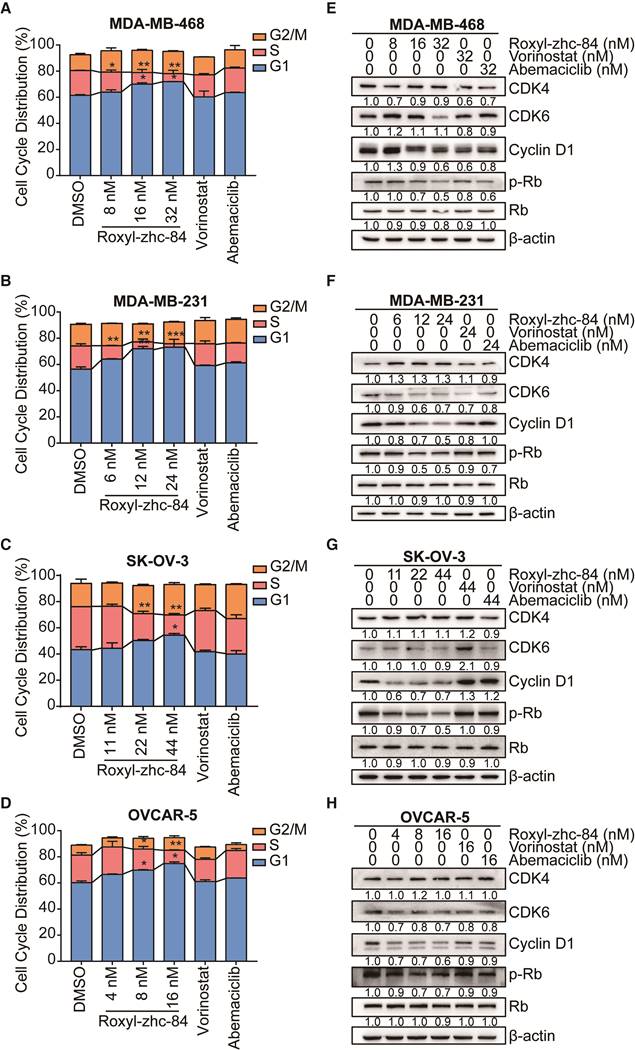
Consistent with the cell cycle results, we found that in vitro exposure to Roxyl-zhc-84 leads to significant downregulation of Rb, CDK6 and cyclin D1 phosphorylation (Figure 3E-H). Importantly, the effect of Roxyl-zhc-84 treatment on cell-cycle-regulatory proteins (p21, p-Rb, cyclin D1) may be responsible for G1-phase arrest. In contrast, vorinostat and abemaciclib did not induce G1 to S phase arrest or alter the expression of cell cycle associated proteins. Of note, the expression of CDK4 was increased in MDA-MB-231 and SK-OV-3, which proliferate faster than the other cell lines, potentially caused by compensated upregulation of CDK inhibition.
Roxyl-zhc-84 significantly inhibits tumour growth in homograft and xenograft mouse models of breast cancer with low toxicity
Next, to evaluate the in vivo antitumour activity of Roxyl-zhc-84, we established both homograft and xenograft transplantation breast cancer mouse models. In the 4T1-firefly luciferase (4T1-Fluc) BALB/c mouse model, tumour-bearing mice were given daily oral administration of Roxyl-zhc-84, vorinostat or abemaciclib for 80 days. Mouse weights were monitored twice per week over 32 days, and there was no significant difference among the 6 groups of mice (Figure 4A). As shown in Figure 4B-C, the luminescence density of tumours was measured by Living Image System. Bioluminescence imaging was performed on representative animals at 10 days and 20 days for Fluc reporter gene expression to estimate the number of viable tumour cells. Tumours treated with Roxyl-zhc-84 therapy demonstrated a significant decrease in the density of Fluc signal, suggesting considerable tumour inhibition. Quantitative analysis of bioluminescence imaging signals demonstrated that Roxyl-zhc-84 showed significantly better inhibition than the other treatments. Oral treatment with Roxyl-zhc-84 significantly reduced tumour volume (Figure 4D). In contrast, tumour volume in DMSO and vorinostat groups were barely impacted, while tumour volume in the abemaciclib-treated group decreased only slightly. Of note, extended Roxyl-zhc-84 treatment significantly prolonged the lifetime of mice compared to treatment with the positive compounds (Figure 4E).
Roxyl-zhc-84 significantly inhibits growth of breast cancer in homograft and xenograft mouse models. (A) Body weight curves of BALB/c mice in each group 1-32 days after injection (5 mice/group). Values were analysed using Igor Pro software. (B) Bioluminescence images of tumour-bearing mice. The 4T1-Fluc allograft model was treated with Roxyl-zhc-84, vorinostat (60 mg/kg) and abemaciclib (60 mg/kg) daily from tumour formation. (C) Histogram of firefly luciferase bioluminescence imaging in (B). (D) Tumour growth curve of 4T1-Fluc tumour-bearing mice in response to different treatments. (E) Survival curve of mice treated with Roxyl-zhc-84, vorinostat (60 mg/kg) and abemaciclib (60 mg/kg). (F) Photographs of MDA-MB-468 xenograft tumours. Mice were sacrificed 40 days after tumour implantation. In the 60 mg/kg Roxyl-zhc-84 group, tumour in one mouse was too small to acquire. (G) Tumour growth curve of MDA-MB-468 tumour-bearing mice in response to different treatments. (H) Immunohistochemistry and HE staining of MDA-MB-468 xenograft tumours. Scale bar, 100 μm. Data are shown as the mean ± s.d. *p<0.05, ***p<0.001.
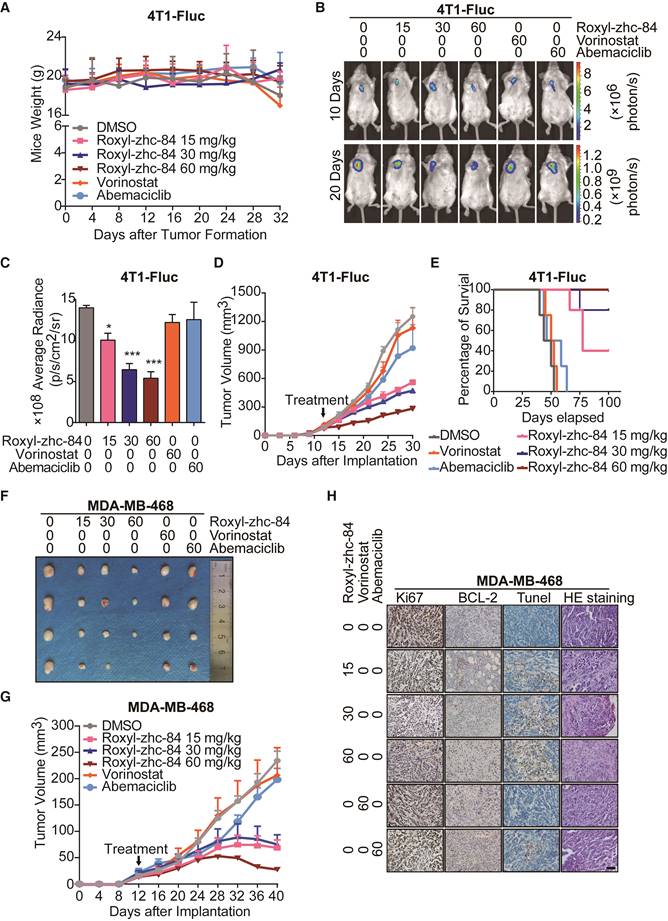
To further confirm the antitumour effect of Roxyl-zhc-84, the aggressive triple-negative breast cancer cell line MDA-MB-468 was utilized to establish a xenograft transplantation mouse model. As shown in Figure 4F-G, Roxyl-zhc-84 exhibited remarkable antitumour activity in MDA-MB-468 tumours, whereas DMSO and positive compounds did not reduce tumour volumes. Next, mice were euthanized for further investigation. Immunohistochemistry staining results showed that the cell proliferation marker Ki67 was downregulated in a dose-dependent manner. The density of the antiapoptotic protein BCL-2 was also downregulated in Roxyl-zhc-84 groups, in contrast to TUNEL staining results (Figure 4H). Hence, pathological analysis indicated that Roxyl-zhc-84 exhibits anti-proliferation and pro-apoptosis effects in vivo. Taken together, our data demonstrate that Roxyl-zhc-84 exerts dramatic antitumour activity in the growth of implanted mouse and human breast tumours in vivo.
Additional inhibition of JAK1 sensitizes solid tumours to Roxyl-zhc-84 treatment
Despite some progress in multiple haematological malignancies, most current HDAC inhibitors have failed to show clinical benefits in almost all types of solid tumours, especially breast cancer. Surprisingly, Roxyl-zhc-84 promoted apoptosis of breast cancer cells in vitro and in vivo. Therefore, our next aim was to investigate the mechanism of Roxyl-zhc-84 to understand how the hybrid inhibitor so remarkably increased sensitivity of the solid tumour to treatment.
STAT3-stimulated antiapoptotic signalling has been reported to account for the limited response to HDACi treatment, and JAK1 is primarily responsible for STAT3 activation [40]. As such, we verified the effect of combination treatment with the HDAC inhibitor vorinostat and the JAK-1 inhibitor filgotinib. Results showed that vorinostat induced activation of p-STAT3 and expression of the antiapoptotic protein BCL-2, while filgotinib inhibited the JAK1-STAT3-BCL2 pathway (Figure 5A), consistent with previous findings. To further confirm the effect of HDAC inhibitor in solid tumour cells is associated with the JAK-STAT3 pathway, gene set enrichment analysis was performed based on GEO datasets. We found that vorinostat-treated SK-OV-3 cells (GSE53603[41]) were significantly enriched in JAK-STAT3 signalling (NES = 1.53, p value<0.05), whereas diffuse large B cell lymphoma cell lines, including OCI-Ly1, OCI-Ly10 and Su-DHL6 (GSE27226[42]), did not exhibit this trend after treatment with panobinostat, another HDAC inhibitor (Figure 5B-C). Moreover, vorinostat extensively increased expression of genes in the JAK-STAT-BCL2 pathway, including BCL2L1, JAK1 and members of the STAT family (Figure 5D).
Conventional HDAC inhibitors activate JAK1-STAT3-BCL2-mediated drug resistance in solid tumours but not in Roxyl-zhc-84-treated tumours. (A) Immunoblot of STAT3, BCL-2, BIM and STAT3 phosphorylation in cells treated with the histone deacetylase inhibitor vorinostat and JAK1 inhibitor filgotinib. (B) GSEA analysis for enrichment of JAK-STAT3 pathway in vorinostat-treated SK-OV-3 cells. (C) HDACi panobinostat-treated DLBCL cell lines. Data were acquired on GEO database (GSE53603, GSE27226). (D) Heatmap showing upregulated genes in the JAK-STAT3 pathway based on data from GSE53603. (E) Dose-response curve showing that Roxyl-zhc-84 (0.1 nM-1 µM) inhibits the activity of recombinant human JAK1 dose-dependently by in vitro kinase assay. (F) Representation of predicted binding modes of Roxyl-zhc-84 (purple) and filgotinib (green) in the ATP pocket of JAK1. Dash lines indicate H-bond interactions between compounds and JAK1. (G) Immunoblot of JAK-1 phosphorylation in MDA-MB-231 and MDA-MB-468 cell lysates. Cells were treated with Roxyl-zhc-84 at different concentrations. Vorinostat is shown as a negative control.
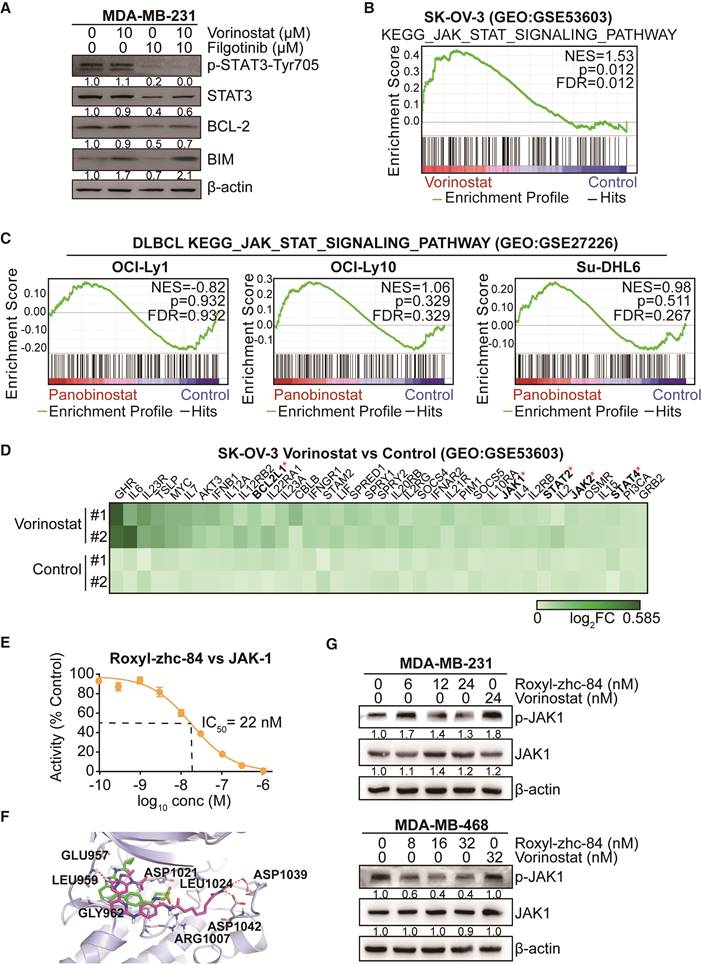
Based on the kinase profiling results indicating that JAK was a strong off-target effect of Roxyl-zhc-84 and to further investigate the function of Roxyl-zhc-84 in JAK1-STAT3-mediated drug resistance, we further assessed IC50 values of Roxyl-zhc-84 on JAK1 in an in vitro kinase assay. Roxyl-zhc-84 (0.1-1000 nM) inhibited the activity of recombinant human JAK1 dose-dependently and exerted additional JAK1 inhibitory activity at nanomolar concentrations (Figure 5E). Moreover, we predicted the binding model of Roxyl-zhc-84 in the ATP pocket of JAK1 by docking analysis (Figure 5F). Roxyl-zhc-84 tightly binds to the ATP-binding site of JAK1 in a binding mode similar to that of filgotinib. The fluorine atom of benzimidazole at proper locations forms a hydrogen bonding interaction with the LEU959 amide NH. Hydrogen bonds were also formed between the hydroxamic acid and the backbone residue of ASP1039 and 1042 carbonyl oxygen. This deep and close binding of Roxyl-zhc-84 to JAK1 may explain the inhibitory activity of Roxyl-zhc-84 for JAK1. We next examined whether Roxyl-zhc-84 influences the JAK-STAT3 pathway. Roxyl-zhc-84 inhibited phosphorylation of JAK1 in MDA-MB-231 and MDA-MB-468 TNBC cell lines (Figure 5G). Our findings provide an explanation for the sensitizing of solid tumours to hybrid drug treatment by Roxyl-zhc-84 via targeting JAK1.
Roxyl-zhc-84 sensitizes solid tumours to treatment significantly more than an HDACi and JAKi combination
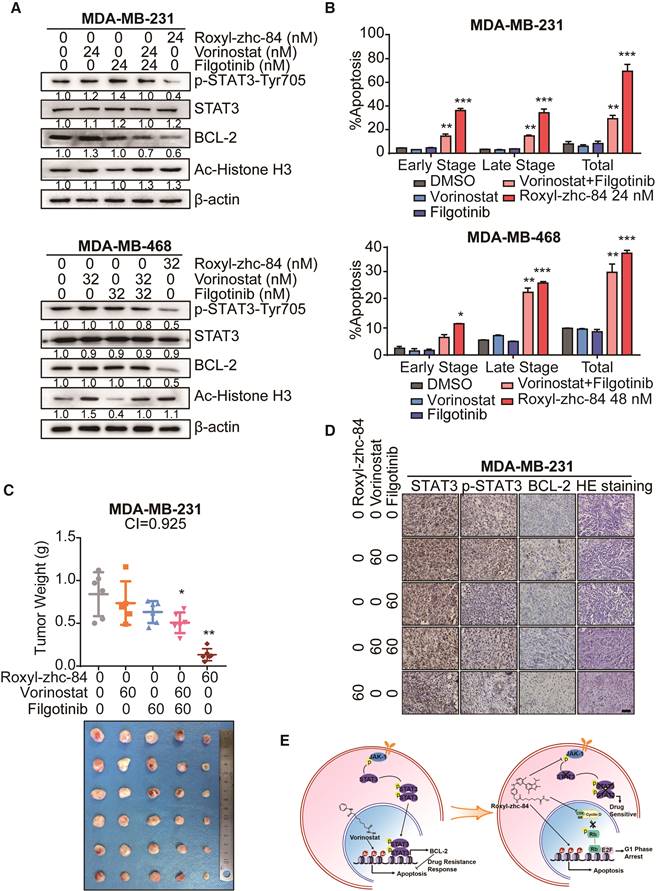
Previous work by Jian Ding et al. [40] demonstrated that JAKis block STAT3-BCL2-mediated antiapoptotic effects, sensitizing solid tumour cells to HDACi treatment. We then assessed whether Roxyl-zhc-84 acts as a superior therapeutic agent compared to an HDACi and JAKi combination. Compared with the synergistic effects of combined vorinostat and filgotinib treatment, Roxyl-zhc-84 distinctly suppressed phosphorylation of JAK1 and STAT3 and markedly downregulated expression of the antiapoptotic protein BCL-2 (Figure 6A). These data confirmed that the hybrid inhibitor blocks STAT3 activation and sensitizes cancer cells to HDAC inhibition. Apoptosis analysis also showed that Roxyl-zhc-84 exerts stronger antitumour effects than vorinostat/filgotinib or vorinostat-filgotinib combinations (Figure 6B). For further investigation, we established a xenograft mouse model of triple-negative breast cancer using the MDA-MB-231 cell line as a representative to test the mechanism described above. In the tested model, vorinostat and filgotinib exhibited ideal synergistic effects (Combination Index = 0.925). Furthermore, Roxyl-zhc-84 significantly sensitized tumours to treatment compared to treatment with vorinostat or filgotinib alone, and its potency was relatively stronger than combination treatment of vorinostat and filgotinib. Tumour weights at the study end-point also indicated that the novel inhibitor Roxyl-zhc-84 exhibits potential treatments effect in solid tumours (Figure 6C). Consistent with observations in vitro, immunohistochemistry staining revealed that intratumoural phosphorylation of STAT3 and expression of BCL-2 were stimulated by vorinostat treatment and effectively reversed by treatment with the hybrid Roxyl-zhc-84 (Figure 6D).
Roxyl-zhc-84 significantly sensitizes tumours to treatment compared to synergy between HDACi and JAKi. (A) Immunoblot of BCL-2 expression, Histone H3 acetylation and STAT3 phosphorylation in cell lysates. Cells were treated with Roxyl-zhc-84, vorinostat or abemaciclib. (B) Apoptosis analysis of Roxyl-zhc-84 was performed in MDA-MB-231 and MDA-MB-468 cells. Cells were treated with vorinostat and abemaciclib as positive control compounds. (C) Weights and photographs of MDA-MB-231 xenograft tumours. Mice were treated with Roxyl-zhc-84 (60 mg/kg), vorinostat (60 mg/kg) or/and filgotinib (60 mg/kg) for 30 days. Combination Index (CI) was determined according to the inhibitory effect on tumour weights. (D) Immunohistochemistry detected expression of STAT3, p-STAT3, and BCL-2. HE staining was performed to show tissue structure. Scale bar, 100 μm. (E) Roxyl-zhc-84 resolved the limited response of traditional HDAC inhibitors in solid tumours via overcoming JAK1-STAT3-BCL2-mediated drug resistance. CDK and JAK1 inhibition sensitizes solid tumours to hybrid drug treatment. Data are shown as the mean ± s.d. *p<0.05, **p<0.01, ***p<0.001.
Discussion
HDAC is a therapeutic target in malignant cancer due to association with cancer cell proliferation, survival, migration and invasion. HDACis permit chromatin remodelling and change the composition of multi protein complexes bound to proximal region of specific gene promoter, which are predicted to exhibit efficacy in a broad spectrum of cancer types. HDACis have been widely explored in different cancer types in both preclinical and clinical studies. However, as previously reported, solid tumours do not respond well to HDACi therapy. The development of combination HDAC inhibitors used with chemotherapy or other targeted therapies to enhance the clinical efficacy for solid tumour is urgently needed. Thus, based on the preliminary synergistic combination tests and rational combination design, we synthesized a series of hybrid derivatives based on HDAC and CDK4/6 inhibitors to identify their efficacy in solid tumours.
Hybrid modifications based on the hydroxamic acid motif, as well as on the linker fragments of vorinostat with the 2-aminopyrimidine group, exert efficacy against solid tumours. Among the tested derivatives, Roxyl-zhc-84 was identified as the most effective inhibitor against various types of cancer. Unlike traditional HDAC inhibitors, Roxyl-zhc-84 exhibits remarkably enhanced anti-tumour activity in both in vitro and in vivo assays. HDAC profiling studies showed that Roxyl-zhc-84 possesses significantly improved HDAC isoform selectivity profiles compared to that of the approved pan-HDAC inhibitor vorinostat. It may be responsible for well-tolerated observation during in vivo test and no significant loss of body weight, indicating that its toxicity is low. Roxyl-zhc-84 displayed prominent cytotoxic activity in various cancer cell lines with IC50 values in the 10-40 nM range, 30 times lower than that for the reference drug vorinostat.
To understand the mechanism of action of Roxyl-zhc-84 on cellular proliferation and HDAC targeting, we identified important mediators related to the HDAC pathway, such as the cell cycle and apoptosis. Roxyl-zhc-84 increased the level of acetylated histone H3, consistent with HDAC inhibition. We demonstrated that Roxyl-zhc-84 induced apoptosis in solid tumour cells, which reduced expression of the antiapoptotic protein BCL-2 and enhanced expression of cleavage of Ccleaved caspase-3. Additionally, as a target of HDAC, expression of p21 protein, which plays an important role in the regulation of G1/S cell cycle, increased in response to Roxyl-zhc-84 treatment. Consistent with the results that suppression of solid tumour cells by induction of G1-phase cell cycle arrest and reduction of DNA synthesis, we observed that Roxyl-zhc-84 downregulated activity of the CDK4-cyclin D1-Rb pathway. Decreased expression levels of both CDK and cyclin D1 after Roxyl-zhc-84 treatment, along with increased p21, led to additive effects of arresting the cell cycle in the G1 phase. Roxyl-zhc-84 exhibited good in vivo anti-tumour efficacies in both 4T1 and MDA-MB-468 mouse models without reducing mouse body weight, significantly prolonging survival of mice. Importantly, Roxyl-zhc-84 exhibited increased efficacy compared to combined HDAC and CDK inhibitors, even more so than HDAC plus CDK inhibitors in mouse models. Therefore, induction of cell cycle arrest enhances the therapeutic effect of HDACi. Meanwhile, consistent with results of the kinase inhibition profile, acceptable off-target of effects of Roxyl-zhc-84 may sensitize solid tumours to hybrid drug treatment.
In a previous study, a JAK1-STAT3 signalling-centred feedback loop restrained the efficacy of traditional HDAC inhibitors in solid tumours. Increased STAT3 activity stimulates antiapoptotic signalling, accounting for the limited response to HDACi treatment. JAK1 rather than JAK2 was primarily responsible for STAT3 activation. JAK1 siRNA has been previously demonstrated to block feedback activation of STAT3. We discovered that our compound Roxyl-zhc-84 reduced JAK1 and STAT3 phosphorylation and remarkably downregulated the antiapoptotic proteins BCL-2, known as a STAT3 target gene. As such, Roxyl-zhc-84 inhibited STAT3 activation-sensitized cancer cells to HDAC inhibition. We further demonstrated that Roxyl-zhc-84 has stronger antitumour effects than vorinostat/filgotinib or vorinostat-filgotinib combination groups in both in vitro and in vivo assays. Consistent with the mechanistic insights discussed above, immunohistochemistry staining revealed that intratumoural STAT3 phosphorylation and BCL-2 were stimulated by vorinostat treatment, while being effectively reversed by hybrid Roxyl-zhc-84 treatment. Therefore, JAK1 inhibition sensitizes solid tumours to hybrid drug treatment.
In summary, we designed and synthesized hybrid compounds with concurrent inhibition of HDAC and CDK with additional JAK1 targeting that represent novel scaffolds for possible use in drug discovery. The compound resolved the limited response of traditional HDAC inhibitors in solid tumours via overcoming JAK1-STAT3-BCL2-mediated drug resistance [43]. CDK and JAK1 inhibition sensitized solid tumours to hybrid drug treatment (Figure 6E). Roxyl-zhc-84 has excellent pharmacokinetic properties, exhibiting low toxicity as shown by the haemogram in mouse experiments, and primary organs were barely affected. Our study complements the current understanding of HDACis. The novel Roxyl-zhc-84 compound could be a potential, well-tolerated drug candidate for solid tumours with the advantages that CDK and JAK1 inhibition sensitize solid tumours to HDAC inhibitor treatment, providing an efficient strategy for multi-targeted antitumour drug discovery. Even more efficient hybrid compounds may be identified by utilizing other possible combinations of different HDAC and CDK inhibitors. Exploration of the therapeutic opportunities of Roxyl-zhc-84 for the treatment of a range of proliferative disorders could be promising.
Abbreviations
ATCC: American Type Culture Collection; AUC: area under the curve; AUMC: the area under the first moment of the plasma concentration-time curve; BINAP: 2,2'-bis (diphenylphosphino)-1,1'-binaphthyl; CDKs: cyclin-dependent kinases; CI: combination index; CL: clearance; Cmax: maximum plasma concentration; CTCL: cutaneous T-cell lymphoma; DIEA: diisopropylethylamine; DMAP: 4-(dimethylamino)pyridine; DMSO: dimethyl sulfoxide; EDCI: 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide; FACS: fluorescence activated cell sorter; F%: oral bioavailability; GSEA: gene set enrichment analysis; HDAC: histone deacetylases; HDACi: HDAC inhibitor; H&E: haematoxylin and eosin; HOBt: 1-hydroxybenzotriazole; HPLC: high-purity liquid chromatography; LC-MS: liquid chromatography tandem mass spectrometry; MM: multiple myeloma; MRT: mean residence time; PFA: paraformaldehyde; PI: propidium iodide; PK: pharmacokinetics; PTCL: peripheral T-cell lymphoma; RPMI 1640: Roswell Park Memorial Institute 1640; SAR: structure-activity relationship; t1/2: half-life; Tmax: time to peak; 4T1-Fluc: 4T1-firefly luciferase; THF: tetrahydrofuran; VRT: variance of residence time; Vss: distribution volume.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by the Project of Science and Technology Assistance in Developing Countries (KY201501006), the National Natural Science Foundation of China (81470354), the Natural Science Foundation of Tianjin (17JCQNJC13500), and the State Key Laboratory of Medicinal Chemical Biology (2018004).
Author Contributions
Zhi Huang and Wei Zhou are co-first authors and contributed equally to this work. Zhi Huang performed the medicinal chemistry experiments. Wei Zhou performed the biological experiments. Yan Fan, Zhi Huang and Wei Zhou designed the study and performed data analysis. Yan Fan oversaw the overall project with valuable help from Yongtao Li, Mei Cao, Tianqi Wang, Yakun Ma, Qingxiang Guo, Xin Wang, Chao Zhang, Chenglan Zhang, Wenzhi Shen, Yanhua Liu, Yanan Chen, Jianyu Zheng and Shengyong Yang. Rong Xiang contributed to reviewing the study.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501:328-37
2. Meunier B. Hybrid molecules with a dual mode of action: dream or reality? Acc Chem Res. 2008;41:69-77
3. Decker M. Hybrid molecules incorporating natural products: applications in cancer therapy, neurodegenerative disorders and beyond. Curr Med Chem. 2011;18:1464-75
4. Singh P, Kaur M, Verma P. Design, synthesis and anticancer activities of hybrids of indole and barbituric acids-identification of highly promising leads. Bioorg Med Chem Lett. 2009;19:3054-8
5. Korner M, Tibes U. Histone deacetylase inhibitors: a novel class of anti-cancer agents on its way to the market. Prog Med Chem. 2008;46:205-80
6. Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673-91
7. Ueki N, Wang W, Swenson C. et al. Synthesis and Preclinical Evaluation of a Highly Improved Anticancer Prodrug Activated by Histone Deacetylases and Cathepsin L. Theranostics. 2016;6:808-16
8. El Bahhaj F, Denis I, Pichavant L. et al. Histone Deacetylase Inhibitors Delivery using Nanoparticles with Intrinsic Passive Tumor Targeting Properties for Tumor Therapy. Theranostics. 2016;6:795-807
9. Miller TA, Witter DJ, Belvedere S. Histone deacetylase inhibitors. J Med Chem. 2003;46:5097-116
10. He LZ, Tolentino T, Grayson P. et al. Histone deacetylase inhibitors induce remission in transgenic models of therapy-resistant acute promyelocytic leukemia. J Clin Invest. 2001;108:1321-30
11. Lyssiotis CA, Lairson LL, Boitano AE. et al. Chemical control of stem cell fate and developmental potential. Angew Chem Int Ed Engl. 2011;50:200-42
12. Piekarz RL, Frye R, Prince HM. et al. Phase 2 trial of romidepsin in patients with peripheral T-cell lymphoma. Blood. 2011;117:5827-34
13. Steele NL, Plumb JA, Vidal L. et al. A phase 1 pharmacokinetic and pharmacodynamic study of the histone deacetylase inhibitor belinostat in patients with advanced solid tumors. Clin Cancer Res. 2008;14:804-10
14. Giannini G, Cabri W, Fattorusso C, Rodriquez M. Histone deacetylase inhibitors in the treatment of cancer: overview and perspectives. Future Med Chem. 2012;4:1439-60
15. Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287-99
16. Kelly WK, O'Connor OA, Krug LM. et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23:3923-31
17. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769-84
18. Johnstone RW, Licht JD. Histone deacetylase inhibitors in cancer therapy: is transcription the primary target? Cancer Cell. 2003;4:13-8
19. Shah MH, Binkley P, Chan K. et al. Cardiotoxicity of histone deacetylase inhibitor depsipeptide in patients with metastatic neuroendocrine tumors. Clin Cancer Res. 2006;12:3997-4003
20. Guha M. HDAC inhibitors still need a home run, despite recent approval. Nat Rev Drug Discov. 2015;14:225-6
21. Huang Y, Dong G, Li H, Liu N, Zhang W, Sheng C. Discovery of Janus Kinase 2 (JAK2) and Histone Deacetylase (HDAC) Dual Inhibitors as a Novel Strategy for the Combinational Treatment of Leukemia and Invasive Fungal Infections. J Med Chem. 2018;61:6056-74
22. Tang C, Li C, Zhang S. et al. Novel Bioactive Hybrid Compound Dual Targeting Estrogen Receptor and Histone Deacetylase for the Treatment of Breast Cancer. J Med Chem. 2015;58:4550-72
23. Zang J, Liang X, Huang Y. et al. Discovery of Novel Pazopanib-Based HDAC and VEGFR Dual Inhibitors Targeting Cancer Epigenetics and Angiogenesis Simultaneously. J Med Chem. 2018;61:5304-22
24. Dong G, Chen W, Wang X. et al. Small Molecule Inhibitors Simultaneously Targeting Cancer Metabolism and Epigenetics: Discovery of Novel Nicotinamide Phosphoribosyltransferase (NAMPT) and Histone Deacetylase (HDAC) Dual Inhibitors. J Med Chem. 2017;60:7965-83
25. Li Y, Guo Q, Zhang C. et al. Discovery of a highly potent, selective and novel CDK9 inhibitor as an anticancer drug candidate. Bioorg Med Chem Lett. 2017;27:3231-7
26. Schwabe T, Grimme S. Towards chemical accuracy for the thermodynamics of large molecules: new hybrid density functionals including non-local correlation effects. Phys Chem Chem Phys. 2006;8:4398-401
27. Li Y, Luo X, Guo Q. et al. Discovery of N1-(4-((7-Cyclopentyl-6-(dimethylcarbamoyl)-7 H-pyrrolo[2,3- d]pyrimidin-2-yl)amino)phenyl)- N8-hydroxyoctanediamide as a Novel Inhibitor Targeting Cyclin-dependent Kinase 4/9 (CDK4/9) and Histone Deacetlyase1 (HDAC1) against Malignant Cancer. J Med Chem. 2018;61:3166-92
28. Lehar J, Krueger AS, Avery W. et al. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol. 2009;27:659-66
29. Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol. 2007;25:84-90
30. Patnaik A, Rosen LS, Tolaney SM. et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016;6:740-53
31. Huwe A, Mazitschek R, Giannis A. Small molecules as inhibitors of cyclin-dependent kinases. Angew Chem Int Ed Engl. 2003;42:2122-38
32. Napolitani G, Rinaldi A, Bertoni F, Sallusto F, Lanzavecchia A. Selected Toll-like receptor agonist combinations synergistically trigger a T helper type 1-polarizing program in dendritic cells. Nat Immunol. 2005;6:769-76
33. Shen W, Chang A, Wang J. et al. TIFA, an inflammatory signaling adaptor, is tumor suppressive for liver cancer. Oncogenesis. 2015;4:e173
34. Niu HY, Zhou W, Xu YX. et al. Silencing PPA1 inhibits human epithelial ovarian cancer metastasis by suppressing the Wnt/beta-catenin signaling pathway. Oncotarget. 2017;8:76266-78
35. Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2008;426:145-59
36. Delano WL. The PyMOL Molecular Graphics System, version 1.7; Schrödinger, LLC: New York, 2014.
37. McConville M, Bradley DF, Zhou K, Schiffrin DJ, O'Neil IA. Selective trioxolane based bifunctional molecular linkers for covalent heme surface functionalisation. Chem Commun. 2014;50:186-8
38. Le Brazidec JY, Pasis A, Tam B. et al. Structure-based design of 2,6,7-trisubstituted-7H-pyrrolo[2,3-d]pyrimidines as Aurora kinases inhibitors. Bioorg Med Chem Lett. 2012;22:4033-7
39. Zhang CH, Zheng MW, Li YP. et al. Design, Synthesis, and Structure-Activity Relationship Studies of 3-(Phenylethynyl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine Derivatives as a New Class of Src Inhibitors with Potent Activities in Models of Triple Negative Breast Cancer. J Med Chem. 2015;58:3957-74
40. Zeng H, Qu J, Jin N. et al. Feedback Activation of Leukemia Inhibitory Factor Receptor Limits Response to Histone Deacetylase Inhibitors in Breast Cancer. Cancer Cell. 2016;30:459-73
41. Konstantinopoulos PA, Wilson AJ, Saskowski J, Wass E, Khabele D. Suberoylanilide hydroxamic acid (SAHA) enhances olaparib activity by targeting homologous recombination DNA repair in ovarian cancer. Gynecol Oncol. 2014;133:599-606
42. Kalac M, Scotto L, Marchi E. et al. HDAC inhibitors and decitabine are highly synergistic and associated with unique gene-expression and epigenetic profiles in models of DLBCL. Blood. 2011;118:5506-16
43. Ziegler S, Pries V, Hedberg C, Waldmann H. Target identification for small bioactive molecules: finding the needle in the haystack. Angew Chem Int Ed Engl. 2013;52:2744-92
Author contact
![]() Corresponding authors: Yan Fan: E-mail: yanfanedu.cn; Rong Xiang: E-mail: rxiangedu.cn.
Corresponding authors: Yan Fan: E-mail: yanfanedu.cn; Rong Xiang: E-mail: rxiangedu.cn.