Impact Factor
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(11):3087-3098. doi:10.7150/thno.24426 This issue Cite
Research Paper
Dysregulation of 5-hydroxytryptamine 6 receptor accelerates maturation of bone-resorbing osteoclasts and induces bone loss
Kyung-Ran Park1, Eun-Cheol Kim2, Jin Tae Hong1, ![]() , Hyung-Mun Yun2,
, Hyung-Mun Yun2, ![]()
1. College of Pharmacy and Medical Research Center, Chungbuk National University, Chungbuk 194-31, Republic of Korea
2. Department of Oral and Maxillofacial Pathology, School of Dentistry, Kyung Hee University, Seoul 130-701, Republic of Korea
Received 2017-12-18; Accepted 2018-3-13; Published 2018-4-30
Abstract

Rationale: Characterizing the regulation of bone-resorbing osteoclasts is central to the understanding of the pathogenesis and treatment of bone diseases, such as osteoporosis and periodontitis. 5-hydroxytryptamine (5-HT) has drawn considerable attention for its role in bone; however, it remains unknown whether the intracellular signaling of 5-HT receptors (5-HTRs) is linked to any of the regulatory mechanisms in osteoclasts. Herein, we report 5-HT6R to be a key regulatory receptor for osteoclastogenesis.
Methods: In order to explore the critical role of 5-HT6R in bone-resorbing osteoclasts, in vitro experiments were performed using mouse whole bone marrow cells isolated from femora and tibiae and In vivo animal experiments were performed using 5-HT6R-deficient (5-HT6RKO-/-) mice, bone resorption mice model, and osteoporosis mice model.
Results: Compared to other 5HTRs, activation of 5-HT6R relatively increased TRAP (tartrate-resistant acid phosphatase) activity during osteoclastogenesis. 5-HT6RKO(-/-) mice and 5-HT6RKO(-/-) osteoclast lineages presented with an abnormal phenotype and impaired osteoclastogenesis and impaired osteoclastogenesis. Activation of 5-HT6R increased the number of TRAP-positive multinuclear osteoclasts, actin ring formation, and expression of early osteoclast markers with osteoclast lineage commitment. Intracellular 5-HT6R signaling was found to be linked to RhoA GTPase activation and was involved in the maturation of osteoclasts. This signaling pathway also showed enhanced bone destruction after lipopolysaccharide (LPS) administration in mice. Furthermore, inhibition of 5-HT6R-mediated RhoA GTPase signaling protected against ovariectomy(OVX)-induced bone loss in mice.
Conclusion: Taken together, our findings place the 5-HT6R system in a new context of osteoclast lineages in both an in vitro and in vivo system, and also it may offer a novel molecular target for the treatment of bone diseases.
Keywords: 5-HT6R, RhoA GTPase, osteoclast, osteoporosis
Introduction
The physiological process of bone metabolism maintains the integrity of the skeleton by removing and replacing old bone [1]. The number of osteoblasts and the degree of osteoblast differentiation have been shown to enhance new bone formation [2]. Osteoclasts, on the other hand, are large multinucleated cells (MNCs) that have a unique ability to resorb mineralized bone and play a key role in physiological skeletal morphogenesis and bone remodeling [3]. Osteoclasts develop from monocyte-lineage hematopoietic precursors during a multistep differentiation process called osteoclastogenesis [3-4]. Osteoclasts form tartrate-resistant acid phosphatase (TRAP)-positive MNCs, which have a reorganized actin cytoskeleton that facilitates attachment to the bone's surface for bone resorption [5-6]. Osteoclastogenic signals are primarily regulated by two essential cytokines, mainly macrophage colony-stimulating factor (M-CSF) and receptor activator of nuclear factor (NF)-κB ligand (RANKL), which belongs to the tumor necrosis factor (TNF) superfamily [3, 7]. Dysregulation of osteoclast function or differentiation results in an osteopetrotic phenotype with a marked increase in bone density. Conversely, increased bone resorption is associated with bone loss in chronic inflammatory diseases such as osteoporosis, arthritis, and metastatic bone lesions [8-10].
5-hydroxytryptamine (5-HT) is a well-known hormone and neurotransmitter that is primarily produced by enterochromaffin cells in the gut (95% of total body 5-HT), which have critical functions in gastroenterology [11-12], as well as in neurons of the brain stem (5% of total body 5-HT), which is important in psychiatric and neurological diseases [13]. The various actions of 5-HT in peripheral organ systems have been separated from those in the central nervous system (CNS), since the blood-brain barrier is impermeable to 5-HT [12, 14]. The diversity of 5-HT activities results from the existence of multiple 5-HT receptors (5-HTRs), which are been divided into seven classes (5-HT1R to 5-HT7R) based on their respective signaling pathways [15].
Several studies have suggested that 5-HT signaling pathways are involved in bone metabolism. 5-HT-specific reuptake inhibitors or selective 5-HT reuptake inhibitors (SSRIs), such as the antidepressants escitalopram (Cipralex) and fluoxetine (Prozac), increase 5-HT levels by blocking the 5-HT transporter (SERT) [16-18]. Adverse side effects of these SSRIs include increased risks of fragility fractures [19] and massive bone acquisition deficits, as shown in mice treated with SSRIs and SERT-knockout mice [20-21]. In stark contrast to these observations, other groups have either shown improved effects or no change in bone density following 5-HT or SSRI treatment [20-22]. Therefore, there have been conflicting reports regarding the effects of 5-HT on bone metabolism.
All 5-HT receptors are G protein-coupled receptors (GPCRs) with the exception of 5-HT3R, which is a ligand-gated cation channel. [23]. Of all 5-HTRs, 5-HT6R is the G protein-coupled receptor most recently cloned [24]. Expression of 5-HT6R has been detected in regions of the brain, including the striatum, nucleus accumbens, hippocampus, and cortex [25]. As such, all studies have to date focused on the brain when evaluating the pharmacological role of 5-HT6R. 5-HT6R has critical roles in neuronal migration, differentiation, and development [26-28], is a clinical target, and shows clinical success in treating neurological diseases [29-33]. To data, 5-HT6R in the peripheral organs has not been studied, apart from our recent report that 5-HT6R activation suppresses osteoblast differentiation and in vivo bone regeneration via the peripheral 5-HT system [34].
In the present study, we explored the critical role of 5-HT6R in both an in vitro and in vivo system, using 5-HT6R-deficient mice (5-HT6RKO-/-), through lipopolysaccharide (LPS) administration, and by ovariectomy-induced osteoporosis. Our data indicate 5-HT6R as a key regulatory receptor in osteoclastogenesis, bone resorption, and osteoporosis.
Results
Identification of 5-HT6R in osteoclast lineages
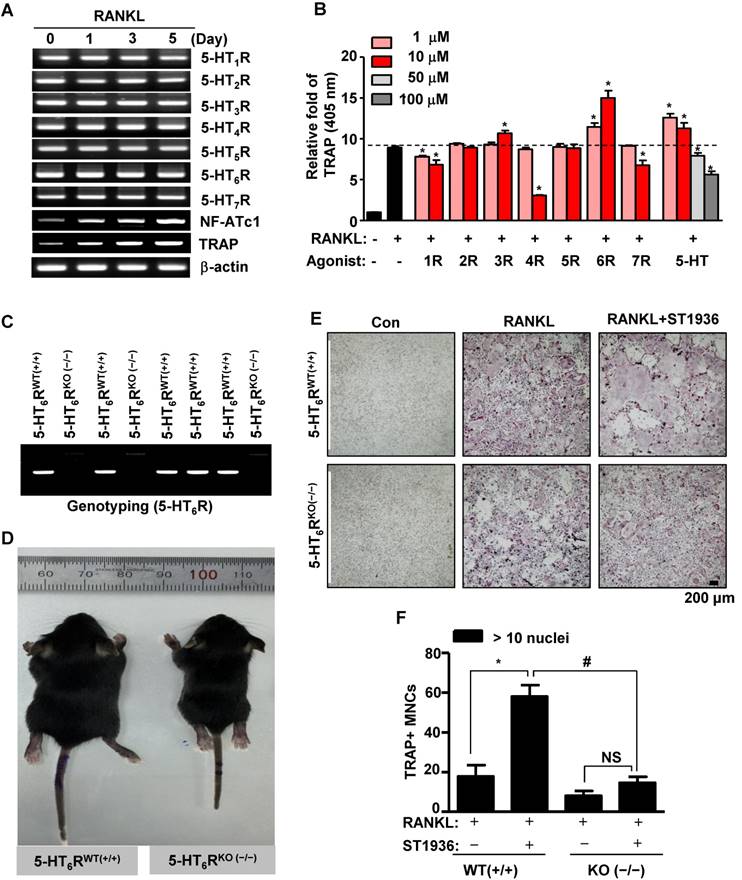
To elucidate bone remodeling by specific 5-HTRs, we first examined the expression of 5-HTRs during RANKL-induced osteoclast differentiation of bone marrow-derived macrophages (BMMs). RT-PCR analysis established that all 5-HTRs were constitutively expressed during osteoclastogenesis (Figure 1A). The prominent expression of all 5-HTRs suggests a systematic requirement of these receptors for the regulation of osteoclast differentiation and function. To explore this possibility, we stimulated all 5-HTRs by 5-HT or by using specific agonists for each 5-HTR. As shown in Figure 1B and Figure S1A-B, agonists for 5-HT1R, 5-HT4R, and 5-HT7R inhibited osteoclast activity, while agonists for 5-HT2R and 5-HT5R did not affect osteoclast activity. Stimulation of all 5-HTRs by 5-HT increased osteoclast activity at concentrations of 10 μM or below. Notably, 5-HT6R was found to increase osteoclast activity, even when stimulated at low concentrations (Figure 1B and Figure S1A-B). Given these findings, we hypothesized that 5-HT6R may be a critical regulator in osteoclasts for maintaining bone remodeling.
Abnormal phenotype and osteoclatogenesis in 5-HT6R-deficient (5-HT6RKO(-/-)) mice
Mice phenotypes were next analyzed using 5-HT6R-deficient (5-HT6RKO(-/-)) mice (Figure 1C). As shown in Figure 1D, the body size of 5-HT6RKO(-/-) mice was much smaller than that of gender-matched wild type (5-HT6RWT(+/+)) littermates. To clarify whether 5-HT6R-mediated signaling regulates RANKL-induced osteoclastogenesis, we examined osteoclast differentiation using ST1936, a specific 5-HT6R agonist in BMMs derived from 5-HT6RKO (-/-) mice (5-HT6RKO(-/-) BMMs). As shown in Figure 1E-F, the formation of RANKL-induced tartrate-resistant acid phosphatase (TRAP)-positive multinucleated osteoclasts (MNCs) in 5-HT6RKO(-/-) BMMs was markedly lower than that in BMMs from wild-type littermates. 5-HT6RKO(-/-) BMMs also profoundly abrogated ST1936-stimulated osteoclast differentiation compared to BMMs from wild-type littermates (Figure 1E-F). Together, these observations indicate that 5-HT6R has a regulatory role in osteoclastogenesis during in vivo bone remodeling.
5-HT6R-deficient (5-HT6RKO(-/-)) mice show an abnormal phenotype and impaired osteoclastogenesis. (A) Expression patterns of 5-HTRs were analyzed by RT-PCR analysis during osteoclastogenesis from primary cultured BMMs. β-actin was used as a loading control. NF-ATc1 and TRAP were used as differentiation markers. (B) BMMs were stimulated with RANKL in the presence of the indicated concentrations of specific 5-HTRs' agonists and 5-HT for 5 days. TRAP activity assays were performed at an absorbance of 405 nm. *, p < 0.05 compared to RANKL. (C) Genotyping was performed to analyze 5-HT6R gene knockout in 5-HT6R-deficient (5-HT6RKO(-/-)) mice. (D) Appearance of wild-type (5-HT6RWT(+/+)) and 5-HT6RKO(-/-) littermate mice. (E-F) Impaired osteoclastogenesis of 5-HT6RKO(-/-) mice in vitro. BMMs isolated from 5-HT6RWT(+/+)) or 5-HT6RKO(-/-) littermate mice were incubated with M-CSF and RANKL for 5 days in the presence of ST1936. TRAP-positive MNCs were detected with TRAP staining (E) and the number of MNCs with more than ten nuclei were counted (F). Scale bar: 200 μm. MNCs: multinucleated cells. NS: not significant. *, p < 0.05 compared to RANKL. #, p < 0.05 compared to RANKL + ST1936. Data shown represent mean ± SEM.
5-HT6R leads to osteoclast lineage commitment and promotes RANKL-induced osteoclastogenesis. (A-E) BMMs were cultured with M-CSF or M-CSF plus RANKL. ST1936 (1 μM) was added during the proliferation (BMM, i), differentiation (preOC, ii and iii) or maturation (OC, iv) stages for the indicated period (arrow) (A). TRAP-positive MNCs were detected with TRAP staining (B) and the number of MNCs with 3-10 nuclei (C), 10-20 nuclei (D), or more than twenty nuclei (E) were counted. Scale bar: 200 μm. MNCs: multinucleated cells. *, p < 0.05 compared to RANKL. (F-G) Osteoclast marker gene expression following RANKL-induced osteoclastogenesis in the presence of ST1936 were measured by RT-PCR (F) and quantitative RT-PCR (G). *, p < 0.05 compared to RANKL. Data shown represent mean ± SEM.
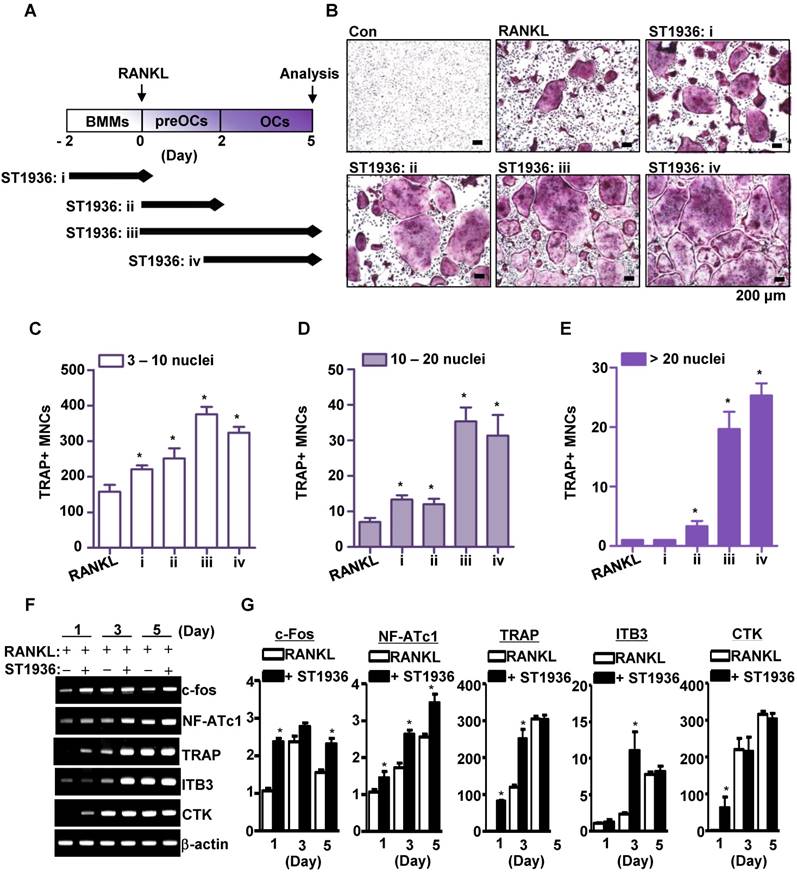
5-HT6R regulates osteoclastogenesis during all stages of cell proliferation, differentiation, and maturation
To determine the stage of osteoclast differentiation at which 5-HT6R acts in further detail, BMMs were treated with ST1936 for various time periods (Figure 2A). As shown in Figure 2B-E, ST1936 treatment during osteoclast differentiation and/or maturation stages (ii, iii, and iv) was necessary and sufficient to increase osteoclastogenesis, while exposure at the proliferation stage (i) increased TRAP-positive MNCs, but not the proportion of larger osteoclasts (Figure 2C, E). Concurrently, there were significant differences in the inducible levels of necessary and/or sufficient activation factors, including c-Fos, nuclear factor of activated T cells 1 (NFATc1), and TRAP for osteoclast differentiation and function (Figure 2F-G). However, the activation of 5-HT6R alone in BMMs without RANKL was found to increase c-Fos and NF-ATc1 gene expression (the initiation factors of osteoclast differentiation), but not that of TRAP, cathepsin K (CTK), and DC-Stamp (functional genes of osteoclasts) (Figure S2A). Furthermore, 5-HT6R alone did not induce TRAP-positive MNCs (Figure S2B-C). These data suggest that 5-HT6R was the driving force behind BMM proliferation into osteoclast lineages and was responsible for the regulation of RANKL-induced osteoclast maturation.
Signaling of 5-HT6R linked to RhoA GTPase is involved in RANKL-induced osteoclatogenesis
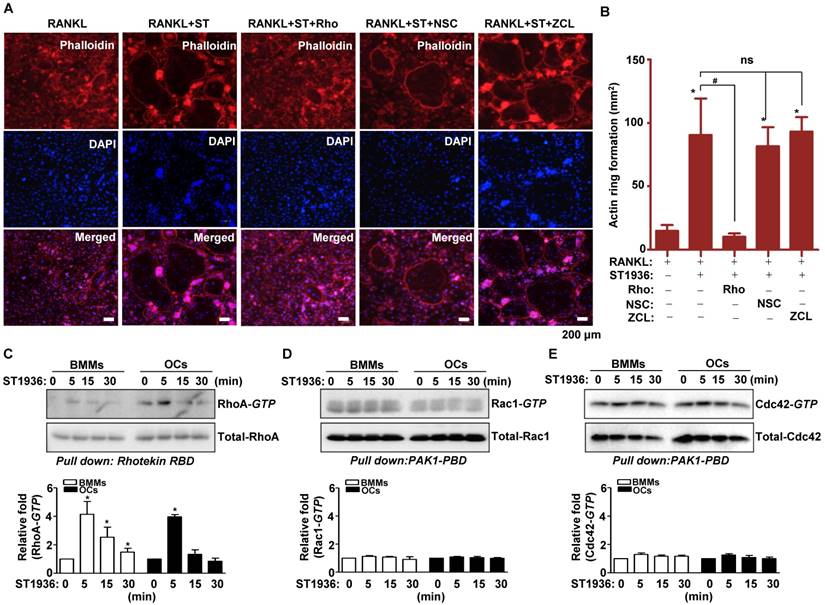
5-HT6R-mediated signaling increased actin ring formation (Figure S3A-B), which is essential for the differentiation and bone resorptive functions of osteoclasts. However, actin dynamics are also dependent on the regulation of the cytoskeletal structure by critical GTPase regulators belonging to the Rho subfamily (RhoA, Rac1, and Cdc42). To gain further insight into the molecular mechanism by which 5-HT6R regulates RANKL-induced osteoclastogenesis, we examined whether GTPases of the Rho subfamily are responsible for the osteoclast behavior induced by 5-HT6R. Therefore, TRAP-positive MNCs and actin ring formation were measured in osteoclasts treated with specific inhibitors of various GTPases of the Rho subfamily. Pretreatment with Rhosin (RhoA GTPase inhibitor) inhibited the 5-HT6R-induced increases in the number of TRAP-positive MNCs (Figure S4A-D) and actin ring formation (Figure 3A-B). Treatment with NSC23766 (Rac1 GTPase inhibitor) and ZCL278 (Cdc42 GTPase inhibitor) did not affect the stimulatory effects of 5-HT6R in osteoclasts (Figure S4A-D and Figure 3A-B). Through pull down assays, we also confirmed that 5-HT6R activation increased RhoA activity in BMMs and osteoclasts (Figure 3C), but not that of Rac1 and Cdc42 (Figure 3D-E). Our data suggest that 5-HT6R increases RANKL-induced osteoclastogenesis through the intracellular small RhoA GTPase pathway.
5-HT6R signaling linked to RhoA GTPase increases bone resorptive function of osteoclasts in mice
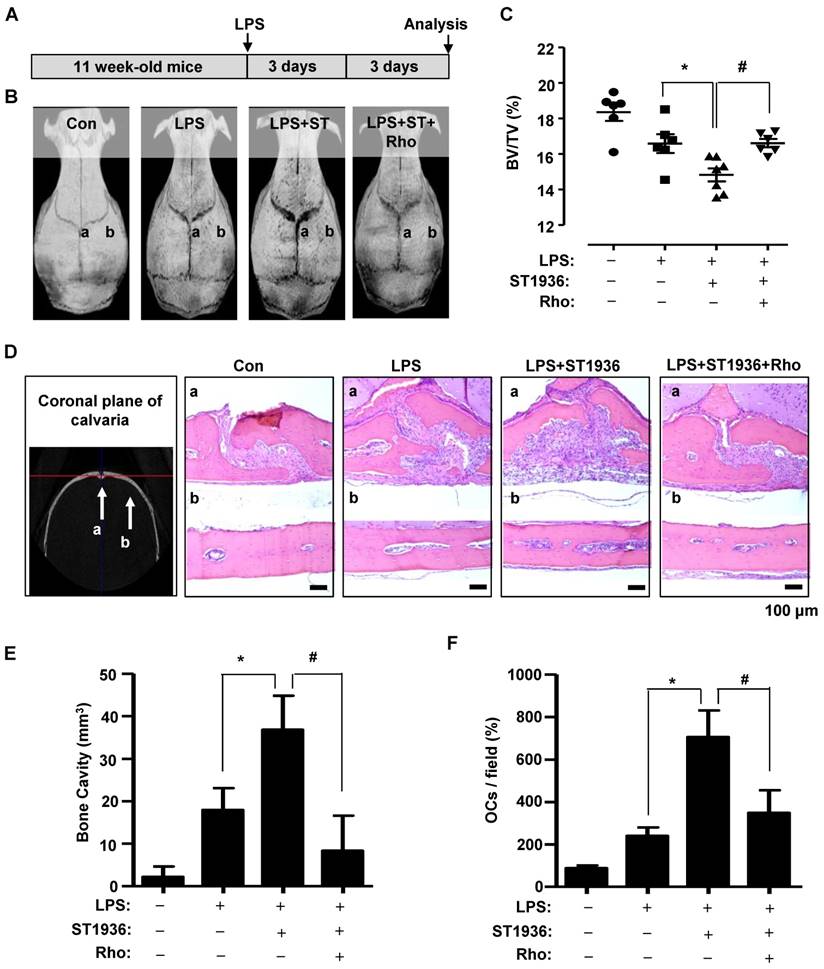
To investigate the in vivo effects of 5-HT6R on the pathological formation of osteoclasts during inflammation-mediated bone resorption, we utilized a lipopolysaccharide (LPS) challenge mouse model (Figure 4A). Bone destruction was visualized by μCT (Figure 4B). As shown in Figure 4B-C, the extent of bone erosion was notably increased in ST1936-treated mice compared to that in LPS-challenged mice. However, Rhosin treatment was found to significantly attenuate bone loss in ST1936-treated mice. Histological evaluation further supported the μCT findings by observation in the region (a and b) of coronally sectioned calvaria (Figure 4B, D). This indicates that bone loss in the calvarial sutures are the main centers for bone formation (Figure 4Da) and along with increased bone resorption inside the cavities of the calvariae (Figure 4Db, E), and an increase in the number of osteoclasts in ST1936-treated mice (Figure 4F). Meanwhile, Rhosin treatment was found to reverse the bone resorptive function of osteoclasts in ST1936-treated mice (Figure 4D-F). These data indicate that the 5-HT6R activates LPS-induced bone destruction via the intracellular small RhoA GTPase pathway.
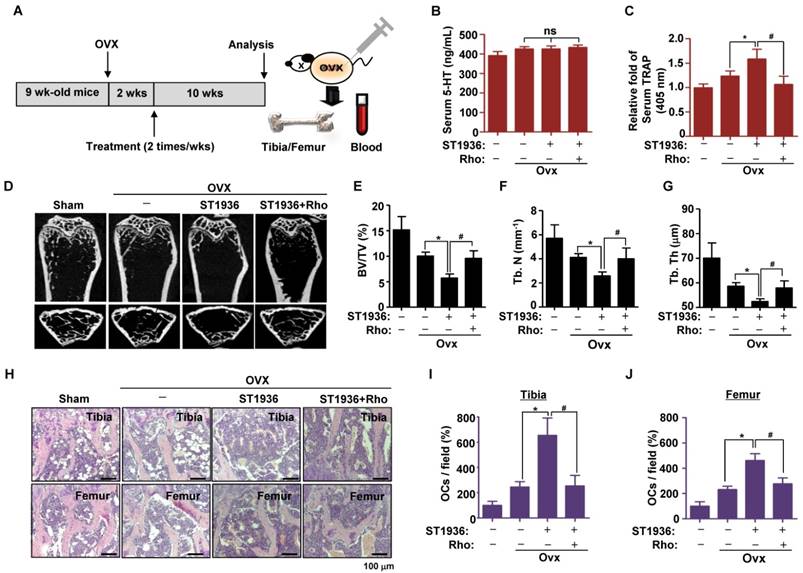
5-HT6R signaling linked to RhoA GTPase develops a high-bone-loss phenotype in ovariectomy-induced osteoporosis mice
To test the potential role of 5-HT6R in bone diseases, we studied the effects of 5-HT6R using an experimental in vivo ovariectomized (OVX) osteoporosis mouse model. Two weeks after ovariectomy, 11-week-old sham-operated or ovariectomized female mice were injected twice a week with compound carrier alone, ST1936, or ST1936 together with Rhosin over the course of 10 weeks (Figure 5A). Since gut-derived serum 5-HT levels are involved in bone metabolism, we first measured the serum concentration of 5-HT. Figure 5B shows there was no significant difference in serum 5-HT levels among OVX mice, while serum TRAP activity in ST1936-treated mice was significantly increased. Rhosin treatment abolished these effects (Figure 5C), suggesting that 5-HT6R does not affect circulating serum 5-HT levels but rather increases osteoclast activity. As shown in Figure 5D, μCT revealed that the femur of ST1936-treated mice displayed a marked decrease in bone mass, with a concomitant decrease in bone volume and trabecular bone number compared to that in OVX mice. Inhibition of RhoA GTPase was found to reverse OVX-induced bone loss in ST1936-treated mice (Figure 5E-G). Histological evaluation of the femur and tibia established that ST1936-treated mice had increased bone marrow cavity space when compared with OVX mice, together with increased osteoclast numbers (Figure 5H-J). Inhibition of RhoA GTPase activity abolished these observed increases (Figure 5H-J). Thus, these results indicate that the 5-HT6R-mediated RhoA GTPase pathway plays a critical role in RANKL-induced differentiation and function of osteoclasts and bone diseases.
5-HT6R increases RANKL-induced osteoclastogenesis via the small RhoA GTPase pathway. (A-B) BMMs were stimulated with RANKL plus 1 μM ST1936 in the absence or presence of 1 μM Rhosin (RhoA GTPase inhibitor), 1 μM NSC23766 (Rac1 GTPase inhibitor), or 1 μM ZCL278 (Cdc42 GTPase inhibitor). F-actin-positive cells (red, (A)), and actin ring formation were measured in actin ring-positive MNCs (B) by confocal microscopy following staining with TRITC-phalloidin (red) and DAPI (blue). Scale bar: 200 μm. Rho: Rhosin; NSC: NSC23766; ZCL: ZCL278. *, p < 0.05 compared to RANKL. #, p < 0.05 compared to RANKL + ST1936. (C-E) BMMs (left) or OC (right) were stimulated with 1 μM ST1936 for the indicated time after being serum-starved for 6 h. RhoA (C), Rac1 (D), and Cdc42 (E) activities were assessed by pull-down assay to detect the GTP-bound form (active form). Total-RhoA, -Rac1, and -Cdc42 were detected as loading controls by western blotting. *, p < 0.05 compared to control. Data shown represent mean ± SEM.
Discussion
To the best of our knowledge, involvement of the 5-HT system in bone metabolism remains controversial. Previously, it was shown that serum 5-HT levels reduce bone formation by inhibiting osteoblast proliferation [35]. In stark contrast, 5-HT treatment has also been found to increase bone formation in mice or rats [20-21]. Other groups have also observed no change in bone density following 5-HT treatment [20]. To study bone remodeling in osteoclasts by specific 5-HTRs, the expression of 5-HTR subtypes was herein analyzed. All known main 5-HTR subtypes (5-HT1R-5-HT7R) were found to be expressed in BMMs, pre-osteoclasts, and mature osteoclasts, indicating systematic roles of 5-HTRs in skeletal modeling, remodeling, and bone mass development. Among the 5-HTR subtypes, activation of 5-HT6R was found to significantly increase the osteoclast differentiation phenotype. In comparison to that of 5-HT6R wild-type (5-HT6RWT(+/+)) mice, the body size of 5-HT6R-deficient (5-HT6RKO(-/-)) mice presented with an abnormal phenotype. In addition, RANKL-induced osteoclast differentiation in 5-HT6RKO(-/-) BMMs was much slower than that in BMMs from 5-HT6RWT(+/+) littermates. Furthermore, ST1936-stimulated osteoclast differentiation was abrogated in 5-HT6RKO(-/-) BMMs. These results provide new evidence that 5-HT6R has a potential role in the regulation of osteoclast differentiation and function in in vivo bone remodeling.
5-HT6R-mediated RhoA GTPase signaling increases bone resorption in mice. (A) Scheme for the assessment of the LPS bone resorption mouse model. (B-C) μCT images are shown (B), and bone volume (BV) per tissue volume (TV) of calvarias (right) were analyzed and plotted as individual points (C). (D-F) Calvarias were coronally sectioned in regions a and b indicated by the arrow (left) and analyzed using H&E staining (right) (D). The differences of bone cavity (E) and the number of osteoclasts (OCs) (F) were analyzed. Scale bar: 100 μm. Rho: Rhosin. *, p < 0.05 compared to LPS. #, p < 0.05 compared LPS+ST1936. Data shown represent mean ± SEM.
A major component of the transcription factor AP-1 is c-Fos, which is induced by RANKL. c-Fos plays an essential role in the initiation of osteoclast differentiation and is responsible for the expression of another key regulator of osteoclastogenesis, namely NFATc1 [36-37]. Microarray analysis revealed that NFATc1 was down-regulated in Fos KO(-/-) osteoclast precursors lacking c-Fos, which led to impaired oteoclast differentiation [38]. Consistent with this notion, 5-HT6R was herein found to markedly increase RANKL-induced c-Fos and NFATc1 expression, together with the generation of TRAP-positive MNCs and actin ring formation. Furthermore, activation of 5-HT6R was shown to potentiate RANKL-induced osteoclast differentiation at both the immature and mature osteoclast stages or even through preactivation of 5-HT6R in BMMs. However, 5-HT6R alone without RANKL did not trigger osteoclast differentiation from BMMs nor did it induce the functional genes of osteoclasts, such as TRAP, CTK, and Dc-Stamp, even though 5-HT6R alone induces c-Fos and NF-ATc1 genes in BMMs without RANKL. These results support our idea that 5-HT6R provides the driving force for osteoclast lineage commitment and is a regulator for the potentiation of osteoclast differentiation and function.
Osteoclasts are highly motile cells that depend on rapid changes in their actin cytoskeleton to accomplish their ordered cycles of movement and attachment during bone resorption [5-6]. Rho GTPase activity is a critical regulator of the actin cytoskeleton in osteoclasts and is required for bone resorption via regulation of actin ring formation [39-41]. Consistent with this notion, 5-HT6R was seen to exhibit markedly increased actin ring formation and osteoclast maturation activity for excavating bone pits via small RhoA GTPase, but not small Cdc42 and Rac1 GTPase. We further demonstrated that 5-HT6R increased bone destruction through bone resorptive osteoclast function in LPS-challenged mice, while inhibition of RhoA GTPase activity was found to block these effects. This observation is consistent with the actin ring structure of resorbing osteoclasts being disrupted by the C3 exoenzyme, which specifically acts by ADP-ribosylating (inactivates) the Rho protein [42-43]. In addition, C3 exoenzyme blocked the effects of RhoVal-14 (constitutively active)-induced and RhoAsn-19 (dominant negative)-inhibited bone resorption in osteoclasts [42, 44]. RhoA GTPase signaling also activates the c-jun promoter and thereby enhances c-Jun expression [45]. These findings suggest that 5-HT6R-mediated RhoA GTPase signaling coordinates intracellular cytoskeletal changes following the transcriptional activation of genetic pathways that contribute to osteoclast differentiation, sealing zone formation, and bone resorption.
Osteoporosis is one of the most common bone diseases characterized by decreased bone mass, increased bone fragility, and increased risk of fractures [1, 46]. Estrogen deficiency is a major factor in the pathogenesis of postmenopausal osteoporosis, resulting in bone fracture in the elderly, especially postmenopausal women [47-48]. The functional roles of 5-HT6R in osteoclastogenesis were herein validated using an osteoporosis disease model. We generated ovariectomized (OVX) mice, a widely used animal model for the development of prevention and treatment strategies for postmenopausal osteoporosis [41, 49]. Yadav et al. showed that decrease of circulating 5-HT by LP533401 caused a major increase in bone formation parameters, such as osteoblast numbers, bone formation rate, and osteocalcin serum levels [35]. In the present study, we observed that serum 5-HT levels did not change through 5-HT6R-mediated RhoA GTPase signaling. This suggests that there is direct regulation of osteoclast bone resorbing function by this system without an influence on circulating serum 5-HT levels. Consistent with an abnormal bone phenotype in 5-HT6R-deficient (5-HT6RKO(-/-)) mice, the 5-HT6R-mediated RhoA GTPase signaling showed significant effects on bone volume and lower trabecular bone number in Ovx mice. Histological assessment further verified that 5-HT6R signaling caused a significant reduction in trabecular bone number and a marked increase in osteoclast number in both femurs and tibias. These data suggest that the pathological dysregulation of 5-HT6R causes bone diseases such as osteoporosis, by directly promoting osteoclast differentiation and function via the RhoA GTPase pathway.
In conclusion, these are the first reported findings that demonstrate the critical role of 5-HT6R-mediated RhoA GTPase signaling in osteoclastogenesis, bone resorption, and osteoporosis, using in vivo 5-HT6RKO(-/-) mice, animal experimental models, and an in vitro system. The crucial roles identified for 5-HT6R, in a new context of osteoclast lineages, suggest that 5-HT6R may be an ideal target for the development of therapeutic agents that could directly regulate bone resorption in bone diseases, such as osteoporosis.
Methods
Generation of 5-HT6R-deficient (5-HT6RKO(-/-)) mice and associated experiments
The mouse strain (B6;129S5-Htr6tm1Lex/Mmucd) was obtained from the Mutant Mouse Regional Resource Center (MMRRC), a NCRR-NIH funded strain repository, which was donated to the MMRRC by Lexicon Genetics Incorporated. The strain was maintained as cryo-archived material for distribution and was recovered prior to use. Genomic DNA isolated from the tail of recovered litters was analyzed by PCR analysis using specific primers. 5-HT6R-deficient (5-HT6RKO(-/-)) mice were generated by breeding heterozygous to heterozygous, and the femur bones of male 5-HT6RWT(+/+) and 5-HT6RKO(-/-) mice were analyzed by micro-computed tomography (μCT) in 2-week-old mice. BMMs isolated from 5-HT6RWT(+/+) and 5-HT6RKO(-/-) mice were cultured for in vitro osteoclast differentiation. All mice used in this study were maintained in accordance with the National Institute of Toxicological Research of the Korea Food and Drug Administration guidelines for the humane care and use of laboratory animals. All animal experiments were performed in accordance with the guidelines prescribed by Kyung Hee University Animal Care Committee and Chungbuk National University Animal Care Committee (CBNUA-436-12-02).
Primary cell isolation and osteoclast differentiation
Mouse whole bone marrow cells were isolated by flushing the femora and tibiae marrow space in 5-week-old mice. Cells were incubated overnight in culture dishes containing α-Modified Eagle Medium (α-MEM) (Gibco Laboratories, Grand Island, NY), supplemented with 10% fetal bovine serum (FBS; Gibco Laboratories) and antibiotics (100 mg/mL penicillin G and 100 IU/mL streptomycin) at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. After discarding adherent cells, floating cells were further incubated with mouse M-CSF (30 ng/mL) in Petri dishes. BMMs became adherent following 3 days of culture, after which cells were differentiated into osteoclasts using mouse RANKL (100 ng/ mL) and M-CSF for 5 days.
Chemical inhibitors and 5-HT receptor ligands
The following 5-HTR ligands and Rho GTPase family inhibitors were purchased from Tocris Bioscience (Bristol, UK): 8-hydroxy-PIPAT oxalate, alpha-methyl-5-hydroxyltryptamine maleate, N-methylquipazine dimaleate, cisapride, 5-carboxamidotrypamine maleate, ST1936 oxalate, AS 19, SB258585, 5-HT hydrochloride. rhosin hydrochloride, NSC 23766, and ZCL 278.
5-HT6R-mediated RhoA GTPase signaling promotes bone destruction in ovariectomy-induced osteoporosis mice. (A) Scheme for the assessment of the ovariectomized (OVX) osteoporosis mouse model. (B-C) Serum 5-HT and TRAP were measured by ELISA. (D-G) μCT images are shown in distal femurs (D). Bone volume (BV) per tissue volume (TV) (E), trabecular number (Tb. N) (F), and trabecular thickness (Tb. Th) (G) were analyzed. (H-J) H&E staining was performed on the tibial and femurs sections (H). The number of osteoclasts (OCs) was analyzed in the tibial (I) and femurs sections (J). Scale bar: 100 μm. Rho: Rhosin. *, p < 0.05 compared to Ovx mice. #, p < 0.05 compared to Ovx + ST1936. Data shown represent mean ± SEM.
TRAP staining and activity analysis
After BMMs were differentiated into osteoclasts for 5 days, cells were fixed with 4% formaldehyde for 15 min. Cells were then washed and stained for TRAP using a leukocyte acid phosphatase cytochemistry kit (Sigma), according to the manufacturer's instructions. TRAP-positive multinucleated cells (MNCs) containing three or more nuclei were counted as mature osteoclasts using a light microscope. Typically, cells were predominantly TRAP-positive and mononuclear 2 days after induction with RANKL, and TRAP-positive and multinuclear another 3 days later. We considered mononuclear TRAP-positive cells to be preosteoclasts and multinuclear cells to be mature osteoclasts, as described previously [41]. The TRAP activity assay was performed as previously described [50].
Actin-ring formation analysis
Osteoclasts were fixed with 4% formaldehyde, permeabilized with 0.1% Triton X-100, and incubated with FITC- or TRITC-phalloidin (Invitrogen) for 30 min. Following washing with PBS, cells were incubated with DAPI (Sigma-Aldrich), washed three times, and subsequently viewed on a fluorescence microscope (Carl Zeiss, Oberkochen, Germany).
Reverse transcription-polymerase chain reaction (RT-PCR) and quantitative real-time PCR analysis
Total RNA from cells was extracted using TRIzol™ reagent (Life Technologies, Gaithersburg, MD) according to the manufacturer's instructions. RNA (1 µg) isolated from each sample was reverse-transcribed using oligo (dT)15 primers with AccuPower® RT PreMix (iNtRON Biotechnology, Gyeonggi-do, South Korea). Next, the resultant cDNA was amplified with AccuPower® PCR PreMix (Bioneer Corporation, Daejeon, South Korea). For mRNA quantification, total RNA was extracted using the RNAqueous® kit and cDNA synthesized from 1 μg of total RNA using the High Capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, CA) according to the manufacturer's protocol. Quantitative real-time PCR was performed using a LightCycler® 1.5 System (Roche Diagnostics GmbH, Mannheim, Germany). Thermocycling conditions consisted of an initial denaturation step of 10 s at 95 ºC, followed by 45 cycles of 95 ºC for 10 s, 60 ºC for 5 s and 72 ºC for 10 s. To calculate relative quantification, the 2-ΔΔCT formula was used, where -ΔΔCT = (CT,target-CT,β-actin) experimental sample - (CT,target-CT,β-actin) control sample. The primer sequences for 5-HTRs are listed in Table S1.
Western blot analysis
Cells were washed twice with ice-cold PBS, and lysed in 20 mM Tris-HCl buffer (pH 7.4) containing a protease inhibitor mixture (0.1 mM PMSF, 5 mg/mL aprotinin, 5 mg/mL pepstatin A, and 1 mg/mL chymostatin). Protein concentration was determined using Bradford reagent (Bio-Rad, Hercules, CA). Equal amounts of lysate (20 μg) were resolved by sodium dodecyl-polyacrylamide gel electrophoresis (SDS-PAGE), and subsequently transferred to a polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, MA). Thereafter, the membrane was blocked with 1 × TBS containing 0.05% Tween 20 (TBST) and 5% skim milk or 2% BSA for 1 h at room temperature. After blocking, the membranes were incubated overnight at 4 °C with the respective primary antibodies, washed with 1 × TBST, and then incubated with diluted horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10,000, Jackson ImmunoResearch, West Grove, PA) for 1 h at room temperature. After three washes, the bound antibodies were detected using an enhanced chemiluminescence (ECL) kit (Millipore, Bedford, MA).
Pull-down assay for RhoA/Rac1/Cdc42 GTPase activity
Activation of RhoA, Rac1, and Cdc42 GTPase was examined with a pull-down assay using a commercially available RhoA/Rac1/Cdc42 Activation Assay Combo Kit (Cell Biolabs, Inc., San Diego, CA). In brief, cell lysates were collected and incubated with Rhotekin RBD Agarose for RhoA or with PAK1 PBD Agarose for Rac1 and Cdc42 at 4 °C with gentle rotation. The resulting complexes were precipitated and eluted by boiling for 5 min at 95 °C in SDS-PAGE sample buffer, followed by western blot analysis using anti-RhoA, anti-Rac1, or anti-Cdc42 antibodies.
Bone resorption animal model for in vivo lipopolysaccharide (LPS) challenge
Development of an inflammation-induced model of bone loss has been previously described [50]. For this study, when mice were 11 weeks of age, subcutaneous tissue over the periosteum of the mouse calvaria was injected with vehicle alone (n = 6), LPS (12.5 mg/kg) alone (n = 6), or LPS in combination with ST1936 (10 mg/kg) (n = 7) in the absence or presence of Rhosin (4 mg/kg) (n = 6) at day 1 and 3. Mice were sacrificed at 6 days after the first injection of LPS. The calvarial bones of the mice were analyzed by μCT. Quantitative analysis of the relative percentage of bone resorption (BV/TV) was performed by μCT imaging. Thereafter, tissues were demineralized in 15% EDTA for 14 days, embedded in paraffin, and sectioned at 5 μm. For histology analysis, sections were stained with hematoxylin and eosin (H&E).
In vivo ovariectomy (Ovx)-induced osteoporosis model
When mice were 9 weeks of age, ovariectomy was performed at the Japan SLC, Inc (Shizuoka, Japan) under anesthesia by intraperitoneal injection of ketamine (Sankyo, Tokyo, Japan) and xylazine (Zenoaq, Fukushima, Japan). Two weeks after ovariectomy (11 weeks of age), mice were divided into four groups: Sham group (n=6), OVX-control group (n = 8), OVX+ST1936 (10 mg/kg) group (n = 8), and OVX + ST1936 + Rhosin (4 mg/kg) group (n = 8). The mice in each group were thereafter injected twice a week. Femur μ-CT analysis for bone volume (BV) per tissue volume (TV), trabecular number (Tb. N), and trabecular thickness (Tb. Th) and histology analysis were carried out essentially as previously described [41].
Micro-computed tomography (μCT)
μCT was performed at the Advanced Institutes of Convergence Technology (Genoss Co., Ltd., Gyeonggi-do Korea). μCT data of calvaria were acquired on a Skyscan 1173 scanner (Bruker-microCT, Kontich, Belgium). Scanning was carried out at 75 kV/106 μA for 500 ms. In total, 800 projections were collected at a resolution of 9.94 μm/pixel. Reconstruction of sections was carried out using software associated with the scanner (Nrecon), with beam hardening correction set to 40%. Realistic 3D-Visualization software (Bruker-microCT, Konitch, Belgium) was used to reconstruct the μCT images in three-dimension, which were acquired as approximately 2000 cross-sections.
Serum collection and serum ELISA assay
Whole blood collected from mice was processed within 2 h of collection using the following protocol. Collected blood was centrifuged at 1000 × g for 15 min at room temperature, after which the serum supernatant was aspirated and transferred to a new tube. This was subsequently aliquoted into Eppendorf tubes and stored at -80 °C until analyzed. An ELISA (5-HT ELISA; Abcam, Cambridge, MA) was used to measure 5-HT ELISA levels in the serum, according to the manufacturer's instructions.
Statistical analysis
The data were analyzed using GraphPad Prism version 5 software (GraphPad Software, Inc., San Diego, CA). Data are presented as mean ± SEM. Statistical significance was evaluated by one-way analysis of variance (ANOVA) and the differences were assessed by the Dunnett's test. A value of P < 0.05 was considered to indicate statistical significance.
Abbreviations
BMMs: bone marrow-derived macrophages; CNS: central nervous system; CTK: cathepsin K; 5-HT: 5-hydroxytryptamine; 5-HT6RKO-/-: 5-HT6R-deficient; 5-HTRs: 5-HT receptors; SERT: 5-HT transporter; GPCRs: G protein-coupled receptors; LPS: lipopolysaccharide; M-CSF: macrophage colony-stimulating factor; μCT: micro-computed tomography; MNCs: multinucleated cells; NFATC1: nuclear factor of activated T cells 1; OVX: ovariectomized; RANKL: receptor activator of nuclear factor (NF)-κB ligand; TRAP: tartrate-resistant acid phosphatase; TNF: tumor necrosis factor.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by the National Research Foundation of Korea [NRF] grant funded by the Korea government (MSIP) (No. MRC, 2017R1A5A2015541, 2015R1D1A1A01059240, and 2012R1A5A2051384)
Author Contributions
K.-R.P. performed conceptualization, data curation, investigation, formal analysis, methodology, software, validation, writing (original draft), and revision. E.-C.K. provided resources. J.T.H. performed formal analysis, methodology, software, validation, project administration, resources, writing (review & editing), and revision. H.-M.Y. performed conceptualization, data curation, investigation, formal analysis, methodology, software, validation, project administration, resources, supervision, writing (original draft, review & editing), and revision.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Marie PJ, Kassem M. Osteoblasts in osteoporosis: past, emerging, and future anabolic targets. Eur J Endocrinol. 2011;165:1-10
2. Khosla S, Riggs BL. Pathophysiology of age-related bone loss and osteoporosis. Endocrinol Metab Clin North Am. 2005;34:1015-30 xi
3. Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4:638-49
4. Vignery A. Macrophage fusion: the making of osteoclasts and giant cells. J Exp Med. 2005;202:337-40
5. Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337-42
6. Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504-8
7. Takayanagi H. Osteoimmunology and the effects of the immune system on bone. Nat Rev Rheumatol. 2009;5:667-76
8. Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13:791-801
9. Mundy GR. Osteoporosis and inflammation. Nutr Rev. 2007;65:S147-51
10. Aoki K, Saito H, Itzstein C, Ishiguro M, Shibata T, Blanque R. et al. A TNF receptor loop peptide mimic blocks RANK ligand-induced signaling, bone resorption, and bone loss. J Clin Invest. 2006;116:1525-34
11. Bornstein JC. Serotonin in the gut: what does it do? Front Neurosci. 2012;6:16
12. Gershon MD, Tack J. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology. 2007;132:397-414
13. Mann JJ, McBride PA, Brown RP, Linnoila M, Leon AC, DeMeo M. et al. Relationship between central and peripheral serotonin indexes in depressed and suicidal psychiatric inpatients. Arch Gen Psychiatry. 1992;49:442-6
14. de Vernejoul MC, Collet C, Chabbi-Achengli Y. Serotonin: good or bad for bone. Bonekey Rep. 2012;1:120
15. Svenningsson P, Tzavara ET, Liu F, Fienberg AA, Nomikos GG, Greengard P. DARPP-32 mediates serotonergic neurotransmission in the forebrain. Proc Natl Acad Sci USA. 2002;99:3188-93
16. Petersen T, Dording C, Neault NB, Kornbluh R, Alpert JE, Nierenberg AA. et al. A survey of prescribing practices in the treatment of depression. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26:177-87
17. Cipriani A, Furukawa TA, Salanti G, Geddes JR, Higgins JP, Churchill R. et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373:746-58
18. Baudry A, Mouillet-Richard S, Schneider B, Launay JM, Kellermann O. miR-16 targets the serotonin transporter: a new facet for adaptive responses to antidepressants. Science. 2010;329:1537-41
19. Eom CS, Lee HK, Ye S, Park SM, Cho KH. Use of selective serotonin reuptake inhibitors and risk of fracture: a systematic review and meta-analysis. J Bone Miner Res. 2012;27:1186-95
20. Cui Y, Niziolek PJ, MacDonald BT, Zylstra CR, Alenina N, Robinson DR. et al. Lrp5 functions in bone to regulate bone mass. Nat Med. 2011;17:684-91
21. Battaglino R, Vokes M, Schulze-Spate U, Sharma A, Graves D, Kohler T. et al. Fluoxetine treatment increases trabecular bone formation in mice. J Cell Biochem. 2007;100:1387-94
22. Gustafsson BI, Westbroek I, Waarsing JH, Waldum H, Solligard E, Brunsvik A. et al. Long-term serotonin administration leads to higher bone mineral density, affects bone architecture, and leads to higher femoral bone stiffness in rats. J Cell Biochem. 2006;97:1283-91
23. Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology. 1999;38:1083-152
24. Branchek TA, Blackburn TP. 5-ht6 receptors as emerging targets for drug discovery. Annu Rev Pharmacol Toxicol. 2000;40:319-34
25. Yoshioka M, Matsumoto M, Togashi H, Mori K, Saito H. Central distribution and function of 5-HT6 receptor subtype in the rat brain. Life Sci. 1998;62:1473-7
26. Duhr F, Deleris P, Raynaud F, Seveno M, Morisset-Lopez S, Mannoury la Cour C. et al. Cdk5 induces constitutive activation of 5-HT6 receptors to promote neurite growth. Nat Chem Biol. 2014;10:590-7
27. Jacobshagen M, Niquille M, Chaumont-Dubel S, Marin P, Dayer A. The serotonin 6 receptor controls neuronal migration during corticogenesis via a ligand-independent Cdk5-dependent mechanism. Development. 2014;141:3370-7
28. Seo J, Tsai LH. Neuronal differentiation: 5-HT6R can do it alone. Nat Chem Biol. 2014;10:488-9
29. Wilkinson D, Windfeld K, Colding-Jorgensen E. Safety and efficacy of idalopirdine, a 5-HT6 receptor antagonist, in patients with moderate Alzheimer's disease (LADDER): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2014;13:1092-9
30. Maher-Edwards G, Zvartau-Hind M, Hunter AJ, Gold M, Hopton G, Jacobs G. et al. Double-blind, controlled phase II study of a 5-HT6 receptor antagonist, SB-742457, in Alzheimer's disease. Curr Alzheimer Res. 2010;7:374-85
31. Maher-Edwards G, Dixon R, Hunter J, Gold M, Hopton G, Jacobs G. et al. SB-742457 and donepezil in Alzheimer disease: a randomized, placebo-controlled study. Int J Geriatr Psychiatry. 2011;26:536-44
32. Asaoka N, Nagayasu K, Nishitani N, Yamashiro M, Shirakawa H, Nakagawa T. et al. Olanzapine augments the effect of selective serotonin reuptake inhibitors by suppressing GABAergic inhibition via antagonism of 5-HT receptors in the dorsal raphe nucleus. Neuropharmacology. 2015;95:261-8
33. Meffre J, Chaumont-Dubel S, Mannoury la Cour C, Loiseau F, Watson DJ, Dekeyne A. et al. 5-HT(6) receptor recruitment of mTOR as a mechanism for perturbed cognition in schizophrenia. EMBO Mol Med. 2012;4:1043-56
34. Yun HM, Park KR, Hong JT, Kim EC. Peripheral serotonin-mediated system suppresses bone development and regeneration via serotonin 6 G-protein-coupled receptor. Sci Rep. 2016;6:30985
35. Yadav VK, Ryu JH, Suda N, Tanaka KF, Gingrich JA, Schutz G. et al. Lrp5 controls bone formation by inhibiting serotonin synthesis in the duodenum. Cell. 2008;135:825-37
36. Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H. et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889-901
37. Kwak HB, Lee BK, Oh J, Yeon JT, Choi SW, Cho HJ. et al. Inhibition of osteoclast differentiation and bone resorption by rotenone, through down-regulation of RANKL-induced c-Fos and NFATc1 expression. Bone. 2010;46:724-31
38. Matsuo K, Galson DL, Zhao C, Peng L, Laplace C, Wang KZ. et al. Nuclear factor of activated T-cells (NFAT) rescues osteoclastogenesis in precursors lacking c-Fos. J Biol Chem. 2004;279:26475-80
39. Hall A. Rho family GTPases. Biochem Soc Trans. 2012;40:1378-82
40. Touaitahuata H, Blangy A, Vives V. Modulation of osteoclast differentiation and bone resorption by Rho GTPases. Small GTPases. 2014;5:e28119
41. Chang EJ, Ha J, Oerlemans F, Lee YJ, Lee SW, Ryu J. et al. Brain-type creatine kinase has a crucial role in osteoclast-mediated bone resorption. Nat Med. 2008;14:966-72
42. Zhang D, Udagawa N, Nakamura I, Murakami H, Saito S, Yamasaki K. et al. The small GTP-binding protein, rho p21, is involved in bone resorption by regulating cytoskeletal organization in osteoclasts. J Cell Sci. 1995;108( Pt 6):2285-92
43. Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389-99
44. Chellaiah MA, Soga N, Swanson S, McAllister S, Alvarez U, Wang D. et al. Rho-A is critical for osteoclast podosome organization, motility, and bone resorption. J Biol Chem. 2000;275:11993-2002
45. Marinissen MJ, Chiariello M, Tanos T, Bernard O, Narumiya S, Gutkind JS. The small GTP-binding protein RhoA regulates c-jun by a ROCK-JNK signaling axis. Mol Cell. 2004;14:29-41
46. Krzeszinski JY, Wei W, Huynh H, Jin Z, Wang X, Chang TC. et al. miR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2. Nature. 2014;512:431-5
47. Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int. 2006;17:1726-33
48. Gambacciani M, Levancini M. Hormone replacement therapy and the prevention of postmenopausal osteoporosis. Prz Menopauzalny. 2014;13:213-20
49. Yadav VK, Balaji S, Suresh PS, Liu XS, Lu X, Li Z. et al. Pharmacological inhibition of gut-derived serotonin synthesis is a potential bone anabolic treatment for osteoporosis. Nat Med. 2010;16:308-12
50. Kim H, Choi HK, Shin JH, Kim KH, Huh JY, Lee SA. et al. Selective inhibition of RANK blocks osteoclast maturation and function and prevents bone loss in mice. J Clin Invest. 2009;119:813-25
Author contact
![]() Corresponding authors: Jin Tae Hong: College of Pharmacy and Medical Research Center, Chungbuk National University, Osongsaengmyeong 1-ro 194-31, Osong-eup, Heungduk-gu, Cheongju, Chungbuk, 28160, Republic of Korea. Tel: +82-43-261-2813. E-mail: jinthongac.kr; Hyung-Mun Yun; Department of Oral and Maxillofacial Pathology, School of Dentistry, Kyung Hee University, 1 Heogi-Dong, Dongdaemun-Gu, Seoul 130-701, Republic of Korea; Telephone: 82-02-961-0691; Fax: 82-02-960-1457; E-mail: yunhmac.kr
Corresponding authors: Jin Tae Hong: College of Pharmacy and Medical Research Center, Chungbuk National University, Osongsaengmyeong 1-ro 194-31, Osong-eup, Heungduk-gu, Cheongju, Chungbuk, 28160, Republic of Korea. Tel: +82-43-261-2813. E-mail: jinthongac.kr; Hyung-Mun Yun; Department of Oral and Maxillofacial Pathology, School of Dentistry, Kyung Hee University, 1 Heogi-Dong, Dongdaemun-Gu, Seoul 130-701, Republic of Korea; Telephone: 82-02-961-0691; Fax: 82-02-960-1457; E-mail: yunhmac.kr