Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(13):3490-3503. doi:10.7150/thno.24157 This issue Cite
Research Paper
Powerful anti-colon cancer effect of modified nanoparticle-mediated IL-15 immunogene therapy through activation of the host immune system
Xiaoxiao Liu1,2,3,*, Yanyan Li4,*, Xiaodong Sun1,*, Yagmur Muftuoglu5, Bilan Wang3, Ting Yu1, Yuzhu Hu1, Lu Ma1, Mingli Xiang1, Gang Guo1, Chao You1, Xiang Gao1, ![]() , Yuquan Wei1
, Yuquan Wei1
1. Department of Neurosurgery and Institute of Neurosurgery, State Key Laboratory of Biotherapy, West China Hospital, West China Medical School, Sichuan University/Collaborative Innovation Center for Biotherapy, Chengdu, 610041, China.
2. Department of Radiation Oncology, Cancer Center, Affiliated Hospital of Xuzhou Medical University; Jiangsu Center for the Collaboration and Innovation of Cancer Biotherapy, Cancer Institute, Xuzhou Medical University, Xuzhou, 221000, China.
3. Department of Pharmacy, West China Second University Hospital of Sichuan University, Chengdu, 610041, China.
4. Department of radiation oncology, Fudan University Shanghai Cancer Center; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, China
5. Stanford University School of Medicine, Stanford, CA 94305, USA
*These authors are considered equal first authors.
Received 2017-12-3; Accepted 2018-5-20; Published 2018-6-6
Abstract

Rationale: Colorectal cancer (CRC) is the third most commonly diagnosed cancer around the world. Over the past several years, immunotherapy has demonstrated considerable clinical benefit in CRC therapy, and the number of immunologic therapies for cancer treatment continues to climb each year. Interleukin-15 (IL15), a potent pro-inflammatory cytokine, has emerged as a candidate immunomodulator for the treatment of CRC.
Methods: In this study, we developed a novel gene delivery system with a self-assembly method using DOTAP and MPEG-PLA (DMA) to carry pIL15, denoted as DMA-pIL15 which was used to treat tumor-bearing mice.
Results: Supernatant from lymphocytes treated with supernatant derived from CT26 cells transfected with DMA-pIL15 inhibited the growth of CT26 cells and induced cell apoptosis in vitro. Treatment of tumor-bearing mice with DMA-pIL15 complex significantly inhibited tumor growth in both subcutaneous and peritoneal models in vivo by inhibiting angiogenesis, promoting apoptosis, and reducing proliferation through activation of the host immune system.
Conclusion: The IL-15 plasmid and DMA complex showed promise for treating CRC clinically as an experimental new drug.
Keywords: colorectal cancer, immunotherapy, interleukin-15, MPEG-PLA, DOTAP
Introduction
Colorectal cancer (CRC) is the third most commonly diagnosed cancer globally. Each year, more than 600,000 people die from colorectal cancer [1]. The majority of CRC cases are diagnosed at later stages, and it is estimated that approximately 40% of CRC patients progress to fatal metastasis with poor survival rates as a result [2]. Despite the well-established treatment modalities (surgery, radiation, and chemotherapy) used in management of carcinoma of the colon, the survival of patients has not improved significantly in the past decades, making it a major public health problem [3, 4]. Therefore, the development of novel, less toxic therapeutic agents is imperative to help reduce the high mortality and morbidity rates associated with CRC [5, 6].
Gene therapy involves the delivery of DNA, RNA, small interfering RNAs, or antisense oligonucleotides [7-9]. Thus far, most of the clinical trials in gene therapy for cancer treatment report remarkable efficacies both in vitro and in vivo [1]. In recent years, increased recognition of the link between inflammation and development of cancer has led to progress in cancer immunotherapy, which is designed to stimulate host immune mechanisms to reject and destroy tumors and circulating tumor cells [11,12]. Of these immune-modulators, interleukin-15 (IL-15), a potent pro-inflammatory cytokine, has emerged as a candidate for the treatment of colon cancer [13, 14].
IL-15 is a member of the four α-helix bundle family of cytokines and was first identified in the supernatant of the monkey epithelial cell line CV-1/EBNA [15]. Since its discovery two decades ago, IL-15 has become one of the most promising therapeutic agents in cancer immunotherapy, with multiple studies attributing antitumor effects to IL-15, along with enhanced cell cytotoxicity by natural killer (NK) and CD8+ T cells [2]. Previous studies found that IL-15 administration inhibits tumor growth and prolongs survival rates in mice with CT26 colon cancer [17]. Despite this success, systemic bolus administration of IL-15 results in a pharmacokinetic profile with multiple peaks and valleys. Furthermore, as a soluble molecule, IL-15 undergoes rapid clearance by the kidneys and must be administrated daily in large doses, potentially resulting in toxicity or suboptimal efficacy [3]. However, the low transfection potency, cytotoxicity of the reported gene vector caused by its high positive charge density, and poor stability of the formulation has largely limited the application of gene therapy for treating cancer. A delivery method with reduced side effects and high transfection potency can address this need, as described here.
In this study, we developed a novel gene delivery system with a self-assembly method by MPEG-PLA and DOTAP and reported that transfected cancer cells expressed and secreted high level of IL-15 cytokine, which serves to activate the host immune system. Furthermore, the potential toxicity is dramatically reduced, and steady expression of IL-15 creates a stronger antitumor immune response. The pharmaceutical properties, in vitro biological activity, in vivo antitumor effects, antitumor mechanisms, and preliminary toxicity evaluation of DOTAP/MPEG-PLA-pIL15 (DMA-pIL15) are presented in this study.
Results
Preparation and characterization of DMA-pIL15
DMA and DMA-pIL15 were produced by the described self-assembly method (Figure 1). As shown in Figure 2, interaction between DOTAP and MPEG-PLA copolymer in the environment of water made them gradually approach each other. DOTAP tried, on the surface of MPEG-PLA copolymer, to find a suitable place for interaction by moving its position and adjusting its conformation. Meanwhile, MPEG-PLA copolymer continuously adjusted its conformation to provide a good site for DOTAP.
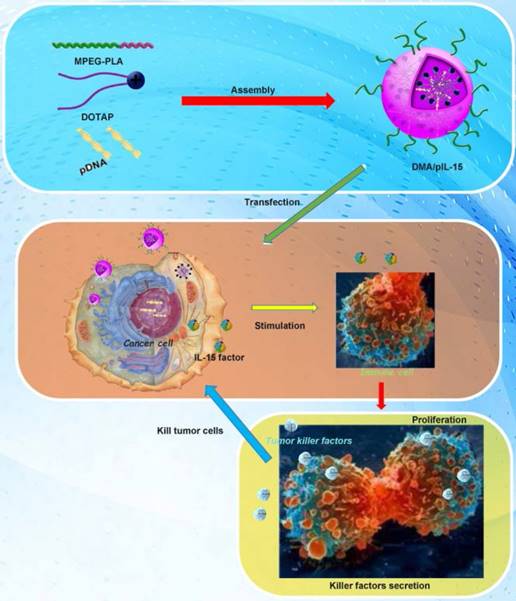
Transfection and function of pIL15 in vitro and in vivo. MPEG-PLA and DOTAP were assembled into a new gene carrier—DOTAP/MPEG-PLA (DMA) micelles—which carry the pIL15 plasmid into cancer cells. Cancer cells then express and secrete IL15, which stimulates host immune cells and promotes proliferation and secretion of immunogenic factors that help kill tumor cells.
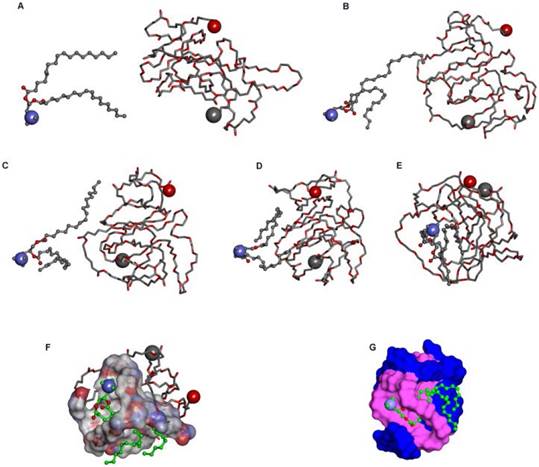
Interaction modes between polymer and DOTAP revealed by molecular dynamics simulations in a water environment and in an aqueous environment after running for 300 ps. (A) The initial conformation of polymer complexed with DOTAP. (B-E) Conformations corresponding to snapshots of the complex collected at 4.990 ps, 9.970 ps, 15.015 ps, and 300 ps, respectively. Polymer is represented with thick stick, while DOTAP is depicted with scaled ball and stick style. Two terminal heavy atoms in the polymer and the head heavy atom in the compound are highlighted using CPK style. (F) Polymer is represented with thick stick, while DOTAP is depicted with scaled ball and stick style. Two terminal heavy atoms in the polymer and the head heavy atom in the compound are highlighted using CPK style. (G) Polymer is described with a solid surface and the part corresponding to [C2H5O] is colored pink. DOTAP is represented in the same way and its carbon atoms are colored green.
The zeta-potential and size were found to be 7 mV and 83.47 nm (PDI: 0.21) for DMA-pIL15, and TEM images depicted the spheroidal morphology of DMA/pIL15, with an estimated size of ~25 nm (Figure 3).
Preparation and physicochemical properties of DMA. (A) Schematic depiction of DMA/pIL15. (B) Zeta-potential of DMA. (C) Morphological characteristics. (D) Particle size of DMA. (E) Gel retardation assay of DNA and complexes: lane 0, DNA marker; lanes 1 to 3, naked pIL15; lanes 4 to 6, N/P ratios of DMA/pIL15 (1.25:1); lanes 7 to 9, N/P ratios of DMA/pIL15 (2.5:1); lanes 10 to 12, N/P ratios of DMA/pIL15 (5:1). Results show that pIL15 is completely incorporated into DMA at a N/P ratio of 5: 1 and that complexes are prepared without free DNA.
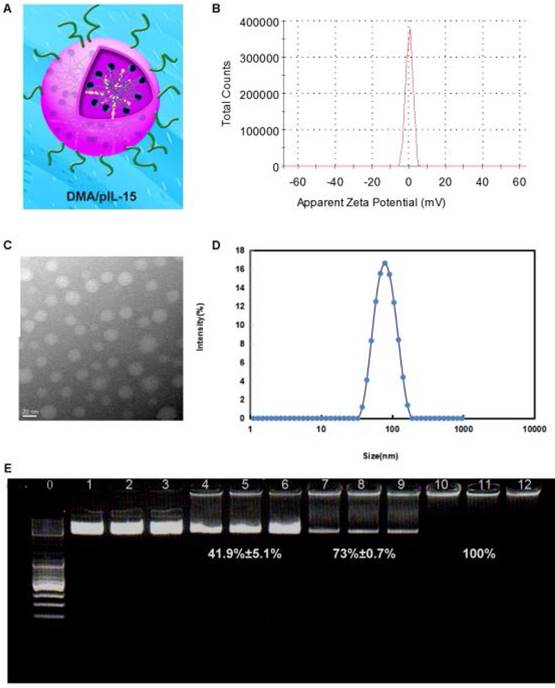
Gel retardation assays were used to characterize the ability of the DMA liposome to carry DNA. As shown in Figure 3E, the free DNA was not entrapped in the liposome and appears as a bright band (lanes 1 to 9), and no other band of free DNA was observed in lanes 10 to 12. This suggests that pIL-15 DNA was completely incorporated into the DMA and that the lipoplexes were prepared successfully without free pIL15 DNA at a N/P ratio of DMA: DNA of 5:1.
High transfection efficiency of DMA-pIL15 in CT26 cells
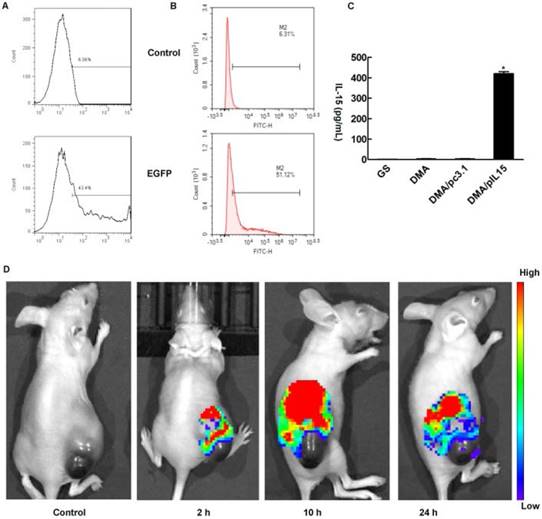
In the in vitro cell transfection assay, DMA produced high GFP transfection efficiency in CT26 cells after incubation for 24 h as shown in Supplementary Figure 1. The specific transfection rate demonstrated by flow cytometry analysis was 43.4%±2.1% and 51.1%±1.7% when DMA was mixed with GFP at a N/P ratio of 5:1 after 24 h with serum-free media and at a N/P ratio of 20:1 after 24 h with 10% serum media during transfection (Figure 4A-B). CT26 cells were transfected with DMA, DMP-pc3.1 or DMA-pIL15, or were treated with GS as a negative control, and we found that the IL-15 expression level in the DMA-pIL15-transfected cell culture supernatant to be significantly higher than that of the control groups after 72 h of incubation in culture by an ELISA test (Figure 4C). In order to test the ability of DMA to deliver plasmid, mice bearing tumors were treated by intravenous administration with plasmid labeled with toto-3 loaded into DMA. In vivo imaging showed high delivery efficiency in the mice treated with intravenously injected DMA-pDNA-toto-3 (Figure 4D).
Measurement of transfection efficiency in vitro and in vivo. DMA containing pEGFP (4 ug) was used to transfect CT26 cells at a weight ratio of 1:10 pEGFP to DMA. Transfection efficiency was measured by flow cytometry (A) at a N/P ratio of 5:1 after 24 h with serum-free medium and (B) at a N/P ratio of 20:1 after 24 h with 10% serum of transfection. (C) Detection of IL-15 in cell supernatant from different groups by ELISA (Mean ± SEM, n=3, *, p < 0.01, DMA/pIL15 versus GS, DMA, DMA/pc3.1). (D) Bio-imaging analysis showing that tumors of DMA and DMA/pcDNA-toto-3 can be visualized by fluorescence due to uptake of DMA containing pcDNA-toto-3.
DMA-pIL15 increased proliferation and the cytotoxic effect of lymphocytes
CT26 cells were transfected with DMA, DMA-pc3.1, or DMA-pIL15, or treated with GS as a negative control for 72 h. Supernatant from these treatment groups was collected for further culture of lymphocytes over 24 h; we then observed the activity of lymphocytes from each group. Expressions of TNF-α and IFN-γ in the serum by lymphocytes in the DMA-pIL15 group were found to be significantly higher than those of all the control groups (Figure 5A). We further analyzed these cultures of lymphocytes and found a greater number of CD4 and CD8 lymphocytes in the DMA-pIL15 group than in the control group (Supplementary Figure 2). Furthermore, the level of CD4/IFN-γ and CD8/IFN-γ was also found to be improved in the group treated with DMA-pIL15 (Supplementary Figure 3).
Lymphocytes stimulated by the supernatant of transgenic tumor cells and cytotoxicity of lymphocytes supernatant. When CT26 cells were transfected with DMA, DMA/pc3.1, or DMA/pIL15, or treated with GS as a negative control, for 72 h, the supernatant from different treatments were used to culture lymphocytes over the course of 24 h. (A) Proliferation of lymphocytes was detected by CCK8. Secretion of IFN-γ and TNF-α in the lymphocyte supernatant was detected by ELISA (mean ± SEM; n=3; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1). When CT26 cells were treated with GS or transfected with DMA, DMA/pc3.1, or DMA/pIL15 for 72 h, the supernatants from different treatments were used to culture lymphocytes for 24 h. Resulting supernatant was then used to culture CT26 cells for 24 h, and the activity of CT26 cells was assessed by using the CCK8 test (B) (mean ± SEM; n=5; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1). Apoptosis of CT26 cells was also detected by flow cytometry 24 h after adding the supernatant of lymphocytes. Results from flow cytometry are shown for (C) untreated CT26 cells, (D) the GS group, (E) the DMA group, (F) the DMA/pc3.1 group, and (G) the DMA/pIL15 group.
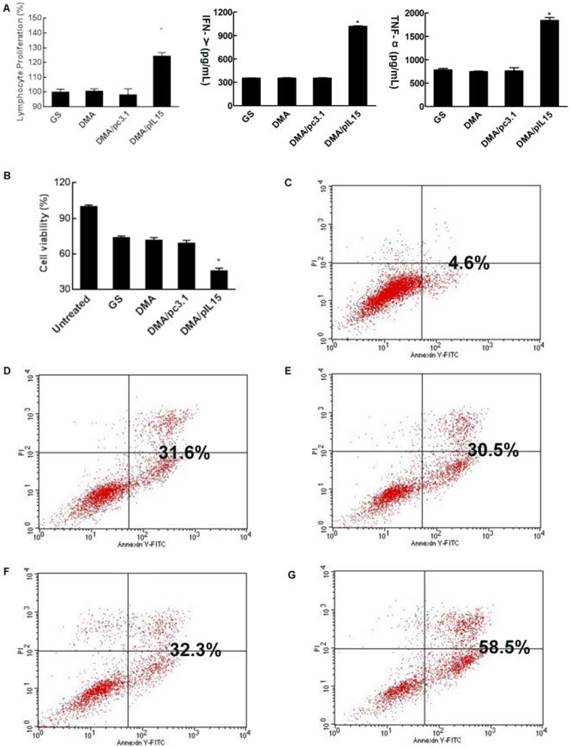
When lymphocytes were treated for 24 h, the supernatants were collected to culture CT26 cells for 24 h. We found that the CT26 cell activity of the DMA-pIL15 group was significantly inhibited in the DMA-pIL15 group when compared with the cell activity of the DMA or DMA-pc3.1 groups. We then analyzed the potential reason behind the growth inhibition of the CT26 cells and found evidence that the level of apoptosis in the DMA-pIL15 group was 58.5% higher than in all the control groups (p<0.05) (Figure 5B-G). The supernatant may have some killer factors from lymphocyte secretion, thus GS, DMA and DMA/pc3.1 also induced obvious cell apoptosis.
In vivo antitumor effect of DMA-pIL15
DMA loaded with pc3.1 and pIL-15 was applied to treat CT26 colon cancer in a Balb/c mouse model via intravenous injection. DMA-pIL15 treatment showed a dramatic anti-tumor effect compared to the other lipoplexes (DMA-pc3.1, DMA, and GS), and otherwise no significant differences were observed among the DMA-pc3.1-treated, DMA-treated, and control groups. DMA/pIL-15 significantly reduced the mass of tumors (with an average tumor mass of 0.52 ±0.09g) compared to the DMA (2.04 g±0.28), DMA/pc3.1 (1.84±0.19 g), and GS (1.97 g±0.23) groups. In addition, mice treated with DMA-pIL15 were also found to have the lowest body weights compared to those of control groups (Figure 6).
Anti-tumor effect of DMA/pIL15 in the subcutaneous model of colon cancer. (A) Tumor growth curves (mean ± SEM; n=6; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1). (B) Tumor mass (mean ± SEM; n=6; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1). (C) Body weight of mice (mean ± SEM; n=6; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1). (D) Images of tumors.
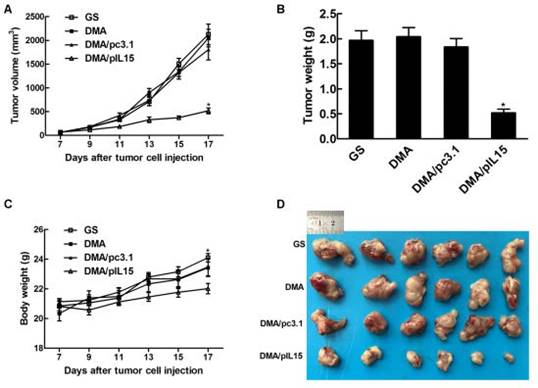
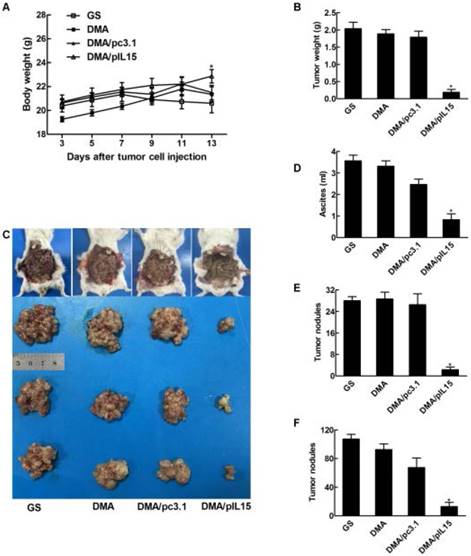
We also observed similar anti-tumor effects in a peritoneal model. Figure 7 shows that the average body weight of mice treated with DMA-pIL15 was significantly lower than that of DMA-pc3.1-treated mice, DMA-treated mice, or untreated tumor-bearing mice, which was caused by tumor weight. The mice treated with DMA-pIL15 had significantly fewer and smaller tumor nodules than mice in the control group. Furthermore, treatment with DMA-pIL15 also controlled the occurrence of ascites compared to that of the controls.
Anti-tumor effect of DMA/pIL15 in the peritoneal model of colon cancer. (A) Body weight of mice (mean ± SEM; n=6; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1). (B) Tumor mass (mean ± SEM; n=6; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1). (C) Images of mice and tumors. (D) Volume of ascites (mean ± SEM; n=6; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1). (E) Number of tumor nodules (≥3 mm) in different groups (mean ± SEM; n=6; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1). (F) Number of tumor nodules (<3 mm) in different groups (mean ± SEM; n=6; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1).
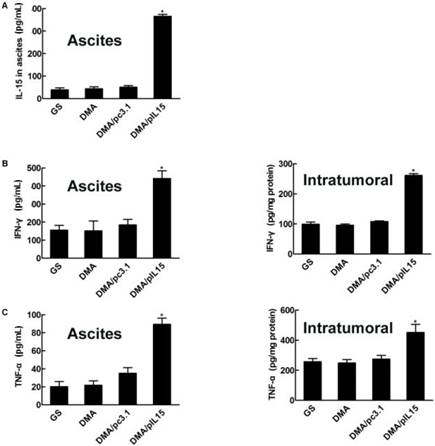
DMA-pIL15 treatment increased the expression of IL-15 and secretion of IFN-γ, TNF-α, and IL12
We then explored the potential anti-tumor role of DMA-pIL15 and measured expression of IL-15 by ELISA. After two rounds of treatment, ascites and tumors were harvested. We found that mice treated with DMA-pIL15 had significantly higher expression of IL-15 than control groups treated with DMA or DMA-pc3.1, and this held true in both models—subcutaneous and intraperitoneal. Anti-tumor effects of DMA-pIL15 could also be explained by the marked increase in expression of IFN-γ, TNF-α, and IL12 in ascites and tumor biopsies after the treatment was completed with DMA-pIL15 (Figure 8).
Expression of IL-15, IFN-γ, TNF-α, and IL-12 in vivo. (A) Ascites were collected after the final treatment, and levels of IL-15 expression were measured in the four groups (GS, DMA, DMA/pc3.1, and DMA/pIL15) of the peritoneal model (mean ± SEM; n=3; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1). Tumor tissue of the subcutaneous model and ascites of the peritoneal model (GS, DMA, DMA/pc3.1 and DMA/pIL15) were collected after the final treatment, and expression of (B) IFN-γ, (C) TNF-α, and (D) IL-12 were measured in the tumor tissue and ascites by ELISA. (For (B-D), mean ± SEM; n=3; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1).
Induction of lymphocytes
Herein, we hypothesized that the antitumor effects induced by IL-15 were due to T cell activation. The effect of DMA-pIL15 on spleen cells was investigated by flow cytometry analysis (Figure 9A) and standard 51Cr-release assay. Spleen cell-mediated cytotoxicity was examined via a 51Cr release assay to determine levels of T cell activation. Results show that treatment with DMA/pIL15 increased spleen cell-mediated cytotoxicity (Figure 9B). At effector to target (E/T) ratios of 200/1 or 50/1, the splenocytes from mice treated with DMA/IL15 showed elevated cytotoxic activity relative to the control groups (p<0.05; Figure 9B). Because lymphocytes are mixed cells, the splenic lymphocytes at E/T=200 showed a low CTL efficacy of ~20%, which was caused by the low percent of the specific effector cells.
Proliferation of lymphocytes and increase in the numbers of cytotoxic T cells in the peritoneal model. Splenic cells from the peritoneal model (GS, DMA, DMA/pc3.1, and DMA/pIL15) were collected after the final treatment. (A) Expression of IL-15 promoted the proliferation of CD8+ T lymphocytes and CD4+ T lymphocytes. (B) Splenic lymphocytes were tested against CT26 cells at different E:T ratios by a standard 4 h 51Cr release assay (mean ± SEM; n=3; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1).
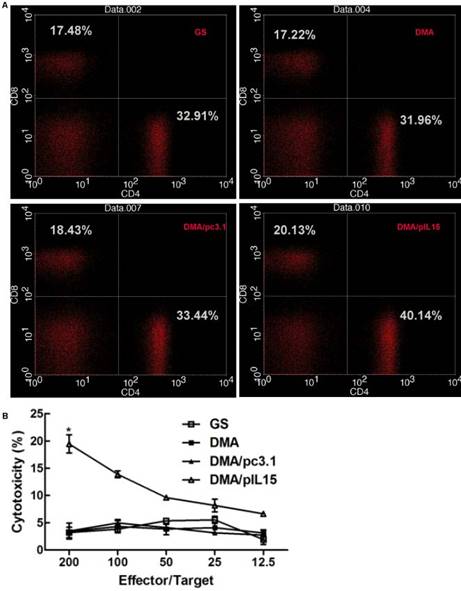
When examining spleen IFN-γ+ cytotoxic T cells of subtypes CD4+ and CD8+, DMA/pIL15-treated mice showed increased CD4+ T cell levels (0.59%) and CD8+ T cell levels (0.41%) relative to the control group (p<0.05; (Supplementary Figure 4). These results suggest that increased T cell activation and expansion of IFN-γ+ cytotoxic T cells were induced by the secretion of IL-15 in tumor cells treated with DMA/pIL15, thus generating the observed antitumor effects.
DMA-pIL15 induces cancer cell apoptosis, inhibits tumor cell proliferation, and suppresses tumor angiogenesis
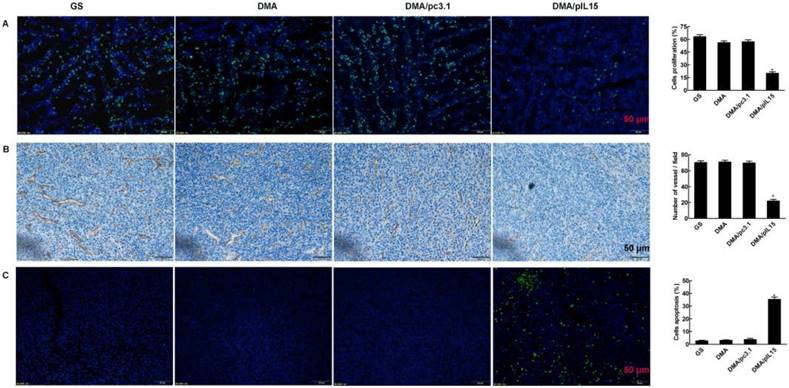
Tumor growth is considered a destruction of the balance between apoptosis and proliferation. To explore whether phenotypic changes occurred in the tumor tissues, the percentage of apoptotic and proliferating cells were examined (Figure 10). The TUNEL assay was used to determine severity of apoptosis. This assay functions by detecting early DNA fragmentation and allowed us to understand whether apoptosis contributes to the antitumor effects of treatment with DMA-pIL15 observed in vivo. In the DMA-pIL15-treated group, 37.1% TUNEL-positive cells were identified, exceeding that of the DMA/pc3.1-treated group. To assess levels of tumor cell proliferation, Ki67 staining was performed. The results showed fewer proliferating cells in tumor tissues treated with DMA-pIL15 than in those treated with DMA-pc3.1 or GS. Moreover, higher numbers of necrotic tumor cells were observed in DMA-pIL15-treated tumor tissues following H&E staining. In addition, the DMA-pIL15 group also demonstrated anti-angiogenesis effects when compared to the DMA-pc3.1, DMA, and GS groups by CD31 staining. Taken together, these evidences suggest that DMA-pIL15 can successfully deliver pIL15 to tumor cells and generate antitumor effects by activating immune cells, inducing tumor cytotoxicity, activating apoptotic mechanisms, and inhibiting tumor cell proliferation and vascular formation.
Detection of cell proliferation, tumor angiogenesis, and cell apoptosis. (A) Cell proliferation was assessed by counting the number of Ki67-positive cells in the field, and less cell proliferation was observed in the DMA/pIL15 group compared to the other groups (mean ± SEM; five high power fields per slide; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1). (B) Angiogenesis was assessed by counting the number of CD31-positive vessels in the field, and reduced angiogenesis was observed in the DMA/pIL15 group compared to the other groups (mean ± SEM; five high power fields per slide; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1). (C) Cell apoptosis was assessed by counting the number of TUNEL-positive cells in the field, and a greater level of apoptosis was observed in the DMA/pIL15 group compared to the other groups (mean ± SEM; five high power fields per slide; *, p < 0.01 DMA/pIL15 versus GS, DMA, DMA/pc3.1).
Assessment of the safety and toxicity of DMA-pIL15
To examine potential toxicity of DMA-pIL15, mouse organs (heart, liver, spleen, lungs, and kidneys) were harvested and stained with H&E for histopathological analysis. Vital organ sections from the DMA-pIL15 group all showed normal histological morphology, and no toxicity was found. In addition, no obvious toxicities were observed in the mice, as determined by hematological index (Supplementary Figures 5-7).
Discussion
Cancer gene therapy, which is the treatment of cancer by employing antisense, small interfering RNA, or other DNA at the tumor site, has attracted considerable attention worldwide [19]. IL-15, a cell growth factor that regulates lymphocyte function and homeostasis, demonstrates strong immunostimulatory activities that make it an exciting candidate for cancer therapy [20]. Unfortunately, the recombinant IL-15 protein used in clinical trials undergoes rapid renal clearance and has a short plasma half-life, diminishing its antitumor effects [21, 22]. Furthermore, systemic administration of IL-15 can increase the risk of potential toxic side effects, including the induction of autoimmunity [4]. Possible solutions to lengthen the time window of IL-15 bioavailability or to administer IL-15 locally were found through gene therapy approaches such as transferring the gene of interest as naked DNA or transfecting it into immune or tumor cells through lipofection, electroporation, or viral transduction. These approaches are currently being investigated as new methods for IL-15 combination therapies. Gene delivery systems help to enhance the specific delivery and expression of therapeutic genes at the site of tumor development, thus improving the efficacy of gene therapy. One aim of the present study was to generate a novel vector that overcomes the barrier of low transfection efficiency of liposome vector systems for IL-15 gene therapy.
In this study, we showed that the DMA-based lipoplex significantly increased gene transfection efficiency of GFP in CT26 colon cells in vitro as detected by flow cytometry and imaging. Treatment of tumor-bearing mice with DMA/pIL-15 significantly inhibited tumor growth, prolonged survival, and reduced tumor burden from the formation of ascites. Intensively increased expression of IL-15 was found in tumor tissue of mice treated with DMA/pIL-15 compared to other groups, a potential benefit from using DMA as a carrier. High levels of IL-15 induced IFN-γ and TNF-α secretion at the tumor site and in peritoneal fluid. Anti-tumor effect was achieved by induction of tumor cell apoptosis, inhibition of tumor cell proliferation, and inhibition of angiogenesis, as detected by immunohistochemical staining. Moreover, the preliminary safety of DMA/pIL-15 was assessed, and no abnormalities were observed in vital organs of all targeted gene-treated groups of mice. Therefore, we conclude that the DMA-based gene delivery system offers promise as an anticancer agent, given its effectiveness in inducing gene expression and targeting therapy as well as its positive safety profile.
Interleukin-15, as an immune modulator, has the potential to both modulate immune suppression and promote immune activation, rendering it a promising therapeutic option. However, substantial dose-dependent toxicity is induced by the initial clinical use of recombinant IL-15 protein, whereas no signs of IL-15 toxicity were observed in our DMA-based gene therapy protocol, albeit in mouse models. Published reports using recombinant IL-15 protein only yielded low levels of IL-15 bioavailability, and the need for high doses of IL-15 to realize the anti-cancer immunity could cause toxicity locally or systemically [24]. However, in this work, we showed a relatively high transfection efficiency of IL-15 with DMA. Furthermore, previous studies in mice demonstrated moderate anti-tumor activity of murine IL-15 when it is administered alone, and this can only be improved in combination with other cytokines or therapy [13]. In our study, incorporation of pIL-15 by DMA resulted in higher intracellular uptake of IL-15 DNA, causing higher levels of transcription and higher levels of secreted IL-15, and improving the anti-tumor efficiency of cytokine gene transfer.
Through our experiments, we observed that IL-15 induces high-level expression of TNF-α and IFN-γ at the tumor site. These results agree with data from other groups demonstrating that the anti-tumor effects of IL15 are executed by enhancing NK cells' cytotoxicity, thereby increasing the production of cytokines such as TNF-α and IFN-γ [25, 26]. Previous reports have also indicated that IL-15 has anti-angiogenic activity. This effect is exerted in an indirect manner by triggering the secretion of IFN-γ, which in turn induces the anti-angiogenic effects [27]. In addition, TNF-α serves as a multi-functional antitumor cytokine playing a key role in cell apoptosis, inflammation, and host immunity [5]. Taken together, information from this and previous studies help explain the increased severity of apoptosis [29-31], reduced tumor cell proliferation, and reduced angiogenesis observed in the tumor tissues treated with DMA/pIL-15 in our study. Our data provide additional evidence that IL-15 gene delivery directly into tumors can alter the local cytokine environment by induction of IFN-γ and TNF-α expression, suggesting that IL-15-mediated tumor regression may partially depend on these cytokines [32, 33]. Furthermore, we also found that DMA/pIL-15 gene therapy heightens host adaptive immunity by increasing the numbers of CD4+ and CD8+ lymphocytes and activating NK cells. Similar to earlier descriptions [34-37], IL-15 proved to be a stimulator both of CD8+ T cells of the adaptive immune system and NK cells of the innate immune system, which proves immunotherapy has a good prospect in tumor therapy [38]. Overall, in this context, we showed that DMA-IL-15 gene therapy enhances both apoptosis of tumor cells as well as the host immune response, leading to a significant reduction in tumor burden and increased survival in both subcutaneous and intra-peritoneal mouse models of colon cancer.
In summary, we developed novel and safe DMA-based lipoplexes loaded with pIL-15 (DMA/pIL-15). The lipoplexes were composed of DMA as the vector carrier and pIL-15 as the therapeutic gene. DMA liposomes enhanced the uptake of pIL-15 by CT26 colon cancer cells, and treatment with DMA/pIL-15 lead to high levels of IL-15 both in tumor cells in vitro and in tumor tissues in vivo. This lead to an efficient anti-tumor effect mediated by the induction of cancer cell apoptosis, inhibition of tumor cell proliferation, and suppression of tumor angiogenesis through improved lymphocyte proliferation and secretion of TNF-α and IFN-γ. Survival of tumor-bearing mice treated with DMA/pIL-15 was significantly prolonged. Preliminary safety evaluation implies that DMA/pIL-15 is a safe and efficient therapeutic agent in gene therapy for breast cancer. Therefore, the DMA-based lipoplexes leading to therapeutic gene expression of IL-15 serve as a promising candidate for the clinical treatment of CRC.
Methods
Materials
Materials were purchased from standard sources: 1,2-dioleoyl-3-trimethylammonium-propane (chloride salt) (DOTAP) (Avanti Polar Lipids Inc., Alabaster, AL, USA); 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) (Sigma, USA); Dulbecco's Modified Eagle's Medium (DMEM) and fetal bovine serum (FBS) (Gibco BRL, USA); methanol and acetic acid (HPLC grade) (Fisher Scientific, UK); and dimethyl sulfoxide (DMSO) and acetone (KeLong Chemicals, China). Antibodies used include: rat anti-mouse CD31 polyclonal antibody (BD PharmingenTM, USA), CD4 antibody (BD PharmingenTM, USA), CD8 antibody (BD PharmingenTM, USA), CD69 antibody (BD PharmingenTM, USA), IFN gamma antibody (BD PharmingenTM, USA), rabbit anti-mouse Ki67 antibody (Abcam, USA), and rhodamine-conjugated secondary antibody (Abcam, USA). Toto-3 was purchased from Thermo Fisher Scientific. Mouse TNF alpha ELISA kit, mouse IFN gamma ELISA kit and mouse IL-15/IL-15R Complex ELISA kit were purchased from eBioscience.
MPEG(2000)-PLA(4000) diblock copolymer with a designed molecular weight of 3,000 Da was synthesized by opening of the l-lactide ring, initiated by MPEG. MPEG (5.0 g) was melted in a 50-mL flask following the addition of anhydrous l-lactide (10 g) and Sn(Oct)2 (1 mL) under nitrogen. The reactant mixture was maintained at 125 °C for 24 h. The crude product was dissolved in tetrahydrofuran and then purified by precipitation in ice-cold diethyl ether followed by filtration. This process was performed in triplicate, and the resultant product was vacuum dried at ambient temperature. The number average molecular weight (Mn) of MPEG-PLA copolymer was 6,010 Da (data not shown). MPEG (with a molecular weight of 2,000 Da) (Sigma-Aldrich Co.) was dried in a one-necked flask under vacuum and stirred at 105 °C for 90 min before use.
Molecular dynamics
Simulated annealing was performed on the optimized structures by using MD so as to obtain a lower energy minimum. Solvent effect was considered implicitly by CHARMM27 [39] in the process of MD simulation. The MD simulations include heating from 0 K to 600 K, simulating at 600 K, cooling from 600 K to 300 K, and running at 300 K. At each stage, the simulated time was set to 100 ps.
Preparation of DOTAP/MPEG-PLA (DMA) and DMA-pIL15
MPEG-PLA (90 mg) and DOTAP (10 mg) were dissolved in acetone (2 mL). The mixture was then placed into a round-bottom flask, and acetone was removed by way of a water bath (55 ℃) under negative pressure conditions (20 min). At last, a 5% glucose solution (GS) (5 mL) was added to prepare the DMA gene carrier. pIL15 was added into DMA solution, and incubated for 20 min. DMA-pIL15 was obtained.
Characterization of DMA-pIL15
The morphological characteristics of DMA-pIL15 were observed using transmission electron microscopy (TEM; FEI Tecnai G2 F20, Hillsboro, OR, US). Prior to analysis, samples were diluted with distilled water, placed on a copper grid, stained with molybdophosphoric acid (1 min), and allowed to dry at room temperature. Mean particle size and zeta potential of DMA were determined by a Zetasizer NanoZS ZEN 3600 (Malvern Instruments, Ltd., Malvern, Worcestershire, U.K.).
Agarose gel electrophoresis of naked plasmid DNA and DMA-IL15 complexes
After DMA-IL15 was prepared, agarose gel electrophoresis was conducted in pH 7.4 TAE buffer containing Gold View to stain nucleic acids. Briefly, DMA was mixed with IL-15 in increasing N/P ratios (0:1, 1.25:1, 2.5:1 and 5:1) to form complexes. Gel retardation assays were conducted on a 1% agarose gel (Invitrogen Corp., Carlsbad, CA, U.S.) in tris-acetate running buffer containing Gold View (120 V for 20 min). Results were documented using a standard imaging system (Bio-Rad Laboratories, Hercules, CA, U.S.).
Cell culture and in vitro gene transfer and expression
The murine colon cancer cell line CT26 was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and were cultured using standard sterile procedure in RPMI 1640 medium (Gibco-BRL, Rockville, IN, USA) containing 10% FBS (Sigma Chemical Co., St. Louis, MO), 100 U/mL penicillin, and 100 μg/mL streptomycin. Cells were maintained with humidity and 5% CO2 at 37 °C.
CT26 cells were seeded on a Costar six-well plate (Corning Incorporated, Corning, NY, USA) at a density of 1.5×105 cells/well in 2 mL of complete RPMI-1640 culture medium. After 24 h of incubation, media was replaced with 800 μL serum-free RPMI-1640 medium per well. DMA/pGFP, DMA/pc3.1, or DMA/pIL15 in a final volume of 200 μL and containing 4 μg plasmid DNA were subsequently added to designated wells and allowed to incubate for 5 h before replacement with RPMI-1640 culture media. GFP-transfected cells were observed under a fluorescent microscope (Olympus IX73, U-HPLGPS, Olympus Corporation, Shinjuku, Tokyo, Japan) following an additional 24-h incubation, and transfection efficiency was determined via FACS flow cytometry (BD Biosciences, San Jose, CA, USA).
In cells transfected with DMA, DMA/pc3.1, DMA/pIL15 or otherwise treated with GS as a negative control, IL-15 expression levels were determined after 72 h using a mIL-15 ELISA kit (eBioscience Inc., San Diego, CA, U.S.) according to the manufacturer's instructions.
Imaging in vivo
In vivo colon cancer mouse models were established by subcutaneous injection of CT26 cells (1×106 cells in 0.2 mL of serum-free RPMI-1640). After two weeks, mice from the experimental group (DMA/pDNA) were intravenously injected with liposomal plasmid DNA (labeled with toto-3; 20 μg) in 200 μL of GS, while the control group received the same volume of GS with the same amount of DMA-pc3.1. Images were taken on a IVIS Lumina imaging system (Caliper, USA).
In vitro lymphocyte stimulation test
CT26 cells were cultured in a six-well plate, at a concentration of 2×105 cells per well. After CT26 cells were transfected with DMA, DMA-pc3.1, DMA-IL15, or treated with GS as a negative control, and cultured for 72 h, supernatant from each group was collected. These were further applied to culture spleen-derived lymphocytes, and lymphocyte proliferation after 24 h in culture was tested using the CCK-8 method. Furthermore, ELISA assays were used to measure the expression of IFN-γ and TNF-α by lymphocytes in each treatment group at 24 h. Expression of IFN-γ was determined by IFN-γ-PE (BD Biosciences) staining for 30 min at 4 °C after samples were fixed and permeabilized with paraformaldehyde and Triton-X 100. In addition, proliferation of lymphocytes from each group after 24 h in culture was also tested by cytometry (BD Biosciences, San Jose, CA, USA) using carboxyfluorescein succinimidyl ester (CFSE) staining. Lymphocytes from each group were stained for T cell markers CD19-APC (BD Biosciences), CD4-APC (BD Biosciences), and CD8-APC (BD Biosciences), as well.
In vitro lymphocyte cytotoxicity test
CT26 cells were transfected with DMA, DMA-pc3.1 or DMA-IL15, or treated with GS as a negative control, for 72 h, and supernatants were collected and used to culture lymphocytes. After culturing lymphocytes for 24 h, supernatants were collected to further culture CT26 cells. Briefly, cells were seeded on 96-well plates (Corning Inc., NY, US) at a density of 3,000 cells per well with the collected supernatants. After the cells were cultured following standard protocols, 20 μL of CCK-8 stock solution was added to each well, and the cells were further cultured at 37 °C for an additional 4 h.
Absorbance values at 570 nm from each well, proportional to the number of viable cells in that well, were read on a Multiskan MK3 microplate reader (Thermo Fisher Scientifc Inc., Waltham, MA, US). Untreated cells were used as a control, and cell viability (%) was determined relative to the control by calculating Atreated/Acontrol×100%. We also measured apoptosis of cancer cells by flow cytometry.
Murine colon cancer model establishment
CT26 colon carcinoma cells were obtained from American Type Culture Collection (Manassas, VA, US), cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS), and maintained at 37 °C with 5% CO2. All animal experiments were performed in accordance with guidelines of the Animal Care Committee of Sichuan University (Chengdu, China) and were approved. Female BALB/c mice (6-8 weeks old) were purchased from Vital River (Beijing, China) and housed in a specific-pathogen-free (SPF) environment at stable room temperature and humidity and were handled in strict accordance with proper, standard animal practices.
In vivo mouse models of colon cancer were established by subcutaneous (s.c.) injection of CT26 cells (1×106 cells in 0.2 mL of serum-free RPMI-1640) or by intraperitoneal (i.p.) injection of CT26 cells (~2×105 cells in 0.2 mL of serum-free RPMI-1640). Mice were randomly allocated into four groups (GS, DMA, DMA-pc3.1, or DMA/pIL15). To assess tumor growth, treatment began seven days (in the subcutaneous model) or three days (in the intraperitoneal model) after inoculation. Experimental group mice (DMA/pIL15) were given liposomal plasmid DNA (10μg) intravenously in 200 μL GS every two days for five total injections, while the other groups received the same amount of DMA or DMA-pc3.1 in the same volume of GS or received GS only as a negative control.
Mice were monitored daily for adverse effects. At the time of sacrifice (48 h after the final dose), tumor tissues, spleen, and other vital organs were harvested, and the total mouse mass, tumor volume, tumor mass, and volume of ascites were recorded. Specifically, tumor nodules were separated from the peritoneal cavity, cleared with 0.9% normal saline, and counted by two researchers using a double-blind method. Finally, a third researcher calculated and recorded the number of tumor nodules in each group. The level of secreted IL-15, IFN-γ, and TNF-α in the tumor tissues and peritoneal fluid were determined using commercially available ELISA kits (eBioscience, Inc., San Diego, CA, U.S.). Tumor tissues and vital organs of mice were also harvested for further analysis.
Flow cytometry analysis and spleen cell-mediated cytotoxicity assay
Spleens from the peritoneal mouse models from the GS, DMA/pc3.1, DMA and DMA/pIL15 groups were harvested, ground with a pestle, and filtrated using a cell strainer (BD Biosciences, San Jose, CA, US) to form separate single-cell suspensions. Cells were stained for T cell markers CD4-APC (BD Biosciences) and CD8-FITC (BD Biosciences). The expression of IFN-γ was determined by IFN-γ-PE (BD Biosciences) staining for 30 min at 4 °C after samples were fixed and permeabilized with paraformaldehyde and Triton-X 100.
A 4 h 51Cr release assay was performed as previously described by other reports [7]. In the peritoneal tumor model, splenocytes from the four groups (GS, DMA, DMA-pc3.1 or DMA-pIL15) were harvested after the mice received two rounds of treatment. Briefly, these splenic lymphocytes, the effector cells, were treated with ammonium-chloride-potassium lysing buffer to deplete erythrocytes. CT26 cells (~1×106), the target cells, were labeled with 100 μCi 51Cr for 1 h at 37 °C and then washed and resuspended at a concentration of 1×105 cells/mL. A total of 200 μL of effector cells and 51Cr-labeled target cells were assigned at different effector:target ratios (200:1, 100:1, 50:1, 25:1, and 12.5:1) to each well of 96-well plates and incubated for 4 h at 37 °C. Supernatant (100 μL) from each sample was then harvested, and the activity was calculated: % cytotoxicity = (experimental release - spontaneous release)/(maximum release - spontaneous release)×100%.
Proliferation, apoptosis, and microvessel density assays of tumor tissues
Tumor tissue proliferation was immunohistochemically analyzed using a rabbit anti-human Ki67 antibody (Novus Biologicals, Littleton, CO, US) with a streptavidin-biotin detection method. Ki67 expression was quantified by counting the number of positive cells in ten randomly selected fields at a 200× magnification. Tumor apoptotic levels were determined using a terminal deoxynucleotidyl transferase-mediated nick end labeling (TUNEL) immunofluorescence kit (Promega, Madison, WI, US) according to the manufacturer's instructions. Quantification of microvessel density was achieved by counting the number of microvessels in five random fields at 200× magnification. A single microvessel was defined as a discrete cluster or single cell stained positive for CD31. All the sections were observed and digitally photographed under a DM 2500 fluorescence microscope (Leica Microsystems CMS GmbH, Wetzlar, Germany). All above sections were observed or counted by two investigators or pathologists in a blinded fashion.
Toxicity assessment
To evaluate the potential toxicity associated with DMA-pIL15 treatment, vital organs tissues (heart, liver, spleen, lung, and kidney) of the treated mice were harvested, fixed in 4% paraformaldehyde solution, embedded in paraffin, and sectioned at 5 μm. Sections were hydrated and stained with hematoxylin and eosin (H&E) for histomorphometric analysis and observed by two pathologists in a blinded manner. Furthermore, blood and serum were also harvested for routine blood tests as well as hepatic and renal function tests. Blood cell counts were performed using an automated veterinary haematological analyser with a pre-programmed murine calibration mode (Hemavet 950FS; Drew Scientific, Waterbury, CT). Hepatic and renal function tests were performed in a Hitachi Automatic Analyzer (Boehringer, Indianapolis, IN).
Statistical analysis
All data were analyzed using GRAPHPAD PRISM software (GraphPad, San Diego, CA). Data from multiple groups were analyzed using ANOVA, and multiple comparisons between the groups were performed using the Newman-Keuls method after ANOVA. Survival data were plotted using Kaplan-Meier curves and analyzed using the log-rank test. Tumor volumes were calculated as (smaller diameter)2(larger diameter)×0.52. Statistical significances of observed differences in tumor volume and vessel density were analyzed using the Student's t test. All values were presented as the mean ± the standard error of measurement. A p value of <0.05 was considered to be statistically significant for all experiments.
Abbreviations
CRC: colorectal cancer; CCK-8: cell counting kit 8; CTL: cytotoxic lymphocyte; DMA: DOTAP and MPEG-PLA; DNA: deoxyribonucleic acid; IL15: Interleukin-15; ELISA: enzyme linked immunosorbent assay; GS: glucose solution; H&E: hematoxylin and eosin; IFN-γ: Interferon-γ; NK: natural killer; RNA: ribonucleic acid; SPF: specific-pathogen-free; TUNEL: transferase mediated deoxyuridine triphosphate biotin nick end labeling; TEM: transmission electron microscopy; TNF-α: tumor necrosis factor-α.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC81502165, NSFC81602075) and the Sichuan University Outstanding Young Scholars Research Fund (2016SCU04A04).
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL. et al. Global cancer statistics. CA Cancer J Clin. 2015;65:87-108
2. Tsikitis VL, Larson DW, Huebner M. et al. Predictors of recurrence free survival for patients with stage II and III colon cancer. BMC Cancer. 2014;14:336
3. Mongan JP, Fadul CE, Cole BF. et al. Brain metastases from colorectal cancer: risk factors, incidence, and the possible role of chemokines. Clin Colorectal Cancer. 2009;8:100-5
4. Shah MA, Renfro LA, Allegra CJ. et al. Impact of patient factors on recurrence risk and time dependency of oxaliplatin benefit in patients with colon cancer: analysis from modern-era adjuvant studies in the adjuvant colon cancer end points (ACCENT) database. J Clin Oncol. 2016;34:843-53
5. Sankpal UT, Nagaraju GP, Gottipolu SR. et al. Combination of tolfenamic acid and curcumin induces colon cancer cell growth inhibition through modulating specific transcription factors and reactive oxygen species. Oncotarget. 2016;7:3186-200
6. Liu Y, Zhang X, Han C. et al. TP53 loss creates therapeutic vulnerability in colorectal cancer. Nature. 2015;520:697-701
7. Liu X, Gao X, Zheng S. et al. Modified nanoparticle mediated IL-12 immunogene therapy for colon cancer. Nanomedicine. 2017;13:1993-2004
8. Yin H, Kanasty RL, Eltoukhy AA. et al. Non-viral vectors for gene-based therapy. Nat Rev Genet. 2014;15:541-55
9. Naldini L. Gene therapy returns to centre stage. Nature. 2015;526:351-60
10. Ginn SL, Alexander IE, Edelstein ML. et al. Gene therapy clinical trials worldwide to 2012 - an update. J Gene Med. 2013;15:65-77
11. Boleij A, Tjalsma H. Gut bacteria in health and disease: a survey on the interface between intestinal microbiology and colorectal cancer. Biol Rev Camb Philos Soc. 2012;87:701-30
12. Fridman WH, Galon J, Dieu-Nosjean MC. et al. Immune infiltration in human cancer: prognostic significance and disease control. Curr Top Microbiol Immunol. 2011;344:1-24
13. Zhang M, Yao Z, Dubois S. et al. Interleukin-15 combined with an anti-CD40 antibody provides enhanced therapeutic efficacy for murine models of colon cancer. Proc Natl Acad Sci USA. 2009;106:7513-8
14. Van den Bergh JM, Van Tendeloo VF, Smits EL. Interleukin-15: new kid on the block for antitumor combination therapy. Cytokine Growth Factor Rev. 2015;26:15-24
15. Grabstein KH, Eisenman J, Shanebeck K. et al. Cloning of a T cell growth factor that interacts with the beta chain of the interleukin-2 receptor. Science. 1994;264:965-8
16. Cheever MA. Twelve immunotherapy drugs that could cure cancers. Immunol Rev. 2008;222:357-68
17. Yu P, Steel JC, Zhang M. et al. Simultaneous blockade of multiple immune system inhibitory checkpoints enhances antitumor activity mediated by interleukin-15 in a murine metastatic colon carcinoma model. Clin Cancer Res. 2010;16:6019-28
18. Di Scala M, Gil-Farina I, Olague C. et al. Identification of IFN-gamma-producing T cells as the main mediators of the side effects associated to mouse interleukin-15 sustained exposure. Oncotarget. 2016;7:49008-49026
19. Hayes GM, Carpenito C, Davis PD. et al. Alternative splicing as a novel of means of regulating the expression of therapeutic genes. Cancer Gene Ther. 2002;9:133-41
20. Chen XL, Bobbala D, Cepero Donates Y. et al. IL-15 trans-presentation regulates homeostasis of CD4(+) T lymphocytes. Cell Mol Immuno. 2014;11:387-97
21. Ochoa MC, Fioravanti J, Rodriguez I. et al. Antitumor immunotherapeutic and toxic properties of an HDL-conjugated chimeric IL-15 fusion protein. Cancer Res. 2013;73:139-49
22. Kishida T, Asada H, Itokawa Y. et al. Interleukin (IL)-21 and IL-15 genetic transfer synergistically augments therapeutic antitumor immunity and promotes regression of metastatic lymphoma. Mol Ther. 2003;8:552-8
23. Jakobisiak M, Golab J, Lasek W. Interleukin 15 as a promising candidate for tumor immunotherapy. Cytokine Growth Factor Rev. 2011;22:99-108
24. Chang CM, Lo CH, Shih YM. et al. Treatment of hepatocellular carcinoma with adeno-associated virus encoding interleukin-15 superagonist. Hum Gene Ther. 2010;21:611-21
25. Berger C, Berger M, Hackman RC. et al. Safety and immunologic effects of IL-15 administration in nonhuman primates. Blood. 2009;114:2417-26
26. Liu RB, Engels B, Arina A. et al. Densely granulated murine NK cells eradicate large solid tumors. Cancer Res. 2012;72:1964-74
27. Koka R, Burkett P, Chien M. et al. Cutting edge: murine dendritic cells require IL-15R alpha to prime NK cells. J Immunol. 2004;173:3594-8
28. Waters JP, Pober JS, Bradley JR. Tumour necrosis factor and cancer. J Pathol. 2013;230:241-8
29. Nastala CL, Edington HD, McKinney TG. et al. Recombinant IL-12 administration induces tumor regression in association with IFN-gamma production. J Immunol. 1994;153:1697-706
30. Brunda MJ, Luistro L, Hendrzak JA. et al. Role of interferon-gamma in mediating the antitumor efficacy of interleukin-12. J Immunother Emphasis Tumor Immunol. 1995;17:71-7
31. Tannenbaum CS, Wicker N, Armstrong D. et al. Cytokine and chemokine expression in tumors of mice receiving systemic therapy with IL-12. J Immunol. 1996;156:693-9
32. Kobayashi H, Dubois S, Sato N. et al. Role of trans-cellular IL-15 presentation in the activation of NK cell-mediated killing, which leads to enhanced tumor immunosurveillance. Blood. 2005;105:721-7
33. Evans R, Fuller JA, Christianson G. et al. IL-15 mediates anti-tumor effects after cyclophosphamide injection of tumor-bearing mice and enhances adoptive immunotherapy: the potential role of NK cell subpopulations. Cell Immunol. 1997;179:66-73
34. Diab A, Cohen AD, Alpdogan O. et al. IL-15: targeting CD8+ T cells for immunotherapy. Cytotherapy. 2005;7:23-35
35. Carson WE, Giri JG, Lindemann MJ. et al. Interleukin (IL) 15 is a novel cytokine that activates human natural killer cells via components of the IL-2 receptor. J Exp Med. 1994;180:1395-403
36. Waldmann TA, Tagaya Y. The multifaceted regulation of interleukin-15 expression and the role of this cytokine in NK cell differentiation and host response to intracellular pathogens. Annu Rev Immunol. 1999;17:19-49
37. Carson WE, Fehniger TA, Haldar S. et al. A potential role for interleukin-15 in the regulation of human natural killer cell survival. J Clin Invest. 1997;99:937-43
38. Kong M, Tang J, Qiao Q. et al. Biodegradable Hollow Mesoporous Silica Nanoparticles for Regulating Tumor Microenvironment and Enhancing Antitumor Efficiency. Theranostics. 2017;7:3276-3292
39. Brooks BR, Brooks CL, MacKerell AD. et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545-1614
Author contact
![]() Corresponding author: Xiang Gao. Tel: +86 28 8542 2136; Fax +86 28 8550 2796; Email: xianggaoedu.cn
Corresponding author: Xiang Gao. Tel: +86 28 8542 2136; Fax +86 28 8550 2796; Email: xianggaoedu.cn