Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Results
Discussion
Methods
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(10):2846-2861. doi:10.7150/thno.23463 This issue Cite
Research Paper
Long non-coding RNAs AC026904.1 and UCA1: a “one-two punch” for TGF-β-induced SNAI2 activation and epithelial-mesenchymal transition in breast cancer
Guo-Yin Li1*, Wei Wang2*, Jian-Yong Sun3*, Bo Xin4, Xiang Zhang1, Ting Wang1, Qian-Feng Zhang5, Li-Bo Yao1, Hua Han1, Dai-Ming Fan6, An-Gang Yang2, Lin-Tao Jia1 ![]() , Lei Wang1
, Lei Wang1 ![]()
1. State Key Laboratory of Cancer Biology, Department of Biochemistry and Molecular Biology, Fourth Military Medical University, Xi'an, China;
2. Department of Immunology, Fourth Military Medical University, Xi'an, China;
3. Department of Thoracic Surgery, Tangdu Hospital, Fourth Military Medical University, Xi'an, China;
4. Department of Oncology, No. 88 Hospital of PLA, Tai'an, China;
5. Department of Obstetrics and Gynecology, Xijing Hospital, Fourth Military Medical University, Xi'an, China;
6. Institute of Digestive Diseases, Xijing Hospital, Fourth Military Medical University, Xi'an, China
*These authors contributed equally to this work.
Received 2017-10-23; Accepted 2018-3-6; Published 2018-4-15
Citation:
Li GY, Wang W, Sun JY, Xin B, Zhang X, Wang T, Zhang QF, Yao LB, Han H, Fan DM, Yang AG, Jia LT, Wang L. Long non-coding RNAs AC026904.1 and UCA1: a “one-two punch” for TGF-β-induced SNAI2 activation and epithelial-mesenchymal transition in breast cancer. Theranostics 2018; 8(10):2846-2861. doi:10.7150/thno.23463. https://www.thno.org/v08p2846.htm
Other stylesAbstract

Transforming growth factor-β (TGF-β) has received much attention as a major inducer of epithelial-mesenchymal transition (EMT) during cancer progression, mainly by activating a set of pleiotropic transcription factors including SNAI2/Slug. However, the involvement of long non-coding RNAs (lncRNAs) in TGF-β-induced Slug activation and EMT remains largely unknown.
Methods: In this study, we used microarray analysis to compare lncRNA expression profiles between TGF-β treated and untreated breast cancer cells. Then, the clinical significance of lncRNAs in breast cancer was investigated by qPCR and Kaplan-Meier survival analysis. The molecular mechanisms and EMT-promoting effects in vitro were analyzed by confocal laser microscopy, Western blotting, chromosome conformation capture (3C), chromatin isolation by RNA purification (ChIRP), ChIP, luciferase reporter assay and transwell migration assay. Lastly, the pro-metastatic effects in vivo were evaluated by bioluminescent imaging and hematoxylin and eosin (H&E) staining.
Results: We observed that TGF-β induced genome-wide changes in lncRNA levels in breast cancer cells, among which AC026904.1 and UCA1 were highly expressed in metastatic breast cancer and closely associated with poor prognosis. Mechanistic study revealed that AC026904.1 and UCA1 were upregulated by non-canonical and canonical TGF-β pathways, respectively. Further analysis showed that AC026904.1 functions as an enhancer RNA in the nucleus, whereas UCA1 exerts a competitive endogenous RNA (ceRNA) activity in the cytoplasm. In addition, the biological functions of these two lncRNAs converged on the activation and maintenance of Slug, constituting a one-two punch in promoting EMT and tumor metastasis.
Conclusion: These findings uncover for the first time that AC026904.1 and UCA1 could cooperatively upregulate Slug expression at both transcriptional and post-transcriptional levels, exerting critical roles in TGF-β-induced EMT. The present work provides new evidence that lncRNAs function as key regulators of EMT and hold great promise to be used as novel biomarkers and therapeutic targets for metastatic breast cancer.
Keywords: EMT, lncRNA, enhancer RNA, ceRNA, breast cancer.
Introduction
Breast cancer is the most common malignant disease and the leading cause of cancer death among females worldwide [1], of which more than 90% mortality is attributable to metastases[2]. Cancer metastasis occurs through a series of sequential steps, which involves dissemination of tumor cells from a primary site and colonization in distant tissues[3]. As epithelial tumor cells are usually tightly connected to each other via E-cadherin-containing adherens junctions, these epithelial barriers must be broken down before the tumor cells can spread out as single cells and invade the stroma. To this end, carcinoma cells may initiate a cell-biological program known as EMT to promote the invasion-metastasis cascade[3, 4]. The full accomplishment of the EMT process is orchestrated by a set of pleiotropically acting transcription factors, including Snail, Slug, ZEB1, ZEB2 and Twist, which organize entrance into a mesenchymal state by repressing epithelial markers and inducing mesenchymal ones. The expression of these key transcription factors is tightly regulated at different steps of transcription, translation and protein stability control by a variety of cell-intrinsic pathways as well as extracellular cues[5]. Nevertheless, a full understanding of this regulatory network is still far from being achieved.
Among the many EMT-inducing stimuli, TGF-β has received much attention as a major inducer of EMT during cancer progression. The multifunctional cytokine TGF-β exerts tumor-suppressive effects by inducing cell cycle arrest and apoptosis in premalignant cells but promotes tumor invasion and metastasis in advanced cancers [6]. Once activated, TGF-β signaling could regulate a wide range of downstream genes including EMT transcription factors, through Smad-dependent and Smad-independent pathways[7, 8]. In particular, several studies suggest that the TGF-β/Slug/E-cadherin axis plays pivotal roles in the process of EMT and tumor metastasis[9, 10]. The expression of Slug, but not its homolog Snail, was found to be strongly correlated with E-cadherin downregulation in breast cancer cells, although both factors possess the ability to repress E-cadherin expression[10]. Clinical data showed that Slug was highly expressed in metastatic breast cancer and closely correlated with poor prognosis of breast cancer[11]. Intriguingly, previous studies revealed that Slug expression could be induced and maintained by both ERK and Smad signaling pathways[12, 13], suggesting that crosstalk between these pathways might be orchestrated by TGF-β to regulate Slug expression. However, the precise molecular mechanisms by which TGF-β signaling activates Slug and induces EMT remain largely unknown. Therefore, understanding the TGF-β/Slug regulatory network may help to develop novel therapeutic strategies against metastatic breast cancer.
LncRNAs are a large heterogeneous class of transcripts longer than 200 nucleotides with limited protein-coding potential[14]. Recent evidence has demonstrated that lncRNAs are pervasively transcribed throughout eukaryotic genomes[15, 16], implicating their significant regulatory roles in complex organisms. Although only a small portion of functional lncRNAs have been well characterized to date, they have been shown to control nearly every aspect of gene expression and a diverse set of cellular processes, such as cell proliferation and migration[17, 18]. Recently, several studies reported that lncRNAs represent some of the most differentially expressed transcripts between primary and metastatic cancers[19, 20], highlighting their potential in enhancing tumor invasion and metastasis. In this study, we investigated lncRNA expression profiles in the context of TGF-β-induced EMT in breast cancer and found genome-wide changes in lncRNA levels upon EMT. We focused on two key lncRNAs, AC026904.1 and UCA1, which were significantly upregulated by non-canonical and canonical TGF-β signaling pathways, respectively. For the first time, we have shown that these two lncRNAs could cooperatively upregulate Slug expression at both transcriptional and post-transcriptional levels, thus promoting EMT and tumor metastasis. The present work provides new evidence that lncRNAs function as key regulators of EMT and hold great promise to be used as novel biomarkers and therapeutic targets for metastatic breast cancer.
Results
AC026904.1 and UCA1 are respectively upregulated by non-canonical and canonical TGF-β signaling
To identify lncRNAs that are regulated by TGF-β and with key roles in mediating TGF-β-induced EMT, we treated epithelial-like breast cancer cells MCF7 with TGF-β continuously for 4 days. Sustained TGF-β exposure caused MCF7 cells to undergo EMT, which was characterized by a spindle-like morphology (Figure 1A), a switch from E-cadherin to N-cadherin expression, and increased expression of Fibronectin and several transcription factors such as Snail, Slug, ZEB1 and Twist1 (Figure 1B and Figure S1). Then we used microarray analysis to compare lncRNA expression profiles between TGF-β treated and untreated cells, and found 346 upregulated and 198 downregulated lncRNAs in the treated cells (Figure 1C). Among the deregulated lncRNAs, we focused on two top-ranked lncRNAs, AC026904.1 and UCA1, and confirmed TGF-β-induced upregulation of these two genes by performing quantitative real-time PCR (qPCR) assays (Figure 1D). Analysis of protein coding potential of lncRNAs using LNCipedia database[21] showed that either AC026904.1 or UCA1 had limited or no coding potential (https://lncipedia.org/; data not shown), supporting the HAVANA annotation of these transcripts as non-coding.
Figure 1
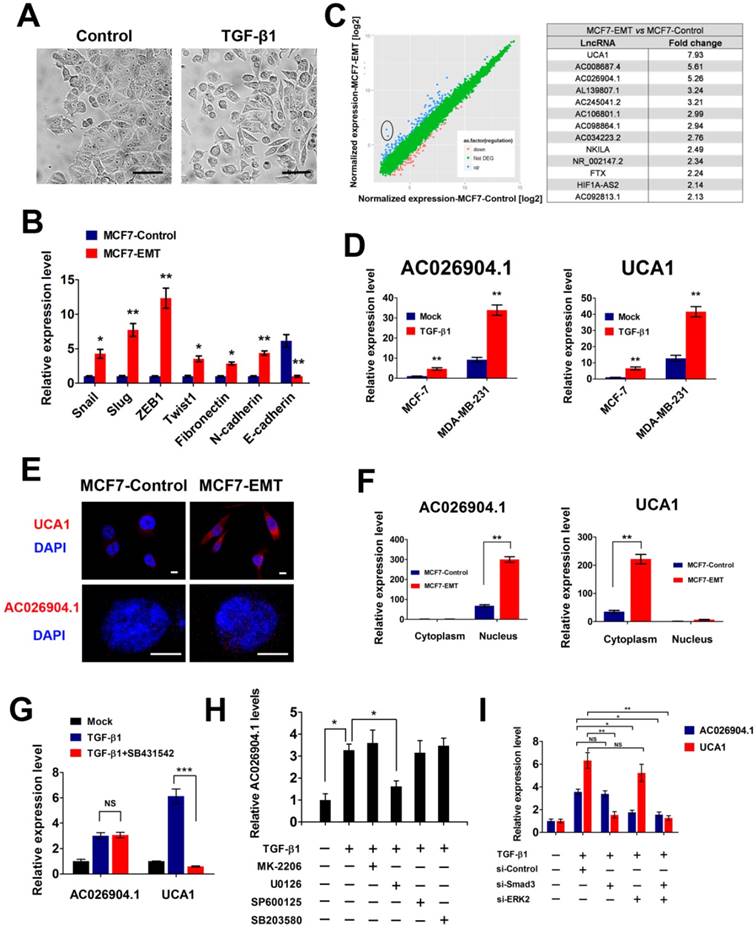
TGF-β upregulates UCA1 and AC026904.1 through Smad and ERK signaling, respectively. (A) Phase-contrast images of MCF7 cells treated with TGF-β1 (10 ng/mL) or control vehicle for 4 days. Scale bar: 50 μm. (B) Relative mRNA levels of EMT markers (mean ± SEM, n = 3) in MCF7 cells treated with TGF-β1 (10 ng/mL) for 4 days. (C) Left panel: Scatter plot of microarray analysis of differentially expressed lncRNAs in TGF-β1 treated and untreated cells. Right panel: Top upregulated lncRNAs in TGF-β-treated MCF7 cells. (D) Relative RNA levels of AC026904.1 and UCA1 (mean ± SEM, n = 3) in MCF7 or MDA-MB-231 cells treated with TGF-β1. (E) Confocal RNA-FISH images showing subcellular localization of UCA1 and AC026904.1 in MCF7 cells underwent EMT. Scale bar: 10μm. (F) Relative RNA levels of AC026904.1 and UCA1 (mean ± SEM, n = 3) in cytoplasmic or nuclear RNA fraction of MCF7 cells treated with TGF-β1. (G) Relative RNA levels of AC026904.1 and UCA1 (mean ± SEM, n = 3) in MDA-MB-231 cells pretreated with SB431542 (10 μM) for 60 min, followed by TGF-β1 for 24 h. (H) Relative RNA levels of AC026904.1 (mean ± SEM, n = 3) in MDA-MB-231 cells pretreated with MK-2206 or U0126 or SP600125 or SB203580, followed by TGF-β1 for 24 h. (I) Relative RNA levels of AC026904.1 and UCA1 (mean ± SEM, n = 3) in MDA-MB-231 cells pretreated with si-Control or si-Smad3 or si-ERK2, followed by TGF-β1 for 24 h.
Because the function of lncRNAs is associated with their unique subcellular localization[22], we next performed RNA fluorescence in situ hybridization (RNA-FISH) to investigate the subcellular distributions of AC026904.1 and UCA1. Interestingly, RNA-FISH data showed that UCA1 was predominantly localized in the cytoplasm (Figure 1E, upper panel) while AC026904.1 was highly enriched in the nucleus (Figure 1E, lower panel). The unique subcellular localization patterns of these two lncRNAs were further confirmed by qPCR assays, although AC026904.1 displayed a relatively weaker signal than UCA1 in breast cancer cells (Figure 1E-F).
Next, we asked whether AC026904.1 and UCA1 are induced by canonical TGF-β/Smad pathway. We found that pretreatment with SB431542 (a specific inhibitor of TGF-β type I receptor kinase[23]) completely abolished the TGF-β-induced upregulation of UCA1 in breast cancer cells, but had no effect on that of AC026904.1 (Figure 1G). Subsequent ChIP-qPCR analyses showed that TGF-β treatment greatly enhanced Smad2/3 binding to the UCA1 promoter in MCF7 cells, reaching a level similar to that in mesenchymal-like breast cancer cells MDA-MB-231 (Figure S2). In contrast, AC026904.1 promoter exhibited only minimal background signal of Smad2/3 binding in all tested cells. These data suggested that, UCA1, but not AC026904.1, was a direct target of canonical TGF-β/Smad pathway. Besides activating Smad, TGF-β has been reported to be able to utilize multiple Smad-independent pathways such as various branches of MAPK, PI3K/AKT, and RhoA signaling pathways to regulate a variety of cellular functions[7]. To identify the particular pathway through which TGF-β upregulates AC026904.1, we used selective inhibitors of these pathways to treat breast cancer cells prior to TGF-β stimulation. Subsequent qPCR analyses showed that TGF-β-induced upregulation of AC026904.1 was largely abrogated by pretreatment with U0126 (a specific inhibitor of MEK/ERK pathway[24]), rather than inhibitors targeting AKT, JNK, or p38 MAPK kinases (Figure 1H). In accordance with previous reports[25], our findings confirmed that TGF-β stimulation rapidly increased ERK phosphorylation in MCF7 cells, and pretreatment with U0126 completely eliminated this effect (Figure S3). Furthermore, we found that specific knockdown of Smad3 strikingly abolished the TGF-β-induced upregulation of UCA1, while activation of AC026904.1 by TGF-β was significantly impaired by ERK2 silencing (Figure 1I). Taken together, these results demonstrated that lncRNA UCA1 and AC026904.1 are upregulated through TGF-β/Smad and TGF-β/ERK pathways respectively in breast cancer.
AC026904.1 and UCA1 are highly expressed in metastatic breast cancer and predict poor outcome in breast cancer patients
To determine the clinical significance of the TGF-β-induced upregulation of AC026904.1 and UCA1, we first examined the expression levels of these two lncRNAs in a cohort of 60 breast cancer tissues comprised of ductal carcinoma in situ (DCIS) and invasive ductal carcinoma (IDC). The results showed that both AC026904.1 and UCA1 are expressed at higher levels in metastatic breast cancer samples (TnN>0M≥0) relative to non-metastatic ones (TnN0M0) (Figure 2A-B). Moreover, RNA-ISH analysis revealed that UCA1 was strongly expressed in the tested IDC samples, while normal breast tissues and DCIS displayed only very weak signals of UCA1 (Figure 2C, upper panel). Interestingly, an inverse expression pattern of E-cadherin and UCA1 was observed in breast cancer tissues (Figure 2C), suggesting that UCA1 may have a potential role in repressing E-cadherin and promoting tumor metastasis.
To investigate whether the expression levels of AC026904.1 and UCA1 are correlated with the invasive and migratory potential of breast cancer cells in vitro, we cultured six breast cancer cell lines including three typical epithelial-like lines (MCF7, T47D, and BT-474) and three mesenchymal-like lines that express increased Vimentin and decreased E-cadherin (MDA-MB-231, MDA-MB-436, and BT-549) (Figure S4). Subsequent qPCR analyses showed that AC026904.1 and UCA1were widely overexpressed in all three mesenchymal-like lines compared with epithelial-like lines (Figure 2D). We further examined whether the expression levels of these two lncRNAs were correlated with the clinical outcome of breast cancer after mastectomy. Kaplan-Meier's analysis in the 60 breast cancer patients revealed that high expression levels of AC026904.1 and UCA1 in breast tumor tissues significantly correlated with a reduction in recurrence-free survival (p < 0.05; Figure 2E-F), implicating the critical roles of these two lncRNAs in the pathogenesis and prognosis of breast cancer.
AC026904.1 and UCA1 cooperatively upregulate Slug expression in breast cancer
To uncover the molecular mechanisms explaining the clinical significance of AC026904.1 and UCA1 in breast cancer, we first knocked down the expression of these two lncRNAs in MDA-MB-231 and BT-549 cells by using two individual siRNA duplexes targeting different sequences of each transcript (Figure S5). Western blotting analysis revealed that silencing of either AC026904.1 or UCA1 remarkably repressed Slug and ZEB1 and upregulated E-cadherin at the protein level (Figure 3A-D). Intriguingly, simultaneous knockdown of AC026904.1 and UCA1 exerted a stronger effect when compared with silencing of either lncRNA (Figure 3E), suggesting that functional cooperation existed between these two lncRNAs. Furthermore, we found that lentiviral-mediated overexpression of UCA1 in MCF7 cells significantly upregulated Slug and ZEB1 and downregulated E-cadherin at the protein level (Figure 3F). These results were further confirmed by immunocytochemical (ICC) assay, which additionally showed that E-cadherin expression was significantly increased upon silencing of either AC026904.1 or UCA1, especially at sites of cell-cell contact in cultured MDA-MB-231 (Figure 3G). Moreover, we found that short hairpin RNA (shRNA)-mediated stable knockdown of either AC026904.1 or UCA1 greatly impaired TGF-β-induced EMT in MCF7 cells, as indicated by changes in cell morphology and EMT marker levels (Figure 3H and Figure S6). Because ZEB1 was reported to be transcriptionally activated by Slug and to act downstream of it[26], we reasoned that AC026904.1 and UCA1 might promote EMT mainly by upregulating Slug expression. Intriguingly, we found a positive correlation between Slug and AC026904.1 levels (R2= 0.6599, p <0.0001), as well as UCA1 levels (R2 = 0.3817, p <0.0001) in breast cancer samples (Figure 3I), suggesting that both AC026904.1 and UCA1 may play a functional role in Slug upregulation.
Figure 2
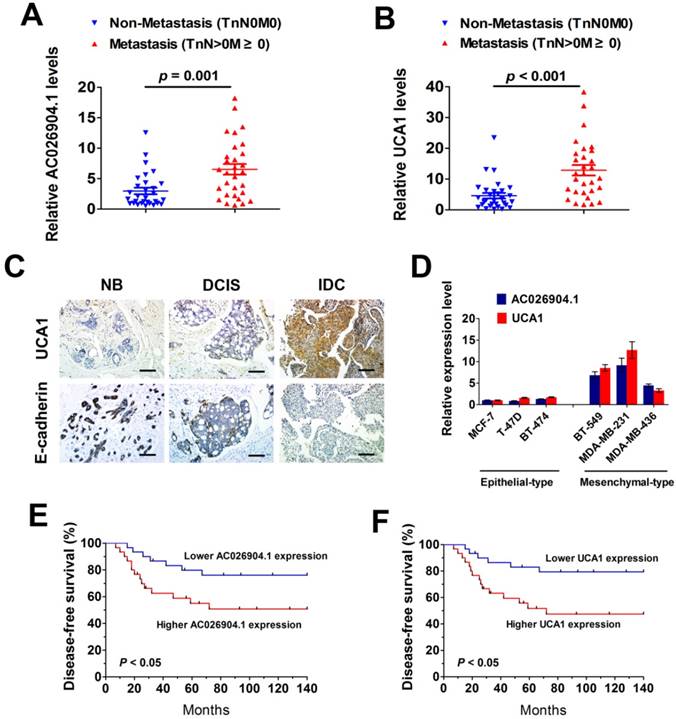
AC026904.1 and UCA1 are highly expressed in metastatic breast cancer and predict poor outcome in breast cancer patients. (A-B) Relative RNA levels of AC026904.1 (A) and UCA1 (B) in metastatic and non-metastatic breast cancer (n = 30 per group). Each data point represents an individual breast cancer sample. (C) Representative ISH staining of UCA1 and IHC staining of E-cadherin in normal breast tissue (NB), ductal carcinoma in situ (DCIS) and invasive ductal carcinoma (IDC). Scale bar: 100 μm. (D) Relative RNA levels of AC026904.1 and UCA1 (mean ± SEM, n = 3) in epithelial-like (MCF7, T47D, and BT-474) and mesenchymal-like (MDA-MB-231, MDA-MB-436, and BT-549) breast cancer cell lines. (E-F) Kaplan-Meier curves representing the probabilities of recurrence-free survival in 60 breast cancer patients stratified according to the expression levels of AC026904.1 (E) and UCA1 (F).
Figure 3
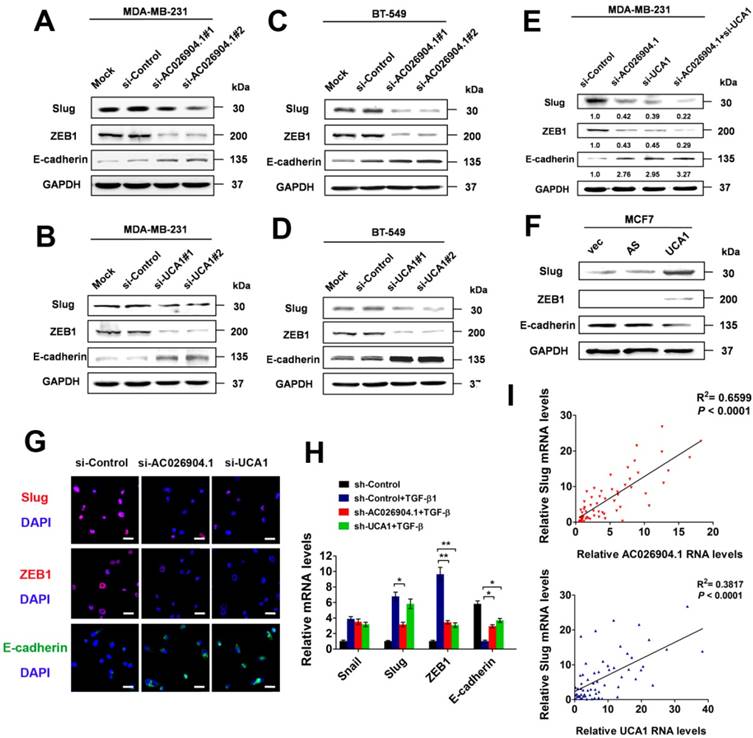
AC026904.1 and UCA1 upregulate Slug expression in breast cancer cells. (A-B) Western blotting analysis of Slug, ZEB1 and E-cadherin protein levels in MDA-MB-231 cells treated with si-Control, or si-AC026904.1 (A), or si-UCA1 (B) for 48 h. Blots were probed with an antibody against GAPDH to ensure equal loading. (C-D) Western blotting analysis of Slug, ZEB1 and E-cadherin protein levels in BT-549 cells treated with si-Control, or si-AC026904.1 (C), or si-UCA1 (D) for 48 h. Blots were probed with an antibody against GAPDH to ensure equal loading. (E) Western blotting analysis of Slug, ZEB1 and E-cadherin protein levels in MDA-MB-231 cells treated with si-Control, or si-AC026904.1, or both. (F) Western blotting analysis of Slug, ZEB1 and E-cadherin protein levels in MCF7 cells overexpressing UCA1 or its antisense control (AS) or empty vector (vec). (G) Immunofluorescence microscopy analysis of Slug, ZEB1 and E-cadherin in MDA-MB-231 cells treated with si-Control or si-AC026904.1 or si-UCA1 for 48 h. Scale bar: 30 μm. (H) Relative mRNA levels of EMT markers (mean ± SEM, n = 3) in stable AC026904.1 or UCA1 knockdown MCF7 cells treated with TGF-β1 (10 ng/mL) for 4 days. (I) Correlation between Slug mRNA levels and AC026904.1 or UCA1 transcript levels. Each data point represents an individual breast cancer sample, and a coefficient of determination (R2) is shown.
AC026904.1 functions as an enhancer RNA to activate Slug expression in cis
Because AC026904.1 is a lncRNA highly enriched in the nucleus (Figure 1E-F), we reasoned that it may act to regulate its neighboring genes[22]. To test this hypothesis, we interrogated a 500 kb window around the AC026904.1 gene locus and surprisingly found that only two protein-coding genes, SNAI2/SLUG and EFCAB1, were contained within this genomic region (Figure 4E, upper panel). RNA interference (RNAi)-mediated AC026904.1 silencing in MDA-MB-231 cells resulted in a concomitant downregulation of the neighboring SLUG gene at the transcriptional level (Figure 4A). This effect was specific, as we did not detect any obvious change in EFCAB1, another protein-coding gene surrounding AC026904.1 (Figure 4A). Additionally, a strong positive correlation between Slug and AC026904.1 RNA levels was also observed in multiple breast cancer cell lines (Figure 4B). These findings led us to speculate that AC026904.1 might function as an enhancer RNA (eRNA)[27] to activate SLUG expression in breast cancer.
Figure 4
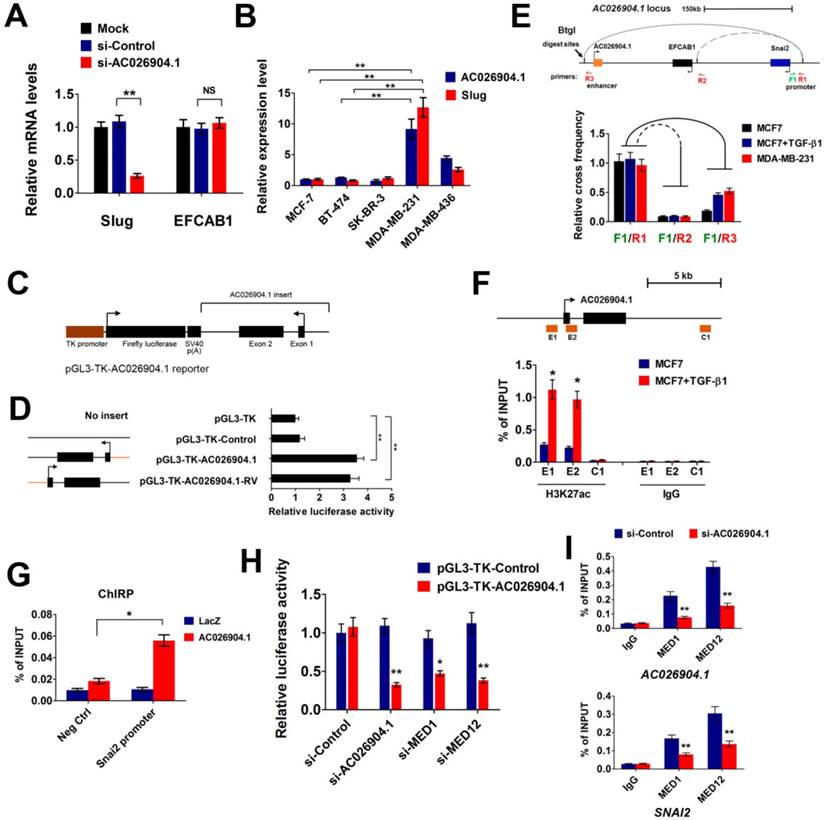
AC026904.1 functions as an eRNA to activate Slug expression in cis. (A) Relative mRNA levels of Slug and EFCAB1 (mean ± SEM, n = 3) in MDA-MB-231 cells treated with si-Control or si-AC026904.1 for 48 h. (B) Relative RNA levels of AC026904.1 and Slug (mean ± SEM, n = 3) in various breast cancer cell lines. (C) AC026904.1 was cloned and inserted downstream of luciferase driven by a TK-promoter in a reporter plasmid as shown. (D) Luciferase reporter assays with the vector containing AC026904.1 or its reversed sequence as indicated in the left panel. X-axis shows the normalized relative luciferase activity (mean ± SEM, n = 3). (E) Evaluation of the enhancer-promoter interaction at the AC026904.1 locus in MDA-MB-231 and MCF7 cells treated or untreated with TGF-β1 by 3C assays. F: forward primer; R: reverse primer. (F) ChIP-qPCR analysis of H3K27ac at the AC026904.1 locus in MCF7 cells treated or untreated with TGF-β1 for 72 h. Immunoglobulin G (IgG) was used as a control. ChIP DNA was analyzed by qPCR (mean ± SEM, n = 3). (G) ChIRP assay using biotin-labeled LacZ or AC026904.1-specific DNA probe and streptavidin beads in MDA-MB-231 cells, followed by qPCR analysis (mean ± SEM, n = 3). Neg Ctrl, negative control. (H) Depletion of AC026904.1 or MED1 or MED12 reduces the luciferase activity in the AC026904.1 luciferase reporter cell lines. (I) ChIP-qPCR analysis of MED1 and MED12 at the AC026904.1 locus or SNAI2 promoter region in MDA-MB-231 cells treated with si-Control or si-AC026904.1 for 48 h. Immunoglobulin G (IgG) was used as a control. ChIP DNA was analyzed by qPCR (mean ± SEM, n = 3).
To determine whether AC026904.1 has an enhancer-like function or not, we applied a method widely used to validate potential enhancers[28]. We constructed a series of reporter vectors with inserts containing AC026904.1 or its reversed sequences, or control DNA without transcriptional potential, and placed them downstream of Firefly luciferase driven by a thymidine kinase (TK) promoter (Figure 4C). Importantly, Dual-Luciferase reporter assay revealed that inclusion of either AC026904.1 or its reversed sequence resulted in similar enhancement of transcriptional activity ranging from 3- to 4-fold (Figure 4D), suggesting that AC026904.1 was an enhancer-like lncRNA with orientation independence. We next investigated whether AC026904.1 contributes to transcriptional activation of SLUG by establishing chromatin loop architecture, using chromosome conformation capture (3C) assays in MDA-MB-231 and TGF-β-stimulated MCF7 cells. We found that sustained TGF-β exposure significantly increased the strength of specific enhancer-promoter looping between AC026904.1 locus and SLUG promoter in MCF7 cells, reaching a level similar to that in MDA-MB-231 cells (Figure 4E).
As eRNA expression can be characterized by high enrichment of active histone modifications, particularly H3K27ac[27], we next performed ChIP assays to examine the H3K27ac level at the AC026904.1 locus. The results showed that H3K27ac modifications were enriched in both the promoter and gene body regions of AC026904.1, but not in a distant control element (Figure 4F). Importantly, this H3K27ac enrichment was further enhanced upon TGF-β stimulation (Figure 4F), in line with TGF-β-induced upregulation of AC026904.1 and Slug in breast cancer cells (Figure 1B-D). Chromatin isolation by RNA purification (ChIRP) assays showed that AC026904.1 transcript was present in the promoter region of SLUG (Figure 4G). Moreover, we found that depletion of the components of the co-activator complex, MED1 and MED12, specifically and potently diminished the AC026904.1-induced activation of transcription in the above-mentioned luciferase reporter assay (Figure 4H). ChIP-qPCR analysis revealed that MED1 and MED12 were recruited to the SLUG gene targeted by AC026904.1 and played a role in regulating the expression of Slug (Figure 4I). Taken together, these results supported the notion that AC026904.1 functions as an eRNA to activate SLUG gene transcription in cis.
UCA1 functions as a competitive endogenous RNA to upregulate Slug expression
Recent studies have demonstrated that cytoplasmic lncRNAs could exert functional roles by titrating microRNAs (miRNAs), sequestering proteins, or interfering with protein post-translational modifications, etc[22]. To uncover the precise mechanism underlying Slug upregulation by cytoplasmic UCA1, we first knocked down UCA1 expression in MDA-MB-231 cells and found that UCA1 depletion had no effect on the RNA level of Slug (Figure 5A), suggesting that UCA1 might promote Slug expression at the post-transcriptional level. Next, we used the DIANA-LncBase algorithm to predict putative functional interactions between UCA1 and miRNAs[29]. Numerous experimentally verified and computationally predicted miRNA recognition elements (MREs) were unraveled in the UCA1 transcript, including a set of miR-1 and miR-203a binding sites (Figure 5B). Intriguingly, both miR-1 and miR-203a were reported to inhibit EMT by repressing Slug expression[30, 31]. Therefore, we speculated that UCA1 might promote Slug expression by competitively inhibiting miR-1 and miR-203 functions in breast cancer.
To test this hypothesis, we used specific miRNA inhibitors to antagonize miR-1 or miR-203a in the MDA-MB-231 cells with stable knockdown of UCA1. As mentioned in Figure 3B, UCA1 silencing resulted in a remarkable downregulation of Slug protein expression. However, this effect was significantly abrogated by antagonizing either miR-1 or miR-203a, particularly when both miRNAs were inhibited simultaneously (Figure 5C). Importantly, inhibition of miR-1 and miR-203a did not lead to a compensatory upregulation of endogenous UCA1 (Figure S7), suggesting that the downregulation of Slug by UCA1 depletion was largely dependent on miR-1 and miR-203a functions. Moreover, transfection of miR-1 or miR-203a mimics into UCA1-overexpressing MCF7 cells partially impaired Slug upregulation, while co-transfection of these two miRNA mimics largely abrogated Slug upregulation induced by UCA1 overexpression (Figure 5D). To validate the direct binding between UCA1 and these two miRNAs, we performed an RNA immunoprecipitation (RIP) assay to pull down the endogenous miRNAs associated with UCA1. Subsequent qPCR analyses showed that the UCA1 RIP in MDA-MB-231 cells was significantly enriched for both miR-1 and miR-203a, compared with miR-21, a non-targeting miRNA (Figure 5E). In particular, mutations of miR-1/miR-203a MREs (henceforth named UCA1-mut1 and UCA1-mut2 respectively) almost completely abolished the specific enrichment of miR-1 or miR-203a by UCA1 (Figure 5E), implicating the direct interactions between UCA1 and these two miRNAs.
Moreover, we performed luciferase reporter assays using vectors containing wild-type (WT) UCA1 or UCA1-mut1 or UCA1-mut2. We found that overexpression of either miR-1 or miR-203a greatly reduced the luciferase activity of WT-UCA1 reporter vector. However, miR-1 overexpression had no effect on the transcriptional activity of UCA1-mut1 reporter vector, similar to the observation that mutation of miR-203a MREs on UCA1 eliminated the repressive effect exerted by miR-203a (Figure 5F). Next, we constructed a luciferase reporter vector containing the 3' untranslated region (3'UTR) of Slug transcript (pGL3-Slug-3'UTR). Overexpression of both miR-1 and miR-203a significantly suppressed the transcriptional activity of pGL3-Slug-3'UTR, while this effect was completely abolished by enforced overexpression of WT-UCA1, rather than the mutated form of UCA1 (Figure 5G). Altogether, these results demonstrated that UCA1 promoted Slug expression at the post-transcriptional level, by directly titrating miR-1 and miR-203a in breast cancer.
Figure 5
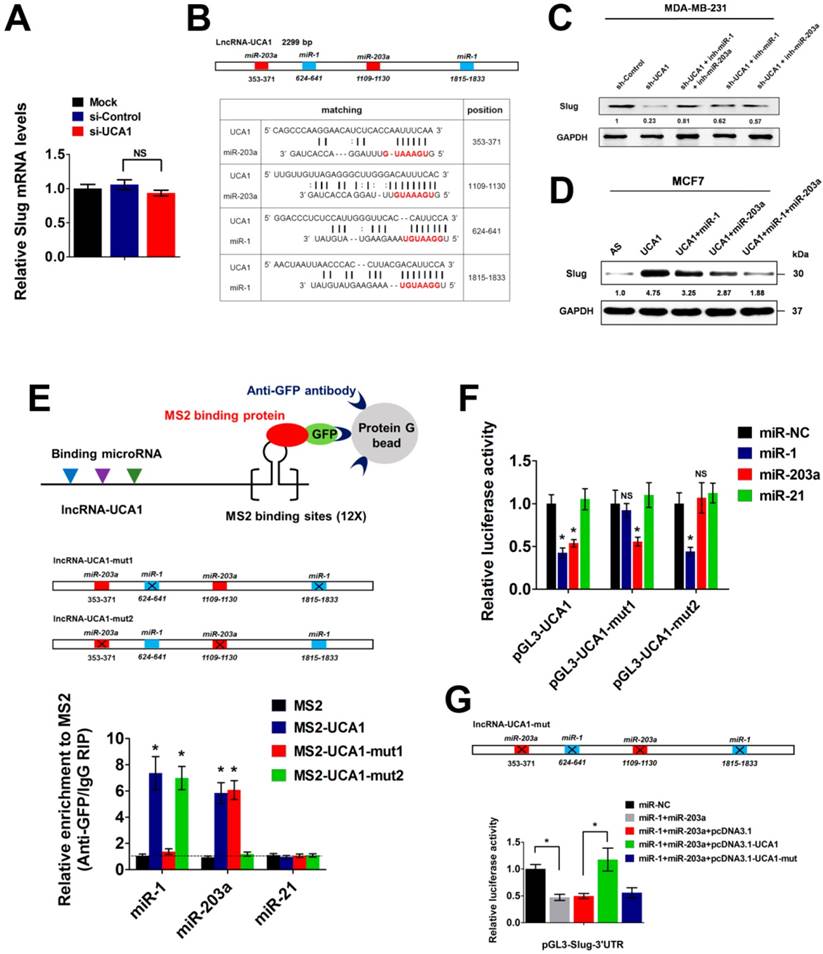
UCA1 functions as a ceRNA to upregulate Slug at post-transcriptional level. (A) Relative mRNA levels of Slug (mean ± SEM, n = 3) in MDA-MB-231 cells treated with si-Control or si-UCA1 for 48 h. (B) Schematic outlining the predicted binding sites of miR-1 and miR-203a on UCA1 transcript. (C) Western blotting analysis of Slug protein levels in stable UCA1 knockdown MDA-MB-231 cells treated with miR-1 inhibitor or miR-203a inhibitor, or both. Blots were probed with an antibody against GAPDH to ensure equal loading. (D) Western blotting analysis of Slug protein levels in UCA1-overexpressing MCF7 cells transfected with miR-1 or miR-203a mimics, or both. AS: antisense control of UCA1. Blots were probed with an antibody against GAPDH to ensure equal loading. (E) MS2-RIP followed by qPCR analysis (mean ± SEM, n = 3) to detect miRNAs endogenously associated with wild-type UCA1 or UCA1-mut1 or UCA1-mut2. (F) Luciferase activity (mean ± SEM, n = 3) in MCF7 cells co-transfected with miRNA mimics and luciferase reporters containing wild-type UCA1 or UCA1-mut1 or UCA1-mut2. (G) Slug 3'-UTR reporter activity (mean ± SEM, n = 3) in MCF7 cells concurrently transfected with miRNA mimics and vectors expressing wild-type or mutant UCA1.
AC026904.1 and UCA1 inhibitions suppress the invasion-metastasis cascade in breast cancer
To further investigate the biological roles of AC026904.1 and UCA1, we used CCK8 assay and transwell assay to evaluate the effects of inhibition of these two lncRNAs on cell proliferation and migration in vitro. We found that stable knockdown of UCA1 caused a slightly reduction in cell proliferation rate, while AC026904.1 depletion did not have any effect on proliferation of MDA-MB-231 cells (Figure S8). However, it is noteworthy that stable knockdown of either AC026904.1 or UCA1 resulted in a remarkable suppression of motility of MDA-MB-231 and BT-549 cells, reducing the number of migrating cells to about 20% of that in the control group (Figure 6A-B and Figure S9), in line with the phenotypic change following Slug depletion[12]. Ectopic expression of Slug largely abrogated the inhibitory effects on cell migration mediated by AC026904.1 or UCA1 knockdown (Figure 6A-B and Figure S9), suggesting that AC026904.1 and UCA1 promote breast cancer cell migration primarily by upregulating Slug expression. We also observed that simultaneous knockdown of AC026904.1 and UCA1 exerted a stronger effect on impairing cell migration ability when compared with silencing of either lncRNA (Figure S10). Furthermore, lentiviral-mediated overexpression of UCA1 greatly enhanced the migration ability of MCF7 cells, and this effect could be abolished by Slug knockdown (Figure S11).
To examine the effects of AC026904.1 and UCA1 on breast cancer metastasis in vivo, we used a well-characterized orthotopic model with MDA-MB-231-luc-D3H2LN cells, which is a highly metastatic breast cancer line stably expressing firefly luciferase[32]. MDA-MB-231-luc-D3H2LN cells with stable knockdown of either AC026904.1 or UCA1 (henceforth named D3H2LN-shAC026904.1 and D3H2LN-shUCA1, respectively) were inoculated into the mammary fat pads of nude mice. Afterward, both primary tumor growth and metastatic potential were monitored by regular measurements using a digital caliper. As expected, we found that depletion of AC026904.1 or UCA1 had little or no effect on the primary tumor growth in nude mice (Figure 6C and Figure S12). On day 28, spontaneous lung metastases were observed in 75% (6/8) of the mice with D3H2LN mammary tumors (Figure 6C), and metastatic colonization was further confirmed by ex vivo imaging after resection (Figure 6D). In sharp contrast, lung metastases were nearly absent in mice with either D3H2LN-shAC026904.1 or D3H2LN-shUCA1 tumors (Figure 6C-D). Moreover, Kaplan-Meier's analysis revealed that knockdown of either AC026904.1 or UCA1 markedly prolonged survival time of the nude mice bearing D3H2LN mammary tumors (Figure 6E). Collectively, these results indicated that AC026904.1 and UCA1 exerted biological effects mainly by promoting the invasion-metastasis cascade in metastatic breast cancer.
Discussion
It is widely accepted that the TGF-β/Slug/E-cadherin axis plays pivotal roles in the process of EMT and tumor metastasis, especially in breast and lung cancer[10, 33]. The expression of Slug, but not its homolog Snail, was found to be strongly correlated with E-cadherin downregulation in breast cancer cells, although both factors possess the ability to repress E-cadherin expression[10]. Clinical data showed that Slug was highly expressed in metastatic breast cancer and closely correlated with poor prognosis of breast cancer[11]. The expression of Slug has been reported to be regulated at multiple levels, including transcriptional control, post-translational control, differential splicing, and miRNA regulation[5]. Nevertheless, the TGF-β-Slug signaling is rather complicated and how distinct regulatory aspects of Slug expression are orchestrated by extracellular cues remains largely obscure. To date, multiple non-Smad pathways, including various branches of MAPK, PI3K/AKT, and RhoA signaling pathways, have been verified to be utilized by TGF-β to regulate a variety of cellular functions[7]. In particular, ERK pathway was reported to be closely related to the TGF-β signaling and play important roles in inducing Slug expression and promoting EMT. Rapid activation of ERK by TGF-β was observed in a variety of cell lines (in accordance with Figure S3) and this activation was initiated by ShcA tyrosine phosphorylation by the type I TGF-β receptor[25]. Importantly, activation of ERK pathway was further demonstrated to be required for TGF-β-induced EMT in vitro[34], and could promote breast cancer cell migration by maintaining Slug expression[12]. Nevertheless, TGF-β alone conferred a mesenchymal-like phenotype to Ras-transformed cells but not to normal cells, suggesting that ERK activation by TGF-β is context-dependent and TGF-β/Smad signaling cooperates with Ras/ERK pathway elaborately to modulate the invasiveness of epithelial tumor cells[35]. In addition, Aomatsu et al. revealed that TGF-β could induce sustained upregulation of Slug through both Smad and non-Smad pathways[13]. These results strongly implicated the existence of crosstalk between canonical and non-canonical TGF-β signaling pathways in EMT[36]. However, the precise mechanism by which TGF-β orchestrates these pathways to regulate Slug expression remains elusive.
Figure 6
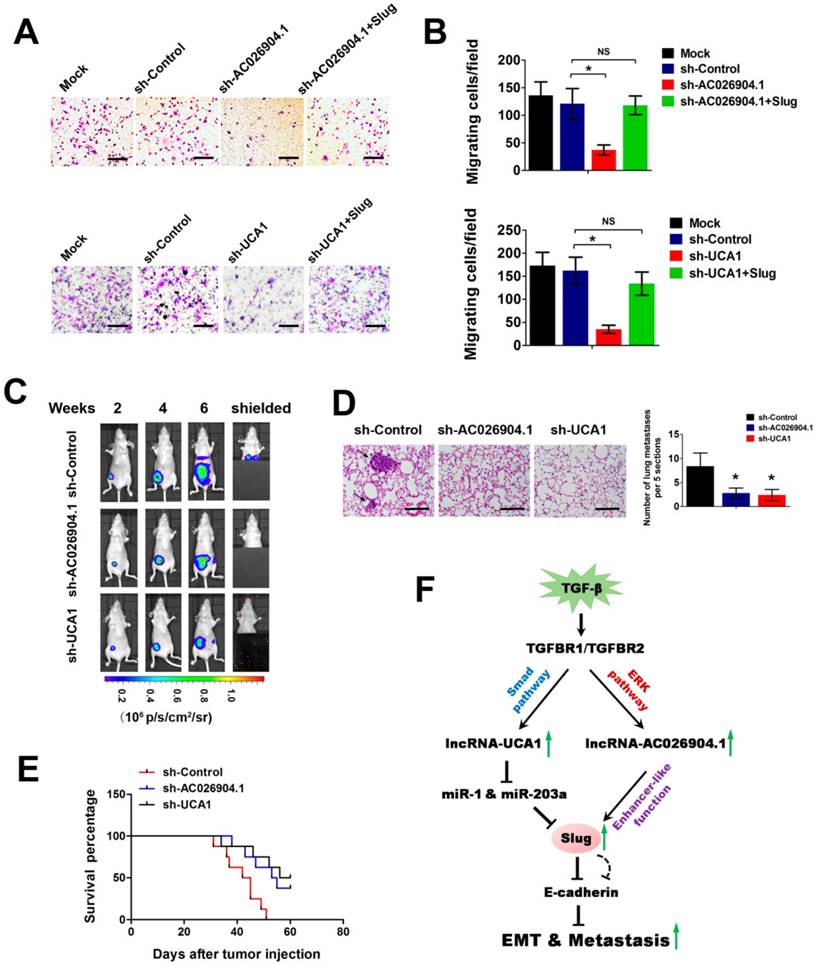
AC026904.1 and UCA1 inhibitions suppress the invasion-metastasis cascade in breast cancer. (A) Transwell migration assay of MDA-MB-231 cells with stable knockdown of AC026904.1 or UCA1, or with concurrent overexpression of Slug. (B) Quantitative analysis of cell migration in (A) (mean ± SEM, n = 3). (C) Bioluminescence imaging in a murine model of metastatic breast cancer. MDA-MB-231-luc-D3H2LN cells with stable knockdown of AC026904.1 or UCA1 were inoculated into the mammary fat pads of nude mice (n = 8 per group). Afterward, both primary tumor growth and metastatic potential were monitored by regular measurements using a digital caliper. (D) Hematoxylin and eosinstaining of lung tissues isolated from the mice in (C). Arrows indicate metastatic foci. Scale bar: 200 μm. The histogram (right panel) indicates the number of metastatic foci in each group (mean ± SEM for six mice per group, 5 sections per mouse). (E) Kaplan-Meier curves showing the survival time of nude mice bearing mammary tumors established with stable AC026904.1 or UCA1 knockdown MDA-MB-231-luc-D3H2LN cells (n = 8 per group). (F) A schematic model of AC026904.1 and UCA1 function during TGF-β-induced EMT. TGF-β activates UCA1 and AC026904.1 through Smad and ERK pathways, respectively. Then, AC026904.1 and UCA1 cooperatively upregulate Slug expression at both transcriptional and post-transcriptional levels.
Accumulating evidence suggests that unique sets of lncRNAs are dynamically regulated by distinct extracellular cues, such as TGF-β[37, 38], EGF[39], and Notch signaling[40] etc., implicating their critical roles in development, homeostasis and disease. For example, Yuan et al. showed that a number of lncRNAs including lncRNA-ATB were activated by TGF-β signaling in hepatocellular carcinoma (HCC). Depletion of lncRNA-ATB abrogated TGF-β-induced EMT and inhibited tumor metastasis, which supports the notion that lncRNA-ATB mimics the prometastatic role of TGF-β[37]. Moreover, Trimarchi et al. uncovered that a fraction of T cell acute lymphoblastic leukemia (T-ALL)-specific lncRNAs were directly controlled by the Notch1/Rpbjκ activator complex, and LUNAR1, one specific Notch-regulated lncRNA, was indispensable for efficient T-ALL growth in vitro and in vivo[40]. In the present work, we found genome-wide changes in lncRNA levels upon TGF-β-induced EMT in breast cancer (Figure 1C). Among these lncRNAs, NKILA was previously reported to be upregulated by classical TGF-β signal pathway in non-small cell lung cancer cells[41]. This consistency suggests that TGF-β could activate common lncRNAs in different cancer types and further confirms the reliability of our microarray data. Deeper studies revealed that two key lncRNAs, AC026904.1 and UCA1, were significantly upregulated by non-canonical and canonical TGF-β pathways respectively. Interestingly, the biological roles of these two lncRNAs converge on Slug upregulation, constituting a “one-two punch” in promoting EMT and tumor metastasis (Figure 6F). To our knowledge, this is the first report which shows that different TGF-β-activated lncRNAs could cooperatively upregulate Slug expression at both transcriptional and post-transcriptional levels. Therefore, our study adds a new dimension to the TGF-β-Slug signaling and provides novel insights into the signaling pathway cooperation in TGF-β-induced EMT.
Although there is growing interest in identifying lncRNAs with potential clinical significance, it remains a big challenge in dissecting their biological roles. Predicting the functionality of specific lncRNAs using bioinformatics tools is often unreliable by now, largely due to the enormous heterogeneity and poor sequence conservation of lncRNA species[14]. However, increasing studies have revealed that the function of lncRNAs is usually associated with their unique subcellular localization patterns: nuclear lncRNAs are enriched for functionalities involving chromatin interactions, transcriptional regulation, and RNA processing, while cytoplasmic lncRNAs can modulate mRNA stability and translation or influence cellular signaling cascades[22]. In this study, we performed RNA-FISH on breast cancer cells and found that UCA1 and AC026904.1 were predominantly localized in the cytoplasm and nucleus, respectively (Figure 1E). Subsequent analysis demonstrated that AC026904.1 functions as an eRNA to activate SLUG gene transcription in cis (Figure 4), while UCA1 promotes Slug expression at the post-transcriptional level, by directly titrating miR-1 and miR-203a (Figure 5). These exciting results nicely fit the hypothesis that the functionality of specific lncRNAs in part begins with their subcellular localization[22].
As an evolutionarily conserved class of non-coding RNAs, miRNAs usually contribute to translational repression and mRNA decay of their cognate mRNA targets through sequence-specific interactions with MREs[42]. Our previous study revealed that miR-101 and EZH2 could reciprocally inhibit each other, thus forming a double-negative feedback loop that regulates the process of hepatocarcinogenesis[43]. Moreover, miR-1 and miR-203a were reported to inhibit EMT by directly repressing Slug mRNA translation[30, 31]. Thus, regulation of miRNA expression and abundance is of great importance in development, homeostasis and disease. To date, mounting evidence has suggested that cytoplasmic lncRNAs can function as ceRNAs and compete miRNAs from their mRNA targets[44]. For example, lncRNA-ATB could upregulate ZEB1 and ZEB2 by competitively binding the miR-200 family, then induces EMT and metastasis in HCC[37]. Several studies also demonstrated that UCA1 could promote tumorigenesis in a variety of cancer types, mainly by titrating miRNAs such as miR-145[45], miR-193a[46], and miR-196a[47] etc. In the present work, we conducted several classical assays often used in exploring ceRNA mechanisms, and uncovered that UCA1 promotes Slug expression by competitively binding miR-1 and miR-203a (Figure 5). Importantly, we found that UCA1 was highly expressed in the cytoplasm of metastatic breast cancer cells, with a magnitude comparable to that of Slug mRNA (Figure 1E and data not shown). These results excluded the possibility of inefficient miRNA titration by lncRNAs[48] and further supported our notion that UCA1 functions as a ceRNA to upregulate Slug expression in breast cancer. Moreover, we also demonstrated that both miR-1 and miR-203a were expressed at lower levels in metastatic breast cancer samples relative to non-metastatic ones (Figure S13), indicating their potential role in suppressing breast cancer metastasis.
Recently, evidence of pervasive RNA transcripttion at active enhancer elements (hence termed eRNA) has become apparent with new advances in sequencing technology. Since then, mechanistic studies have delivered a detailed understanding of eRNAs, showing that they can function either locally to regulate the expression of neighboring protein-coding genes or distally at regulatory elements of other chromosomal regions, mainly through enhancer-promoter interactions. Unlike non-transcribing enhancers on a genome-wide scale, eRNA-producing enhancers exhibit higher binding affinities for transcriptional coactivators and higher enrichment of active histone modifications such as H3K27ac[27]. In the present work, we found that AC026904.1 contributes to transcriptional activation of SLUG by establishing chromatin loop architecture (Figure 4D-E). We also uncovered that H3K27ac modifications are enriched in both the promoter and gene body regions of AC026904.1 in breast cancer cells, which can be further enhanced upon TGF-β stimulation (Figure 4F). Moreover, ENCODE data in the UCSC Genome Browser (http://genome.ucsc.edu/) showed that H3K27ac modifications are highly enriched at the AC026904.1 locus in NHLF and HSMM cell lines, both of which exhibit mesenchymal-like morphology. But in epithelial-like NHEK cell line, the same locus displays much weaker enrichment of H3K27ac modification (Figure S16). These data indicate that the activation status of AC026904.1 locus is closely related to the mesenchymal-like phenotype in different cells, further supporting our notion that AC026904.1 functions as an eRNA to activate SLUG expression in TGF-β-induced EMT. Interestingly, a recent work demonstrated that deregulation of the Ras-ERK signaling could modulate the enhancer landscape in MEF cells[49], which is consistent with our finding that AC026904.1 is activated through the TGF-β/ERK pathway (Figure 1). We reasoned that TGF-β signaling could induce a unique set of eRNAs involving AC026904.1 to promote the invasion-metastasis cascade in metastatic breast cancer, which certainly deserves more attention in the near future.
Methods
Cell culture and treatments
MDA-MB-231-luc-D3H2LN cell line was purchased from Caliper Life Sciences (Hopkinton, MA). All other breast cancer cell lines were obtained from ATCC (Manassas, VA) and maintained as recommended. Where indicated, cells were treated with 10 ng/mL of recombinant TGF-β1 (R&D Systems, Minneapolis, MN) for the indicated time. TGF-β1 was replenished every 2 days with a fresh medium change. To inhibit respective pathways, cells were incubated with vehicle (DMSO), 10 μM SB431542, 1 μM MK-2206, 1 μM U0126, 10 μM SP600125, or 5 μM SB203580 (Selleck Chemicals, Houston, TX) for 1 h at 37°C prior to the experiments.
Transfection and lentiviral transduction
Transfection was performed using Lipofectamine 2000 reagent (Invitrogen). The siRNAs, miRNA mimics and miRNA inhibitors were chemically synthesized by Shanghai GenePharma Company (Shanghai, China). For lentiviral transduction of breast cancer cells, cells were infected with recombinant lentiviruses and were selected with puromycin (1 μg/mL) or blasticidin (10 μg/mL) for 7 days prior to use of homogenous pool of the infected cells for further assays. Target sequences of siRNAs and shRNAs are listed in Supplementary Material.
Microarray analysis
The Human Transcriptome Array 2.0 (HTA 2.0; Affymetrix, Santa Clara, CA) was employed in this study. HTA 2.0 covers global profiling of full-length transcripts, containing more than 40,000 non-coding and 245,000 coding transcripts in human genome. Each transcript is accurately identified by probes target specific exons or splice junctions. Total RNAs were prepared from MCF7 cells, with three biological replicates for each group. The differentially expressed lncRNAs with statistical significance were identified through volcano plot filtering, with a predefined threshold (fold change ≥ 1.5 and p-value < 0.05). The cDNA labeling, microarray hybridization and bioinformatics analysis were performed by the National Engineering Center for Biochip (Shanghai, China). The obtained data were submitted to GEO (accession number: GSE101809).
Patients and specimens
A total of 60 human breast tumor samples identified as DCIS and IDC were obtained from Chinese patients at Xijing Hospital (Xi'an, China), and non-neoplastic (NB) samples were obtained from the perilesional mammary tissue from patients submitted to resection of benign or malignant lesions. The use of clinical specimens in this study was approved by the Ethic Committee at Xijing Hospital, and informed consent was obtained from all patients. Stratified analysis of progression-free survival (PFS) was performed as described previously[50].
Quantitative real-time PCR (qPCR) and western blotting
Total RNA was extracted using TRIzol reagent (Invitrogen). First-strand cDNA was synthesized from 500 ng of total RNA using the PrimeScript RT Reagent Kit (TaKaRa) for detecting mRNA and lncRNA levels, or miScript Reverse Transcription Kit (Qiagen) for detecting miRNA levels. Real-time PCR was performed in triplicate using SYBR Premix Ex Taq (TaKaRa) on CFX96 Real-Time PCR Detection System (Bio-Rad). Expression levels of mRNAs, lncRNAs and miRNAs were normalized to GAPDH, 18S rRNA and U6 snRNA, respectively. Western blotting was performed as described previously[50]. The primers for qPCR and antibodies used for western blotting are listed in Supplementary Material.
In situ hybridization (ISH) and immunohistochemistry (IHC)
Adherent cells were detected by Stellaris RNA FISH assay using fluorophore-labeled probes (Biosearch Technologies, Novato, CA) following the manufacturer's protocol. Briefly, cells were washed with PBS, fixed for 10 min at room temperature, washed twice with PBS, and permeabilized in 70% (v/v) ethanol for 1 h at 4°C. Then, 1 μL of respective probe stock (12.5 μM) was diluted in 100 μL hybridization buffer and added to the permeabilized cells. Cells were incubated in a dark chamber at 37°C overnight. After washing with the wash buffer (2× SSC with 10% (v/v) formamide), DAPI nuclear stain in PBS (100 ng/mL) was added to counterstain the nuclei. Images were collected using a Nikon A1R confocal laser scanning microscope system. ISH and IHC staining in tissue samples were performed as described previously[43].
RNA immunoprecipitation (RIP)
RIP assays were carried out using the EZ-Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore, Billerica, MA) according to the manufacturer's instructions. Briefly, the wild-type sequence of UCA1 or its mutant sequence was subcloned into plasmid pSL-MS2-12X containing twelve MS2-binding sites. The resultant construct was co-transfected with pMS2-GFP into MCF7 cells, followed by immunoprecipitation using anti-GFP antibody or IgG as a control. The precipitated RNA fraction was reverse-transcribed and analyzed by qPCR. The detailed information on antibodies and primers used for RIP are shown in Supplementary Material.
Luciferase reporter assay
Luciferase reporter assay was performed as described previously[50]. Briefly, cells at 60-90% confluence in 48-well plates were transfected using Lipofectamine 2000 (Invitrogen). 100 ng of firefly luciferase reporter gene construct and 5 ng of the pRL-TK Renilla luciferase construct (for normalization) were co-transfected per well. Cell extracts were prepared 48 h after transfection, and the luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega). The experiment was carried out in triplicate wells and repeated at least twice.
Transwell migration assay
Briefly, 3×104 cells suspended in medium without serum or growth factors were plated in the top chamber (24-well insert; pore size, 8 μm; Corning Costar), and medium supplemented with serum was used as a chemoattractant in the lower chamber. The cells were incubated for 24 h and cells that did not migrate through the pores were removed by a cotton swab. Cells on the lower surface of the membrane were stained with crystal violet, air dried and photographed.
Orthotopic model of spontaneous breast cancer metastasis
A total of 3×106 MDA-MB-231-luc-D3H2LN cells were injected into the abdominal mammary fat pad of female nude mice aged 6-8 weeks. Tumor growth and metastasis were assessed weekly by bioluminescent imaging on the Xenogen In Vivo Imaging System (IVIS, Caliper Life Science, Hopkinton, MA). In some experiments, the lower portion of each animal was shielded before reimaging to minimize the bioluminescence from the primary tumor so that the signals from the metastatic regions could be observed in vivo. The imaging time was 10 s for detection of primary tumors and 1 min for metastases. All animal experiments were approved by the Institutional Animal Care and Use Committee of Fourth Military Medical University.
Chromatin immunoprecipitation (ChIP) and chromatin isolation by RNA purification (ChIRP)
ChIP assays were performed as described previously[43]. The ChIRP experiment was performed essentially as per the original protocol as described previously[51]. The ChIP primers and ChIRP probes are shown in Supplementary Material.
Quantitative chromosome conformation capture (3C) assay
3C assays were performed as described previously[52]. Briefly, the crosslinked chromatin was digested with specific restriction enzymes overnight. The crosslinking was reversed and ligated DNA was purified. The primers for qPCR are listed in Supplementary Material.
Statistical analysis
Statistical analysis was performed using SPSS 11.0 for Windows. All data were presented as mean ± SEM. Progression-free survival was analyzed with the Kaplan-Meier method and the statistical probability (p-value) was generated by log-rank test. The significance of associations between Slug and lncRNA expression values was judged via a test statistic based on Pearson product-moment correlation coefficient. Two-tailed Student's t-test was used to evaluate the statistical significance of differences between two groups of data in luciferase assay, qPCR and transwell migration assay. Differences were considered significant when p< 0.05 (*), p< 0.01 (**) or p< 0.001 (***).
Abbreviations
ceRNA: competitive endogenous RNA; ChIP: chromatin immunoprecipitation; ChIRP: chromatin isolation by RNA purification; DCIS: ductal carcinoma in situ; eRNA: enhancer RNA; EMT: epithelial-mesenchymal transition; IDC: invasive ductal carcinoma; lncRNA: long non-coding RNA; miRNA: microRNA; MREs: miRNA recognition elements; qPCR: quantitative real-time PCR; RIP: RNA immunoprecipitation; RNAi: RNA interference; RNA-FISH: RNA fluorescence in situ hybridization; shRNA: short hairpin RNA; T-ALL: T cell acute lymphoblastic leukemia; TGF-β: transforming growth factor-β.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We would like to thank Dr. Ying-Feng Lei and Ms Xiao-Ying Liu for technical supports, as well as other lab members for helpful discussion.
Author contributions
LW and LJ designed and supervised the study. GL, WW, JS, XZ, TW performed experiments and GL, WW, JS analyzed data. JS, BX and QZ provided clinical samples and clinical information. LJ, AY, HH, DF and LY provided administrative, technical, and material support. LW wrote and revised the manuscript.
Financial support
This study was supported by grants from the National Natural Science Foundation of China [81602517 to L.W., 81630069 to A.Y., 81472631 to L.J.].
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
2. Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563-572
3. Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275-292
4. Zheng H, Kang Y. Multilayer control of the EMT master regulators. Oncogene. 2014;33:1755-1763
5. De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97-110
6. Massague J. TGFbeta in Cancer. Cell. 2008;134:215-230
7. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577-584
8. Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156-172
9. Slabakova E, Pernicova Z, Slavickova E, Starsichova A, Kozubik A, Soucek K. TGF-beta1-induced EMT of non-transformed prostate hyperplasia cells is characterized by early induction of SNAI2/Slug. Prostate. 2011;71:1332-1343
10. Hajra KM, Chen DY, Fearon ER. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002;62:1613-1618
11. Martin TA, Goyal A, Watkins G, Jiang WG. Expression of the transcription factors snail, slug, and twist and their clinical significance in human breast cancer. Ann Surg Oncol. 2005;12:488-496
12. Chen H, Zhu G, Li Y, Padia RN, Dong Z, Pan ZK. et al. Extracellular signal-regulated kinase signaling pathway regulates breast cancer cell migration by maintaining slug expression. Cancer Res. 2009;69:9228-9235
13. Aomatsu K, Arao T, Sugioka K, Matsumoto K, Tamura D, Kudo K. et al. TGF-beta induces sustained upregulation of SNAI1 and SNAI2 through Smad and non-Smad pathways in a human corneal epithelial cell line. Invest Ophthalmol Vis Sci. 2011;52:2437-2443
14. Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145-166
15. Hangauer MJ, Vaughn IW, McManus MT. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 2013;9:e1003569
16. Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT. et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316:1484-1488
17. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904-914
18. Schmitt AM, Chang HY. Long Noncoding RNAs in Cancer Pathways. Cancer Cell. 2016;29:452-463
19. Sorensen KP, Thomassen M, Tan Q, Bak M, Cold S, Burton M. et al. Long non-coding RNA expression profiles predict metastasis in lymph node-negative breast cancer independently of traditional prognostic markers. Breast Cancer Res. 2015;17:55
20. Tahira AC, Kubrusly MS, Faria MF, Dazzani B, Fonseca RS, Maracaja-Coutinho V. et al. Long noncoding intronic RNAs are differentially expressed in primary and metastatic pancreatic cancer. Mol Cancer. 2011;10:141
21. Volders PJ, Helsens K, Wang X, Menten B, Martens L, Gevaert K. et al. LNCipedia: a database for annotated human lncRNA transcript sequences and structures. Nucleic Acids Res. 2013;41(Database issue):D246-D251
22. Chen LL. Linking Long Noncoding RNA Localization and Function. Trends Biochem Sci. 2016;41:761-772
23. Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD. et al. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65-74
24. Duncia JV, Santella JR, Higley CA, Pitts WJ, Wityak J, Frietze WE. et al. MEK inhibitors: the chemistry and biological activity of U0126, its analogs, and cyclization products. Bioorg Med Chem Lett. 1998;8:2839-2844
25. Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J. et al. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007;26:3957-3967
26. Wels C, Joshi S, Koefinger P, Bergler H, Schaider H. Transcriptional activation of ZEB1 by Slug leads to cooperative regulation of the epithelial-mesenchymal transition-like phenotype in melanoma. J Invest Dermatol. 2011;131:1877-1885
27. Lam MT, Li W, Rosenfeld MG, Glass CK. Enhancer RNAs and regulated transcriptional programs. Trends Biochem Sci. 2014;39:170-182
28. Banerji J, Rusconi S, Schaffner W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences. Cell. 1981;27:299-308
29. Paraskevopoulou MD, Georgakilas G, Kostoulas N, Reczko M, Maragkakis M, Dalamagas TM. et al. DIANA-LncBase: experimentally verified and computationally predicted microRNA targets on long non-coding RNAs. Nucleic Acids Res. 2013;41(Database issue):D239-D245
30. Liu YN, Yin JJ, Abou-Kheir W, Hynes PG, Casey OM, Fang L. et al. MiR-1 and miR-200 inhibit EMT via Slug-dependent and tumorigenesis via Slug-independent mechanisms. Oncogene. 2013;32:296-306
31. Ding X, Park SI, McCauley LK, Wang CY. Signaling between transforming growth factor beta (TGF-beta) and transcription factor SNAI2 represses expression of microRNA miR-203 to promote epithelial-mesenchymal transition and tumor metastasis. J Biol Chem. 2013;288:10241-10253
32. Jenkins DE, Hornig YS, Oei Y, Dusich J, Purchio T. Bioluminescent human breast cancer cell lines that permit rapid and sensitive in vivo detection of mammary tumors and multiple metastases in immune deficient mice. Breast Cancer Res. 2005;7:R444-R454
33. Shih JY, Yang PC. The EMT regulator slug and lung carcinogenesis. Carcinogenesis. 2011;32:1299-1304
34. Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia. 2004;6:603-610
35. Oft M, Peli J, Rudaz C, Schwarz H, Beug H, Reichmann E. TGF-beta1 and Ha-Ras collaborate in modulating the phenotypic plasticity and invasiveness of epithelial tumor cells. Genes Dev. 1996;10:2462-2477
36. Derynck R, Muthusamy BP, Saeteurn KY. Signaling pathway cooperation in TGF-beta-induced epithelial-mesenchymal transition. Curr Opin Cell Biol. 2014;31:56-66
37. Yuan JH, Yang F, Wang F, Ma JZ, Guo YJ, Tao QF. et al. A long noncoding RNA activated by TGF-beta promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. 2014;25:666-681
38. Zhuang J, Shen L, Yang L, Huang X, Lu Q, Cui Y. et al. TGFbeta1 promotes gemcitabine resistance through regulating the LncRNA-LET/NF90/miR-145 signaling axis in bladder cancer. Theranostics. 2017;7:3053-3067
39. Sas-Chen A, Aure MR, Leibovich L, Carvalho S, Enuka Y, Korner C. et al. LIMT is a novel metastasis inhibiting lncRNA suppressed by EGF and downregulated in aggressive breast cancer. EMBO Mol Med. 2016;8:1052-1064
40. Trimarchi T, Bilal E, Ntziachristos P, Fabbri G, Dalla-Favera R, Tsirigos A. et al. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell. 2014;158:593-606
41. Lu Z, Li Y, Wang J, Che Y, Sun S, Huang J. et al. Long non-coding RNA NKILA inhibits migration and invasion of non-small cell lung cancer via NF-kappaB/Snail pathway. J Exp Clin Cancer Res. 2017;36:54
42. Kiriakidou M, Nelson PT, Kouranov A, Fitziev P, Bouyioukos C, Mourelatos Z. et al. A combined computational-experimental approach predicts human microRNA targets. Genes Dev. 2004;18:1165-1178
43. Wang L, Zhang X, Jia LT, Hu SJ, Zhao J, Yang JD. et al. c-Myc-mediated epigenetic silencing of MicroRNA-101 contributes to dysregulation of multiple pathways in hepatocellular carcinoma. Hepatology. 2014;59:1850-1863
44. Karreth FA, Pandolfi PP. ceRNA cross-talk in cancer: when ce-bling rivalries go awry. Cancer Discov. 2013;3:1113-1121
45. Xue M, Pang H, Li X, Li H, Pan J, Chen W. Long non-coding RNA urothelial cancer-associated 1 promotes bladder cancer cell migration and invasion by way of the hsa-miR-145-ZEB1/2-FSCN1 pathway. Cancer Sci. 2016;107:18-27
46. Nie W, Ge HJ, Yang XQ, Sun X, Huang H, Tao X. et al. LncRNA-UCA1 exerts oncogenic functions in non-small cell lung cancer by targeting miR-193a-3p. Cancer Lett. 2016;371:99-106
47. Pan J, Li X, Wu W, Xue M, Hou H, Zhai W. et al. Long non-coding RNA UCA1 promotes cisplatin/gemcitabine resistance through CREB modulating miR-196a-5p in bladder cancer cells. Cancer Lett. 2016;382:64-76
48. Denzler R, Agarwal V, Stefano J, Bartel DP, Stoffel M. Assessing the ceRNA hypothesis with quantitative measurements of miRNA and target abundance. Mol Cell. 2014;54:766-776
49. Nabet B, O BP, Reyes JM, Shieh K, Lin CY, Will CM. et al. Deregulation of the Ras-Erk Signaling Axis Modulates the Enhancer Landscape. Cell Rep. 2015;12:1300-1313
50. Wang L, Guo ZY, Zhang R, Xin B, Chen R, Zhao J. et al. Pseudogene OCT4-pg4 functions as a natural micro RNA sponge to regulate OCT4 expression by competing for miR-145 in hepatocellular carcinoma. Carcinogenesis. 2013;34:1773-1781
51. Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44:667-678
52. Hagege H, Klous P, Braem C, Splinter E, Dekker J, Cathala G. et al. Quantitative analysis of chromosome conformation capture assays (3C-qPCR). Nat Protoc. 2007;2:1722-1733
Author contact
![]() Corresponding author: Lei Wang, Department of Biochemistry and Molecular Biology, Fourth Military Medical University, 169 Changle West Road, Xi'an 710032, China; Tel.: +86 29 84774516, Fax: +86 29 83253816, Email: rnawanglnet; and Lin-Tao Jia, Email: Jialthedu.cn
Corresponding author: Lei Wang, Department of Biochemistry and Molecular Biology, Fourth Military Medical University, 169 Changle West Road, Xi'an 710032, China; Tel.: +86 29 84774516, Fax: +86 29 83253816, Email: rnawanglnet; and Lin-Tao Jia, Email: Jialthedu.cn
Citation styles
APA
Li, G.Y., Wang, W., Sun, J.Y., Xin, B., Zhang, X., Wang, T., Zhang, Q.F., Yao, L.B., Han, H., Fan, D.M., Yang, A.G., Jia, L.T., Wang, L. (2018). Long non-coding RNAs AC026904.1 and UCA1: a “one-two punch” for TGF-β-induced SNAI2 activation and epithelial-mesenchymal transition in breast cancer. Theranostics, 8(10), 2846-2861. https://doi.org/10.7150/thno.23463.
ACS
Li, G.Y.; Wang, W.; Sun, J.Y.; Xin, B.; Zhang, X.; Wang, T.; Zhang, Q.F.; Yao, L.B.; Han, H.; Fan, D.M.; Yang, A.G.; Jia, L.T.; Wang, L. Long non-coding RNAs AC026904.1 and UCA1: a “one-two punch” for TGF-β-induced SNAI2 activation and epithelial-mesenchymal transition in breast cancer. Theranostics 2018, 8 (10), 2846-2861. DOI: 10.7150/thno.23463.
NLM
Li GY, Wang W, Sun JY, Xin B, Zhang X, Wang T, Zhang QF, Yao LB, Han H, Fan DM, Yang AG, Jia LT, Wang L. Long non-coding RNAs AC026904.1 and UCA1: a “one-two punch” for TGF-β-induced SNAI2 activation and epithelial-mesenchymal transition in breast cancer. Theranostics 2018; 8(10):2846-2861. doi:10.7150/thno.23463. https://www.thno.org/v08p2846.htm
CSE
Li GY, Wang W, Sun JY, Xin B, Zhang X, Wang T, Zhang QF, Yao LB, Han H, Fan DM, Yang AG, Jia LT, Wang L. 2018. Long non-coding RNAs AC026904.1 and UCA1: a “one-two punch” for TGF-β-induced SNAI2 activation and epithelial-mesenchymal transition in breast cancer. Theranostics. 8(10):2846-2861.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.