Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(10):2830-2845. doi:10.7150/thno.23209 This issue Cite
Research Paper
Targeting intracellular MMPs efficiently inhibits tumor metastasis and angiogenesis
Yaqi Lv1, Xiangmei Zhao1, Lidan Zhu1, Sijia Li1, Qingqing Xiao1, Wei He1 ![]() , Lifang Yin1,2
, Lifang Yin1,2 ![]()
1. Department of Pharmaceutics, School of Pharmacy, China Pharmaceutical University, Nanjing, 210009, P.R. China
2. Key Laboratory of Druggability of Biopharmaceutics, China Pharmaceutical University, Nanjing, 210009, PR China
Received 2017-10-7; Accepted 2018-2-27; Published 2018-4-15
Citation:
Lv Y, Zhao X, Zhu L, Li S, Xiao Q, He W, Yin L. Targeting intracellular MMPs efficiently inhibits tumor metastasis and angiogenesis. Theranostics 2018; 8(10):2830-2845. doi:10.7150/thno.23209. https://www.thno.org/v08p2830.htm
Other stylesAbstract

Treatment for metastatic cancer is a great challenge throughout the world. Commonly, directed inhibition of extracellular matrix metalloproteinases (MMPs) secreted by cancer cells can reduce metastasis. Here, a novel nanoplatform (HPMC NPs) assembled from hyaluronic acid (HA)-paclitaxel (PTX) prodrug and marimastat (MATT)/β-casein (CN) complexes was established to cure a 4T1 metastatic cancer model via targeting CD44 and intracellular, rather than extracellular, MMPs.
Methods: HPMC NPs were prepared by assembling the complexes and prodrug under ultrasonic treatment, which the interaction between them was evaluated by förster resonance energy transfer, circular dichroism and fluorescence spectra. The developed nanoplatform was characterized via dynamic light scattering and transmission electron microscopy, and was evaluated in terms of MMP-sensitive release and stability. Subsequently, the cellular uptake, trafficking, and in vitro invasion were studied by flow cytometry, confocal laser microscopy and transwell assay. MMP expression and activity was determined by western blotting and gelatin zymography. Finally, the studies of biodistribution and antitumor efficacy in vivo were performed in a mouse 4T1 tumor breast model, followed by in vivo safety study in normal mouse.
Results: The interaction between the prodrug and complexes is strong with a high affinity, resulting in the assembly of these two components into hybrid nanoparticles (250 nm). Compared with extracellular incubation with MATT, HPMC NP treatment markedly reduced the expression (100%) and activity (50%) of MMPs in 4T1 cells and in the tumor. HPMC NPs exhibited 1.4-fold tumor accumulation, inhibited tumor-growth by >8-fold in volume with efficient apoptosis and proliferation, and suppressed metastasis (>5-fold) and angiogenesis (>3-fold). Overall, HPMC NPs were efficient in metastatic cancer therapy. Conclusions: According to the assembly of polymer prodrug and protein-drug complexes, this study offers a new strategy for constructing nanoparticles for targeted drug delivery, biomedical imaging, and combinatorial treatment. Importantly, via inhibition of intracellular MMPs, metastasis and angiogenesis can be potently blocked, benefiting the rational design of nanomedicine for cancer treatment.
Keywords: prodrug, complexes, targeted co-delivery, intracellular matrix metalloproteinases, metastatic cancer
Introduction
Increasing evidence demonstrates that matrix metalloproteinases (MMPs) play a critical role in tumor metastasis because the MMPs break down the extracellular matrix (ECM) [1, 2] and, as a result, destroy the tumor microenvironment (TME) and promote the migration of cancer cells [3, 4]. Accordingly, inhibiting the expression and activity of MMPs in the TME can block metastasis. Numerous reports have indicated that direct downregulation of extracellular MMPs in the TME can indeed inhibit metastasis. Nonetheless, MMPs are predominantly secreted by cancer cells [1, 5]; therefore, it is unknown if suppressing the expression of intracellular MMPs in cancer cells will inhibit metastasis. So far, few reports concentrate on this issue.
Marimastat (MATT) is a potent MMP inhibitor and acts by mimicking the substrate of the MMPs to work with MMPs in a competitive and reversible pattern [6, 7]. MATT enables the downregulation of MMPs, especially MMP-1, MMP-2, MMP-7, MMP-9 and MMP-14, by 50%, even at nanomolar concentrations less than 5 nM. Furthermore, our recent findings indicate that this drug is a robust inhibitor for tumor angiogenesis. However, MATT is not a cytotoxic agent and consequently is unable to efficiently suppress tumor growth. Paclitaxel (PTX), one of several cytoskeletal drugs that target tubulin, is an efficient chemotherapeutic agent against various malignancies, such as breast cancer, ovarian cancer, and non-small-cell lung cancer [8, 9]. Based on these two drugs' active mechanisms, we assumed that their combination is a promising approach for treating metastatic cancer.
However, both drugs are poorly water-soluble compounds, and co-delivery of them is challenging. Here, we used the following strategies to address the solubility issue: (i) Hyaluronic acid (HA) is a natural and biodegradable polymer and, importantly, can target CD44 receptors overexpressed on cancer cells [10-12]. Therefore, a HA-PTX prodrug was synthesized for enhanced solubility and CD44 targeting. (ii) β-Casein (CN) is a calcium-sensitive phosphoprotein and has a molecular weight (MW) of 24 kDa, an isoelectric point (pI) of 5, and a diameter of 5 nm [13]. Interestingly, β-CN is a substrate of certain MMPs, such as MMP-3, MMP-7, MMP-10, MMP-12, MMP-14 and MMP-16 [5, 14] and, accordingly, is a potential MMP-sensitive material. Based on its properties, MATT/CN complexes were prepared to solubilize MATT and improve its delivery to cells.
Nevertheless, co-delivery of the HA-PTX prodrug and MATT/CN complexes with conventional carriers is challenging due to their extremely high MW and water-solubility. As is well known, as small molecule drugs or nanoparticles enter the blood, the protein in the blood binds with the drug or adsorbs onto the nanoparticles. Furthermore, hydrophobic molecules are able to trigger aggregation of globular proteins, forming nanoparticles [15, 16]. Inspired by these findings, we hypothesized that due to the hydrophobicity of PTX in the prodrug HA-PTX, the prodrug and protein complexes could self-aggregate into nanoparticles and, accordingly, improve delivery of the two drugs.
In this study, a novel nanoplatform assembled from HA-PTX prodrug and MATT/CN complexes (HPMC NPs) was established to target CD44 and intracellular MMPs (Figure 1). Instead of directly inhibiting the extracellular MMPs in TME, we hypothesized that via delivery of an MMP inhibitor, MATT, to cancer cells, the MMP secretion into the TME would decrease, and therefore, tumor metastasis would also decrease. The detailed hypothesis for the present HPMC NPs is as follows (Figure 1): after systemic injection, HPMC NPs accumulate in the tumor site, enter the cancer cells via CD44-mediated endocytosis, are destroyed in lysosomes, and release MATT/CN complexes and HA-PTX prodrug. HA-PTX prodrug is hydrolysed for PTX release and resultant cancer-cell killing and, meanwhile, intracellular MMPs decompose MATT/CN complexes and trigger MATT release for inhibiting MMP expression. Overall, we assume that via targeting CD44 receptors for enhanced uptake and intracellular MMPs rather than extracellular MMPs, the developed HPMC NPs would enable inhibition of metastasis and efficient cancer cell killing, finally achieving combined treatment for metastatic cancer.
Methods
Materials
PTX with more than 98% purity was purchased from Yew Biotechnology Co. Ltd. (Jiangsu, China). Taxol (marked product of PTX) was purchased from Bristol-Myers Squibb (China) Investment Co. Ltd. (Shanghai, China). β-CN (No. C6905, more than 98% purity), IR 783 probe (No. 543292, 90% purity), Fluorescein isothiocyanate (FITC) (No. F7250, 98% purity), Rhodamine B (Rho, No. 83689), Rhodamine B isothiocyanate (RITC, No. 283924) and 3-(4,5)-dimethylthiahiazo(-z-y1)-3,5-diphenyltetrazoliumromide (MTT) (No. M5655, 98% purity) were obtained from Sigma-Aldrich Co. Ltd. (St. Louis, MO, USA). 4T1, HELF MDA-MB-435, and MCF-7 cells were purchased from Nanjing KeyGEN Biotech Co., Ltd. (Nanjing, China). Fetal bovine serum (FBS), RPMI-1640, DMEM, Trypsin and Penicillin-Streptomycin Solution were obtained from Wisent Inc. (Nanjing, China). DAPI, Lyso-tracker red, and the Annexin V-FITC/PI staining kit were obtained from the Beyotime Institute of Biotechnology (Haimen, China). Lyso-tracker green was purchased from Yeasen Biotech Co., Ltd. (Shanghai, China). Recombinant MMP-3 was purchased from Cloud-Clone Crop (Houston, TX, USA). Alexa Fluor® 488-conjugated MMP-3 antibody, CD31 antibody, and CD68 antibody were purchased from Abcam (Britain).
Figure 1
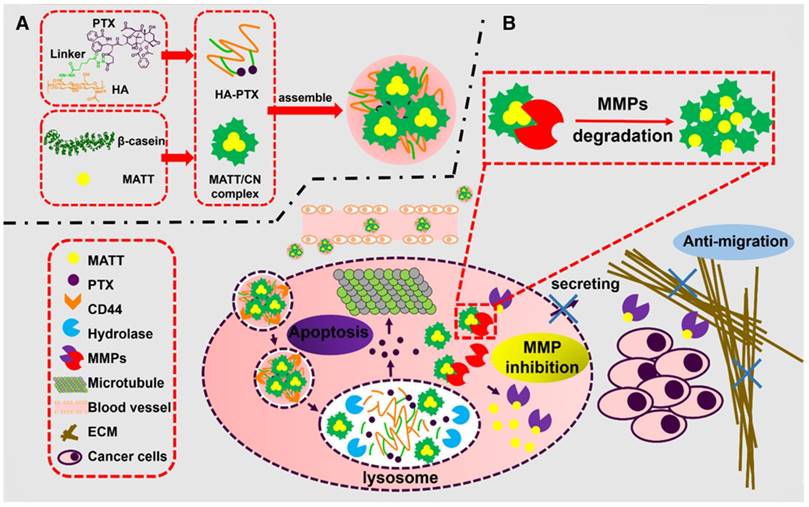
Schematic illustration of (A) the preparation of HPMC NPs and (B) the proposed activation procedure: Following systemic injection, HPMC NPs accumulate in the tumor site, enter cancer cells via CD44-mediated endocytosis, are destroyed in lysosomes, and release MATT/CN complexes and HA-PTX prodrug. The prodrug associates with microtubules and induces apoptosis; meanwhile, the complexes release MATT triggered by intracellular MMPs to inhibit MMP expression and secretion to the extracellular circulation, which decreases ECM degradation and the resultant migration of cancer cells. Ultimately, combined treatment for metastatic cancer is achieved.
Preparation and characterization
MATT/CN complexes were first prepared by the following procedure: 7.2 mg of CN was dissolved in 6 mL of water, stirred to give a homogeneous solution and cooled down below 4 °C, followed by the addition of MATT aqueous solution (0.8 mL, 3.75 mg/mL) and treatment using an ultrasonic probe (Scientz Biotechnology Co., Ltd., Ningbo, China) at 360 W for 10 min. HA-PTX prodrug was synthesized as described in our previous report [10]. HPMC NPs were prepared by mixing 3 mg of HA-PTX dissolved in 500 μL of water with the prepared MATT/CN complex solution under gentle stirring for 1 min and ultrasonic treatment for 10 min at 360 W. The temperature during the preparation period was controlled below 4 °C. HA-PTX/CN NPs without MATT loading and other dye-labelled NPs were prepared following the same procedure.
Particle size and size distribution were measured with a Malvern Zetasizer 3000HS system according to the dynamic laser scattering (DLS) principle (Malvern Instruments Ltd., UK). The samples were diluted 50-fold in water to obtain a suitable concentration for measurement.
Transmission electron microscopy (TEM) examination was performed on a JEM-1230 TEM (Tokyo, Japan) at an acceleration voltage of 200 kV. One drop of diluted sample (100-fold) was deposited on a copper mesh, and excessive sample was removed followed by drying at room temperature. Next, one drop of 2% uranyl acetate (w/v) was added and stained for 30 s. Finally, the sample was dried at room temperature after removing the extra acid.
Förster resonance energy transfer (FRET)
FITC and Rho were used as the donor and the acceptor, respectively, and were conjugated to HA-PTX and MATT/CN complexes, respectively. FITC-HA-PTX at a fixed concentration of 1 mg/mL was assembled with Rho-MATT/CN complexes at different concentrations to form FITC-HA-PTX/Rho-MATT/CN NPs with mass ratios of 2:1, 1:1 and 1:2, respectively. The emission spectra at 500 nm to 700 nm were recorded using a fluorescence spectrometer (SHIMADZU RF-5301PC, Japan) at an excitation wavelength of 450 nm (donor, FITC). The split widths of the excitation and emission were 5 nm and 15 nm, respectively.
To confirm the assembly, the prepared nanoparticles were observed by confocal microscopy (LSM700, Carl Zeiss, Germany) after being applied to a round glass cover slip.
CD spectroscopy and fluorescence measurements
Circular dichroism (CD) spectra were measured with a J-810 fluorescence spectrometer (Tokyo, Japan) equipped with a temperature-controlling unit and a quartz cuvette, and the ellipticity was expressed in millidegrees. The wavelength ranged from 190-250 nm. The bandwidth was 1 nm. The scanning speed was 100 nm/min. The cell length was 0.1 cm, and the temperature was 25 °C. The protein concentration of CN in each sample was 0.25 mg/mL.
Fluorescence spectra were measured with a fluorescence spectrometer (SHIMADZU RF-5301PC, Japan). The emission wavelength was scanned from 300-500 nm excited by a wavelength of 295 nm. Both the excitation and emission slit widths were set as 5 nm. The protein concentration of CN in each sample was 0.1 mg/mL.
Affinity study
MATT/CN complexes were prepared with final concentrations of 0.05, 0.1, 0.2, 0.5, and 1 mg/mL for MATT. HA-PTX/CN NPs were prepared with final concentrations of 0.25, 0.5, 1, 2, and 3 mg/mL for HA-PTX. For the preparation of HPMC NPs, the concentrations of HA-PTX in various formulations were 0.25, 0.5, 1, 2 and 3 mg/mL with a fixed MATT concentration at 0.5 mg/mL. After a 10-fold dilution, each sample was divided into 3 tubes and incubated at 15 °C, 25 °C, and 35 °C in a water bath. The fluorescence intensity was measured under the conditions as described above after equilibrating for at least 5 min. The protein concentration for determination was 0.1 mg/mL.
The affinity between two materials was measured to determine the quenching type according to the Stern-Volmer equation:
F0/F = 1 + Ksv[Q] = 1 + Kqτ0[Q] (1)
where Ksv and Kq are the Stern-Volmer quenching rate constant and fluorescence quenching rate constant, respectively, F0 and F are the fluorescence intensities in the absence and presence of quencher, respectively, τ0 is the average lifetime of the protein without quencher, and [Q] is the concentration of the quencher.
The binding constant (Ka) and the number of binding sites (n) of a static quenching were calculated as:
log[(F0-F)/F] = logKa + nln[Q] (2)
where Ka and n can be calculated as the intercept and slope of the double logarithm regression curve of log[(F0-F)/F] versus log [Q], respectively.
MMP sensitivity
MMP-3 was added to each sample at a final concentration of 5 μg/mL followed by incubation at 37 °C and shaking at a speed of 100 rpm for 24 h. At specific time intervals, samples were withdrawn for size measurement by DLS.
Drug release
The release of Rho and PTX from Rho-labelled HPMC NPs was tested using a dialysis method performed in a shaker (SHA-C, Jintan, China) at 37 °C with a shaking speed of 100 rpm for 24 h. The MW cut-off of the dialysis bag was 3,500. MMP-3 was added to each sample at a final concentration of 5 μg/mL. At specific time intervals, samples were withdrawn from outside of the bag. The Rho fluorescence were measured using a microplate reader (POLARstar Omega, Germany) at excitation and emission wavelengths of 544 nm and 615 nm, respectively. The PTX content in sample was assayed by an HPLC method described in a previous report [17].
Stability
Sample (1 mL) was mixed with 3 mL of 1640 medium containing 10% FBS or PBS (pH 7.4) and was then incubated at 37 °C. At specific time intervals, particle size and PDI of sample were measured with a Malvern Zetasizer 3000HS system.
Determination of intracellular MMP expression
Cells (2×105) were seeded in 6-well plates and incubated for attachment. After a 48 h incubation, cells were harvested to analyse MMP expression and activity.
First, cells were cultured in RIPA buffer on ice for 15 min, followed by homogenization and centrifugation at 9,000 ×g at room temperature for 10 min. The total protein amount was determined by a BCA protein assay kit (Beyotime, China) and separated on a 10% SDS-polyacrylamide gel (SDS-PAGE). Then, the protein was transferred to a nitrocellulose membrane and incubated in a blocking solution containing a 5% skim milk powder at 37 °C for 2 h. After being rinsed with phosphate buffer solution containing Tween 20 (PBST) 2-3 times, the membrane was incubated with primary antibody at 4 °C for 2 h and a secondary antibody at 4 °C overnight. Finally, target proteins were determined by staining with a chemiluminescence kit (KeyGEN Biotech., China), and blotting pictures were taken by an Odyssey Infrared Imaging System (LICOR Biotechnology, Lincoln, NE). β-Actin or GADPH was used as a loading control.
For gelatin zymography [18], 10 μL of protein extracted from cells was analysed by SDS-PAGE containing a 0.4% gelatin. First, the gels were rinsed with 2.5% Triton X-100 and then incubated in 8% separation solution for 4 h at 25 °C followed by staining with Coomassie Blue for 2 h. Then, the gels were destained with 25% methanol and 10% acetic acid for 1 h. Clear bands on the dark background indicated gelatin degradation.
Cellular uptake
4T1 cells (1×105) were seeded in 12-well plates and incubated for 48 h at 37 °C. Dye-labelled MATT/CN complexes and HPMC NPs were added and incubated for 4 h at 37 °C at a dye concentration of 1 μg/mL or 10 μg/mL. Then, cells were digested from the plates, washed with PBS three times, and resuspended in 500 μL of PBS for flow cytometry analysis (Accuri C6, BD, USA).
To study the co-delivery of HA-PTX and MATT/CN complexes, cells were incubated with dual-labelled HPMC NPs at a dye concentration of 800 ng/mL for a fixed duration at 37 °C and were analyzed by flow cytometry after digestion, rinsing and resuspension.
To further study the CD44-mediated targeted delivery, the cells were pre-cultured with 10 mg/mL of HA or PBS for 1 h and then were incubated with FITC-HMPC NPs for 4 h at 37 °C. The uptake was measured by flow cytometry, while the intracellular distribution was examined by CLSM (LSM700, Carl Zeiss, Germany) after the nuclei were stained with DAPI.
Intracellular location
To study the intracellular localization, 2×105 cells were cultured on round glass cover slips and incubated with the fluorophore-conjugated samples at a concentration of 800 ng/mL for Rho or FITC in serum-free medium for 4 h at 37 °C. After washing with PBS three times, the cells were treated with 1 mL of Lyso-tracker red or green for 2 h to stain late endosomes and lysosomes, fixed with 4% paraformaldehyde and incubated with 1 mL of DAPI for 10 min to mark the nucleus. Then, images were taken by CLSM (LSM700, Carl Zeiss, Germany).
To study the co-localization with MMPs, cells were subsequently treated with Rho-labelled nanoparticles for 4 h at a Rho concentration of 800 ng/mL, washed with cold PBS three times, fixed for 15 min, permeabilized for 5 min, blocked with a blocking reagent for 15 min, and incubated with Alexa Fluor® 488-conjugated MMP-3 antibody overnight at 4 °C. After, CLSM examination was performed (LSM700, Carl Zeiss, Germany).
MMP inhibition in vitro
4T1 (2×105) cells were seeded in 6-well plates and incubated for 48 h for attachment. Then, the cells were treated with different nanoparticles. After a 24 h incubation, the cells were harvested, and MMP expression and activity were examined. Western blotting and gelatin zymography were performed by the same method as mentioned above. Quantitative integrated optical density (IOD) of western blotting and quantitative analysis of gelatinase activity from zymographs were calculated by Gel pro 4.0.
For casein zymography, 10 μL of proteins extracted from 4T1 cells was analysed by SDS-PAGE containing 1 mg/mL casein. Then, the gels were removed and incubated in 1× renaturating buffer for 2 h at room temperature. After being washed twice, the gels were incubated in 20 mL 1× enzyme reaction buffer at room temperature overnight followed by staining with Coomassie Blue for 2 h and destaining with 25% methanol and 10% acetic acid for 1 h. Clear bands on the dark background indicated casein degradation. Quantitative analysis of caseinase activity from zymographs was performed by Gel pro 4.0.
Invasion inhibition
A transwell assay was performed in 24-well transwell chambers (Corning Life Sciences, Inc., America) containing filters with 8 μm pores. First, the 4T1 cells were seeded in the upper chambers coated with 5 μg/mL of diluted Matrigel® (BD Biosciences, America) at a density of 1×105 cells/well for 48 h and then cultured with preparations for another 24 h at 37 °C at PTX and MATT concentrations of 2 μg/mL and 5 μg/mL, respectively. After incubation, the cells on the upper chamber were removed with a cotton swab while the migrated cells were fixed with 4% paraformaldehyde and stained with 0.1% crystal violet for imaging with optical microscopy (Olympus IX53, Japan). The optical density (OD) ratio was determined at 570 nm after crystal violet was dissolved in 33% acetic acid.
In vivo tumor targeting and biodistribution
Balb/C mice (18-22 g) were purchased from the College of Veterinary Medicine Yangzhou University (License No: SCXK (Su) 2012-0004, Yangzhou, China). The animals used in the experiments received care in compliance with the Principles of Laboratory Animal Care and the Guide for the Care and Use of Laboratory Animals. All of the animal experiments were performed in accordance with a protocol approved by the China Pharmaceutical University Institutional Animal Care and Use Committee.
IR783-labelled nanoparticles (0.2 mL) were injected into the 4T1 tumor-bearing BALB/c mice via tail vein at a fixed IR783 dose of 1 mg/kg. At 1, 2, 4, 6, and 8 h post injection, the mice were anaesthetized and fluorescence images were taken with an in vivo imaging system (IVIS Spectrum, PerkinElmer, USA). At 8 h post-injection, the mice were sacrificed to harvest the main organs for ex vivo imaging and fluorescence intensity measurement.
Antitumor efficacy
4T1 tumor-bearing BALB/c mice were randomly divided into four groups treated with saline (negative control), free MATT, HA-PTX or HPMC NPs (n = 5). Each mouse was injected intravenously every 3 days with preparations at a dose of 2 mg/kg for PTX and 5 mg/kg for MATT. The tumor size and mouse weight were recorded every 3 days in the treatment period. After 15 days post-treatment, the mice were sacrificed to sample tumors and other main organs for further examination.
The lungs in each group of mice were harvested and immersed in Bouin's solution for 72 h. The number of tumor nodules on the lungs was calculated to assess lung metastasis from breast cancer.
The tumors were fixed in 4% paraformaldehyde and embedded in paraffin to prepare 5 µm thick sections. Then, an immunohistochemical staining test was performed according to the standard instructions. Haematoxylin-eosin (H&E) staining was performed for histological analysis. CD31 staining was used to quantify the vessel density in tumors. TUNEL and Ki67 immunohistochemical staining were employed to measure cell apoptosis and proliferation in tumors.
MMP expression assay in vivo
MMP expression was assayed by western blotting. Briefly, the tumors were treated in RIPA buffer on ice for 10-20 min, homogenized and centrifuged at 9,000 ×g for 10 min at 25 °C for a total protein assay using a BCA protein assay kit and separation on a 10% SDS-PAGE. Then, the protein was subsequently transferred to a nitrocellulose membrane, incubated in blocking solution with a 5% skimmed milk powder at 37 °C for 2 h, rinsed with PBS containing Tween 20 2-3 times, followed by incubation with primary antibody at 4 °C for 2 h and a secondary antibody at 4 °C overnight. Ultimately, immunoreactive proteins were analysed, and blotting pictures were imagined in an Odyssey Infrared Imaging System (LICOR Biotechnology, Lincoln, NE) after staining with a chemiluminescence kit. β-Actin was used as an internal control to normalize the protein expression. Integrated optical density (IOD) was detected by a gel optical density analysis software Gel pro 4.0.
MMP activity assay in vivo
MMP activity was determined by gelatin zymography under conditions described in a previous report [18]. In brief, 10 μL of separated proteins from tumors was assayed on an 8% SDS-PAGE with 0.4% gelatin, followed by washing twice with 2.5% Triton X-100, treatment in 8% separation solution for 4 h at room temperature, staining with Coomassie Blue for 2 h and destaining in 25% methanol for 1 h and 10% acetic acid for 1 h. Areas with clear bands on the dark background indicated gelatin degradation. Relative MMP activity was calculated based on the ratio between the density of the active band and the total densities of the bands for the active and pro-MMP. Quantitative analysis of gelatinase activity from zymographs was conducted by Gel pro 4.0.
Safety study in vivo
Normal BALB/c mice were randomly divided into 5 groups (3 animals/each) and were injected with CN, MATT, HA-PTX, or HPMC NPs via the tail vein every 3 days five times. Saline was used as control. The dose for CN was equivalent to that for HPMC NPs, whereas for treatment with drug-loading preparations, the doses were 2 mg/kg and 5 mg/kg for PTX and MATT, respectively. At 16 days post-administration, blood of each mouse was collected for hematologic analysis and the mice were sacrificed to isolate the major tissues for H&E and CD68 immunohistochemical staining.
Statistical analysis
The data are expressed as mean ± standard deviation. One-way analysis of variance was performed to assess the statistical significance of the differences between samples. P < 0.05 indicates significant differences.
Results
Preparation and characterization
First, HA-PTX prodrugs and MATT/CN complexes were prepared. The prodrug was synthesized using PTX-NHS and HA-adipic acid dihydrazide (ADH) as intermediates, as described in our previous report [10]. HA-PTX prodrugs were obtained via a nucleophilic reaction between HA-ADH and PTX-NHS amino groups. MATT/CN complexes were fabricated by mixing protein and MATT solutions followed by ultrasonic treatment. The fluorescence spectra demonstrated that the addition of MATT quenched the intrinsic fluorescence of CN, with a trend of increasing fluorescence quenching for rising drug concentration from 0 to 1 mg (Figure 2A), demonstrating that the local environment of tryptophan (Trp) residues was disturbed [19]. Far-UV CD spectra displayed that with an increase in drug concentration, the negative maximum of protein was reduced (Figure 2C), which therefore implied a decreased α-helical content and change in protein secondary structure [20]. The results indicated the formation of MATT/CN complexes.
Next, we assembled HA-PTX prodrugs and MATT/CN complexes into HPMC NPs. Mixing these two components induced significant fluorescence quenching and alteration in the protein's secondary structure, and these changes became profound with increasing prodrug concentration (Figure 2B, D), an indicator of interaction between the prodrugs and complexes. The physical properties of these nanoparticles are shown in Table S1. To further quantify the interaction, an affinity study based on fluorescence quenching was performed (Table S2). A comparison indicated that the affinity (Ka) followed the following order: HPMC NPs > MATT/CN complexes > HA-PTX/CN NPs. These findings suggested that the formation of MATT/CN complexes enforced the interaction with HA-PTX.
Figure 2
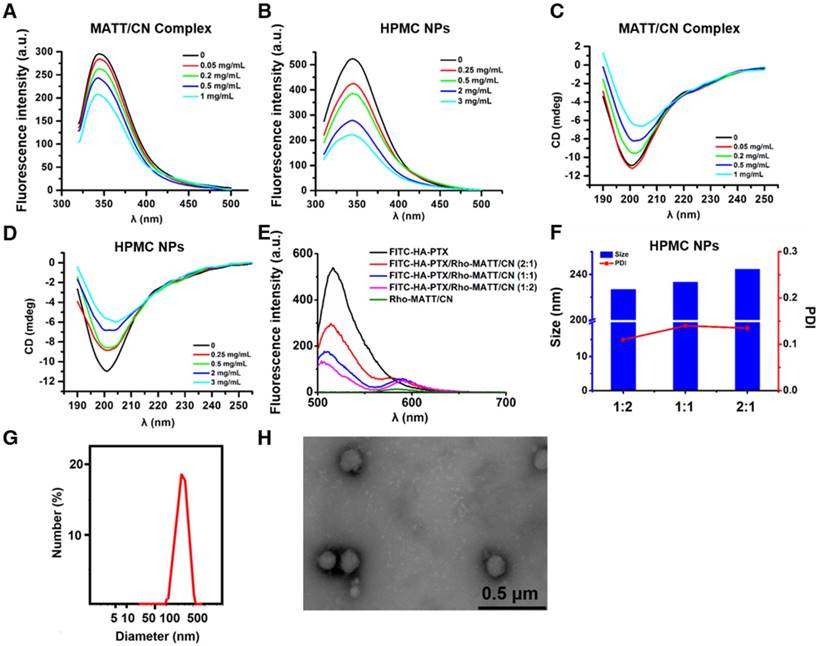
(A-B) Fluorescence emission and (C-D) far UV CD spectra of MATT/CN complex and HPMC NPs prepared with different drug concentrations. (E) Fluorescence emission spectra of dual-labelled HPMC NPs with FITC-HA-PTX/Rho-MATT/CN mass ratios of 2:1, 1:1 and 1:2, respectively (FITC/Rho, w/w). Excitation wavelength: 450 nm. Characterization of HPMC NPs: (F) size and PDI of HPMC NPs with FITC-HA-PTX/Rho-MATT/CN mass ratios of 2:1, 1:1 and 1:2, respectively, (G) size distribution and (H) TEM image of HPMC NPs.
FRET is a well-defined energy transfer process between a fluorescent donor and acceptor within 1-10 nm distance and is frequently used to test the interplay between nanoparticles and external or internal stimuli by monitoring the changes of fluorescence from both donor and acceptor [21-23]. Here, we used FRET to confirm that HA-PTX prodrugs and MATT/CN complexes assembled together, in which FITC was utilized as a donor conjugated to HA-PTX while Rho was employed as an acceptor and conjugated to MATT/CN complexes (Figure 2E). FITC-HA-PTX showed strong fluorescence intensity at 515 nm while there was no signal at the emission wavelength (575 nm) of the acceptor Rhodamine (Rho) (Rho-MATT/CN complexes). Significantly, combining FITC-HA-PTX with Rho-MATT/CN complexes resulted in fluorescence reduction at 515 nm but increased fluorescence at 575 nm, and these changes increased as the ratio of Rho-MATT/CN complexes increased. These results implied a profound FRET effect with the combination of these two complexes. Confocal laser scanning microscope (CLSM) observation on the combination at a mass ratio of 2:1 exhibited yellow fluorescence throughout and, accordingly, indicated the overlap of these two probes (Figure S1).
Collectively, the fluorescence and CD spectra and affinity and FRET studies revealed that the interaction between HA-PTX prodrugs and MATT/CN complexes was strong, and these two components can assemble together into nanoparticles. Finally, HPMC NPs were prepared at a prodrug/complex mass ratio of 2:1 for further study, in which MATT loading in MATT/CN complexes was 42% (drug weight compared with the weight of CN). The HPMC NPs had a diameter of 220-240 nm with a spherical morphology (Figure 2F-H), despite the difference in prodrug/complex ratio. The particle size and PDI of MATT/CN complexes or HPMC NPs remained unchanged after incubation in medium containing 10% serum or PBS (pH 7.4) for 36 h at 37 °C, indicating their stability (Figure S2A-B).
Cellular uptake and trafficking
First, the uptake of FITC-labelled or Rho-labelled nanoparticles in 4T1 cells was investigated. The uptake of HPMC NPs assayed by flow cytometry were concentration-dependent at determined concentrations after a 4 h incubation (Figure S3A-B), and HPMC NPs were distributed in the cytoplasm (Figure S3C-D). Critically, the uptake of HPMC NPs was significantly higher than that of complexes at a high FITC concentration (Figure 3A), indicating that the targeting effect stemmed from HA ligand-CD44 receptor affinity.
To further demonstrate the HA-CD44-mediated cellular uptake, FITC-labelled HPMC NP was incubated with 4T1 cells pre-treated with 10 mg/mL HA to saturate CD44 receptors. CLSM observation demonstrated that the location of FITC-HPMC NPs in pre-treated cells was significantly less than that in untreated cells (Figure 3B). Measurement by flow cytometry further confirmed the results (Figure 3C). Again, these results demonstrated that HA-CD44 affinity improved the uptake of HMPC NPs.
To study the intracellular trafficking of HPMC NPs, the nanoparticles were labelled with FITC or Rho, and lysosomes were stained with lyso-tracker markers. CLSM observation performed at a 4 h incubation post-administration displayed FITC fluorescence (FITC-HPMC NPs) in perinuclear regions of the cells and yellow spots from overlap of the lyso-tracker red and FITC signals in merged images (Figure 3D). Similar results were obtained from the intracellular trafficking of Rho-HPMC NPs (Figure 3E). These findings suggested that HPMC NPs were taken up well by cancer cells via a lysosomal pathway.
Figure 3
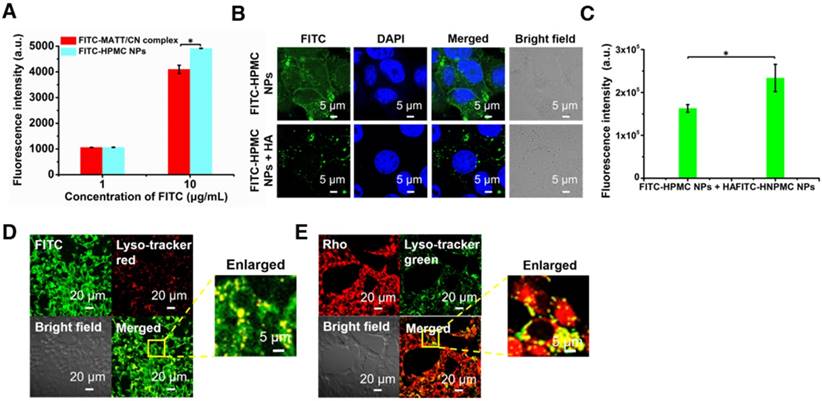
Cellular uptake and trafficking. (A) Cellular uptake of FITC-labelled nanoparticles in 4T1 cells measured by flow cytometry. Uptake of FITC-HPMC NPs in 4T1 cells with or without pre-treatment of 10 mg/mL HA for 1 h at 37 °C; (B) CLSM observation and (C) flow cytometry measurement. The nuclei were stained with DAPI (blue). The data are presented as mean ± s.e.m. (n = 3, *P < 0.05). (D-E) Co-localization of FITC or Rho-HPMC NPs with lysosomes at a dye concentration of 800 ng/mL. The lysosomes were stained with lyso-tracker red (red) or lyso-tracker green (green). The cells were incubated with dye-labelled nanoparticles for 4 h at 37 °C before measurement.
High MMP expression in cancer cells
The expression MMP-3 in different cell lines was examined by western blotting assay. Definitely, MMP-3 in 4T1 and MDA-MB-435 cancer cells was highly expressed (Figure S4A-B).
MMP-2 and MMP-9 play a critical role in tumor metastasis; therefore, their expression and activity in different cells was detected by western blotting assay and gelatin zymography. Indeed, significantly higher expression and activity of the two MMPs was displayed in cancer cells, especially in 4T1 and MDA-MB 435 cell lines, compared with non-cancer cells, such as HELF (Figure S5A-B).
Intracellular co-delivery, co-localization with MMPs and sensitive release
To confirm the co-delivery of HA-PTX prodrugs and MATT/CN complexes, dual-labelled HPMC NPs were prepared, incubated with 4T1 cells for 4 h and followed by a flow cytometry assay and CLSM examination. Fluorescence intensity of both Rho and FITC increased simultaneously (Figure 4A), and yellow fluorescence in perinuclear areas in merged images was observed (Figure 4B), demonstrating co-delivery of the prodrugs and complexes using HPMC NPs.
CN is a substrate of various MMPs, such as MMP-3, MMP-7, and MMP-8 [3, 4, 14]. Hence, it was hypothesized that the intracellular MMPs would tend to associate with the uptaken HPMC NPs and, as a result, break them down and facilitate drug release into the cytoplasm. First, co-localization of Rho-labelled HPMC NPs (red) with intracellular MMP-3 that was marked in green with Alexa Fluor® 488-conjugated MMP-3 antibody was studied after a 4 h incubation (Figure 4C). Dramatically, intense yellow fluorescence was observed throughout in the merged image (enlarged view), an indicator of their co-localization.
Figure 4
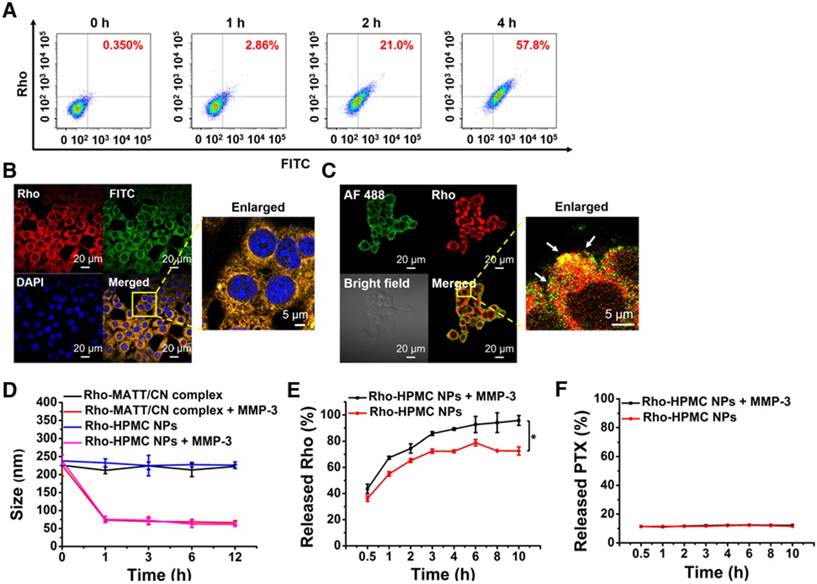
Co-delivery of HPMC NPs, co-localization with intracellular MMP-3, and MMP sensitivity. (A) Intracellular uptake of dual-labelled HPMC NPs at 0 h, 1 h, 2 h and 4 h measured by flow cytometry after incubation at 37 °C at a FITC or RITC concentration of 800 ng/mL. Dual fluorophore-labelled HPMC NPs were prepared by conjugating FITC (green) to HA-PTX and RITC (red) to CN. (B) CLSM observation of intracellular dual-labelled HPMC NPs at 4 h post incubation at a FITC or RITC concentration of 800 ng/mL. (C) Co-localization of Rho-labelled HMPC NPs (red) and Alexa Fluor 488 (green)-labelled MMP-3 in 4T1 cells observed by CLSM. Cells were cultured with Rho-HPMC NPs at a Rho concentration of 800 ng/mL for 4 h at 37 °C. MMP sensitivity: (D) size change of Rho-MATT/CN complexes or Rho-HPMC NPs after culturing with pH 6.5 PBS containing MMP-3 and (E) Rho or (F) PTX release profiles in pH 6.5 PBS containing a 5 µg/mL MMP-3 at 37 °C. The data are presented as mean ± s.e.m. (n = 3, *P < 0.05).
Figure 5
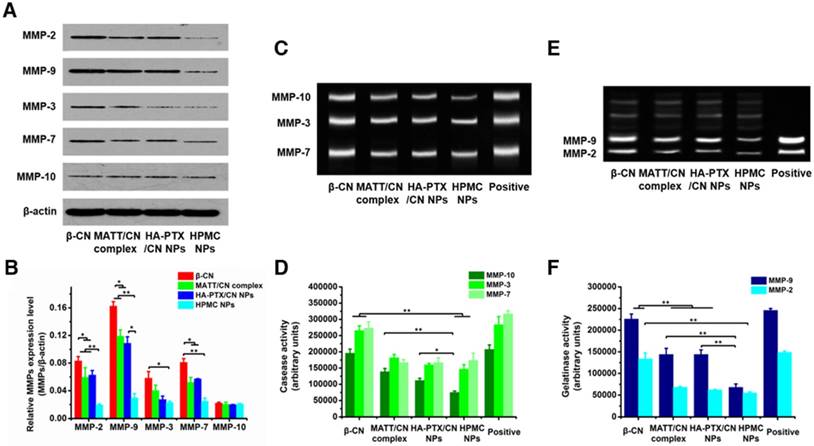
Inhibition of MMP expression and in vitro activities. (A) Western blotting analysis of MMP-2, MMP-3, MMP-7, MMP-9 and MMP-10 expression in 4T1 cells treated with preparations for 24 h at PTX and MATT concentrations of 2 μg/mL and 5 μg/mL, respectively. β-Actin was used as a loading control. Dark bands indicate MMP expression. (B) Quantitative analysis of the expression of MMP-2, MMP-3, MMP-7, MMP-9 and MMP-10. (C) MMP activity of MMP-3, MMP-7 and MMP-10 in 4T1 cells analysed by casein zymography. Bright bands on the dark background indicate MMP activity. (D) Quantitative analysis of caseinase activity using a computer-supported image analysis program. (E) MMP activity of MMP-2 and MMP-9 in 4T1 cells analysed by gelatin zymography. Bright bands on the dark background indicate MMP activity. (F) Gelatinase activity from zymograms using a computer-supported image analysis program. The data are presented as mean ± s.e.m. (n = 3, *P < 0.05, **P < 0.01).
Next, the in vitro sensitivity of HPMC NPs to MMP-3 was tested. As shown in Figure 4D, the addition of MMP-3 resulted in a dramatic size reduction of Rho-complexes or Rho-HPMC NPs at 1 h post incubation, and a promotion of Rho release (Figure 4E) compared with that of PBS, therefore demonstrating the digestion of CN and breakdown of HPMC NPs. On the other hand, MMP-3 imposed little influence on PTX release (Figure 4F).
Overall, these findings suggested that the uptaken HPMC NPs could be decomposed by MMPs and release their active components.
In vitro inhibition of MMP expression and activity and cell migration
MMP expression and activity in 4T1 cells incubated with various preparations for 24 h were analysed by western blotting and gelatin zymography. HPMC NP treatment downregulated the expressions of MMP-2, -9, -3 and -7 by 100-300% compared with MATT/CN complexes or HA-PTX/CN NPs (Figure 5A-B), yet treatment with all of these preparations had little effect on the MMP-10 level. Caseinase zymography (Figure 5C-D) and gelatin zymography (Figure 5E-F) demonstrated that these preparations reduced MMP activity compared with control; of note, HPMC NPs inhibited the activities of MMP-9 and -7 with higher efficiency (50-100%) than the MATT/CN complexes and HA-PTX/CN NPs. Generally, the MATT/CN complex, a protein-drug complex, has difficulty entering cells due to its high MW (24 kDa) and poor membrane penetration. As a result, this complex mainly suppressed the extracellular instead of intracellular expression and activity of MMPs. Collectively, intracellular delivery of MMP inhibitor was capable of potently decreasing MMP expression and activity.
High expression of MMPs in tumors inducing ECM degradation would promote the migration of cancer cells [24]. Here, an in vitro anti-metastasis test was performed using a Matrigel transwell model (Figure S6A-B). Incubation with HA-PTX/CN NPs, MATT/CN complexes or HPMC NPs inhibited the migration of 4T1 cells by 30%, 60% and 70%, respectively. However, MATT loading preparations (MATT/CN complexes or HPMC NPs) exhibited a more profound suppression than HA-PTX/CN NPs with no MATT loading whose inhibition effect resulted from its cytotoxicity. MATT/CN complexes predominantly inhibited extracellular MMPs to exert an anti-migration effect because these complexes were poorly taken up by cells; by contrast, HPMC NPs entered cells with high efficiency via CD44 mediation and targeted intracellular MMPs. Accordingly, via inhibiting intracellular MMPs, the migration of cancer cells was also reduced.
Biodistribution and tumor targeting
To evaluate tumor accumulation, the nanoparticles were labelled with a probe, IR783, for in vivo imaging at predetermined time intervals after dosing. These nanoparticles were distributed in the main tissues after injection and, in particular, the fluorescence in the tumor from IR783-HPMC NPs was most robust among them (Figure 6A-B). A quantified assay demonstrated that the accumulation of IR783-HPMC NPs was approximately 1.4-fold as great as the other two-type nanoparticles, a demonstration of enhanced tumor targeting (Figure 6C).
In vivo antitumor activity
To assess the antitumor activity, 4T1 tumor-bearing mice were intravenously injected with free MATT, HA-PTX prodrug or HPMC NPs at a MATT dose of 5 mg/kg or a PTX dose of 2 mg/kg. Compared with saline, treatment with MATT or HA-PTX prodrug reduced the tumor volume by 8-10-fold at day 15 post dosing (Figure 7A). Importantly, administration of HPMC NPs displayed a 5.5-fold reduction in tumor growth at day 15, along with a 5-7-fold decrease compared with the other two drug loading preparations. Measurements of tumor weight and digitally imaging the tumor at the end of treatment confirmed the result of tumor growth (Figure 7B-C). No significant loss in animal body weight after treatment with these preparations was observed after 5 injections, which indicated low toxicity during treatment (Figure 7D). Altogether, HPMC NPs exhibited potent inhibition of tumor growth.
To ascertain the antitumor efficacy, we further detected the necrosis, apoptosis and proliferation of cancer cells in tumors at the end of treatment. H&E staining indicated that HPMC NPs induced the most severe necrosis and change of morphology among these preparations (Figure 7E). Furthermore, TUNEL and Ki67 assays demonstrated that HPMC NPs enabled the greatest number of positive cells compared with other formulations (Figure 7F-H). Quantitative analysis indicated that treatment with HPMC NPs induced a 53% apoptotic rate (Figure 7G) and 8% proliferation rate (Figure 7I), which were approximately 20% higher and >30% lower than those after use of MATT or HA-PTX prodrug alone. These results suggested that HPMC NPs were efficient in killing cancer cells in vivo.
Figure 6
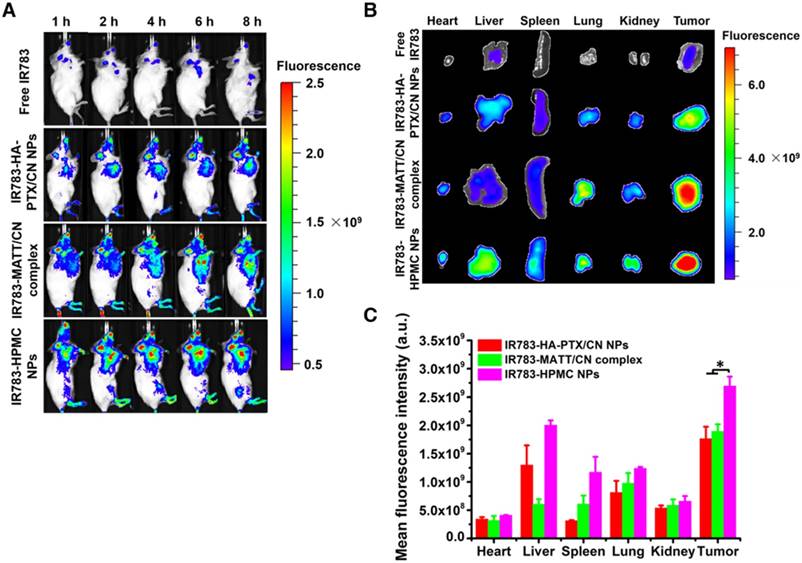
Biodistribution and tumor targeting. (A) Fluorescence imaging of 4T1 tumor-bearing Balb/C mice after intravenous injection with IR783 labelled nanoparticles at an IR783 dose of 1 mg/kg, according to body weight. (B) Ex vivo fluorescence images of the heart, liver, spleen, kidney, lung, and tumors harvested from the mice at 8 h after administration and (C) mean fluorescence intensity of the IR 783 signal in each preparation group. The data are presented as mean ± s.e.m. (n = 3, *P < 0.05).
Figure 7
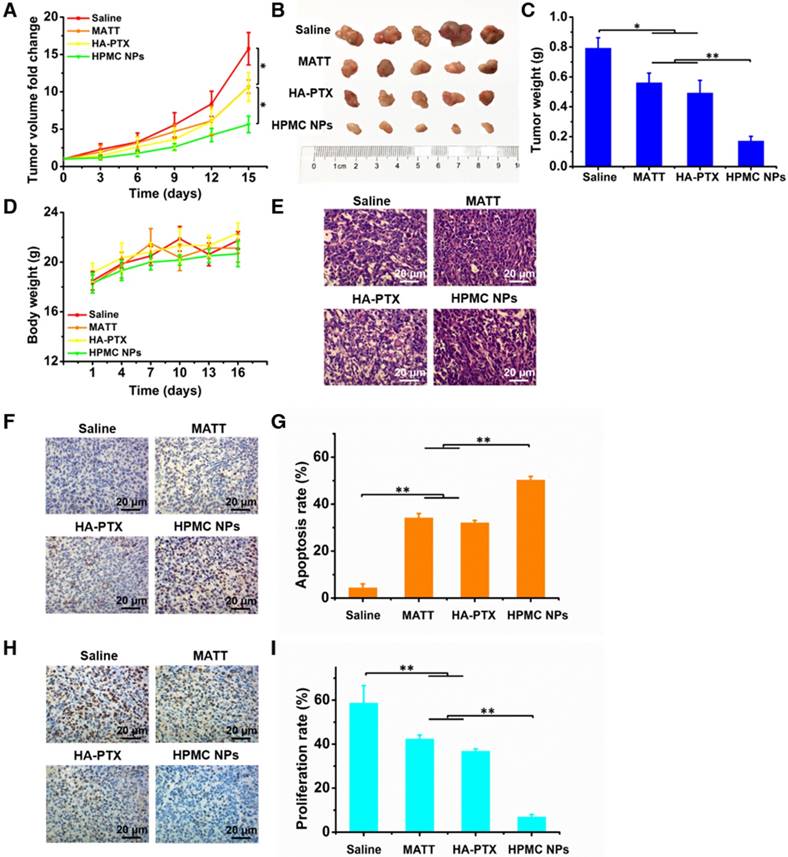
In vivo antitumor activity and cell apoptosis and proliferation in tumors. Preparations (0.2 mL) were administered to 4T1 tumor-bearing Balb/C mice via tail vein injection every 3 days at a PTX dose of 2 mg/kg or MATT dose of 5 mg/kg, according to body weight. (A) Tumor volume fold change in 4T1 tumor-bearing mice. The comparison of the tumor volume fold change between groups was performed on day 15. (B) Digital photos and (C) tumor weights of tumor tissue harvested from the 4T1 tumor-bearing mice on day 16. (D) Body weight changes of 4T1 tumor-bearing mice during the therapy period. (E) H&E analysis of tumor tissues harvested from 4T1 tumor-bearing mice on day 16 post-administration. The nuclei were stained blue, and the cytoplasm and extracellular matrix were stained red. The scale bar is 20 μm. (F) TUNEL and (H) Ki67 staining of tumor tissue sections from tumor tissues harvested from 4T1 tumor-bearing mice on day 16 after treatment. Quantitative analysis of tumor cell (G) apoptosis and (I) proliferation in tumors. Cell apoptosis and proliferation rate were quantified by five representative fields of cell nuclei under an optical microscope. The data are presented as mean ± s.e.m. (n = 5, *P < 0.05, **P < 0.01).
In vivo inhibition of angiogenesis and lung metastasis
The TME, mainly consisting of ECM, cancer cells, blood vessels and non-cancer cells, is a sanctuary for cancer cells. Destroying the TME, in particular ECM degradation by MMPs, would drive the migration of cancer cells and angiogenesis [25]. Here, the inhibition of angiogenesis and tumor metastasis to the lung was assessed at day 16 post dosing. In the saline group, microvessels stained brown with CD31 antibody were observed throughout; by contrast, the treatments with drug-loading preparations greatly reduced the number of microvessels (Figure 8A-B). Significantly, HPMC NPs exhibited exceedingly profound antiangiogenesis over MATT or HA-PTX prodrug, with >3-fold less microvasculature than the other two preparations.
The 4T1 breast tumor model is a highly metastatic cancer, frequently metastasizing to the lung or liver [26]. In this study, metastasis of 4T1 tumors to the lung in 4T1 tumor-bearing mice sampled at day 16 post treatment was assessed. The nodules in lungs treated with HPMC NPs were dramatically lower than that of HA-PTX prodrug or MATT, exhibiting a 5-6-fold decrease in nodule number (Figure 8C-D).
Taken together, HMPC NPs enabled the effective inhibition of angiogenesis and tumor metastasis.
In vivo inhibition of MMP expression and activity
MMPs, in particular MMP-2 and -9, possess a potent ability to degrade ECM [27], promoting tumor metastasis and angiogenesis. Here, first, the expressions of MMP-2 and -9 were determined by western blotting assay. The band signals of the two MMPs from HPMC NPs were significantly weaker than in the other groups (Figure 9A). Quantitative measurement indicated that treatment with HPMC NPs downregulated these two MMPs by >150% compared with the saline group (Figure 9B-C) while use of MATT, an MMP inhibitor, alone decreased the levels of MMP-2 and -9 by 45% and 18%, respectively. These results implied that HMPC NPs markedly reduced the expressions of MMP-2 and -9 in tumors.
Next, the activities of MMP-2 and -9 in tumors were evaluated by gelatin zymography. Compared with saline, treatment with preparations inhibited the activities of these two MMPs greatly (Figure 9D-E). Importantly, HPMC NP reduced the activities of MMP-2 and -9 by >50% and >100%, independently, over saline and >15% and >25%, respectively, compared to the other two preparations, implying enhanced suppression in the activity of MMPs.
Figure 8
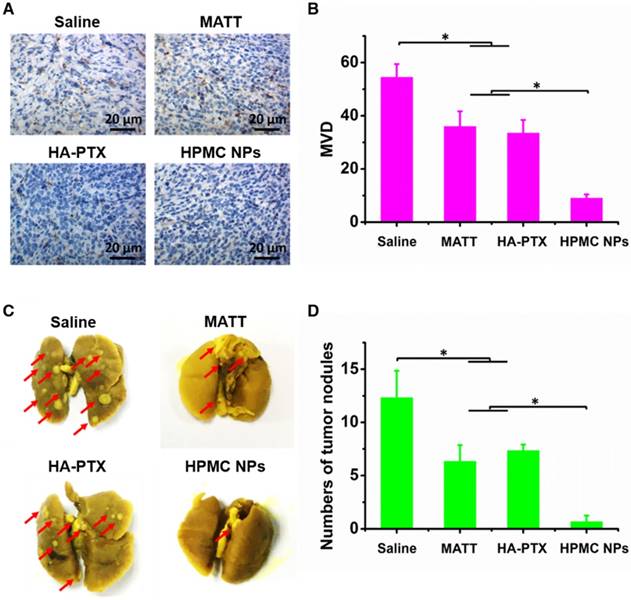
Inhibition of angiogenesis and lung metastasis. (A) Immunochemistry of CD31 staining for microvessels (brown) in tumor tissues harvested from 4T1 tumor-bearing mice at day 16 after treatment. (B) Quantitative analysis of microvascular density (MVD). MVD was quantified by five representative fields of cell nuclei under an optical microscope. (C) Digital photos of lungs harvested from 4T1 tumor-bearing mice at day 16 after treatment. The arrows indicated the tumor nodules in the lungs. (D) Quantitative analysis of tumor nodules in lungs by counting. The data are presented as mean ± s.e.m. (n = 5, *P < 0.05).
Figure 9
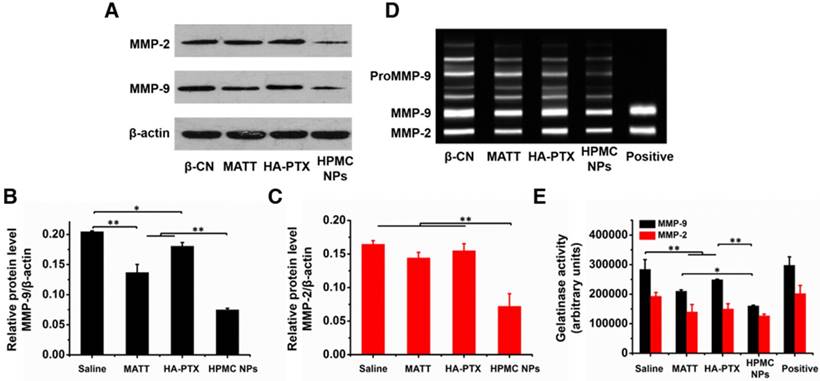
Inhibition of MMP expression and activities in vivo. (A) Western blotting analysis of MMP-2 and MMP-9 expressions in tumors harvested from 4T1 tumor-bearing mice at day 16 after treatment. β-Actin was used as a loading control. Dark bands indicate MMP expression. Quantitative analysis of the expressions of (B) MMP-9 and (C) MMP-2. (D) Activities of MMP-2 and MMP-9 in tumors analysed by gelatin zymography. Bright bands on the dark background indicate MMP activity. (E) Quantitative analysis of gelatinase activities of MMP-2 and MMP-9 from zymograms using a computer-supported image analysis program. The data are presented as mean ± s.e.m. (n = 3, *P < 0.05, **P < 0.01).
Safety study in vivo
Hematologic examination demonstrated that the blood indexes of treated mice showed no significant difference compared with the saline group and that their values were within the normal limits (Table S3). CD68 is a kind of protein present in activated macrophages and can be used to assess immune response. CD68 immunohistochemical staining indicated that the repeated injection of CN did not induce inflammation in the main tissues (Figure S7A). In addition, no damages to these tissues were observed following systemic administration of drug-loaded preparations (Figure S7B). Therefore, HPMC NPs were safe for intravenous injection.
Discussion
Prodrugs based on HA and protein-drug complexes were able to assemble into nanoparticles and improve drug delivery. It is well known that as materials, especially hydrophobic or lipid compounds (particles) [28, 29], enter the blood or are exposed to physiological fluids, various proteins associate with the materials and form protein coronas [30]. Inspired by this mechanism, we speculated that a HA-PTX prodrug can interact with MATT/CN complexes and aggregate into nanoparticles. Truly, according to studies of fluorescence and CD spectra, affinity and FRET, we verified that the interaction between these two materials is strong, rendering the formation of hybrid nanoaggregates with a diameter of ~250 nm. Numerous reports demonstrate that proteins can bind drugs or nanoparticles; however, until now, there have been few reports that polymer-based prodrug and protein-drug complexes are capable of aggregating into nanoparticles for targeted co-delivery. As a result, this study presents a novel approach for constructing nanoplatforms for targeted drug delivery, biomedical imaging, and combined therapy.
Intracellular delivery of MMP inhibitors allows for the efficient inhibition of MMP expression and activity and consequent anti-metastasis activity. Commonly, metastasis is blocked via directly delivering MMP inhibitors to the TME and inactivating extracellular MMPs mainly secreted by cancer cells [1, 31, 32]. Nevertheless, using an MMP inhibitor alone only enables modest efficiency in metastatic cancer because this inhibitor is not a cytotoxic agent and is unable to potently kill the cancer cells. Therefore, a combination use of a cytotoxic agent with an MMP inhibitor is critical for treatment of metastatic cancer. However, to spatially target to the TME and cancer cells, a complicated delivery system has to be designed, which is an extremely challenging work. In this study, by targeting intracellular MMPs using HPMC NPs, the expression and activity of MMP-2 and -9 were decreased by >100% and >15%, respectively, compared with that of extracellular inhibition with free MATT. Additionally, treatment with HPMC NPs reduced angiogenesis >3-fold and lung metastasis >5-fold compared with MATT treatment. In addition, in vitro experiments demonstrated that intracellular delivery of MATT with HPMC NPs is significantly superior in suppressing MMP expression and activity to extracellular delivery (Figure 5). These findings demonstrate a potent inhibition of MMPs in tumors by targeting intracellular rather than extracellular MMPs. To our best knowledge, this is the first report regarding intracellular delivery of an MMP inhibitor for suppressing MMPs and blocking cancer metastasis. We believe that these findings could lead to a new opportunity for understanding MMP functions and, most importantly, greatly facilitate the design of a drug delivery system to treat metastatic cancer. Accordingly, by intracellularly co-delivering an MMP inhibitor and a cytotoxic drug with a common carrier, effective treatment of metastatic cancer may be achieved.
HPMC NPs, co-delivering HA-PTX prodrug and MATT/CN complexes, are robust for curing metastatic cancer. 4T1 tumors are highly metastatic and, subsequently, their clinical treatment outcome is extremely poor [33]. Here, HPMC NPs enabled significant suppression of tumor-growth (approximately >10-fold reduction in tumor volume compared with saline treatment) in 4T1 tumor-bearing mice with efficient apoptosis (~53%) and proliferation (~8%) in tumors and dramatic anti-metastasis and anti-angiogenesis. The improved anti-tumor efficacy is dominantly ascribed to the following factors: (i) enhanced tumor accumulation (~1.4-fold increase), (ii) efficient cellular uptake via CD44 mediation and resultant intracellular delivery of a cytotoxic agent, PTX, and MMP inhibitor, MATT; and (iii) potent inhibition of MMP expression and activity. These results indicated that intracellularly co-delivering chemotherapy and an MMP inhibitor is also a promising strategy for treating metastatic cancer, differing from the traditional combination preparations that independently deliver a MMP inhibitor to extracellular conditions in the TME and a cytotoxic agent to cancer cells. We believe that HPMC NPs represent a new approach to treat metastatic cancer.
Conclusions
In this study, a new hybrid nanoplatform based on a HA-chemotherapy prodrug and a protein-drug complex is established. We reason that this is a common strategy that can be adopted to assemble other polymer prodrug and protein complexes into nanoparticles. Importantly, we uncover that by targeting intracellular instead of extracellular MMPs with HPMC NPs, MMP expression and activity can be significantly inhibited, thereby enabling effective anti-metastasis and anti-angiogenesis effects. This finding would assist in the rational design of new nanomedicines for treating cancer, in particular metastatic tumors. Additionally, co-delivery of HA-PTX prodrug and MATT/CN complexes is a promising approach for cancer treatment in terms of inhibiting tumor growth, cancer cell killing, MMP expression and activity, angiogenesis and metastasis. Altogether, based on the assembly of polymer prodrug and protein-drug complexes, this work builds a new approach for constructing nanoparticles for combined disease treatment, drug delivery, and biomedical imaging. Furthermore, we demonstrate that inhibiting intracellular MMPs robustly inhibits cancer metastasis, which can help design rational nanomedicines for cancer treatment.
Abbreviations
CLSM: confocal laser scanning microscopy; CN: β-casein; DLS: dynamic laser scattering; ECM: extracellular matrix; FBS: fetal bovine serum; FITC: fluorescein isothiocyanate; FRET: Förster resonance energy transfer; HA: hyaluronic acid; HCT: hematocrit; HGB: hemoglobin; HPLC: high performance liquid chromatography; HPMC NPs: nanoparticles assembled from HA-PTX prodrug and MATT/CN complexes; H&E: haematoxylin-eosin; IOD: integrated optical density; MATT: marimastat; MMPs: matrix metalloproteinases; MTT: 3-(4,5)-dimethylthiahiazo (-z-y1)-3,5-diphenyltetrazoliumromide; MW: molecular weight; OD: optical density; PBS: phosphate buffer solution; PBST: phosphate buffer solution containing tween 20; PDI: polydispersity index; pI: isoelectric point; PLT: platelet; PTX: paclitaxel; RBC: red blood cell; Rho: rhodamine B; RITC: rhodamine B isothiocyanate; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; TEM: transmission electron microscopy; TME: tumor microenvironment; WBC: white blood cell.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (Nos. 81673377, 81473152, and 81402869), and the Fostering Plan of University Scientific and Technological Innovation Team and Key Members of the Outstanding Young Teacher of Jiangsu Qing Lan Project (2014 and 2016). We also thank Xiaonan Ma and Minhui Sun from the Cellular and Molecular Biology Center of China Pharmaceutical University for providing technical assistance.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Vandenbroucke RE, Libert C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat Rev Drug Discov. 2014;13:904-27
2. Isaacson KJ, Martin Jensen M, Subrahmanyam NB, Ghandehari H. Matrix-metalloproteinases as targets for controlled delivery in cancer: An analysis of upregulation and expression. J Control Release. 2017;259:62-75
3. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52-67
4. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161-74
5. Cathcart J, Pulkoski-Gross A, Cao J. Targeting matrix metalloproteinases in cancer: bringing new life to old ideas. Genes Dis. 2015;2:26-34
6. Behrends M, Wagner S, Kopka K, Schober O, Schafers M, Kumbhar S. et al. New matrix metalloproteinase inhibitors based on gamma-fluorinated alpha-aminocarboxylic and alpha-aminohydroxamic acids. Bioorg Med Chem. 2015;23:3809-18
7. Rasmussen HS, McCann PP. Matrix metalloproteinase inhibition as a novel anticancer strategy: a review with special focus on batimastat and marimastat. Pharmacol Ther. 1997;75:69-75
8. Rowinsky EK, Onetto N, Canetta RM, Arbuck SG. Taxol: the first of the taxanes, an important new class of antitumor agents. Semin Oncol. 1992;19:646-62
9. Luo D, Carter KA, Miranda D, Lovell JF. Chemophototherapy: an emerging treatment option for solid tumors. Adv Sci. 2017;4:1600106
10. Xu C, He W, Lv Y, Qin C, Shen L, Yin L. Self-assembled nanoparticles from hyaluronic acid-paclitaxel prodrugs for direct cytosolic delivery and enhanced antitumor activity. Int J Pharm. 2015;493:172-81
11. Hasan A, Khattab A, Islam MA, Hweij KA, Zeitouny J, Waters R. et al. Injectable hydrogels for cardiac tissue repair after myocardial infarction. Adv Sci. 2015;2:1500122
12. Li L, Schmitt M, Matzke-Ogi A, Wadhwani P, Orian-Rousseau V, Levkin PA. CD44v6-peptide functionalized nanoparticles selectively bind to metastatic cancer cells. Adv Sci. 2017;4:1600202
13. Elzoghby AO, El-Fotoh WS, Elgindy NA. Casein-based formulations as promising controlled release drug delivery systems. J Control Release. 2011;153:206-16
14. Zitka O, Kukacka J, Krizkova S, Huska D, Adam V, Masarik M. et al. Matrix metalloproteinases. Curr Med Chem. 2010;17:3751-68
15. Sheng Z, Hu D, Zheng M, Zhao P, Liu H, Gao D. et al. Smart human serum albumin-indocyanine green nanoparticles generated by programmed assembly for dual-modal imaging-guided cancer synergistic phototherapy. ACS Nano. 2014;8:12310-22
16. Chen Q, Wang X, Wang C, Feng L, Li Y, Liu Z. Drug-induced self-assembly of modified albumins as nano-theranostics for tumor-targeted combination therapy. ACS Nano. 2015;9:5223-33
17. Xin X, Pei X, Yang X, Lv Y, Zhang L, He W. et al. Rod-shaped active drug particles enable efficient and safe gene delivery. Adv Sci. 2017;4:1700324
18. Ikeda M, Maekawa R, Tanaka H, Matsumoto M, Takeda Y, Tamura Y. et al. Inhibition of gelatinolytic activity in tumor tissues by synthetic matrix metalloproteinase inhibitor: application of film in situ zymography. Clin Cancer Res. 2000;6:3290-6
19. He W, Wang Y, Lv Y, Xiao Q, Ye L, Cai B. et al. Denatured protein stabilized drug nanoparticles: tunable drug state and penetration across the intestinal barrier. J Mater Chem B. 2017;5:1081-97
20. Pan H, Qin M, Meng W, Cao Y, Wang W. How do proteins unfold upon adsorption on nanoparticle surfaces? Langmuir. 2012;28:12779-87
21. Kneidl B, Peller M, Winter G, Lindner LH, Hossann M. Thermosensitive liposomal drug delivery systems: state of the art review. Int J Nanomedicine. 2014;9:4387-98
22. Jares-Erijman EA, Jovin TM. FRET imaging. Nat Biotechnol. 2003;21:1387-95
23. Chen C, Li C, Shi Z. Current Advances in Lanthanide-doped upconversion nanostructures for detection and bioapplication. Adv Sci. 2016;3:1600029
24. Sinno M, Biagioni S, Ajmone-Cat MA, Pafumi I, Caramanica P, Medda V. et al. The matrix metalloproteinase inhibitor marimastat promotes neural progenitor cell differentiation into neurons by gelatinase-independent TIMP-2-dependent mechanisms. Stem Cells Dev. 2013;22:345-58
25. Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298-306
26. Schroeder A, Heller DA, Winslow MM, Dahlman JE, Pratt GW, Langer R. et al. Treating metastatic cancer with nanotechnology. Nat Rev Cancer. 2012;12:39-50
27. Shuman Moss LA, Jensen-Taubman S, Stetler-Stevenson WG. Matrix metalloproteinases: changing roles in tumor progression and metastasis. Am J Pathol. 2012;181:1895-9
28. Nussbaumer MG, Duskey JT, Rother M, Renggli K, Chami M, Bruns N. Chaperonin-dendrimer conjugates for siRNA delivery. Adv Sci. 2016;3:1600046
29. Li Y, Wu Z, He W, Qin C, Yao J, Zhou J. et al. Globular protein-coated paclitaxel nanosuspensions: interaction mechanism, direct cytosolic delivery, and significant improvement in pharmacokinetics. Mol Pharm. 2015;12:1485-500
30. Nel AE, Madler L, Velegol D, Xia T, Hoek EMV, Somasundaran P. et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8:543-57
31. Arkadash V, Yosef G, Shirian J, Cohen I, Horev Y, Grossman M. et al. Development of high affinity and high specificity inhibitors of matrix metalloproteinase 14 through computational design and directed evolution. J Biol Chem. 2017;292:3481-95
32. Winer A, Janosky M, Harrison B, Zhong J, Moussai D, Siyah P. et al. Inhibition of breast cancer metastasis by presurgical treatment with an oral matrix metalloproteinase inhibitor: a preclinical proof-of-principle study. Mol Cancer Ther. 2016;15:2370-7
33. Dart A. Metastasis: Breaching barriers. Nat Rev Cancer. 2017;17:270
Author contact
![]() Corresponding author: E-mail: lifangyin_com (L.F. Yin), weiheedu.cn (W. He). Department of Pharmaceutics, School of Pharmacy, China Pharmaceutical University, Nanjing, 210009, P.R. China
Corresponding author: E-mail: lifangyin_com (L.F. Yin), weiheedu.cn (W. He). Department of Pharmaceutics, School of Pharmacy, China Pharmaceutical University, Nanjing, 210009, P.R. China
Citation styles
APA
Lv, Y., Zhao, X., Zhu, L., Li, S., Xiao, Q., He, W., Yin, L. (2018). Targeting intracellular MMPs efficiently inhibits tumor metastasis and angiogenesis. Theranostics, 8(10), 2830-2845. https://doi.org/10.7150/thno.23209.
ACS
Lv, Y.; Zhao, X.; Zhu, L.; Li, S.; Xiao, Q.; He, W.; Yin, L. Targeting intracellular MMPs efficiently inhibits tumor metastasis and angiogenesis. Theranostics 2018, 8 (10), 2830-2845. DOI: 10.7150/thno.23209.
NLM
Lv Y, Zhao X, Zhu L, Li S, Xiao Q, He W, Yin L. Targeting intracellular MMPs efficiently inhibits tumor metastasis and angiogenesis. Theranostics 2018; 8(10):2830-2845. doi:10.7150/thno.23209. https://www.thno.org/v08p2830.htm
CSE
Lv Y, Zhao X, Zhu L, Li S, Xiao Q, He W, Yin L. 2018. Targeting intracellular MMPs efficiently inhibits tumor metastasis and angiogenesis. Theranostics. 8(10):2830-2845.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.