Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results and Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Results and Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(15):4062-4071. doi:10.7150/thno.24683 This issue Cite
Research Paper
DNA nanotriangle-scaffolded activatable aptamer probe with ultralow background and robust stability for cancer theranostics
Yanli Lei, Zhenzhen Qiao, Jinlu Tang, Xiaoxiao He, Hui Shi ![]() , Xiaosheng Ye, Lv'an Yan, Dinggeng He, Kemin Wang
, Xiaosheng Ye, Lv'an Yan, Dinggeng He, Kemin Wang ![]()
State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Biology, College of Chemistry and Chemical Engineering, Hunan University, Key Laboratory for Bio-Nanotechnology and Molecular Engineering of Hunan Province, Changsha 410082, China.
Received 2018-1-1; Accepted 2018-6-16; Published 2018-7-16
Citation:
Lei Y, Qiao Z, Tang J, He X, Shi H, Ye X, Yan L, He D, Wang K. DNA nanotriangle-scaffolded activatable aptamer probe with ultralow background and robust stability for cancer theranostics. Theranostics 2018; 8(15):4062-4071. doi:10.7150/thno.24683. https://www.thno.org/v08p4062.htm
Other stylesAbstract

Activatable aptamers have emerged as promising molecular tools for cancer theranostics, but reported monovalent activatable aptamer probes remain problematic due to their unsatisfactory affinity and poor stability. To address this problem, we designed a novel theranostic strategy of DNA nanotriangle-scaffolded multivalent split activatable aptamer probe (NTri-SAAP), which combines advantages of programmable self-assembly, multivalent effect and target-activatable architecture.
Methods: NTri-SAAP was assembled by conjugating multiple split activatable aptamer probes (SAAPs) on a planar DNA nanotriangle scaffold (NTri). Leukemia CCRF-CEM cell line was used as the model to investigate its detection, imaging and therapeutic effect both in vitro and in vivo. Binding affinity and stability were evaluated using flow cytometry and nuclease resistance assays.
Results: In the free state, NTri-SAAP was stable with quenched signals and loaded doxorubicin, while upon binding to target cells, it underwent a conformation change with fluorescence activation and drug release after internalization. Compared to monovalent SAAP, NTri-SAAP displayed greatly-improved target binding affinity, ultralow nonspecific background and robust stability in harsh conditions, thus affording contrast-enhanced tumor imaging within an extended time window of 8 h. Additionally, NTri-SAAP increased doxorubicin loading capacity by ~5 times, which further realized a high anti-tumor efficacy in vivo with 81.95% inhibition but no obvious body weight loss.
Conclusion: These results strongly suggest that the biocompatible NTri-SAAP strategy would provide a promising platform for precise and high-quality theranostics.
Keywords: DNA self-assembly, multivalent effect, activatable imaging, split aptamer, cancer theranostics
Introduction
The development of theranostic agents that can simultaneously support sensitive imaging and precise therapy against diseases such as cancer is expected to significantly facilitate advances in personalized medicine [1]. Nucleic acid aptamers, which are single-stranded RNA or DNA with highly selective binding affinity to various targets including cancer markers and cells, provide a promising molecular tool for cancer theranostics [2]. Conventionally, aptamer-incorporated theranostic probes rely solely on initiative enrichment of reporters and drug payloads in target domains, thus achieving differential signal intensity and weakened toxicity [3-5]. In view of their constant signaling and indiscriminate drug release, the above always-on probes are inevitably accused of high background, poor imaging contrast, delayed diagnosis and toxicity to normal tissues [6]. Alternatively, activatable aptamer probes with target-triggered signal change and drug response architectures have been designed, benefiting from the “induced-fit effect” of aptamers [7, 8]. For example, our group developed a smart split aptamer-based activatable theranostic probe, which successfully realized cancer-activated in vivo imaging and in situ drug release [9]. Because the probe could only be lit up by targets, the imaging sensitivity was greatly improved and the nonspecific toxicity was effectively reduced. However, due to their unsatisfied affinity, small payload, poor stability and fast excretion under physiological conditions, the reported monovalent activatable aptamer probes do not meet clinical requirements. Hence, there is still an urgent need to explore novel activatable theranostic agents with robust in vivo applicability.
The multivalent effect is commonly used in life processes to enhance affinity and selectivity for molecular recognition, which is a synergistic effect resulting from the simultaneous interaction of ligands clustering on one entity with multiple receptor sites on another [10,11]. Inspired by nature, multiple identical aptamers have been modified on scaffolds such as metallic nanoparticles (NPs) and liposomes, or embedded in DNA nanostructures via rolling circle amplification (RCA) and hybridization chain reaction (HCR) to improve avidity [12-15]. Nevertheless, most of the developed multivalent theranostic platforms still adopt the always-on design pattern, which will easily lead to limited sensitivity, time-consuming diagnosis and side effects due to lack of target activation [5,16-18]. Encouragingly, our group has recently developed an activatable theranostic platform based on assembly of multiple activatable aptamers on spherical Au@Ag/Au NPs [19]. By adopting a target-responsive architecture, the platform realized contrast-enhanced image-guided site-specific cancer thermotherapy. But, this multivalent strategy is limited by imprecise control of aptamer density and spatial orientation, which is crucial for binding improvement. Besides, using non-biodegradable metallic NPs as the scaffold will also pose potential biotoxicity.
As an alternative, structural DNA nanotechnology with programmable self-assembly and spatial addressability has demonstrated great potential for precise organization of functional molecules [20, 21]. By varying the length of the intervening duplex, an optimal ligand density on the DNA scaffold could be manipulated, thus achieving balance between unfavorable steric obstruction and effective local density. In particular, for planar DNA nanostructures, multiple ligands could be decorated on one face with uniform orientation such that a maximized local density and availability of ligands might be possible for multivalent binding with dramatic enhancement in avidity [22, 23]. Additionally, DNA is inherently biocompatible [20, 21], thus clearing a critical obstacle of self-toxicity for the clinical use of DNA nanostructures. We thereupon envisage that if a planar DNA nanostructure could scaffold multiple activatable aptamers, a novel multivalent theranostic platform would be developed with superb functionality, good biocompatibility and robust in vivo applicability.
Scheme 1
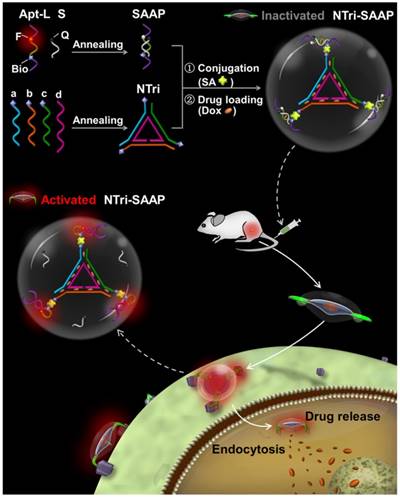
Assembly structure and working principle of NTri-SAAP for cancer theranostics. By conjugating DNA nanotriangle (NTri) scaffold and multiple split activatable aptamer probes (SAAPs) via streptavidin-biotin (SA-Bio) interaction, the NTri-SAAP is assembled. The CG base pair regions allow doxorubicin (Dox) intercalation. In the free state, the NTri-SAAP is a flat, double helix-jointed structure with quenched fluorescence and loaded drugs. Once it encounters the target cell, SAAP can disassemble due to aptamer binding-triggered shape change, thus leading to signal activation and partial drug release. The remaining drugs can then be further freed after internalization.
Herein, to develop a planar multivalent theranostic system, DNA nanotriangle (NTri), one of the simplest planar DNA structures with high assembling efficiency, was employed as the model scaffold. By arming the NTri with multiple split activatable aptamer probes (SAAPs) in a site and orientation controllable manner, a novel theranostic strategy of NTri-scaffolded multivalent SAAP (NTri-SAAP) has been put forward. As illustrated in Scheme 1, the SAAP monomer, which supported an activatable theranostic in our previous study [9], consists of a long DNA strand (Apt-L) and a short one (S). The Apt-L strand comprises two cancer-targeted split aptamer fragments and a linker DNA co-labeled with a fluorophore (F) and biotin (Bio). The S strand is complementary to the linker and attached by a quencher (Q), thus resulting in quenched fluorescence in the free state after hybridization of Apt-L with S. The NTri scaffold is assembled from three Bio-modified exterior strands (a, b, and c) and an inner strand (d). Through the specific streptavidin-biotin (SA-Bio) interaction, SAAP monomers can be precisely positioned on the three corners of NTri as target binding and signaling domain. And, to ensure sufficient rotational flexibility, three non-pairing thymidine residues were added, overhanging on the terminals of the NTri, just like the hinge domain of an antibody. Meanwhile, by utilizing the abundant CG base pairs designed in double helix regions, anticancer drugs such as doxorubicin (Dox) can be loaded via intercalation [3, 5]. The assembled NTri-SAAP thus displays a roughly flat structure like a “triangle UFO”, keeping the signal quenched and drugs packed in the absence of targets. Upon binding to target cancer cells, the flexible SAAPs undergo conformational changes to form recognition shapes, thus forcing disassembly of Apt-L/S hybrid regions with activated fluorescence emission and slight drug release from SAAP. Later, the activated NTri-SAAP is internalized into target cells and frees Dox due to forces from the intracellular pH, ionic and nuclease environment [3]. As an activatable design, the NTri-SAAP will reduce background, enhance image contrast, speed diagnosis and eliminate side effects. As a planar multivalent binding system, the NTri-SAAP is expected to improve affinity and stability substantially for uses in biofluids, by virtue of the increased local ligand density and reduced steric constraint. And, as a DNA-composed theranostic platform, the NTri-SAAP would surely be biocompatible.
Figure 1
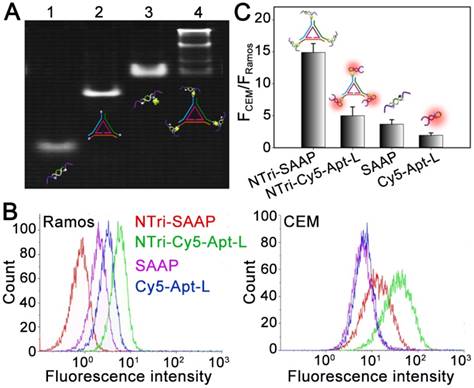
Characterization of the formation and activation of NTri-SAAP. (A) Native PAGE analysis of the stepwise addressability of NTri-SAAP. (Lane 1, SAAP; Lane 2, NTri; Lane 3, SAAP and SA; Lane 4, SAAP, SA and NTri). (B) Flow cytometry assays of CEM or Ramos cells after incubation with different probes in PBS with Mg2+ supplied. (C) The corresponding histogram of the fluorescence ratios of CEM to Ramos cells for probes in (B). Error bars represent standard deviations from three repeated experiments.
Results and Discussion
Construction and characterization of the multivalent NTri-SAAP system
The planar nanotriangle (NTri) DNA scaffold was constructed based on a rational design according to previous studies [24, 25]. Specifically, it was prepared from three exterior DNA strands (a, b and c) and an inner DNA strand (d) based on programmable self-assembly [26]. As listed in Table S1, each exterior strand could hybridize to d with 21 bases, thus affording two turns of B-form DNA. And, a 5-base pair extending arm at each corner was designed to facilitate incorporation of functional domains. The stepwise assembly of NTri was confirmed by native polyacrylamide gel electrophoresis (PAGE) with a high yield of >90% (Figure S1). Then, the NTri was functionalized with SAAP monomers via a streptavidin-biotin (SA-Bio) interaction, to develop a multivalent DNA nanostructure (NTri-SAAP) with controllable spatial orientation and density of ligands. The SAAP monomer was constructed using the sequence of a previously-developed split activatable aptamer probe targeting human leukemia CCRF-CEM cells, which was demonstrated to be activated by tyrosine kinase-7 (PTK 7) on the target cell surface with consequential fluorescence emission and drug release [9]. Importantly, the high density of PTK 7 on the CEM cell surface (~28 nm between adjacent receptors) makes it possible to realize multivalent binding with a multivalent ligand system, which may greatly improve avidity [10, 27]. The formation of NTri-SAAP was verified by gel retardation assay. As shown in Figure 1A, distinct band lag was observed in Lane 4, revealing successful self-assembly of NTri-SAAP with increased mass and geometrical size. The four bands with different gradients were guessed to correspond to three assembly products (monovalent, divalent, and trivalent NTri-SAAP) and residual SAAP (with SA). And, luminance analysis disclosed that multivalent probes accounted for 73.2% of the total products, appearing as the main product. Then, atomic force microscopy (AFM) and dynamic light scattering (DLS) analyses were performed, which further supported the formation of NTri-SAAP (Figure S2 and Figure S3). The former revealed an increased size from NTri (~9×2 nm) to NTri-SAAP (~32×5 nm), and the latter confirmed the average hydrodynamic diameters of NTri and NTri-SAAP as ~10.2 nm and ~33.7 nm, respectively. These values are in close agreement with the predicted sizes of the model structures. Additionally, fluorescence spectral measurements disclosed that the NTri-SAAP was successfully assembled with quenched signals, while the presence of SA and/or NTri did not affect the fluorescence of Cy5 dye (Figure S4).
To inspect the feasibility of the NTri-SAAP-based multivalent activatable strategy, flow cytometry assays of target CEM cells were performed by using human lymphoma Ramos cells as a negative control. Figure 1B displays the fluorescence responses of these two cell lines to different probes, including Cy5-Apt-L (monovalent always-on probe), SAAP (monovalent activatable probe), NTri-Cy5-Apt-L (multivalent always-on probe) and NTri-SAAP (multivalent activatable probe). To make a reasonable comparison, the concentrations of these probes were regulated to have the same amount of Cy5. It was shown that after a simple wash-free incubation, both SAAP monomer and multivalent NTri-SAAP were activated by target cells so as to have higher signal responses to CEM cells than Ramos cells. In contrast, NTri-SAAP realized a much higher activation efficiency with dramatically lowered background and greatly improved affinity (Figure S5). Compared with always-on probes, such as Cy5-Apt-L and NTri-Cy5-Apt-L, SAAP had a lower background due to the activatable design. But interestingly, the nonspecific response to Ramos cells was further reduced by NTri-SAAP. Fluorescence spectral tests revealed that NTri-SAAP was activated more slowly than SAAP by cDNA, a complementary DNA strand acting as a target mimic (Figure S6). We thus speculated that it might be steric effects that decreased nonspecific activation and further contributed to the ultralow background. As for the improved binding affinity, studies confirmed that it could not be achieved by simply mixing SAAP and NTri without SA conjugation (Figure S7), and the NTri-SAAP as a whole unit was bound to the CEM cell surface (Figure S8). It was thus believed that the increase in positive signal derived from the multivalent binding system of NTri-SAAP, in which an increased local ligand density could effectively facilitate target binding events. For always-on probes, a similar multivalent effect was also found, but the difference was that the multivalent NTri-Cy5-Apt-L elevated both positive signals and background in comparison with Cy5-Apt-L monomers. Nevertheless, due to the multivalent effect, these two multivalent systems both displayed higher signal-to-background ratios (S/B) than the corresponding monomers for in vitro CEM cell analysis (Figure 1C). And from another point of view, the activatable probes showed higher contrast than always-on probes. This was consistent with previous reports [6, 28]. Specifically, the S/B value of NTri-SAAP was ~4 times and ~3 times higher than that of SAAP and NTri-Cy5-Apt-L, respectively. The results substantially supported the feasibility of the NTri-SAAP strategy for sensitive analysis of cancer cells.
Evaluation of the binding performance of the multivalent NTri-SAAP system
The poor binding stability of conventional aptamer probes in biofluid is still a critical obstacle to their in vivo application. One important cause is the low level of Mg2+ in biofluid, which acts as a chaperone in conformational folding of aptamers [29, 30]. We thereupon investigated the binding property of NTri-SAAP in several representative buffers, including binding buffer, PBS with Mg2+ supplied (+Mg), and PBS without Mg2+ supplied (-Mg). Among them, binding buffer prepared by adding Mg2+, BSA, yeast-tRNA and glucose into PBS, was employed as an ideal buffer for aptamer binding, because it is used in cell-SELEX processes routinely. As illustrated in Figure 2, SAAP was susceptible to the environment. After removal of cofactors, its binding ability dropped sharply. Especially in PBS (-Mg), affinity loss was observed with an ~91% drop in S/B. In contrast, NTri-SAAP kept relatively stable avidity to CEM cells in different buffers. Even in the harshest PBS (-Mg), its S/B value could still retain ~35% of that in binding buffer. It was obvious that NTri-SAAP had a better binding stability than SAAP, which might result from the collective action of the multivalent effect and weakened conformational energy penalty for increased mass [30]. The stable binding performance would surely facilitate the use of NTri-SAAP in real biological systems.
Binding affinity against target cancer cells is also an important parameter to evaluate for theranostic probes. As shown in Figure 2C, the binding affinity of NTri-SAAP against CEM cells was further measured using flow cytometry, in comparison with SAAP. It was disclosed that multivalent NTri-SAAP could afford a higher binding affinity, having a calculated equilibrium dissociation constant (Kd) in the nanomolar-to-picomolar range (Kd = 3.41 ± 0.88 nM), which was ~2.5 times that of monovalent SAAP (Kd = 8.58 ± 0.93 nM, the inset in Figure 2C).
Figure 2
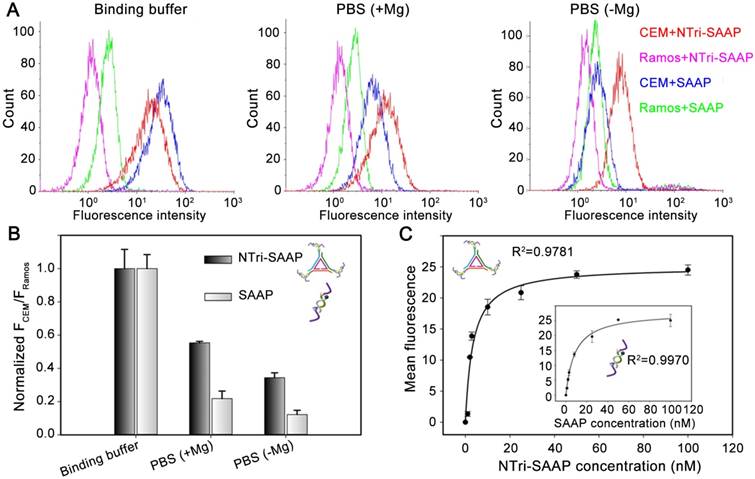
Binding performance investigation of NTri-SAAP by flow cytometry, in comparison with SAAP monomer. (A) Binding stability of NTri-SAAP in different buffers. (B) Corresponding histogram of the normalized fluorescence ratios of CEM cells to Ramos cells for probes and buffers in (A). (C) Binding affinity comparison of NTri-SAAP and SAAP (the inset) against CEM cells.
Selectivity and sensitivity investigation of the multivalent NTri-SAAP system
Next, the specificity of NTri-SAAP was investigated. NTri-Control, which was assembled from NTri scaffold and control probes with little affinity to target CEM cells, was utilized as a negative control probe. Two cancer cell lines (Ramos and U266) and one normal cell line (L02) were used as negative control cells. Flow cytometry analysis indicated that NTri-SAAP had a much higher fluorescence response to CEM cells than NTri-Control, due to its substantial activation based on recognition-induced conformational change (Figure 3A). By contrast, for the other three control cell lines, the signal intensities of NTri-SAAP were slightly lower than that of NTri-Control, because the latter had a slightly higher background [9]. The results confirmed that the NTri-SAAP strategy could afford high sequence specificity and selective binding to target cells. Confocal microscopy subsequently verified that the fluorescence activation of NTri-SAAP was triggered by target membrane proteins, thus leading to distinct red halos around cells (Figure 3B). This finding was in line with the previous report [9, 31].
To further evaluate the sensitivity of the NTri-SAAP strategy, a quantitative analysis of CEM cells was implemented using flow cytometry (Figure S9). Samples with cell numbers ranging from 0 to 290,000 were obtained by serial dilution. It was observed that the number of events detected in the upright region positively correlated with the number of prepared CEM cells. A quasi-linear correlation with a regression equation of log(y) = 1.4092 × log(x) - 2.1938 was acquired, indicating a good linear correlation in the range of 58 to 290,000 cells (Figure 3C). The lowest number of CEM cells detected was 58 in 150 μL binding buffer, which was comparable to other activatable aptamer-based approaches [9, 28, 32].
Drug loading and release characteristics of the multivalent NTri-SAAP system
The drug loading capacity of NTri-SAAP was investigated by taking Dox as a drug model. Previous reports have shown that Dox could preferentially incorporate into double-stranded CG base pairs with quenched fluorescence [3, 5, 9]. Hence, fluorescence spectroscopy was used to evaluate the drug loading capacity of multivalent NTri-SAAP, NTri scaffold and SAAP monomer as carriers (Figure 4A and Figure S10). It was found that the fluorescence intensity of Dox decreased gradually with an increase in probe concentration, which suggested effective drug loading for all three carriers. However, benefiting from the combination of NTri and SAAP, the loading capacity of NTri-SAAP was revealed to be ~6 times that of SAAP and ~2 times that of NTri. We next inspected the stability of Dox-loaded NTri-SAAP (NTri-SAAP-Dox) at physiological temperature, in comparison with Dox-loaded SAAP (SAAP-Dox). As presented in Figure 4B, NTri-SAAP-Dox and SAAP-Dox both displayed desirable stabilities in PBS and 10% fetal bovine serum with little drug leaking, suggesting a potential in vivo drug delivery application. But, when exposed to cell lysate or a high dose of nuclease, Dox was successfully released with restored fluorescence due to degradation of DNA carriers (Figure S11), which implied potential drug freeing after cell internalization. Additionally, under the same condition, SAAP-Dox was observed to be more susceptible to nuclease attack, showing much quicker fluorescence restoration than NTri-SAAP-Dox. It was guessed that larger DNA structures are more stable [33, 34]. Overall, NTri-SAAP was superior to SAAP in both drug loading and nuclease resistance, thus promising better performance in theranostics.
Figure 3
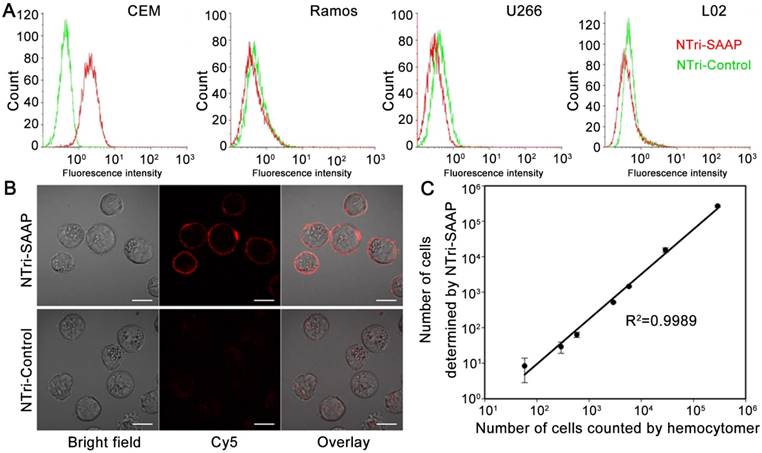
Specificity and sensitivity investigation of the multivalent NTri-SAAP system. (A) Flow cytometry assays of different cells, incubated with NTri-SAAP or NTri-Control. (B) Confocal microscopy images of CEM cells incubated with NTri-SAAP or NTri-Control, showing the binding of NTri-SAAP on cell membrane surface (scale bar: 10 μm). (C) Calibration curve illustrating the relationship between the amount of CEM cells counted by hemocytometer and the number of CEM cells detected with NTri-SAAP. Error bars represent standard deviations from three repeated experiments.
To investigate the cellular internalization-induced drug release of NTri-SAAP-Dox, CEM cells treated with NTri-SAAP-Dox, pure Dox and NTri-SAAP were analyzed on a confocal microscope. Hoechst 33342 was utilized as a nuclear staining reference (Figure 4C). It was revealed that, similar to other multivalent systems [10, 35], NTri-SAAP with or without Dox loading could aggregate on the target cell surface for ligand binding-induced receptor clustering, and then enter into cells through receptor-mediated endocytosis after a longer period of incubation. Due to pH/ionic perturbation or nuclease degradation in cytoplasm [3], Dox could be released from NTri-SAAP-Dox, restoring fluorescence. Note that the weak signal of Dox might be a result of incomplete release or diffusion into the nucleus with quenched fluorescence. In addition, a few drugs that were released on the cell surface due to target activation could also permeate into cells just like free Dox. This implied that the NTri-SAAP strategy could achieve efficient drug delivery to target cells.
Selective cytotoxicity of the Dox-loaded multivalent NTri-SAAP system
MTS assays were conducted to assess the selective cytotoxicity of NTri-SAAP-Dox. As displayed in Figure 5A, NTri-SAAP-Dox realized a much higher toxicity to target CEM cells than nontarget Ramos cells. In contrast, free Dox presented a similarly high toxicity to both cell lines due to lack of selectivity. As a negative control, NTri-Control loaded with Dox (NTri-Control-Dox) was also used to treat cells; its toxicity to CEM cells was found to be significantly alleviated. More importantly, pure NTri-SAAP showed negligible cytotoxicity. The results implied that the drug delivery profile of NTri-SAAP-Dox based on recognition-induced activation and endocytosis would benefit targeted therapy, thus contributing to an ideal vehicle with inherent biocompatibility. Furthermore, the cytotoxiccity of NTri-SAAP-Dox was also found to be dose-dependent (Figure 5B). Consistent with their drug loading efficiencies, NTri-SAAP-Dox showed stronger suppression of cell viability over SAAP-Dox. The high selectivity and therapeutic efficacy of the NTri-SAAP strategy suggested its potential uses in precise therapy.
Figure 4
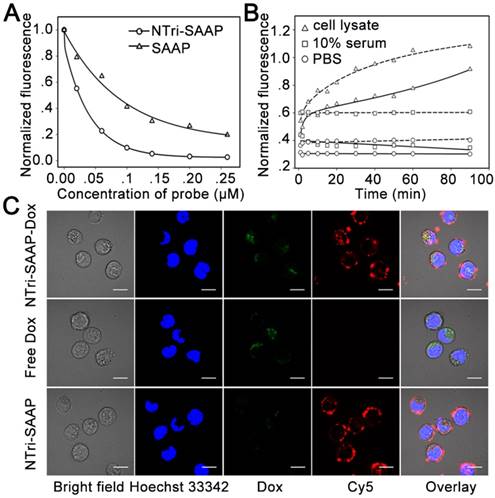
Characterization of drug loading and release. (A) Drug loading capacity comparison of NTri-SAAP with SAAP based on fluorescence quenching of Dox. (B) Time-lapse fluorescence monitoring of Dox diffused from NTri-SAAP (solid line) or SAAP (dotted line) at 37 °C in cell lysate, 10% fetal bovine serum and PBS. (C) Confocal microscopy images of CEM cells after incubation with NTri-SAAP-Dox, free Dox, and pure NTri-SAAP (scale bar: 10 μm).
Figure 5
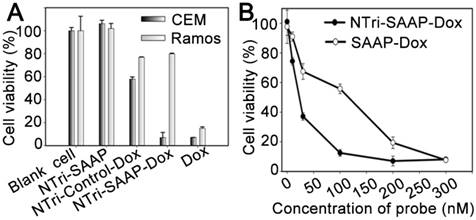
Cytotoxicity of Dox-loaded NTri-SAAP measured by MTS assays. (A) Viability of target CEM cells and nontarget Ramos cells after different treatments (Dox, 2.0 μM). (B) Viability of CEM cells treated by NTri-SAAP-Dox or SAAP-Dox, at different doses. Error bars represent standard deviations from three repeated experiments.
In vivo contrast-enhanced imaging with the multivalent NTri-SAAP system
To ascertain whether the NTri-SAAP strategy could improve the performance of activatable probes in live animals, monovalent SAAP* and multivalent NTri-SAAP* were injected into CEM tumor-bearing nude mice via tail vein for comparison. The NTri-SAAP* was assembled from NTri scaffold and SAAP*, which has a structure similar to SAAP but was proved to be more stable in biofluid due to proper enhancement of the hybridization ability in the double helix region [9]. As shown in Figure 6, both monomer SAAP* and multivalent NTri-SAAP* were effectively activated in CEM tumor areas after injection, thus revealing clear tumor figures with elevated fluorescence relative to surrounding muscle areas. Specifically for SAAP*, obvious fluorescence enhancement was seen at the tumor site as early as 10 min post-injection. And, as a result of nuclease degradation, nonspecific disorganization and target activation, fluorescence signals in the whole body including the tumor site gradually increased over time and reached the brightest level at 60 min. Then, the signals gradually decreased due to the continuous clearance of probes. As for the NTri-SAAP* group, the same trend of signal changes was also observed. However, unlike SAAP*, NTri-SAAP* realized a much lower fluorescence background in the whole body within 8 h of observation, thus contributing to prominent fluorescent contrast of CEM tumor at most post-injection time points. Moreover, even by adjusting the color scale to deduct more background signals to obtain clear tumor figures, nonspecific fluorescence interference, especially the influence of metabolic organs caused by SAAP*, was still much larger than that of NTri-SAAP* (Figure S12). This again demonstrated the advantage of the NTri-SAAP* strategy in ultralow background and nuclease resistance. Of course, the activation rate of NTri-SAAP* in CEM tumors was measured to be slower than that of SAAP*, which was consistent with the in vitro study where NTri-SAAP* needed a longer time to become activated. In addition, benefiting from its high avidity and robust stability, the fluorescence of NTri-SAAP* could still be imaged in the CEM tumor site at 8 h, while the SAAP* bound to CEM tumor cells were almost eliminated along with unmeasurable fluorescence before 6 h post-injection. Further, ex vivo fluorescence imaging of the tumor and major organs was performed at 8 h post-injection. The results showed that the NTri-SAAP* mainly accumulated in tumor and liver (Figure S13), which further confirmed its long retention time in tumor and suggested improved anti-tumor efficacy. And, it should also be noted that, different from the predominant renal clearance of SAAP*, NTri-SAAP was excreted more through the hepatic route due to its increased particle size, thus showing prolonged elimination life (Figure S14) [36]. Hence, it could be concluded that the NTri-SAAP strategy did greatly improve tumor imaging quality.
Figure 6
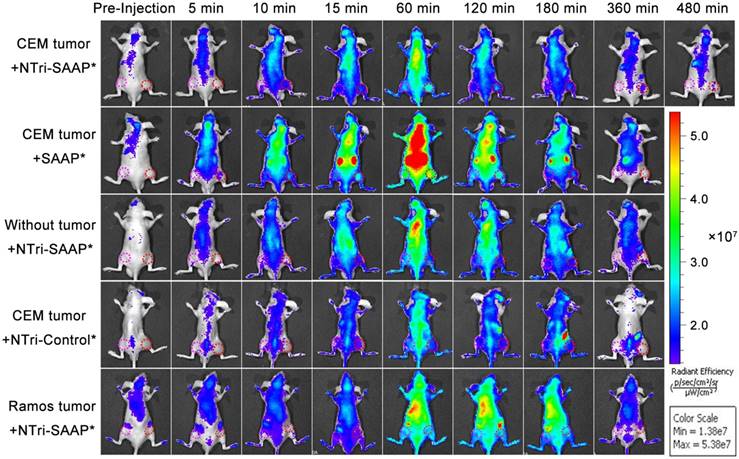
In vivo time-lapse fluorescence imaging of nude mice bearing different tumors after intravenous injection with different probes. Red circles indicate tumor sites and pink circles indicate muscle sites.
To verify the imaging specificity of the NTri-SAAP strategy, NTri-Control* was used as the negative probe control, without tumor-bearing and Ramos tumor-bearing mice were used as the negative tumor control. As anticipated, under the same condition, no obvious activated fluorescence was observed either in tumor sites or in similar sites of normal mice for all the control groups, thus displaying undifferentiated fluorescence intensities between tumor sites and muscle sites (Figure 6 and Figure S15). In comparison, for both the NTri-SAAP*-treated group and the SAAP*-treated group, higher fluorescence signals were acquired in tumor sites, and the former showed higher image contrasts within an extended time window. The results thus implied that the NTri-SAAP strategy could afford in vivo cancer imaging with high specificity.
In vivo anti-tumor effect of Dox-loaded multivalent NTri-SAAP system
We next evaluated the anti-tumor efficacy of the Dox-loaded multivalent NTri-SAAP system by utilizing CEM tumor-bearing nude mice. Drug treatments were started when the tumors reached a volume of ~100 mm3 (appointed as Day 1). All the mice were divided into four groups treated by free Dox, NTri-SAAP*-Dox, NTri-SAAP*, and PBS, respectively, for comparison. Dox and NTri-SAAP*-Dox were injected intravenously at a dose equivalent to 2 mg/kg of doxorubicin every other day, and the amount of bare NTri-SAAP* was given according to that of NTri-SAAP*-Dox. Tumor sizes and body weights were monitored every other day until the mice were sacrificed at day 13. As seen in Figure 7, compared with PBS, bare NTri-SAAP* negligibly affected tumor growth. Conversely, both NTri-SAAP*-Dox and free Dox exhibited significant inhibition of tumor growth, and NTri-SAAP*-Dox gave a higher anti-tumor efficacy with 81.95% inhibition. This might be attributed to the targeted drug delivery function of NTri-SAAP*-Dox, as well as its prolonged residence in tumor due to its specific binding and increased particle size, in line with the results revealed by in vivo imaging. Furthermore, compared with the PBS group, no obvious loss of body weight was observed in the other three groups during treatment, thus indicating a good biocompatibility of the NTri-SAAP strategy.
Figure 7
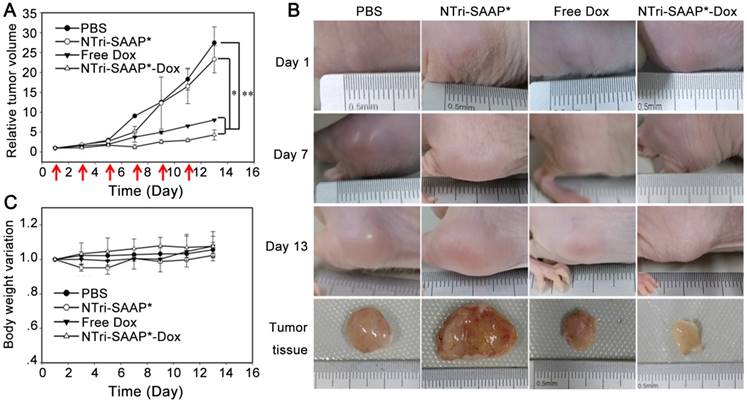
In vivo anti-tumor effect evaluation of the Dox-loaded multivalent NTri-SAAP system. (A) Tumor growth profiles of mice under different treatments for 13 days. (Red arrows indicate the treatment time points; *P<0.05, **P<0.01). (B) Representative photographs of mice in (A) for varied time periods. (C) Body weight variation of mice in (A).
Conclusions
In summary, a novel theranostic strategy of DNA nanotriangle-scaffolded multivalent split activatable aptamer probe (NTri-SAAP) was first developed. Benefiting from programmable self-assembly of DNA nanotechnology, SAAPs were assembled on the biocompatible planar NTri with controllable density, location and orientation. Taking advantage of the collective action of multivalent effect and activatable architecture, the assembled NTri-SAAP realized greatly-improved target binding affinity, ultralow nonspecific background and robust stability in harsh conditions. Especially for in vivo application, contrast-enhanced specific tumor imaging could be observed and the tumor imaging window was extended to over 8 h due to improved biostability. Furthermore, by virtue of the high Dox loading capacity and targeted drug delivery, NTri-SAAP rendered highly efficient cancer treatment both in vitro and in vivo with 81.95% tumor inhibition efficacy, but no observable side effects. Collectively, the strategy combined advantages from aspects including controllable assembly and biocompatibility of DNA scaffolds, flexible design and specific recognition of aptamer probes, as well as multivalent effect and target-activatable architecture. Due to its high affinity, low background and good stability, NTri-SAAP might have great promise in precise and high-quality cancer theranostics.
Abbreviations
Apt-L: split aptamer-contained long DNA strand; AFM: atomic force microscopy; Bio: biotin; Dox: doxorubicin; DLS: dynamic light scattering; F: fluorophore; HCR: hybridization chain reaction; Kd: equilibrium dissociation constant; NTri: DNA nanotriangle; NTri-SAAP: DNA nanotriangle-scaffolded multivalent split activatable aptamer probe; NTri-SAAP-Dox: Dox-loaded NTri-SAAP; NPs: nanoparticles; PAGE: polyacrylamide gel electrophoresis; PTK 7: tyrosine kinase-7; Q: quencher; RCA: rolling circle amplification; SAAP: split activatable aptamer probe; S: complementary short DNA strand; SA: streptavidin; S/B: signal-to-background ratio.
Supplementary Material
Supplementary table, figures and experimental section.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grants 21735002, 21575037, 21521063, 21675046, 21778016, 21775036, and 21305038) and the Hunan Provincial Natural Science Foundation (Grant 2015JJ3044).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lim EK, Kim T, Paik S. et al. Nanomaterials for theranostics: Recent advances and future challenges. Chem Rev. 2015;115:327-394
2. Sun HG, Zu YL. Aptamers and their applications in nanomedicine. Small. 2015;11:2352-2364
3. Zhu GZ, Zheng J, Song EQ. et al. Self-assembled, aptamer-tethered DNA nanotrains for targeted transport of molecular drugs in cancer theranostics. Proc Natl Acad Sci USA. 2013;110:7998-8003
4. Li XS, Kim J, Yoon JY. et al. Cancer-associated, stimuli-driven, turn on theranostics for multimodality imaging and therapy. Adv Mater. 2017;29:1606857-1606880
5. Zhang HM, Ma YL, Xie Y. et al. A controllable aptamer-based self-assembled DNA dendrimer for high affinity targeting, bioimaging and drug delivery. Sci Rep. 2015;5:10099
6. Wang YM, Wu Z, Liu SJ. et al. Structure-switching aptamer triggering hybridization chain reaction on the cell surface for activatable theranostics. Anal Chem. 2015;87:6470-6474
7. Wu X, Chen J, Wu M. et al. Aptamers: active targeting ligands for cancer diagnosis and therapy. Theranostics. 2015;5:322-344
8. Munzar JD, Ng A, Corrado M. et al. Complementary oligonucleotides regulate induced fit ligand binding in duplexed aptamers. Chem Sci. 2017;8:2251-2256
9. Lei YL, Tang JL, Shi H. et al. Nature-inspired smart DNA nanodoctor for activatable in vivo cancer imaging and in situ drug release based on recognition triggered assembly of split aptamer. Anal Chem. 2016;88:11699-11706
10. Zhang ZQ, Eckert MA, Ali MM. et al. DNA-scaffolded multivalent ligands to modulate cell function. ChemBioChem. 2014;15:1268-1273
11. Sheng W, Chen T, Tan WH. et al. Multivalent DNA nanospheres for enhanced capture of cancer cells in microfluidic devices. ACS Nano. 2013;7:7067-7076
12. Zhu J, Huang H, Dong SW. et al. Progress in aptamer-mediated drug delivery vehicles for cancer targeting and its implications in addressing chemotherapeutic challenges. Theranostics. 2014;4:931-944
13. Wu YR, Sefah K, Liu HP. et al. DNA aptamer-micelle as an efficient detection delivery vehicle toward cancer cells. Proc Natl Acad Sci USA. 2010;107:5-10
14. Tang JL, Yu YR, Shi H. et al. Polyvalent and thermosensitive DNA nanoensembles for cancer cell detection and manipulation. Anal Chem. 2017;89:6637-6644
15. Zhou GB, Lin MH, Song P. et al. Multivalent capture and detection of cancer cells with DNA nanostructured biosensors and multibranched hybridization chain reaction amplification. Anal Chem. 2014;86:7843-7848
16. Jang B, Choi Y. Photosensitizer-conjugated gold nanorods for enzyme-activatable fluorescence imaging and photodynamic therapy. Theranostics. 2012;2:190-197
17. Ye XS, Shi H, He XX. et al. Cu-Au alloy nanostructures coated with aptamers: a simple, stable and highly effective platform for in vivo cancer theranostics. Nanoscale. 2016;8:2260-2267
18. Zhang ZQ, Ali MM, Eckert MA. et al. A polyvalent aptamer system for targeted drug delivery. Biomaterials. 2013;34:9728-9735
19. Ye XS, Shi H, He XX. et al. Au@Ag/Au nanoparticles assembled with activatable aptamer probes as smart “nanodoctors” for image-guided cancer thermotherapy. Nanoscale. 2014;6:8754-8761
20. Rinker S, Ke YG, Liu Y. et al. Self-assembled DNA nanostructures for distance dependent multivalent ligand-protein binding. Nat Nanotechnol. 2008;3:418-422
21. Kumar V, Palazzolo S, Bayda S. et al. DNA nanotechnology for cancer therapy. Theranostics. 2016;6:710-725
22. Lee H, Lytton-Jean AK, Chen Y. et al. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat Nanotechnol. 2012;7:389-393
23. Zhao XC, Lis JT, Shi H. A systematic study of the features critical for designing a high avidity multivalent aptamer. Nucleic Acid Ther. 2013;23:238-242
24. Jasinski DL, Khisamutdinov EF, Lyubchenko YL. et al. Physicochemically tunable polyfunctionalized RNA square architecture with fluorogenic and ribozymatic properties. ACS Nano. 2014;8:7620-7629
25. Khisamutdinov EF, Jasinski DL, Guo P. RNA as a boiling-resistant anionic polymer material to build robust structures with defined shape and stoichiometry. ACS Nano. 2014;8:4771-4781
26. Li HL, He BF, Liu XP. et al. Regulation on toll-like receptor 4 and cell barrier function by Rab26 siRNA-loaded DNA nanovector in pulmonary microvascular endothelial cells. Theranostics. 2017;7:2537-2554
27. Chen Y, Munteanu AC, Huang YF. et al. Mapping receptor density on live cells by using fluorescence correlation spectroscopy. Chem A European J. 2009;15:5327-5336
28. Shi H, He XX, Wang KM, Wu X. et al. Activatable aptamer probe for contrast-enhanced in vivo cancer imaging based on cell membrane protein-triggered conformation alteration. Proc Natl Acad Sci USA. 2011;108:3900-3905
29. Andrusishina IN. Diagnostic values of calcium and magnesium forms determined in human serum and saliva. J Elem. 2010;15:425-433
30. Kuai HL, Zhao ZL, Mo LT. et al. Circular bivalent aptamers enable in vivo stability and recognition. J Am Chem Soc. 2017;139:9128-9131
31. Shangguan DH; Li Y, Tang ZW. et al. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc Natl Acad Sci USA. 2006;103:11838-11843
32. Yin JJ, He XX, Wang KM. et al. Label-free and turn-on aptamer strategy for cancer cells detection based on a DNA-silver nanocluster fluorescence upon recognition-induced hybridization. Anal Chem. 2013;85:12011-12019
33. Mei Q, Wei XX, Su FY. et al. Stability of DNA origami nanoarrays in cell lysate. Nano Letters. 2011;11:1477-1482
34. Keum J-W, Bermudez H. Enhanced resistance of DNA nanostructures to enzymatic digestion. Chem Commun. 2009;45:7036-7038
35. Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands as probes of signal transduction. Angew Chem Int Ed. 2006;45:2348-2368
36. Yu M, Zheng J. Clearance pathways and tumor targeting of imaging nanoparticles. ACS Nano. 2015;9:6655-6674
Author contact
![]() Corresponding authors: Kemin Wang, email: kmwangedu.cn; Hui Shi, email: huishi_2009edu.cn.
Corresponding authors: Kemin Wang, email: kmwangedu.cn; Hui Shi, email: huishi_2009edu.cn.
Citation styles
APA
Lei, Y., Qiao, Z., Tang, J., He, X., Shi, H., Ye, X., Yan, L., He, D., Wang, K. (2018). DNA nanotriangle-scaffolded activatable aptamer probe with ultralow background and robust stability for cancer theranostics. Theranostics, 8(15), 4062-4071. https://doi.org/10.7150/thno.24683.
ACS
Lei, Y.; Qiao, Z.; Tang, J.; He, X.; Shi, H.; Ye, X.; Yan, L.; He, D.; Wang, K. DNA nanotriangle-scaffolded activatable aptamer probe with ultralow background and robust stability for cancer theranostics. Theranostics 2018, 8 (15), 4062-4071. DOI: 10.7150/thno.24683.
NLM
Lei Y, Qiao Z, Tang J, He X, Shi H, Ye X, Yan L, He D, Wang K. DNA nanotriangle-scaffolded activatable aptamer probe with ultralow background and robust stability for cancer theranostics. Theranostics 2018; 8(15):4062-4071. doi:10.7150/thno.24683. https://www.thno.org/v08p4062.htm
CSE
Lei Y, Qiao Z, Tang J, He X, Shi H, Ye X, Yan L, He D, Wang K. 2018. DNA nanotriangle-scaffolded activatable aptamer probe with ultralow background and robust stability for cancer theranostics. Theranostics. 8(15):4062-4071.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.