Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
Conflicts of Interest
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(18):4957-4968. doi:10.7150/thno.27868 This issue Cite
Research Paper
Efficacy of chronic BACE1 inhibition in PS2APP mice depends on the regional Aβ deposition rate and plaque burden at treatment initiation
Matthias Brendel1, Anna Jaworska2,3, Felix Overhoff1, Tanja Blume1,2, Federico Probst1, Franz-Josef Gildehaus1, Peter Bartenstein1,4, Christian Haass2,4,5, Bernd Bohrmann6, Jochen Herms2,4, Michael Willem5, Axel Rominger1,4,7 ![]()
1. Department of Nuclear Medicine, University Hospital, LMU Munich; Munich, Germany
2. DZNE - German Center for Neurodegenerative Diseases, Munich, Germany
3. Laboratory of Neurodegeneration, International Institute of Molecular and Cell Biology, Warsaw, Poland
4. Munich Cluster for Systems Neurology (SyNergy), Munich, Germany
5. Biomedical Center (BMC), Ludwig-Maximilians-University Munich, 81377 Munich, Germany
6. F. Hoffmann-La Roche, Basel, Switzerland
7. Department of Nuclear Medicine, Inselspital, University Hospital Bern, Bern, Switzerland.
Received 2018-6-13; Accepted 2018-8-14; Published 2018-9-9
Abstract

Beta secretase (BACE) inhibitors are promising therapeutic compounds currently in clinical phase II/III trials. Preclinical [18F]-florbetaben (FBB) amyloid PET imaging facilitates longitudinal monitoring of amyloidosis in Alzheimer's disease (AD) mouse models. Therefore, we applied this theranostic concept to investigate, by serial FBB PET, the efficacy of a novel BACE1 inhibitor in the PS2APP mouse, which is characterized by early and massive amyloid deposition.
Methods: PS2APP and C57BL/6 (WT) mice were assigned to treatment (PS2APP: N=13; WT: N=11) and vehicle control (PS2APP: N=13; WT: N=11) groups at the age of 9.5 months. All animals had a baseline PET scan and follow-up scans at two months and after completion of the four-month treatment period. In addition to this longitudinal analysis of cerebral amyloidosis by PET, we undertook biochemical amyloid peptide quantification and histological amyloid plaque analyses after the final PET session.
Results: BACE1 inhibitor-treated transgenic mice revealed a progression of the frontal cortical amyloid signal by 8.4 ± 2.2% during the whole treatment period, which was distinctly lower when compared to vehicle-treated mice (15.3 ± 4.4%, p<0.001). A full inhibition of progression was evident in regions with <3.7% of the increase in controls, whereas regions with >10% of the increase in controls showed only 40% attenuation with BACE1 inhibition. BACE1 inhibition in mice with lower amyloidosis at treatment initiation showed a higher efficacy in attenuating progression to PET. A predominant reduction of small plaques in treated mice indicated a main effect of BACE1 on inhibition of de novo amyloidogenesis.
Conclusions: This theranostic study with BACE1 treatment in a transgenic AD model together with amyloid PET monitoring indicated that progression of amyloidosis is more effectively reduced in regions with low initial plaque development and revealed the need of an early treatment initiation during amyloidogenesis.
Keywords: BACE1 inhibitor, transgenic AD mouse model, [18F]-florbetaben PET, amyloid deposition rate
Introduction
Alzheimer's disease (AD) is the most common form of neurodegenerative dementia, and has an enormous impact on societies with aging populations [1]. The core histopathological features for diagnosis of AD consist of neurofibrillary tangles and amyloid plaques [2], which can now be assessed by molecular imaging in vivo [3, 4]. The principal component of amyloid plaques, the amyloid β-peptide (Aβ), is a heterogeneous cleavage product of the β-amyloid precursor protein (APP). After initial ectodomain shedding of APP by the β-site amyloid precursor protein cleaving enzyme 1 (BACE1), which has many other known physiological substrates, the remaining APP C-terminal fragment (CTF-β) is subsequently cleaved by the intramembrane protease γ-secretase, thereby releasing free Aβ. Alternately, the smaller peptide fragment p3 is released following shedding by ADAM10. Additional fragments are formed via the η-secretase ectodomain shedding pathway, which is followed by a BACE1 or an ADAM10 cleavage to produce longer (Aη-α) or shorter (Aη-β) forms of the native Aη peptide [5]. However, the rate limiting step for production of the Aβ peptide is the activity of the β-secretase enzyme, which is widely considered to have a crucial role in the initiation of AD pathology. Current symptomatic therapeutic options for AD include acetylcholinesterase inhibitors [6] and NMDA receptor antagonists [7], both of which provide some transient amelioration of cognitive symptoms, but without any disease-modifying effects [8, 9]. With the advent of highly potent and BBB-permeable inhibitors of BACE1, this enzyme has emerged as a primary drug target for reducing Aβ deposition in the AD brain. Therefore, the development and in vivo characterization of BACE1 inhibitors as therapeutic agents is of highest interest, with the caveat that untoward side effects can occur because of the wide spectrum of identified BACE1 substrates, especially in the central nervous system [10]. Most of the hitherto available BACE1 inhibitors also block the activity of BACE2, a close homologue of BACE1, which may cause additional on-target side effects [11]. Nonetheless, several BACE1 inhibitors are in human clinical trials testing for efficacy and safety in individuals with pre-symptomatic or manifest AD [12]. However, these trials have not so far imparted cognitive improvement in AD patients, indicating the necessity of much earlier and sustained BACE1 inhibitor treatment to efficiently prevent the accumulation of Aβ in the brain [13].
Amyloid positron-emission-tomography (PET) has in recent years emerged as valuable tool for assessment of cortical amyloidosis in vivo, and has been designated as a gatekeeper for inclusion of patients in anti-amyloid treatment trials. For such trials it is crucial to determine the dose-response of BACE1 inhibition and to define the optimal stage of AD for initiation of effective intervention with specific BACE1 inhibitors [12]. By the same measure, Aβ-PET also serves as a non-invasive tool for monitoring the amyloid plaque burden in transgenic mice, yielding excellent correlations with histological or biochemical assessments [14-16]. In this regard, longitudinal preclinical trials of BACE1 inhibition using Aβ-PET as an endpoint are of great translational value, and are readily validated by ex vivo histological examinations. As in clinical studies, baseline Aβ-PET results can be used to construct comparable experimental animal groups, and to investigate preconditions for individual differences in the progression of pathology longitudinally [15].
Given this background, we aimed to apply a theranostic concept for monitoring by [18F]-florbetaben Aβ-PET the progression of amyloidosis in living PS2APP mice treated for four months with the small molecule BACE1 inhibitor RO5508887 [17]. We used serial and regional PET analyses for identifying the determinants of efficacy of BACE1 inhibition. Multimodal histological and biochemical readouts obtained post mortem served to substantiate and extend the conclusions drawn from PET.
Methods
Study design
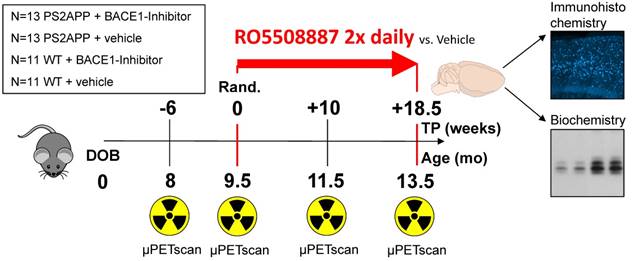
Groups of 26 female PS2APP-Swe (TG) and 22 female C57BL/6 (WT) mice were randomly assigned to either treatment (TG-BSI; WT-BSI) or vehicle (TG-VEH; WT-VEH) groups at the age of 9.5 months. A baseline [18F]-florbetaben-PET scan (Aβ-PET) was performed at this time, followed by initiation of daily oral RO5508887 treatment or vehicle, for a period of four months. Follow-up Aβ-PET-scans were acquired after 10 weeks (at 11.5 months of age), and 18.5 weeks of treatment (at 13.5 months of age), whereupon the study was terminated. An additional pre-baseline PET scan at 8 months (-6 weeks) had been performed to investigate the natural longitudinal Aβ deposition rates in all mice prior to their randomization. Thus, each mouse underwent a total of four [18F]-florbetaben-PET scans over a period of 18 weeks. After completing the final scan, mice were killed and the brains removed for histological and biochemical analyses. Dose-titration experiments had been conducted prior to the chronic treatment in a separate group of 24 PS2APP mice. Figure 1 illustrates the study design.
Illustration of the study design. PS2APP (TG) and WT mice were scanned by Aβ-PET starting at 8 months of age (Time-point (TP) -6). Treatment/vehicle randomization was realized after the 9.5 month scan (TP 0). Shortly after the terminal scan at 13.5 months (TP +18.5) all brains were split after perfusion and randomized hemispheres were used for terminal immunohistochemistry (methoxy-X04 plaque staining) and biochemistry analyses (protein assays).
Animals
All experiments were performed in compliance with the National Guidelines for Animal Protection, Germany, with approval of the local animal care committee (Regierung Oberbayern), and overseen by a veterinarian. The transgenic B6.PS2APP (line B6.152H) mouse is homozygous for both the human presenilin (PS) 2, N141I mutation and the human amyloid precursor protein (APP) K670N, M671L mutation. The APP and PS2 transgenes are driven by mouse Thy-1 and mouse prion promoters, respectively. This line had been created by co-injection of both transgenes into C57BL/6 zygotes [18]. Homozygous B6.PS2APP mice show first appearance of amyloid plaques in the cortex and hippocampus at only five to six months of age [19].
BACE1 inhibitor
The previously evaluated BACE1 inhibitor RO5508887 was used at a defined dose of 100 mg/kg/d, which proved to be suitable in the chronic treatment arm of the same evaluation study using APPV717I transgenic mice [17]. Treatment was performed orally two times per day (11-13 h apart) per os (gavage). Acute in vivo treatment was performed for a dose finding in the PS2APP-Swe transgenic mouse model (N=24) with mutations causing increased BACE1 affinity and higher substrate turnover compared to normal APP, where RO5508887 proved to substantially block the enzyme at an acute dose of 30 mg/kg [17]. Therefore, the compound was administered once per os in PS2APP-Swe mice (60/90/120 mg/kg). For comparison, control PS2APP-Swe mice were treated with vehicle only (that is: 5% ethanol (VWR, Germany), 10% Solutol (MilliporeSigma, Germany) dissolved in sterile water). Brains were collected 2 and 4 h after acute treatment and stored at -80 °C until analysis. Two mice were used per time-point and dose/vehicle. DEA and RIPA lysates served to monitor the changes in BACE1-dependent APP processing affecting the levels of soluble Aβ as measured by Western blot analysis (Figure S1).
Aβ-PET
Aβ-PET image acquisition, reconstruction and analysis followed a standardized protocol, as previously published [14, 16, 20], with some refinements and adaptions as described in detail in Supplementary Material. After measuring tracer uptake in cerebral cortex and white matter templates, standardized-uptake-value-ratios (SUVRCTX/WM) values were calculated for all groups of mice at each Aβ-PET scan timepoint. Individual longitudinal changes were calculated as a function of time (Δ%- SUVRCTX/WM).
Regional deposition rates were assessed by voxel-wise PET analyses. To this end, mean parametric images (SUVRWM) were calculated for the TG-BSI and TG-VEH groups at each time point (N =12-13 mice). The %-difference images were obtained by parametric image algebra in PMOD ((Timepoint-2 - Timepoint-1)/Timepoint-1 × 100%). Next, the %-difference map of vehicle-treated animals between baseline and terminal PET scan was used for defining volumes-of-interest (VOIs) depicting 2%-stepwise increments of Aβ deposition rate over the 18.5 weeks of treatment. Adjacent 2%-step VOIs were combined to define regions of low (2-10%) and high (>10%) amyloid deposition rates.
Biochemistry
Aβ extraction from brain and quantification
Brains were removed from the cranium and dissected into left and right hemispheres. One randomly selected hemisphere was snap-frozen by immersion in liquid nitrogen and stored at -80 °C. Soluble proteins were extracted with DEA buffer (50 mM NaCl, 0.2 % diethylamine, pH 10 + protease inhibitor (P8340, Sigma-Aldrich)). Membrane proteins were extracted with RIPA buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate + protease inhibitor). RIPA-insoluble material was dissolved in 70% formic acid. Quantification of Aβ was performed according to the manufacturer's protocol using the MSD® MULTI-SPOT Human (6E10) Aβ Triplex Assay (Meso Scale Diagnostics, Rockville, USA) with a MSD SECTOR Imager 4200. Quantitative data were analyzed statistically by two-tailed Student's t-test (N =12 mice).
Protein analysis
Proteins were separated under denaturing conditions using discontinuous SDS-PAGE. Equal amounts of proteins denatured in Laemmli buffer were loaded onto the gel and 10 µL portions of the SeeBlue Plus2 Prestained Standard (Invitrogen) served as a molecular weight marker. Electrophoresis was performed in Tris-glycine buffer (25 mM Tris, 190 mM glycine in ddH2O) using the Mini-PROTEAN system (BIORAD) on activated PVDF membranes. Low molecular weight proteins (< 20 kDa) were separated using precast gradient Tricine Protein Gels (10-20%, 1 mm, Novex) in Tris-tricine buffer using the XCell SureLock Mini-Cell system (Novex). After separation by SDS-PAGE, proteins were transferred onto membranes using the tank/wet Mini Trans-Blot cell system (BIORAD). CTFs, Aη and Aβ were detected after transfer on Nitrocellulose membranes (Protran BA85; GE Healthcare), while other proteins were blotted on PVDF (Immobilon-P, Merck Millipore). Upon completion of the transfer and prior to blocking, proteins transferred to nitrocellulose membranes were additionally denatured by boiling the membranes in PBS (140 mM NaCl, 10 mM Na2HPO4, 1.75 mM KH2PO4, 2.7 mM KCl in ddH2O, pH 7.4) for 5 min. After cooling to room temperature, the nitrocellulose membranes as well as the PVDF membranes were blocked in I-Block solution (0.2% Tropix I-Block (Applied Biosystems) in PBS-T (0.1% Tween20 in PBS) for 1 h at room temperature or overnight at 4 °C (with agitation). Transferred proteins were detected using immunodetection and enhanced chemiluminescence (ECL). First, blocked membranes were incubated with primary antibodies diluted in I-Block solution overnight at 4 °C (with agitation). After removal of the antibody, membranes were washed 3x in PBS-T buffer (10 min each, at room temperature, with agitation) and subsequently incubated with a horseradish peroxidase (HRP)-coupled secondary antibody (obtained from Santa Cruz Biochemicals). Secondary antibodies were diluted in I-Block solution and membranes were incubated for 1 h at RT (with agitation) followed by three washes in PBS-T. For ECL detection, membranes were incubated with HRP substrate (ECL, GE Healthcare or ECL Plus, Thermo Scientific) for 1 min at RT and signals were captured by exposure of X-ray film (Super RX Medical X-Ray, Fujifilm), which were subsequently developed using an automated film developer (CAWOMAT 2000 IR, CAWO). Signal intensities were quantified using ImageJ analysis software and plotted as relative differences of the treatment group compared to control.
Antibodies
DEA soluble protein was analysed for APP processing products with the following antibodies: 22C11 for detecting the APP N-terminus (used at 1 µg/mL, obtained from MilliporeSigma, Darmstadt, Germany), 6A1 for detecting sAPPβ-swe cleaved by BACE1 (used at 1 µg/mL; obtained from Tecan/ IBL International, Hamburg, Germany). 2D8 is an in-house rat monoclonal antibody against the N-terminus of Aβ, detecting also Aη-α (used at 2 µg/mL).
Histochemical analyses
Fibrillary β-amyloid plaques in brain specimens were stained with the fluorescent dye methoxy-X04 (0.01 mg/mL in PBS at pH 7.4 for 15 min) [21]. Full details are described in Supplementary Material. Plaque load was calculated as the summed area of all plaques relative to the frontal cortex area. Plaque density was calculated as the number of plaques relative to frontal cortex volume [20]. These analyses were performed by an operator blind to the Aβ-PET results.
Statistics
Group comparisons of VOI-based Aβ-PET results and biochemistry were performed with multivariate analysis of variance (MANOVA) using IBM SPSS Statistics (Version 23.0). Histology data (plaque load and plaque density) were compared between treated and untreated transgenic mice by multivariate analysis of covariance (MANCOVA) using Aβ-PET baseline estimates as a covariate. Treatment effects in low and high amyloid accumulating regions were compared with a t-test. For correlation analyses, Pearson's coefficients of correlation (R) were calculated. Plaque size distributions were compared with a Chi-square test followed by the Kolmogorov-Smirnov test with Prism V5.04 software (GraphPad Software, San Diego, CA, USA). A Shapiro-Wilk test was performed to verify normal distribution of sample values. A threshold of p<0.05 was deemed significant for rejection of the null hypothesis.
Results
Chronic BACE-I treatment effectively inhibits fibrillary Aβ deposition as monitored by PET
We applied a theranostic concept of longitudinal Aβ-PET during chronic BACE-1 inhibition in PS2APP mice to study treatment effects over 18.5 weeks and to investigate the value of treatment monitoring. Multimodal readouts by terminal Aβ peptide level, and terminal plaque load to immunohistochemistry served for further characterization of the effect of chronic treatment with RO5508887 on amyloidosis in PS2APP mice.
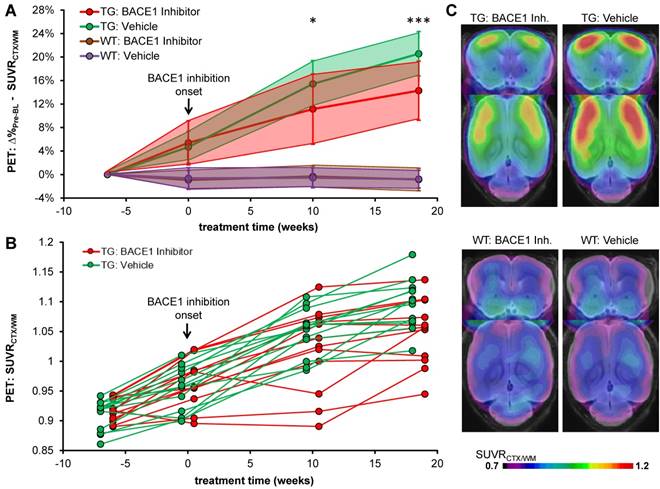
Treatment and serial PET imaging proved well-tolerated, with only a single dropout in the TG-BSI group. All modalities revealed a significantly reduced progression of amyloidosis in TG-BSI mice when compared to the TG-VEH group. Particularly, PET revealed a significantly lower progression of amyloidosis as early as 10 weeks into the treatment regimen (+6.7% in BSI vs. +10.3% in VEH, p < 0.05) and an even greater effect at 18.5 weeks of treatment (+8.4% in BSI vs. +15.2% in VEH, p < 0.001; Figure 2). WT mice presented with stable PET values either in the treatment or the vehicle groups.
Longitudinal Aβ-PET imaging of BACE1 inhibition. (A) Plots show means (±SD) of the cortical amyloid signal relative to the baseline imaging for treated and vehicle groups of TG and WT mice. (B) Individual progression of the cortical amyloid signal in TG mice. (C) Signal intensities of the frontal cortical amyloid signal in TG and WT mice at the terminal Aβ-PET scan (18.5 weeks). Coronal and axial slices illustrate group averages upon a T1w MRI template. * p < 0.05; *** p < 0.001
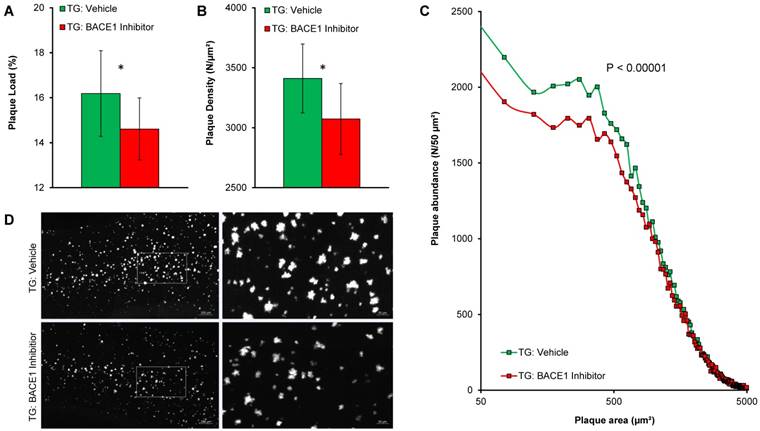
Terminal immunohistochemistry confirmed PET findings by the direct validation of fibrillary Aβ and indicated lower plaque load (-9.7%, p < 0.05; Figure 3A) as well as plaque density (-9.9%, p < 0.05; Figure 3B) in BACE1-treated TG mice when compared to TG vehicle controls. The major effect was observed on de novo plaque deposition, as the fraction of small plaques was apparently lower in the same contrast of treated versus control mice (Figure 3C-D).
Immunohistochemical validation of Aβ-PET results. Plots show frontal cortical mean (±SD) of plaque load (A) and plaque density (B) by methoxy-X04 immunohistochemistry in TG-VEH (green) and TG-BSI (red) mice. (C) Histogram plotting of plaque size revealed significantly fewer small plaques in the TG-BSI animals (red) in comparison to the TG-VEH group (green), which was also explored in the visual analysis of sagittal cortical slices of methoxy-X04 staining (D). Error bars indicate SD. * p < 0.05.
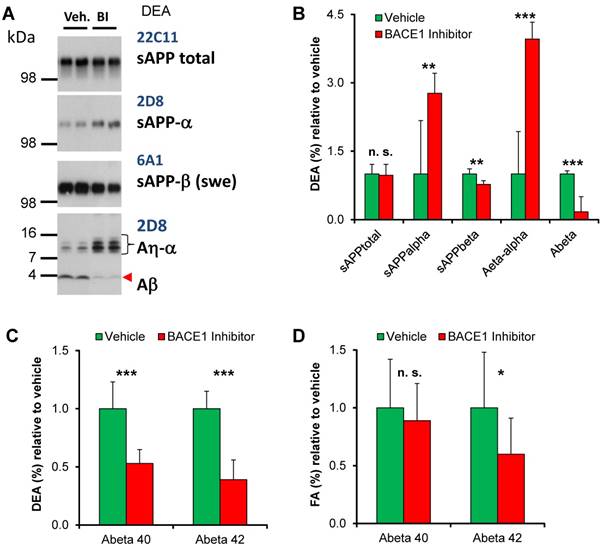
Acute and chronic BACE1 treatment lowers Aβ deposition
Brains of the PSAPP2-swe transgenic mice were collected 4 h after the last BACE1 inhibitor or vehicle treatment to allow monitoring of both the steady-state enzyme activity after inhibitor application and the long-term effect of the chronic treatment. BACE1 inhibition results in reduction of the amyloidogenic pathway as indicated by lower levels of sAPP-β (Swe) and increased sAPP-α, while total sAPP remained unchanged. BACE1 inhibition resulted in a clear reduction of soluble Aβ and consequently an increase of Aη-α in the DEA fraction, which is enriched in soluble fragments, of the TG-BSI mice compared to TG-VEH controls 4 h after acute treatment (Figures 4A-B). Furthermore, soluble Aβ (DEA) and deposited Aβ (FA) were quantified by ELISA (Figures 4C-D). Compared to the TG-VEH group, we saw a 47% reduction of soluble Aβ40 and a 61% reduction of Aβ42, respectively, in the group treated with the BACE inhibitor RO550887 (Figure 4C). However, the TG-BSI animals showed only a slight reduction of Aβ40 levels (-10% in TG-BSI vs. TG-VEH, p = 0.45), but significantly reduced Aβ42 levels after chronic treatment over 18 weeks in the fraction of deposited Aβ (-40% in TG-BSI vs. TG-VEH, p < 0.05) (Figure 4D).
Biochemical analyses. (A) In Western blots, BACE1 inhibition was observed to result in reduction of the amyloidogenic pathway, as indicated by lowered levels of sAPP-β (Swe) and increased sAPP-α levels. The lower panel indicates that BACE1 inhibition caused reduced levels of soluble Aβ and an increase of Aη-α in brains of the TG-BSI mice compared to TG-VEH controls 4 h after acute treatment. (B) Quantification of APP processing products indicated significant increase for sAPP-α and Aη-α, but significant reduction for the BACE1-dependent products sAPP-β and Aβ, while total sAPP shedding remained unchanged (vehicle (n=6); BI/BACE inhibitor treated (n=5)). (C) Lower DEA levels of soluble Aβ40 and Aβ42 were observed in TG-BSI mice relative to TG-VEH controls. (D) Insoluble FA levels likewise indicated a significant reduction of Aβ42, but only a small tendency for reduced Aβ40 levels. Error bars indicate SD, * p < 0.05; ** p < 0.005; *** p < 0.001; n.s.= not significant.
In summary, all readouts and modalities indicated a significant chronic treatment effect by RO5508887 on progression of amyloidosis in PS2APP mice.
Effectiveness of chronic BACE1 inhibition treatment depends on regional Aβ deposition rates
In a separate analysis, we used the theranostic Aβ-PET approach to investigate regional longitudinal differences in amyloidosis during chronic treatment. We aimed to reveal possible relationships between the “naïve” regional deposition rate and the subsequent regional chronic treatment effect. Individual PET results in TG-VEH mice revealed the highest “naïve” regional increase in amyloidosis (Δ%-SUVRWM) in the bilateral frontal cortex (primary somatosensory areas), followed by primary motor areas and auditory and visual cortices. Lower progression of amyloidosis was observed in temporal cortex areas (e.g., gustatory, insular), as well as in the bilateral thalamus and hippocampus (Figure S2).
Interestingly, we found a linear relationship (R>0.99; p<0.001) between the regional amyloid progression rate in TG-VEH, i.e., naïve progression, and the corresponding progression in TG-BSI mice (Figure 5A). This relationship indicated a threshold for regional deposition rate of 3.7% / 18.5 weeks in TG-VEH, below which complete blocking of further signal increase could be achieved with RO5508887 treatment of TG mice. A polynomial function could be fitted to a plot of the individual treatment effect relative to naïve progression in the TG-VEH group, thus revealing very high treatment effect in regions with low naïve progression, which declined to a trough, with only 40% attenuation of amyloidosis in those regions with naïve progression exceeding about 15% (Figure 5B). This finding was verified by voxel-wise mapping of the %-treatment effect, which showed the highest relative effect (nearly 100%) of BACE1 inhibition in brain regions with inherently low Aβ-accumulation (e.g., gustatory and insular areas, thalamus; Figure 5C).
Analysis of high and low Aβ accumulating regions. (A) Progression of the Aβ-PET signal in TG-VEH (x-axis) against TG-BSI (y-axis) mice. Values were obtained from regions with equal Aβ accumulation in TG-VEH mice. Dotted line shows halted fibrillary Aβ accumulation (0%) in TG-BSI mice and the nearly linear correlation of single regions between TG-VEH and TG-BSI mice calculates a complete stop of further fibrillary Aβ accumulation in regions of ≤ +3.7% signal increase in TG-VEH. Low and high Aβ accumulating regions were defined by 2-10% and > 10% signal increase in TG-VEH mice over 18.5 months. (B) Consecutively, an exponential increase of the treatment effect in TG-BSI relative to TG-VEH mice was found with decreasing regional Aβ accumulation, as observed in drug-naïve TG mice. (C) A 3D analysis of the forebrain confirmed these findings by indicating the highest relative treatment effect in lateral neocortical brain regions with a low Aβ accumulation. The absolute reduction of the Aβ deposition rate was significantly larger in high Aβ accumulating regions (D), whereas the relative therapy effect was significantly lower in high Aβ accumulating regions (E).
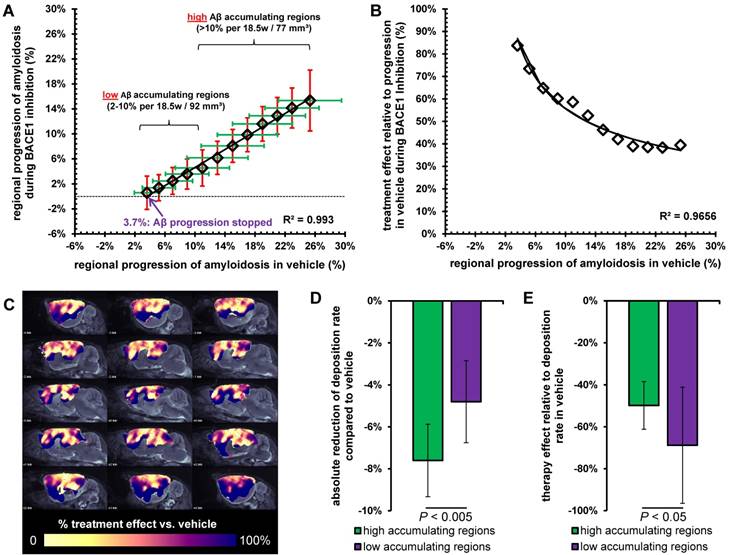
Concerning the absolute reduction in Aβ deposition with treatment, we found a decrease of 7.6 ± 1.7% in high accumulating regions (≥ 10%; total volume: 77 mm³) and a lesser decrease of 4.8 ± 2.0% in low accumulating regions (<10%; total volume: 92 mm³; p < 0.005, Figure 5D). However, the relative treatment effect was significantly lower in high accumulating brain regions (49 ± 11%) when compared to brain regions with low accumulation, in which there was a relative treatment effect of 69 ± 28% (p < 0.05, Figure 5E). Kinetics of accumulation in individual animals to serial PET measurements in high and low Aβ accumulating regions are presented in Figure S3.
We find significant regional differences in the response to chronic BACE1 inhibition treatment, which is characterized by a linear relationship relative to vehicle treatment. Regions with more pronounced fibrillary Aβ deposition are characterized by greater resistance to BACE1 inhibition, whereas a 100% blocking was achieved in regions with low deposition rates, all relative to findings in the vehicle group. The finding of individual deposition rate thresholds for complete blocking of Aβ deposition by a theranostic concept might guide individual dose regimens for BACE1 inhibitors in future clinical studies. Optimal dosing for blockade in high accumulating regions must be assessed in the light of a possible trade-off between side effects and clinical efficacy.
BACE1 inhibition treatment is more effective during early amyloid build-up
The serial Aβ-PET design made possible an assessment of baseline plaque load in brains of individual mice prior to initiation of the treatment. We used this technique to predict differences in longitudinal fibrillary Aβ deposition from the baseline level, based on our earlier findings of such an association [15]. Indeed, present findings in TG-VEH mice indicated an inverse association between baseline Aβ burden and the relative increase from 9.5 to 13.5 months of age (R = -0.53, p < 0.05; Figure 6A). Thus, TG-VEH mice starting at a low baseline Aβ-level tended to catch up their initially delayed Aβ accumulation. TG-BSI mice revealed the highest treatment-related difference relative to the TG-VEH group when their initial Aβ-level was low, as the accelerated increase observed in vehicle animals was prevented, leading to a reversal in the direction of the correlation (R = +0.27, p = n.s.; Figure 6A). In those TG-BSI mice with high Aβ-levels to baseline PET, the treatment effect was hardly discernible from the TG-VEH group. Findings in representative mice with a nearly equal baseline amyloidosis are illustrated in Figure 6B. To confirm the “regular” inverse correlation between baseline Aβ-level and the subsequent deposition rate, we longitudinally imaged another vehicle-treated TG group (N=12), which confirmed the above results (RBL/∆%-BL-TER = -0.58, p < 0.05).
Treatment effect dependency from baseline amyloidosis. (A) Response prediction by means of Aβ-PET. The percentage increase from baseline to termination SUVRCTX/WM is depicted as a function of the individual baseline value. For the TG-BSI, a low baseline value predicted a lesser increase in amyloidosis during treatment, whereas TG-VEH mice with a low baseline value increased tremendously. The treatment effect in TG-BSI relative to TG-VEH was markedly lower when amyloidosis was already established. The correlations (R) between the percentage increase and the baseline value are indicated. (B) Aβ-PET signal intensities of individual exemplary mice for serial baseline, follow-up and terminal imaging time-points. The TG-BSI mouse with a low baseline (#20) indicates a very low Aβ accumulation whereas the TG-VEH mouse with a low baseline (#08) shows high Aβ accumulation. TG-BSI and TG-VEH with a high baseline show only minor differences in their sequential Aβ accumulation. Coronal and axial slices are illustrated upon a T1w MRI template.
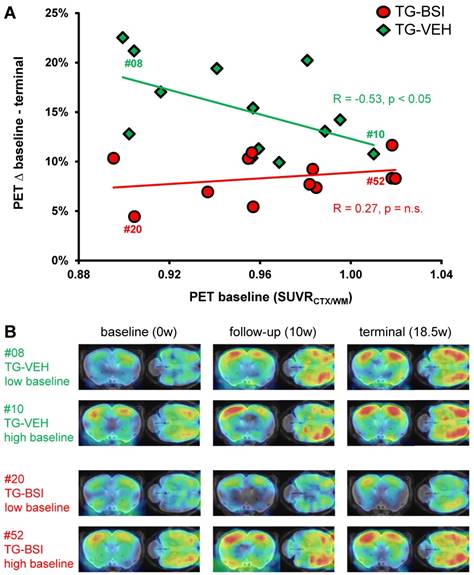
The finding that baseline PET results predict treatment efficacy indicates that therapeutic BACE1 inhibition should be obtained early in the pathology time course, as present amyloid deposits proved more resistant to later treatment initiation.
Discussion
In this adequately powered longitudinal PET study of cerebral amyloidosis in a transgenic AD mouse model we investigated regional effects of a chronic disease-modifying therapy based on BACE1 inhibition by applying a theranostic concept. The molecular imaging study entailed corroborative histopathological as well as biochemical analyses, thus encompassing three different readout modalities for monitoring amyloidogenesis.
Serial [18F]-florbetaben Aβ-PET in PS2APP mice provides a robust method for monitoring anti-amyloid treatment effects
Serial Aβ-PET excellently captured the treatment effects of BACE1 inhibition versus vehicle and allowed in vivo assessment of the therapeutic target in individual mice. A recent study using a multi-tracer design succeeded in monitoring BACE1 effects in an amyloid mouse model using another Aβ-PET ligand ([18F]-florbetapir), although the similarly altered signal in treated WT mice led the authors to conclude that the observed binding effects were not specific treatment effects on amyloid deposition [22]. In contrast, we found a highly stable background signal in WT mice in treatment and vehicle groups using the amyloid ligand [18F]-florbetaben, which encourages the use of the mouse model in combination with the earlier-validated quantification method [16]. A recent PET study showed the ability to detect non-fibrillar Aβ forms in vivo during treatment of transgenic ArcSwe mice with another BACE1 inhibitor [23], whereas our PET ligand binds only fibrillary parts of the plaque. Future monitoring regimes could therefore entail dual PET with selective tracers to capture both components of amyloidosis during BACE1 inhibition.
Linear dependence of BACE1 inhibition treatment effect from Aβ deposition rates can be captured by individual serial imaging
In the present study with the BACE1 inhibitor RO5508887 at a daily dose of 100 mg/kg, we obtained only 49% attenuation of progression in brain regions with high Aβ deposition, suggesting that a higher dose might have been more efficacious. The APP-Swe transgenic mouse model, given its high rate of Aβ synthesis, presents a clear limitation arising from the need for relatively high inhibitor dosing. However, more recent data have indicated a dose-dependent impairment of synaptic plasticity and cognitive functions or reduced muscle spindle numbers in adult mice with chronic BACE1 inhibitor treatment, which could be attributed to impairing function of the BACE1 substrate Neuregulin-1 [24, 25]. Thus, there is need for caution in selecting dosage regimens for drugs targeting BACE1 aiming to achieve a therapeutic window with optimal trade-off between safety and efficacy. The current data provide evidence that regional efficacy of RO5508887 treatment linearly correlates with the regional naïve deposition rate revealed by Aβ-PET in vehicle-treated animals. Thus, prior knowledge of the individual Aβ deposition rate might inform the estimation of an optimal dosage of BACE1 inhibitor to slow or even arrest further progression of cortical fibrillary amyloidosis. This issue is even more important when considering the clinical finding that Aβ accumulation rates in amyloid-positive humans are very heterogeneous [26, 27]. Such a treatment design and combinatorial treatments together, e.g., with an Aβ immunotherapy strategy might require dosage titration of BACE1 inhibitors in individual patients according to their individual amyloidosis rates at initiation of therapy. Harkening back on the potential for cognitive side effects, it could well be that there are patients for whom the BACE1 inhibitor dosage predicted to be effective for slowing amyloidosis would bring an excessively high risk for adverse effects. On the other hand, some patients might require only low doses of BACE1 inhibitors, either because of their earlier disease stage, or an intrinsically lower Aβ progression. In this regard, recent detailed PET study of amyloid accumulation rates of humans with preclinical AD revealed a strong global and regional variation, which has major implications for anti-amyloid trials [28]. Our current findings of high and low amyloid accumulating regions in PS2APP mice are in line with the human findings, which emphasizes that our novel preclinical approach for dose-titration by considering amyloid deposition rates in multiple brain regions might be translatable to human investigations. Serial Aβ-PET therefore has the potential to serve as a readout for determining the individual dosage as well as for monitoring effectiveness of treatment in individual patients in the context of personalized medicine.
Early treatment initiation ensures high efficacy in BACE1 inhibition treatment
Current results constitute compelling evidence that efficacy of RO5508887 in individual TG mice receiving a predefined dose of the medication depends on their fibrillary individual Aβ levels at therapy initiation, as is revealed by baseline Aβ-PET. By selecting the present dose of 100 mg/kg/d RO5508887 on the basis of earlier findings [17] and by single dose validation in the current model, we observed a greater benefit in those mice having a relatively low plaque load at baseline, which matches findings in our previous study with a gamma secretase modulator in Swedish mutant mice [15]. Thus, the relationship emerges as a general principle, irrespective of the molecular target in a treatment intervention. We now show that BACE1 inhibitor-treated mice with high and low baseline amyloid levels progressed equally, whereas the course in vehicle mice with low Aβ to baseline PET clearly had a more pronounced increase of amyloidosis between 9.5 and 13.5 months of age. This concept is coherent in the light of our immunohistochemistry findings, since the major impact of BACE1 treatment was clearly observed in lowering the fraction of small plaques. Thus, this treatment evoked the anticipated molecular effect on de novo plaque formation [17]. Findings of a recent chronic treatment study using a different BACE1 inhibitor and a different amyloid mouse model are in line with our terminal immunohistochemistry results as their images indicated a similar main treatment effect on small plaques [22]. In summary, we find that the present dosage of RO5508887 can substantially lower the progression of amyloidosis in those brain regions and in those individual animals with low pre-existing plaque load. Due to the logistic difficulty and cost of serial PET imaging, we only investigated one preselected dosage and only one starting age. However, higher doses and persistent inhibition achievable, e.g., by oral drug administration with food would be necessary if the treatment initiation were at a disease stage with established plaque deposition. It remains to be elucidated if sufficient RO5508887 doses for such cases can be administered without provoking treatment-limiting side effects. While our present results in a double transgenic β-amyloid mouse model are not immediately translatable to clinical AD, we provide compelling evidence that Aβ-PET can potentially inform a crucial decision for clinical AD trials: at what stage of AD should BACE inhibitors be administered? With precisely this in mind, cortical Aβ levels are being examined in current phase II/III trials of BACE inhibitors [12]. Very recently, two phase III trials with Lanabecestat were discontinued early as they would probably not have met their primary endpoints [29]. Full details on the reasons behind the decisions are not yet released, but there is a general opinion that BACE1 inhibitors are unlikely to be effective in individuals who already have symptoms of AD dementia [29]. Our current results of lesser efficacy in mice with established amyloidosis give the first PET-based in vivo evidence for this contention. It will be of enormous interest if the efficacy in human treatment indeed proves to be predictable from the state of cortical amyloidosis at initiation of treatment. The lack of behavioral testing in this study presents a limitation since we can make no claims about beneficial or detrimental effects on cognition in the treated mice.
Conclusion
We demonstrate that serial Aβ-PET in PS2APP mice provides a robust theranostic approach for monitoring BACE1 inhibition treatment effects in this adequately powered longitudinal study. Regional analyses indicated a linear dependence of treatment efficacy upon Aβ deposition rates in the vehicle-treated group. Serial Aβ-PET could therefore serve as a readout for titrating the optimal individual dosage as well as for monitoring treatment effectiveness in the context of personalized medicine. Finally, the correlation between baseline and the individual treatment effects in conjunction with terminal histological results revealed the need of an early BACE1 treatment initiation for prevention of de novo amyloidogenesis.
Abbreviations
Aβ: amyloid β-peptide; Aβ-PET: [18F]-florbetaben β-amyloid positron-emission-tomography; AD: Alzheimer's disease; APP: β-amyloid precursor protein; BACE1: β-site amyloid precursor protein cleaving enzyme 1; BBB: blood brain barrier; CTF-β: C-terminal fragment; DEA: diethanolamine; ECL: enhanced chemiluminescence; FBB: [18F]-florbetaben; HRP: horseradish peroxidase; MANOVA: multivariate analysis of variance; MANCOVA: multivariate analysis of covariance; MRI: magnetic resonance imaging; NMDA: N-Methyl-D-Aspartat; PET: positron-emission-tomography; PBS: phosphate buffered saline; PS: presenilin; PVDF: polyvinylidendifluorid; R: Pearson's coefficients of correlation; RT: room temperature; SD: standard deviation; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; SUVR: standardized-uptake-value-ratio; TG: transgenic PS2APP-Swe mice; TG-BSI: treatment group of transgenic PS2APP-Swe mice; TG-VEH: vehicle group of transgenic PS2APP-Swe mice; TP: time-point; VOI: volume-of-interest; WT: C57BL/6 mice; WT-BSI: treatment group of C57BL/6 mice; WT-VEH: vehicle group of C57BL/6 mice.
Supplementary Material
Supplementary methods and figures.
Acknowledgements
We thank Karin Bormann-Giglmaier, Rosel Oos, Heike Hampel and Veronika Müller for excellent technical assistance. Florbetaben precursor was kindly provided by Piramal Imaging. We thank Jaroslaw Dzbek for help in data processing. We acknowledge Inglewood Biomedical Editing for professional manuscript editing.
Funding
The study was financially supported by the SyNergy Cluster (J.H., P.B., C.H., and A.R.) and by the European Research Council under the European Union's Seventh Framework Program (FP7/2007-2013)/ ERC Grant Agreement No. 321366-Amyloid (advanced grant to C.H.). A. J. was supported by the Foundation for Polish Science within the International PhD Project 'Studies of nucleic acids and proteins - from basic to applied research', co-financed from European Union - Regional Development Fund; MPD/2009-3/2.
Author contributions
MB: conceived of the study, performed manuscript writing, performed PET imaging and analyses; AJ: performed manuscript writing, performed immunohistochemical analyses; FO: performed PET imaging and analyses; TB: performed immunohistochemical analyses; FP: performed PET imaging and analyses; FG: performed radiochemistry analyses and synthesis of the tracer; PB: interpretation of PET imaging; CH: conceived the study and analyzed the biochemical results, interpretation of biochemical results; BB: conceived the study and analyzed the treatment results; JH: conceived the study and analyzed the immunohistochemical results, interpretation of immunohistochemical results; MW: conceived the study and analyzed the results and performed biochemical analyses, interpretation of biochemical results, performed manuscript writing; AR: conceived the study and analyzed the PET results; all authors drafted the work, revised it critically for important intellectual content, and approved the final version.
Conflicts of Interest
PB received speaking honoraria from Piramal Imaging. CH is an advisor of Hoffmann-La Roche. BB is an employee of Hoffmann-La Roche. AR received speaking honoraria from Piramal Imaging. All other authors declare no conflicts of interest.
References
1. Ziegler-Graham K, Brookmeyer R, Johnson E, Arrighi HM. Worldwide variation in the doubling time of Alzheimer's disease incidence rates. Alzheimers Dement. 2008;4:316-23
2. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239-59
3. Dubois B, Feldman HH, Jacova C, Hampel H, Molinuevo JL, Blennow K. et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. The Lancet Neurol. 2014;13:614-29
4. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH. et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263-9
5. Willem M, Tahirovic S, Busche MA, Ovsepian SV, Chafai M, Kootar S. et al. eta-Secretase processing of APP inhibits neuronal activity in the hippocampus. Nature. 2015;526:443-7
6. Sun X, Jin L, Ling P. Review of drugs for Alzheimer's disease. Drug Disov Ther. 2012;6:285-90
7. Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med. 2003;348:1333-41
8. Herrmann N, Li A, Lanctot K. Memantine in dementia: a review of the current evidence. Expert Opin Pharmacother. 2011;12:787-800
9. Dhillon S. Rivastigmine transdermal patch: a review of its use in the management of dementia of the Alzheimer's type. Drugs. 2011;71:1209-31
10. Yan R, Vassar R. Targeting the beta secretase BACE1 for Alzheimer's disease therapy. Lancet Neurol. 2014;13:319-29
11. Yan R, Fan Q, Zhou J, Vassar R. Inhibiting BACE1 to reverse synaptic dysfunctions in Alzheimer's disease. Neurosci Biobehav Rev. 2016;65:326-40
12. Vassar R. BACE1 inhibitor drugs in clinical trials for Alzheimer's disease. Alzheimers Res Ther. 2014;6:89
13. Egan MF, Kost J, Tariot PN, Aisen PS, Cummings JL, Vellas B. et al. Randomized trial of Verubecestat for mild-to-moderate Alzheimer's disease. N Engl J Med. 2018;378:1691-703
14. Brendel M, Jaworska A, Griessinger E, Rotzer C, Burgold S, Gildehaus FJ. et al. Cross-sectional comparison of small animal [18F]-florbetaben amyloid-PET between transgenic AD mouse models. PloS One. 2015;10:e0116678
15. Brendel M, Jaworska A, Herms J, Trambauer J, Rotzer C, Gildehaus FJ. et al. Amyloid-PET predicts inhibition of de novo plaque formation upon chronic gamma-secretase modulator treatment. Mol Psychiatry. 2015;20:1179-87
16. Overhoff F, Brendel M, Jaworska A, Korzhova V, Delker A, Probst F. et al. Automated spatial brain normalization and hindbrain white matter reference tissue give improved [(18)F]-Florbetaben PET quantitation in Alzheimer's model mice. Front Neurosci. 2016;10:45
17. Jacobsen H, Ozmen L, Caruso A, Narquizian R, Hilpert H, Jacobsen B. et al. Combined treatment with a BACE inhibitor and anti-Abeta antibody gantenerumab enhances amyloid reduction in APPLondon mice. J Neurosci. 2014;34:11621-30
18. Richards JG, Higgins GA, Ouagazzal AM, Ozmen L, Kew JN, Bohrmann B. et al. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J Neurosci. 2003;23:8989-9003
19. Ozmen L, Albientz A, Czech C, Jacobsen H. Expression of transgenic APP mRNA is the key determinant for beta-amyloid deposition in PS2APP transgenic mice. Neurodegener Dis. 2009;6:29-36
20. Rominger A, Brendel M, Burgold S, Keppler K, Baumann K, Xiong G. et al. Longitudinal assessment of cerebral beta-amyloid deposition in mice overexpressing Swedish mutant beta-amyloid precursor protein using 18F-florbetaben PET. J Nucl Med. 2013;54:1127-34
21. Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP. et al. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol. 2002;61:797-805
22. Deleye S, Waldron AM, Verhaeghe J, Bottelbergs A, Wyffels L, Van Broeck B. et al. Evaluation of muPET outcome measures to detect disease modification induced by BACE inhibition in a transgenic mouse model of Alzheimer's disease. J Nucl Med. 2017;58:1977-83
23. Meier SR, Syvanen S, Hultqvist G, Fang XT, Roshanbin S, Lannfelt L. et al. Antibody-based in vivo PET imaging detects amyloid-beta reduction in Alzheimer transgenic mice after BACE-1 inhibition. J Nucl Med. 2018 In press
24. Filser S, Ovsepian SV, Masana M, Blazquez-Llorca L, Brandt Elvang A, Volbracht C. et al. Pharmacological inhibition of BACE1 impairs synaptic plasticity and cognitive functions. Biol Psychiatry. 2015;77:729-39
25. Cheret C, Willem M, Fricker FR, Wende H, Wulf-Goldenberg A, Tahirovic S. et al. Bace1 and Neuregulin-1 cooperate to control formation and maintenance of muscle spindles. EMBO J. 2013;32:2015-28
26. Brendel M, Hogenauer M, Delker A, Sauerbeck J, Bartenstein P, Seibyl J. et al. Improved longitudinal [(18)F]-AV45 amyloid PET by white matter reference and VOI-based partial volume effect correction. Neuroimage. 2015;108:450-9
27. Villemagne VL, Burnham S, Bourgeat P, Brown B, Ellis KA, Salvado O. et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013;12:357-67
28. Guo T, Dukart J, Brendel M, Rominger A, Grimmer T, Yakushev I. Rate of β-amyloid accumulation varies with baseline amyloid burden: Implications for anti-amyloid drug trials. Alzheimers Dement. 2018 In press
29. Burki T. Alzheimer's disease research: the future of BACE inhibitors. The Lancet. 2018;391:2486
Author contact
![]() Corresponding author: Prof. Dr. Axel Rominger, Department of Nuclear Medicine, University Hospital Bern, Switzerland. Phone: +41 31 632 2610; Fax: +41 31 632 7663; E-Mail: axel.romingerch
Corresponding author: Prof. Dr. Axel Rominger, Department of Nuclear Medicine, University Hospital Bern, Switzerland. Phone: +41 31 632 2610; Fax: +41 31 632 7663; E-Mail: axel.romingerch