Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Conclusion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(13):3584-3596. doi:10.7150/thno.25409 This issue Cite
Research Paper
Tumor-targeted hybrid protein oxygen carrier to simultaneously enhance hypoxia-dampened chemotherapy and photodynamic therapy at a single dose
Zhenyu Luo1,3,‡, Hao Tian1,2,‡, Lanlan Liu1,3,‡, Zhikuan Chen1,3, Ruijing Liang1, Ze Chen1, Zhihao Wu1, Aiqing Ma1,2, Mingbin Zheng1,2, ![]() , Lintao Cai1,
, Lintao Cai1, ![]()
1. Guangdong Key Laboratory of Nanomedicine, Shenzhen engineering Laboratory of nanomedicine and nanoformulations, CAS Key Lab for Health Informatics, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, P. R. China
2. Department of Chemistry, Guangdong Medical University, Dongguan 523808, PR China
3. University of Chinese Academy of Sciences, Beijing 100049, PR China
‡ These authors contributed equally to this work.
Received 2018-2-7; Accepted 2018-5-1; Published 2018-6-7
Abstract

Hypoxia is a characteristic feature of solid tumors and an important causation of resistance to chemotherapy and photodynamic therapy (PDT). It is challenging to develop efficient functional nanomaterials for tumor oxygenation and therapeutic applications.
Methods: Through disulfide reconfiguration to hybridize hemoglobin and albumin, tumor-targeted hybrid protein oxygen carriers (HPOCs) were fabricated, serving as nanomedicines for precise tumor oxygenation and simultaneous enhancement of hypoxia-dampened chemotherapy and photodynamic therapy. Based on encapsulation of doxorubicin (DOX) and chlorin e6 (Ce6) into HPOCs to form ODC-HPOCs, the mechanism and therapeutic efficacy of oxygen-enhanced chemo-PDT was investigated in vitro and in vivo.
Results: The precise oxygen preservation and release of the HPOC guaranteed sufficient tumor oxygenation, which is able to break hypoxia-induced chemoresistance by downregulating the expressions of hypoxia-inducible factor-1α (HIF-1α), multidrug resistance 1 (MDR1) and P-glycoprotein (P-gp), resulting in minimized cellular efflux of chemodrug. Moreover, the oxygen supply is fully exploited for upgrading the generation of reactive oxygen species (ROS) during the photodynamic process. As a result, only a single-dose treatment of the HPOCs-based chemo-PDT exhibited superior tumor suppression. The combination therapy was guided by in vivo fluorescence/photoacoustic imaging with nanoparticle tracking and oxygen monitoring.
Conclusion: This well-defined HPOC as a versatile nanosystem is expected to pave a new way for breaking multiple hypoxia-induced therapeutic resistances to achieve highly effective treatment of solid tumors.
Keywords: hybrid protein nanoparticle, targeted oxygen carrier, tumor hypoxia, chemotherapy, photodynamic therapy
Introduction
Cancer nanotheranostics—nanomedicines with integrated diagnostic and therapeutic functions—have received tremendous attention in recent years, especially for tumor-targeted drug delivery and modulation of tumor microenvironment, as well as precise imaging-guided therapy [1-4]. However, the success of anticancer therapy is usually limited by the complex tumor microenvironment and induced therapeutic resistances. Hypoxic microenvironments are typically scattered in the central region of solid tumors, owing to the insufficient oxygen supply of abnormal tumor vasculature and high oxygen consumption of tumor cells with vigorous metabolism [5]. Effects of hypoxia on tumor biology include tumor angiogenesis, vasculogenesis, invasiveness and metastasis, as well as resistance to therapies through multiple mechanisms [6-9]. In chemotherapy, hypoxic cells are resistant to most anticancer drugs, which require oxygen to increase the cytotoxicity of the DNA lesions [10]. Moreover, hypoxia regulates the gene/protein expressions involved in drug resistance and induces poor drug delivery [11-13]. Beyond that, hypoxia is the major barrier of photodynamic therapy (PDT) that converts oxygen into cytotoxic reactive oxygen species (ROS) in the presence of light and photosensitizer, in which the hypoxic tumor fails to provide photosensitizers with an adequate oxygen source and sustained oxygen supply [14-16]. Lately, by ingenious combinations of chemotherapy and photodynamic therapy, chemophototherapy is emerging as a potential treatment option for solid tumors. It is valuable for improving drug bioavailability and the photodynamic reaction to achieve superior efficacy [17, 18].
Considering the critical roles of hypoxia in resistance to tumor therapy, tumor-driven oxygenation might be the best validated strategy to achieve highly efficient therapies. For instance, hyperbaric oxygen chambers have been clinically utilized to force more oxygen into the blood and the tumor when patients undergo cancer therapies [19-22]. However, even with high oxygen tension, hyperbaric oxygen treatment provides insufficient local oxygenation of tumors with poor vascular architecture and limited blood supply, and induces oxygen toxicity to the lung and central nervous system [23, 24]. Recently, a variety of oxygen generators (MnO2, CaO2, etc.) [25, 26] and oxygen carriers (perfluorocarbon, hemoglobin (Hb), etc.) [27, 28] have been explored for overcoming tumor hypoxia, which showed promising therapeutic outcomes. Among these oxygen carriers, Hb is an iron-rich protein in red blood cells that delivers oxygen to tissues. It can reversibly bind four oxygen molecules to form oxygenated Hb (HbO2). Unfortunately, the free blood-circulating Hb is not an excellent oxygen donor due to its short circulation time, potential side effects and poor stability, resulting in low-efficiency oxygen delivery to tumor regions [29, 30]. Thus, it remains challenging to design a Hb-based nanoplatform with minimized side-effects as well as improved stability and specificity of tumor oxygenation.
To construct a Hb-based nanoplatform for tumor-targeted oxygen delivery, human serum albumin (HSA), the most abundant plasma protein showing complete biocompatibility, non-toxicity and low immunogenicity, as well as in vivo stability and stimuli-responsive properties, was chosen as a building block for fabricating a protein-based hybrid nanoparticle as an oxygen carrier and nanodelivery system [31, 32]. More importantly, HSA has been proved to have an active tumor-targeting ability, which was mediated by 60-kDa glycoprotein (gp60)/caveolae endothelial transcytosis and binding to SPARC (secreted protein, acidic and rich in cysteine) [33]. Nevertheless, considering that the large quantity of endogenous HSA in blood circulation may saturate most HSA-binding proteins, it is necessary to evaluate the practical tumor targeting effect when HSA-based nanomedicine is implemented in vivo [34].
Herein, an engineered hybrid protein oxygen carrier (HPOC) was developed through a disulfide-bond reconfiguration strategy to integrate the advantages of two natural proteins, oxygen-carrying Hb and tumor-targeting HSA. The resulting HPOC is capable of loading oxygen, chemodrug (doxorubicin, DOX) and photosensitizer (chlorin e6, Ce6), forming ODC-HPOC to actualize chemotherapy and PDT, which are simultaneously enhanced with sufficient oxygen supply in different ways. On one hand, the targeted oxygen supply would modulate the hypoxic tumor microenvironment by altering the hypoxia-induced gene/protein expressions associated with drug resistance, consequently enhancing the chemotherapy. On the other hand, the laser-triggered PDT takes advantage of the tumor-targeted oxygen supply to efficiently generate ROS and enhance PDT. For in vivo imaging, the near-infrared (NIR) fluorescence of Ce6 was utilized to trace ODC-HPOC, and the photoacoustic response of ODC-HPOC could be imaged in real time to map the HbO2/Hb distribution for monitoring tumor oxygenation. This versatile nanoplatform with tumor-targeted delivery of oxygen and drugs could break tumor hypoxia-induced multiple therapeutic resistances and achieve efficient combination therapy outcomes.
Methods
Materials
Bovine hemoglobin (Hb), human serum albumin (HSA) and glutathione (GSH) were obtained from Hefei Bomei Biotechnology (China). Chlorin e6 (Ce6) and doxorubicin (DOX) were purchased from J&K Scientific Ltd. (China). 2',7'-dichlorofluorescin diacetate (DCFH-DA) was purchased from Sigma-Aldrich (USA). Hypoxyprobe kit was bought from Hypoxyprobe, Inc. (USA). Singlet oxygen sensor green (SOSG), 4',6-Diamidino-2-Phenylindole (DAPI) were bought from Thermo Fisher Scientific (USA). Anti-P-gp antibody, and goat anti-rabbit polyclonal immunoglobulin G Alexa Fluor® 488-conjugated secondary antibody were purchased from Abcam (USA). Goat anti-rabbit secondary antibody and Alex 488-conjugated goat anti-mouse secondary antibody were purchased from KPL (USA). Hypoxyprobe-1 Plus kit was bought from Hypoxyprobe Inc. (USA). Phospho-Histone H2A.X (Ser139) (20E3) Rabbit mAb was pruchased from Cell Signaling Technology (USA). Fetal bovine serum, dulbecco's modified eagle medium (DMEM), phosphate buffered saline (PBS) and penicillin-streptomycin were purchased from Gibco Life Technologies (USA). 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) was bought from Promega (USA). All other chemicals used in this study were of analytical reagent grade and used without further purification. Superpure water (18.25 MΩ cm, 25 °C) was used to prepare all solutions. BALB/c athymic nude mice were maintained under aseptic conditions in a small animal isolator. All food, water, bedding and cages were autoclaved before use.
Formulation of the ODC-HPOCs
20 mg HSA was reduced with 6.7 mg GSH in 2 mL deionized water at 37 °C for 1 h, then the solution was dialyzed (membrane cutoff MW: 3 kDa) for 12 h to remove excess GSH. 3.6 mg Hb, 0.2 mg DOX and 0.2 mg Ce6 were added in the reduced HSA solution at pH 8.5 and mixed well. Then, 3 mL ethanol was added into the solution to precipitate the ODC-HPOCs. The suspension was kept under thorough stirring at room temperature for 30 min to form disulfide bonds. After that, the suspension was dialyzed (membrane cutoff MW: 100 kDa) with deionized water at 4 °C for 24 h to remove ethanol, free DOX, Ce6 and Hb. The concentration of DOX and Ce6 were determined by UV/vis absorption spectroscopy and fluorescence spcetroscopy. Fresh ODC-HPOCs were isolated from the aqueous suspension medium by Beckman OptimaTM MAX-XP Ultracentrifuge (38430×g, 30 min) (Beckman, USA). The non-entrapped DOX and Ce6 in the supernatant were quantified to calculate the encapsulation efficiency (EE) and loading efficiency (LE) using the formula: EE (%) = (weight of loaded drug)/(weight of initially added drug)×100; LE (%) = (weight of loaded drug)/(total weight of NPs)×100 [28]. The same procedures were used to prepare C-HSA in the absence of Hb and DOX, as well as to prepare OC-HPOC in the absence of DOX. The ODC-HPOC was deoxygenated by pure argon stream for storage, and was oxygenated by pure oxygen stream before experiments. For oxygenation or deoxygenation operations, pure oxygen or argon gas ran through the nanoparticle solutions filled in an airtight cell for 1 h, followed by monitoring the absorption spectra to confirm the oxy- or deoxy- state [35].
Characterization of the ODC-HPOCs
The number of free sulfhydryl of HSA, reduced HSA and ODC-HPOCs was determined using the Ellman's method [36]. Size distribution of nanoparticles was acquired by dynamic light scattering (DLS) using a Malvern Zetasizer (Nano ZS, Malvern, USA) at 25 °C. The morphology and structure were obtained through transmission electron microscopy (F20, TECHNI, USA). Size exclusion chromatography (SEC) was performed on a protein purification system (AKTAprime plus, GE, USA) using PBS as the mobile phase. The eluent was monitored at 280 nm. To investigate the drug release profile, 1 mL of ODC-HPOCs was transfered into a dialysis bag (MW: 3500), which was submerged into 2 L of PBS at 37 °C. The dialysate was collected to determine DOX/Ce6 content by fluorescence spectrometry. The O2-releasing properties of HbO2 and ODC-HPOCs were obtained by measuring the concentration of oxygen in solutions using a dissolved oxygen detector (Mettler Toledo, Switzerland). Briefly, HbO2 or ODC-HPOCs (containing 4 mg mL-1 Hb) was prepared under pH 7.4 at 25 °C. 1 mL of the samples were oxygenated to dissolved oxygen concentration of 35 mg L-1, while the 10 mL PBS (pH 7.4) was deoxygenated (< 1 mg L-1). Then the two solutions were mixed and sealed, and the concentration of dissolved oxygen was recorded. For measuring oxygen release in oxygen-enriched solution, the 10 mL PBS was also oxygenated (35 mg L-1). The oxygen equilibrium curves of ODC-HPOCs and HbO2 were determined automatically by a Hemox-Analyzer (TSC Scientific Corporation, USA).
Cell culture
The human breast cell line MCF-7 was cultured in DMEM supplemented with 10% (v/v) fetal bovine serum, 1% (v/v) penicillin, and 1% (v/v) streptomycin. For normoxic and hypoxic cell types, cells were incubated in a humidified incubator (37 °C, 5% CO2) in normal (21% O2) and hypoxic (2.5 % O2) conditions, respectively.
In vitro cell uptake
For in vitro studies, 104 MCF-7 cells were seeded on a glass-bottomed culture dish. For quantitative measurement, MCF-7, LO2 and 293T cells were seeded in 12-well plates under normoxic conditions (21% O2). The density of various cells was consistent before experiments. After 12 h incubation, DOX+Ce6, DC-HPOCs or ODC-HPOCs (all the groups contained 10 μg mL-1 DOX and 10 μg mL-1 Ce6) was incubated with cells for 2 h at 37 °C. Then cells were washed three times by PBS, and then fixed with 4% formaldehyde solution (1 mL) for 20 min. The nucleus dye DAPI was used as a positive control to stain nuclei. Images of cells were acquired using a TCS SP5 laser confocal microscope (Leica, Germany). For quantitative measurement, after ODC-HPOCs incubation, cells were washed thrice with phosphate buffered saline (PBS) and then digested by trypsin and harvested by centrifugation. The fluorescence histograms of Ce6 were recorded by flow cytometry (Becton Dickinson, San Jose, CA, USA).
Quantitative real-time polymerase chain reaction (PCR) analysis for HIF-1α and MDR1, and western blot assay for P-gp
According to reference, MCF-7 cells (105 cells per well) were seeded in cell culture flasks (75 cm2) and cultured for 48 h in normoxia or hypoxia [38]. Then, one group of hypoxic cells was incubated with ODC-HPOCs (all groups contained 10 μg mL-1 DOX and 10 μg mL-1 Ce6). After 2 h incubation, total RNA was isolated using Trizol reagent (Life Technologies, USA), followed by reverse transcription to synthesize cDNA using a cDNA RT kit (Toyobo, Japan), according to the manufacturer's instructions. QRT-PCR was then performed by using Thunder Bird SYBR qPCR mix (Toyobo, Japan) with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an endogenous control for normalization. The primers for HIF-1α gene were 5'-CCACAGGACAGTACAGGATG-3' (forward) and 5'-TCAAGTCGTGCTGAATAATACC' (reverse). The primers for MDR1 gene were 5'-GAGGAAGACATGACCAGGTATGC-3' (forward) and 5'-GTGTTAAGCTCCCCAACATCG-3' (reverse). For western blot assay, cells were lysed on ice by RIPA buffer for 30 min and then total protein concentration in cell lysates was quantified by a BCA protein assay kit (Thermo Fisher, USA). Equal amount of protein was electrophoresed in 10% SDS-PAGE gel and then transferred onto a polyvinylidene fluoride (PVDF) membrane. For immunoblotting, the membrane was incubated overnight with anti-P-gp antibody, followed by incubation with goat anti-rabbit secondary antibody for 1 h, and then visualized with enhanced chemiluminescence system (Bio-Rad, USA). For measurement of HIF-1α, MDR1 and P-gp in vivo, mice were intravenously injected with DOX+Ce6 or ODC-HPOCs (both DOX and Ce6 dose were 2.5 mg kg-1), and tumor cells were collected and analyzed with the same procedure.
In vitro reactive oxygen species (ROS) detection
For in vitro detection of ROS generation, MCF-7 cells were incubated with DCFH-DA (10 µM) along with Ce6, C-HPOCs or OC-HPOCs (contained 10 μg mL-1 Ce6) for 2 h, according to the manufacturer's suggested protocol. After 100 mW cm-2 660 nm laser irradiation for 2 min, cells were washed with PBS trice and nuclei were stained with DAPI. Finally, cells were then observed under a TCS SP5 laser confocal microscope or analyzed using flow cytometry.
In vitro chemotherapy and PDT evaluation
The MCF-7 cells were seeded into 96-well plates (104 per well) in 150 μL of medium overnight. For treatment under hypoxic condition, MCF-7 cell culture was conducted in a hypoxic incubator, and the O2 concentration was set under 2.5%. The medium was replaced by the medium with DOX+Ce6, DC-HPOCs or ODC-HPOCs at a series of concentrations. After 2 h incubation, the plate was placed on a 37 °C Labnet AccublockTM digital dry bath. For the laser-treated group, the 96-well plate was exposed to 100 mW cm-2 660 nm laser for 2 min. Before the cells were taken out of the incubator for laser irradiation, the plates were sealed to avoid gas exchange to outer environment. The cells were incubated for another 24 h and the cell viability was investigated by MTS assay.
Animals and tumor model
Female BALB/c nude mice (4-6 weeks old) were purchased from Vital River Laboratory Animal Technology Co. Ltd. (China). Animals received care in accordance with the Guidance Suggestions for the Care and Use of Laboratory Animals. All the animals received care complying with the guidelines in the Guide for the Care and Use of Laboratory Animals, and the procedures for animal experiments were approved by the Institutional Ethical Committee of Animal Experimentation of Shenzhen Institutes of Advanced Technology (Chinese Academy of Sciences). The methods were carried out strictly in accordance with governmental and international guidelines on animal experimentation. All efforts were made to minimize the number of animals and suffering during experiments according to the request of Biosafety and Animal Ethics. MCF-7 cells (1×106) were administered by subcutaneous injection into the flank region of the mice. Tumor volume was calculated as (tumor length)×(tumor width)2/2.
In vivo FL/PA imaging and biodistribution analysis
The nude mice were randomly divided into three groups (three per group). Mice were intravenously injected with free DOX+Ce6 or ODC-HPOCs (the doses of DOX and Ce6 were both 2.5 mg kg-1). Images and fluorescence (FL) semiquantitative analysis of Ce6 were taken at 0, 2, 6, and 24 h after injection using the IVIS spectrum imaging system (Xenogen, USA) with a 640 nm excitation wavelength and a 680 nm filter to collect the FL signals of Ce6. For DOX ex vivo imaging, mice were sacrificed at 24 h post-injection. The organs including heart, liver, spleen, lung, kidneys and tumor were collected for DOX imaging and semiquantitative biodistribution analysis with a 500 nm excitation wavelength and a 600 nm filter. For in vivo photoacoustic (PA) imaging, the ODC-HPOCs (DOX and Ce6 dose were both 2.5 mg kg-1)-injected mice were monitored using a multi-spectral optoacoustic tomography (MSOT) system (Endra Nexus 128, USA) at 750 and 850 nm. FL spectrometry was used for in vivo quantification of DOX [39] and Ce6 [40]. To extract DOX and Ce6, the major organs and tumors of BALB/c mice/nude mice were homogenized in 5 mL of dimethyl sulfoxide (DMSO), and centrifuged at 7493×g for 15 min. The DOX/Ce6 content of each sample was determined by FL spectrometry. To detect the pO2 of liver, kidney and tumor, an optical pO2 measurement device (OxyLite, Oxford Optronix, UK) was used by embedding the fiber sensor into the tissue at 1 mm depth.
Immunohistochemistry
Tumor-bearing mice were intravenously injected with DOX+Ce6 and ODC-HPOCs (the doses of DOX and Ce6 were both 2.5 mg kg-1). After 5 h, the mice were intravenously injected with pimonidazole hydrochloride (60 mg kg-1) (Hypoxyprobe-1 Plus kit), which was reductively activated in hypoxic cells and forms stable adducts with sulfhydryl groups in proteins, peptides, and amino acids. The tumors were dissected 1 h later and sliced by frozen sectioning. For the detection of pimonidazole, tumor sections were incubated with mouse antipimonidazole primary antibody (dilution 1:200, Hypoxyprobe-1 Plus kit) and then Alex 488-conjugated goat anti-mouse secondary antibody (dilution 1:200, KPL, USA) following the vendor's instructions. Cell nuclei were stained with DAPI (dilution 1:1000). The images were taken with a TCS SP5 laser confocal microscope.
For detection of singlet oxygen, the mice were intraperitoneally injected with singlet oxygen sensor green (SOSG, 50 μM) at 5.5 h after injection of C-HSA or OC-HPOCs (the dose of Ce6 was 2.5 mg kg-1), and 0.5 h later mice were irradiated with 660 nm laser for 20 min. Then, the tumors of the mice were extracted and cryosectioned onto slides. The fluorescence of activated SOSG in tumor was observed using a TCS SP5 laser confocal microscope (Ex/Em = 488/525nm). For γH2AX-marked DNA damage investigation, mice were intravenously injected with ODC-HPOCs (or PBS as control). After 6 h, the tumors of mice were exposed to 660 nm laser for 20 min, and the tumors were extracted and cryosectioned onto slides 24 h later. The cryosections were then mounted onto Superfrost Plus glass slides (Fisher Scientific, TX, USA) and fixed as a previous reference described [41]. To investigate DNA damage, tumor sections were incubated with Phospho-Histone H2A.X (Ser139) (1:200, Cell signaling technology, USA) at room temperature for 2 h. The sections were then washed and incubated with goat anti-rabbit polyclonal immunoglobulin G Alexa Fluor® 488-conjugated secondary antibody (1:1000, Abcam, USA) for another 1 h. The fluorescence images of tumor sections were recorded using a confocal laser scanning microscope.
In vivo chemotherapy and PDT
The mice (six per group) were intravenously injected with PBS, free DOX+Ce6 (the doses of DOX and Ce6 were both 2.5 mg kg-1), and ODC-HPOCs (the doses of DOX and Ce6 were both 2.5 mg kg-1), respectively. For laser treatment groups, the tumors of mice were irradiated at 6 h post-injection by the 660 nm laser for 20 min. Tumor volumes of the mice were recorded. To further detect the therapeutic effect in vivo, excised tumors at 24 h after treatment were stained with hematoxylin and eosin. On day 15, the hearts, liver, spleen, lung and kidneys were collected for biosafety analysis via H&E staining. The hearts were also homogenated for cardiac function (cTnI) evaluation according to the manufacturer's instruction (JianCheng Biotech, China).
Statistical analysis
Data are reported as mean ± SD. The differences among groups were determined using one-way ANOVA analysis and Student's t-test; (*) P < 0.05, (**) P < 0.01, (***) P < 0.001
Results
Preparation of HPOCs and ODC-HPOCs
The process of fabricating hybrid protein oxygen carriers (HPOCs) and oxygen, DOX and Ce6 co-loaded HPOCs (ODC-HPOCs) are shown in Figure 1A. HSA molecule is composed of 1 sulfhydryl group (Cys-34) and 17 pairs of disulfide bonds [42]. GSH was used as a reductive agent to cleave the 17 disulfide bonds (S-S) of HSA, producing free sulfhydryl groups (-SH). Hb possesses two sulfhydryl group (-SH) from Cys-93(β) residues in the central cavity, which enabled us to make a disulfide bridge in the proteins through intermolecular conjugations [43]. The Hb and reduced HSA were hybridized by disulfide conjugations (S-S) to form HPOCs, with a 1:6 (Hb:HSA) molar ratio, which was quantified by the absorption spectra. During the disulfide bonds rebuilding process, therapeutic drugs (DOX and Ce6) were encapsulated into the HPOCs, forming the DC-HPOCs. After oxygenation, DC-HPOCs entrapped the oxygen to form ODC-HPOCs and were applied to the oxygen-enhanced chemotherapy and PDT. The EE of DOX and Ce6 in the ODC-HPOCs were 93.1±3.4% and 97.2±4.5%, respectively. The LE of DOX and Ce6 in the ODC-HPOCs were 0.78±0.03% and 0.81±0.04%, respectively.
Preparation and characterization of the ODC-HPOCs. (A) Schematic illustration of the fabrication of the ODC-HPOCs. GSH as reductive agent cleaved the disulfide bonds of HSA to produce sulfhydryl groups, and the reduced HSA was hybridized with Hb by rebuilding the disulfide bonds to form the DOX and Ce6 loaded hybrid protein oxygen carriers (DC-HPOCs), which was further oxygenated to form ODC-HPOCs. (B) TEM images of ODC-HPOCs. (C) Weight analysis of ODC-HPOCs by size exclusion chromatography (SEC). The decreased elution time of HbO2/HSA indicated the formation of hybrid protein. (D) Oxygen release profile of ODC-HPOCs and HbO2 by measuring dissolved oxygen in deoxygenated (red line) and oxygenated (black line) PBS solutions. (E) Oxygen equilibrium curves of ODC-HPOCs and free HbO2. Blue/red regions indicate venous/arterial blood circumstances.
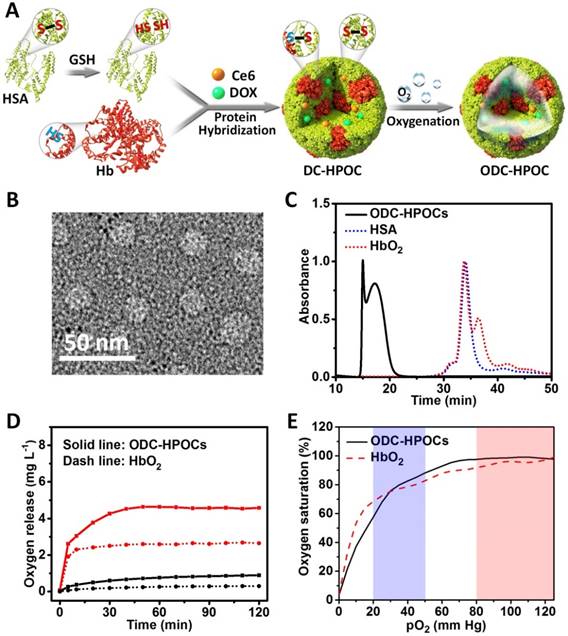
Characterization of the ODC-HPOCs
Transmission electron microscopy (TEM) demonstrated that the ODC-HPOCs were well-defined, with a typical spherical shape with good monodispersity. And, DLS analysis showed that the mean diameter of the ODC-HPOCs was 30 nm, while the mean diameter of free Hb and HSA molecules were approximately 7 nm (Figure 1B and Figure S1). The SEC of ODC-HPOCs confirmed a peak with short elution time (17.2 min) as compared to free Hb (34.1 min) or HSA (33.8 min) (Figure 1C), demonstrating that the molecules of Hb and HSA were hybridized to form a larger protein nanoparticle. Tested with Ellman's method, the free sulfhydryl groups in each Hb, HSA and reduced HSA were 1.26, 0.58 and 10.13 [36]. The number decreased to 1.53 after the Hb/HSA hybridization where the disulfide-bond reconfigured to form a stable hybrid protein nanoparticle. UV-Vis absorption spectroscopy of ODC-HPOCs showed the characteristic absorption peaks of HSA (280 nm), Hb (405 nm), DOX (490 nm) and Ce6 (640 nm) (Figure S2). We have tested the release profiles of Ce6 and DOX from ODC-HPOCs in physiological condition. 44.6%, 66.6% and 86.0% of DOX was released from ODC-HPOCs at 6 h, 12 h and 48 h. Ce6 showed a slower release profile, with 32.5%, 45.3% and 67.7% release at 6 h, 12 h and 48 h (Figure S3). In order to describe the mechanism of DOX/Ce6 release from the ODC-HPOCs, three classical models (first-order, Higuchi and Ritger-Peppas models) were applied. Simulated equations and correlation coefficients were calculated. The correlation coefficients clearly indicate that both DOX and Ce6 release from the ODC-HPOCs can be best described using first-order models (correlation coefficients > 0.96), which was developed to explain drug release from albumin-based particles (Table S1) [37]. The pO2 in tumor was detected by an optical pO2 measurement device (OxyLite, Oxford Optronix, UK), indicating that the pO2 of the central region of tumor is ~0.52 mm Hg (0.89 mg L-1 dissolved O2). Then, deoxygenated phosphate buffered saline (PBS) (dissolved O2 < 1 mg L-1) was prepared to simulate tumor hypoxia, and the oxygen release profile of ODC-HPOC was measured by detecting the increasing curve of dissolved O2 concentration in the deoxygenated PBS. Compared to HbO2, ODC-HPOC released a larger amount of oxygen in deoxygenated PBS, suggesting its upgraded oxygen carrying ability than free Hb (Figure 1D). Otherwise, both ODC-HPOCs and HbO2 barely released oxygen in oxygenated PBS (35 mg L-1 dissolved O2). With protein hybridization, the oxygen carrying ability of HPOCs was better preserved with protection of the nanosystem. In addition, we tested the oxygen equilibrium curves of HbO2 and ODC-HPOCs by measuring their oxygen saturation over different pO2 from 0 to 125 mm Hg. As the result in Figure 1E indicates, ODC-HPOCs under hypoxic condition (pO2 < 20 mm Hg) maintained lower saturation (lower oxygen affinity) than HbO2. Therefore, in the oxygen release experiment (Figure 1D), ODC-HPOC more easily released its carried oxygen in deoxygenated PBS than free HbO2. Moreover, the high oxygen affinity may ensure the ODC-HPOCs retain oxygen before arriving at the hypoxic tumor region [44].
ODC-HPOCs for overcoming hypoxia-induced chemoresistance and enhancing PDT in vitro
We then tested the tumor-targeting ability of ODC-HPOC in vitro by studying its cellular uptake. The MCF-7 breast cancer cells were incubated with DC-HPOCs (deoxygenated to eliminate oxygen loading), ODC-HPOCs or DOX+Ce6 under 2.5% O2 to simulate the hypoxic tumor microenvironment, and then were observed through fluorescence confocal microscopy. With comparison to the free DOX+Ce6, the intracellular Ce6 fluorescence of DC-HPOCs and ODC-HPOCs was greatly enhanced, which confirmed the active targeting ability of the HPOCs (Figure 2A). For further analyzing the active targeting ability of the ODC-HPOCs, we quantitatively measured the cellular uptake of Ce6 by flow cytometry after MCF-7 (human breast cancer cells), LO2 (human hepatic cells) and 293T (human embryonic kidney cells) were incubated with ODC-HPOCs. The result revealed that the Ce6 uptake of MCF-7 cells was significantly more than that of the LO2 and 293T cells (Figure S4). Moreover, the DOX fluorescence of the ODC-HPOCs groups were significantly higher than those of DC-HPOCs, while the Ce6 uptake efficiency was identical. As suggested in previous reports, tumor chemoresistance is correlated with upregulated HIF-1α, which activates the expressions of MDR1 and P-gp, and mediates the efflux of chemodrugs (such as DOX and cisplatin [45, 46], while Ce6 is not included [47]) from cancer cells. Therefore, we investigated the oxygen interference on hypoxia-induced chemoresistance by measuring the expressions of HIF-1α, MDR1 and P-gp in MCF-7 cancer cells. In the hypoxic cells, the HIF-1α expression was 7-fold higher than that in normoxic cells, and the difference of MDR 1 expression between hypoxic and normoxic cells was even higher (19.5-fold). However, when hypoxic cells were incubated with ODC-HPOCs, both the expressions of HIF-1α and MDR1 were downregulated to a similar level as in normoxic cells (Figure 2B-C). Western blotting analysis revealed that the expression of P-gp was downregulated to 57% of that of the hypoxic group after 2 h incubation with ODC-HPOCs, and continuously downregulated to 48% at 6 h. At 24 h, the P-gp profile was barely detectable (Figure 2D and Figure S5). At 24 h post the chemotherapy in vitro, there was no obvious killing effect on cell viability of the free DOX+Ce6 (98.7%) and DC-HPOCs (92.1%) groups at the highest concentration (0.80 μg mL-1 DOX, 0.85 μg mL-1 Ce6). However, the ODC-HPOCs-treated group exhibited the lowest cell viability (7.5%) (Figure 2E). The results indicated that the HPOCs enhanced cellular uptake by tumor targeting and reduced efflux by oxygen interference, resulting in high intracellular DOX accumulation.
HPOCs for breaking hypoxia-induced chemoresistance and enhancing PDT by sufficient oxygen supply in vitro. (A) Cellular uptake of DOX+Ce6, DC-HPOCs and ODC-HPOCs in hypoxic condition (2.5% O2). (B-D) MCF-7 cells were incubated with ODC-HPOCs in normoxic and hypoxic culture conditions. The mRNA levels of HIF-1α (B) and MDR1 (C) gene were determined by QRT-PCR and normalized to the expression of GAPDH. The protein levels of P-gp were determined by western blotting with GAPDH as a loading control (D). Statistical P-values: ***P < 0.001. (E) Cell viability of MCF-7 cells treated with DOX+Ce6, DC-HPOCs and ODC-HPOCs without laser irradiation. (F) Cell viability of MCF-7 cells treated with DOX+Ce6, DC-HPOCs and ODC-HPOCs with laser irradiation. (G) Schematic diagram of the O2-enhanced chemotherapy and PDT with tumor-targeted ODC-HPOCs. The active targeting of ODC-HPOCs increased the cellular uptake of oxygen, DOX and Ce6, and then the released oxygen inhibited DOX efflux for highly effective chemotherapy by downregulating the expressions of HIF-1α, MDR1 and P-gp, and provided sufficient oxygen to boost ROS generation in laser-triggered PDT.
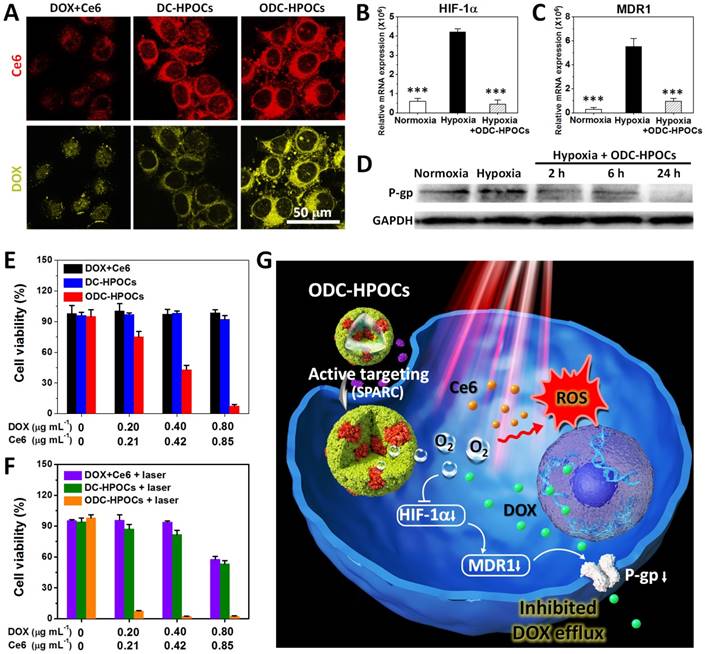
The oxygen in ODC-HPOCs not only played a critical role in breaking hypoxia-induced chemoresistance, but also served as a source of ROS generation in laser-triggered PDT. To further study whether the sufficient oxygen supply acted on PDT simultaneously, intracellular ROS generation was measured after PDT using confocal microscopy. Because ROS generation was included in DOX-induced cytotoxicity [48], free Ce6, C-HPOCs (Ce6-loaded HPOCs without DOX) and OC-HPOCs (O2/Ce6-loaded HPOCs without DOX) were set as the experimental groups. After laser irradiation, intracellular ROS generation between Ce6 and C-HPOCs groups had no significant differences, while the ROS generation amount of OC-HPOCs was 17.5 times more than that of C-HPOCs and 58.1 times more than that of free Ce6, due to the targeted accumulation of intracellular Ce6 and the sufficient oxygen supply (Figure S6).
We further investigated the in vitro chemo-photodynamic combination therapy with the ODC-HPOCs plus 660 nm laser. Compared to the DOX+Ce6+laser (57.7%) and DC-HPOCs+laser (53.4%) groups, the cell viability decreased to 2.5% in the ODC-HPOCs+laser group at the highest concentration (0.80 μg mL-1 DOX, 0.85 μg mL-1 Ce6) (Figure 2F). Overall, the active targeting of the ODC-HPOCs improved intracellular accumulation of DOX/Ce6, and then the released oxygen inhibited DOX efflux by downregulating the expressions of HIF-1α, MDR1 and P-gp. The PDT was also enhanced by oxygen supply with massive ROS generation. The hybrid protein oxygen carriers in overcoming hypoxia-induced chemoresistance and enhancing PDT obtained a highly efficient combination killing effect (Figure 2G).
Tumor-targeted accumulation and oxygen delivery of ODC-HPOCs
Inspired by the above results, an in vivo investigation was performed to illustrate targeted drugs/oxygen delivery of ODC-HPOCs. The biodistribution of ODC-HPOCs was examined with an in vivo imaging system by measuring Ce6 fluorescence with emission in the NIR region. As a comparison, mice were injected with free DOX+Ce6. The signal of free Ce6 vanished within 6 h, which was related to its fast clearance from circulation. After the injection of ODC-HPOCs, the fluorescence of Ce6 distributed widely throughout the mouse body at 2 h, and remained relatively strong at the tumor site for 6 h, which demonstrated that ODC-HPOC was able to target to and accumulate in the tumor. Strong fluorescence signals from kidneys and urocyst of the mice indicated rapid renal excretion of ODC-HPOCs with small sizes [2] (Figure 3A-B). Meanwhile, DOX/Ce6 in major organs and tumor of mice was separately extracted and quantified. After 24 h post-injection, there was about 5% ID/g Ce6 and DOX accumulated in the tumor, which was obviously higher than the Ce6 and DOX accumulated in normal tissues (Figure S7). The higher DOX accumulation than Ce6 in the heart may be due to the propensity of cardiac muscle to accumulate DOX [49]. Benefiting from the transcytosis of HSA in vascular endothelial cells, which is mediated by gp60 activation [33], the ODC-HPOCs could intelligently pass through the vessels and diffuse in the tumor. Subsequently, the enhanced permeability and retention (EPR) effect enables extravasation of the ODC-HPOCs into the tumor interstitial space [50]. Moreover, the ODC-HPOCs with small size have more advantages for deep tumor penetration (far away from tumor blood vessels) than larger ones because of their reduced diffusional hindrance [51]. Apparently, ODC-HPOCs are efficient in tumor-targeted oxygen delivery, while natural red blood cells of larger size fail to deeply penetrate the tumor interstitium [52]. To further visualize the real-time tumor penetration and oxygenation of ODC-HPOCs, photoacoustic imaging was used to measure HbO2 at 850 nm and Hb at 750 nm. As shown in Figure 3C and Figure S8A, there were no obvious changes within 24 h after mice were intravenously injected with PBS. However, after ODC-HPOCs were injected, about 25% more HbO2 signal was detected at 2 h and was extravasated from the vessels and dispersed in the surrounding tissue. The amount of HbO2 was maintained at a high level for at least 6 h, suggesting that ODC-HPOCs modulated tumor hypoxia by donating bound oxygen deep in the tumor. In addition, the Hb signal in the ODC-HPOCs-injected tumor was slightly reduced at 2 h and 6 h, and presented in the whole tumor at 24 h (Figure 3C and Figure S8B). Due to the targeted oxygen delivery of the ODC-HPOCs, the hypoxic condition of MCF-7 solid tumor was modulated to an oxygen-enriched condition for a long time. Therefore, the hypoxia-induced chemoresistance and low photodynamic efficiency could be ameliorated. In addition, an optical pO2 measurement device was used to detect the pO2 of liver and kidney (where there was a relatively high accumulation of nanoparticles) after intravenous injection of ODC-HPOCs. As shown in Figure S9, the injection of ODC-HPOCs did not affect the pO2 of liver (~30 mm Hg) and kidney (~40 mm Hg) at all timepoints. As indicated in Figure 1D, at a pO2 of 30-50 mm Hg, the ODC-HPOCs remained saturated with captured O2; thus, the nonspecific oxygen release to normal tissues could be negligible.
In vivo targeting and O2 interference after intravenous injection of ODC-HPOCs. (A) Time-lapse Ce6 fluorescence images of nude mice after intravenous administration of DOX+Ce6 and ODC-HPOCs. (B) Quantified analysis of Ce6 FL intensity around the tumor at indicated time points. (C) Representative photoacoustic images of HbO2 (λ = 850 nm) and Hb (λ = 750 nm) in tumor after intravenous injection of ODC-HPOCs.
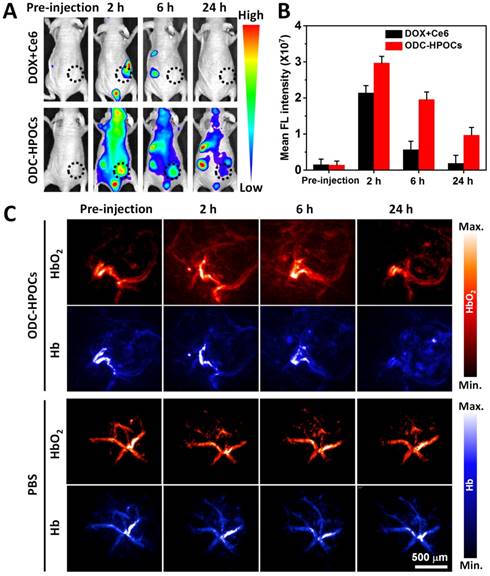
Oxygen-enhanced chemo-PDT of ODC-HPOCs in vivo at a single dose
To investigate the oxygen-interfered DOX accumulation in tumor, semiquantitative fluorescence measurement was carried out based on ex vivo imaging of major organs and tumors collected from mice at 24 h post-injection [53]. As shown in Figure 4A, DOX was mainly located in liver, kidney and tumors for the ODC-HPOCs group. In the tumor, the DOX fluorescence intensity of the ODC-HPOCs group was 4.5-fold stronger than that of the DOX+Ce6 group (Figure S10). The tumors were further sliced into sections and stained with hypoxyprobe (green) for mapping the distribution of hypoxia. The result indicated that tumor hypoxia presented about 150 μm beneath the tumor margin, and the DOX signals rarely presented in hypoxic regions. Compared to that of the free DOX group, the area of hypoxia in the ODC-HPOCs group was dramatically decreased and scattered, which offered proof of the hypoxia attenuation by targeted oxygen supply. Moreover, the hypoxia-reduced tumor region was occupied by DOX (Figure 4B). Moreover, we performed statistical analysis of the whole tumor images for measuring the fluorescence of hypoxia/DOX quantitatively. The results indicated that, with ODC-HPOCs injection, the tumor hypoxia was significantly attenuated and DOX accumulation was improved (Figure 4C). The Ce6 fluorescence signals had less significant difference between the DOX+Ce6 and ODC-HPOCs groups, due to the independence of Ce6 in P-gp-mediated efflux (Figure S11). The results demonstrated that the intratumoral DOX accumulation, with targeted DOX delivery and oxygen-interfered inhibition of cellular DOX efflux, could be superior to conventional chemotherapy. The HIF-1α and MDR1 (genes) expressions in vivo were measured by quantitative real-time PCR analysis, and P-gp (protein) expression was measured by western blotting analysis. Compared to the treatment with free DOX+Ce6 injection, the ODC-HPOCs injection with sufficient oxygen delivery downregulated 90% of the HIF-1α expression and 88% of MDR1 expression (Figure 4D-E). The expression of P-gp, which mediates efflux of DOX, was downregulated by 83% in the ODC-HPOCs-injected group, compared to the free DOX+Ce6-injected group (Figure 4F and Figure S12).
Combined chemo-PDT of ODC-HPOCs by dampening chemoresistance and enhancing PDT at a single dose. (A) DOX accumulation in major organs and tumor. (B) Hypoxia mapping and DOX distribution in tumor at 24 h after intravenous injection of ODC-HPOCs. Hypoxic regions are circled by white dots, and white dashed lines indicate the distance from the interior tumor region. Scale bar, 50 μm. (C) Quantitative measurement of total fluorescence intensity of hypoxia and DOX in whole tumor tissue fluorescence images (n=7). (D-F) Two days after treatment with DOX+Ce6 and ODC-HPOCs, tumors were harvested. The mRNA levels of HIF-1α (D) and MDR1gene (E) were determined by QRT-PCR and normalized to the expression of GAPDH. The protein levels of P-gp were determined by western blotting (F). (G) Singlet oxygen measurement in vivo after administration of C-HSA and OC-HPOCs with laser irradiation at 6 h. (H) DNA damage (γH2AX as marker) in tumor tissue at 24 h after the enhanced chemotherapy (ODC-HPOCs without laser) and the combined chemo-PDT (ODC-HPOCs+laser). (I) Tumor growth curves of different groups after treatments. Statistical P-values: *P < 0.05, **P < 0.01. (J) Biosafety evaluation by H&E staining of the major organs at the end of the experiment. Scal bar, 200 μm.
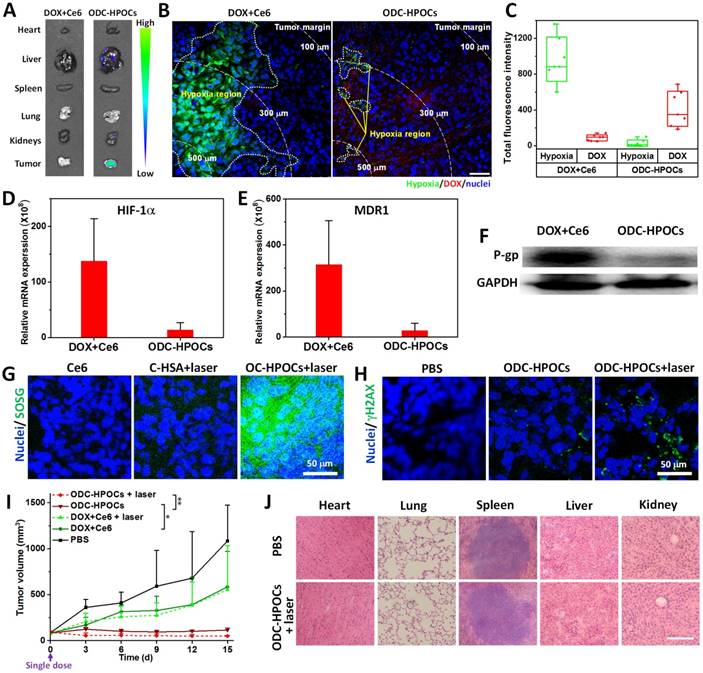
Meanwhile, the targeted delivered Ce6 in ODC-HPOCs could take advantage of the sufficient tumor oxygenation for PDT in vivo. SOSG was used to measure singlet oxygen in tumor after the laser irradiation. To avoid DOX-induced ROS generation [47], C-HSA (Ce6-loaded HSA nanoparticles) and OC-HPOCs (O2/Ce6-loaded HPOCs without DOX) was employed to investigate the in vivo PDT effect. After laser irradiation, strong SOSG fluorescence was observed in the tumor treated with OC-HPOCs, while the C-HSA failed to generate enough singlet oxygen, not to mention free Ce6 (Figure 4G). The differences between OC-HPOCs and C-HSA reflected the vital role of sufficient oxygen supply for PDT in hypoxic tumor. To evaluate the synergistic efficacy of chemo-PDT, DNA damage in tumor cells was measured using a fluorescent marker (γH2AX). DNA damage was observed in the ODC-HPOCs group, suggesting the considerable therapeutic effect of the oxygen-interfered chemotherapy. When the elevated chemotherapy and PDT were joined together (ODC-HPOCs+laser), the DNA damage of tumor cells was enhanced, which was indicated by the increased green fluorescence (Figure 4H).
The in vivo antitumor efficacy of ODC-HPOCs-based combination chemo-PDT was evaluated by using MCF-7 tumor-bearing nude mice. In this experiment, mice were treated with a single dose in five groups: PBS, DOX+Ce6 (chemotherapy), DOX+Ce6+laser (chemotherapy+PDT), ODC-HPOCs (enhanced chemotherapy) and ODC-HPOCs+laser (co-enhanced chemo-PDT). The tumor growth curves are shown in Figure 4I. A remarkable tumor growth inhibition was observed in the ODC-HPOCs group, which improved the tumor inhibition rate to 89.5% in comparison to the DOX+Ce6 (46.0%) and DOX+Ce6+laser (48.9%) groups at day 15. Also, compared to literature reports of chemotherapy with 3 injections [54], or 8-fold DOX content in nanoparticles at one dose [55], chemotherapy of ODC-HPOCs, which was undertaken with a single injection at rather low DOX concentration, also exhibited highly efficient tumor inhibition due to the targeted oxygen supply to break down chemoresistance. However, after treatment with ODC-HPOCs without laser, tumor relapse showing a slight increase in tumor volume was observed from day 9 to day 15. The efficacy of ODC-HPOCs+laser was even more potent than the enhanced chemotherapy alone, demonstrating the efficacy of the co-enhanced chemo-PDT. Lately, it was reported that oxygen-enhanced chemo-PDT effectively modulates the immuno-suppresive tumor microenvironment, which might be the fundamental mechanism for achieving potent therapeutic outcomes [56]. DC-HSA (DOX/Ce6-loaded HSA nanoparticles) was set as the control nanoparticle for showing the comparison in chemotherapy and chemo-phototherapy without oxygen supplying (Figure S13). The HSA nanoparticle-based chemotherapy (DC-HSA) or chemo-PDT (DC-HSA+laser) exhibited more intensive tumor inhibition than free DOX+Ce6 and DOX+Ce6+laser, which was attributed to the tumor-targeted drug delivery by HSA nanoparticles. Nevertheless, without sufficient oxygen supply, the DC-HSA failed to kill all the tumor cells and showed tumor recurrence.
We also evaluated the biosafety of ODC-HPOCs at day 15 after treatments. Compared with the PBS group, hematoxylin and eosin (H&E) staining of the major organs showed that no apparent histological changes were found from ODC-HPOCs+laser, which indicated the great biocompatibility and biosafety of ODC-HPOCs (Figure 4J). Further, the biosafety of ODC-HPOCs was evaluated by heart, liver and kidney function index (heart function index: cardiac troponin I (cTnI); liver function index: aspartate aminotransferase (AST), alanine aminotransferase (ALT); kidney function index: creatinine (CRE), blood urea nitrogen (BUN)) of the mice in the PBS and ODC-HPOCs groups by blood biochemistry tests [57]. Conformably, there was no difference in concentration of all those markers between the PBS and ODC-HPOCs groups (Figure S14). These results presented the co-enhanced antitumor efficacy of chemotherapy and PDT based on ODC-HPOCs without toxicity to normal tissue.
Conclusion
In summary, we utilized disulfide bond reconfiguration technology to fabricate well-defined hybrid protein oxygen carriers with loading of chemodrug and photosensitizer (ODC-HPOCs), serving as a universal nanoplatform for overcoming tumor hypoxia-induced chemoresistance as well as low photodynamic efficiency. A single dose of ODC-HPOCs plus laser achieved oxygen-enhanced chemo-PDT with potent tumor suppression, which could be contributed to a series of advantages: (i) The tumor-targeted ODC-HPOCs with small sizes allowed outstanding performance in tumor accumulation of O2, DOX and Ce6. (ii) The high oxygen affinity of our oxygen carriers allowed oxygen preservation during circulation, and oxygen release in the hypoxic tumor, resulting in precise and sufficient tumor oxygenation. (iii) The drastic alteration of tumor hypoxia broke the hypoxia-induced chemoresistance through inhibiting the expressions of HIF-1α, MDR1 and P-gp; meanwhile, the abundant oxygen enhanced the ROS generation in PDT, and the enhanced chemo-PDT managed to offer a single-dose treatment with minimized dosage of DOX and Ce6. (iv) Real-time dual-modal fluorescence/photoacoustic imaging mapped and monitored the drug accumulation and tumor oxygenation, resulting in imaging-guided therapy. (v) The ODC-HPOC was composed of biosafe and biocompatible materials without side effects to normal tissues, offering a potential for clinical translation. Overall, this work provides new insight into the design of tumor-targeted oxygen nanocarriers to achieve more precise and effective therapy in hypoxic tumors, and might be valuable for additional therapies involving hypoxia modulation (radiotherapy, immunotherapy, etc.).
Abbreviations
ALT: alanine aminotransferase; AST: aspartate aminotransferase; BUN: blood urea nitrogen; cTnI: cardiac function; Ce6: chlorin e6; CRE: creatinine; DOX: doxorubicin; DLS: dynamic light scattering; EE: encapsulation efficiency; GSH: glutathione; Hb: hemoglobin; HSA: human serum albumin; HPOCs: hybrid protein oxygen carriers; HIF-1α: hypoxia-inducible factor-1α; LE: loading efficiency; MDR1: multidrug resistance 1; NIR: near-infrared; P-gp: P-glycoprotein; PBS: phosphate buffered saline; PCR: polymerase chain reaction; PDT: photodynamic therapy; ROS: reactive oxygen species; SOSG: singlet oxygen sensor green; SEC: size exclusion chromatography; SPARC: secreted protein, acidic and rich in cysteine; TEM: transmission electron microscopy.
Acknowledgements
This work was supported by the Key International S&T Cooperation Project (2015DFH50230), National Natural Science Foundation of China (81671758, 81701816, 81501591, 31571013, 51502333 and 81501580), Guangdong Natural Science Foundation of Research Team (2016A030312006), Natural Science Foundation of Guangdong Province (2017A030313079 and 2017A030313726), Dongguan project on Social science and Technology Development (2015108101019), Shenzhen Science and Technology Program (JSGG20160331185422390, and JCYJ20160429191503002).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wang Y, Xie Y, Li J. et al. Tumor-penetrating nanoparticles for enhanced anticancer activity of combined photodynamic and hypoxia-activated therapy. ACS Nano. 2017;11:2227-38
2. Chen Q, Feng L, Liu J. et al. Intelligent albumin-MnO2 nanoparticles as pH-/H2O2-responsive dissociable nanocarriers to modulate tumor hypoxia for effective combination therapy. Adv Mater. 2016;28:7129-36
3. Lee JY, Carugo D, Crake C. et al. Nanoparticle-loaded protein-polymer nanodroplets for improved stability and conversion efficiency in ultrasound imaging and drug delivery. Adv Mater. 2015;27:5484-92
4. Hou Z, Zhang Y, Deng K. et al. UV-emitting upconversion-based TiO2 photosensitizing nanoplatform: near-infrared light mediated in vivo photodynamic therapy via mitochondria-involved apoptosis pathway. ACS Nano. 2015;9:2584-99
5. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393-410
6. Rouschop KM, van den Beucken T, Dubois L. et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120:127-41
7. Schug ZT, Vande Voorde J, Gottlieb E. The metabolic fate of acetate in cancer. Nat Rev Cancer. 2016;16:708-17
8. Chang Q, Jurisica I, Do T. et al. Hypoxia predicts aggressive growth and spontaneous metastasis formation from orthotopically grown primary xenografts of human pancreatic cancer. Cancer Res. 2011;71:3110-20
9. Nakazawa MS, Keith B, Simon MC. Oxygen availability and metabolic adaptations. Nat Rev Cancer. 2016;16:663-73
10. Deavall DG, Martin EA, Horner JM. et al. Drug-induced oxidative stress and toxicity. J Toxicol. 2012;2012:645460
11. Owen MR, Stamper IJ, Muthana M. et al. Mathematical modeling predicts synergistic antitumor effects of combining a macrophage-based, hypoxia-targeted gene therapy with chemotherapy. Cancer Res. 2011;71:2826-37
12. Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721-32
13. Qi X, Xu W, Xie J. et al. Metformin sensitizes the response of oral squamous cell carcinoma to cisplatin treatment through inhibition of NF-kappaB/HIF-1alpha signal axis. Sci Rep. 2016;6:35788
14. Qian C, Yu J, Chen Y. et al. Light-activated hypoxia-responsive nanocarriers for enhanced anticancer therapy. Adv Mater. 2016;28:3313-20
15. Li M, Gao Y, Yuan Y. et al. One-step formulation of targeted aggregation-induced emission dots for image-guided photodynamic therapy of cholangiocarcinoma. ACS Nano. 2017;11:3922-32
16. Wang S, Yuan F, Chen K. et al. Synthesis of hemoglobin conjugated polymeric micelle: A ZnPc carrier with oxygen self-compensating ability for photodynamic therapy. Biomacromolecules. 2015;16:2693-700
17. Nahabedian MY, Cohen RA, Contino MF. et al. Combination cyto-toxic chemotherapy with cisplatin or doxorubicin and photodynamic therapy in murine tumors. J Natl Cancer Inst. 1988;80:739-43
18. Luo D, Carter KA, Miranda D. et al. Chemophototherapy: An emerging treatment option for solid tumors. Adv Sci. 2017:4
19. Stepien K, Ostrowski RP, Matyja E. Hyperbaric oxygen as an adjunctive therapy in treatment of malignancies, including brain tumours. Med Oncol. 2016;33:101
20. Lu Z, Ma J, Liu B. et al. Hyperbaric oxygen therapy sensitizes nimustine treatment for glioma in mice. Cancer Med. 2016;5:3147-55
21. Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437-47
22. Barker HE, Paget JT, Khan AA. et al. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. 2015;15:409-25
23. Chen Y, Zhang Y, Wang Z. et al. Effects of adenosine metabolism in astrocytes on central nervous system oxygen toxicity. Brain Res. 2016;1635:180-9
24. Dinu D, Chu C, Veith A. et al. Mechanistic role of cytochrome P450 (CYP)1B1 in oxygen-mediated toxicity in pulmonary cells: a novel target for prevention of hyperoxic lung injury. Biochem Biophys Res Commun. 2016;476:346-51
25. Huang C, Chia W, Chung M. et al. An implantable depot that can generate oxygen in situ for overcoming hypoxia-induced resistance to anticancer drugs in chemotherapy. J Am Chem Soc. 2016;138:5222-25
26. Fan W, Bu W, Shen B. et al. Intelligent MnO2 nanosheets anchored with upconversion nanoprobes for concurrent pH-/H2O2-responsive UCL imaging and oxygen-elevated synergetic therapy. Adv Mater. 2015;27:4155-61
27. Song X, Feng L, Liang C. et al. Ultrasound triggered tumor oxygenation with oxygen-shuttle nanoperfluorocarbon to overcome hypoxia-associated resistance in cancer therapies. Nano Lett. 2016;16:6145-53
28. Luo Z, Zheng M, Zhao P. et al. Self-monitoring artificial red cells with sufficient oxygen supply for enhanced photodynamic therapy. Sci Rep. 2016;6:23393
29. Fleming IN, Manavaki R, Blower PJ. et al. Imaging tumour hypoxia with positron emission tomography. Br J Cancer. 2015;112:238-50
30. Li T, Jing X, Huang Y. Polymer/hemoglobin assemblies: biodegradable oxygen carriers for artificial red blood cells. Macromol Biosci. 2011;11:865-75
31. Sailor MJ, Park JH. Hybrid nanoparticles for detection and treatment of cancer. Adv Mater. 2012;24:3779-802
32. Chen Q, Liu Z. Albumin carriers for cancer theranostics: a conventional platform with new promise. Adv Mater. 2016;28:10557-66
33. Elsadek B, Kratz F. Impact of albumin on drug delivery-new applications on the horizon. J Control Release. 2012;157:4-28
34. Li R, Zheng K, Yuan C. et al. Be active or not: the relative contribution of active and passive tumor targeting of nanomaterials. Nanotheranostics. 2017;1:346-57
35. Duan L, Yan X, Wang A. et al. Highly loaded hemoglobin spheres as promising artificial oxygen carriers. ACS Nano. 2012;6:6897-904
36. Riener CK, Kada G, Gruber HJ. Quick measurement of protein sulfhydryls with Ellman's reagent and with 4,4'-dithiodipyridine. Anal Bioanal Chem. 2002;373:266-76
37. Gupta PK, Hung CT, Perrier DG. Albumin microspheres. 1. Release characteristics of adriamycin. Int J Pharm. 1986;33:137-46
38. Yi H, Liu L, Sheng N. et al. Synergistic therapy of doxorubicin and miR-129-5p with self-cross-linked bioreducible polypeptide nanoparticles reverses multidrug resistance in cancer cells. Biomacromolecules. 2016;17:1737-47
39. Zheng M, Yue C, Ma Y. et al. Single-step assembly of DOX/ICG loaded lipid-polymer nanoparticles for highly effective chemo-photothermal combination therapy. ACS Nano. 2013;7:2056-67
40. Chen Q, Wang X, Wang C. et al. Drug-induced self-assembly of modified albumins as nano-theranostics for tumor-targeted combination therapy. ACS Nano. 2015;9:5223-33
41. Ma Y, Ross AC. Toll-like receptor 3 ligand and retinoic acid enhance germinal center formation and increase the tetanus toxoid vaccine response. Clin Vaccine Immunol. 2009;16:1476-84
42. Sheng Z, Hu D, Zheng M. et al. Smart human serum albumin-indocyanine green nanoparticles generated by programmed assembly for dual-modal imaging-guided cancer synergistic phototherapy. ACS Nano. 2014;8:12310-22
43. Mueser TC, Rogers PH, Arnone A. Interface sliding as illustrated by the multiple quaternary structures of liganded hemoglobin. Biochemistry. 2000;39:15353-64
44. Wang Y, Wang L, Yu W. et al. A PEGylated bovine hemoglobin as a potent hemoglobin-based oxygen carrier. Biotechnol Prog. 2017;33:252-60
45. Comerford KM, Wallace TJ, Karhausen J. et al. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62:3387-94
46. Samanta D, Gilkes DM, Chaturvedi P. et al. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc Natl Acad Sci USA. 2014;111:E5429-38
47. Merlin JL, Gautier H, Barberi-Heyob M. et al. The multidrug resistance modulator SDZ-PSC 833 potentiates the photodynamic activity of chlorin e6 independently of P-glycoprotein in multidrug resistant human breast adenocarcinoma cells. Int J Oncol. 2003;22:733-9
48. Minotti G, Menna P, Salvatorelli E. et al. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. 2004;56:185-229
49. Stewart DJ, Grewaal D, Green RM. et al. Concentrations of doxorubicin and its metabolites in human autopsy heart and other tissues. Anticancer Res. 1993;13:1945-52
50. Tang L, Yang X, Yin Q. et al. Investigating the optimal size of anticancer nanomedicine. Proc Natl Acad Sci USA. 2014;111:15344-9
51. Li H, Du J, Du X. et al. Stimuli-responsive clustered nanoparticles for improved tumor penetration and therapeutic efficacy. Proc Natl Acad Sci USA. 2016;113:4164-9
52. Chang TM. Therapeutic applications of polymeric artificial cells. Nat Rev Drug Discov. 2005;4:221-35
53. Zhao P, Zheng M, Luo Z. et al. NIR-driven smart theranostic nanomedicine for on-demand drug release and synergistic antitumour therapy. Sci Rep. 2015;5:14258
54. Zhu J, Zheng D, Zhang M. et al. Preferential cancer cell self-recognition and tumor self-targeting by coating nanoparticles with homotypic cancer cell membranes. Nano Lett. 2016;16:5895-5901
55. Liang M, Fan K, Zhou M. et al. H-ferritin-nanocaged doxorubicin nanoparticles specifically target and kill tumors with a single-dose injection. Proc Natl Acad Sci USA. 2014;111:14900-5
56. Yang G, Xu L, Chao Y. et al. Hollow MnO2 as a tumor-microenvironment-responsive biodegradable nano-platform for combination therapy favoring antitumor immune responses. Nat Comm. 2017;8:902
57. Ji M, Li P, Sheng N. et al. Sialic acid-targeted nanovectors with phenylboronic acid-grafted polyethylenimine robustly enhance siRNA-based cancer therapy. ACS Appl Mater Inter. 2016;8:9565-76
Author contact
![]() Corresponding authors: L.C. (lt.caiac.cn) and M.Z. (mb.zhengac.cn).
Corresponding authors: L.C. (lt.caiac.cn) and M.Z. (mb.zhengac.cn).