Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2018; 8(11):2927-2938. doi:10.7150/thno.25317 This issue Cite
Research Paper
Histone deacetylase 6 modulates macrophage infiltration during inflammation
Bing Yan1*, Songbo Xie2 ![]() *, Yang Liu1*, Wenxuan Liu1, Dengwen Li1, Min Liu2, Hongbo R. Luo3, Jun Zhou1,2
*, Yang Liu1*, Wenxuan Liu1, Dengwen Li1, Min Liu2, Hongbo R. Luo3, Jun Zhou1,2 ![]()
1. State Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Bioactive Materials of the Ministry of Education, Tianjin Key Laboratory of Protein Science, College of Life Sciences, Nankai University, Tianjin 300071, China.
2. Shandong Provincial Key Laboratory of Animal Resistance Biology, Collaborative Innovation Center of Cell Biology in Universities of Shandong, Institute of Biomedical Sciences, College of Life Sciences, Shandong Normal University, Jinan, Shandong 250014, China.
3. Department of Pathology, Harvard Medical School and Department of Lab Medicine, Children's Hospital Boston and Dana-Farber/Harvard Cancer Center, Boston, MA 02115, USA.
*These authors contributed equally to this work.
Received 2018-2-3; Accepted 2018-3-16; Published 2018-4-18
Abstract

Mice with histone deacetylase 6 (HDAC6) deficiency grow and develop normally but exhibit impaired immune response. The molecular mechanisms for this phenotype remain largely elusive.
Methods: A mouse acute peritonitis model was used to study the infiltration of neutrophils and monocyte-derived macrophages. In vitro cell motility assays were performed to analyze monocyte/macrophage recruitment. Fluorescence microscopy and flow cytometry were performed to examine the phagocytic ability of macrophages. Immunofluorescence microscopy was used to investigate protein localization, protrusion formation, and microtubule acetylation.
Results: HDAC6 deficiency does not affect neutrophil infiltration, but instead attenuates the infiltration of monocyte-derived macrophages into the peritoneal cavity. HDAC6 plays a specific role in monocyte/macrophage recruitment. Loss of HDAC6 suppresses the phagocytic capacity of macrophages challenged with E. coli. Lipopolysaccharide stimulation results in the translocation of HDAC6 and cortactin from the cytosol to the cell periphery, promotes the formation of filopodial protrusions, and enhances microtubule acetylation around the microtubule-organizing center, all of which are abrogated by HDAC6 deficiency.
Conclusion: These findings implicate HDAC6 in the innate immune response and suggest that it may serve as a promising target for the treatment of macrophage-associated immune diseases.
Keywords: HDAC6, inflammation, macrophage, infiltration, migration, acetylation
Introduction
Inflammation, a protective innate immune response to invading pathogens or tissue injury, is a 'double-edged sword' that can also cause a number of immune disorders, such as septic shock, rheumatoid arthritis, atherosclerosis, and neurodegenerative diseases, when over-activated or inappropriately activated [1-4]. The process of inflammation begins with an influx of neutrophils, followed by recruitment of monocytes that subsequently differentiate into inflammatory macrophages or dendritic cells [5]. Neutrophils and monocytes/macrophages serve as the key effector cells at sites of inflammation, where they recognize and phagocytose pathogens, as well as necrotic cells, contributing to the clearance of the infection [5, 6]. In addition, they activate the adaptive immune response through communication and coordination with other immune cells. A growing body of evidence reveals that recruitment of inflammatory immune cells, including neutrophils and monocytes/macrophages, also plays a key role in tumorigenesis, as these cells can be hijacked by cancers to promote cancer development [7-9]. Therefore, targeting infiltration of inflammatory immune cells may represent an effective therapeutic strategy for the treatment of cancer, in addition to inflammatory and immune disorders. However, the molecular mechanisms that contribute to infiltration of these cells remain poorly understood.
Actin filaments and microtubules are central to a wide variety of cellular functions, including maintenance and alteration of cell shape, as well as cell division, cell motility, and intracellular transport. In response to inflammatory cues, the actin filaments and microtubules of innate immune cells undergo dynamic remodeling to promote extravasation and phagocytosis. Neutrophils move faster than monocytes/macrophages and display a unique asymmetric polarized morphology during migration [10], suggesting that cytoskeletal remodeling differs between neutrophils and other types of cells. However, the molecular mechanisms by which actin filaments and microtubules are differentially orchestrated in different immune cell types are unknown.
Histone deacetylase 6 (HDAC6) has recently been identified as an important regulator of cytoskeletal dynamics. It modifies two key cytoskeletal substrates: α-tubulin and cortactin, a filamentous actin (F-actin)-associated protein essential for Arp2/3-mediated actin network formation [11, 12]. Emerging evidence reveals that HDAC6 promotes inflammation by regulating expression of genes involved in inflammatory responses [13-15]. Despite evidence supporting the importance of HDAC6 in diverse cellular functions, HDAC6 knockout (KO) mice grow and develop normally; however, these mice exhibit impaired immune response [16]. The molecular mechanisms for this phenotype are largely unclear. In this study, we established an acute inflammation model in wild-type and HDAC6 knockout mice and examined the role of HDAC6 in the regulation of myeloid cell infiltration and phagocytosis during inflammation.
Results
HDAC6 deficiency suppresses the infiltration of myeloid cells into inflammatory lesions
In response to inflammatory cues, monocytes and neutrophils originating in the bone marrow emigrate from the circulation into sites of infection, where monocytes differentiate into macrophages. To investigate the role of HDAC6 in these processes, we first examined the effects of loss of HDAC6 on myeloid lineage cell differentiation. Neutrophils exclusively express the Ly6G antigen and express higher levels of Gr-1 antigen (a composite epitope of the Ly6C and Ly6G antigens) relative to monocytes/macrophages. In contrast, CD11b is a marker of all myeloid lineage cells, including granulocytic neutrophils, monocytes, and macrophages [17]. Therefore, we quantified subpopulations of cells labeled with Gr-1 and CD11b monoclonal antibodies in murine bone marrow samples isolated from wild-type (WT) and HDAC6 knockout mice using flow cytometry (Figure 1A). There was no significant difference in the ratio of neutrophils (CD11b+/Gr-1high) to monocytes (CD11b+/Gr-1intermediate) in WT and HDAC6 KO mice (Figure 1B-C), indicating that HDAC6 deficiency does not affect myeloid cell development.
HDAC6 regulates the infiltration of monocytes/macrophages, but not neutrophils, during inflammation. (A-C) Flow cytometric analysis of neutrophil (CD11b+/Gr-1high) and monocyte (CD11b+/Gr-1intermediate) subpopulations in myeloid cells isolated from the bone marrow of wild-type (WT) and HDAC6 knockout (KO) mice (A). Quantification of neutrophils (B) and monocytes (C) in (A). (D-F) Flow cytometric analysis of recruited neutrophils (Ly6G+/Ly6B+) and monocytes/macrophages (Ly6G-/Ly6B+) present in the peritoneal cavities of mice with E. coli-induced acute peritonitis (D). Quantification of neutrophils (E) and monocytes/macrophages (F) in (D). (G-I) Flow cytometric analysis of recruited neutrophils (CD11b+/Gr-1high) and monocytes/macrophages (CD11b+/Gr-1low) present in the peritoneal cavities of mice with LPS-induced acute peritonitis (G). Quantification of neutrophils (H) and monocytes/macrophages (I) in panel G. *P < 0.05; ns, not significant.
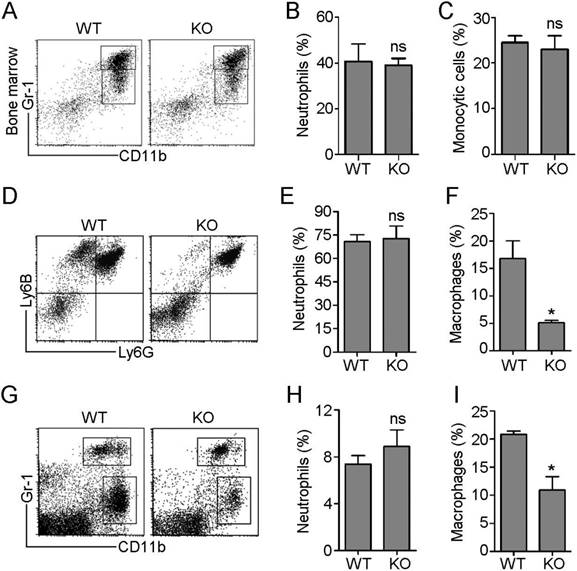
Next, we established an E. coli-induced acute peritonitis model to examine the effects of HDAC6 deficiency on peripheral neutrophil and monocyte/macrophage infiltration. We quantified recruited neutrophils (Ly6G+/Ly6B+) and monocytes/macrophages (Ly6G-/Ly6B+) present in the peritoneal cavity (Figure 1D) in WT and HDAC6 KO animals. The percentage of recruited neutrophils in the WT mice group was similar to that of the KO mice group (Figure 1E), but HDAC6 deficiency resulted in a significant decrease in the percentage of recruited monocytes/macrophages (Figure 1F). To confirm these results, we established a second acute peritonitis model by inducing inflammation using bacterial lipopolysaccharide (LPS). In this model, HDAC6 deficiency was also associated with suppression of peritoneal infiltration of monocytes/macrophages, but not neutrophils (Figure 1G-I). Together, these data reveal a critical role for HDAC6 in promoting monocyte/macrophage infiltration during inflammation.
The deacetylase activity of HDAC6 is required for macrophage migration
To further explore the role of HDAC6 in monocyte/macrophage infiltration, we isolated primary monocytes from bone marrow and differentiated them into macrophages. We then compared the migration ability of macrophages derived from WT and HDAC6 KO mice using transwell assays. We found that macrophages derived from KO mice migrated less than those from WT mice (Figure 2A-B). Next, we examined whether HDAC6 deacetylase activity is required for macrophage migration. Macrophages were treated with tubacin, a selective HDAC6 inhibitor, and inhibition of deacetylase activity was confirmed by immunoblotting (Figure 2C). Consistent with the effects of tubacin on α-tubulin acetylation (Figure 2C), tubacin exposure resulted in a dose-dependent decrease in murine macrophage migration (Figure 2D-E), suggesting that the deacetylase activity of HDAC6 is important for macrophage migration.
HDAC6 is critical for macrophage migration. (A) Representative images of bone marrow-derived macrophages subjected to transwell assays. Scale bar, 50 μm. (B) Quantification of migrated cells from experiments performed as described in (A). (C) Immunoblot analysis of RAW264.7 macrophages treated with tubacin. (D-E) Representative images of RAW264.7 macrophages subjected to transwell assays (D) and quantification of migrated cells (E). Scale bar, 100 μm. (F-G) Representative images of transwell assays using RAW264.7 macrophages treated with tubacin and LPS (F) and quantification of migrated cells (G). Scale bar, 100 μm. (H-J) Representative images of transwell assays using RAW264.7 macrophages treated with tubacin in the presence of M-CSF or serum (H). Quantification of migrated cells induced with M-CSF (I) or serum (J). Scale bar, 100 μm. **P < 0.01; ***P < 0.001.
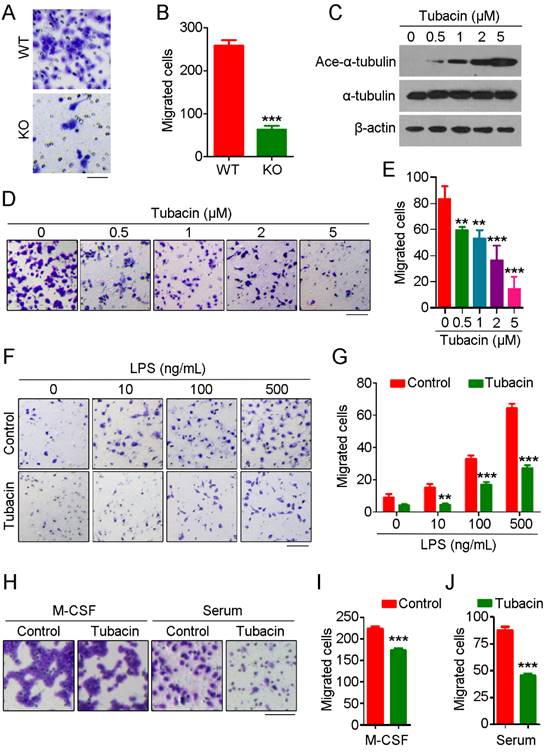
To extend our results showing a role for HDAC6 in macrophage migration, we next investigated whether HDAC6 is important for macrophage migration in response to chemotactic gradients of immune-related signals. As the concentration of LPS increased, the number of migrated macrophages also increased; strikingly, tubacin exposure attenuated LPS-induced macrophage migration (Figure 2F-G). We also examined the response to other chemotactic factors. Tubacin exposure also attenuated macrophage migration in response to macrophage colony-stimulating factor (M-CSF) or serum (Figure 2H-J), suggesting that HDAC6 plays a ubiquitous role in regulation of macrophage migration. Collectively, these findings reveal that HDAC6 positively regulates macrophage migration via its deacetylase activity.
HDAC6 does not affect neutrophil migration
Although we observed no significant difference in neutrophil infiltration between WT and HDAC6 KO mice in the acute peritonitis model experiments, substantial variation was observed among individuals in the KO group in independent replicates, with percentages of recruited neutrophils ranging from 42% to 80%. To rule out the possibility of artifacts resulting from individual variation, we isolated neutrophils from bone marrow and prelabeled WT-derived and KO-derived cells with carboxyfluorescein succinimidyl ester (CFSE) and Dil fluorescent dyes, respectively. These cells were then mixed at a 1:1 ratio and implanted in the mouse acute peritonitis model via tail vein injection. Confirming our previous results (Figure 1), the percentage of CFSE-labeled neutrophils in the peritoneal lavage fluid was similar to that of Dil-labeled neutrophils (Figure 3A-B), suggesting that HDAC6 does not regulate neutrophil infiltration.
HDAC6 does not affect neutrophil migration. (A) Neutrophils from wild-type and HDAC6 knockout mice were labeled with CFSE and Dil, respectively, mixed and injected into acute peritonitis mouse models. Cells infiltrating into the peritoneal cavity were analyzed by flow cytometry. (B) Experiments were performed as described in (A), and the relative migration efficiency was determined. (C) Transwell assays comparing migration of neutrophils from wild-type and HDAC6 knockout mice. Scale bar, 100 μm. (D) Experiments were performed as described in (C), and migrated cells were quantified. (E-G) Neutrophils from wild-type and HDAC6 knockout mice were induced with fMLP, and movement tracks were recorded using EZ-TAXIScan (E). Migration velocity (F) and directionality (G) were quantified. (H-J) Neutrophils from wild-type mice were treated with tubacin, and fMLP-induced movement tracks (H), velocity (I), and directionality (J) were analyzed. (K-M) Neutrophils from wild-type and HDAC6 knockout mice were induced with LBT4 and the movement tracks (K), velocity (L), and directionality (M) were analyzed. *P < 0.05; ***P < 0.001; ns, not significant.
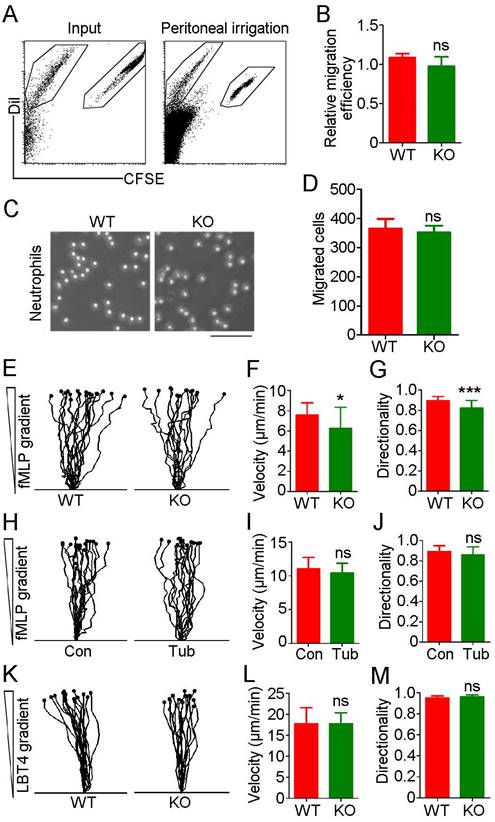
To further rule out a role for HDAC6 in neutrophil migration, we used N-formyl-methionyl-leucyl-phenylalanine (fMLP), a potent neutrophil chemoattractant. No significant difference in neutrophil migration was observed between the WT and KO groups (Figure 3C-D). Analysis of neutrophil chemotactic movement using the EZ-TAXIScan optical device revealed that HDAC6 deficiency had a slight impact on the velocity and directionality of neutrophil migration in response to an fMLP gradient (Figure 3E-G). However, inhibition of HDAC6 activity with tubacin did not affect migration velocity or directionality (Figure 3H-J). In addition, loss of HDAC6 had no effect on velocity or directionality of neutrophil migration induced by exposure to leukotriene B4 (LTB4), another chemoattractant involved in inflammation (Figure 3K-M). Taken together, these data suggest that HDAC6 activity is dispensable for neutrophil infiltration.
HDAC6 deficiency significantly impairs the phagocytic ability of macrophages
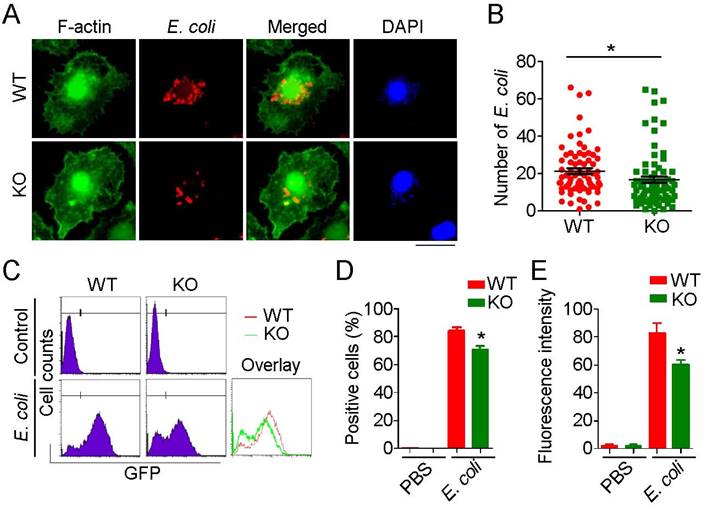
Recruited macrophages are the major phagocytic effector cells engaged in clearance of invading pathogens. Because phagocytosis requires dynamic remodeling of the cytoskeleton and HDAC6 has been linked to cytoskeletal regulation, we investigated the role of HDAC6 in regulation of cytoskeletal remodeling during phagocytosis. E. coli expressing mCherry were added to the culture medium of primary macrophages, and the phagocytosis was visualized by immunostaining. HDAC6 deficiency markedly impaired the phagocytic ability of macrophages, as fewer E. coli were observed within HDAC6 KO macrophages (Figure 4A-B). To quantify these results, we used flow cytometry to measure E. coli uptake. Compared to the WT group, macrophages isolated from KO mice phagocytosed fewer GFP-expressing E. coli (Figure 4C-E), indicating a role of HDAC6 in macrophage phagocytosis.
Loss of HDAC6 impairs the phagocytic ability of macrophages. (A) Fluorescence staining showing phagocytosis of E. coli by macrophages from wild-type or HDAC6 knockout mice. Scale bar, 20 μm. (B) Experiments were performed as described in (A), and the numbers of E. coli within macrophages were quantified. (C-E) Flow cytometric analysis of GFP-labeled E. coli phagocytosed by macrophages isolated from wild-type or HDAC6 knockout mice (C). GFP-positive cells (D) and fluorescence intensity (E) were quantified. *P < 0.05.
HDAC6 deficiency abrogates cortactin translocation and formation of filopodial protrusion in macrophages
Cortactin is an important substrate of HDAC6 that binds F-actin to facilitate assembly and branching of actin filaments, structures crucial for cell migration and phagocytosis [18]. Thus, we investigated whether HDAC6 regulates macrophage infiltration and phagocytosis through regulation of cortactin. Immunostaining revealed that HDAC6 and cortactin colocalized in the cytosol of resting macrophages and that both proteins translocated to the cell periphery in response to LPS challenge (Figure 5A). In contrast, in HDAC6-deficient cells, LPS exposure did not induce cortactin translocation (Figure 5B).
Effects of HDAC6 on LPS-induced cortactin localization and filopodial protrusion formation. (A) Macrophages from wild-type mice were treated with PBS or LPS and immunostained with antibodies against HDAC6 and cortactin. Arrowheads indicate the cell membrane. Scale bar, 20 μm. (B) Macrophages from HDAC6 knockout mice were treated with PBS or LPS and immunostained with antibodies against F-actin and cortactin. Scale bar, 20 μm. (C) Macrophages from wild-type or HDAC6 knockout mice were treated with PBS or LPS and immunostained with antibodies against F-actin. Scale bar, 20 μm. (D) Experiments were performed as in (C), and the percentages of cells with filopodial protrusions were quantified. ***P < 0.001; ns, not significant.
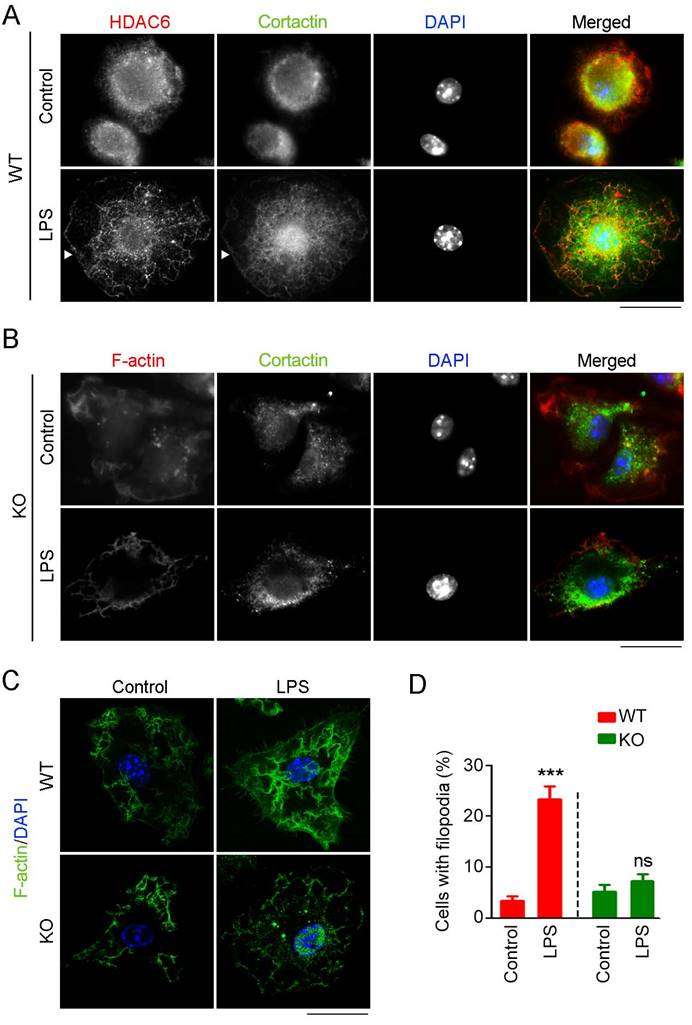
Because the acetylation status of cortactin regulates its binding to F-actin [19], we next examined whether loss of HDAC6 affects formation of F-actin-dependent structures. In WT-derived macrophages, LPS induced the formation of filopodial protrusions, but this activity was abolished by HDAC6 KO (Figure 5C-D). Taken together, these findings reveal that LPS stimulates translocation of HDAC6 to the cell periphery, where it deacetylates cortactin and promotes its binding to F-actin, thereby facilitating the formation of filopodial protrusions.
LPS is unable to promote microtubule acetylation in HDAC6-deficient macrophages
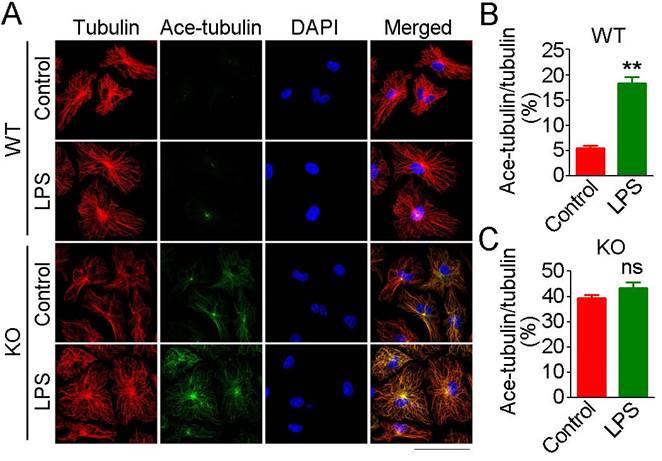
In addition to actin filaments, the microtubule cytoskeleton is also essential for cell migration and phagocytosis [20]. Therefore, we next investigated the effects of loss of HDAC6 on microtubule acetylation in macrophages. In WT-derived macrophages, only microtubules at the microtubule-organizing center (MTOC) were acetylated under basal conditions, and LPS exposure resulted in a significant increase in microtubule acetylation at the MTOC (Figure 6A-B). HDAC6 deficiency was associated with a substantial and global increase in microtubule acetylation under basal conditions, but LPS exposure did not significantly enhance microtubule acetylation (Figure 6A-C). These results implicate HDAC6 as the master regulator of microtubule acetylation in macrophages and suggest that microtubule acetylation is saturated in the absence of HDAC6, thereby preventing LPS-induced acetylation.
Effects of HDAC6 on LPS-induced microtubule acetylation. (A) Macrophages from wild-type or HDAC6 knockout mice were treated with PBS or LPS and immunostained with antibodies against α-tubulin and acetylated α-tubulin. Scale bar, 40 μm. (B-C) Experiments were performed as in (A), and the fluorescent intensity ratios of acetylated α-tubulin to α-tubulin in wild-type derived macrophages (B) and HDAC6 knockout-derived macrophages (C) were determined. **P < 0.01; ns, not significant.
Discussion
Despite the demonstrated roles of HDAC6 in diverse cellular activities, such as cell motility [21, 22], immunity [13, 23], viral infection [24], and degradation of misfolded proteins [25, 26], HDAC6-deficient mice survive and thrive [16], indicating that HDAC6 is not critical for embryonic and postnatal development under normal physiological conditions. These observations, together with the known role of HDAC6 in secretion of inflammatory cytokines [27], led us to investigate whether HDAC6 is important for the inflammatory response under pathological conditions.
In line with the finding that HDAC6 does not affect lymphocyte development [16], we demonstrate that HDAC6 does not affect proliferation of immune cells, as HDAC6-deficient mice exhibit no significant difference in the ratio of neutrophils to monocytes in bone marrow compared to wild-type mice. Neutrophils are the first leukocytes recruited to sites of inflammation. They subsequently secrete chemotactic factors that initiate monocyte/macrophage recruitment, contributing to a switch from neutrophil influx to monocyte/macrophage influx [10, 28]. HDAC6 deficiency does not affect infiltration of neutrophils, but instead, loss of HDAC6 results in a decrease in numbers of monocytes/macrophages recruited to the peritoneal cavity in a model of inflammation. These results are consistent with differing roles for HDAC6 in regulating infiltration of neutrophils and monocytes. Monocyte/macrophage recruitment may be impaired due to either cell-intrinsic or cell-extrinsic mechanisms.
Macrophages act as the principal effector cells during inflammation and play a key role in the initiation, resolution, and tissue repair and regeneration phases of the inflammatory response. Multiple lines of evidence have identified a critical role for HDAC6 in regulation of inflammatory signaling. As mentioned above, HDAC6 is known to regulate expression of inflammatory cytokines [27], in addition to other pro-inflammatory factors, such as monocyte chemoattractant protein-1, a protein released by neutrophils that is critical for recruitment of monocytes during acute inflammation [29]. HDAC6 also attenuates interleukin 10 expression in macrophages and interleukin 17 expression in γδ T cells [14, 15]. The underlying mechanism by which HDAC6 regulates inflammatory gene expression may be through modulation of p38 mitogen activated protein kinase, nuclear factor κB, and activator protein 1 pathways [27]. These data raise the question of whether HDAC6 affects the production of these and other extrinsic chemotactic factors in neutrophils during inflammation.
Due to the complexity of mechanisms involved in HDAC6-mediated inflammatory factor expression and difficulty in recapitulating this system in our model, in this study we did not explore the contribution of HDAC6-mediated production of extrinsic inflammatory factors to monocyte/macrophage recruitment. Instead, we focused on the cell-intrinsic effects of HDAC6 on macrophage migration. Using in vitro transwell assays, we demonstrate that HDAC6 activity is required for macrophage migration, confirming the importance of HDAC6 in cell-intrinsic inflammatory responses. Cortactin and α-tubulin are important substrates of HDAC6 that are important for HDAC6-mediated cell migration [21, 26, 30, 31]. In this study, we reveal that LPS challenge induces translocation of HDAC6 and cortactin from the cytosol to the cell periphery, consistent with previous findings from NIH3T3 cells challenged with epidermal growth factor [19]. HDAC6-mediated deacetylation of cortactin enhances binding of cortactin to F-actin, which, in turn, activates the actin-nucleating complex Arp2/3 and promotes actin filament network formation [18, 32] .
Our study extends these results by revealing that loss of HDAC6 suppresses formation of filopodia, spike-like actin protrusions critical for macrophage migration. Based on these data, we postulate that in response to LPS, HDAC6 is translocated to the cell periphery, where it deacetylates cortactin, promotes its interaction with F-actin, and thereby facilitates actin-related cellular activities, including cell migration and phagocytosis (Figure 7). In addition, LPS-induced translocation of HDAC6 to the cell periphery may also be the underlying cause of increased microtubule acetylation surrounding the MTOC. Thus, translocation of HDAC6 to the cell periphery may increase the dynamics of microtubules at the cell periphery, positively regulating cell migration and phagocytosis (Figure 7). However, it is important to note that HDAC6 has a number of other substrates, such as heat shock protein 90, some of which are also involved in cell migration [33-35]. Therefore, alternative or complementary molecular mechanisms may be identified in the future.
Proposed model for HDAC6-mediated macrophage infiltration and phagocytosis. In wild-type unstimulated macrophages, HDAC6 localizes to the cytosol. In response to inflammatory cues, HDAC6 partially translocates to the cell periphery, where it deacetylates α-tubulin and cortactin, thereby enhancing dynamics of microtubule tips and promoting actin assembly. Changes in cytoskeletal dynamics promote macrophage infiltration and phagocytosis. In the absence of HDAC6, cytoskeletal dynamics are not affected, rendering macrophages insensitive to inflammatory cues.
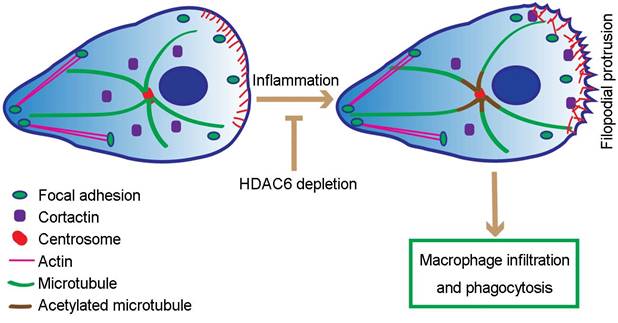
Currently, several HDAC6 inhibitors are under clinical investigation for treatment of cancers, such as multiple myeloma and breast cancer [36, 37]. Our study has uncovered a critical role for HDAC6 in macrophage infiltration, suggesting that these inhibitors may also be beneficial for the treatment of macrophage-associated immune disorders. Sustained recruitment of monocytes/macrophages is associated with a number of chronic inflammatory diseases, including atherosclerosis, rheumatoid arthritis, and autoimmune disorders. Based on the critical role for HDAC6 in regulation of macrophage recruitment shown here, HDAC6-targeted therapies may have significant potential for treatment of these and other chronic inflammatory diseases.
Methods
Materials
LPS, 4'-6-diamidino-2-phenylindole (DAPI), tubacin, M-CSF, fMLP, LTB4, and antibodies against β-actin and acetylated α-tubulin were purchased from Sigma-Aldrich. Antibodies against cortactin (Cell Signaling Technology), α-tubulin (Abcam), horseradish peroxidase-conjugated secondary antibodies (Amersham Biosciences), and fluorescein- or rhodamine-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) were obtained from the indicated sources.
Cell culture
RAW264.7 murine macrophages were purchased from the American Type Culture Collection. RAW264.7 cells and primary macrophages and neutrophils were cultured with the RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin, in a humidified atmosphere with 5% CO2 at 37 °C.
Isolation of primary macrophages and neutrophils
Primary bone marrow-derived macrophages and neutrophils were generated as described previously [38]. In brief, eight-to-twelve-week-old male mice were sacrificed by cervical dislocation, and the femurs and tibias were separated. Bone marrow cells were collected by flushing with cold PBS, and cells were then filtered through a 40-µm cell strainer (BD Biosciences). Red blood cells were lyzed by hypotonic NaCl and the remaining cells were washed with PBS once. For macrophage differentiation, 30 ng/mL of M-CSF was added to the culture medium. Culture medium was replaced each day to remove non-adherent cells, and on day 6, monocytes were differentiated into macrophages. For primary neutrophil isolation and purification, cells were isolated by density gradient centrifugation.
Mouse acute peritonitis model
All mouse experiments were approved by the Animal Care and Use Committee of Nankai University, and all experiments conformed to the relevant regulatory standards. HDAC6 heterozygous mice were obtained from Tso-Pang Yao (Duke University, Durham, NC, USA) and intercrossed to generate wild-type and HDAC6 knockout KO littermates as described previously [39]. Eight-to-Twelve-week-old male mice were intraperitoneally injected with 5×108 colony forming unit/mL of E. coli suspensions or 200 μL of LPS (100 mg/mL) to induce acute peritonitis. At 5 h post injection, mice were anesthetized with ether exposure. Sterile phosphate-buffered saline (PBS) was injected into the abdomen, followed by external massage of the abdomen for 1 min. The peritoneal lavage was collected for flow cytometric analysis.
Phagocytosis
The pET-28a-GFP and pET-28a-mCherry plasmids were transformed into BL-21 E. coli. Bacteria were collected and resuspended with PBS. The suspension was added to the primary bone marrow-derived macrophages and phagocytosis was examined by immunofluorescence staining or flow cytometric analysis. For immunofluorescence staining, E. coli expressing mCherry were incubated with macrophages for 1 h and phagocytosis was examined by immunostaining with fluorescein-labeled phalloidin. For flow cytometric analysis, E. coli expressing GFP were incubated with macrophages for 1 h, and phagocytosis was examined by analyzing GFP intensity.
Western blotting
Western blotting was performed as described previously [40]. In brief, proteins were loaded into 10% SDS-PAGE gel, separated, and transferred onto PVDF membranes (Millipore). The membranes were blocked in Tris-buffered saline containing 5% fat-free dry milk and 0.2% Tween 20. The membranes were subsequently incubated with primary antibodies and horseradish peroxidase-conjugated secondary antibodies. The proteins of interest were visualized with enhanced chemiluminescence detection reagent according to the manufacturer's instructions (Pierce Biotechnology).
Flow cytometric analysis
Cells isolated from bone marrow or peritoneal lavage were labeled with the indicated antibodies for 30 min at 4 °C. Anti-mouse CD11b-PE, Gr-1-APC and Ly6B-biotin (Biolegend), and Ly6G-fluorescein (BD Bioscience) were used in this study. Biotinylated antibodies were detected with APC-streptavidin (eBioscience). Cells were washed with PBS twice before analysis using a Caliber flow cytometer (BD Biosciences).
Immunofluorescence microscopy
Immunofluorescence microscopy was performed as described previously [41, 42]. Cells were fixed with 4% paraformaldehyde for 30 min at room temperature, permeabilized with 0.2% Triton X-100 for 15 min. Cells were then blocked with 2.5% bovine serum albumin for 20 min at room temperature. Cells were incubated with primary antibodies for 2 h, subsequently with fluorescein and/or rhodamine-conjugated secondary antibodies for 2 h. Nuclei were stained with DAPI. Coverslips were mounted with 90% glycerol in PBS and visualized with a Zeiss fluorescence microscope.
Transwell assays
Transwell assays were carried out as prescribed previously [43, 44]. In brief, cell suspension in serum-free medium was added to the upper inserts, and the inserts were then placed in a 24-well plate containing complete medium or the indicated chemotactic factors. After 16 h of incubation, the inserts were gently taken out and cells on the inner surface of the inserts were wiped by cotton swabs. Cells on the underside of inserts were fixed with 4% paraformaldehyde and stained with crystal violet solution. Five different fields were photographed and calculated.
Time-lapse chemotaxis assays
Time-lapse chemotaxis assays were carried out by using an EZ-Taxiscan chamber (Effector Cell Institute) as described [38]. Primary neutrophils were loaded into the EZ-Taxiscan channel with chemotactic factors in the chemoattractant well at 37 °C. Phase-contrast images of migrating cells were recorded every 30 s for 30 min. The movement track, velocity, and directionality of cells were determined by using ImageJ software.
Statistical analysis
All the experiments were repeated independently three times and the results are presented as mean ± standard errors (SEM). Student's t-tests and one-way analysis of variance (ANOVA) were carried out for statistical analysis.
Abbreviations
CFSE: carboxyfluorescein succinimidyl ester; DAPI: 4',6-diamidino-2-phenylindole; F-actin: filamentous actin; fMLP: N-formyl-methionyl-leucyl-phenylalanine; HDAC6: histone deacetylase 6; KO: knockout; LPS: lipopolysaccharide; LTB4: leukotriene B4; MTOC: microtubule-organizing center; PBS: phosphate-buffered saline; WT: wild-type.
Acknowledgements
We thank Dr. Tso-Pang Yao for providing the HDAC6 heterozygous mice. This work was supported by grants from the National Key R&D Program of China (2017YFA0503502), the National Natural Science Foundation of China (31728005, 31701216, and 31741039), Shandong Provincial Natural Science Foundation (ZR2017MC008), and China Postdoctoral Science Foundation (2016M600553).
Author contributions
BY, SX, YL, WL, DL, and ML carried out experiments and analyzed the data; BY, HRL, and JZ conceived the experiments and interpreted the data; SX and JZ wrote the paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161-6
2. Montecucco F, Liberale L, Bonaventura A, Vecchie A, Dallegri F, Carbone F. The role of inflammation in cardiovascular outcome. Curr Atheroscler Rep. 2017;19:11
3. Foley JF. Focus issue: understanding mechanisms of inflammation. Sci Signal. 2013;6:eg2
4. Tabas I, Glass CK. Anti-inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339:166-72
5. Chrobok NL, Sestito C, Wilhelmus MM, Drukarch B, van Dam AM. Is monocyte- and macrophage-derived tissue transglutaminase involved in inflammatory processes? Amino Acids. 2017;49:441-52
6. Crane MJ, Hokeness-Antonelli KL, Salazar-Mather TP. Regulation of inflammatory monocyte/macrophage recruitment from the bone marrow during murine cytomegalovirus infection: role for type I interferons in localized induction of CCR2 ligands. J Immunol. 2009;183:2810-7
7. Wang JS, Zhang Q, Cui F, Hou L, Zhao SZ, Xia H. et al. Genome-wide analysis of gene expression provides new insights into cold responses in thellungiella salsuginea. Front Plant Sci. 2017;8:713
8. Zarif JC, Taichman RS, Pienta KJ. TAM macrophages promote growth and metastasis within the cancer ecosystem. Oncoimmunology. 2014;3:e941734
9. Eckert AW, Wickenhauser C, Salins PC, Kappler M, Bukur J, Seliger B. Clinical relevance of the tumor microenvironment and immune escape of oral squamous cell carcinoma. J Transl Med. 2016;14:85
10. Henderson RB, Hobbs JA, Mathies M, Hogg N. Rapid recruitment of inflammatory monocytes is independent of neutrophil migration. Blood. 2003;102:328-35
11. Cai L, Makhov AM, Schafer DA, Bear JE. Coronin 1B antagonizes cortactin and remodels Arp2/3-containing actin branches in lamellipodia. Cell. 2008;134:828-42
12. Ran J, Yang Y, Li D, Liu M, Zhou J. Deacetylation of alpha-tubulin and cortactin is required for HDAC6 to trigger ciliary disassembly. Sci Rep. 2015;5:12917
13. Serrador JM, Cabrero JR, Sancho D, Mittelbrunn M, Urzainqui A, Sanchez-Madrid F. HDAC6 deacetylase activity links the tubulin cytoskeleton with immune synapse organization. Immunity. 2004;20:417-28
14. Yan B, Liu Y, Bai H, Chen M, Xie S, Li D. et al. HDAC6 regulates IL-17 expression in T lymphocytes: implications for HDAC6-targeted therapies. Theranostics. 2017;7:1002-9
15. Wang B, Rao YH, Inoue M, Hao R, Lai CH, Chen D. et al. Microtubule acetylation amplifies p38 kinase signalling and anti-inflammatory IL-10 production. Nat Commun. 2014;5:3479
16. Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S, Kneissel M. et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol. 2008;28:1688-701
17. Zhan X, Fang Y, Hu S, Wu Y, Yang K, Liao C. et al. IFN-gamma differentially regulates subsets of Gr-1(+)CD11b(+) myeloid cells in chronic inflammation. Mol Immunol. 2015;66:451-62
18. Weaver AM, Karginov AV, Kinley AW, Weed SA, Li Y, Parsons JT. et al. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation. Curr Biol. 2001;11:370-4
19. Zhang X, Yuan Z, Zhang Y, Yong S, Salas-Burgos A, Koomen J. et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007;27:197-213
20. Fenteany G, Glogauer M. Cytoskeletal remodeling in leukocyte function. Curr Opin Hematol. 2004;11:15-24
21. Li D, Xie S, Ren Y, Huo L, Gao J, Cui D. et al. Microtubule-associated deacetylase HDAC6 promotes angiogenesis by regulating cell migration in an EB1-dependent manner. Protein Cell. 2011;2:150-60
22. Li D, Sun X, Zhang L, Yan B, Xie S, Liu R. et al. Histone deacetylase 6 and cytoplasmic linker protein 170 function together to regulate the motility of pancreatic cancer cells. Protein Cell. 2014;5:214-23
23. Cheng F, Lienlaf M, Wang HW, Perez-Villarroel P, Lee C, Woan K. et al. A novel role for histone deacetylase 6 in the regulation of the tolerogenic STAT3/IL-10 pathway in APCs. J Immunol. 2014;193:2850-62
24. Zhang L, Ogden A, Aneja R, Zhou J. Diverse roles of HDAC6 in viral infection: Implications for antiviral therapy. Pharmacol Ther. 2016;164:120-5
25. Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727-38
26. Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008;18:291-7
27. Youn GS, Lee KW, Choi SY, Park J. Overexpression of HDAC6 induces pro-inflammatory responses by regulating ROS-MAPK-NF-kappaB/AP-1 signaling pathways in macrophages. Free Radic Biol Med. 2016;97:14-23
28. Doherty DE, Downey GP, Worthen GS, Haslett C, Henson PM. Monocyte retention and migration in pulmonary inflammation. Requirement for neutrophils. Lab Invest. 1988;59:200-13
29. Siddiqui JA, Partridge NC. CCL2/monocyte chemoattractant protein 1 and parathyroid hormone action on bone. Front Endocrinol (Lausanne). 2017;8:49
30. Kaluza D, Kroll J, Gesierich S, Yao TP, Boon RA, Hergenreider E. et al. Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J. 2011;30:4142-56
31. Deakin NO, Turner CE. Paxillin inhibits HDAC6 to regulate microtubule acetylation, Golgi structure, and polarized migration. J Cell Biol. 2014;206:395-413
32. Uruno T, Liu J, Zhang P, Fan Y, Egile C, Li R. et al. Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat Cell Biol. 2001;3:259-66
33. Schulz R, Marchenko ND, Holembowski L, Fingerle-Rowson G, Pesic M, Zender L. et al. Inhibiting the HSP90 chaperone destabilizes macrophage migration inhibitory factor and thereby inhibits breast tumor progression. J Exp Med. 2012;209:275-89
34. Zhao X, Zhang LL, Zhao ZT, Wang WC, Leavitt SD, Lumbsch HT. A molecular phylogeny of the lichen genus Lecidella focusing on species from mainland China. PloS One. 2015;10:e0139405
35. Zhang L, Liu N, Xie S, He X, Zhou J, Liu M. et al. HDAC6 regulates neuroblastoma cell migration and may play a role in the invasion process. Cancer Biol Ther. 2014;15:1561-70
36. Laubach JP, San-Miguel JF, Hungria V, Hou J, Moreau P, Lonial S. et al. Deacetylase inhibitors: an advance in myeloma therapy? Expert Rev Hematol. 2017;10:229-37
37. Tu Y, Hershman DL, Bhalla K, Fiskus W, Pellegrino CM, Andreopoulou E. et al. A phase I-II study of the histone deacetylase inhibitor vorinostat plus sequential weekly paclitaxel and doxorubicin-cyclophosphamide in locally advanced breast cancer. Breast Cancer Res Treat. 2014;146:145-52
38. Sakai J, Li J, Subramanian KK, Mondal S, Bajrami B, Hattori H. et al. Reactive oxygen species-induced actin glutathionylation controls actin dynamics in neutrophils. Immunity. 2012;37:1037-49
39. Gao YS, Hubbert CC, Lu J, Lee YS, Lee JY, Yao TP. Histone deacetylase 6 regulates growth factor-induced actin remodeling and endocytosis. Mol Cell Biol. 2007;27:8637-47
40. Yang Y, Ran J, Liu M, Li D, Li Y, Shi X. et al. CYLD mediates ciliogenesis in multiple organs by deubiquitinating Cep70 and inactivating HDAC6. Cell Res. 2014;24:1342-53
41. Luo Y, Ran J, Xie S, Yang Y, Chen J, Li S. et al. ASK1 controls spindle orientation and positioning by phosphorylating EB1 and stabilizing astral microtubules. Cell Discov. 2016;2:16033
42. Yang Y, Liu M, Li D, Ran J, Gao J, Suo S. et al. CYLD regulates spindle orientation by stabilizing astral microtubules and promoting dishevelled-NuMA-dynein/dynactin complex formation. Proc Natl Acad Sci USA. 2014;111:2158-63
43. Tala Xie S, Sun X Sun X, Ran J Zhang L. et al. Microtubule-associated protein Mdp3 promotes breast cancer growth and metastasis. Theranostics. 2014;4:1052-61
44. Gao J, Sun L, Huo L, Liu M, Li D, Zhou J. CYLD regulates angiogenesis by mediating vascular endothelial cell migration. Blood. 2010;115:4130-7
Author contact
![]() Corresponding author: Jun Zhou, College of Life Sciences, Nankai University, Tianjin 300071, China. Telephone: +86-22-2350-4946; Fax: +86-22-2350-4946; E-mail: junzhouedu.cn or Songbo Xie, College of Life Sciences, Shandong Normal University, Jinan, Shandong 250014, China. Telephone: +86-531-8618-2516; Fax: +86-531-8618-2518; E-mail: xiesongboedu.cn
Corresponding author: Jun Zhou, College of Life Sciences, Nankai University, Tianjin 300071, China. Telephone: +86-22-2350-4946; Fax: +86-22-2350-4946; E-mail: junzhouedu.cn or Songbo Xie, College of Life Sciences, Shandong Normal University, Jinan, Shandong 250014, China. Telephone: +86-531-8618-2516; Fax: +86-531-8618-2518; E-mail: xiesongboedu.cn