Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(18):4969-4984. doi:10.7150/thno.26193 This issue Cite
Research Paper
Sonodynamic therapy-induced foam cells apoptosis activates the phagocytic PPARγ-LXRα-ABCA1/ABCG1 pathway and promotes cholesterol efflux in advanced plaque
Huan Wang1#, Yang Yang1,2#, Xin Sun1, Fang Tian1, Shuyuan Guo1,2, Wei Wang1,2, Zhen Tian3, Hong Jin4, Zhiguo Zhang5, Ye Tian1,2,3 ![]()
1. Department of Cardiology, The First Affiliated Hospital, Cardiovascular Institute, Harbin Medical University, Harbin, 150001, P. R. China.
2. Heilongjiang Academy of Medical Sciences, Harbin, 150001, P. R. China.
3. Department of Pathophysiology and the Key Laboratory of Cardiovascular Pathophysiology, Harbin Medical University, the Key Laboratory of Cardiovascular Medicine Research (Harbin Medical University), Ministry of Education, Harbin, 150086, P. R. China.
4. Karolinska Institute, Department of Medicine, Solna, Stockholm, Sweden.
5. Laboratory of Photo- and Sono-theranostic Technologies and Condensed Matter Science and Technology Institute, Harbin Institute of Technology, Harbin, 150080, P. R. China.
#these authors contributed equally to this work.
Received 2018-3-20; Accepted 2018-9-10; Published 2018-9-29
Citation:
Wang H, Yang Y, Sun X, Tian F, Guo S, Wang W, Tian Z, Jin H, Zhang Z, Tian Y. Sonodynamic therapy-induced foam cells apoptosis activates the phagocytic PPARγ-LXRα-ABCA1/ABCG1 pathway and promotes cholesterol efflux in advanced plaque. Theranostics 2018; 8(18):4969-4984. doi:10.7150/thno.26193. https://www.thno.org/v08p4969.htm
Other stylesAbstract

In advanced atherosclerotic plaques, defective efferocytosis of apoptotic foam cells and decreased cholesterol efflux contribute to lesion progression. In our previous study, we demonstrated that 5-aminolevulinic acid (ALA)-mediated sonodynamic therapy (SDT) could induce foam cells apoptosis via the mitochondrial-caspase pathway. In the current research, we sought to explore ALA-SDT-induced apoptosis of phagocytes and the effects of cholesterol efflux and efferocytosis in advanced apoE-/- mice plaque.
Methods: apoE-/- mice fed western diet were treated with ALA-SDT and sacrificed at day 1, day 3, day 7 and day 28 post treatment. THP-1 macrophage-derived foam cells were treated with ALA-SDT. 5 hours later, the supernatant was collected and added to fresh foam cells (phagocytes). Then, the lipid area, efferocytosis, cholesterol efflux, anti-inflammatory reactions and PPARγ-LXRα-ABCA1/ABCG1 pathway were detected in plaque in vivo and in phagocytes in vitro.
Results: We found that ALA-SDT induced foam cells apoptosis coupled with efferocytosis and upregulation of Mer tyrosine kinase (MerTK) both in vivo and in vitro. The lipid content in plaque decreased as early as 1 day after ALA-SDT and this tendency persisted until 28 days. The enhancement of phagocytes cholesterol efflux was accompanied by an approximately 40% decrease in free cholesterol and a 24% decrease in total cholesterol in vitro. More importantly, anti-inflammatory factors such as TGFβ and IL-10 were upregulated by ALA-SDT treatment. Finally, we found that PPARγ-LXRα-ABCA1/ABCG1 pathway was activated both in vivo and in vitro by ALA-SDT, which could be blocked by PPARγ siRNA.
Conclusions: Activation of PPARγ-LXRα-ABCA1/ABCG1 pathway induced by ALA-SDT treatment engages a virtuous cycle that enhances efferocytosis, cholesterol efflux and anti-inflammatory reactions in advanced plaque in vivo and in phagocytes in vitro.
Keywords: atherosclerosis, foam cells, sonodynamic therapy, phagocyte, cholesterol efflux
Introduction
Atherosclerosis is a chronic and maladaptive inflammatory disease, which is initiated by the accumulation of modified lipoproteins such as oxidized low density lipoprotein (oxLDL) in the arterial wall. These retained lipoproteins activate endothelium, and the activated endothelial cells in turn recruit circulating monocytes that further differentiate into macrophages [1-3]. Macrophages then uptake the subendothelial lipoproteins and become foam cells. As we know, a balance between cholesterol influx in and efflux out of foam cells exists under the normal physiological condition [4]; however, in advanced atherosclerotic plaques, this balance is tilted and cholesterol efflux decreases [5, 6]. This continuous accumulation of free cholesterol in foam cells causes endoplasmic reticulum (ER) stress, which evokes inflammatory responses that ultimately lead to foam cells apoptosis [5, 6]. If apoptotic bodies are not efficiently cleared by efferocytosis, secondary necrosis will occur and result in atherosclerosis progression [5, 6]. Therefore, the homeostasis of cholesterol influx and efflux has a critical importance in atherogenesis. Although reducing cholesterol influx with statin treatment has achieved clinical success, most clinical cardiovascular events still happen due to the pre-established plaque burden at the onset of therapy [7, 8]. Thus, enhancement of cholesterol efflux from atheroma has emerged as the most promising therapeutic strategy to promote atheroma regression [9, 10].
PPARγ-LXRα-ABCA1/ABCG1 pathway in macrophages plays an important role in regulating cholesterol efflux. Both PPARγ and LXRα are transcription factors and can be activated by the components of oxLDL. Activated LXRα directly increases the levels of the membrane ATP-binding cassette transporters, ABCA1 and ABCG1, which pump cholesterol out of macrophages [11-15]. Activated PPARγ, when in the presence of apoptotic cells, further increases the expression level of LXRα and synergizes the cholesterol efflux function of LXRα-ABCA1/ABCG1 [16-18]. More than 50% cholesterol exits out of macrophages through ABCA1, and in advanced plaques, the decreased expression of ABCA1 leads to an 80% reduction in cholesterol efflux [6]. In addition to regulating cholesterol homeostasis, PPARγ and LXRα also show anti-inflammatory functions by promoting the release of anti-inflammatory factors [13, 19-21]. Moreover, the activated LXRα enhances phagocytosis of apoptotic cells by inducing the expression of the cell surface receptor, Mer tyrosine kinase (MerTK) [22]. All of these results suggest that activation of PPARγ and LXRα would improve outcomes of atherosclerotic patients. Indeed, PPARγ and LXRα agonists that promote macrophage cholesterol efflux could enhance apoptotic cell efferocytosis and reduce atherosclerosis in vivo [23-25]. However, in view of their systemic side effects such as heart failure and hepatosteatosis, their clinical applications are limited [22, 26-28]. Therefore, locally activating the PPARγ-LXRα-ABCA1/ABCG1 pathway in atherosclerotic plaques might be a promising therapeutic strategy against atherosclerosis.
Sonodynamic therapy (SDT) is an emerging approach that involves a combination of low-intensity ultrasound and specialized chemical agents known as sonosensitizers [29]. The superior features of ultrasonic radiation, including safety, local treatment with little effects on normal tissues, and a good tissue-penetrating ability without obvious attenuation of its energy, make SDT a very attractive modality from a clinical perspective [29, 30]. 5-aminolevulinic acid (ALA) is a natural precursor of a sonosensitizer, protoporphyrin IX (PpIX), and it is metabolized to PplX in cellular mitochondria [31-33]. In our previous study, we showed that ALA-SDT induces the apoptosis of THP-1 macrophage-derived foam cell via the mitochondria-caspase pathway, which is different from the cell death of foam cells induced by the chronic ER stress in advanced atheroma [34]. And, we found that ALA-SDT inhibited atherosclerosis progression in both rabbit and apoE-/- mouse models [35]. In the present study, we investigated the potential mechanism of effects of ALA-SDT on atherosclerosis in vivo by using a western diet-fed apoE-/- mouse model. The results showed that ALA-SDT activated the PPARγ-LXRα-ABCA1/ABCG1 pathway, enhanced efferocytosis and cholesterol efflux, induced anti-inflammatory response, and eventually ameliorated atherosclerosis.
Methods
Materials are described in Supplementary Material
Mice
Male C57BL/6J apoE-/- mice were purchased from Qingzilan Technology Co., Ltd., (Nanjing, China) and housed in a specific pathogen-free environment on a 12 h/12 h light/dark cycle at 25 °C. From the age of 5 weeks, the mice were fed western diet (Qingzilan Technology Co., Ltd., Nanjing, China) for 16 weeks to induce advanced atherosclerotic plaques in the aortic root. All the animal protocols were approved by the Ethics Committee of Harbin Medical University (ECHMU).
Sonication device and treatment protocol
The ultrasonic generator, transducer and power amplifier used in this study were designed and assembled by the Harbin Institute of Technology (Harbin, China). 21-week-old mice were randomly separated into four groups: control group, ALA group, ultrasound (US) group and SDT group. ALA and US groups were mainly omitted in the results due to the lack of significant difference between control group and ALA group or US group (Figure S1). For the SDT group, the mouse thorax area was exposed to ultrasonic radiation for 15 min at 2 h post-injection of ALA (60 mg/kg) into the caudal vein (Figure 1A). The mouse was placed above a water column with a height of 30 cm, and the bottom of the water column was connected to the ultrasonic transducer (diameter: 3.5 cm; center frequency: 1 MHz; duty factor: 10%; repetition frequency: 100 Hz; the diameter of ultrasound distributed zone at 30 cm from transducer: 1.0 cm). The ultrasonic intensity was 0.5 W/cm2 (86.6 kPa). After treatment, the mice were fed a normal diet until the scheduled follow-ups at 1 d (n=15), 3 d (n=15), 7 d (n=15) or 28 d (n=15).
Tissue harvest
Following anesthetization, the heart and the aorta were carefully removed. The top half of the heart was excised, snap-frozen and serially cross-sectioned. Representative cryosections of aortic valves displaying similar lesion morphology were prepared for TUNEL staining or immunofluorescence staining (n=9). The intimal lesion area, representing the plaque area, was measured. The average of the target area values of the three aortic valves representing one section was calculated. The aorta was stripped of its adventitial fat, weighed, stored in liquid nitrogen and used for western blot assays.
Oil Red O staining
Cryosections of the aortic valves were stained with Oil Red O to detect lipids in the plaques and were then counterstained with hematoxylin (n=6). Images were captured using an Olympus microscope. The red area relative to the plaque area was calculated using Image-Pro Plus 6.0 software (Media Cybernetics, USA).
TUNEL staining and efferocytosis assay
Efferocytosis was defined as the merging of TUNEL-positive/CD68-positive images. Free apoptotic cells were defined as TUNEL-positive alone images [36, 37]. The nuclei were counterstained with DAPI. The phagocytotic rate was determined in situ by counting the number of free apoptotic cells versus efferocytotic macrophages in each lesion section.
Double-immunofluorescence assay
Double-immunofluorescence staining was performed with cryosections. The positive double-staining area relative to the plaque area was measured using Image-Pro Plus 6.0 software.
Cell culture and foam cell formation
Human THP-1 cells obtained from American Type Culture Collection (ATCC; Manassas, VA, USA) were seeded on 35-mm Petri dishes at a density of 0.5×106 cells/mL in RPMI 1640 medium containing 10% FBS, 10 IU/mL penicillin and 10 µg/mL streptomycin and maintained at 37 °C in a humidified atmosphere containing 5% CO2. The THP-1 cells were differentiated into macrophages by adding 100 ng/mL PMA for 72 h. The macrophages were transformed into foam cells by incubating with 50 µg/mL oxLDL in serum-free RPMI 1640 medium containing 0.3% BSA for 48 h.
Treatment protocol
For the SDT group, the foam cells were incubated in the dark in the presence of 1.0 mM ALA for 6 h at 37 °C and then exposed to ultrasound at 0.4 W/cm2 for 5 min [34]. The Petri dish was placed in a water bath above the ultrasonic transducer, and the sonication device was used as described previously (Figure 1B). In the SDT + GW9662 group, the phagocytes were pretreated with 10 μM of the PPARγ antagonist GW9662 for 1 h.
In vitro efferocytosis detection by flow cytometry
Five hours after SDT treatment, the supernatants from each group were collected and added to fresh foam cells (phagocytes) for 4 h. To detect efferocytosis, the phagocytes were washed twice after incubation with supernatants from the different groups. Then, two dishes of the apoptotic cells (AC) induced by SDT were moved to one dish of phagocytes. The AC were labeled with 100 ng/mL pHrodoTM Red SE for 30 min before incubating them with the phagocytes to label those phagocytes that had ingested the AC (“ingesting phagocytes”; IPs) [38]. After the phagocytes were incubated with the apoptotic cells for 1 h, the non-ingested AC were removed from the phagocytes via thorough rinsing. Then, the phagocytes were collected, incubated with Percp-anti-CD11b for 1 h in the dark and then washed with PBS. Samples were assessed using a FACSCalibur flow cytometer within an hour and the data were analyzed with FlowJo software. This assessment was performed in triplicate.
Figure 1
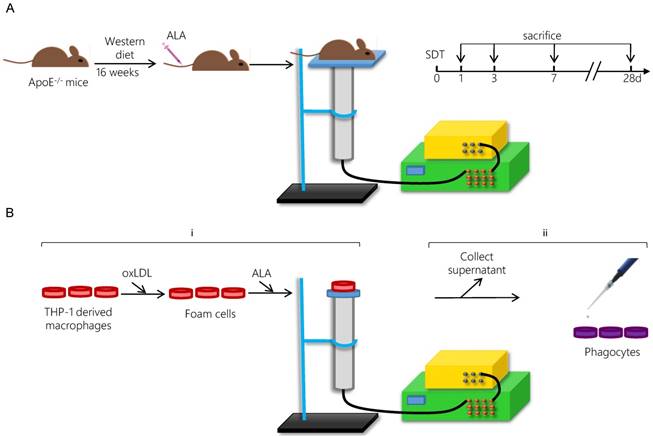
Schematic diagram of the experimental procedure applying ALA-SDT treatment in vivo (A) and in vitro (B).
In vitro Oil Red O staining
After incubation with supernatants from each treatment group for 4 h, the phagocytes were incubated for an additional 12 h in serum-free medium containing 0.2% BSA with or without 15 μg/mL apoA1 and 50 μg/mL HDL. Then, the foam cells were fixed and stained with Oil Red O, counter-stained with hematoxylin, and photographed under a microscope [39].
Quantitative measurement of intracellular cholesterol content
Phagocytes were processed as described in the Methods section for in vitro Oil Red O staining. The total and free cholesterol levels in the phagocytes were analyzed using total and free cellular cholesterol continuous cycle enzymatic colorimetric kits according to the manufacturers' instructions.
Enzyme-linked immunosorbent assay (ELISA)
The supernatants of foam cells from each group after incubation with phagocytes for 4 h and those from the SDT group after incubation with phagocytes for 0-12 h were collected. The levels of the anti-inflammatory factors IL-10 and TGFβ in the supernatants were detected using ELISA kits according to manufacturer's instructions (n=6). This assessment was performed in triplicate.
Immunoblotting
SDS-PAGE and immunoblotting were performed according to standard procedures. The protein samples were loaded onto 10% or 15% SDS-PAGE for electrophoresis and blotting. The membranes were incubated overnight at 4 ℃ with primary antibodies. After washing, the membranes were incubated in a secondary IgG antibody conjugated to horseradish peroxidase for 1 h, and the protein levels were detected using an electro-chemiluminescence detection system (Amersham Pharmacia Biotech, Uppsala, Sweden). Densitometric values were quantified using Quantity One software (Bio-Rad, Hercules, CA, USA).
Transfection of small interfering RNA (siRNA)
To knockdown PPARγ in phagocytes, cells were transfected with siRNA against PPARγ (GenePharma Co., Ltd., Shanghai, China) using X-tremeGENE siRNA Transfection Reagent according to the manufacturer's guidelines. The siRNA oligonucleotides were as follows: PPARγ S1, 5′ -CUGGCCUCCUUGAUGAAUATT-3′; PPARγ S2, 5′-ACUCCACAUUACGAAGACATT-3′; and negative control, 5′-UUCUCCGAACGUGUCACGUTT-3′.
Statistical analysis
All data are presented as the mean value ± SD. The difference among groups was analyzed via one-way analysis of variance (ANOVA) followed by the Student-Newman-Keuls test. Statistical evaluation was performed using SAS 9.1 (SAS Institute Inc., Cary, NC) software. Differences were considered to be statistically significant when P<0.05.
Results
Accumulation and distribution of ALA-PpIX in vivo
The typical emission spectrum of ALA-PpIX at 635 nm under excitation of 405 nm violet light was detected using a fluorescence spectrometer (Figure S2A). In apoE-/- mouse aortic lesion, the fluorescence intensity of ALA-PpIX peaked at 2 h after intravenous administration of ALA (Figure S2B), in accordance with our previous result detected in rabbit carotid artery plaque [40].
To further confirm in which cell type ALA-PpIX accumulated in the advanced plaque, the apoE-/- mouse aortic roots were stained with macrophage and smooth muscle cells marker. As shown in Figure S3A, ALA-PpIX exclusively accumulated in the macrophages in the plaque. The colocalizaiton of ALA-PpIX and TOM20 further verified that ALA-PpIX specifically accumulated in the mitochondria of macrophages (Figure S3B). Figure S3C showed that ALA-PpIX did not accumulate in the smooth muscle cells in the plaque.
ALA-SDT treatment enhances cholesterol efflux both in vivo and in vitro
Figure 2
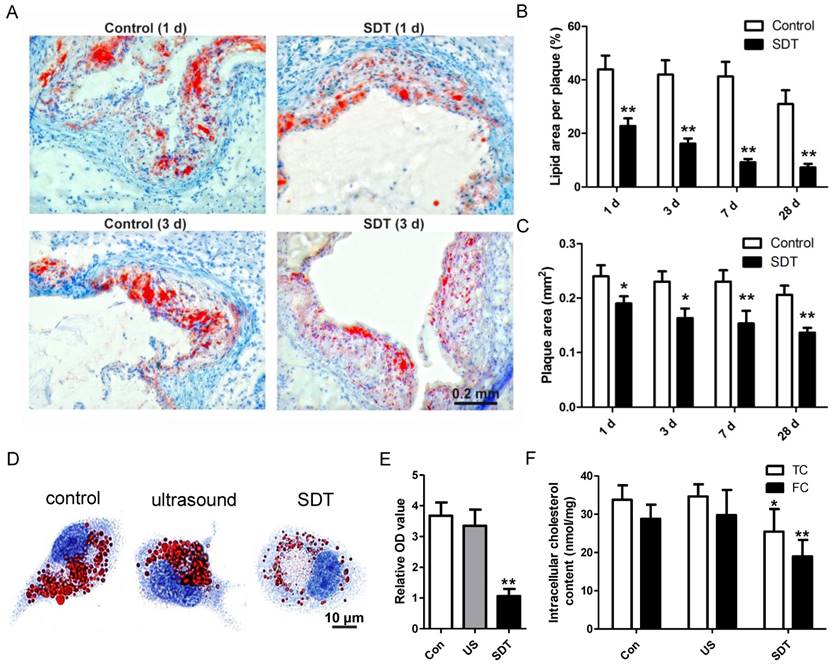
ALA-SDT treatment enhances cholesterol efflux both in vivo and in vitro. (A) Representative microscopic photographs of lipid in mice plaques stained by Oil Red O. (B) Quantitative analysis of lipid area per plaque. (n=6). (C) Quantitative analysis of plaque area. (n=6). (D-E) After incubation with supernatants from foam cells treated by control, ultrasound or SDT for 4 h, the phagocytes were incubated for an additional 12 h in serum-free media containing apoA1 and HDL. (D) Representative microscopic photographs of phagocytes stained with Oil Red O. (E) Quantification of the relative OD value in phagocytes in (D). (F) Quantitative analysis of intracellular total cholesterol (TC) and free cholesterol (FC) levels in phagocytes in (D). The assessment was performed in triplicate. Data are expressed as mean ± SD; *P<0.05 vs. control group, **P<0.01 vs. control group.
Atherosclerosis is a lipoprotein-initiated disease and the necrotic lipid core is a typical marker of advanced atheromatous plaque. In our previous data, we found that ALA-mediated photodynamic therapy (PDT) reduced plaque area by 59% at 4 weeks after PDT in a rabbit model [40]. Thus, we wondered whether ALA-SDT could reduce lipoprotein retention in the advanced atheroma. Oil Red O staining results demonstrated that compared with the control mice, ALA-SDT drastically reduced lipid area in the plaque as early as 1d after the treatment, and the lipid area further gradually shrank along with the time course (Figure 2A-B). The same trend of plaque area changes after ALA-SDT treatment was observed (Figure 2C). Accumulation of lipid in plaques is determined by both cholesterol influx and cholesterol efflux; therefore, the decreased lipid retention in the plaque after ALA-SDT treatment should be due to a relative increase in cholesterol efflux.
ALA-SDT-enhanced cholesterol efflux was further validated by in vitro assay. After co-incubation with supernatants from each treatment group, the foam cells (phagocytes) were incubated in apoA1- and HDL-containing serum-free media. Then, some phagocytes were stained by Oil Red O to observe the intracellular lipid droplets, and other phagocytes were processed to assess intracellular total cholesterol and free cholesterol content. Oil Red O results indicated that compared with control and ultrasound treatment groups, ALA-SDT treatment significantly decreased the lipid droplet in phagocytes (Figures 2D-E). And, the intracellular cholesterol assay result showed that ALA-SDT decreased approximately 40% free cholesterol and 24% total cholesterol compared with those of the Control group (Figure 2F), suggesting that ALA-SDT enhances cholesterol efflux.
Effects of ALA-SDT on reactive oxygen species generation in vivo and in vitro
Our previous results showed that reactive oxygen species (ROS) produced by ALA-SDT induced foam cell mitochondrial pathway apoptosis in vitro [34]. To further detect the effect of ALA-SDT on ROS generation in vivo, the apoE-/- mouse aortic roots were stained with ROS indicator DCFH-DA after ALA-SDT treatment. As shown in Figure S4A-B, ALA-SDT induced a significant increase of ROS production in the plaque; about a four-fold increase compared with the control group.
Next, we detected ROS production by ALA-SDT in vitro. Firstly, water containing 0.5 mM or 1 mM PpIX was exposed to ultrasound. Ultrasound alone increased ROS production detected by DCFH-DA (Figures S4C) and singlet oxygen generation detected by SOSG (Figure S4D). With the existence of PpIX, ultrasound radiation produced even higher ROS detected by DCFH-DA (Figure S4C), but not the singlet oxygen generation detected by SOSG (Figure S4D). And, we couldn't detect any absorbance changes for DPBF (Figure S4E). Then, ROS production by ALA-SDT in foam cells in vitro was detected by DCFH-DA. In accordance with the in vivo result, ALA-SDT induced a time-dependent increase of ROS production in the foam cells in vitro, which peaked at 3 h after the treatment with around a four-fold increase compared with the control group (Figure S4F-G).
ALA-SDT induces foam cells mitochondria pathway apoptosis and decreases ER stress in mouse atherosclerotic plaque
We previously showed that ALA-SDT treatment resulted in THP-1 macrophage-derived foam cell acute apoptosis via the mitochondrial pathway. [34]. To test whether ALA-SDT could induce the same effect in advanced plaque in vivo, TUNEL staining of the aortic root of apoE-/- mice was performed. Comparing to the control mice's plaques, ALA-SDT significantly induced apoptosis within atherosclerotic plaques (Figure 3A). The apoptosis rate in ALA-SDT-treated mice peaked at 1 d after ALA-SDT treatment, and then slowly decreased over time (Figure 3B). At 28 d after treatment, the percentage of apoptotic cells in ALA-SDT-treated mice's plaques approximately returned to the same level as that in the control mice.
In advanced plaques, apoptosis of foam cells is usually induced by chronic ER stress and results in inflammatory responses [41, 42]. To test whether ALA-SDT could induce a different apoptosis pathway as observed in vitro [34] other than chronic ER stress, we further detected the released cytochrome C, cleaved caspapase-3 and C/EBP homologous protein (CHOP) expression in plaques. As shown in Figure 3C-D, the percentage of cytochrome c-positive macrophages in plaques was significantly higher in the SDT 1 d mice compared to the control 1 d and other ALA-SDT-treated mice sacrificed at different time points. The percentage of cleaved caspase 3-positive macrophages also showed the same pattern among different groups (Figure 3E-F). Furthermore, we found that marker of ER stress CHOP-positive macrophages extensively decreased in all ALA-SDT-treated mice compared with the control mice (Figure 3G-H). Western blotting analysis further supported the results of immunofluorescence staining. Blotting results showed that, compared to control mice, the protein levels of cytochrome C and cleaved caspase 3 in atherosclerotic plaques was greatly elevated after ALA-SDT treatment, while CHOP protein expression drastically diminished (Figure 3I-L).
These results suggest that ALA-SDT treatment results in acute mitochondria pathway apoptosis of foam cells, and also considerably reduces ER stress in advanced plaques. The decrease of CHOP-positive macrophages is possibly due to the increase of foam cell mitochondria pathway apoptosis after ALA-SDT treatment.
Figure 3
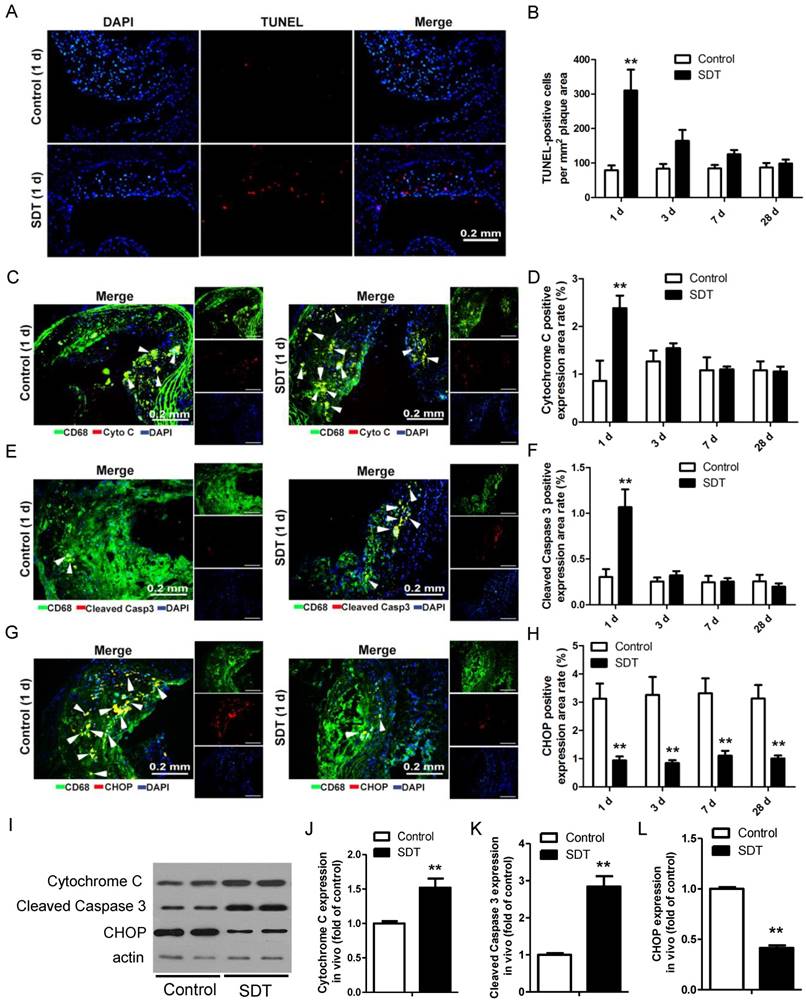
ALA-SDT treatment induces foam cell mitochondria pathway apoptosis and decreases ER stress in mouse advanced atherosclerotic plaque. (A) Representative fluorescence micrographs of aortic root stained with TUNEL (red) for apoptotic cells and counterstained with DAPI (blue) for the nuclei. (B) Quantification of TUNEL-positive apoptotic cells per mm2 plaque area (n=9). (C, E, G) Representative fluorescence micrographs of aortic root double stained with CD68 and cytochrome c (C), cleaved caspase 3 (E) or CHOP (G) (arrow) and counterstained with DAPI for nuclei. (D, F, H) Quantification of corresponding double-positive expression area in the aortic root of each group in (C, E, G) (n=9). (I) Aorta were collected 1 day after ALA-SDT treatment and cytochrome c, cleaved caspase 3 and CHOP expressions were detected by western blot. (J-L) Quantification of the protein expression of cytochrome c (J), cleaved caspase 3 (J) and CHOP (L) in aorta 1 day after treatment (n=6). Data are expressed as mean ± SD; **P<0.01 compared with control group.
Figure 4
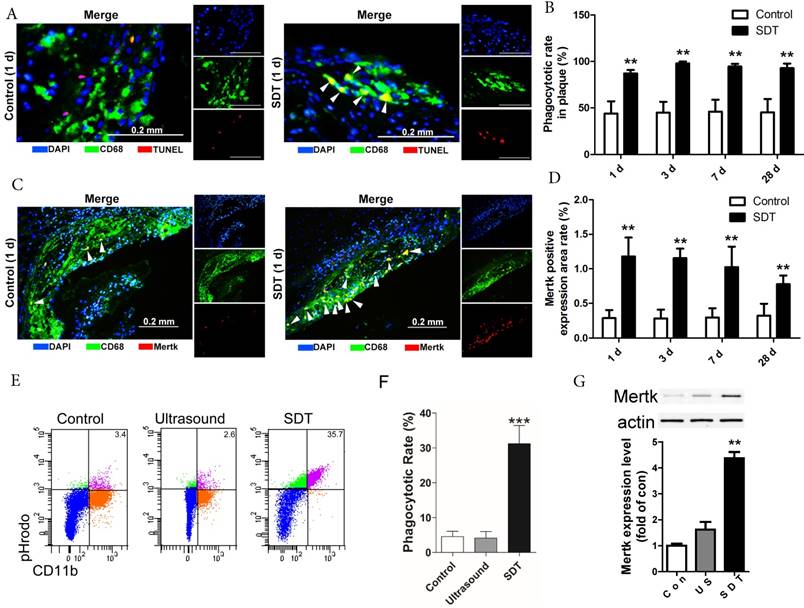
ALA-SDT treatment increases efferocytosis both in vivo and in vitro. (A) Representative fluorescence micrographs of aortic root double-stained with TUNEL (red) and CD68 (green). Efferocytosis was defined as the merging of TUNEL-positive/CD68-positive (yellow, arrow) images. (B) Quantitative analysis of phagocytotic rate in plaques after ALA-SDT treatment (n=9). (C) Representative fluorescence micrographs of aortic root double-stained with CD68 and MerTK (yellow, arrow). (D) Quantitative analysis of MerTK-positive expression area rate in plaques (n=9). (E-G) Five hours after SDT treatment, the supernatants of foam cells were collected and added to fresh foam cells (phagocytes) for 4 h. Then, the phagocytes were washed twice and fresh apoptotic cells (AC) labeled with pHrodo TM Red SE were added to phagocytes. 1 h later, the non-ingested AC were removed from the phagocytes. (E-F) Phagocytes were incubated with Percp-anti-CD11b and detected by flow cytometry. Representative flow cytograms (E) and quantification (F) revealing phagocytosis of phagocytes in different groups. (G) MerTK expression in phagocytes after co-incubation with supernatants of foam cells from different treatment groups was detected by western blot. The assessment was performed in triplicate. Data are expressed as mean ± SD; **P<0.01 vs. control group, ^^P<0.01 vs. ultrasound group.
ALA-SDT treatment increases efferocytosis both in vivo and in vitro
In advanced atherosclerotic lesions, almost all of phagocytes are loaded with free cholesterol and vulnerable to cell death, reducing their phagocytic ability [36, 37, 43]. In this setting, apoptotic foam cells cannot be efficiently cleared out by efferocytosis and undergo secondary necrosis, leading to expansion of the necrotic core. In our results, apoptotic cells drastically elevated in atherosclerotic plaques after ALA-SDT treatment (Figure 3A), accompanied by efferocytosis defined as the merging of TUNEL-positive/CD68-positive cells (Figure 4A), while in control atherosclerotic plaque, free apoptotic cells were detected defined as TUNEL-positive alone cells, indicating defective phagocytosis (Figure 4A). Surprisingly, although the apoptosis induced by ALA-SDT treatment peaked at 1 d after the treatment and then went down to background levels at 28 d (Figure 3B), the efferocytosis rate remained at a high level (~100%) throughout the 28 days following the treatment (Figure 4B). This result indicates that ALA-SDT breaks the detrimental environment in advanced atheroma and changes towards a beneficial balance.
The Mer receptor tyrosine kinase (MerTK) is critical for the engulfment and efficient clearance of apoptotic cells, and MerTK-deficient mice showed inefficient removal of apoptotic cells [37, 44, 45]. Next, we investigated whether ALA-SDT-induced efferocytosis was associated with altered MerTK expression. As shown in Figure 4C-D, MerTK-positive macrophages were dramatically elevated in atherosclerotic plaques of ALA-SDT-treated mice and sustained until 28 d after the treatment, enhancing the phagocytic capability of macrophages to clear apoptotic cells. This result provides a potential evidence for the continuous efferocytosis in the plaque after ALA-SDT treatment.
To further validate the effects of ALA-SDT on phagocytosis, we carried out an in vitro co-incubation assay. The same number of pHrodoTM Red SE-labeled apoptotic cells were incubated with phagocytes that were pre-incubated with supernatant from different treatment cells. The results revealed that the phagocytes treated with supernatant from the ALA-SDT group stimulated the most phagocytosis (Figure 4E-F). In addition, the protein levels of MerTK in ALA-SDT-treated phagocytes increased (Figure 4G).
The above results indicate that ALA-SDT treatment increases efferocytosis both in vivo and in vitro, partially due to the increase of MerTK expression in phagocytes.
ALA-SDT increases ABCA1 and ABCG1 expression both in vivo and in vitro
The observation that ALA-SDT decreased the lipid burden in atherosclerotic lesions made us hypothesize that ALA-SDT might modulate cholesterol efflux. To test this hypothesis, we stained atherosclerotic plaques with antibodies against ABCA1 and ABCG1, which are two members of the ABC family of transporter proteins and are mainly responsible for the efflux of phospholipid and cholesterol [15]. After ALA-SDT treatment, macrophages with positive staining of ABCA1 and ABCG1 were significantly elevated in atherosclerotic lesions (Figure 5A-D), suggesting that ALA-SDT-promoted cholesterol efflux is related to the increase in ABCA1 and ABCG1 expressions. Consistently, the levels of ABCA1 and ABCG1 detected by western blotting were notably enhanced after ALA-SDT treatment both in vivo (Figure 5E-G) and in vitro (Figure 5H-J).
Figure 5
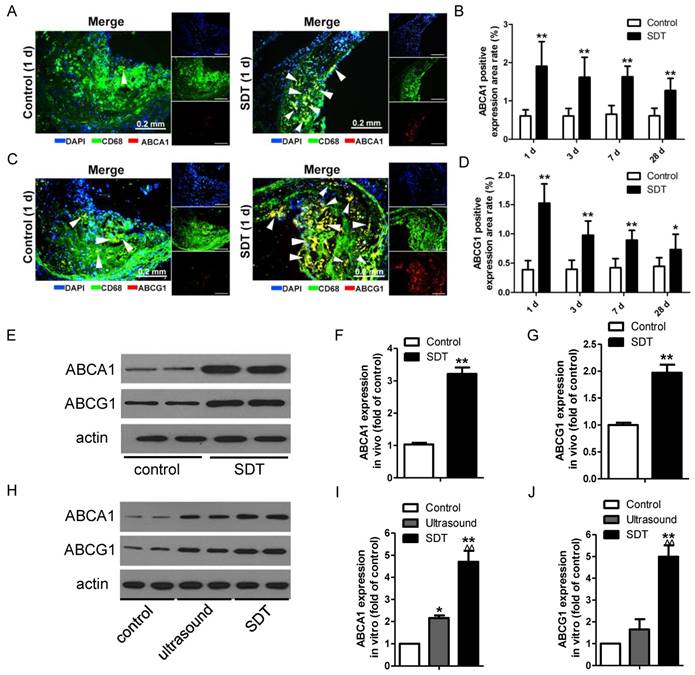
ALA-SDT treatment increases ABCA1 and ABCG1 expression both in vivo and in vitro. (A, C) Representative fluorescence micrographs of aortic root double-stained with CD68 and ABCA1 (A) or ABCG1 (C) (arrows). (B, D) Quantification of positive area of ABCA1 (B) and ABCG1 (D) in atherosclerotic lesions (n=9). (E) ABCA1 and ABCG1 expressions in atherosclerotic lesions were detected by western blot. (F-G) Quantification of ABCA1 (F) and ABCG1 (G) expressions in atherosclerotic lesions (n=6). (H) ABCA1 and ABCG1 expressions in phagocytes after co-incubation with supernatants of foam cells from different treatment groups were detected by western blot in vitro. (I-J) Quantification of ABCA1 (I) and ABCG1 (J) expressions in (H). The assessment was performed in triplicate. Data are expressed as mean ± SD; *P<0.05 vs. control group, **P<0.01 vs. control group, ^^P<0.01 vs. ultrasound group.
Figure 6
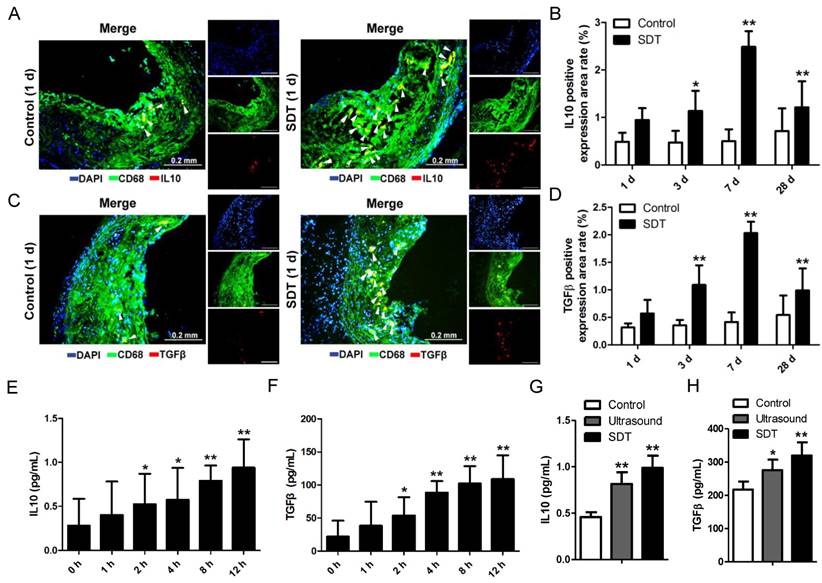
ALA-SDT treatment activates anti-inflammation reactions both in vivo and in vitro. (A, C) Representative fluorescence micrographs of aortic root double-stained with CD68 and IL10 (A) or TGFβ (C) (arrow). (B, D) Quantitative analysis of IL10-positive (B) or TGFβ-positive (D) expression area rate in mice plaques in vivo (n=9). (E-F) IL10 (E) and TGFβ (F) levels in culture medium of phagocytes after co-incubation with supernatants of foam cells treated with ALA-SDT were detected by ELISA in vitro. Data are expressed as mean ± SD; *P<0.05 and **P<0.01 vs. 0 h. (G-H) IL10 (G) and TGFβ (H) levels in culture medium of phagocytes after co-incubation with supernatants of foam cells from different treatment groups for 12 h were detected by ELISA in vitro. The assessments were performed in triplicate. Data are expressed as mean ± SD; *P<0.05 vs. control group, **P<0.01 vs. control group.
ALA-SDT treatment activates anti-inflammatory reactions both in vivo and in vitro
Lipoprotein retention in plaque initiates a vicious circle leading to inflammation enhancement. Thus, we wondered whether the increase of cholesterol efflux after ALA-SDT treatment was accompanied by a decrease of inflammation. In the advanced plaque in vivo, compared with control mice, the IL10 (Figure 6A-B) and TGFβ (Figure 6C-D) positive expression rates significantly increased after ALA-SDT treatment and peaked at 7 d after the treatment. This result suggests that ALA-SDT activates anti-inflammatory reactions in the advanced atheroma.
We further assessed the IL10 and TGFβ levels in supernatants of phagocytes in vitro. When co-incubated with medium from ALA-SDT-treated foam cells, the levels of IL10 and TGFβ released from fresh phagocytes increased with time (Figure 6E-F). At 12 h post incubation, the level of IL10 was triple that of basal, and that of TGFβ was five-fold that of basal (Figure 6E-F). Also, compared with the control group, the ALA-SDT-treated phagocytes released significantly higher levels of IL10 and TGFβ (Figure 6G-H).
ALA-SDT treatment induces PPARγ and LXRα expression both in vivo and in vitro
PPARγ and LXRα are two key upstream transcription factors regulating cholesterol efflux in macrophages. Strikingly, ALA-SDT induced an increase of the positive area rates of PPARγ and LXRα in the atherosclerotic plaque, which peaked at 1 d after the treatment, corresponding to the ABCA1 and ABCG1 results, then gradually went down (Figure 7A-D). For LXRα, the expression remained higher than that of the control group even at 28 d after the treatment (Figure 7D). Consistent with the results detected by immunofluorescent staining, the protein levels of PPARγ and LXRα were dramatically increased in the ALA-SDT-treated plaques (Figure 7E-G) and phagocytes pre-incubated with media from the ALA-SDT group (Figure 7H-J).
Figure 7
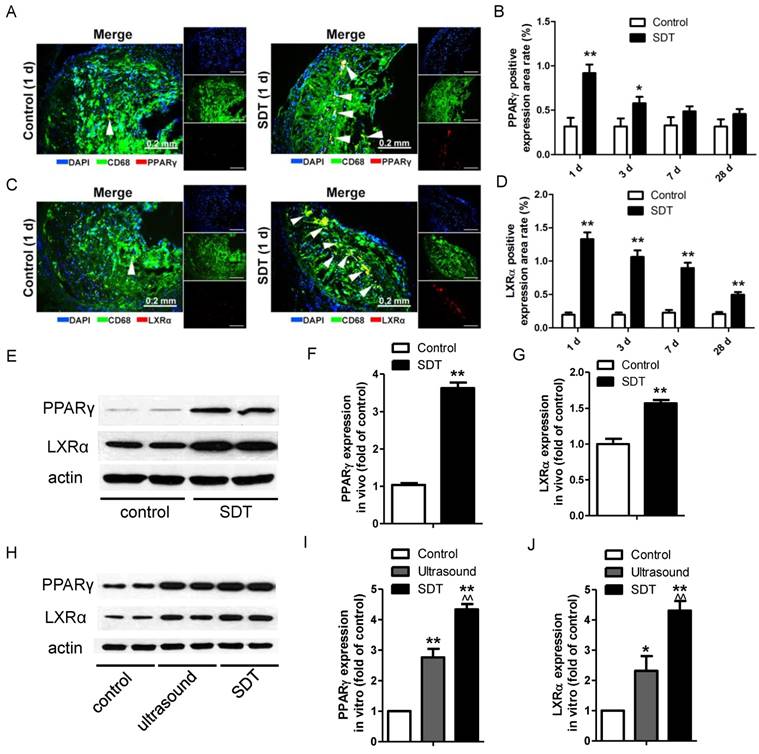
ALA-SDT treatment induces PPARγ and LXRα expression. (A, C) Representative fluorescence micrographs of aortic root double-stained with CD68 and PPARγ (A) or LXRα (C) (arrows). (B, D) Quantification of positive area of PPARγ (B) and LXRα (D) expression in mice plaques (n=9). (E) PPARγ and LXRα expressions in atherosclerotic lesions were detected by western blot. (F-G) Quantification of PPARγ (F) and LXRα (G) expressions in atherosclerotic lesions (n=6). (H) PPARγ and LXRα expressions in phagocytes after co-incubation with supernatants of foam cells from different treatment groups were detected by western blot in vitro. (I-J) Quantification of PPARγ (I) and LXRα (J) expressions in (H). The assessment was performed in triplicate. Data are expressed as mean ± SD; *P<0.05 vs. control group, **P<0.01 vs. control group, ^^P<0.01 vs. ultrasound group.
Promotion of cholesterol efflux induced by ALA-SDT is PPARγ dependent
Engulfed apoptotic cells provide natural ligands for PPARγ and stimulate cholesterol efflux out of phagocytes via the PPARγ-LXRα-ABCA1/ABCG1 pathway [15]. To further verify the function of PPARγ on cholesterol efflux induced by ALA-SDT, PPARγ was knocked down with small interfering RNA (siRNA) in phagocytes. Our results demonstrated that PPARγ was the initiator of this pathway, as knockdown of PPARγ strongly inhibited the increased expression of LXRα, ABCA1 and ABCG1 in phagocytes co-incubated with ALA-SDT-treated supernatant (Figure 8A-B). Pre-knockdown of PPARγ in phagocytes also reversed the reduction of total and free cholesterol contents induced by ALA-SDT (Figure 8C-D). Moreover, PPARγ antagonist GW9662 inhibited the ALA-SDT-induced cholesterol efflux effect in phagocytes, as indicated by Oil Red O staining (Figure 8E-F). Meanwhile, GW9662 blocked the phagocytosis and anti-inflammatory reactions in phagocytes induced by ALA-SDT treatment (Figure 8G-I). The above results suggest that ALA-SDT treatment enhances cholesterol efflux, phagocytosis and anti-inflammatory reactions by activating PPARγ in phagocytes.
Figure 8
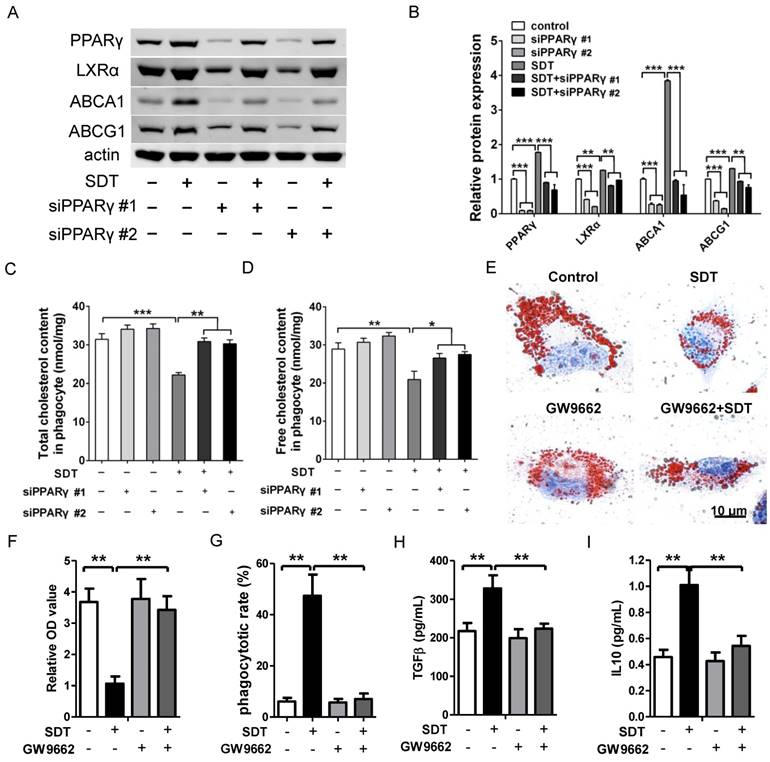
Promotion of cholesterol efflux induced by ALA-SDT is PPARγ dependent. (A-D) PPARγ was knocked down in phagocytes before exposing them to the supernatant of foam cells treated with ALA-SDT in vitro. (A-B) Expressions of PPARγ, LXRα, ABCA1 and ABCG1 in phagocytes were detected by western blot. (C-D) Quantification of total cholesterol (C) and free cholesterol (D) contents in phagocytes. (E-I) Phagocytes were pretreated with 10 μM of the PPARγ antagonist GW9662 for 1 h before exposing them to the supernatant of foam cells treated with ALA-SDT in vitro. (E-F) After incubation with supernatants from foam cells treated by ALA-SDT for 4 h, the phagocytes were incubated for an additional 12 h in serum-free medium containing apoA1 and HDL. (E) Representative microscopic photographs of phagocytes stained with Oil Red O. (F) Quantification of the relative OD value in phagocytes in (E). (G) Comparison of phagocytotic rate in phagocytes preincubated with GW9662. (H-I) Comparison of TGFβ (H) and IL10 (I) levels in culture medium of phagocytes preincubated with GW9662 after co-incubation with supernatants of foam cells stimulated by ALA-SDT. The assessment was performed in triplicate. Data are expressed as mean ± SD; *P<0.05, **P<0.01, ***P<0.001.
Discussion
During the progression of atherosclerosis, continuous accumulation of lipid in foam cells induces chronic ER stress that eventually leads to foam cell apoptosis. Due to the defective phagocytosis in advanced atherosclerotic plaques, apoptotic foam cells cannot be efficiently cleared out and release intracellular cholesterol into plaques. Released cholesterol further enhances inflammatory responses and contributes to lesion progression. In this study, we administered ALA-SDT to western diet-fed apoE-/- mice and THP-1 macrophage-derived foam cells, and demonstrated that the activation of PPARγ-LXRα-ABCA1/ABCG1 pathway following ALA-SDT-induced foam cell apoptosis stimulates effective phagocytosis and enhances cholesterol efflux and anti-inflammatory responses.
Ultrasound is emerging as a promising modality in various medical areas, such as cancer and cardiovascular diseases [29, 30]. Low-frequency ultrasound provides an additive effect to antiplatelet, antithrombotic and fibrinolytic drug therapies [46]. In a rabbit balloon angioplasty and stenting model, a chlorin derivative (PAD-S31) activated by transdermal ultrasonic irradiation inhibited neointimal hyperplasia [47]. Ultrasound activated PpIX, a sonosensitizer and metabolite of ALA, and induced mitochondria-specific damage, which led to foam cell apoptosis [31-33]. In our previous study, we treated THP-1 macrophage-derived foam cells with ALA-SDT and found that ALA-SDT induced foam cell mitochondrial pathway apoptosis due to the generation of ROS [34], which was further confirmed by our current study in the advanced plaque of apoE-/- mice. The specific accumulation of ALA-PpIX in the mitochondria of macrophages in the advanced plaque and the generation of ROS upon ultrasound irradiation makes the cell death of foam cells quite different from that which naturally occurs in late-stage atherosclerotic plaques. The distinctions between these two types of cell death of foam cells are described as follows: 1) the former is mediated by the mitochondrial pathway, which is the preferred cell death mode in normal tissues since it does not trigger an inflammatory response, whereas the latter is induced by chronic ER stress and results in inflammatory responses [41, 42]; 2) the former is acute apoptosis that occurs as rapidly as within 5 h in vitro and 1 d in vivo (Figure 3B), whereas the latter is a chronic cell death that can take as long as years. Additionally, no systemic side effect of ALA-SDT treatment was detected in vivo (Figure S5).
ALA-SDT not only induced foam-cell apoptosis, but also enhanced the clearance of apoptotic foam cells. In advanced atherosclerotic lesions, almost all phagocytes are loaded with free cholesterol and are vulnerable to death, which reduces their phagocytic ability [36, 37, 43]. In this setting, apoptotic foam cells cannot be efficiently cleared out by efferocytosis and undergo secondary necrosis, which leads to expansion of the necrotic core. Hence, both the increase of foam cell apoptosis and enhancement of efferocytosis triggered by ALA-SDT would significantly inhibit the progression of atherosclerosis. The increase of phagocytosis efficiency after ALA-SDT treatment is probably due to the apoptosis of the disabled phagocytes in the advanced plaques and recruitment of healthy circulating monocytes by locally elevated ATP [35]. The MerTK receptor on macrophages mediates the engulfment of apoptotic cells, and its shedding in advanced plaques contributes to the accumulation of apoptotic cells and the formation of a necrotic core [37, 44, 45]. The up-regulation of the MerTK receptor on phagocytes following ALA-SDT results in the efferocytosis enhancement (Figure 4).
Figure 9
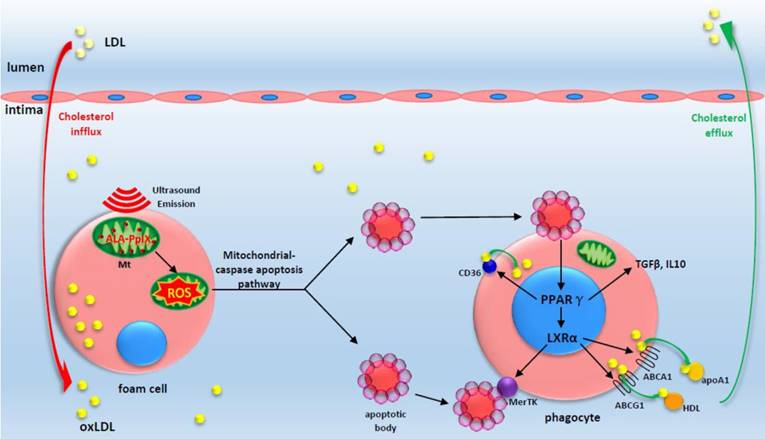
A proposed theoretical model of the anti-atherosclerosis mechanism of ALA-SDT. ALA-PpIX accumulates in mitochondria of foam cells in the advanced plaque. Once exposed to ultrasound, the foam cells undergo apoptosis via mitochondria-caspase pathway. This process activates the phagocytic PPARγ-LXRα-ABCA1/ABCG1 pathway and results in the enhancement of efferocytosis, cholesterol efflux as well as anti-inflammatory responses, thus eventually ameliorating atherosclerosis. The red arrow indicates cholesterol influx into the plaque; the green arrow indicates cholesterol efflux out of the plaque by apoA1 and HDL.
Numerous studies suggest that defective cholesterol efflux plays a critical role in atherogenesis. In other words, acceleration of cholesterol export from atherosclerotic plaque may result in amelioration of atherosclerosis. In the present study, we found that ALA-SDT enhanced cholesterol efflux, which was concurrent with the regression of lesion size. The plaque lipid efflux was detected as early as 1 d after ALA-SDT treatment (Figure 2B). However, the rapid lipid export from atherosclerotic plaque was not attributed to changes in blood cholesterol levels, which display no obvious difference between the control and SDT groups (Figure S6). To date, the only successful model of rapid regression of atherosclerosis was established by Fisher et al. They transplanted a segment of plaque-containing aorta from a hyperlipidemic apoE-/- mouse into a wild-type recipient and found atherosclerotic regression 3 d after transplantation [10]. Their results confirmed that lipid efflux occurs surprisingly rapidly once the plaque milieu is improved. In the current study, we observed rapid ROS production in advanced plaque by ALA-SDT treatment, followed by an increase of ABCA1 and ABCG1 expression and a decrease of lipid area at one day after the treatment. A previous study showed that subendothelial lipoprotein-matrix interaction hinders cholesterol efflux by inhibiting movement of lipid particles as well as emigration of macrophages [3]. We wondered whether ultrasonic oscillation may physically disrupt the lipid-matrix interaction and facilitate cholesterol efflux. However, ultrasound treatment alone didn't decrease the lipid area or plaque area (Figure S1A-B). In addition, it didn't induce a significant increase of apoptosis or an increase in ROS production in vivo (Figure S1C-D). These results indicate that the ROS production from the combination of ALA and ultrasound plays a determinant role in plaque regression by ALA-SDT treatment. Whether ultrasonic oscillation can break the physical interaction between lipoprotein particles and matrix and contributes to the cholesterol efflux and macrophage emigration still needs additional exploration.
The important roles of inflammation in promoting atherosclerosis are now well documented. In advanced plaques, chronic ER stress-induced apoptotic foam cells elicit an inflammatory response [21, 48]. ALA-SDT drastically improved the inflammatory environment, increasing the expressions of IL10 and TGFβ (Figure 6). This is probably due to the fact that ALA-SDT induces apoptosis of foam cells mainly via the mitochondrial pathway, enhances the clearance of apoptotic foam cells and increases the recruitment of circulating monocytes [35]. Indeed, ER stress marker protein CHOP, which is strongly correlated with plaque rupture [21], was clearly decreased after ALA-SDT (Figure 3H), suggesting ALA-SDT corrects the defective phagocytosis in advanced plaque and destroys the inflammatory initiator.
Regarding the potential mechanism of these beneficial consequences of ALA-SDT treatment, nuclear receptor PPARγ plays critical roles. First, the activation of PPARγ in phagocytes upon recognition of PtdSer on apoptotic cells induces the expression of LXRα [15], and LXRα promotes efficient clearance of apoptotic cells by inducing expression of the MerTK gene, a receptor tyrosine kinase that is critical for phagocytosis [22]. In our study, we verified that ALA-SDT treatment increased MerTK expression in the plaque and the upregulation of MerTK was sustained for 28 d (Figure 4D). Secondly, the activation of PPARγ and LXRα interferes with NF-κB and STAT activity and performs potent anti-inflammatory functions in macrophages [13, 19-21]. In this study, PPARγ-dependent anti-inflammatory responses were detected in vivo and in vitro (Figure 6 and Figure 8). Lastly, PPARγ and LXRα play key roles in cholesterol efflux from macrophages by inducing ABCA1 and ABCG1 expression. In the present study, we detected activation of PPARγ-LXRα-ABCA1/ABCG1 pathway and a rapid decrease in plaque lipids after ALA-SDT treatment (Figure 2A). Because of the limitations of lipid staining and cholesterol analysis, we cannot distinguish intracellular lipids from extracellular lipids in the lesions. Nevertheless, in the in vitro study, we found that 40% more intracellular free cholesterol was exported out of cells 12 h after ALA-SDT intervention compared with the control group (Figure 2F). The macrophage scavenger receptor CD36 mediates apoptotic cells and oxLDL engulfment and is a target gene of PPARγ [49]. Loss of CD36 does not ameliorate atherosclerosis in hyperlipidemic mice but increases aortic sinus lesion areas [50]. In this study, an upregulation of CD36 was found both in vivo (Figure S7A-D) and in vitro (Figure S7E-F) after ALA-SDT treatment, indicating that ALA-SDT simultaneously enhances the cholesterol influx and efflux abilities of phagocytes in the plaque, contributing to the clearance of lipid deposit outside the phagocytes, and the net effect is an increase in lipid efflux. Based on these results, we infer that the intracellular and extracellular lipids in the plaque decreased concurrently after ALA-SDT treatment. However, there was no obvious cholesterol efflux detected in the ultrasound group compared with the control group in vitro (Figure 2F). A potential explanation might be a balance of cholesterol influx and efflux in the ultrasound group.
ALA-SDT-mediated rapid cholesterol export may exert more beneficial effects than chronic interventions, such as oral PPARγ and LXR activators, since it occurs locally without off-target toxicity [51]. Indeed, ALA-SDT somehow activates the PPARγ-LXRα-ABCA1/ABCG1 pathway, which at least partially results in the enhancement of apoptotic cell efferocytosis and cholesterol efflux and the improvement of the inflammatory environment in advanced plaques (Figure 9). We are not quite sure how ALA-SDT activates the PPARγ-LXRα-ABCA1/ABCG1 pathway. In our in vitro study, we demonstrated that the PPARγ-LXRα-ABCA1/ABCG1 pathway is activated when phagocytes are incubated with media from the ALA-SDT-treated foam cells (Figure 7H), indicating that the PtdSer-containing apoptotic particles or cytokines secreted by ALA-SDT-induced apoptotic cells activate this pathway in phagocytes. However, other possibilities cannot be excluded. For instance, ALA-SDT somehow directly activates this pathway.
In summary, ALA-SDT ameliorates atherosclerosis by inducing foam cell mitochondria pathway apoptosis, enhancing efferocytosis, and improving cholesterol efflux and the inflammatory environment (Figure 9). The advantage of local treatment by ALA-SDT is the avoidance of off-target and systemic side effects. Additionally, no endothelium damage was detected after ALA-SDT treatment [35]. In a pilot clinical trial including patients with atherosclerotic peripheral artery disease, a combination of SDT and atorvastatin treatment abrogated the progression of atherosclerosis within four weeks, and the efficacy lasted until 40 weeks follow-up [35]. Thus, ALA-SDT might become a novel therapeutic strategy for atherosclerosis.
Abbreviations
ABCA1: ATP-binding cassette transporter A1; ABCG1: ATP-binding cassette transporter G1; ALA: 5-aminolevulinic acid; ApoA1: apolipoprotein A1; BSA: bovine serum albumin; CD36: cluster of differentiation 36; CD68: cluster of differentiation 68; CHOP: C/EBP homologous protein; ER: endoplasmic reticulum; HDL: high density lipoprotein; IL-10: interleukin 10; LXRα: liver X receptor alpha; MerTK: Mer tyrosine kinase; oxLDL: oxidized low density lipoprotein; PDT: photodynamic therapy; PPARγ: peroxisome proliferator-activated receptor gamma; PpIX: protoporphyrin IX; ROS: reactive oxygen species; SDT: sonodynamic therapy; TGFβ: transforming growth factor beta; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling; US: ultrasound.
Supplementary Material
Supplementary figures.
Acknowledgements
This study was supported by the Major Research Equipment Development Project of National Natural Science of China (81727809), the State Key Project of National Natural Science of China (81530052), the National Natural Science Foundation of China (81400339, 81701848) and the Major State Basic Research Development Program of China (2017YFB0403800). This study was also supported by Research Project of the First Affiliated Hospital of Harbin Medical University (2015B006).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gistera A, Hansson GK. The immunology of atherosclerosis. Nat Rev Nephrol. 2017;13:368-80
2. Cochain C, Zernecke A. Macrophages and immune cells in atherosclerosis: recent advances and novel concepts. Basic Res Cardiol. 2015;110:34
3. Moreno PR, Sanz J, Fuster V. Promoting mechanisms of vascular health: circulating progenitor cells, angiogenesis, and reverse cholesterol transport. J Am Coll Cardiol. 2009;53:2315-23
4. Boren J, Williams KJ. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. 2016;27:473-83
5. McLaren JE, Michael DR, Ashlin TG, Ramji DP. Cytokines, macrophage lipid metabolism and foam cells: implications for cardiovascular disease therapy. Prog Lipid Res. 2011;50:331-47
6. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341-55
7. Cui D, Thorp E, Li Y, Wang N, Yvan-Charvet L, Tall AR. et al. Pivotal advance: macrophages become resistant to cholesterol-induced death after phagocytosis of apoptotic cells. J Leukoc Biol. 2007;82:1040-50
8. McMahan CA, McGill HC, Gidding SS, Malcom GT, Newman WP, Tracy RE. et al. PDAY risk score predicts advanced coronary artery atherosclerosis in middle-aged persons as well as youth. Atherosclerosis. 2007;190:370-7
9. Williams KJ, Feig JE, Fisher EA. Rapid regression of atherosclerosis: insights from the clinical and experimental literature. Nat Clin Pract Cardiovasc Med. 2008;5:91-102
10. Williams KJ, Feig JE, Fisher EA. Cellular and molecular mechanisms for rapid regression of atherosclerosis: from bench top to potentially achievable clinical goal. Curr Opin Lipidol. 2007;18:443-50
11. Schumacher T, Benndorf RA. ABC Transport Proteins in Cardiovascular Disease-A Brief Summary. Molecules. 2017;22:589
12. Westerterp M, Bochem AE, Yvan-Charvet L, Murphy AJ, Wang N, Tall AR. ATP-binding cassette transporters, atherosclerosis, and inflammation. Circ Res. 2014;114:157-70
13. Duan SZ, Usher MG, Mortensen RM. Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature. Circ Res. 2008;102:283-94
14. Babaev VR, Yancey PG, Ryzhov SV, Kon V, Breyer MD, Magnuson MA. et al. Conditional knockout of macrophage PPARgamma increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:1647-53
15. Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol. 2007;7:964-74
16. Ricote M, Valledor AF, Glass CK. Decoding transcriptional programs regulated by PPARs and LXRs in the macrophage: effects on lipid homeostasis, inflammation, and atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:230-9
17. Li AC, Glass CK. The macrophage foam cell as a target for therapeutic intervention. Nat Med. 2002;8:1235-42
18. Choudhury RP, Lee JM, Greaves DR. Mechanisms of disease: macrophage-derived foam cells emerging as therapeutic targets in atherosclerosis. Nat Clin Pract Cardiovasc Med. 2005;2:309-15
19. Bensinger SJ, Tontonoz P. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature. 2008;454:470-7
20. Bouhlel MA, Derudas B, Rigamonti E, Dievart R, Brozek J, Haulon S. et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137-43
21. Li Y, Gerbod-Giannone MC, Seitz H, Cui D, Thorp E, Tall AR. et al. Cholesterol-induced apoptotic macrophages elicit an inflammatory response in phagocytes, which is partially attenuated by the Mer receptor. J Biol Chem. 2006;281:6707-17
22. N AG, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N. et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245-58
23. Argmann CA, Sawyez CG, McNeil CJ, Hegele RA, Huff MW. Activation of peroxisome proliferator-activated receptor gamma and retinoid X receptor results in net depletion of cellular cholesteryl esters in macrophages exposed to oxidized lipoproteins. Arterioscler Thromb Vasc Biol. 2003;23:475-82
24. Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA. et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci U S A. 2002;99:7604-9
25. Naik SU, Wang X, Da Silva JS, Jaye M, Macphee CH, Reilly MP. et al. Pharmacological activation of liver X receptors promotes reverse cholesterol transport in vivo. Circulation. 2006;113:90-7
26. Linsel-Nitschke P, Tall AR. HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat Rev Drug Discov. 2005;4:193-205
27. Groot PH, Pearce NJ, Yates JW, Stocker C, Sauermelch C, Doe CP. et al. Synthetic LXR agonists increase LDL in CETP species. J Lipid Res. 2005;46:2182-91
28. Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol. 2010;10:36-46
29. Shibaguchi H, Tsuru H, Kuroki M, Kuroki M. Sonodynamic cancer therapy: a non-invasive and repeatable approach using low-intensity ultrasound with a sonosensitizer. Anticancer Res. 2011;31:2425-9
30. Costley D, Mc Ewan C, Fowley C, McHale AP, Atchison J, Nomikou N. et al. Treating cancer with sonodynamic therapy: a review. Int J Hyperthermia. 2015;31:107-17
31. Wang X, Wang P, Tong W, Liu Q. Comparison of pharmacokinetics, intracellular localizations and sonodynamic efficacy of endogenous and exogenous protoporphyrin IX in sarcoma 180 cells. Ultrasonics. 2010;50:803-10
32. He Y, Xia X, Xu C, Yu H, Bai D, Xiang J. et al. 5-Aminolaevulinic acid enhances ultrasound-induced mitochondrial damage in K562 cells. Ultrasonics. 2010;50:777-81
33. Lv Y, Fang M, Zheng J, Yang B, Li H, Xiuzigao Z. et al. Low-intensity ultrasound combined with 5-aminolevulinic acid administration in the treatment of human tongue squamous carcinoma. Cell Physiol Biochem. 2012;30:321-33
34. Wang H, Yang Y, Chen H, Dan J, Cheng J, Guo S. et al. The predominant pathway of apoptosis in THP-1 macrophage-derived foam cells induced by 5-aminolevulinic acid-mediated sonodynamic therapy is the mitochondria-caspase pathway despite the participation of endoplasmic reticulum stress. Cell Physiol Biochem. 2014;33:1789-801
35. Sun X, Guo S, Yao J, Wang H, Peng C, Li B. et al. Rapid Inhibition of Atherosclerotic Plaque Progression by Sonodynamic Therapy. Cardiovasc Res. 2018 doi:10.1093/cvr/cvy139
36. Schrijvers DM, De Meyer GR, Kockx MM, Herman AG, Martinet W. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:1256-61
37. Thorp E, Cui D, Schrijvers DM, Kuriakose G, Tabas I. Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe-/- mice. Arterioscler Thromb Vasc Biol. 2008;28:1421-8
38. Miksa M, Komura H, Wu R, Shah KG, Wang P. A novel method to determine the engulfment of apoptotic cells by macrophages using pHrodo succinimidyl ester. J Immunol Methods. 2009;342:71-7
39. Xue JH, Yuan Z, Wu Y, Liu Y, Zhao Y, Zhang WP. et al. High glucose promotes intracellular lipid accumulation in vascular smooth muscle cells by impairing cholesterol influx and efflux balance. Cardiovasc Res. 2010;86:141-50
40. Peng C, Li Y, Liang H, Cheng J, Li Q, Sun X. et al. Detection and photodynamic therapy of inflamed atherosclerotic plaques in the carotid artery of rabbits. J Photochem Photobiol B. 2011;102:26-31
41. Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184-90
42. Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965-75
43. Michlewska S, Dransfield I, Megson IL, Rossi AG. Macrophage phagocytosis of apoptotic neutrophils is critically regulated by the opposing actions of pro-inflammatory and anti-inflammatory agents: key role for TNF-alpha. FASEB J. 2009;23:844-54
44. Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum BC, Henson PM. et al. A soluble form of the Mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood. 2007;109:1026-33
45. Garbin U, Baggio E, Stranieri C, Pasini A, Manfro S, Mozzini C. et al. Expansion of necrotic core and shedding of Mertk receptor in human carotid plaques: a role for oxidized polyunsaturated fatty acids? Cardiovasc Res. 2013;97:125-33
46. Atar S, Luo H, Birnbaum Y, Nagai T, Siegel RJ. Augmentation of in-vitro clot dissolution by low frequency high-intensity ultrasound combined with antiplatelet and antithrombotic drugs. J Thromb Thrombolysis. 2001;11:223-8
47. Arakawa K, Hagisawa K, Kusano H, Yoneyama S, Kurita A, Arai T. et al. Sonodynamic therapy decreased neointimal hyperplasia after stenting in the rabbit iliac artery. Circulation. 2002;105:149-51
48. Li Y, Schwabe RF, DeVries-Seimon T, Yao PM, Gerbod-Giannone MC, Tall AR. et al. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis. J Biol Chem. 2005;280:21763-72
49. Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93:229-40
50. Moore KJ, Kunjathoor VV, Koehn SL, Manning JJ, Tseng AA, Silver JM. et al. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J Clin Invest. 2005;115:2192-201
51. Cuchel M, Rader DJ. Macrophage reverse cholesterol transport: key to the regression of atherosclerosis? Circulation. 2006;113:2548-55
Author contact
![]() Corresponding author: Ye Tian, Department of Cardiology, The First Affiliated Hospital, Cardiovascular Institute, Harbin Medical University, 23 Youzheng Street, Harbin 150001 (China), Tel.: +86-451-85555943; Fax: +86-451-87530341; E-Mail: yetianhrbmu.edu.cn
Corresponding author: Ye Tian, Department of Cardiology, The First Affiliated Hospital, Cardiovascular Institute, Harbin Medical University, 23 Youzheng Street, Harbin 150001 (China), Tel.: +86-451-85555943; Fax: +86-451-87530341; E-Mail: yetianhrbmu.edu.cn
Citation styles
APA
Wang, H., Yang, Y., Sun, X., Tian, F., Guo, S., Wang, W., Tian, Z., Jin, H., Zhang, Z., Tian, Y. (2018). Sonodynamic therapy-induced foam cells apoptosis activates the phagocytic PPARγ-LXRα-ABCA1/ABCG1 pathway and promotes cholesterol efflux in advanced plaque. Theranostics, 8(18), 4969-4984. https://doi.org/10.7150/thno.26193.
ACS
Wang, H.; Yang, Y.; Sun, X.; Tian, F.; Guo, S.; Wang, W.; Tian, Z.; Jin, H.; Zhang, Z.; Tian, Y. Sonodynamic therapy-induced foam cells apoptosis activates the phagocytic PPARγ-LXRα-ABCA1/ABCG1 pathway and promotes cholesterol efflux in advanced plaque. Theranostics 2018, 8 (18), 4969-4984. DOI: 10.7150/thno.26193.
NLM
Wang H, Yang Y, Sun X, Tian F, Guo S, Wang W, Tian Z, Jin H, Zhang Z, Tian Y. Sonodynamic therapy-induced foam cells apoptosis activates the phagocytic PPARγ-LXRα-ABCA1/ABCG1 pathway and promotes cholesterol efflux in advanced plaque. Theranostics 2018; 8(18):4969-4984. doi:10.7150/thno.26193. https://www.thno.org/v08p4969.htm
CSE
Wang H, Yang Y, Sun X, Tian F, Guo S, Wang W, Tian Z, Jin H, Zhang Z, Tian Y. 2018. Sonodynamic therapy-induced foam cells apoptosis activates the phagocytic PPARγ-LXRα-ABCA1/ABCG1 pathway and promotes cholesterol efflux in advanced plaque. Theranostics. 8(18):4969-4984.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.