Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(13):3611-3628. doi:10.7150/thno.25613 This issue Cite
Research Paper
Distinct immune signatures in directly treated and distant tumors result from TLR adjuvants and focal ablation
Michael Chavez1,†, Matthew T. Silvestrini1,†, Elizabeth S. Ingham1, Brett Z. Fite1, Lisa M. Mahakian1, Sarah M. Tam1, Asaf Ilovitsh1, Arta M. Monjazeb2, William J. Murphy3, Neil E. Hubbard4, Ryan R. Davis5, Clifford G. Tepper5, Alexander D. Borowsky4, Katherine W. Ferrara1 ![]()
1. University of California, Davis, Department of Biomedical Engineering, 451 Health Sciences Drive, Davis, CA 95616, USA
2. University of California, Davis, Department of Radiation Oncology, 4501 X Street, Sacramento, CA 95817
3. University of California, Davis, Department of Dermatology, 2921 Stockton Blvd., Institute for Regenerative Cures, Suite 1630, Sacramento, CA 95817, USA
4. University of California, Davis, Center for Comparative Medicine, Davis, CA 95616, USA
5. University of California, Davis School of Medicine, Department of Biochemistry and Molecular Medicine, 4645 Second Avenue, Sacramento, CA 95817, USA
† Authors contributed equally to this work
Received 2018-2-19; Accepted 2018-5-15; Published 2018-6-7
Citation:
Chavez M, Silvestrini MT, Ingham ES, Fite BZ, Mahakian LM, Tam SM, Ilovitsh A, Monjazeb AM, Murphy WJ, Hubbard NE, Davis RR, Tepper CG, Borowsky AD, Ferrara KW. Distinct immune signatures in directly treated and distant tumors result from TLR adjuvants and focal ablation. Theranostics 2018; 8(13):3611-3628. doi:10.7150/thno.25613. https://www.thno.org/v08p3611.htm
Other stylesAbstract

Both adjuvants and focal ablation can alter the local innate immune system and trigger a highly effective systemic response. Our goal is to determine the impact of these treatments on directly treated and distant disease and the mechanisms for the enhanced response obtained by combinatorial treatments.
Methods: We combined RNA-sequencing, flow cytometry and TCR-sequencing to dissect the impact of immunotherapy and of immunotherapy combined with ablation on local and systemic immune components.
Results: With administration of a toll-like receptor agonist agonist (CpG) alone or CpG combined with same-site ablation, we found dramatic differences between the local and distant tumor environments, where the directly treated tumors were skewed to high expression of F4/80, Cd11b and Tnf and the distant tumors to enhanced Cd11c, Cd3 and Ifng. When ablation was added to immunotherapy, 100% (n=20/20) of directly treated tumors and 90% (n=18/20) of distant tumors were responsive. Comparing the combined ablation-immunotherapy treatment to immunotherapy alone, we find three major mechanistic differences. First, while ablation alone enhanced intratumoral antigen cross-presentation (up to ~8% of CD45+ cells), systemic cross-presentation of tumor antigen remained low. Combining same-site ablation with CpG amplified cross-presentation in the draining lymph node (~16% of CD45+ cells) compared to the ablation-only (~0.1% of CD45+ cells) and immunotherapy-only cohorts (~10% of CD45+ cells). Macrophages and DCs process and present this antigen to CD8+ T-cells, increasing the number of unique T-cell receptor rearrangements in distant tumors. Second, type I interferon (IFN) release from tumor cells increased with the ablation-immunotherapy treatment as compared with ablation or immunotherapy alone. Type I IFN release is synergistic with toll-like receptor activation in enhancing cytokine and chemokine expression. Expression of genes associated with T-cell activation and stimulation (Eomes, Prf1 and Icos) was 27, 56 and 89-fold higher with ablation-immunotherapy treatment as compared to the no-treatment controls (and 12, 32 and 60-fold higher for immunotherapy-only treatment as compared to the no-treatment controls). Third, we found that the ablation-immunotherapy treatment polarized macrophages and dendritic cells towards a CD169 subset systemically, where CD169+ macrophages are an IFN-enhanced subpopulation associated with dead-cell antigen presentation.
Conclusion: While the local and distant responses are distinct, CpG combined with ablative focal therapy drives a highly effective systemic immune response.
Keywords: ablation, RNA sequencing, immunotherapy, T-cell receptor sequencing, ultrasound
Introduction
Checkpoint inhibition strategies result in impressive clinical responses in a subset of patients, yet patient response remains limited in tumors lacking a robust T-cell infiltrate [1]. Response requires effective cross-priming of T-cells by the innate immune system [2, 3]. Immunotherapy protocols incorporate local adjuvants such as CpG (toll-like receptor 9 (TLR9) agonist) to enhance the innate immune response and cross-priming. Local adjuvants can be highly effective [4], particularly when initiated while tumors are small [5]. Treatment of larger tumors can incorporate surgery or debulking with focal therapy. Many clinical cancer treatment protocols involve focal ablative therapy (e.g., 47-60% of cancer patients receive radiation therapy (RT) over the course of their treatment [6]). Therefore, combinations of focal therapy and immune adjuvants must be optimized, particularly in large lesions. Such focal therapies have the potential to offer rapid response by inducing immunogenic cell death, releasing tumor-associated antigen [7-9] and endogenous danger signals [10], increasing tumor infiltrating lymphocytes and antigen presenting cells [11, 12], and reducing tumor burden and circulating immunosuppressive cytokines [13]. We endeavor to identify mechanisms by which adjuvant and ablation-activated innate immune responses can be combined to enhance efficacy.
Transcriptomic analyses have increasingly been adopted to understand immunotherapeutic mechanisms [14-19], and these analyses yield important insights into T-cell activity as well as the role of specific pathways (e.g., the Wnt pathway) and signals (e.g., β-catenin, Wnt7b, Fzd3) in T-cell infiltration. Further, they facilitate insight into the relevant signaling cascades in the bulk tumor environment. For example, recent studies have shown that chemotherapy and certain focal ablative therapies trigger signaling between the innate and adaptive immune systems, in part due to the secretion of type I interferons (IFNs) and subsequent expression of interferon-stimulated genes (ISGs) [20-22]. More than 150 ISGs have been reported in this context, many of which are traditionally thought to be expressed in response to a viral infection, demonstrating the unique immune environment created by some effective therapies. Type I IFN and TLR9 agonists in particular have been shown to alter macrophage phenotype in the context of systemic sclerosis [23]; yet, less is known about their effects on macrophage phenotype in the context of cancer ablative therapies.
Previously, we demonstrated that ablation drastically diminishes viable tumor tissue 24 hours after treatment [24]; however, it is still unknown how these dead tumor cells within the ablated region are processed and presented. Five distinct macrophage subpopulations have now been identified: classically-activated macrophages (M1), alternatively-activated macrophages (M2), tumor-associated macrophages (TAM), CD169+ macrophages, and T-cell receptor positive (TCR+) macrophages [25]. CD169+ macrophages are a class of professional antigen-presenting cells (APCs) known to be enhanced by Type I IFN and proven to be uniquely capable of cross-priming independent of dendritic cells (DCs) [26, 27]. We evaluate changes in macrophage and DC number and gene expression to determine how such cell types are altered by CpG and by ablative therapy.
High-intensity focused ultrasound and high-dose hypofractionated RT are focal ablative techniques for the minimally invasive treatment of solid tumors. Each stimulates some degree of immunization to tumor-associated antigens following treatment [28, 29]; but each alone has proven insufficient to overcome tumor-mediated immunosuppression, thus limiting abscopal effects and the ability to treat metastatic disease [30, 31]. Thermal treatment with magnetic resonance-guided focused ultrasound (MRgFUS) is particularly attractive as such treatment is non-invasive and spatially delineated, resulting in a precisely controlled temperature increase. Under image guidance, a fraction of the tumor is ablated and heat-fixed, while remaining tumor cells undergo an immunogenic cell death over 1-2 days. Moreover, MRgFUS can be repeated on a schedule that can be optimized for each patient without concern for radiation-mediated toxicities, and such treatments have the potential to speed cell death as compared with RT. While both focal therapies create observable increases in tumor infiltration of macrophages and CD8+ and CD4+ lymphocytes after treatment, synergistic focal-immunotherapy protocols are required to create an effective, systemic anti-tumor response [32-34]. Although RT is currently the most prevalent clinical focal therapy protocol and the most frequently explored in combination with immunotherapy [33, 34], the radiation dose cloud from RT harms surrounding normal tissues and may negatively affect the immune infiltrate. Recently, the combination of MRgFUS ablation and immunomodulatory adjuvants has performed well in pre-clinical studies [24]. Thermal dosing can be monitored with magnetic resonance thermometry to mediate controlled cell death to predefined tumor volumes.
By combining RNA and T-cell receptor sequencing (RNA-seq and TCR-seq, respectively) with flow cytometry, immunohistochemistry (IHC) and quantitative PCR (qPCR), we show for the first time, the distinct effects of adjuvants and ablation on the local and distant immune response and their impact in driving a T-cell response. We evaluated MRgFUS ablation, immunotherapy alone, and combined ablative-immunotherapy (AI) in three models of multi-focal cancer: the B16-F10/B16-OVA model of melanoma, which provides the opportunity to assess antigen presentation associated with immunogenic cell death [35], the neu exon deletion line (NDL), a syngeneic, Erbb2/Her2-driven orthotopic model of epithelial, focal, mammary adenocarcinoma [36], and the MMTV-PyMT transgenic model of breast cancer [37]. In our studies, we created AI protocols by combining the checkpoint inhibitor, anti-PD-1, with CpG and MRgFUS ablation. Without anti-PD-1, the adaptive immune response is muted in either case in our models and, therefore, we included anti-PD-1 in each local treatment protocol; however, anti-PD-1 is delivered systemically and, therefore, does not drive the differences between the directly treated and distant sites. Through each model, we developed mechanistic insights for treatments that bridge a local innate immune response and a distant adaptive response.
Results
We first evaluated the systemic efficacy of combining ablation with a potent immunotherapeutic regimen and compared the outcome with that achieved with immunotherapy alone. Next, we determined the impact of ablation and agonists on the T-cell population and TCR clones through flow cytometry, RNA-seq, and TCR-seq. We then explored the mechanisms by which thermal dosing contributes to cross-priming and synergizes with a TLR agonist including: the dynamics of tumor antigen release and subsequent cross-presentation and cross-priming, the local release of type I IFNs and expression of ISGs, and myeloid cell expansion and phenotypic changes.
Combining ablation with immunotherapy enhances anti-tumor response and generates a robust abscopal response
We first combined MRgFUS ablation and immunotherapy to produce a combined ablative-immunotherapy (AI) treatment in a murine model of primary and distant site mammary adenocarcinoma. FVB mice bilaterally transplanted with NDL tumors (i.e., HER2+) in the fourth and ninth mammary fat pads were administered immunotherapy one week prior to the partial ablation of the treated tumor, where CpG was intratumorally injected into a single tumor and anti-PD-1 was intraperitoneally injected (Figure 1A). Treated tumors are denoted as either immunotherapy-treated (I-T) or thermal-ablative immunotherapy-treated (AI-T), and contralateral tumors are denoted as either immunotherapy-contralateral (I-C) or thermal-ablative immunotherapy-contralateral (AI-C). Across the cohort of 118 NDL tumor mice (divided as 45 AI, 35 immunotherapy-only and 38 no treatment control (NTC) mice), tumor volume was similar for the AI and immunotherapy-only treated mice at the start of treatment.
The NDL tumor growth reduction resulting from the combined AI treatment was greater than that achieved with immunotherapy alone at the 38-day time point (Figure 1B provides tumor growth data for the RNA-seq study, which is representative of the entire study). Tumor growth was monitored across RNA-seq, TCR-seq and qPCR studies. 100% (n=20/20) of AI-T tumors and 90% (n=18/20) of AI-C tumors were responsive to AI treatment, where a responder was defined as tumor growth at least one standard deviation smaller than that of the NTC tumors over the course of treatment. The average tumor growth during this period (Day 21 to Day 38) was -0.013 ± 0.025 cm3 (n=20), 0.07 ± 0.15 cm3 (n=20) and 0.33 ± 0.16 cm3 (n=27) for AI-T, AI-C and NTC tumors, respectively. The fraction of viable tumor cells in AI-T tumors was significantly lower than that of the I-T, I-C, or NTC cohorts (Figure 1C). Furthermore, viable tumor cells were significantly reduced in AI-C, but not I-C, tumors as compared to NTC tumors. (Figure 1C).
In the PyMT model, AI-T tumor growth was also suppressed as compared with that of I-T and NTC tumors (Figure S1A). We further assessed cell viability in AI-T and I-T B16-F10 melanoma tumors at day 19 (4 days after ablation). Both treatments significantly reduced the number of viable tumor cells (Figure S1B), and, on average, AI treatment resulted in fewer viable tumor cells as compared with immunotherapy alone. Also, in the B16-OVA model, at 4 days after ablation, tumor volume was reduced by AI treatment in 4 of 5 mice with an average reduction of -0.05 cm3 from the start of treatment (Figure S1C). At this time point, while tumor growth was reduced by immunotherapy alone compared with the NTC, only 1 of 5 and 0 of 4 mice regressed in the immunotherapy-only and ablation-only cohorts, respectively.
Both directly treated and contralateral tumors with associated stroma and immune cells were then analyzed using bulk RNA-seq of the tumor tissue. Pairwise comparison of treated and NTC tumors revealed a maximum differential expression of 10,181 genes between any two treatment groups (the greatest change was between AI-T and NTC). 961 genes were differentially expressed between AI-T vs I-T tumors and 451 genes were differentially expressed between AI-C vs I-C tumors. We performed a principal component analysis (PCA) to elucidate transcriptome-wide differences between biological groups (Figure 1D). Expression at the directly treated and distant tumors clustered into distinct groups, suggesting their dramatically different cell populations. PCA further revealed that half of the I-C tumors cluster with NTC tumors (Figure 1D, black circle), demonstrating that immunotherapy alone did not notably alter the tumor's transcriptome in a fraction of the distant lesions. The lack of consistent response in I-C tumors extends to hierarchical clustering of genes corresponding to important cancer pathways. For example, the WNT signaling (276 genes expressed in this model, GO:0060070), MAPK cascade (113 genes, GO:0000165), and Cell Cycle (584 genes, GO:0007049) (Figure S2A-C) ontologies indicate that some I-C tumors remained minimally affected by treatment. Interestingly, IHC of NDL tumors revealed a similar effect, where AI treatment dramatically reduced tumor volume in the distant lesion by day 38 as compared to the NTC but immunotherapy alone did not (Figure 1E). Similarly, cancer-related gene expression was reduced following AI treatment in genes including Wnt7b, the signaling protein essential to canonical Wnt signaling, Erbb2, a tumor associated antigen in HER2+ breast cancer, Psrc1, the essential regulator of cellular progression through mitosis, and S100a14 and Faim2, genes associated with inhibition of apoptosis (Figure S2D-H). qPCR of Wnt7b on isolated cancer cells demonstrates that this pattern largely reflects differences in cancer cell number, not changes in expression, due to the potency of AI treatment (Figure S2I).
Figure 1
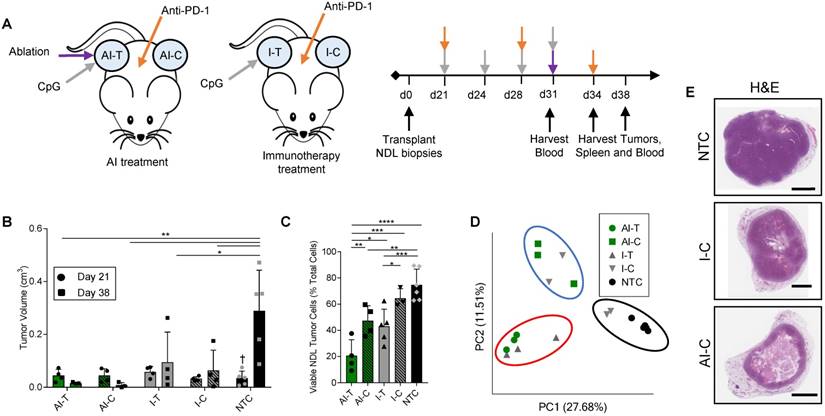
Thermal ablation with immunotherapy alters the transcriptome and improves the abscopal response in NDL tumor mice. (A) Treatment protocol administered to FVB mice orthotopically transplanted with NDL tumors in the fourth and ninth mammary fat pad (n=118). Treatment began on day 21 after implantation and assays were conducted on day 31, 34, or 38. Groups included: ablation-immunotherapy (AI) (n=45), immunotherapy-alone (I) (n=35), and no treatment control (NTC, n=38), where treated tumors are denoted as either AI-T or I-T, and contralateral tumors are denoted as either AI-C or I-C. (B) Representative volumes of treated and contralateral tumors as measured by ultrasound for AI (n=4, 8 tumors), I (n=4, 7 tumors), and NTC (n=5, 10 tumors). (C) Tumor cells were isolated and viability quantified by flow cytometry. Groups included: AI (n=8 tumors) or I (n=8 tumors), and NTC (n=6 tumors). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 (ANOVA with Fishers LSD test), and †<0.05 compared to Day 38 NTC (unpaired t-test with Welch's correction). All data are plotted as mean ± SD. (D) Principal component analysis (PCA) from RNA-seq analysis of treated and contralateral tumors as measured by ultrasound for AI-T (n=3, 3 tumors), AI-C (n=3, 3 tumors), I-T (n=3, 3 tumors), I-C (n=4, 4 tumors) and NTC (n=5, 5 tumors). (E) H&E staining in NDL contralateral and NTC tumors at day 38. Scale bars are 2 mm.
AI treatment enhances the T-cell response in distant tumors
The tumor T-cell infiltrate was investigated with flow cytometry and RNA-seq one week after ablation (Day 38). Flow cytometry (Figure S3A) demonstrated that the frequencies of CD3+, CD4+, and CD8+ T-cells were significantly increased in the AI-C tumors as compared to all other treatment groups, particularly when viewed as a percent of the live cells (Figure 2A-C). The results were confirmed with IHC of CD8+ T-cells at the Day 38 time point in both NDL and PyMT tumors (Figure S4). By flow cytometry, the frequency of IFN-γ-secreting CD8+ T-cells was increased in contralateral tumors treated with both immunotherapy protocols (I-C and AI-C) compared to the NTC (Figure 2D).
Expression of Eomes, Prf1 and Icos, genes associated with T-cell activation and stimulation, was 27, 56 and 89-fold higher in the AI-C cohort as compared to NTC (compared with 12, 32 and 60-fold increases, respectively, for the I-C treatment as compared with the NTC). Expression of these genes was thus nearly double the level in AI-C as compared with the I-C tumors; however, the direct AI-C vs. I-C comparison was not significant due to the variability in the I-C group (Figure S5A). Genes associated with apoptosis (Casp8, Casp9, Faf1) were upregulated in AI-C tumors, but not in I-C tumors, as compared to the NTC (Figure S5B). Genes associated with response to reactive oxygen species (ROS), including Txnip and Cat, were also significantly upregulated in the AI-C, as compared with the I-C, tumors (Figure 2E). Chemotaxis annotation (GO:0006935, p<0.03) was also significantly upregulated in AI-C, as compared with the I-C tumor. Exhaustion markers were similarly enhanced with AI treatment or immunotherapy alone (Figure S5A). Other cell populations (e.g., B- and T-regulatory cells) were low in number and not differentially impacted by ablation (data not shown).
Figure 2
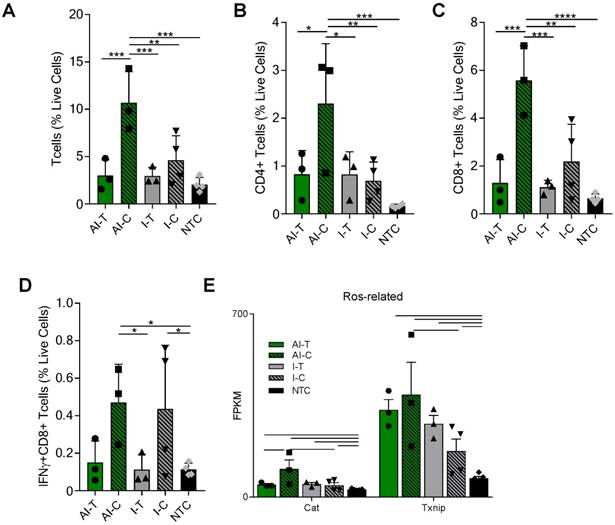
AI treatment enhances adaptive immune response in distant tumors. Groups for both flow cytometry and RNA-seq of NDL tumor mice at day 38 as treated in Figure 1A included: ablation-immunotherapy (AI), immunotherapy-alone (I), and no treatment control (NTC), where treated tumors are denoted as AI-T or I-T, and contralateral tumors are denoted as AI-C or I-C. (A-D) Flow cytometry results for (A) CD3+, (B) CD4+, (C) CD8+ T-cells, and (D) Interferon-gamma secreting CD8+ T-cells. AI (n=6 tumors), immunotherapy-alone (n=7 tumors), and NTC (n=4 tumors). * p <0.05, ** p <0.01, *** p <0.001, ****p<0.0001 (ANOVA with Fisher's LSD test). (E) RNA-seq results (samples as in Figure 1D) for genes associated with response to reactive oxygen species (ROS). All data plotted are mean ± SD. For RNA-seq results, bars represent significance of at least p < 0.01 as defined by CuffDiff.
To characterize the effects of ablation and immunotherapy on T-cell diversity, we sequenced the CDR3 region of the TCR beta chain of tumor-infiltrating T-cells treated with AI, immunotherapy-only, or NTC. Further, we assessed systemic T-cells in the blood from a subset of mice treated with the AI therapy acquired prior to treatment (at day 21, AI-Pre) and at the time of tumor harvesting (at day 38, AI-Post). Characterization of these CDR3 rearrangements and their frequency provide insight into the expansion of specific T-cells in response to antigen recognition.
Pairwise analysis of the proportion of shared TCR amino acid sequences (TCR overlap) between all samples revealed treatment-specific patterns (Figure 3A). While TCR overlap between mice is minimal before treatment (Figure 3A, black box), immunotherapy induces similar CDR3 sequences to arise between mice (Figure 3A, green and grey boxes). Treatment with immunotherapy increased the TCR overlap between tumors of different mice by two-fold as compared to the NTC cohort, independent of the inclusion of ablation within the protocol.
Most notably, the addition of ablation to the immunotherapy protocol dramatically increased the number of unique CDR3 rearrangements in the distant tumor when compared to all other treatment groups (Figure 3B). Further, TCR overlap between tumors and blood is greater after treatment than before treatment in both the directly treated and distant tumors with the mean overlap exceeding 0.2 for blood and distant tumors after treatment (Figure 3C). Thus, the expansion of TCR-specific clones in response to therapy is systemic as both AI-T and AI-C have increased TCR overlap in the blood samples after treatment (AI-Post).
Figure 3
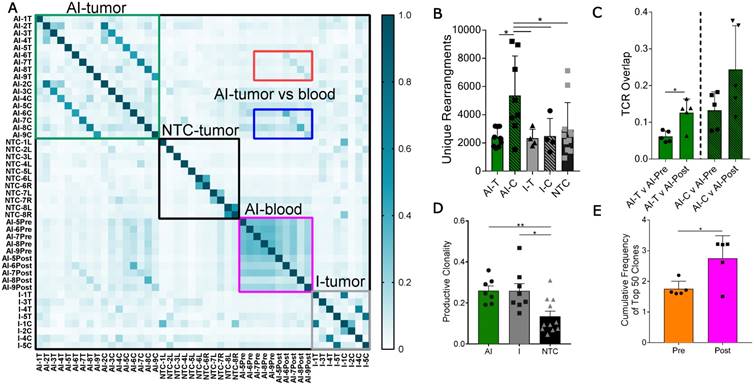
Thermal ablation increases the number of unique CDR3 rearrangements and TCR overlap between tumor and blood. Groups for T-cell receptor (TCR) sequencing of NDL tumor mice at day 38, as treated in Figure 1A, included: ablation-immunotherapy (AI, n=17 tumors), immunotherapy-alone (I, n=8 tumors), and no treatment control (NTC, n=11 tumors), where treated tumors are denoted as AI-T or I-T, and contralateral tumors are denoted as AI-C or I-C. Blood was harvested before (AI-Pre, n=5) or after AI treatment (AI-Post, n=5). Data represent two separate experiments and numbers indicate mouse number. (A) Pairwise comparison of the proportion of shared TCR clones (TCR overlap) across all samples, including comparisons between AI tumors (green box), NTC tumors (black box), blood before and after AI (pink box), and immunotherapy-only tumors (grey box). Pre- and post-blood compared to the AI-T (red box) and AI-C (blue box) tumors are also highlighted. (B) Number of unique TCR primary sequences in all tumors. (C) TCR overlap between tumors and blood from the same mouse after treatment. (D) Tumor productive clonality; data are from pooled treated and contralateral tumors in each group. Data are mean ± SD. * p <0.05, ** p <0.01 (ANOVA with Fisher's LSD test). (E) Therapeutic expansion of specific clones in blood. All data are mean ± SD. * p <0.05 (unpaired t-test with Welch's correction).
In addition, immunotherapy, with or without ablation, moved the TCR repertoire of infiltrating T-cells to a more monoclonal space (a productive clonality closer to 1) when compared to the polydisperse (a productive clonality closer to 0) repertoire of NTC (Figure 3D). The variability of the cumulative frequency of the top 50 clones is smaller for the AI treatment than for immunotherapy alone (Figure S6A). Similarly, TCR expansion is evident in circulating T-cells after, as compared with before, treatment (Figure 3E). AI treatment increased the cumulative frequency of the top systemic clones by 1%, a large change when considering the diverse TCR repertoire in the blood (Figure S6B). Taken together, the results demonstrate that the combination of ablation and immunotherapy enhanced the distant anti-tumor response, and T-cell expansion and activation were factors in the enhanced response.
Thermal ablation alone mediates tumor antigen release and cross-presentation and cross-priming; AI treatment greatly enhances lymphatic cross-presentation
We next set out to identify the mechanisms responsible for increased T-cell activation and expansion after thermal ablation. First, we characterized the dynamics of tumor antigen release, cross-presentation and cross-priming by the immune system after thermal ablation. To do this, we injected B16 cells expressing ovalbumin (B16-OVA) at one site and a secondary B16 tumor line not expressing the cognate antigen (B16-F10) into a distant site in the same wildtype immune-competent C57BL/6J mouse. A single treatment of either MRgFUS partial ablation or hyperthermia was applied to a discrete, central region within the B16-OVA tumor 13 days after tumor cell transplantation (Figure 4A), and temperature profiles of the heated intratumoral volume were recorded using MR thermometry. To assess tumor antigen uptake and presentation by the immune system, we quantified the fraction of antigen presenting cells (APCs) displaying the OVA peptide, SIINFEKL, bound to the H-2Kb of MHC class I molecules in tumors, local and distant inguinal lymph nodes, spleen and blood ~2 days after treatment via flow cytometry (Figure S7A-B).
Figure 4
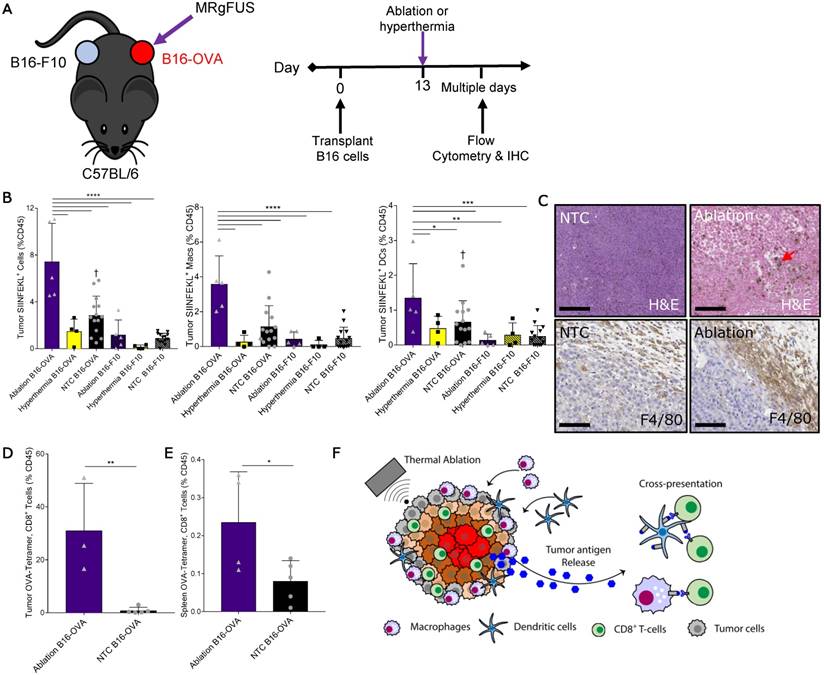
Thermal ablation induces tumor antigen cross-presentation and cross-priming in vivo. (A) B16-F10 and B16-OVA tumors subjected to MRgFUS hyperthermia (n=4), MRgFUS ablation (n=5) or no treatment control (NTC, n=15) to assess tumor antigen presentation by antigen-presenting cells (APCs). (B) Flow cytometry result obtained ~2 days after ablation or hyperthermia; SIINFEKL bound to H2-Kb of MHC class I molecules on APCs in the tumor was analyzed. * p <0.05, ** p <0.01, *** p <0.001, **** p <0.0001, † p<0.01 compared to NTC B16-F10 groups (ANOVA with Fishers LSD test). (C) Representative images of tumor sections from NTC (n=4) and ablation-treated (n=2) mice stained for H&E and F4/80 at 48 h. Red arrow indicates hemosiderin laden macrophages within the ablated region. Scale bars are 100 µm. (D-E) Flow cytometry result from the B16-OVA unilateral tumor model 7 days after ablation; OVA specific CD8+ T-cells measured via tetramer binding in the (D) tumor and (E) spleen for NTC (n=5) and ablation (n=5) cohorts. * p <0.05, and ** p <0.01 (unpaired t-test). (F) Schematic of tumor antigen release, uptake and presentation by APCs after MRgFUS ablation. All data are mean ± SD.
Thermal ablation was sufficient to locally enhance the tumor fraction of leukocytes, macrophages and DCs presenting the OVA SIINFEKL peptide (Figure 4B), reaching ~8% of the intratumoral CD45+ cells. This effect was temperature dependent, as hyperthermia at maximum temperatures of ~50 ºC did not provide an immunocompetent response. To further characterize the SIINFEKL+, CD45+ population in tumors directly treated with ablation, we stratified these cells into CD11b and CD11c subsets (Figure S7C). The greatest fraction of the SIINFEKL+ cells was CD11b+CD11c-. SIINFEKL was not enhanced in the lymph nodes of ablation-only treated mice at the 48-h time point, where the fraction of SIINFEKL+ CD45 cells was ~0.1% (Figure S7D). SIINFEKL presentation by tumor leukocytes was no longer significantly enhanced 7 days after ablation (Figure S7E); however, the fraction of SIINFEKL+ macrophages in the spleen was elevated as a result of ablation 7 days after treatment (Figure S7F).
Tumor IHC at 48 h confirmed that thermal ablation increased the numbers of infiltrating macrophages (red arrow) within the treated region, and the expression of the myeloid cell marker, F4/80, in the tumor periphery was dramatically elevated as compared to the control (Figure 4C). To verify cross-priming after ablation-mediated tumor antigen release, we quantified the fraction of CD8+ T-cells whose T-cell receptors (TCRs) were specific for the MHC-SIINFEKL complex 7 days after treatment via flow cytometry using a SIINFEKL-specific MHC-I tetramer (Figure S8). We found that thermal ablation significantly increased the fraction of OVA-specific CD8+ T-cells in the tumor (Figure 4D) and spleen (Figure 4E) of ablation-treated mice. Taken together, thermal ablation induces an increase in antigen release, cross-presentation, and cross-priming but in the absence of immunotherapy is insufficient to produce a significant increase in tumor-specific APCs in lymph nodes or in the distant tumor (Figure 4F).
Figure 5
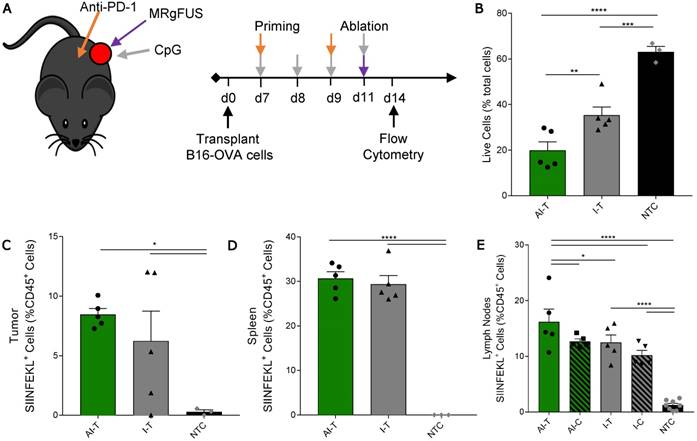
Quantification of SIINFEKL+ cells in B16-OVA model after AI treatment. (A) B16-OVA tumor cells were injected unilaterally into C57BL/6 mice (day 0). Groups included: ablation-immunotherapy (AI, n=5), immunotherapy-alone (I, n=5), and no treatment control (NTC, n=4), where treated tumors and their associated draining lymph nodes are denoted as either AI-T or I-T, and contralateral (distant) lymph nodes are denoted as either AI-C or I-C. Tumors, spleen, draining and contralateral (distant) lymph nodes were harvested for flow cytometric quantification at ~3 days after ablation. (B) Viable tumor cells quantified by flow cytometry. (C-E) Fraction of SIINFEKL+ leukocytes in the (C) tumor, (D) spleen, and (E) lymph nodes. *p<0.05, **p<0.01, *** p <0.001, **** p <0.0001 (ANOVA with Fisher's LSD test). All data plotted are mean ± SEM.
We then studied a unilateral B16-OVA model to determine whether immunotherapy priming increases the frequency of leukocytes displaying the SIINFEKL peptide after ablation (Figure 5A). As in other studies, the AI treatment was more effective: the fraction of viable tumor cells in AI-T tumors was significantly lower than that of both the I-T and NTC cohorts (Figure 5B). The frequency of SIINFEKL+ leukocytes was increased in the tumor and spleen of AI-T and I-T tumors as compared with that of the NTC (Figure 5C-D). Further, AI treatment significantly elevated the fraction of SIINFEKL+ leukocytes in the draining lymph node compared to that of the I-T and NTC cohorts (Figure 5E); this is greatly enhanced compared to the ablation-only results (Figure S7D). Taken together, AI treatment amplified cross-presentation in the draining lymph node (~16% of CD45+ cells) compared to the ablation only (~0.1% of CD45+ cells) and the immunotherapy-only cohorts (~10% of CD45+ cells).
AI treatment induces type I IFN release and stimulates ISGs to potentiate inflammatory chemokine and cytokine signaling
Next, we sought to determine the impact of the adjuvant CpG and/or ablation on the innate immune response. The direct injection of CpG into the directly treated tumors resulted in similarly enhanced expression of TLR-related signaling components with and without ablation (Figure S9A). Despite similarly elevated expression of TLR pathway components (e.g., Myd88 and Tnf) (Figure S9A), ablation resulted in a more robust upregulation of many downstream inflammatory and innate immune response ontologies in AI-T, as compared to I-T tumors (Table S1). Gene set enrichment analysis using DAVID demonstrated differential upregulation of an interferon (IFN) signature in the AI-T, as compared to I-T tumors, including the defense response to virus ontology (GO:0051607, p<1.50×10-4) [38]. Similarly, DAVID identified Herpes simplex infection (mmu05168), Measles (mmu05162), Influenza A (mmu05164), and Malaria (mmu05144) KEGG pathways as upregulated (p<0.05) in AI-T. The interferon-stimulated genes (ISGs) (Figure 6A) that made up these annotations encode for functions including transmembrane receptors associated with primary immune response, dsRNA-activated enzymes, an RNA-binding protein, the Irf7 transcription factor and associated regulators, an extracellular matrix pattern-recognition molecule for microbial pathogens, and an adhesive glycoprotein that mediates cell-to-cell and cell-to-matrix interactions.
Figure 6
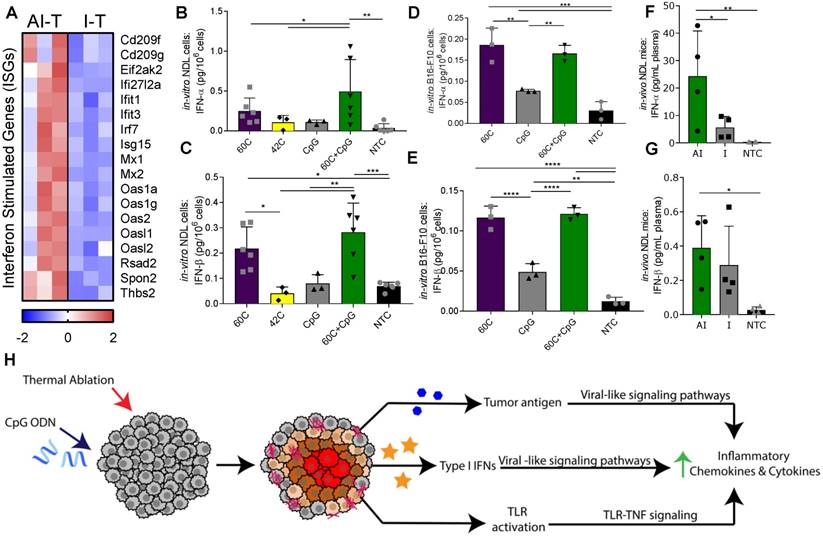
Ablative-immunotherapy treatment alters type I interferon gene expression and secretion. In vivo results for gene expression (day 38 in Figure 1A) and in vitro and ex vivo results (day 31 in Figure 1A) for IFN production. (A) Groups for RNA-seq of NDL tumor mice as treated in Figure 1A included: ablation-immunotherapy (AI), immunotherapy-alone (I), and no treatment control (NTC), where treated tumors are denoted as AI-T or I-T. RNA-seq result for expression of interferon-stimulated genes (ISGs) associated with a viral immune response in AI-T (n=3) and I-T (n=3) tumors. (B-E) In-vitro production of IFN-α and IFN-β from (B-C) NDL and (D-E) B16-F10 cells in response to heat and CpG. (F-G) In vivo blood plasma concentration of (F) IFN-α (n=12) and (G) IFN-β (n=12) from NDL tumor bearing mice, all assayed by ELISA 6 h after ablation. *p<0.05, **p<0.01, ***p<0.001 (ANOVA with Fisher's LSD test). (H) Schematic of innate immune response due to the combined effects of CpG and thermal ablation.
Based on the increased ISG expression observed in the AI-T tumors, we asked whether thermal exposure with or without CpG results in the release of type I IFN protein from NDL (Figure 6B-C) and B16-F10 (Figure 6D-E) tumor cells in vitro. We found a significant increase in expression of IFN-α after exposure to thermal-ablative temperatures (60 ºC) combined with CpG, but not by individual treatments nor exposure to hyperthermia (42 ºC) in NDL cells (Figure 6B). In B16-F10 cells, exposure to both 60 ºC alone and 60 ºC+CpG resulted in enhanced release of IFN-α (Figure 6D). IFN-β production resulted from exposure to the combination of 60 ºC and CpG or to 60 ºC alone in both NDL (Figure 6C) and B16-F10 (Figure 6E) cells. Next, we set out to characterize the release of type I IFN from tumors after thermal ablation. In bilateral NDL tumor-bearing mice, the IFN-α and IFN-β concentrations in blood were significantly increased by AI treatment at 6 h after the ablation time point (assayed at Day 31) (Figure 6F-G). Exposure of cultured NDL cells to IFN-β resulted in significantly elevated expression of the ISG Ifi27l2a (Figure S9B) as was also observed in Figure 6A. Taken together, the results indicate that exposure to heat induces type I IFN release, as compared with CpG alone across multiple cell types, and further, AI treatment enhances the type I IFN protein concentration in the blood and local ISGs in vivo. Thus, we hypothesize that the ablation-enhanced cytokine and chemotactic signaling results from the addition of ablation-mediated ISGs to the TLR immunotherapy protocol (Figure 6H).
As a result of the released antigen and ISGs, many genes were upregulated in the AI-T, as compared with I-T tumors. These differentially upregulated signals in the AI-T tumors included genes associated with the TNF pathway (KEGG mmu04668), including MMPs and cytokines (Figure 7A, 1st -3rd blocks) and annotations for chemokine signaling (mmu04062, p<9.61×10-4) and lymphocyte chemotaxis (GO:0048247, p<9.99×10-5) (Figure 7A, 4th and 5th blocks). Additionally, interleukin 1 receptors, Il1r1 and Il1rl1 (Figure 7A, 6th block) and other components of pathways associated with leukocyte migration during an inflammatory response (GO:0003, p<1.75×10-4), positive regulation of inflammatory response (GO:0050729, p<1.98×10-7) and cytokine-mediated signaling pathway (GO:0019221, p<0.002) were upregulated in AI-T, as compared with I-T, tumors (Table S1). Expression of genes associated with macrophage cytokines (e.g., Ccl2, Ccl6, Ccl7, Ccl9, Clc12) and other signals associated with monocyte recruitment were upregulated in AI-T, as compared with I-T, tumors. Ccl2 expression is associated with both the TLR-TNF and the ISG-related anti-viral pathways described above.
Figure 7
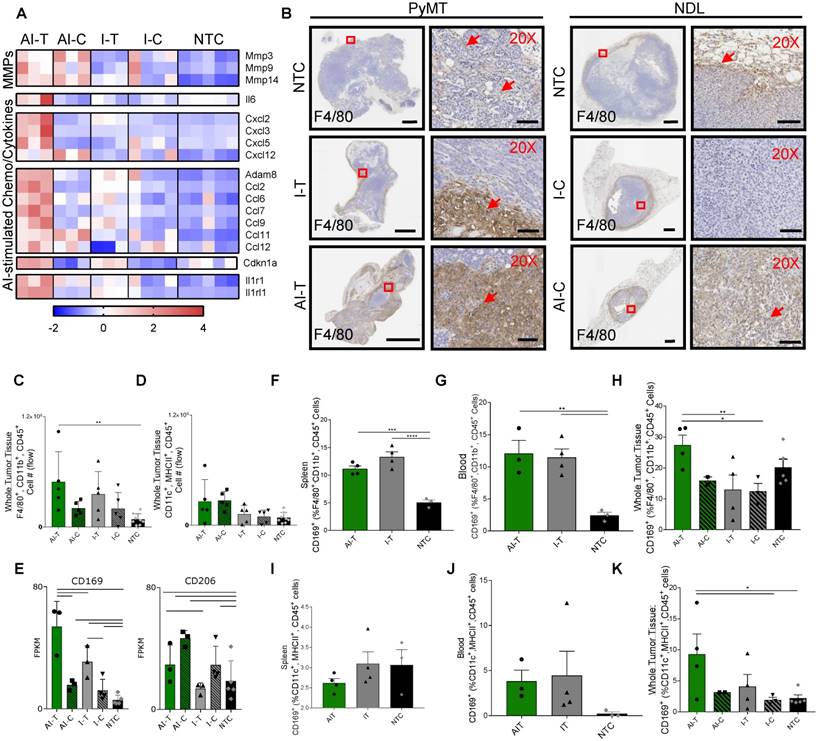
Ablative-immunotherapy enhances intratumoral macrophages and shifts the macrophage phenotype. Treatment as in Figure 1A for NDL (beginning on day 21 after implantation) and PyMT (beginning when tumors reached 0.5 cm in the largest diameter) mice. Groups included: ablation-immunotherapy (AI), immunotherapy-alone (I), and no treatment control (NTC), where treated tumors are denoted as AI-T or I-T, and contralateral tumors are denoted as AI-C or I-C. (A) RNA-seq results at day 38 (samples as in Figure 1D) for matrix metalloproteinases (block 1), Il6 (block 2), TNF pathway cytokines (block 3), lymphocyte chemotaxis cytokines (4th and 5th blocks) and Il1r1 and Il1rl1 (6th block). Heatmaps are normalized by each gene using z-scores of the fragments per kilobase of transcript per million mapped reads (FPKM) values across all presented samples. (B) F4/80 IHC in treated and contralateral tumors in the PyMT and NDL models, respectively, as compared to NTC tumors (red arrows indicate F4/80 infiltration). Scale bars are 2 mm for whole tumors and 100 µm for 20X images. (C-D) Flow cytometry quantitation of the number of tumor-infiltrating (C) macrophages and (D) dendritic cells one week after ablation (day 38): AI (n=10 tumors), immunotherapy-alone (n=10 tumors), and NTC (n=8 tumors). **p<0.01 (ANOVA with Fisher's LSD test). (E) RNA-seq result for expression of the CD169 macrophage markers: Cd169 and Cd206 at day 38. For RNA-seq, bars represent significance of at least p < 0.01 as defined by CuffDiff. (F-H) Quantification of macrophages in the (F) spleen, (G) blood and (H) tumors expressing CD169 72 h after ablation (day 34, n=4 animals per group). (I-K) Quantification of dendritic cells in the (I) spleen, (J) blood and (K) tumors expressing CD169 72 h after ablation (day 34, n=4 animals per group) *p<0.05, **p<0.01, *** p <0.001, **** p <0.0001 (ANOVA with Fisher's LSD test). All data plotted are mean ± SEM.
Both AI treatment and immunotherapy alone enhance macrophages in the treated tumors and dendritic cells in the distant tumors
Given the increased expression of chemokines and cytokines induced by AI treatment, we characterized the myeloid phenotype in the tumor immune infiltrate. IHC of NDL and PyMT tumors demonstrated the recruitment of infiltrating F4/80+ macrophages with both AI and immunotherapy-only treatment (Figure 7B). For example, macrophage invasion resulting from treatment is evident in the AI-T, as compared with I-T tumors in PyMT mice, and in AI-C, as compared with the I-C tumors in NDL mice. Flow cytometry (Figure S10A) verified the increase in the number of tumor-infiltrating macrophages in directly treated tumors for I-T and AI-T tumors (Figure 7C). Alternatively, the total number of intratumoral DCs was not significantly elevated compared to the NTC at this time point (Figure 7D), suggesting that the F4/80+ expression seen in IHC represents a macrophage subset. Genes associated with macrophages (Adgre1, Cd68, Cd11b) were dramatically enhanced in treated versus distant tumors; however, they were not differentially enhanced in AI-T as compared with I-T tumors (Figure S10B-D).
Dendritic cell-associated genes (Cd11c, Cd86, H2-ab1 and Cd40) were greatly enhanced in the distant as compared with the directly treated tumors; however, these genes were not differentially enhanced in AI as compared with immunotherapy-only tumors (Figure S10E). Yet, Batf3, required for effective dendritic cross-priming of anti-tumor CD8+ T-cells in other focal therapy regimes, was significantly enhanced in AI-C but not I-C, tumors [39].
Combining ablation with immunotherapy recruits CD169+ macrophages and dendritic cells within the tumor
Among the markers associated with each of the 5 sub-classes of macrophages in [25], only gene expression associated with the CD169+ macrophage (Cd169 and Cd206) was enhanced by AI as compared with immunotherapy-only treatment or with the NTC tumors (Figure 7E). Alternatively, genes typically associated with M2 and tumor-associated macrophages (e.g., Cd163) were reduced in the treated tumors as compared with the NTC tumor (Figure S10F). Cd169 expression is known to be correlated with type I IFN levels [40] and mediates sialic acid-dependent binding to lymphocytes, facilitating direct signaling between tumor-infiltrating macrophages and T-cells [27]. To determine whether ablation increased the fraction of macrophages expressing CD169, we employed flow cytometry 72 h after AI and immunotherapy-only treatment (Figure S11). CD169 was elevated in macrophages (F4/80+, CD11b+, CD45+ cells) in the spleen and blood for both AI and immunotherapy-only treatment (Figure 7F-G). This CD169+ macrophage population was also significantly increased in AI-T tumors compared to I-T and I-C tumors (Figure 7H), suggesting that, while CpG enhanced the circulating population, ablation increased the tumoral recruitment of CD169+ macrophages.
We next investigated the impact of thermal ablation on DCs in the context of CD169 expression. While DCs (MHCII+, CD11c+, CD45+ cells) expressing CD169 were not significantly altered in the spleen or blood compared to the NTC (Figure 7I-J), the fraction of CD169+ DCs in AI-T tumors was elevated compared to that of NTC tumors (Figure 7K). The fraction of DCs displaying CD169 (~9%) was smaller than the CD169+ macrophage fraction (~28%).
Lastly, we set out to elucidate the co-expression of CD169 and tumor antigen on APCs using a unilateral B16-OVA model using the protocol in Figure 5A. Direct treatment of a single B16-OVA tumor with immunotherapy was sufficient to increase the total fraction of CD169+ cells to ~9% of leukocytes in distant lymph nodes 72 h after treatment (Figure S12A). Interestingly, AI treatment significantly increased the fraction of nodal cells displaying both SIINFEKL and CD169 (Figure S12B). Of those double-positive cells, AI treatment significantly increased both SIINFEKL+, CD169+ DCs (Figure S12C) and SIINFEKL+, CD169+ macrophages (Figure S12D) compared to immunotherapy alone and NTC.
Discussion
The treatment of solid tumors, especially those that are non-T-cell inflamed, is challenging as it can be difficult to overcome immunosuppression dictated by the local tumor microenvironment and cytokine milieu. In the mouse models we have studied, we have previously demonstrated that the effect of ablation alone on distant immune populations and distant tumor growth and survival is not significant [24]. Yet, therapeutic efficacy is enhanced by a well-designed protocol combining ablation with immunotherapy as compared with immunotherapy alone [24]. We have therefore focused on this direct comparison between ablation-immunotherapy and immunotherapy-only protocols, and specifically characterized the mechanistic differences.
The AI treatment protocol applied here begins by priming the immune system with CpG and anti-PD-1, and this combination expands macrophage and T-cell populations and TCR clones (Figure 8, step 1). MRgFUS is then applied to raise the temperature within a focal region of the tumor to ~60 ºC, creating a small heat-fixed lesion surrounded by a hyperthermic tumor rim in which heat-mediated death occurs and type I IFN and antigen are released over a period of hours (Figure 8, step 2). The CD169+ myeloid population (macrophages and DCs) is enhanced within the tumor following ablation (Figure 8, step 3). We then observe clonal expansion and an increase in antigen-specific CD8+ T-cells with AI treatment (Figure 8, step 4).
Figure 8
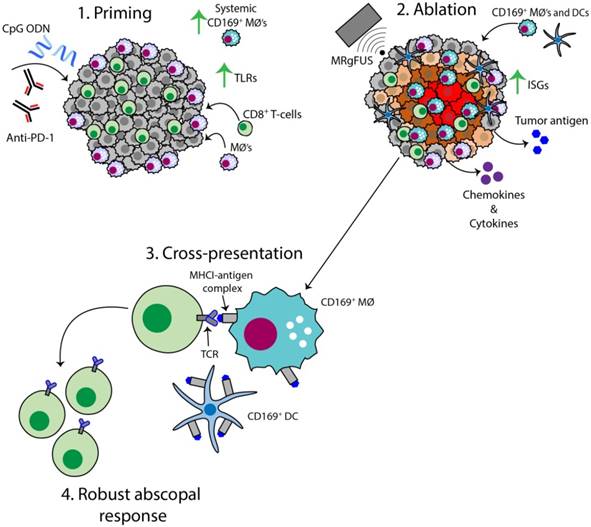
Schematic of the AI-induced immune effects. Illustration depicting the immune effects through the multi-stage therapeutic protocol of AI treatment. First, immunotherapy bolsters the number of tumor-infiltrating CD8+ T-cells and macrophages, enhances TLR expression and increases the systemic CD169+ subpopulation (Step 1). Second, MRgFUS ablation releases tumor antigen, inflammatory chemokines and cytokines, enhances interferon stimulating genes (ISGs) and alters local macrophage phenotype (Step 2). Cross-presentation and cross-priming, mediated by macrophages and DCs, occurs to drive the clonal expansion of tumor-specific CD8+ T-cells (Step 3) and results in an effective abscopal response (Step 4).
Mechanistic differences for the enhanced response from AI treatment in the directly treated tumor
We found three major mechanistic differences when ablation was added to immunotherapy. First, thermal dosing mediated the release of tumor antigen; macrophages and DCs processed and presented this antigen to CD8+ T-cells, which generated specific TCR clones. Our data demonstrate that partial tumor ablation with MRgFUS (without immunotherapy) promoted recruitment and infiltration of myeloid cells to the tumor nest and locally released tumor antigen in an immunocompetent manner. These professional cells provided cross-priming to MHC-I restricted cytotoxic lymphocytes (CTLs) one week after ablation. Further, the addition of CpG to same-site ablation significantly enhanced cross-presentation of tumor antigen in the draining lymph nodes compared to ablation-only and immunotherapy-only cohorts.
Second, when ablation was combined with CpG, type I IFN release from tumor cells was greater as compared with immunotherapy or heat treatment alone. This is the first such report demonstrating the release of type I IFN from tumor cells due to thermal ablation. We find that this type I IFN production by tumor cells varies between tumor lines and should be further investigated to determine whether this is a major source of the differences in abscopal response between patients. The IFN release induced a viral-like immune response in the AI-T tumors, including the elevation of ISGs involved in polynucleotide sensing and the binding of non-self RNA. The combination of type I IFN resulting from ablation and introduction of a TLR9 agonist can be synergistic and induce further type I IFN production through Irf7 signaling (Figure 6A). With our AI treatment protocols, the Irf7 transcription factor was significantly enhanced in AI-T, as compared with I-T, tumors. The enhanced TLR-TNF and IFN-related signaling were expected to upregulate MMPs, cytokines and chemokines associated with lymphocyte recruitment and indeed, these changes were observed with RNA-seq. By IHC and flow cytometry, we observed that inflammatory myeloid cells invaded the AI-T and AI-C tumors. Further, downstream genes for lymphocyte chemotaxis were elevated in AI-T tumors only, demonstrating the potential of MRgFUS ablation to convert the local innate immune response into an adaptive immune response.
Third, we found that the TLR9 agonist polarizes macrophages and DCs towards a CD169 subset systemically. Of the 5 macrophage phenotypes described in [25], only systemic CD169+ macrophages, a class of APCs known to be enhanced by type I IFN and proven to be capable of cross-priming, were expanded [26, 27]. Genes associated with M2 macrophage markers were reduced. CD169+ cells were elevated in the spleen and blood of treated mice, and the fraction of macrophages was 3 to 5-fold greater than DCs. After ablation, the fraction of CD169+ DCs and macrophages was elevated at the site of the local injury, with a greater fraction of macrophages observed in this subset. One previous report has indicated that type I IFN and TLR9 agonists in particular enhance the expression of CD169 in the context of systemic sclerosis [23]. Given the recent reports of successful immunotherapy protocols incorporating CpG as a TLR9 agonist, recognition of the potential role of CD169+ myeloid cells in cross-priming is important [4]. Our results demonstrate that AI treatment increases the fraction of tumor antigen-presenting CD169+ DCs and macrophages in distant lymph nodes 3 days after ablation. These data suggest that macrophages and DCs expressing CD169+ are regulators of systemic T-cell activation after thermal ablation, which may be an important contributor for bolstering the abscopal effect of TLR agonists. This is the first such report describing the role of CD169+ myeloid cells after thermal ablation and the differential role of macrophages and DCs, including the Batf3 subtype, will be the focus of future work.
AI treatment enhances T-cell number, TCR diversity and T-cell-mediated distant response
As a result of the enhanced priming at the directly treated site, the systemic T-cell populations were altered. We explored TCR expression through TCR-seq. T-cell antigen specificity is conferred through the TCR, a membrane-bound protein found on the surface of T lymphocytes. We found that the unique rearrangements in the tumor were increased by AI treatment. Comparing the TCR of circulating T-cells in the blood before and after treatment also demonstrated differences in expression with greater similarity between tumor and blood observed after treatment. This result is encouraging for sampling treatment response in blood after therapy.
The enhanced T-cell response is also supported by greatly enhanced T-cell numbers and by enhanced genes associated with T-cell activation and stimulation. Eomes, Prf1 and Icos expression was up to 89-fold higher in the AI-C cohort as compared to the NTC and nearly double the level in AI-C as compared with the I-C tumors. While we observed significantly enhanced expression of Fas, Tnf, Casp8, and Casp9 in all directly treated tumors, in the distant tumors, only AI treatment differentially enhanced Faf1, Casp8 and Casp9. Casp8 activation is reported to have the greatest impact on IFN-induced apoptosis; the increase in caspase expression observed here has the potential to sensitize cells to apoptosis [41].
Bulk RNAseq analysis was validated by flow cytometry and qPCR
We chose to combine a bulk RNA-seq analysis with selected qPCR of sorted cells. This provided the opportunity to compare gene expression over multiple treatment groups and to assess both the relative frequency of cellular phenotypes and specific aspects of individual tumor, immune and stromal cells. Our results, with the cancer cell, macrophage and T-cell population differences across treatment groups validated by flow cytometry, demonstrate the power of such analysis in differentiating the impact of therapeutic components. Treatment-related changes in cancer cell, T-cell and macrophage markers were evident in the unsorted RNA-seq and flow cytometry datasets. The principal component, functional annotation, and hierarchical clustering analyses clearly demonstrated that the directly treated and distant tumors had distinct gene expression patterns across many cellular functions. Further, a fraction of the immunotherapy-treated animals did not significantly respond to treatment and, therefore, clustered with the NTC. While the analyses detected the expected effects of the TLR agonist, CpG, and checkpoint inhibitor, anti-PD-1, shared in all treated mice, important mechanistic differences between the AI and immunotherapy-only treatments were also detected by RNA-seq.
With administration of a TLR agonist (CpG) alone or CpG combined with same-site ablation, we found dramatic differences between the local and distant tumor environments. With both ablation-immunotherapy and immunotherapy alone, directly treated tumors were skewed to high expression of F4/80, Cd11b and Tnf, and the distant tumors to enhanced Cd11c, Cd3 and Ifng. These extensive differences were evident through the RNA-seq analysis.
Thermal ablation can enhance the speed and spatial precision of treatment as compared with RT
Thermal ablation mediates rapid, immunogenic cell death (1-2 days) that is capable of driving a tumor-specific immune response. Thermal dosing with MRgFUS can be tightly controlled in a spatiotemporal manner and repeated within the same treatment volume. Protocols combining immunotherapy with RT are now widespread [30, 33]; however, mitotic catastrophe and subsequent cell death in irradiated cells can require up to one week to develop. Further, radiation is known to induce “bystander effects”, or chromosomal damage consistent with ionizing radiation in non-irradiated (non-targeted), neighboring cells, as previously demonstrated in lymphocytes, fibroblasts, keratinocytes and endothelial cells [42]. Approximately 10% of the RT dose is expected to impact tissue 10 cm away from the target site [43]. Deciphering radiation-induced bystander effects in real-time is challenging, as the local microenvironment of the irradiated site undergoes a complex wound healing process. Thus, to ensure tumor control and patient safety, the number of radiation treatments at a single location is restricted. MRgFUS provides greater flexibility in structuring such treatment protocols.
In summary, dramatic differences between local and systemic tumor immune infiltrates result from adjuvants applied with or without ablation. A multi-step protocol in which immunotherapy expands T-cells and macrophages, followed by partial ablation, enhances antigen presentation and type I IFN release, alters the myeloid phenotype to enhance cross-priming, and results in a robust adaptive immune response.
Methods
Study design
The research objective of these studies was to evaluate the immunological and transcriptome alterations created by combining MRgFUS ablation with immunotherapy. We hypothesized that tumor debulking with thermal ablation would induce a potent, local innate immune response to potentiate an abscopal immune response. A combination of RNA-seq, TCR-seq, flow cytometry, ELISA (enzyme-linked immunosorbent assay), qPCR and IHC were performed to investigate the immune phenotype at different points throughout the treatment protocol. All animals and cells used in these studies were randomized to biological groups, and were performed with the appropriate controls, which are reported in the figure legends. Tumors were continuously measured with ultrasound, and therapy was started once tumors reached 5 mm in the largest diameter. All data in the figures are presented as means ± SD.
Cell lines
B16-F10 cells were purchased from American Type Culture Collection (ATCC, #CRL-6475), and B16-OVA cells, genetically modified to express chicken ovalbumin (OVA), were a generous gift from the William Murphy Laboratory (UC Davis, Davis, CA). Cells were cultured in high-glucose DMEM (Gibco, #11995) containing 10% FBS and 1% penicillin-streptomycin in a 37 ºC humidified chamber containing 5% CO2. B16-F10 and B16-OVA cells were passaged at a density of 1-3×106 into 175 cm2 tissue culture flasks (VWR, Radnor PA). Once cells reached 85-90% confluency, approximately 4 days after plating, they were collected and resuspended in 1:1 Matrigel (Corning, #356234): PBS without calcium and magnesium (PBS-/-) for injection into mice.
Study approval
All animal experiments were conducted with approval from the University of California, Davis, (Davis, California) Institutional Animal Care and Use Committee (IACUC).
Animal models
A total of 193 mice were studied (118 bilateral NDL, 24 B16-F10/B16-OVA, 23 unilateral B16-OVA, 4 CAG OVA, 10 B16-F10 bilateral and 14 PyMT tumor-bearing mice) with the study breakdown in Table S2. For all relevant groups, anti-PD-1 was injected intraperitoneally (i.p.) at a dose of 200 µg, as in [44] and CpG was injected intratumorally (i.t.) into a single tumor at a dose of 100 µg, as in [45]. All same day treatments with ablation and immunotherapy involved injections administered immediately following MRgFUS ablation.
Therapeutic and sequencing protocols in the NDL or PyMT model
The neu exon deletion line (NDL), a syngeneic model of mammary adenocarcinoma, was obtained from the Alexander Borowsky Laboratory (UC Davis, Davis, CA). Four- to five-week-old FVB/n female mice purchased from Charles River (Wilmington, MA) were bilaterally transplanted with NDL tumor biopsies (~1 mm3) into the fourth and ninth inguinal mammary fat pads. Approximately 21 days later, mice were randomized into treatment groups with average tumor sizes ranging between 4-6 mm in the longest dimension as measured by ultrasound (Sequoia, Siemens Preclinical Solutions).
NDL mice treated with immunotherapy received injections of CpG into a single tumor on days 21, 24, 28, and 31 after tumor transplantation; anti-PD-1 was injected on days 21, 28 and 35 after tumor transplantation (see Figure 1A). In the NDL protocols, MRgFUS ablation was performed in a single tumor on day 31 after a course of immunotherapy as in [24]. The IFN level was assayed in blood on day 31 (6 h after ablation) by ELISA. NDL mice treated with immunotherapy with and without ablation were euthanized on day 34 or 38; the tumors were harvested and processed for RNA-seq, TCR-seq, flow cytometry, qPCR or IHC. Additionally, blood was harvested and processed on day 31, 34 or 38, and spleens were harvested and processed on day 34 for flow cytometry quantification.
FVB/N-Tg(MMTV-PyVT)634Mul/J transgene female mice (5 weeks old) were purchased from The Jackson Laboratory. Once tumors reached approximately 5 mm in the largest dimension, they were randomized into treatment groups (~10 weeks old). With the exception of the starting time point, the treatment protocol administered was the same as that in NDL mice in Figure 1A. Timing corresponds to days after the start of the treatment. CpG was administered into a single tumor on days 0, 3, 7, and 10; anti-PD-1 was injected on days 0, 3 and 14; and thermal ablation was administered on day 10.
In vivo tumor antigen release studies in B16-F10 and B16-OVA models
C57BL/6J female mice (7 weeks old, 15-25 g) were purchased from The Jackson Laboratory (Bar Harbor, ME).
Ablation or hyperthermia alone and controls (Figure 4A): A B16-F10/B16-OVA dual tumor model and appropriate controls were studied. Mice were subcutaneously (s.c.) injected with B16-F10 (3×105 B16-F10 cells per 50 μL Matrigel:PBS-/-) and B16-OVA cells (6×105 B16-OVA cells per 50 μL Matrigel:PBS-/-) in the left and right flank, respectively. Negative control mice bearing bilateral B16-F10 control tumors were injected s.c. with B16-F10 cells (2.5×105 cells per 50 μL Matrigel:PBS-/-) into the left and right flanks. C57BL/6-Tg(CAG-OVA)916Jen/J (CAG-OVA) mice (6 weeks old, 15-25 g) were purchased from The Jackson Laboratory and studied as positive controls.
For the ablation- and hyperthermia-only groups, MRgFUS was directed to the B16-OVA tumor on day 13 after tumor cell injection (approximately 5 mm in the largest diameter). The mice were euthanized either on day 15 or 20 post tumor cell implant (2 or 7 days after treatment). Tumors and their respective draining inguinal lymph nodes, as well as the spleen and blood were dissected and harvested for flow cytometry or IHC.
Ablation with immunotherapy (Figure 5A): For B16 studies incorporating immunotherapy, a unilateral B16-OVA tumor model was studied, where mice received s.c. injections (4-6×105 cells per 50 μL Matrigel:PBS-/-) into the left flank only. Immunotherapy was administered before and with thermal ablation (CpG, days 7, 8, 9, and 11 post tumor cell implantation; anti-PD-1, days 7 and 9 post tumor cell implant). For the AI cohort, ablation was administered on day 11. Tumors, both inguinal lymph nodes (right and left side), spleen and blood were harvested on day 14 after tumor cell implant for flow cytometry. In this study, the draining lymph node is defined as the inguinal node adjacent to the treated B16-OVA tumor (AI-T or I-T) and the distant lymph node is the inguinal lymph node on the contralateral side with no tumor (AI-C or I-C).
MRgFUS ablation protocol
MRgFUS ablation and hyperthermia were performed with a MR-guided pre-clinical HIFU system (Image Guided Therapy) using a 16-element annular array transducer at 3 MHz (Imasonic SAS), on a Bruker BioSpec 7 T small animal MR system (Bruker Biospin), as previously described [46]. The thermal ablation treatment plan was designed to heat a discrete region (2-3 mm) within a tumor volume to temperatures above 60 °C, where a thermal dose in cumulative equivalent minutes at 43 °C (CEM43) of more than 5,000 was achieved.
Reagents
TLR9 agonist, CpG 1826 (class B), was purchased from InvivoGen (San Diego, CA). Anti-PD-1 (CD279) mAb (rat IgG2a, clone RMP1-14), was purchased from Bio X Cell (West Lebanon, NH). iTAG MHC murine tetramer H-2 kb OVA conjugated to phycoerythrin (PE) was purchased from MBL International (#T03000, Woburn, MA).
High-throughput TCR-β sequencing
TCR-β CDR3 regions were amplified and sequenced on the ImmunoSEQ platform (Adaptive Biotechnologies, Seattle, WA, USA). Data from the productive reads were uploaded to the ImmuneACCESS provided by Adaptive Biotech. Diversity metrics, sample overlaps, and frequency tables were downloaded from the ImmunoSeq Analyzer tool and processed in MATLAB and in VDJTools [47].
In vitro and in vivo IFN-α and IFN-β ELISA studies
NDL or B16-F10 tumor cells were plated in 12-well tissue-culture treated plates at a concentration of 3.5×105 or 3×105 cells/well, respectively. Cells were cultured overnight in 1 mL high-glucose DMEM (Gibco, #11995) containing 10% FBS and 1% penicillin-streptomycin in a 37 ºC humidified chamber containing 5% CO2. The next day, the cell media was removed and replaced with 500 μL of pre-warmed media. The biological groups were the following: 37 ºC (n=6), 37 ºC+CpG (n=3), 42 ºC (n=3), 60 ºC (n=6), or 60 ºC+CpG (n=6). These samples were then placed in the respective water bath (37 ºC, 42 ºC, or 60 ºC) for 1 min, removed, and allowed to cool to room temperature for 5 min, followed by addition of CpG (5 μg/ 500 μL) to the appropriate dishes. Cells were then incubated continuously for 24 h in a 37 ºC humidified chamber containing 5% CO2. After 24 h, the cell culture medium was collected, centrifuged (300 ×g, 10 min, 4 ºC), and the supernatant was collected for IFN-α and IFN-β ELISA quantification (#42115 and 42410, respectively, PBL Assay Science, Piscataway, NJ). IFN-α and IFN-β supernatant concentrations were normalized to total cell number for each sample.
For the type-1 IFN in vivo study, blood was harvested via retro-orbital collection from bilateral NDL mice treated as in Figure 1A, 6 h after ablation on Day 31 post tumor implant. Blood was processed to plasma by centrifugation at 1000 ×g for 10 min at 4 ºC and frozen at -80 ºC until IFN-α and IFN-β protein measurement via ELISA as above.
Statistical analysis
Statistical analyses were performed using Prism 7 software (GraphPad Software Inc.). Results are presented as mean ± SD, unless otherwise indicated. For analysis of three or more groups, a one-way ANOVA test was performed followed by a Fisher's LSD test without multiple comparisons correction in GraphPad Prism. Analysis of differences between two normally distributed test groups was performed using an unpaired t-test assuming unequal variance. P values less than 0.05 were considered significant.
Abbreviations
AI: ablative-immunotherapy; AI-C: thermal-ablative immunotherapy-contralateral; AI-T: thermal-ablative immunotherapy-treated; AI-Post: blood of mice after ablative-immunotherapy treatment; AI-Pre: blood of mice before ablative-immunotherapy treatment; ANOVA: analysis of variance; APCs: antigen-presenting cells; B16-OVA: B16 cells expressing ovalbumin; CAG-OVA: C57BL/6-Tg(CAG-OVA)916Jen/J mice; CpG: cytosine-phosphodiester-guanine oligodeoxynucleotide; DCs: dendritic cells; ELISA: enzyme-linked immunosorbent assay; FPKM: fragments per kilobase of transcript per million mapped reads; I: immunotherapy-alone; IFNs: interferons; IFN-α: interferon-alpha; IFN-β: interferon-beta; IHC: immunohistochemistry; I-C: immunotherapy-contralateral; I-T: immunotherapy-treated; ISGs: interferon-stimulated genes; i.t.: intratumorally; i.p.: intraperitoneally; M1: classically-activated macrophages; M2: alternatively-activated macrophages; MMPs: matrix metalloproteinases; MRgFUS: magnetic resonance-guided focused ultrasound; NDL: neu exon deletion line; NTC: no treatment control; OVA: ovalbumin; PCA: principal component analysis; PyMT mice: FVB/N-Tg(MMTV-PyVT)634Mul/J transgene female mice; qPCR: quantitative PCR; RNA-seq: RNA sequencing; RT: radiation therapy; TAM: tumor-associated macrophages; TCR: T-cell receptor; TCR-seq: T-cell receptor sequencing; TLR: toll-like receptor; TLR9: toll-like receptor-9.
Supplementary Material
Supplementary materials and methods, figures and tables.
Acknowledgements
We gratefully acknowledge the support of the Focused Ultrasound Foundation and National Institutes of Health (NIH) NIHR01CA199658, NIHR01CA211602, NIHR01CA210553, and NIHR01CA134659. Real-time quantitative TaqMan PCR assays were performed by the UCD, Real-time PCR Research and Diagnostics Core Facility with technical assistance from Ms. Edlin Escobar and Dr. Emir Hodzic. This project is also supported by the University of California Davis Flow Cytometry Shared Resource Laboratory with funding from the NCI P30 CA093373 (Cancer Center), and NIH NCRR C06-RR12088, S10 OD018223, S10 RR12964 and S10 RR 026825 grants and with technical assistance from Ms. Bridget McLaughlin and Mr. Jonathan Van Dyke. The UC Davis Comprehensive Cancer Center Genomics Shared Resource is supported by Cancer Center Support Grant (P30 CA093373) from the National Cancer Institute.
Author contributions
M.C. and M.T.S. designed and conducted experiments, performed the analysis and wrote the manuscript; E.S.I. designed and conducted experiments and performed analysis; L.M.M. and S.M.T. maintained mouse models, provided assistance with design of experiments and MRgFUS ablations. B.Z.F and A. I. conducted MRgFUS ablation. N.E.H., A.M.M., W.J.M., and A.D.B. designed experiments. K.W.F. planned and initiated the project, designed experiments, wrote the manuscript and supervised the project.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gajewski TF. The Next Hurdle in Cancer Immunotherapy: Overcoming the Non-T-Cell-Inflamed Tumor Microenvironment. Seminars in Oncology. 2015;42:663-71
2. Topalian SL, Drake CG, Pardoll DM. Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell. 2015;27:450-61
3. Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3-potential mechanisms of action. Nat Rev Immunol. 2015;15:45-56
4. Sagiv-Barfi I, Czerwinski DK, Levy S, Alam IS, Mayer AT, Gambhir SS. et al. Eradication of spontaneous malignancy by local immunotherapy. Science Translational Medicine. 2018:10
5. Baird JR, Monjazeb AM, Shah O, McGee H, Murphy WJ, Crittenden MR. et al. Stimulating Innate Immunity to Enhance Radiation Therapy-Induced Tumor Control. International Journal of Radiation Oncology*Biology*Physics. 2017;99:362-73
6. Schaue D, McBride WH. Opportunities and challenges of radiotherapy for treating cancer. Nature reviews Clinical oncology. 2015;12:527-40
7. van den Bijgaart RJ, Eikelenboom DC, Hoogenboom M, Futterer JJ, den Brok MH, Adema GJ. Thermal and mechanical high-intensity focused ultrasound: perspectives on tumor ablation, immune effects and combination strategies. Cancer immunology, immunotherapy: CII. 2017;66:247-58
8. Reits EA, Hodge JW, Herberts CA, Groothuis TA, Chakraborty M, Wansley EK. et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. The Journal of experimental medicine. 2006;203:1259-71
9. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annual review of immunology. 2013;31:51-72
10. Hu Z, Yang XY, Liu Y, Morse MA, Lyerly HK, Clay TM. et al. Release of endogenous danger signals from HIFU-treated tumor cells and their stimulatory effects on APCs. Biochemical and biophysical research communications. 2005;335:124-31
11. Lu P, Zhu XQ, Xu ZL, Zhou Q, Zhang J, Wu F. Increased infiltration of activated tumor-infiltrating lymphocytes after high intensity focused ultrasound ablation of human breast cancer. Surgery. 2009;145:286-93
12. Xu ZL, Zhu XQ, Lu P, Zhou Q, Zhang J, Wu F. Activation of tumor-infiltrating antigen presenting cells by high intensity focused ultrasound ablation of human breast cancer. Ultrasound in medicine & biology. 2009;35:50-7
13. Zhou Q, Zhu XQ, Zhang J, Xu ZL, Lu P, Wu F. Changes in circulating immunosuppressive cytokine levels of cancer patients after high intensity focused ultrasound treatment. Ultrasound in medicine & biology. 2008;34:81-7
14. Linnemann C, Heemskerk B, Kvistborg P, Kluin RJ, Bolotin DA, Chen X. et al. High-throughput identification of antigen-specific TCRs by TCR gene capture. Nat Med. 2013;19:1534-41
15. Mlecnik B, Bindea G, Kirilovsky A, Angell HK, Obenauf AC, Tosolini M. et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci Transl Med. 2016;8:327ra26
16. Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP. et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Transl Med. 2017:9
17. Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S. et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2016;165:35-44
18. Kristensen VN, Lingjaerde OC, Russnes HG, Vollan HK, Frigessi A, Borresen-Dale AL. Principles and methods of integrative genomic analyses in cancer. Nat Rev Cancer. 2014;14:299-313
19. Braun DA, Burke KP, Van Allen EM. Genomic Approaches to Understanding Response and Resistance to Immunotherapy. Clinical Cancer Research. 2016;22:5642-50
20. Fuertes MB, Woo S-R, Burnett B, Fu Y-X, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends in Immunology. 2013;34:67-73
21. Lim JYH, Gerber SA, Murphy SP, Lord EM. Type I interferons induced by radiation therapy mediate recruitment and effector function of CD8+ T cells. Cancer Immunology, Immunotherapy. 2014;63:259-71
22. Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J. et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20:1301-9
23. York MR, Nagai T, Mangini AJ, Lemaire R, van Seventer JM, Lafyatis R. A macrophage marker, siglec-1, is increased on circulating monocytes in patients with systemic sclerosis and induced by type i interferons and toll-like receptor agonists. Arthritis & Rheumatism. 2007;56:1010-20
24. Silvestrini M, Ingham ES, Mahakian LM, Kheirolomoom A, Liu Y, Fite BZ, Tam SM, Tucci S, Watson KD, Wong A, Monjazeb AM, Hubbard NE, Murphy WJ, Borowsky AD, Ferrara KW. Priming is key to effective incorporation of image-guided thermal ablation into immunotherapy protocols. JCI Insight. 2017;2:e90521
25. Chavez-Galan L, Olleros ML, Vesin D, Garcia I. Much More than M1 and M2 Macrophages, There are also CD169(+) and TCR(+) Macrophages. Front Immunol. 2015;6:263
26. Bernhard CA, Ried C, Kochanek S, Brocker T. CD169+ macrophages are sufficient for priming of CTLs with specificities left out by cross-priming dendritic cells. Proc Natl Acad Sci U S A. 2015;112:5461-6
27. Asano K, Nabeyama A, Miyake Y, Qiu C-H, Kurita A, Tomura M. et al. CD169-Positive Macrophages Dominate Antitumor Immunity by Crosspresenting Dead Cell-Associated Antigens. Immunity. 2011;34:85-95
28. Nesslinger NJ, Sahota RA, Stone B, Johnson K, Chima N, King C. et al. Standard treatments induce antigen-specific immune responses in prostate cancer. Clin Cancer Res. 2007;13:1493-502
29. Schaue D, Comin-Anduix B, Ribas A, Zhang L, Goodglick L, Sayre JW. et al. T-cell responses to survivin in cancer patients undergoing radiation therapy. Clin Cancer Res. 2008;14:4883-90
30. Formenti SC, Demaria S. Combining Radiotherapy and Cancer Immunotherapy; A Paradigm Shift. Journal of the National Cancer Institute. 2013
31. Wu F. High intensity focused ultrasound ablation and antitumor immune response. J Acoust Soc Am. 2013;134:1695-701
32. Chen Z, Shen S, Peng B, Tao J. Intratumoural GM-CSF microspheres and CTLA-4 blockade enhance the antitumour immunity induced by thermal ablation in a subcutaneous murine hepatoma model. International journal of hyperthermia: the official journal of European Society for Hyperthermic Oncology, North American Hyperthermia Group. 2009;25:374-82
33. Vacchelli E, Bloy N, Aranda F, Buque A, Cremer I, Demaria S. et al. Trial Watch: Immunotherapy plus radiation therapy for oncological indications. Oncoimmunology. 2016;5:e1214790
34. Shabason JE, Minn AJ. Radiation and Immune Checkpoint Blockade: From Bench to Clinic. Seminars in radiation oncology. 2017;27:289-98
35. Curran MA, Kim M, Montalvo W, Al-Shamkhani A, Allison JP. Combination CTLA-4 blockade and 4-1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production. PLoS One. 2011;6:e19499
36. Cardiff RD, Hubbard NE, Engelberg JA, Munn RJ, Miller CH, Walls JE. et al. Quantitation of fixative-induced morphologic and antigenic variation in mouse and human breast cancers. Lab Invest. 2013;93:480-97
37. Fluck MM, Schaffhausen BS. Lessons in Signaling and Tumorigenesis from Polyomavirus Middle T Antigen. Microbiology and Molecular Biology Reviews. 2009;73:542-63
38. Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J. et al. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biology. 2007;8:R183
39. Vanpouille-Box C, Alard A, Aryankalayil MJ, Sarfraz Y, Diamond JM, Schneider RJ. et al. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nature Communications. 2017;8:15618
40. Akiyama H, Ramirez NP, Gibson G, Kline C, Watkins S, Ambrose Z. et al. Interferon-Inducible CD169/Siglec1 Attenuates Anti-HIV-1 Effects of IFN-alpha. J Virol. 2017
41. Thyrell L, Erickson S, Zhivotovsky B, Pokrovskaja K, Sangfelt O, Castro J. et al. Mechanisms of Interferon-alpha induced apoptosis in malignant cells. Oncogene. 2002;21:1251-62
42. Suzuki K, Yamashita S. Radiation-Induced Bystander Response: Mechanism and Clinical Implications. Adv Wound Care (New Rochelle). 2014;3:16-24
43. Elias WJ, Khaled M, Hilliard JD, Aubry J-F, Frysinger RC, Sheehan JP. et al. A magnetic resonance imaging, histological, and dose modeling comparison of focused ultrasound, radiofrequency, and Gamma Knife radiosurgery lesions in swine thalamus Laboratory investigation. Journal of Neurosurgery. 2013;119:307-17
44. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T. et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577-81
45. Marabelle A, Kohrt H, Sagiv-Barfi I, Ajami B, Axtell RC, Zhou G. et al. Depleting tumor-specific Tregs at a single site eradicates disseminated tumors. J Clin Invest. 2013;123:2447-63
46. Wong AW, Fite BZ, Liu Y, Kheirolomoom A, Seo JW, Watson KD. et al. Ultrasound ablation enhances drug accumulation and survival in mammary carcinoma models. J Clin Invest. 2016;126:99-111
47. Shugay M, Bagaev DV, Turchaninova MA, Bolotin DA, Britanova OV, Putintseva EV. et al. VDJtools: Unifying Post-analysis of T Cell Receptor Repertoires. PLOS Computational Biology. 2015;11:e1004503
Author contact
![]() Corresponding author: Katherine W. Ferrara, PhD, Distinguished Professor, Department of Biomedical Engineering, University of California, Davis, One Shields Ave, Davis, CA 95616. Phone: (530)754-9436; FAX: (530)754-5739; kwferraraedu
Corresponding author: Katherine W. Ferrara, PhD, Distinguished Professor, Department of Biomedical Engineering, University of California, Davis, One Shields Ave, Davis, CA 95616. Phone: (530)754-9436; FAX: (530)754-5739; kwferraraedu
Citation styles
APA
Chavez, M., Silvestrini, M.T., Ingham, E.S., Fite, B.Z., Mahakian, L.M., Tam, S.M., Ilovitsh, A., Monjazeb, A.M., Murphy, W.J., Hubbard, N.E., Davis, R.R., Tepper, C.G., Borowsky, A.D., Ferrara, K.W. (2018). Distinct immune signatures in directly treated and distant tumors result from TLR adjuvants and focal ablation. Theranostics, 8(13), 3611-3628. https://doi.org/10.7150/thno.25613.
ACS
Chavez, M.; Silvestrini, M.T.; Ingham, E.S.; Fite, B.Z.; Mahakian, L.M.; Tam, S.M.; Ilovitsh, A.; Monjazeb, A.M.; Murphy, W.J.; Hubbard, N.E.; Davis, R.R.; Tepper, C.G.; Borowsky, A.D.; Ferrara, K.W. Distinct immune signatures in directly treated and distant tumors result from TLR adjuvants and focal ablation. Theranostics 2018, 8 (13), 3611-3628. DOI: 10.7150/thno.25613.
NLM
Chavez M, Silvestrini MT, Ingham ES, Fite BZ, Mahakian LM, Tam SM, Ilovitsh A, Monjazeb AM, Murphy WJ, Hubbard NE, Davis RR, Tepper CG, Borowsky AD, Ferrara KW. Distinct immune signatures in directly treated and distant tumors result from TLR adjuvants and focal ablation. Theranostics 2018; 8(13):3611-3628. doi:10.7150/thno.25613. https://www.thno.org/v08p3611.htm
CSE
Chavez M, Silvestrini MT, Ingham ES, Fite BZ, Mahakian LM, Tam SM, Ilovitsh A, Monjazeb AM, Murphy WJ, Hubbard NE, Davis RR, Tepper CG, Borowsky AD, Ferrara KW. 2018. Distinct immune signatures in directly treated and distant tumors result from TLR adjuvants and focal ablation. Theranostics. 8(13):3611-3628.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.