Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(13):3474-3489. doi:10.7150/thno.23804 This issue Cite
Research Paper
Drug-delivering-drug platform-mediated potent protein therapeutics via a non-endo-lysosomal route
Xiaofei Xin1, Chao Teng1, Xiaoqing Du1, Yaiqi Lv1, Qingqing Xiao1, Yubing Wu1, Wei He1, ![]() , Lifang Yin1,2,
, Lifang Yin1,2, ![]()
1. Department of Pharmaceutics, School of Pharmacy, China Pharmaceutical University, Nanjing, 210009, PR China.
2. Key Laboratory of Druggability of Biopharmaceutics, China Pharmaceutical University, Nanjing, 210009, PR China.
Received 2017-11-12; Accepted 2018-4-21; Published 2018-6-6
Citation:
Xin X, Teng C, Du X, Lv Y, Xiao Q, Wu Y, He W, Yin L. Drug-delivering-drug platform-mediated potent protein therapeutics via a non-endo-lysosomal route. Theranostics 2018; 8(13):3474-3489. doi:10.7150/thno.23804. https://www.thno.org/v08p3474.htm
Other stylesAbstract

Protein therapeutics is playing an increasingly critical role in treatment of human diseases. However, current vectors are captured by the digestive endo-lysosomal system, which results in an extremely low fraction (<2%) of protein being released in the cytoplasm. This paper reports a drug-delivering-drug platform (HA-PNPplex, 200 nm) for potent intracellular delivery of protein and combined treatment of cancer.
Methods: The platform was prepared by loading functional protein on pure drug nanoparticles (PNPs) followed by hyaluronic acid coating and was characterized by dynamic light scattering, transmission electron microscopy, and gel electrophoresis. In vitro, cellular uptake, trafficking, and cytotoxicity were evaluated by flow cytometry and confocal laser microscopy. Protein expression was assayed by western blot. In vivo, blood circulation and biodistribution were studied using a fluorescence imaging system, antitumor efficacy was assessed in a caspase 3-deficient tumor model, and biocompatibility was determined by comparison of hemolytic activity and proinflammatory cytokines and tissue histology.
Results: HA-PNPplex delivered the functional protein, caspase 3, to cells via bypassing endo-lysosomes and raised the caspase-3 level 6.5-fold in caspase 3-deficient cells. Promoted tumor accumulation (1.5-fold) and penetration were exhibited, demonstrating a high tumor-targeting ability of HA-PNPplex. HA-PNPplex rendered a 7-fold increase in caspase 3 in tumor and allowed for a 100% tumor growth inhibition and >60% apoptosis, implying significant antitumor activities.
Conclusions: This platform gains cellular entry without entrapment in the endo-lysosomes and enables efficient intracellular protein delivery and resultant profound cancer treatment. This platform, with extremely high drug-loading, is a valuable platform for combined cancer therapy with small-molecule drugs and proteins. More importantly, this work offers a robust and safe approach for protein therapeutics and intracellular delivery of other functional peptides, as well as gene-based therapy.
Keywords: rod-like pure drug particles, caspase 3, caveolae-mediated endocytosis, intracellular protein delivery, endo-lysosomes
Introduction
Since insulin was commercially marketed in 1982, an increasing number of proteins and peptides, with greater than 200 products [1] in the past 3 decades, have been approved for treatment of diverse diseases including cancer, infection, diabetes, and inflammatory disease [2, 3]. Undoubtedly, protein therapeutics, such as protein replacement therapies and human antibodies [4, 5], play an essential role in treatment of human diseases. Moreover, protein-based therapy is considered safer than gene therapy due to the absence of random or permanent genetic alterations. Nonetheless, protein delivery is extremely difficult because proteins are susceptible to enzymatic degradation, have large size, short circulation half-lives and poor-membrane permeability [6, 7]. Nanomaterials, such as nanogels [8], cationic lipid particles [9], nanocapsules [10, 11], and virus-like particles [12], have been reported to improve protein delivery. Several vectors, such as PULsin (PUL) and BioPORTER, have been marketed for protein delivery [13, 14]. Indeed, these conventional nanoparticles enhance the therapy outcomes of proteins. However, intracellular protein delivery has remained a challenge. To obtain intracellular targeting by proteins, delivery into the cytoplasm is a prerequisite. Unfortunately, during internalization, these nanoparticles will be captured by the endo-lysosomal system where numerous digestive enzymes inhabit acidic conditions at pH of less than 4.5 [15-17]; therefore, dramatic degradation of proteins occurs, which results in <2% of proteins escaping into cytoplasm [10, 18, 19]. In addition, only when these nanoparticles are entrapped in a specific compartment that shares both early and late endosomal characteristics can escape occur in an exceedingly short duration [20]. Accordingly, escape from the endo-lysosomal system is a critical rate-limiting step for intracellular protein delivery; and hence, 100% escape of endosome-trapped proteins into cytoplasm has been claimed and is highly demanded [21].
Scheme 1
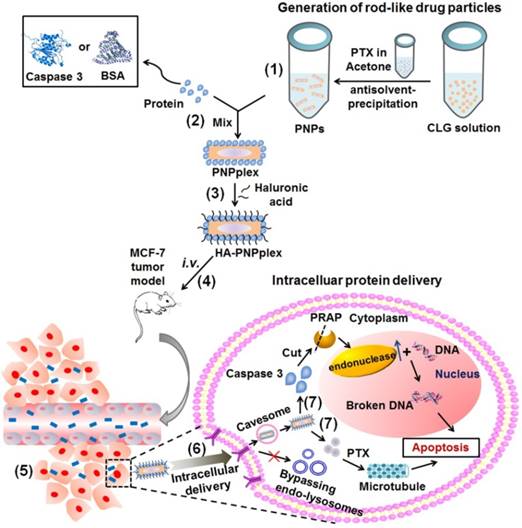
Design and proposed active mechanism for the intracellular protein delivery platform based on rod-like pure drug nanoparticles (PNPs) with cellular entry bypassing endo-lysosomes. (1) Preparation of PNPs via antisolvent-precipitation. (2) Proteins, such as caspase 3 or BSA, are loaded on the PNPs surface to prepare PNPplex through electrostatic interaction. (3) Hyaluronic acid (HA) is used to further coat PNPplex and form HA-PNPplex, aiming to shield the positive charge and target CD44 receptors. The proposed procedure in vivo: (4) HA-PNPplex are administered via intravenous injection (i.v.), (5) accumulate in the tumor site, (6) enter cancer cells through CD44-receptor mediation via the caveosome pathway without entrapment in endo-lysosomes, and finally (7) release protein and drug in cytoplasm for disease therapy.
In nature, living cells are capable of taking up certain materials without incorporation into endosomes, e.g., cellular uptake via the caveolar pathway [22]. Recently, we demonstrated that rod-like pure drug nanoparticles (PNPs) of paclitaxel (PTX) 160 nm in length entered cancer cells via caveolae-mediated pathway without entrapment in endo-lysosomes [23]. Additionally, these PNPs possess a significantly extended in vivo blood-circulation time and penetrate well inside the tumor [23, 24]. Importantly, using PNPs as vectors, miRNA, lethal-7a, was delivered to cells with high efficiency and safety, bypassing the endo-lysosomal system [25]. In this study, based on these previous findings, we hypothesized that PNPs can achieve potent intracellular delivery of functional proteins via a non-endo-lysosomal pathway for cancer treatment (Scheme 1). This drug-delivering-drug (DDD) platform for protein delivery consists of PNPs, functional protein, and hyaluronic acid (HA) and is designed as follows (Scheme 1): Protein is loaded on positively charged PNPs via electrostatic interaction, named PNPs/protein complexes (PNPplex); and subsequently, HA coats these PNPplex (named HA-PNPplex) to cover the charge and target CD44-receptors. Caspase 3 has been identified as a biomedically important enzyme and the dominant mediator of apoptosis in mammalian cells [26, 27] and, thus, was selected for the demonstration of therapeutic efficacy in a caspase 3-deficient MCF-7 tumor-bearing model [28, 29]. Meanwhile, fluorescence-labeled bovine serum albumin (BSA) was utilized as a model protein to examine the intracellular delivery of proteins. This work provides a robust platform for intracellular protein delivery and malignant cancer treatment.
Methods
Materials and cells
PTX (99% purity) was obtained from Yew Biotechnology Co., Ltd. (Jiangsu, China). Taxol (marketed product of PTX) was from Bristol-Myers Squibb (China) Investment Co., Ltd. (Shanghai, China). Beta-lactoglobulin (β-LG, No. L3908, 90% purity), BSA (A2153, 96% purity), fluorescein isothiocyanate isomer I (FITC, 98% purity), rhodamine B isothiocyanate (RITC, 98% purity), IR783 (90% purity), 3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazoliumbromide (MTT, 98% purity), and polyethylenimine (PEI, 408727, 25000 Da) were purchased from Sigma-Aldrich Co., Ltd. (St. Louis, MO, USA). Human recombinant caspase 3 was purchased from Cloud-Clone Corp. (USA). PULsin (PUL) was purchased from Polyplus-transfection® SA (New York. USA). HA was purchased from Shandong Furuida Pharmaceutical Co., Ltd. (6,600 Da, Shandong, China). The cell lines and Triton X-100 were purchased from Nanjing Key GEN Biotech Co., Ltd. (Nanjing, China). Fetal bovine serum (FBS), RPMI-1640, Dulbecco's Modified Eagle Medium (DMEM), lyso-tracker green or red, and trypsin were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA, USA). 4,6-diamino-2-phenyl indole (DAPI), Annexin V-FITC/PI, coomassie blue fast staining solution, 30% acrylamide-bisacrylamide solution (30% Acr-Bis), blocking buffer and Hematoxylin & Eosin Staining (H&E staining) Kits were obtained from the Beyotime Institute of Biotechnology (Haimen, China). Caveolae Marker (Alexa Fluor® 488, ab185043), early endosome marker (ab109009) and late endosome marker (ab179815) were from Abcam Trading Co., Ltd. (Shanghai, China). Alexa Fluor® 488- cholera toxin subunit B (CTB) was from Molecular Probes (Waltham, MA, USA). Mouse interferon-α (INF-α) ELISA kits and mouse interleukin-12 (IL-12) ELISA kits were purchased from Enzyme-linked Biotechnology Co. Ltd. (Shanghai, China). FITC phalloidine was from Shanghai Yeasen Biotechnology Co., Ltd. (Shanghai, China). The In Situ Cell Death Detection Kit was purchased from F. Hoffmann-La Roche Ltd. (Basel, Switzerland).
Preparation and characterization
Cationic beta-lactoglobulin (CLG) was synthesized via conjugation of ethylenediamine to β-LG, as described in a previous report [25]. The PNPs were prepared via an antisolvent-precipitation method, using CLG as a stabilizer [24]. Briefly, after mixing the CLG solution (10 mL, 1 mg/mL) and acetone (1 mL, 20 mg PTX) under stirring conditions, the sample was treated with an ultrasonic probe (20-25 kHz, Scientz Biotechnology Co., Ltd., Ningbo, China) at 400 W for 15 min and evaporated under reduced pressure to remove the residual acetone. The temperature in the entire procedure was controlled below 4 °C in an ice-water bath.
PNPplex and HA-PNPplex were prepared as follows: PNPs were mixed with an equal volume solution of caspase 3 or BSA (30 µg/mL) and incubated at room temperature for 30 min to form PNPs/protein complexes (PNPplex) through spontaneous electrostatic interaction. HA was further added to the solution of PNPplex to prepare PNPplex/HA complexes (HA-PNPplex), in which the amount of HA was optimized by examining different mass ratios of CLG/protein/HA. The stability of HA-PNPplex regarding changes in particle sizes and polydispersity index (PDI) was tested via incubation in a 10% serum at 37 °C for 24 h. Using a similar method, FITC, RITC or IR783-labeled nanoparticles (FITC/RITC/IR783-PNPplex/HA-PNPplex) were prepared by dissolving the dye with PTX in acetone together as the organic phase prior to mixing with CLG protein. RITC-labeled nanoparticles (RITC-PNPplex/HA-PNPplex) and dual-labeled PNPplex or HA-PNPplex were fabricated by assembling RITC-BSA on PNPs and FITC-PNPplex/HA-PNPplex, respectively. PUL/caspase 3 complexes (PULplex) were prepared according to the manufacturer's instructions. The encapsulation efficiency (EE) and drug loading (DL) of protein were calculated using following formulas:
EE (%) = Amount of protein encapsulated/Amount of protein added × 100%
DL (%) = Amount of protein encapsulated/Amount of CLG × 100%
The particle size, PDI and zeta-potential were determined using a 90Plus Particle Size Analyzer (Brookhaven Instruments, Holtsville, NY) at 25 °C according to the dynamic light scattering (DLS) principle. The examination of TEM was conducted using a JEM-1230 transmission electron microscope (TEM, Tokyo, Japan) at 200 kV. Briefly, one drop of the sample was deposited on a copper mesh and dried at 25 °C, followed by removing the sample excess with filter paper and staining with a 2% (w/w) phosphotungstic acid for 30 s. After removing the superfluous phosphotungstic acid, the mesh was desiccated at 25 °C for 5 min.
To confirm the PNPplex formation, native polyacrylamide gel electrophoresis (PAGE) was performed for the PNPs/caspase 3 and PNPs/BSA complexes. Briefly, first, 3% acrylamide/bisacrylamide (0.25 M Tris-HCl, pH 6.8) and 12.7% acrylamide/bisacrylamide (0.75 M Tris-HCl, pH 8.8) were cast as stacking and separating gels, respectively. Then, the complexes with a fixed 1 μg of caspase 3 or BSA and positive control (1 μg of naked caspase 3 or BSA) were loaded into the respective wells of the gel and run at 20 mA until the sample reached the separating gel, followed by running at 30 mA for 1 h and marking with Coomassie Brilliant Blue overnight. Finally, the results were recorded and analyzed using a Bio-Rad high-sensitivity chemiluminescence imaging system (Chemidoc XRS+, USA).
To assess PTX release, the sample was transferred into a 3.5 kDa dialysis bag and incubated in release media with different pH values. The test was conducted in an incubator (SHA-C, Jintan, China) with a shaking speed of 100 rpm/min at 37 °C. At predetermined times, samples were withdrawn and the PTX content was quantified by a high performance liquid chromatography (HPLC) system (SHIMAZU LC-10AT, Kyoto, Japan) described in a previous report [25]. Using a similar method, the study of protein release from PNPplex and HA-PNPplex that encapsulated RITC-BSA was conducted. The preparations were placed into a 300 kDa dialysis bag, and the RITC-BSA content was assayed using a fluorescence spectrometer (SHIMADZU RF-5301PC, Japan).
Fluorescence resonance energy transfer and circular dichroism spectroscopy
In fluorescence resonance energy transfer (FRET) determination, FITC and RITC were employed as the donor and acceptor, respectively. The dual-labeled HA-PNPplex (FITC-RITC-HA-PNPplex) were fabricated by assembling FITC-BSA on RITC-labeled PTX nanoparticles followed by HA coating. The emission spectra of FITC-BSA and dual-labeled HA-PNPplex from 500-700 nm were scanned at an excitation wavelength of 492 nm (donor) using a fluorescence spectrometer (SHIMADZU RF-5301PC, Japan). In addition, the split width of the excitation and emission were 5 nm and 15 nm, respectively.
Circular dichroism (CD) spectra were recorded using a J-810 spectrometer (Tokyo, Japan) equipped with a temperature-controlling unit and a quartz cuvette, and ellipticity was expressed in millidegrees. The parameters for measurement were as follows: bandwidth, 1 nm; response, 1 s; wavelength range, 250-190 nm; scanning speed, 100 nm/min; cell length, 0.1 cm; temperature, 25 °C; protein concentration, 0.1 mg/mL.
Cellular uptake and trafficking
The MCF-7 and Caco-2 cells were seeded on 12-well plates at a density of 2×105 cells/well and cultured for 48 h at 37 °C. After pretreatment with cellular uptake inhibitors, including cytochalasin D (Cy-D, 10 μg/mL), nystatin (10 μM), chlorpromazine (CPZ, 10 μg/mL), methyl-β-cyclodextrin (M-CD, 2.5 mM), monensin (MON, 200 nM) and nystatin (10 μM) plus M-CD (2.5 mM) for 0.5 h at 37 °C, the cells were cultured with nanoparticles in a serum-free culture medium at 37 °C for 4 h. After that, the cells were washed three times with phosphate buffer saline (PBS) and collected via trypsinization for fluorescence determination using flow cytometry (FCM, BD FACSCalibur, USA) and examination via confocal laser scanning microscopy (CLSM, LSM700, Carl Zeiss, Germany), respectively.
To examine the location in caveolae, Caco-2 cells were incubated with RITC-labeled nanoparticles at a 500 ng/mL of RITC in DMEM for 4 h at 37 °C followed by fixing with 4% paraformaldehyde for 10 min, permeabilizing in 0.1% Triton X-100 for 5 min, blocking in a blocking buffer (1% BSA/10% normal goat serum/0.3 M glycine in 0.1% PBS-Tween) for 15 min, staining with a Caveolae Marker (Alexa Fluor® 488) at a working dilution of 1 in 50 for 2 h at room temperature, and rinsing, in sequence. Finally, CLSM observation was performed.
To study the co-localization in CTB, the cells were incubated with Alexa Fluor® 488-CTB at 20 μg/mL and RITC-labeled nanoparticles at 500 ng/mL RITC in DMEM for 3 h at 37 °C. Then, the cells were rinsed and observed using CLSM.
For the co-localization in actin, the cells were cultured with RITC-labeled nanoparticles at 500 ng/mL of RITC at 37 °C for 4 h. Then, the cells were rinsed and fixed with 4% (w/w) paraformaldehyde (10 min), permeabilized in 0.5% Triton X-100 for 5 min, and incubated with a 200 nM FITC phalloidine in PBS containing 1% BSA for 30 min at room temperature.
The co-localization of nanoparticles with lysosomes was observed using CLSM. MCF-7 and Caco-2 cells were seeded on 30-mm glass-bottomed cell culture dishes at a density of 1×105 cells/dish and cultured for 48 h. Then, the cells were cultured with RITC-labeled nanoparticles with a RITC concentration of 500 ng/mL in a serum-free culture medium for 4 h at 37 °C, stained with 1 mL of Lyso-tracker green for 1 h at 37 °C, and washed three times with PBS.
To examine co-localization in early or late endosomes, the cells pretreated with RITC-labeled nanoparticles were fixed with 4% paraformaldehyde for 10 min, permeabilized in 0.1% Triton X-100 for 10 min, blocked in a blocking buffer for 30 min, incubated in a working dilution of 1 in 500 for early endosomes and 1 in 250 for late endosomes overnight at 4 °C, incubated with Alexa Fluor® 488-labeled secondary antibody for 1 h and rinsed with PBS.
To further confirm the co-delivery of PNPs and proteins to cells, cellular uptake of dual-labeled nanoparticles was performed using a method that is similar to the method described above, except that the lysosomes were not marked by staining with Lyso-tracker red or green.
Real-time CLSM
Caco-2 cells cultured in a confocal dish at a density of 1×105 for 48 h were incubated with RITC-HA-PNPplex at 500 ng/mL of RITC. The cellular uptake at 37 °C was monitored by real-time CLSM (LSM700, Carl Zeiss, Germany).
Western blot assay
MCF-7 cells were seeded in 6-well culture plates at a density of 2×105 cells/well for 48 h in advance. The cells were incubated with diverse nanoparticles at a fixed PTX concentration of 10 μg/mL or caspase 3 concentration of 76 nM for 6 h at 37 °C, followed by incubating for another 48 h in fresh media, lysing in cold lysis buffer, centrifuging at 10,000 ×g for 10 min, and then the supernatants were collected for protein determination using the BCA protein assay kit (KeyGEN Biotech., China) and protein separation using SDS-PAGE. After that, the separated proteins were transferred onto a PVDF membrane (Millipore, USA), incubated with a blocking solution (5% fat-free dried milk) at room temperature for 1 h, treated with a monoclonal antibody against caspase 3 overnight at 4 °C, cultured with goat anti-rabbit secondary antibody for 1 h, and stained with an ECL chemiluminescence kit (KeyGEN Biotech., China). Finally, the sample was imaged under a G: Box ChemiXR5 (Syngene, Cambridge, UK) with β-actin as an internal reference for normalization.
In vitro cytotoxicity and apoptosis
Cell viability was assayed using the MTT method. The 4T1 and MCF-7 cells were seeded on 96-well plates at a density of 5,000 cells/well and cultured for 48 h. After removing the culture media, the cells were incubated with 200 μL of RPMI 1640 or DMEM supplemented with 10% FBS containing various nanoparticles for 48 h at 37 °C, followed by incubation with 20 µL of MTT at a concentration of 5 mg/mL for 4 h and addition of 200 µL of dimethyl sulfoxide (DMSO) for 4 h, and absorbance measurement at 570 nm using a Microplate Reader (Multiskan FC, USA).
Cell apoptosis was conducted using Annexin V-FITC/PI apoptosis detection kits and FCM. The cells were treated with different nanoparticles at a fixed PTX concentration of 10 μg/mL or caspase 3 at 76 nM for 48 h at 37 °C. Then, the cells were collected, processed according to the manufacturer's protocol, and analyzed using FCM.
Pharmacokinetic study
The animals used in all experiments received care in compliance with the Principles of Laboratory Animal Care and the Guide for the Care and Use of Laboratory Animals. Animal experiments followed a protocol approved by the China Pharmaceutical University Institutional Animal Care and Use Committee.
The pharmacokinetic study was performed in rats. Sixteen rats were randomly allocated into four groups (4 rats per group) and were intravenously injected with Taxol, free RITC-BSA and RITC-HA-PNPplex at a PTX dose of 10 mg/kg or a RITC dose of 166 µg/kg, according to the rat's body weight. At predetermined time points, 0.5 mL of blood was sampled from the orbital vein and immediately centrifuged for 10 min at 5,000 ×g to obtain the plasma. The fluorescence intensity of RITC-BSA was directly quantified by a multi-function microplate reader (POLARstar Omega, BMG LABTECH, USA). The plasma concentration of RITC-BSA was calculated by a standard curve of RITC-BSA vs fluorescence intensity. The plasma concentration of PTX was determined using a similar method described in a previous report [30]. The pharmacokinetic parameters were calculated based on a statistical moment theory using the Microsoft Excel 2007 program.
In vivo imaging and biodistribution
MCF-7 tumor-bearing Balb/C mice were used in studies of biodistribution and antitumor activities. Briefly, MCF-7 tumor tissues without degeneration and necrosis were acquired, cut into 5×5×5 mm3 pieces, clipped using eye tweezers and finally inoculated under armpit skin. The mice with a tumor volume of approximately 10×10×5 mm3 were selected for the subsequent experiments.
The biodistribution of diverse IR783-labeled nanoparticles was analyzed in the MCF-7 tumor-bearing mice. Eighteen mice were randomly divided into 3 groups with 6 mice in each group and then intravenously injected via the tail vein with 0.2 mL of these nanoparticles at an IR783 dose of 2.5 mg/kg, according to the animal's body weight. At predetermined time points, the mice were sacrificed to collect their major organs, followed by imaging in vivo to detect the fluorescence intensity using an imaging system (IVIS Spectrum, PerkinElmer, USA) at an excitation wavelength of 768 nm and emission wavelength of 789 nm. To further investigate the accumulation of nanoparticles in the tumors, the tumors were excised from the mice at 2 h post dosing dual-labeled nanoparticles. One part of tumors was frozen and cut into sections with a microtome (Leica CM1860, Germany), and finally observed using CLSM. The other part of tumors was washed with PBS, successively fixed with 2.5% glutaraldehyde and 1% osmium tetroxide for 2 h at 4 °C, dehydrated with serially diluted aqueous ethanol solutions at 4 °C, washed with acetone, embedded in epoxy resin, sectioned into 80-100 μm thick slices and stained with 2% uranyl acetate, and observed using TEM.
Therapeutic efficacy
The MCF-7 tumor-bearing Balb/C nude mice were randomly divided into nine groups (6 mice/group). The mice were intravenously injected via the tail vein with 0.2 mL of nanoparticles every 3 days 5 times at a fixed PTX dose of 10 mg/kg and caspase 3 dose of 156 μg/kg, according to the animal's body weight. The tumor volume and body weight were determined at predetermined time intervals. At day 16 post injection, the mice were killed to harvest the major tissues. The sectioned tumors suffered from hematoxylin-eosin staining (H&E staining), TUNEL assay and Ki67 staining, respectively. The level of caspase 3 in tumor was assessed via Western blot (WB) analysis.
Immunogenicity assay and biocompatibility
Fresh mouse blood cells were harvested by centrifuging mouse blood at 1,500 ×g for 15 min and, then, washed and resuspended with a PBS solution. CLG or PEI were added to the erythrocytes and incubated at 37 °C for 1 h. After being centrifuged at 1,500 ×g for 15 min, the supernatant was collected to compare hemolytic activity between CLG and PEI.
The immunogenicity of CLG was examined by measuring the IL-12 and INF-α concentration in plasma, using saline and PEI as the negative and positive controls, respectively. Briefly, ICR mice were injected with CLG at doses of 1 mg/kg, 5 mg/kg, 10 mg/kg, 50 mg/kg and 100 mg/kg, respectively, or PEI at doses of 1 mg/kg, 2.5 mg/kg and 5 mg/kg, respectively, according to the animal's body weight. At 2 h, 12 h and 24 h post injection, plasma was collected to measure the concentrations of IL-12 and INF-α using ELISA kits according to the manufacturer's instructions.
Statistical analysis
The results are expressed as the mean ± standard deviation. Significant differences were set as P < 0.05. Statistical analysis and graphing were performed with Origin Pro 8.0 software (OriginLab Corp.). For the experiments with two groups, one-way analysis of variance was performed to compare the difference. In experiments with multiple groups, Student-Newman-Keuls-test was conducted for multiple comparisons upon verification of normal data distribution.
Results
Preparation and characterization
Three steps were involved in preparation of HA-PNPplex: preparation of PNPs (pure PTX nanoparticles), loading of functional protein, and HA coating. First, we prepared the PNPs with CLG as a stabilizer and optimized their formulations, where CLG was adsorbed onto the PNPs via hydrophobic interaction to prevent their aggregation [24, 25]. The particle size of PNPs did not exceed 200 nm even at a PTX-loading of up to 500% (the weight of drug compared with CLG), demonstrating that PNPs possessed an extremely high drug-loading capacity (Figure 1A). The PNPs with a PTX-loading of 200% (20 mg in formulation) were selected for further study, which had a particle size of 165.1 ± 3.4 nm (PDI: 0.155) in length and a positive charge of 21.38 ± 1.61 mV (Figure S1A). Next, functional protein, caspase 3 or BSA, was loaded on PNPs via electrostatic interaction to prepare PNPplex. Native PAGE studies displayed protein bands that were not evident at a CLG/protein mass ratio of greater than 32:1 (Figure 1B). PNPplex with this ratio had a particle size of 148 ± 1.3 nm (PDI: 0.230) and a surface potential of 16.10 ± 1.07 mV (Figure 1C, Figure S1B and Figure S2A). Finally, HA was utilized to coat PNPplex and form HA-PNPplex. The increase of the amount of HA with a fixed CLG/caspase 3 mass ratio of 32:1 resulted in an increase of particle size from 127.8 ± 2.4 nm (PDI: 0.151) to 202.6 ± 1.2 nm (PDI: 0.176) and a decrease of zeta potential from 15.00 ± 3.73 mV to 6.99 ± 0.30 mV, which is indicative that the positive charge of PNPplex was shielded by HA coating (Figure S2A). Among these formulations, the HA-PNPplex with a CLG/protein/HA mass ratio of 32:1:1.5 had minimum potential, which would be more stable and less hostile in vivo and, accordingly, was chosen for further experiments (Figure S1C and Figure S2A). TEM examination of HA-PNPplex presented rod-like particles ~200 nm in length, which correlated with the DLS results (Figure 1D).
To ascertain the loading of functional protein in HA-PNPplex, measurement of FRET was performed (Figure 1E), where FITC was utilized as a donor conjugated to BSA (FITC-BSA), and RITC was employed as an acceptor loaded in HA-PNPplex. The maximal fluorescence intensity at 520 nm of FITC (donor) from dual-labeled HA-PNPplex was significantly reduced, whereas the fluorescence at 590 nm was increased (acceptor) compared with FITC-BSA, which demonstrates an effective energy transfer from the donor to acceptor in HA-PNPplex. These results confirmed that functional protein was encapsulated well in HA-PNPplex. Furthermore, proteins loaded in PNPplex or HA-PNPplex exhibited significantly decreased maximal negative signal in CD spectra, which implied their perfect compaction after encapsulation (Figure 1F-G) [31]. In addition, the similarity in CD spectra between naked protein and released protein from HA-PNPplex illustrated that the process of encapsulation did not jeopardize the protein activity (Figure 1H).
Figure 1
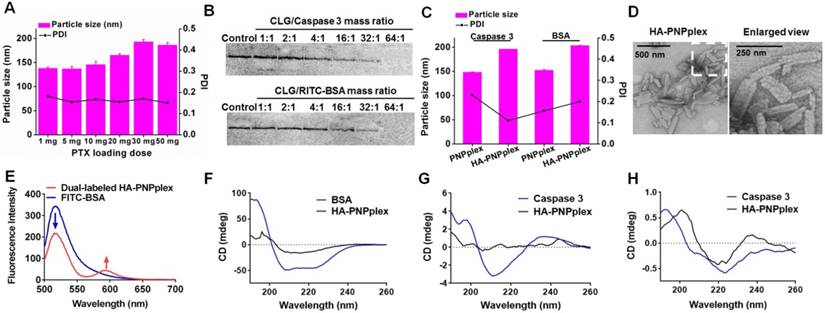
Preparation and characterization. (A) Impact of PTX-loading ranging from 1 mg to 50 mg on the particle size of PNPs. (B) Native PAGE study of caspase 3 or RITC-BSA loaded in PNPplex. The mass ratio of CLG and caspase 3 or RITC-BSA was increased from 1 to 64. (C) Particle size and PDI of the nanoparticles loading caspase 3 or BSA. (D) TEM image of HA-PNPplex. (E) Fluorescence emission spectra for FRET assay. FITC and RITC were used as the donor and acceptor, respectively. Dual-labeled HA-PNPplex (FITC-RITC-HA-PNPplex) were prepared by assembling FITC-BSA on RITC-PNPs followed by HA coating. Excitation wavelength: 492 nm. FRET effect is demonstrated via the reduction fluorescence intensity of donor at 520 nm with increased fluorescence of acceptor at 590 nm. CD spectra of functional proteins: (F-G) encapsulated proteins and (H) released caspase 3. The control was naked functional protein.
The EE% and DL% of protein in PNPplex and HA-PNPplex were 60% and 85%, respectively. The particle size and PDI were not changed after storing in 10% serum for 24 h, indicating that the formulations had sufficient stability for further study (Figure S2B). Release study performed in media at pH 5.0 or 6.8 depicted that PTX was released in a sustained pattern, with <40% being released over 24 h (Figure S2C-D). The release of functional protein exhibited a relative fast pattern (Figure S2E-F), consequently demonstrating the protein could be disassociated from the nanoparticles. In addition, the protein's content was not altered over 12 h, confirming the stability.
Cellular uptake via the non-endo-lysosomal route and trafficking
First, the endocytic pathways of PNPplex and HA-PNPplex in cells pretreated with various inhibitors were studied. The uptakes of PNPplex and HA-PNPplex in Caco-2 cells were declined by ~70% by nystatin, 50% by M-CD and 75% by nystatin plus M-CD (Figure 2A). Moreover, the CLSM observations of accumulation of these two nanoparticles in Caco-2 or MCF-7 cells pretreated with these two inhibitors indicated a significantly reduced fluorescence intensity compared with other groups (Figure 2B, Figure S3 and Figure S4). It is well known that nystatin and M-CD block caveolar endocytosis, which renders materials able to enter cells without entrapment in the endo-lysosomal system [32, 33]. Consequently, the cellular uptakes of PNPplex and HA-PNPplex were essentially dependent on caveolar endocytosis, bypassing the endo-lysosomal system. Further study indicated that this caveolae-mediated uptake of these two nanoparticles was time-related in a fixed duration (Figure S5A-B). In addition, PNPplex and HA-PNPplex are distinct at their surface, but they have similarities in the endocytic pathway, which demonstrates that the determinant for the pathway is the shape rather than surface properties.
Next, the co-localization of PNPplex or HA-PNPplex with caveolae was demonstrated (Figure 2C-E). After a 4-h treatment, a significant yellow fluorescence from co-localizing red nanoparticles with a caveolae marker Alexa Fluor® 488 (green) was displayed in the merged image and enlarged areas (Figure 2C), which is indicative of the entrapment of PNPplex or HA-PNPplex in caveolae. Especially, the involvement of actin cytoskeleton and cholera toxin subunit B (CTB) for caveolar endocytosis is indispensable. Hence, we further investigated the co-localization of RITC-labeled nanoparticles (red) with CTB or actin stained in green. Noticeably, significant yellow spots in the merged images and their enlarged views were observed and, in turn, demonstrated the caveolae-mediated internalization of these two nanoparticles (Figure 2D-E). Furthermore, little yellow fluorescence in early or late endosomes and lysosomes stained in red or green was exhibited in the merged images at 4 h of incubation with RITC-PNPplex or HA-PNPplex (Figure S6), which implied that the uptake of these two nanoparticles indeed was not detained by the endo-lysosomal system. Real-time CLSM further ascertained the intracellular delivery of RITC-HA-PNPplex (red, Movie S1). In addition, CLSM observation of cells pretreated with FITC and RITC dual-labeled nanoparticles for 4 h presents a robust yellow fluorescence in cytoplasm (Figure S5C), which suggests the integrity of PNPplex or HA-PNPplex in cellular trafficking.
Figure 2
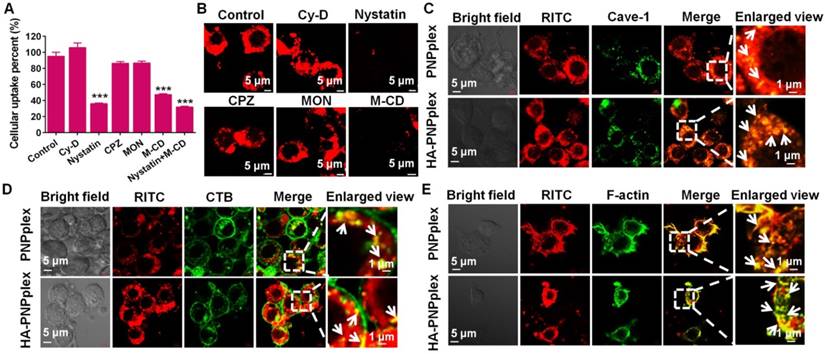
Endocytic pathway and caveolae trafficking. (A-B) Cellular uptake of RITC-labeled HA-PNPplex in Caco-2 cells pretreated with different endocytic pathway inhibitors for 0.5 h at 37 °C with a RITC concentration of 500 ng/mL. (A) Cellular uptake percent was detected using FCM. ***P < 0.001 versus the control (n = 5). (B) CLSM observation of intracellular accumulation. (C-E) Caveolae trafficking examination. Co-localization of RITC-PNPplex or RITC-HA-PNPplex with (C) Alexa Fluor 488-labeled Cave-1, (D) Alexa Fluor 488-labeled CTB and (E) FITC-labeled F-actin observed using CLSM. White arrows show co-localization of the nanoparticles with caveolae or CTB and F-actin related to caveolar trafficking. Caco-2 cells were incubated with the RITC-labeled nanoparticles at a RITC concentration of 500 ng/mL for 4 h at 37 °C.
Collectively, these results confirmed the intracellular delivery of PNPplex or HA-PNPplex without detainment by the endo-lysosomes.
Intracellular protein delivery
MCF-7 cells are a caspase 3-deficient cell line [28, 29]. Intracellularly delivered caspase 3 in MCF-7 cells after the 48-h treatment with formulations was determined via WB assay. Significant band signals from PNPplex and HA-PNPplex followed by PUL/caspase 3 complexes (PULplex) were displayed, whereas other groups exhibited small signals (Figure 3A). Quantitative assay demonstrated an ~4.5-, 6.5-, and 2-fold increase in caspase-3 level for PNPplex, HA-PNPplex and PULplex prepared with a marketed formulation for protein delivery, respectively, compared with the saline group, along with a 2.8- and 1.9-fold increase for HA-PNPplex and PNPplex over PULplex (Figure 3B). Importantly, HA-PNPplex enabled an ~1.5-fold increase in the caspase-3 level compared with PNPplex. These data demonstrated that PNPplex were able to efficiently deliver caspase 3 to cells, and the decoration of HA targeting CD44-receptors significantly boosted this intracellular delivery.
Figure 3
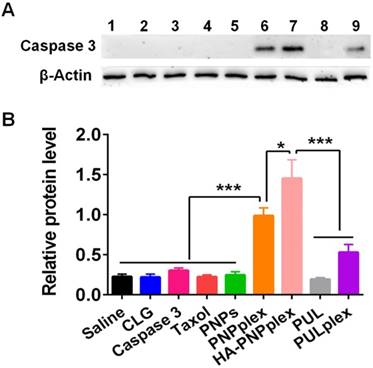
Intracellular protein delivery. Intracellular delivery of caspase 3 in MCF-7 cells after 48 h incubation at a fixed PTX concentration of 10 μg/mL and caspase-3 concentration of 76 nM. (A) WB assay: (1) saline, (2) CLG, (3) naked caspase 3, (4) Taxol, (5) PNPs, (6) PNPplex, (7) HA-PNPplex, (8) PUL, (9) PULplex. (B) Quantitative analysis (n = 3, *P < 0.05 and ***P < 0.001).
In vitro cytotoxicity and apoptosis
The cytotoxicity to MCF-7 (a caspase 3-deficient cell line) and 4T1 (a normal caspase 3-expression cell line) were assessed using the MTT assay. In MCF-7 cells, the viability of nanoparticles (PNPplex and HA-PNPplex) loading caspase 3 was dramatically lower than that of PNPs at all incubated concentrations (Figure 4A). Furthermore, these two nanoparticles exhibited promoted toxicity against 4T1 cells at >2 µg/mL PTX (Figure 4B). Significantly, both HA-PNPplex and PNPplex possessed an essentially robust toxicity to cancer cells over free caspase 3 (Figure 4C-D). A further comparison displayed that HA-PNPplex were more profound in killing cancer cells, especially MCF-7 cells, over PNPplex (Figure 4A-D), because the HA coating, which targets CD44 receptors, was able to enhance cellular uptake. Indeed, the stabilizer, CLG, which was employed for stabilizing PTX nanoparticles, generated negative toxicity to cells compared with the marketed formulation, PUL (Figure 4E). Altogether, the caspase 3-loaded particles exhibited enhanced toxicity against cancer cells.
Figure 4
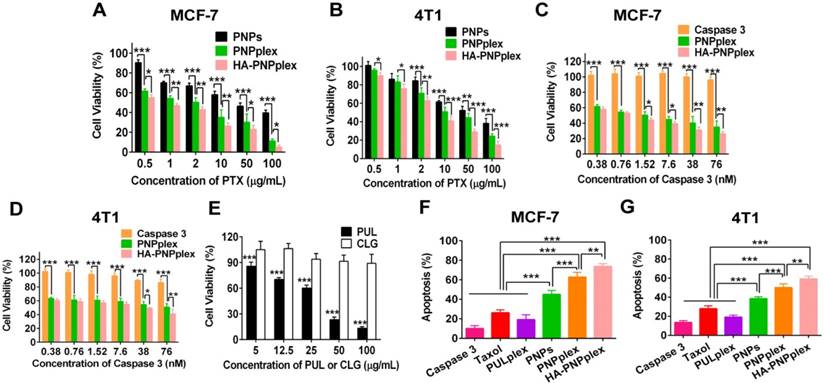
Cell viability and apoptosis. Cytotoxicity of preparations against (A) MCF-7 without expression of the endogenous caspase 3 and (B) 4T1 with normal caspase 3 expression cells after 48 h of incubation with PTX at concentrations ranging from 0.5 to 100 µg/mL (n = 5, *P < 0.05, **P < 0.01 and ***P < 0.001). Cytotoxicity of naked caspase 3 and other preparations against (C) MCF-7 and (D) 4T1 cells after 48 h incubation with caspase 3 at concentrations ranging from 0.38 to 76 nM (n = 5, *P < 0.05, **P < 0.01 and ***P < 0.001). (E) Comparison of cell viability between PUL and CLG with their concentrations from 5 to 100 µg/mL after 48 h of incubation in MCF-7 cells (n = 5, ***P < 0.001). Cell apoptosis percentage determined by FCM in (F) MCF-7 and (G) 4T1 cells induced by diverse formulations after 48 h of incubation at a fixed PTX concentration of 10 μg/mL or caspase 3 of 76 nM (n = 3, **P < 0.01 and ***P < 0.001).
To further study the cytotoxicity, apoptosis in MCF-7 and 4T1 cells after 48 h of incubation with different formulations was examined via FCM using the Annexin V-FITC/PI apoptosis detection kits. A comparative study with the PULplex prepared from marketed PUL revealed that HA-PNPplex and PNPplex had a 3-4-fold higher apoptosis (%) in MCF-7 and 4T1 cells, respectively, with the apoptotic rate (%) of ~74% for MCF-7 cells and 59% for 4T1 cells (Figure 4F, G and Figure S7). Therefore, these results demonstrated that HA-PNPplex and PNPplex were potent in inducing apoptosis in cancer cells, in particular in caspase 3-deficient cells, which was further promoted by HA-PNPplex. In addition, PNPs also induced efficient apoptosis compared with the controls, mainly due to their extremely high loading of PTX.
Pharmacokinetics, in vivo imaging and biodistribution
Blood circulation plays a critical role in accumulation of nanoparticles in a diseased site following intravenous injection. Therefore, the pharmacokinetic study of HA-PNPplex was performed with free drugs, Taxol (marketed product of PTX) and FITC-BSA, as controls. Significantly, HA-PNPplex prolonged the t1/2 by 3-fold, decreased the CL by >10-fold, and increased the AUC values as well compared with the controls (Figure 5A-B and Table 1-2), an indicator of marked improvement in blood circulation.
To study the biodistribution, whole-body and ex vivo NIR fluorescence imaging were performed in MCF-7 tumor-bearing Balb/C nude mice after intravenous injection of free IR783, IR783-PNPplex and IR783-HA-PNPplex at various time points. Free IR783 were mainly confined to liver, kidney and lung and dramatically decreased with elapsing time (Figure 5C and Figure S8C, F), and the fluorescence signal was undetectable at 24 h post injection, because IR783 has a short life-time (t1/2 = 185 min) and is easily metabolized by liver [34]. In contrast, strong fluorescence signals located in tumor sites from both PNPplex and HA-PNPplex at 2 h post injection were displayed, and the signals became significantly more intense at 4 h and 8 h and were still noticeable even at 24 h, thereby demonstrating potent tumor accumulation with prolonged retention (Figure 5C-D and Figure S8A-B, D-E). Additionally, a comparison indicated that the fluorescence intensity of HA-PNPplex in tumor at 2 h and 4 h was approximately 1.5-fold and 1.3-fold greater than that of PNPplex, respectively, whereas the promotion became less profound 8 h later, which indicates that HA decoration benefits tumor accumulation and, on the other side, the rod-like PNPplex themselves possess a better ability for tumor targeting. Interestingly, the accumulation of PNPplex and, particularly HA-PNPplex, in tumor at 4 h post dosing was markedly greater than in other mononuclear phagocyte system (MPS) organs (liver, spleen and lung) (Figure S8). Overall, these results suggested that PNPplex accumulated well in tumor and, importantly, the HA coating on PNPplex (HA-PNPplex) further enhanced this accumulation.
Figure 5
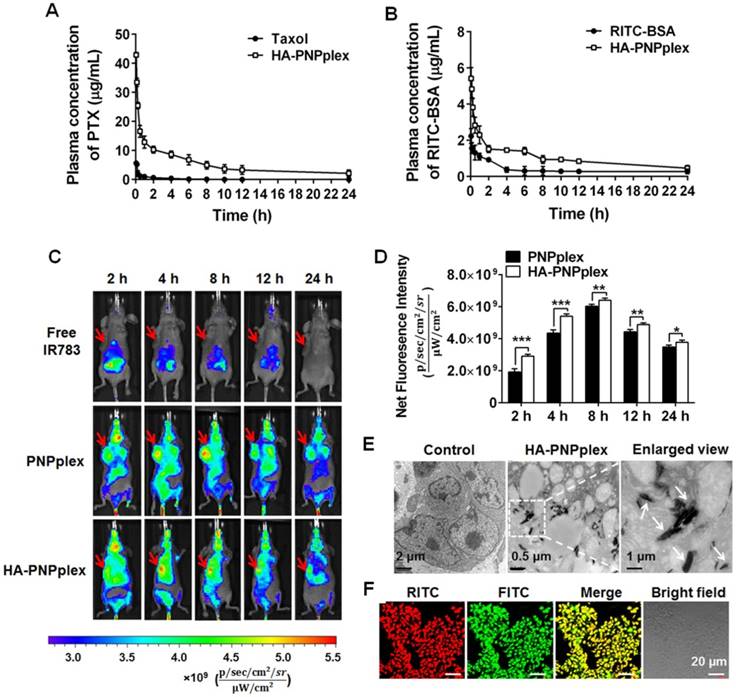
Pharmacokinetics, tumor targeting and penetration. Mean plasma concentration-time curves of (A) PTX and (B) RITC-BSA in different preparations (n = 4). (C) In vivo imaging of MCF-7 tumor-bearing nude Balb/C mice at different durations after injection of the IR 783-labeled nanoparticles at an IR783 dose of 2.5 mg/kg. Free IR783 was used as the control. (D) Quantification assay of tumor accumulation at different durations post injection (n = 3, *P < 0.05, **P < 0.05 and ***P < 0.001). In vivo penetration of HA-PNPplex into the tumor examined by (E) TEM and (F) CLSM after injection of dual-labeled HA-PNPplex at a FITC dose of 0.25 mg/kg and a RITC dose of 0.05 mg/kg. The tumors were collected at 2 h post injection.
Table 1
Pharmacokinetic parameters of PTX after intravenous injection of preparations at a PTX dose of 10 mg/kg (n = 4)
| Formulations | Cmax (mg/L) | Tmax (h) | AUC0-∞ (mg·h/L) | t1/2 (h) | CL (L·h-1·kg-1) |
|---|---|---|---|---|---|
| Taxol | 5.65 ± 0.22 | 0.17 ± 0.00 | 5.10 ± 0.35 | 2.32 ± 0.35 | 3.92 ± 0.27 |
| HA-PNPplex | 43.20 ± 1.12 | 0.17 ± 0.00 | 102.6 ± 4.12 | 7.50 ± 0.35 | 0.32 ± 0.05 |
Table 2
Pharmacokinetic parameters of RITC-BSA after intravenous injection of preparations at a RITC dose of 166 µg/kg (n = 4)
| Formulations | Cmax (mg/L) | Tmax (h) | AUC0-∞ (µg·min/L) | t1/2 (min) | CL (L·min-1·kg-1) |
|---|---|---|---|---|---|
| RITC-BSA | 1.95 ± 0.05 | 0.08 ± 0.00 | 378.26 ± 67.82 | 160.16 ± 12.00 | 1.32 ± 0.12 |
| HA-PNPplex | 6.44 ± 0.78 | 0.08 ± 0.00 | 1396.12 ± 78.65 | 618.62 ± 18.00 | 0.07 ± 0.01 |
Next, we conducted TEM observation of a sectioned tumor collected at 2 h post injection of HA-PNPplex to investigate tumor penetration (Figure 5E). Significantly, elongated particles with a size of 150-200 nm inside tumor were present. Furthermore, CLSM visualization of frozen sectioned tumor harvested at 2 h post administration of dual-labeled HA-PNPplex exhibited a homogenized yellow fluorescence in the merged image (Figure 5F), which demonstrates that HA-PNPplex were distributed in the entire tumor. Above all, these results implied that HA-PNPplex were capable of penetrating inside tumor with a uniform distribution.
Therapeutic efficacy
The MCF-7 tumor model without expression of the endogenous caspase 3 [28, 29] was selected to assess antitumor activity regarding changes in tumor volume, tumor weight and body weight after dosing preparations with saline as the negative control, and Taxol and PULplex as the positive controls (Figure 6). Saline treatment did not inhibit tumor-growth, ending with a 6-fold increase in tumor volume at day 16, neither did other preparations without drug-loading (PUL and CLG) (Figure 6A-B). Taxol (marketed product of PTX) reduced by 2-fold in the tumor volume, while caspase 3 had little suppression due to its poor membrane penetration. Critically, HA-PNPplex and PNPplex, both loaded with caspase 3, exhibited a significant inhibition on tumor-growth, decreasing the tumor volume by >3-fold and 2-fold compared with PNPs (pure PTX particles) and PULplex (marketed PUL loading caspase 3), respectively, and reducing the volume by 5-fold and >4-fold in comparison with saline. Notably, HA-PNPplex decreased the tumor volume by 1-fold compared with PNPplex, along with negative tumor-growth (100% inhibition), demonstrating that HA decoration indeed assisted in improving cancer treatment. The determination of tumor weight confirmed these results (Figure 6C). Furthermore, treatment with HA-PNPplex or PNPplex did not affect animals' body weight, while a significant loss in body-weight was displayed from the Taxol treatment during the administration duration (Figure 6D), which implies safety following repeat administration of these two preparations. These results indicated that the tumor-growth in MCF-7 tumor-bearing mice was significantly inhibited by PNPplex, in particular HA-PNPplex.
Next, we examined the cell apoptosis and proliferation in tumor using histological TUNEL and Ki67 analysis at the end of the experiment to ascertain antitumor activities. Positive cells are shown in TUNEL-stained sections treated with HA-PNPplex, followed by those treated with PNPplex and PNPs, respectively (Figure 6F). Quantified determination indicated that HA-PNPplex showed the highest apoptosis rate of 62%, which was an ~36%, 43%, 24% and 14% increase over Taxol, PULplex, PNPs and PNPplex, respectively (Figure 6E). The measurement of cell proliferation verified the apoptosis (Figure 6F-G). The pathological test of the tumor collected at the end of the experiment also depicted the greatest cancer cell remission in the HA-PNPplex group among all groups (Figure 6F). The findings demonstrated that HA-PNPplex and PNPplex allowed for competent cancer cell killing in tumors and, meanwhile, HA-PNPplex had promoted efficiency.
It was hypothesized that the improved antitumor efficacies were directly related to the intratumoral delivery of caspase 3 with HA-PNPplex and PNPplex. Accordingly, the detection of caspase-3 level in tumor collected at day 16 post dosing was performed using WB assay (Figure 6H-I). WB determination showed bright band signals in groups treated with HA-PNPplex and PNPplex, while an extremely weak signal appeared in the PULplex-treated group, and little signal was displayed in other groups (Figure 6H). Quantified analysis indicated that HA-PNPplex and PNPplex raised the caspase-3 level by 2-fold and 7-fold compared with PULplex and other controls (Figure 6I). In addition, HA-PNPplex rendered an approximately 30% promotion in caspase 3 over PNPplex. These results were well consistent with the in vitro determination of caspase-3 level in MCF-7 cells (Figure 3). Collectively, both PNPplex and HA-PNPplex can deliver caspase 3 to the tumor, with a higher potency from the latter.
Overall, HA-PNPplex exerted efficient therapeutic effects against MCF-7 breast cancer model with a caspase-3 deficiency in terms of inhibition on tumor-growth, apoptotic rate and cancer cell killing in tumors.
Immunogenicity assay and biocompatibility
Positively charged CLG is an efficient stabilizer for the prepared pure drug nanoparticles [35, 36]; however, it is an exogenous material [36]. Therefore, its systematic injection may generate potential immunotoxicity to human body. Here, the hemolytic activity and immunogenicity of CLG were examined. Little hemolytic activity and hematotoxicity was shown after 1 h of incubation of CLG at even 10 mg/mL with erythrocytes compared with saline (Figure 7A). In contrast, the incubation with a typical cationic vector, polyethyleneimine (PEI), at only 0.1 mg/mL induced dramatic hemolysis. Further immunogenic assay demonstrated that the level of proinflammatory cytokines of interleukin-12 (IL-12) and interferon-α (INF-α) in plasma did not increase over a 24-h period following injection with CLG at a dose of up to 100 mg/kg in comparison with saline treatment, whereas these two cytokines were extremely elevated post injection with PEI at doses of 1-5 mg/kg (Figure 7B-C). These results implied that systemic administration of CLG did not induce an immune response and was safe.
Figure 6
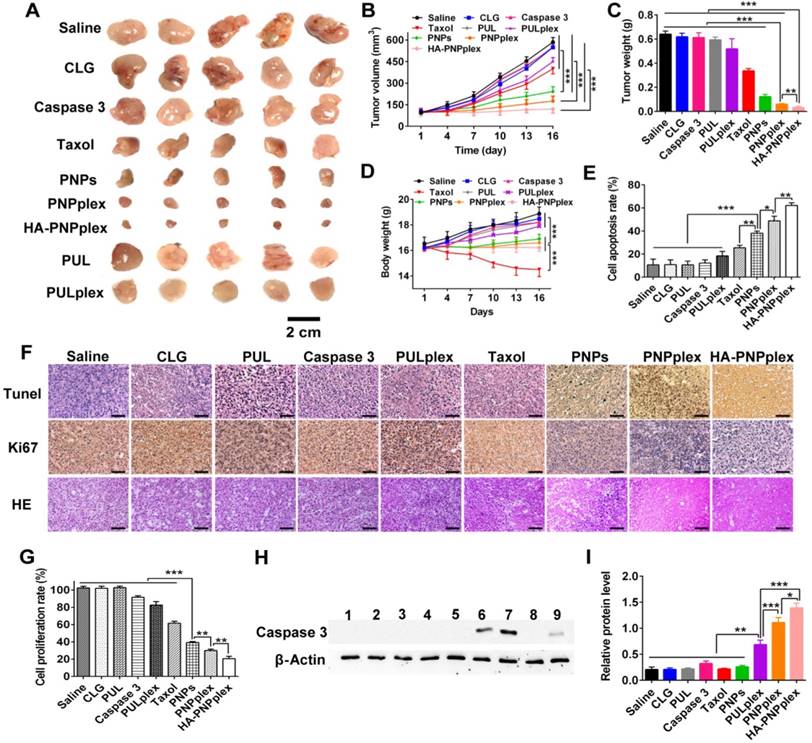
Antitumor efficiency. (A) Image of representative tumors collected from mice at the end of the experiment. (B) Tumor growth curves (n = 6, ***P < 0.001). (C) Tumor weight variation (n = 6, ***P < 0.001 and **P < 0.01). (D) Body weight change curves (n = 6, ***P < 0.001). (E) Quantitative analysis of cell apoptosis (n = 3, *P < 0.05, **P < 0.01 and ***P < 0.001). (F) Ki67, TUNEL, and H&E staining analysis of the tumor collected on day 16. The brown-stained cells represent the positive cells in the TUNEL and Ki67 assay. Nuclei are stained blue, while extracellular matrix and cytoplasm are stained red in H&E analysis. The scale bar is 20 µm. (G) Quantitative analysis of cell proliferation (n = 3, **P < 0.01 and ***P < 0.001). (H) Intratumoral delivery of caspase 3 determined using WB assay. Formulations: 1, saline; 2, CLG; 3, naked caspase 3; 4, Taxol; 5, PNPs; 6, PNPplex; 7, HA-PNPplex; 8, PUL; 9, PULplex. The internal control for normalizing protein expression was β-actin. (I) Quantitative analysis of caspase 3 in tumor (n = 3, ***P < 0.001, **P < 0.01 and *P < 0.05).
To probe the toxicity of preparations to healthy organs, main tissues in mice were collected for H&E staining at day 17 following the test of therapeutic efficacy. Taxol administration exhibited an evident toxicity such as body weight loss (Figure 6D), vacuolar degeneration in liver and kidney and inflammation in lung (Figure 7D). In addition, dosing the marked vector (PUL) or its caspase-3 loading formulation (PULplex) incurred significant inflammation in liver, kidney and lung. Notably, neither activity abnormity nor loss of animal's body weight was observed post administration of PNPs, PNPplex or HA-PNPplex, therefore demonstrating high biocompatibility and tolerability.
Figure 7
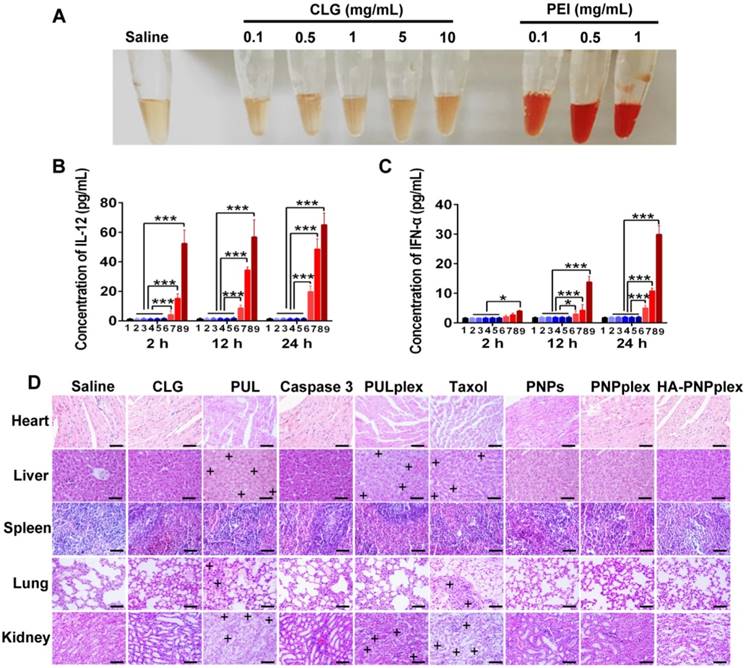
Safety determination. (A) Comparison of hemolytic activity between CLG (0.1 to 10 mg/mL) and PEI (a cationic vector, 0.1 to 1 mg/mL). (B) IL-12 and (C) INF-α concentrations in mouse plasma after injection of CLG and PEI for 2 h, 12 h and 24 h, respectively (n = 5). 1: saline; 2: CLG, 1 mg/kg; 3: CLG, 5 mg/kg; 4: CLG, 10 mg/kg; 5: CLG, 50 mg/kg; 6: CLG, 100 mg/kg; 7: PEI, 1 mg/kg; 8: PEI, 2.5 mg/kg; 9: PEI, 5 mg/kg; *P < 0.05, ***P < 0.001 versus control. (D) Representative tissue histology. Formulations were administered to MCF-7 tumor-bearing nude Balb/C mice via tail vein injections every 3 days at a PTX dose of 10 mg/kg or a caspase-3 dose of 156 μg/kg, according to the animal's body weight. The tissues were collected at day 17 post repeat dosing. Nuclei are stained blue while extracellular matrix and cytoplasm are stained red in H&E analysis. “+” represents the positive area. The scale bar is 20 µm.
Discussion
The developed DDD platform is robust for intracellular delivery of biopharmaceuticals. Undeniably, protein therapies have been considered as an efficient approach for various human diseases. However, intracellular delivery of biopharmaceuticals is greatly hindered by the fact that the current vectors tend to be entrapped in the endo-lysosomal system, and only 1-2% of the drug can escape to the cytoplasm [20]. Pure drug nanoparticles, also named nanocrystals, certainly are a type of nanoparticle [37]. Nonetheless, few reports indicate that active drug particles can serve as vectors for protein delivery. Here, using rod-like PTX particles as vectors followed by HA coating for CD44-receptor targeting, the functional protein, BSA or caspase 3, was delivered to cells via a non-endo-lysosomal route, indicating that almost all loaded proteins was intracellularly delivered to the cells. Furthermore, our previous report demonstrated that these drug particles without the HA coating also entered cells via a non-lysosomal pathway and enabled an efficient and safe miRNA replacement therapy [25]. As a result, the dominant property for these drug particles gaining competent intracellular delivery relies on their rod-like shape rather than their surface properties. Based on these findings, we concluded that the potential advantages of the developed DDD platform over current approaches include (i) internalization without capture by the endo-lysosomal system and the resultant potent cytosol release; (ii) perfect tumor accumulation and penetration (Figure 5 and Figure S8); (iii) high drug loading and facilitation of combined therapy with chemotherapeutics and proteins; and, (iiii) facile preparation without a complex setup. We believe that this work offers a completely new approach for protein therapies, as well as delivery of other biopharmaceuticals. Further work regarding intracellular delivery of antigen for malignant cancer immunotherapy is ongoing.
A new strategy for codelivery of chemotherapeutics and proteins is demonstrated. Combination therapy through codelivery of cytotoxic agents and proteins is emerging as a promising strategy for the clinical synergistic cancer treatment. Various vectors have been developed for this codelivery. However, their applications are limited by diverse disadvantages including poor encapsulation of proteins, short circulation time, difficulties in preparation, etc. [38] In this study, we utilize active drug particles as carriers for loading proteins and co-delivery, without the need to consider the encapsulation of other small-molecule drugs, and with loading of protein on these drug particles only via a simple mixing procedure. Consequently, the total drug loading in optimized HA-PNPplex is extremely high, up to 205% (the weight of drug compared with a stabilizer, CLG), which is significantly higher than that in conventional vectors having less than 10% drug-loading [39] and, thus, benefits maximizing the synergistic effect between the two drugs. Second, we previously reported that the rod-like drug particles possessed significantly prolonged blood-circulation duration and accumulated well in the tumor [23, 24]. Based on these features, we believe that the developed HA-PNPplex are a valuable platform for combined cancer therapy with small-molecule drugs and proteins.
The platform enabled potent treatment against the MCF-7 breast cancer model with caspase-3 deficiency. Treatment of breast cancer is a significant challenging due to the development of drug resistance and metastasis [40]. Here, we found that HA-PNPplex allowed for a 100% inhibition of tumor growth after 6 injections and >60% apoptosis in cancer cells. Generally, caspase 3 is a central downstream executer of apoptotic pathways [41], and its negative expression significantly decreases the sensitivity of cancer cells to chemotherapy, and vice versa [42]. Our in vitro and in vivo experiments demonstrated that the HA-PNPplex treatment elevated caspase-3 level up to 6.5-fold in MCF-7 cells (Figure 3) and up to 7-fold in the tumor (Figure 6H-I). In addition, we recently found that upregulation of caspase 3 was able to sensitize cancer cells to chemotherapy with PTX [43]. Therefore, this exciting antitumor effect on breast cancer is ascribed to the efficient intracellular delivery of caspase 3 by HA-PNPplex and consequent synergy between caspase 3 and PTX. In addition, efficient tumor-targeting ability of HA-PNPplex contributed to the profound antitumor activities as well. Significantly, because of their rational design, high-drug loading, easy preparation and robust stability, these safe HA-PNPplex can also be adopted to cure other malignant cancers and would be tractable for clinical translation.
Conclusions
Here, a DDD platform based on PNPs, which is characterized by extremely high drug-loading, high stability and a simple preparation procedure, was established for protein therapeutics. Most importantly, this platform delivers functional proteins to cells via a non-endo-lysosomal pathway, greatly elevating the efficiency of protein delivery (6.5-fold). Few reports indicate that pure drug particles can efficiently serve for protein delivery; therefore, we believe that this is a pioneering work that opens a new direction for drug delivery, especially protein delivery. Significantly, HA-PNPplex enabled profound therapeutic efficacy against the MCF-7 breast cancer (100% inhibition) due to potent intracellular delivery of caspase 3 (7-fold) and efficient tumor-targeting ability (1.5-fold). Overall, this platform, which enters cells via bypassing the endo-lysosomal system, allows for potent protein delivery and resultant robust cancer therapy. This platform provides a completely new strategy for protein therapies and delivery for other biomacromolecules. Further work on antigen presentation for cancer immunotherapy is underway.
Abbreviations
Acr-Bis: acrylamide-bisacrylamide; AUC: area under the concentration curve; BSA: bovine serum albumin; β-LG: beta-lactoglobulin; CLG: cationic beta-lactoglobulin; CD: circular dichroism; Cy-D: cytochalasin D; CPZ: chlorpromazine; CLSM: confocal laser scanning microscopy; CTB: cholera toxin subunit B; Cmax: maximum plasma concentration; CL: clearance; DDD: drug-delivering-drug; DL: drug loading; DLS: dynamic light scattering; DMSO: dimethyl sulfoxide; DMEM: dulbecco's Modified Eagle Medium; DAPI: 4,6-diamino-2-phenyl indole; EE: encapsulation efficiency; FITC: fluorescein isothiocyanate isomer I; FBS: fetal bovine serum; FRET: fluorescence resonance energy transfer; FCM: flow cytometry; HA: hyaluronic acid; HA-PNPplex: HA-coated PNPplex; HPLC: high performance liquid chromatography; H&E staining: hematoxylin & eosin staining; INF-α: interferon-α; IL-12: interleukin-12; MTT: 3-(4,5-Dimethylthiazol-2yl)-2,5-diphenyltetrazoliumbromide; M-CD: methyl-β-cyclodextrin; MON: monensin; MPS: mononuclear phagocyte system; PNPs: pure drug nanoparticles; PTX: paclitaxel; PNPplex: PNPs/protein complexes; PEI: polyethylenimine; PBS: phosphate buffer saline; PAGE: polyacrylamide gel electrophoresis; PDI: polydispersity index; PUL: PULsin; PULplex: PUL/protein complexes; RITC: rhodamine B isothiocyanate; TEM: transmission electron microscopy; Tmax: time to reach Cmax; t1/2: biological half-life; WB: western blot.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Nos. 81673377, 81473152, and 81402869), the Natural Science Foundation of Jiangsu Province (No. BK20140671), the Ministry of Science and Technology of China (2014ZX09507004-002), and the Fostering Plan of University Scientific and Technological Innovation Team and Key Members of the Outstanding Young Teacher of Jiangsu Qing Lan Project (2014, 2016). We thank Xiaonan Ma, Minhui Sun and Yingjian Hou from the Public platform of State Key Laboratory of Natural Medicines of China Pharmaceutical University for their technical assistance.
Supplementary Material
Supplementary figures.
Supplementary movie S1.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mitragotri S, Burke PA, Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discov. 2014;13:655-72
2. Gu Z, Biswas A, Zhao MX, Tang Y. Tailoring nanocarriers for intracellular protein delivery. Chem Soc Rev. 2011;40:3638-55
3. Yu M, Wu J, Shi J, Farokhzad OC. Nanotechnology for protein delivery: Overview and perspectives. J Control Release. 2016;240:24-37
4. Yin H, Kauffman KJ, Anderson DG. Delivery technologies for genome editing. Nat Rev Drug Discov. 2017;16:387-99
5. Wang M, Yang X, Zhang P, Cai L, Yang X, Chen Y. et al. Sustained delivery growth factors with polyethyleneimine-modified nanoparticles promote embryonic stem cells differentiation and liver regeneration. Adv Sci. 2016;3:1500393
6. Dowdy SF. Overcoming cellular barriers for RNA therapeutics. Nat Biotechnol. 2017;35:222-9
7. Frokjaer S, Otzen DE. Protein drug stability: a formulation challenge. Nat Rev Drug Discov. 2005;4:298-306
8. Kawasaki R, Sasaki Y, Katagiri K, Mukai SA, Sawada S, Akiyoshi K. Magnetically guided protein transduction by hybrid nanogel chaperones with iron oxide nanoparticles. Angew Chem. 2016;55:11377-81
9. Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH. et al. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol. 2015;33:73-80
10. Mout R, Ray M, Tay T, Sasaki K, Yesilbag Tonga G, Rotello VM. General strategy for direct cytosolic protein delivery via protein-nanoparticle co-engineering. ACS Nano. 2017;11:6416-21
11. Yan M, Du J, Gu Z, Liang M, Hu Y, Zhang W. et al. A novel intracellular protein delivery platform based on single-protein nanocapsules. Nat Nanotechnol. 2010;5:48-53
12. Kaczmarczyk SJ, Sitaraman K, Young HA, Hughes SH, Chatterjee DK. Protein delivery using engineered virus-like particles. Proc Natl Acad Sci. 2011;108:16998-7003
13. Weill CO, Biri S, Adib A, Erbacher P. A practical approach for intracellular protein delivery. Cytotechnology. 2008;56:41-8
14. Zassler B, Blasig IE, Humpel C. Protein delivery of caspase-3 induces cell death in malignant C6 glioma, primary astrocytes and immortalized and primary brain capillary endothelial cells. J Neuro-Oncol. 2005;71:127-34
15. Vasir JK, Labhasetwar V. Biodegradable nanoparticles for cytosolic delivery of therapeutics. Adv Drug Deliv Rev. 2007;59:718-28
16. Luo M, Wang H, Wang Z, Cai H, Lu Z, Li Y. et al. A STING-activating nanovaccine for cancer immunotherapy. Nat Nanotechnol. 2017;12:648-54
17. Tang Y, Zeng Z, He X, Wang T, Ning X, Feng X. siRNA crosslinked nanoparticles for the treatment of inflammation-induced liver injury. Adv Sci. 2017;4:1600228
18. Fu A, Tang R, Hardie J, Farkas ME, Rotello VM. Promises and pitfalls of intracellular delivery of proteins. Bioconjugate Chem. 2014;25:1602-8
19. Stewart MP, Sharei A, Ding X, Sahay G, Langer R, Jensen KF. In vitro and ex vivo strategies for intracellular delivery. Nature. 2016;538:183-92
20. Gilleron J, Querbes W, Zeigerer A, Borodovsky A, Marsico G, Schubert U. et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat Biotechnol. 2013;31:638-46
21. Akishiba M, Takeuchi T, Kawaguchi Y, Sakamoto K, Yu H-H, Nakase I. et al. Cytosolic antibody delivery by lipid-sensitive endosomolytic peptide. Nat Chem. 2017;9:751-61
22. Behzadi S, Serpooshan V, Tao W, Hamaly MA, Alkawareek MY, Dreaden EC. et al. Cellular uptake of nanoparticles: journey inside the cell. Chem Soc Rev. 2017;46:4218-44
23. He W, Xin X, Li Y, Han X, Qin C, Yin L. Rod-shaped drug particles for cancer therapy: the importance of particle size and participation of caveolae pathway. Part Part Syst Charact. 2017;34:1600371-n /a
24. Li Y, Wu Z, He W, Qin C, Yao J, Zhou J. et al. Globular protein-coated paclitaxel nanosuspensions: interaction mechanism, direct cytosolic delivery, and significant improvement in pharmacokinetics. Mol Pharmaceut. 2015;12:1485-500
25. Xin X, Pei X, Yang X, Lv Y, Zhang L, He W. et al. Rod-shaped active drug particles enable efficient and safe gene delivery. Adv Sci. 2017;4:1700324
26. Kothakota S, Azuma T, Reinhard C, Klippel A, Tang J, Chu K. et al. Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science (New York, NY). 1997;278:294-8
27. Wang Y, Gao W, Shi X, Ding J, Liu W, He H. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99-103
28. Tsang WP, Kwok TT. Let-7a microRNA suppresses therapeutics-induced cancer cell death by targeting caspase-3. Apoptosis. 2008;13:1215-22
29. Yang X-H, Sladek T L, Liu X, Butler B R, Froelich C J, Thor A D. Reconstitution of caspase 3 sensitizes MCF-7 breast cancer cells to doxorubicin- and etoposide-induced apoptosis. Cancer Research. 2001;61:348-354
30. Zhang L, Yang X, Lv Y, Xin X, Qin C, Han X. et al. Cytosolic co-delivery of miRNA-34a and docetaxel with core-shell nanocarriers via caveolae-mediated pathway for the treatment of metastatic breast cancer. Sci Rep. 2017;7:46186
31. Chang H, Lv J, Gao X, Wang X, Wang H, Chen H. et al. Rational design of a polymer with robust efficacy for intracellular protein and peptide delivery. Nano lett. 2017;17:1678-84
32. Parton RG, Richards AA. Lipid rafts and caveolae as portals for endocytosis: new insights and common mechanisms. Traffic. 2003;4:724-38
33. Mout R, Ray M, Tonga GY, Lee YW, Tay T, Sasaki K. et al. Direct cytosolic delivery of CRISPR/Cas9-ribonucleoprotein for efficient gene editing. ACS Nano. 2017;11:2452-8
34. Li W, Peng J, Tan L, Wu J, Shi K, Qu Y. et al. Mild photothermal therapy/photodynamic therapy/chemotherapy of breast cancer by Lyp-1 modified Docetaxel/IR820 co-loaded micelles. Biomaterials. 2016;106:119-33
35. He W, Lu Y, Qi J, Chen L, Hu F, Wu W. Food proteins as novel nanosuspension stabilizers for poorly water-soluble drugs. Int J Pharm. 2013;441:269-78
36. Chen L, Remondetto GE, Subirade M. Food protein-based materials as nutraceutical delivery systems. Trends in Food Sci Technol. 2006;17:272-83
37. Lu Y, Qi J, Dong X, Zhao W, Wu W. The in vivo fate of nanocrystals. Drug Discov Today. 2017;22:744-50
38. He CL, Tang ZH, Tian HY, Chen XS. Co-delivery of chemotherapeutics and proteins for synergistic therapy. Adv Drug Deliv Rev. 2016;98:64-76
39. Shen Y, Jin E, Zhang B, Murphy CJ, Sui M, Zhao J. et al. Prodrugs forming high drug loading multifunctional nanocapsules for intracellular cancer drug delivery. J Am Chem Soc. 2010;132:4259-65
40. Marcotte R, Sayad A, Brown Kevin R, Sanchez-Garcia F, Reimand J, Haider M. et al. Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell. 2016;164:293-309
41. Friedrich K, Wieder T, Von Haefen C, Radetzki S, Janicke R, Schulze-Osthoff K. et al. Overexpression of caspase-3 restores sensitivity for drug-induced apoptosis in breast cancer cell lines with acquired drug resistance. Oncogene. 2001;20:2749-60
42. Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714-26
43. Mohammad IS, He W, Yin L. A smart paclitaxel-disulfiram nanococrystals for efficient MDR reversal and enhanced apoptosis. Pharm Res. 2018;35:77
Author contact
![]() Corresponding authors: Tel.: +86 2583271018; fax: +86 2583271018. E-mail: lifangyin_com, weiheedu.cn
Corresponding authors: Tel.: +86 2583271018; fax: +86 2583271018. E-mail: lifangyin_com, weiheedu.cn
Citation styles
APA
Xin, X., Teng, C., Du, X., Lv, Y., Xiao, Q., Wu, Y., He, W., Yin, L. (2018). Drug-delivering-drug platform-mediated potent protein therapeutics via a non-endo-lysosomal route. Theranostics, 8(13), 3474-3489. https://doi.org/10.7150/thno.23804.
ACS
Xin, X.; Teng, C.; Du, X.; Lv, Y.; Xiao, Q.; Wu, Y.; He, W.; Yin, L. Drug-delivering-drug platform-mediated potent protein therapeutics via a non-endo-lysosomal route. Theranostics 2018, 8 (13), 3474-3489. DOI: 10.7150/thno.23804.
NLM
Xin X, Teng C, Du X, Lv Y, Xiao Q, Wu Y, He W, Yin L. Drug-delivering-drug platform-mediated potent protein therapeutics via a non-endo-lysosomal route. Theranostics 2018; 8(13):3474-3489. doi:10.7150/thno.23804. https://www.thno.org/v08p3474.htm
CSE
Xin X, Teng C, Du X, Lv Y, Xiao Q, Wu Y, He W, Yin L. 2018. Drug-delivering-drug platform-mediated potent protein therapeutics via a non-endo-lysosomal route. Theranostics. 8(13):3474-3489.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.