Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2018; 8(10):2709-2721. doi:10.7150/thno.20576 This issue Cite
Review
Role of Extracellular Vesicles in Viral and Bacterial Infections: Pathogenesis, Diagnostics, and Therapeutics
Meryl Rodrigues1,2, Jia Fan1, Christopher Lyon1, Meihua Wan3 ![]() , Ye Hu1,2
, Ye Hu1,2 ![]()
1. Virginia G. Piper Biodesign Center for Personalized Diagnostics, Arizona State University Biodesign Institute, Tempe, Arizona, 85287
2. School of Biological and Health Systems Engineering, Arizona State University, Tempe, Arizona, 85287
3. Department of Integrated Traditional Chinese and Western Medicine, West China Hospital of Sichuan University, Chengdu, Sichuan, China, 610041
Received 2017-4-14; Accepted 2018-1-15; Published 2018-4-9
Abstract

Extracellular vesicles (EVs), or exosomes, are nanovesicles of endocytic origin that carry host and pathogen-derived protein, nucleic acid, and lipid cargos. They are secreted by most cell types and play important roles in normal cell-to-cell communications but can also spread pathogen- and host-derived molecules during infections to alter immune responses and pathophysiological processes. New research is beginning to decipher how EVs influence viral and bacterial pathogenesis. In this review, we will describe how EVs influence viral and bacterial pathogenesis by spreading pathogen-derived factors and how they can promote and inhibit the immune response to these pathogens. We will also discuss the emerging potential of EVs as diagnostic and therapeutic tools.
Keywords: extracellular vesicles, exosomes, pathogenesis, immune system, diagnostic, therapeutic
Introduction
Extracellular vesicles (EVs), particularly exosomes, have gained attention for their potential as disease biomarkers and therapeutic agents. Exosomes are EVs released by the endocytic pathway that range from 30-100 nm in diameter, and contain host (and pathogen)-derived nucleic acid, protein and lipid cargos. Microvesicles (also known as shedding vesicles, ectosomes or microparticles), are a distinct type of EV that forms by the outward budding of the plasma membrane and are 100-1000 nm in diameter. These EV types are distinct in their subcellular site of origin and physical parameters, but the size overlap of exosomes and microvesicles and differences in EV isolation and handling procedures can lead to confusion, despite ongoing efforts by the International Society for Extracellular Vesicles (ISEV) to standardize and harmonize these methods [1, 2]. This review will focus on the role of exosomes in chronic viral and bacterial disease, but due to the potential for confusion between EV types among studies we will refer to vesicles analyzed in all cited studies, except those focusing on the exosome biosynthesis pathway itself, as EVs to adhere to the ISEV nomenclature guidelines.
Most cells secrete EVs, but EVs produced during pathogen infections can reveal differences in their composition to reflect their origin from infected cells and the overall state of the infection. Studies have revealed multiple ways by which viruses and bacteria can manipulate EV synthesis to enhance their transmission and pathogenesis (reviewed in [3]). Conversely, EVs produced by immune cells play an important role in host responses to infection. One early example of this role was the finding that EVs from B lymphocytes contained class II major histocompatibility (MHCII)-antigen complexes that could activate CD4+ T cells in an antigen-specific manner [4]. Subsequent studies found that EVs of dendritic cells contained class I major histocompatibility (MHCI)-peptide complexes that could stimulate cytotoxic CD8+ T cell responses, identified cell-dependent and independent mechanisms for the antigen presenting activity, and identified receptor and cytokine/chemokine effects to regulate multiple cells involved in the adaptive immune response [5-7]. Notably, EVs derived from cells infected with either viral or bacterial pathogens demonstrate several mechanisms to mediate the immune system, including effects to inhibit host EV effects to promote adaptive immune responses, indicating that better understanding of these mechanisms is important to improve therapeutic approaches used to treat these pathogens. EVs carrying pathogen-derived factors are also of interest as biomarkers of infection, since these factors should be more stable than soluble factors in circulation that are exposed to circulating hydrolase activities. EVs are also stable in circulation, are capable of packaging a broad array of biomolecules and small molecule drugs, and exhibit potential as selective targeted biogenic carriers [8-14]. Based on these properties, this review describes current knowledge of EV actions to promote disease and regulate host immunity, and the potential of these EVs as disease biomarkers and future therapeutic agents.
Exosomes
Exosome Biogenesis: Exosomes are formed in a multi-step process, where regulated invagination of early endosome membranes results in the accumulation of intraluminal vesicles (ILVs) and the formation of multivesicular bodies (MVBs) that eventually fuse with the plasma membrane to release exosomes into the extracellular compartment (Figure 1). Soluble factors (e.g., nucleic acids, proteins, carbohydrates, and other factors), are captured from the cytosol during endosomal membrane invagination, but these components can be preferentially enriched by interaction with endosomal membrane factors, including the endosomal sorting complexes required for transport (ESCRTs) [15], which recognize ubiquitinylated proteins. Despite much progress, there are still significant gaps in our understanding of the mechanisms responsible for sorting proteins into internal vesicles of multivesicular compartments and, hence, to exosomes. Exosome and microvesicle production are, thus, fundamentally different in that microvesicles bud directly from the plasma membrane (Figure 1) [16].
(A) Exosome biogenesis begins with the invagination of the plasma membrane to generate early endosomes. These endosomes can then invaginate to form intraluminal vesicles (ILVs). This process creates multi-vesicular bodies (MVBs) that can then fuse with the plasma membrane to release mature ILVs, now called exosomes, into the extracellular space. (B) Exosomes contain proteins, nucleic acids (including mRNAs, miRNAs, and DNA fragments), and lipids, and these cargos can reflect selective incorporation during exosome formation in a process controlled by lipid raft proteins, ESCRT accessory proteins (e.g., ALIX and TSG101) and tetraspanin proteins. The cytosolic release of these contents upon exosome uptake can alter the phenotype of the recipient cells.
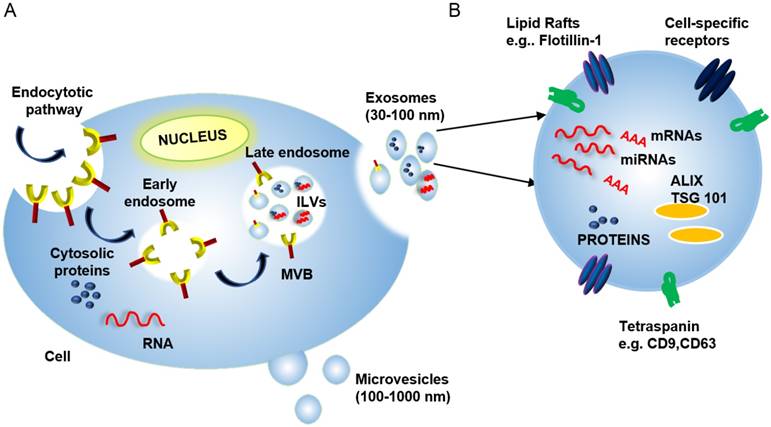
Exosome composition: Mature exosomes range from 30-100 nm in diameter, contain host- and pathogen-derived proteins and nucleic acids, and play important roles in health and disease [13, 17, 18]). Exosomes primarily contain endosome-related proteins: annexins involved in intracellular membrane fusion and transport, lipid raft-associated proteins, and ESCRT accessory proteins (Table 1) (reviewed in [3, 19, 20]). Exosome membranes are enriched for several broadly expressed tetraspanin proteins, including CD9, CD63, and CD81, which interact with multiple proteins (e.g., integrins and MHC molecules) to regulate the organization of large molecular complexes and membrane subdomains. Cell-specific exosome enrichment for several transmembrane proteins—including α- and β-integrins, immunoglobulin family members, and cell-surface peptidases—may also regulate preferential exosome interactions with their target cells (reviewed in [21]). Exosomes also contain RNA cargos, and while exosome RNA content reflects the cell of origin and its physiological state, evidence indicates that exosome RNA content is also influenced by selective packaging mechanisms that can, in some cases, enrich specific RNA species in mature exosomes (Figure 1) [22].
A list of the different proteins presents on the EV surface and its cargos.
| Class | Proteins | Function |
|---|---|---|
| Tetraspanins | CD9, CD63, CD81 | Cell signaling and cell adhesion |
| Histocompatibility complexes | MHCI, MHCII | Antigen presentation |
| Annexins | Annexin I, II, IV, V, VI | Intracellular membrane fusions and transport |
| Heat shock proteins | HSP 70, HSP90 | Mediates folding |
| Ubiquitylated cargo-binding proteins | TSG101 | ESCRT pathway and tumor suppressor |
| Lipid raft protein | FLOT1 | Scaffolding protein for vesicle formation |
| PDCD6-interacting protein | ALIX | ESCRT pathway |
| Transmembrane proteins | Integrin-α, integrin-β | Cell adhesion |
Exosome trafficking and uptake: Secreted exosomes can specifically interact with cells close to their release site, or at distant sites after transport through the circulation, in a process regulated by exosome membrane factors (e.g., integrins, annexins, galectins and intercellular adhesion molecule 1) [16]. Exosome uptake can alter cell expression via RNA transfer (mRNAs, miRNAs, and lncRNAs), cytosolic proteins and receptors, and cell-specific adhesion molecules to influence cell function and later exosome uptake [23]. Exosome-derived factors can directly affect gene transcription and translation, influence signaling cascades to alter transcript and protein modifications, and regulate protein localization and key enzymatic reactions, with interactions among factors supplied by the exosome and recipient cell determining which molecular mechanisms predominate [16].
EV regulation of immune responses. EVs derived from immune cells carry proteins that can regulate important aspects of host immunity, including T-cell activation (e.g., MHCI and MHCII, lymphocyte function-associated antigen-1, and intercellular adhesion molecule-1, depending on the parent cell) [24]. MHC I and MHC II and immunomodulatory proteins are enriched on EVs of antigen-presenting cells (APCs; e.g., dendritic cells (DCs) and macrophages) [4] and these EVs appear capable of activating T cells by transferring antigens or MHC-antigen complexes to conventional APCs, or by directly presenting MHC-antigen complexes to T cells as APC surrogates [5, 25-28].
For the first mechanism, evidence suggests that immature DCs that do not support robust immune responses secrete EVs that can transfer MHC-antigen complexes, or antigens, to mature DCs to activate CD4+ and CD8+ T-cell responses [28, 29]. In the so-called “cross-dressing” model, intact MHC-antigen complexes are transferred from inactive to active DC populations [27-30], whereas in the “cross-presentation” mechanism, mature DCs present peptides derived from captured EVs on their own MHC molecules [25, 26].
There is also evidence that APC-derived EVs can directly activate CD4+ and CD8+ T cells [31-33], and stimulate both previously activated and memory T cells [13, 34]. APC-derived EVs are also able to directly activate naïve CD8+ T cells in vitro [5], although they appear to be 10- to 20-fold less efficient than APCs, suggesting that EVs may not have a direct effect on naïve T cell activation in vivo. Similar studies indicate that EVs of B cells can also directly present antigens to induce T cell responses [13], although with less efficiency than their parental B cells. Receptor aggregation between interacting T cells and DCs also creates an extended “immunological synapse” where DC-derived EVs can directly interact with adjacent T cells in a LFA-1 (lymphocyte function-associated antigen 1) dependent manner to promote their activation [6].
However, while evidence indicates that EVs can directly and indirectly regulate in vitro CD4+ and CD8+ T cell responses, it is unclear to what extent they affect these responses in vivo.
EV roles in infectious diseases: EVs released by infected cells contain pathogen- and host-derived factors, and play key roles in pathogen-host interactions, including pathogen uptake and replication and regulation of the host immune response (reviewed in [7, 35]). For example, studies have shown that multiple viruses—including human immunodeficiency virus 1 (HIV-1), hepatitis viruses B, C and E (HBV, HCV, and HEV), and multiple members of the human herpesvirus (HHV) family—utilize exosome ESCRT machinery for viral transmission [36, 37]. HIV-1, in particular, has developed several exosome-mediated strategies to manipulate the behavior of its target cells [38], including a Nef-regulated mechanism that alters EV protein trafficking in CD4+ T cells. Hepatitis A virus (HAV), HCV and HEV employ the exosome biogenesis machinery to produce enveloped virions that allow the virus to avoid immune surveillance (reviewed in [39]). HHVs, which are responsible for a broad range of important pathologies, employ endosomes to evade anti-viral immune responses through several distinct mechanisms that differ among these viruses [37]. In the following sections, we describe how several viruses subvert the exosome biogenesis machinery for their replication and infectivity. Much less is known about how bacteria employ the EVs of their host to favor their growth and survival, and most studies focus on the behavior of Mycobacterium tuberculosis (Mtb), an important intracellular pathogen. We will therefore summarize current knowledge on how Mtb regulates cellular and systemic processes to favor active and latent Mtb infections, and how these processes overlap with those of other bacterial pathogens (Figure 2).
Overview of the EV incorporation of pathogen-derived factors by the EVs of their host cells, including pathogen receptors and regulatory factors, to promote infection and pathogenesis. (A) EVs of HIV-infected cells express the HIV receptor target proteins CCR5 and CXCR4 and regulatory factors, including the HIV protein Nef and TAR RNA, among others. (B) EVs of HCV-infected cells express E2 and CD81, which promote HCV uptake, as well as viral RNA and host proteins (e.g., CD63) that promote HCV infections. (C) EVs of HBV-infected cells contain EBV RNA and proteins (e.g., LMP1) and host proteins (e.g., EGFR and FGF2) that promote EBV infectivity and pathogenesis. (D) EVs of Mtb-infected cells contain Mtb-derived glycolipids (LAM) and lipoproteins (LpqH) that regulate innate and acquired immune responses to promote infection.
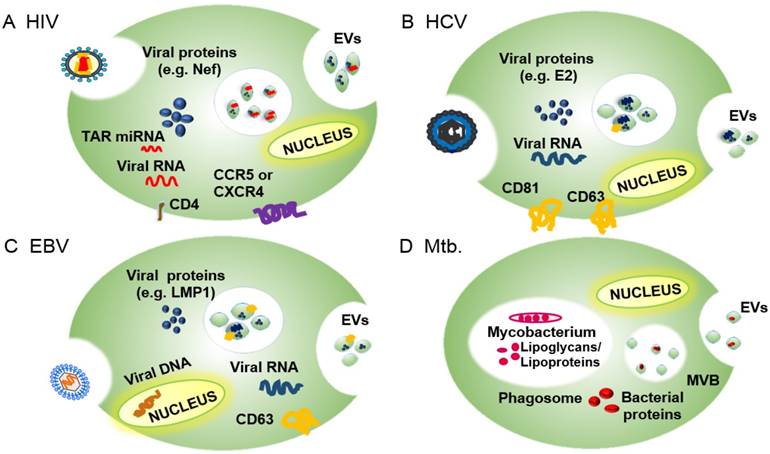
EVs facilitate viral and bacterial pathogenesis
Viral and bacterial pathogens can subvert exosome functions to promote pathogen replication, survival, or pathology. Cells employ EVs to transfer regulatory factors that modulate the response of local and distant cells and systemic responses. In cells with active viral or bacterial infections, the exosome machinery can also package pathogen-derived factors that alter the phenotype of EV recipient cells. Many pathogen factors that are packaged into EVs interact with ESCRT proteins or related factors, suggesting that pathogens have evolved to exploit this intercellular transport and signaling pathway, using it to promote infection and repress anti-pathogen host responses. We discuss several examples of these interactions in the following sections.
Viruses
Functional overlaps between exosome biogenesis and viral budding: Mechanisms involved in exosome and enveloped virus budding share common features. Many retroviruses are reported to interact with ESCRT complex and ESCRT-related proteins involved in exosome biogenesis through conserved protein motifs, referred to as late domains since their deletion or mutation leads to the arrest of virus assembly at late stages of virion synthesis [36]. For example, HIV-1 virion interactions with TSG101, ALIX, and other host proteins are similar to those employed to package host proteins during exosome formation [40]. Similarities between the exosome and HIV-1 packaging mechanisms led to the statement of “the Trojan exosome hypothesis”, which proposes that HIV-1 evolved to utilize exosome biogenesis proteins to package its capsid, while also exploiting exosome uptake mechanisms to allow cell infection in the absence of viral envelope proteins that normally direct HIV-1 uptake [40]. Mounting evidence suggests that several human viruses hijack proteins involved in exosome biogenesis to package their capsids. Human herpesvirus (HHV) family members exploit proteins that regulate exosome biogenesis for their virion production. HHV-1 (herpes simplex virus 1; HSV-1) structural proteins contain potential TSG101 (ESCRT-I complex) and ALIX (ESCRT-I associated) binding motifs. Dominant-negative and siRNA expression studies also indicate that HHV-1 does not employ TSG101 or ALIX, but requires functional expression of CHMP (ESCRT-III complex) and VPS4 (ESCRT-III associated) proteins for the formation of its virion envelope [36, 37]. HHV-5 (human cytomegalovirus; HCMV) also appears to utilize a similar packaging mechanism, since inhibition of either CHMP1A or VPS4, but not ALIX, interferes with its virion packaging. Neither HHV-4 (Epstein-Barr virus; EBV) nor HHV-8 (Kaposi Sarcoma-associated herpesviruses) appear to require exosomal protein interactions for their secretion [37].
Despite these functional interactions, it is not clear how all of these viruses employ exosome proteins in their packaging and secretion. While components of multiple viruses have been shown to associate with MVBs, there is little evidence that these viruses localize within MVBs and are secreted by MVB fusion with the plasma membrane. Only one HHV study appears to provide data consistent with an MVB release mechanism, reporting that HHV-6 virions localize within MVBs in infected cells [37], although these virions (~200 nm) would be much larger than exosomes (30-100 nm).
All members of the hepatitis virus family are reported to employ exosome-related proteins to form enveloped virions. HBV envelope proteins colocalize with MVB proteins ALIX and VPS4B, and dominant-negative versions of either of these proteins block the release of enveloped HBV virions [41]. HCV interacts with Hrs (ESCRT-0 complex) to promote apparent MVB uptake of viral capsids [42] and EVs isolated from HCV-infected hepatoma cell lines and sera of patients with chronic HCV infections contain HCV core and envelope proteins and full-length HCV RNA [43]. HAV and HEV are shed as naked viral particles in feces but circulate as membrane-enclosed virions, which are less infectious but are masked by the host's immune response [37]. Recent evidence suggests that production of these circulating enveloped virions requires interaction with the exosomal sorting components CHMP2a (ESCRT-III complex), ALIX, and VSP4 (ESCRT-I and -III associated) for HAV [44, 45] or Hrs (ESCRT-0 complex) for HEV [46]. Similar to HHV, however, there is scarce evidence for MVB-mediated release of hepatitis family viruses, with only one study indicating that ~50 nm enveloped virions are detectable in MVBs of HEV-infected cells [46].
EVs can alter virus antigenicity and infectivity: Results suggest that some persistent viruses (e.g., HCV and HAV) employ EVs as a strategy to escape negative selective pressure from neutralizing antibodies and other immune responses that act to promote viral clearance [47-49]. MVB-mediated encapsulation may also allow a virus to spread beyond its normal range of cell hosts through the normal EV uptake process, as demonstrated by the ability of EVs containing HAV capsids to infect target cells using EV surface proteins instead of the EV-masked viral receptor proteins (reviewed in [3]).
EVs can spread viral docking receptors to promote viral infectivity: HIV normally binds to CD4 and the chemokine receptors CCR5 or CXCR4 on target cells to mediate infection, and cells lacking these receptors, or with receptor mutations, are resistant to HIV infection. EVs secreted by HIV-infected cells contain CCR5 or CXCR4, however, and their uptake by cells lacking these receptors facilitates HIV infection of these otherwise HIV-resistant cells [50, 51]. The widespread EV markers CD81 and CD63 colocalize with subgenomic HCV RNA and appear to promote its packaging into EVs. The HCV envelope glycoprotein E2 also colocalizes with CD81 and cells that internalize EVs containing this complex are more susceptible to HCV infection [49, 52]. Interaction with this complex may also promote HCV uptake by EVs [53, 54]. Studies suggest that EV proteins may facilitate viral-receptor-independent transmission of HCV and HAV, and presumably other EV-enveloped viruses, to uninfected cells [43, 49].
Regulatory actions of virus-associated EVs on host cells: EVs derived from virus-infected cells can also transfer viral proteins to influence viral pathogenesis. EVs from HIV-infected cells contain the HIV-1 protein Nef, which regulates endocytosis, cytoskeletal rearrangement, and organelle trafficking to increase the number of EVs released from HIV-infected cells [55, 56], and may thus promote EV-mediated HIV infectivity. Nef-induction of EV-associated ADAM17 also appears to promote HIV-infection of resting CD4+ T cells [57], while ADAM17 and TNFα together can activate latent HIV-1 infections in primary CD4+ T lymphocytes and macrophages [17]. Finally, EVs carrying Nef appear to exert complex effects to regulate HIV-1 infection and pathogenesis through actions on uninfected cells [58-60], including the ability to alter the functions of import immune responses.
Human gammaherpesviruses, such as EBV, have complex effects to promote both viral infection and cancer. EVs derived from EBV-infected cells exploit the endosomal-exosomal pathway to enclose both EBV- and host-derived regulatory factors [61, 62]. In EBV-infected cells, an interaction between the EV protein CD63 and the viral protein LMP1 appears to promote LMP1 incorporation into EVs [63, 64] and EVs that contain LMP1 can deliver important signaling proteins to uninfected cells [62]. LMP1 also induces the expression of both epidermal growth factor receptor (EGFR) and fibroblast growth factor 2 (FGF2), which are also packaged in LMP-1-marked EVs [65, 66], suggesting that EV-mediated transfer of these receptors may stimulate the growth of recipient cells with a potential to promote EBV-mediated tumor development.
Effects of virus-associated EVs to inhibit anti-viral responses: Viruses employ several EV-mediated strategies to attenuate host immune responses. EVs of HIV-infected macrophages deliver Nef to recipient cells to alter their immune function. Nef is associated with intracellular sorting and trafficking pathways that promote the lysosomal degradation of CD4 and MHCI to reduce their surface expression [59, 67], rendering cells that express Nef less susceptible to cytotoxic immune responses. Evidence also indicates that Nef+ EVs facilitate HIV pathogenesis by conditioning their target cells to undergo apoptosis, promoting CD4+ T cell depletion and HIV-mediated immune suppression to reduce immune clearance of HIV-infected cells [60, 68, 69].
The major EBV oncoprotein LMP1, which is carried by EVs of EBV-infected cells, plays an important role in EBV infection (reviewed in [3]). LMP1 expression has an important function to activate B cells; however, recent work suggests that EVs carrying LMP1 may also promote B cell activation and proliferation [70] and can inhibit proliferation of T cells and the ability of natural killer (NK) cells to exert cytotoxic effects. [61, 65]. EBV also encodes a number of miRNAs that can modify the transcriptome of infected cells, and non-infected cells via EV transfer [71, 72]. EBV-infected cells release EVs containing EBV miRNAs that suppress EBV target genes, including CXCL11, an immunoregulatory gene involved in antiviral activity [73]. EVs released by EBV-infected cells also contain the host-derived protein galectin-9, which is known to induce apoptosis of EBV-specific CD4+ T cells through an interaction with immunoglobulin mucin-3, and to negatively regulate both macrophage and T cell activation [74].
Viruses thus appear to employ multiple EV-based mechanisms to suppress the clearance of their host cells by the immune system to promote continued viral infections; however, the in vivo relevance of these mechanisms is not clear.
Viral transfer through immune cell EVs: In addition to actions to inhibit the antiviral activity of immune cells, virus-derived EVs can also use these cells to promote viral transfer to new host cells. HIV-1 virions captured by immature DCs and exocytosed in association with the DC cell's EVs can trans-infect CD4+ T cells [75]. The Trojan horse hypothesis of HIV-1 trans-infection [40, 76] takes this further, suggesting that HIV-1 virions are retained in the MVB compartment of mature DCs and trans-infect CD4+ T cells in lymph nodes by following the same trafficking pathway that DC EVs use to disseminate antigens [40, 76, 77].
Bacteria
Regulatory actions of bacterial-associated EVs on host cells: Bacterial pathogens can be classified based on the nature of their interactions with their host, including whether they prefer or require an intracellular or extracellular niche to initiate and maintain active infections. Both extracellular and intracellular bacteria can display complicated lifecycles, but intracellular bacteria have several unique options to subvert cellular processes, including the EV pathway, to promote their growth and survival. Mycobacterium tuberculosis is perhaps the best studied of these pathogens with respect to its EV effects due to its significant worldwide impact on public health, although several other intracellular pathogens are responsible for significant human diseases. M. tuberculosis (Mtb) evades the innate immune response by stably infecting phagocytic cells, such as macrophages, which are primarily responsible for clearance of microbial pathogens. Mtb lipoproteins and lipoglycans inhibit phagosome maturation, generating a stable intracellular niche for the engulfed Mtb bacilli and blocking MHCII-antigen complex cycling to the cell surface to inhibit the host response to Mtb-derived antigens [78, 79]. Mtb-derived factors can also promote EV release and it is hypothesized that some mycobacterial proteins contain signals that direct them to MVBs to promote their incorporation into EV [80].
Mtb-related EVs play important roles in regulating the phenotypes of both infected and uninfected cells and likely contribute to the overall pathogenesis of Mtb infections [78, 79]. EVs of Mtb-infected macrophages can stimulate non-infected macrophages to secrete chemokines to induce the migration of naïve T-cells and macrophages [81]. Mice intranasally injected with EVs from Mtb and M. bovis BCG, revealed increased TNFα and IL-12 production, as well as the recruitment of macrophages and neutrophils to the lung [82], suggesting that these EVs could recruit non-infected target cells to promote disease progression. Macrophages infected with M. avium also revealed increased EV secretion, which lead to a pro-inflammatory response in non-infected macrophages while simultaneously downregulating a number of IFN-γ-inducible genes in naïve cells to inhibit the inducible expression of MHC-II and the CD64 immunoglobulin receptor [83]. Macrophages infected with M. avium and M. smegmatis exhibited increased EV secretion and increased EV expression of HSP70 to promote in vitro macrophage activation and TNFα expression [84]. EVs of Mtb-infected cells also contain the 19 kDa lipoprotein LpqH, which can promote inflammation and stimulate in vitro macrophage activation and TNF-α expression via the Toll-like receptor/MyD88 pathway (reviewed in [85]). These results suggest that EVs from mycobacterium-infected cells can both activate and recruit immune cells, and may therefore influence innate and acquired immune responses during mycobacterial infection [82], although the relative impact of such putative effects on the overall immune response is not clear.
Effects of bacterial-associated EVs to inhibit anti-bacterial immune responses: EVs released by macrophages infected with Mtb bacilli contain protein cargos that regulate both innate and adaptive immune responses. EVs from Mtb-infected macrophages contain the glycolipid lipoarabinomannan (LAM) that inhibits T cell receptor signaling and T cell activation responses, which may induce immune suppressive mechanisms that promote the survival of Mtb-infected cells to maintain active Mtb infections [86, 87]. EVs from Mtb-infected cells can partially suppress the ability of macrophages to respond to INF-γ to inhibit macrophage APC function [88]. EVs of macrophages infected with M. avium can also downregulate a number of IFN-γ-inducible genes in naïve cells to inhibit the inducible expression of MHC-II and the CD64 immunoglobulin receptor [83].
EVs as diagnostic markers: EV expression of pathogen-derived factors and changes in the EV abundance of specific host-derived factors can serve as diagnostic biomarkers, indicators of disease progression, and/or capture targets for the enrichment of pathogen-derived EVs for further analysis. Changes in EV composition during disease progression make them excellent biomarker candidates. The first blood-based EV test for cancer diagnosis became commercially available in the US in January 2016, marking a major step in the maturation of EVs as diagnostic factors [89]. The study of EVs for diagnosis of infectious disease is relatively new but shows great promise, particularly for intracellular bacterial pathogens, such as mycobacteria. Diagnosis of these pathogens normally requires culture or molecular analysis of pathogen samples derived from the site of infection and can misdiagnose patients with low pathogen loads. EVs containing pathogen-derived factors are actively secreted from most cells to accumulate in the circulation; some studies indicate that infection increases EV release rates [81, 90] although it is not clear if this increase is common to all infections. Most current approaches that diagnose active tuberculosis cases use sputum samples as the primary diagnostic specimen. However, there are limitations associated with sputum diagnostics and the World Health Organization has issued a call for new approaches that can diagnose active tuberculosis cases using minimally invasive patient samples, such as peripheral blood samples [91].
Several studies have now indicated the potential of EVs from minimally or non-invasive biological samples to detect such infections. Serum EV concentrations in mice infected with M. bovis BCG correlated and exhibited similar kinetics with M. bovis BCG mycobacterial load, suggesting the potential utility of serum EVs as diagnostic biomarkers for disease burden [81]. A subsequent study used liquid-chromatography and tandem mass spectrometry to identify 41 mycobacterial proteins in EVs derived from Mtb-infected J774 cells [80] and in 2014, analysis of serum EVs isolated from patients with active tuberculosis cases detected numerous mycobacterial proteins, indicating that Mtb-derived EVs can function as markers of active disease [92]. Mycobacterial RNA was also detected in EVs derived from Mtb-infected macrophages [93], implying the potential for analyzing Mtb RNA in EVs as a diagnostic marker for active tuberculosis cases.
Recognition and quantitation of pathogen-derived EVs: Despite increasing scientific and clinical interest in the potential of EVs for disease diagnosis, there are few standard procedures for their isolation, detection, characterization and quantification. The ISEV has emphasized the development and harmonization of standard protocols for specimen handling, isolation and analysis to facilitate comparison of results achieved within this fast-growing field [1, 2]. EVs are too small to analyze by conventional optical detection methods, and their low refractive index and heterogeneous size and composition complicate such analyses, but recent advances now allow nanoparticle quantitation, which is useful for general EV analyses.
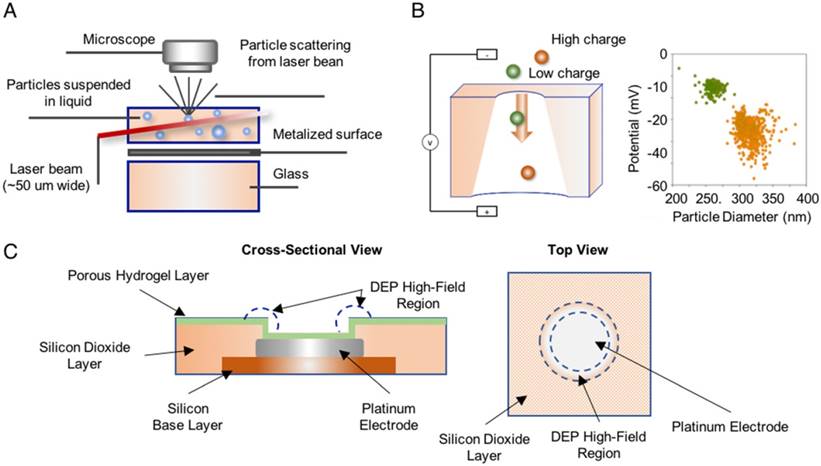
Nanoparticle tracking analysis (NTA) employs a laser beam to illuminate all vesicles in a sample suspension, a light microscope to record the scattered light, and software to measure vesicle sizes as determined by the Brownian motion track of each particle [94, 95]. These instruments are commercially available and can measure the number and absolute size distribution of vesicles in a solution, and quantitate EVs based on their unique size profile. Resistive pulse sensing (RPS) can determine the absolute size distribution of vesicles in sample suspensions via the Coulter principle [96], and at least one company has developed an instrument to exploit this approach. This system consists of two fluid cells divided by a non-conductive nanoporous membrane. A particle moving through one of these nanopores in response to a voltage applied across the cell membrane alters the ion flow, resulting in a brief “resistive pulse”, which is recorded for calculation against a reference standard made with beads of known diameter and concentration [94, 96]. Finally, alternating current electrokinetic (ACE) microarrays can isolate EVs from plasma samples to allow on-chip immunofluorescent detection of EV proteins and to provide mRNA for RT-PCR analysis [97], providing a potential means to isolate and analyze total EV populations without a separate EV isolation step (Figure 3). None of these approaches can quantitate disease-specific EV populations from the general EV population.
Methods used to detect and quantify EVs. (A) Nanoparticle tracking analysis (NTA) analyzes laser scattering in purified vesicle suspensions to track the Brownian motion of the vesicles and determine their absolute size (reproduced with permission from [95], copyright 2011 Elsevier). (B) Resistive pulse sensing (RPS) measures the “resistive pulse” rate produced when purified vesicle suspensions migrate across a membrane in response to applied voltage to determine vesicle concentration relative to a reference standard (adapted with permission from [96], copyright 2012 American Chemical Society). (C) Alternating current electrokinetic (ACE) microarrays use the electrical properties of EVs to isolate these particles from plasma sample and then detect the immunofluorescent signal from target EV proteins. The schematic shows a cross-section and a top-down view of a single electrode well on the array (reproduced with permission from [97], copyright 2017 American Chemical Society).
Transmission electron microscopy can visualize EVs and analyze size and morphology, but this method is labor intensive and requires the use of procedures that are expensive and impractical for clinical use. More importantly, while it can potentially visualize specific EV sub-types after immune-gold staining, it cannot cope with the challenge of identifying and quantitating disease-associated EVs amidst the diverse population of serum EVs, particularly during early disease progression when these EVs should be extremely rare in the highly abundant circulating EV population.
Other optical approaches have attempted to address this issue. Nanoparticles are too small for direct detection by conventional flow cytometry, but one company has developed a high-resolution flow cytometer that can directly detect EVs. This specialized machine requires high-power lasers and high-performance photomultiplier tubes, detection of light scattering at customized angles, and the application of fluorescence-based thresholding to distinguish particles of interest from noise [98, 99]. Standard flow cytometers can, however, analyze multiplex bead-based platforms to detect and analyze aggregate signal derived from multiple EVs after they are bound to micrometer polystyrene capture beads [100]. Stimulated emission depletion (STED) microscopy can also be used to measure multiple markers on single EVs, but this approach does not appear suitable for the analysis of rare EV populations without a prior isolation step.
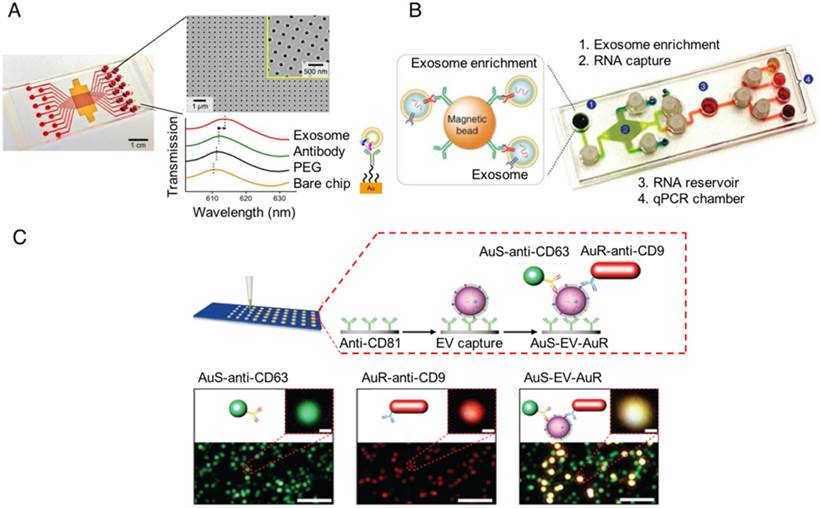
Several groups have recently reported robust on-chip isolation and detection methods to study and profile EVs. In 2014, a group reported the development of a nanoplasmonic exosome (nPLEX) sensor consisting of an affinity ligand-modified gold film that contained an array of periodic nanoholes, in which EV binding produced a spectral shift proportional to the number of targeted EVs bound on the array [101]. In 2015, a second group reported the development of an immunomagnetic exosome RNA (iMER) platform for on-chip EV enrichment, RNA isolation, reverse transcription and real-time analysis of distinct RNA targets, which they used for treatment-induced mRNA in glioblastoma multiforme patients [102]. In 2017, we published a nanoplasmonic enhanced scattering (nPES) method where EVs are bound to a chip by a pan-specific EV antibody, and hybridized with antibody-labeled nanoparticles specific to a second common EV protein and a disease-specific EV marker so that target EVs produce a shifted nPES signal in direct proportion to their number [103]. All these technologies should allow one to modify the EV targets analyzed by changing the affinity of the detection antibody or ligand, and thus should be readily adaptable for any disease for which there is a disease-specific EV biomarker available (Figure 4). None of these approaches are yet available for clinical applications, but they demonstrate the potential of new chip technologies to rapidly profile disease-specific EVs from human samples after minimal sample preparation.
On-chip designs for isolation and detection of disease-specific exosomes. (A) An image of the nanoplasmonic exosome (nPLEX) sensor chip integrated into a multichannel microfluidic cell for independent and parallel analysis (adapted with permission from [101], copyright 2014 Nature America). This device uses specific affinity ligands to capture EVs on an array containing nanoholes, which are blocked by EV binding to produce a spectral shift that indicates the number of target EVs bound to the array. (B) Photograph of the microfluidic immunomagnetic exosomal RNA (iMER) platform iMER prototype with a schematic of its input material (adapted with permission from [102], copyright 2015 Macmillan Publishers Limited). The iMER chip binds antibody-labeled magnetic beads reacted with serum EVs, isolates their RNA, and performs RT-PCR to analyze target mRNAs. (C) Schematic and data from a nanoplasmonic-enhanced scattering analysis (nPES) assay (adapted with permission from [103], copyright 2017 Macmillan Publishers Limited). Serum EVs bound to the nPES chip by an EV-specific antibody produce red or green light when they bind only the EV- or the disease-specific nPES probe (lower left and middle images; single probe controls), but produce an intense yellow signal when they bind both nanoprobes (lower right image; experimental well).
Potential roles for EVs as disease therapeutics: Many of the EV features that allow EVs to regulate the pathogenesis of infectious pathogens may also allow them to act as effective agents for the development of novel therapeutic approaches. For example, the ability of EVs to selectively deliver molecules to specific recipient cell types has significant potential for the delivery of small molecules and other therapeutic agents. There are a number of advantages associated with using EVs as the basis for a pathogen-specific therapeutic system (reviewed in [35, 104-107]). EVs can incorporate various pathogen-derived factors, including receptors involved in cell targeting, suggesting it should be possible to modify EVs to target specific cells of interest via incorporation of receptors that target a cell type or cell population of interest. EVs are also efficiently internalized by APCs, implying that they may be useful in the direct delivery of antigens or MHC-antigen complexes and costimulatory molecules to directly promote robust immune responses against a pathogen. EVs package various materials and maintain the integrity of these materials as they freely circulate, suggesting that EVs can be employed to package and shuttle various therapeutic agents (e.g., siRNAs, small molecule therapeutics, and other agents) with similar efficiency (reviewed in [108]).
APC-derived EVs can exhibit anti-pathogen activities, as previously discussed, through their ability to prime immune responses. Mice injected with EVs from macrophages treated with Mtb culture filtrate protein produced antigen-specific CD4+ and CD8+ T cell activation responses similar to those of mice immunized with M. bovis BCG, the only accepted means of vaccination for Mtb [109]. Notably, however, the mice injected with Mtb-derived EVs did not exhibit an antigen-specific increase in TH2 cells that can limit the effectiveness of a vaccine response, unlike the M. bovis BCG-immunized mice. This finding matched results from previous studies reporting that antigenic EVs primarily induced cytotoxic TH1 immune responses that supported the clearance of intracellular pathogens [110]. Mice vaccinated with Mtb-derived EVs and M. bovis BCG revealed similar protection when subsequently challenged with a Mtb inoculation. Taken together these data indicate that vaccination with Mtb-derived EVs was as efficient as vaccination with M. bovis BCG bacilli, if not more so, indicating the potential for pathogen-free vaccine approaches using non-infectious EVs containing pathogen-derived antigens [109]. However, it is not clear if the MHC of such EVs must match the MHC of the vaccine recipient to promote an optimal pathogen clearance response and avoid inducing autoimmune responses that might occur when a small number of cells incorporate EV-derived MHC complexes that do not match those of the vaccine recipient. This would present a significant challenge to large-scale production and administration of an EV-based vaccine.
Liposomes are currently a favored drug delivery system to carry therapeutics to target tissues and cells, but have several drawbacks. These can include poor in vivo stability and retention; problems with drug loading, leakage and release; and difficulty directing therapeutic liposomes to specific cell and tissue targets (reviewed in [105]). By contrast, EVs can selectively package diverse factors during their biogenesis, and can selectively target multiple different cells and tissue types by virtue of specific membrane factor interactions (reviewed in [111, 112]). Selective tagging of therapeutic agents to utilize packing factors involved in EV biogenesis or simple mass action may allow cultured cells to efficiently package therapeutic agents into EVs for therapeutic applications. Producing therapeutic EVs in cells with known cell specificities or modifying EVs to carry cell-specific targeting factors may also yield better specificity and uptake rates than current liposomes and nanocarriers [113, 114], although EVs can still demonstrate some nonspecific accumulation in highly vascularized tissues, including the lung and liver [114], likely due to EV size, as previously observed for other particles [115].
EVs have already been used for in vitro delivery of exogenous nucleic acids to target cells. For example, one study found that EVs released by cultured THP-1 monocytes efficiently transferred miR150 to recipient cells, where it regulated gene expression and cell function [116]. Researchers have also exploited the RNA transport capacity of EVs to deliver short interfering RNAs (siRNA) to post-translationally silence recipient cell target genes during in vitro cancer studies, and one in vivo study pre-transfected EV donor cells with suicide genes and injected the resulting EVs into an orthotopic mouse cancer model to target schwannoma tumors [117]. Most studies using EVs as delivery vehicles have concentrated on cancer models, so the potential for EV therapy in infectious diseases is still unclear; however, similar approaches should be feasible for the treatment of chronically infected cells once researchers have identified or designed EV factors that exhibit specific interactions with infected host cells.
Conclusion
Mounting evidence indicates pathogen-derived EV factors play important roles in several human diseases, and a better understanding of these mechanisms may provide new insights for future therapeutic development. Several reports indicate that immunomodulatory molecules present in or on EVs can affect pathogen responses through actions to activate or suppress immune responses, and it is possible that increased knowledge of these mechanisms will improve pathogen treatment approaches, including the potential use of EVs to develop more effective vaccines and immunotherapies. Pathogen-specific EV factors are also of great interest as novel disease biomarkers, due to their close association with disease and their potential for greater diagnostic sensitivity and specificity due to their stability in blood and urine. Specific EV biomarkers and means to analyze specific EV subsets have been lacking to date, but new research, including the recent development of several approaches to analyze specific EV populations, appear poised to allow the clinical translation of EV diagnostic assays. The stability, cell-targeting and packaging properties of EVs also recommend them as vectors to deliver new therapeutics, although there are still several questions that must be addressed to allow their translation to therapeutic applications. These include how to control the purity of EV preparations, how to evaluate and control the co-expression of different molecules on these EVs, and what are the best administration routes to achieve targeted delivery and desired effects for different applications. Further research is needed to address these and other issues and to evaluate whether specific EVs can be used for diagnostic and therapeutic approaches, but the inherent properties of these particles appear likely to lend themselves to these approaches.
Abbreviations
APCs: antigen-presenting cells; DCs: dendritic cells; EBV: Epstein-Barr virus; EGFR: epidermal growth factor receptor; ESCRTs: endosomal sorting complexes required for transport; EVs: extracellular vesicles; FGF2: fibroblast growth factor 2; HAV, HBV, HCV, and HEV: hepatitis viruses A, B, C and E; HCMV: human cytomegalovirus; HHV: human herpesvirus; HIV-1: human immunodeficiency virus-1; HSV-1: herpes simplex virus 1; ILVs: intraluminal vesicles; iMER: immunomagnetic exosome RNA; ISEV: International Society for Extracellular Vesicles; LAM: lipoarabinomannan; LFA-1: lymphocyte function-associated antigen 1; lncRNAs: long non-coding RNAs; MHCI and MHCII: class I and II major histocompatibility complexes; miRNAs: microRNAs; mRNAs: messenger RNAs; Mtb: Mycobacterium tuberculosis; MVBs: multivesicular bodies; NK: natural killer; nPLEX: nPES: nanoplasmonic enhanced scattering; nPLEX: nanoplasmonic exosome; NTA: nanoparticle tracking analysis; RPS: resistive pulse sensing; ACE: alternating current electrokinetic; siRNA: short interfering RNAs; STED: stimulated emission depletion.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lener T, Gimona M, Aigner L, Borger V, Buzas E, Camussi G. et al. Applying extracellular vesicles based therapeutics in clinical trials - an ISEV position paper. Journal of Extracellular Vesicles. 2015:4
2. Witwer KW, Buzas EI, Bemis LT, Bora A, Lasser C, Lotvall J. et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. 2013:2
3. Raab-Traub N, Dittmer DP. Viral effects on the content and function of extracellular vesicles. Nat Rev Microbiol. 2017
4. Raposo G, Nijman HW, Stoorvogel W, Leijendekker R, Harding CV, Melief CJM. et al. B lymphocytes secrete antigen-presenting vesicles. J Exp Med. 1996;183:1161-72
5. Hwang IY, Shen XF, Sprent J. Direct stimulation of naive T cells by membrane vesicles from antigen-presenting cells: Distinct roles for CD54 and B7 molecules. P Natl Acad Sci USA. 2003;100:6670-5
6. Nolte-'t Hoen ENM, Buschow SI, Anderton SM, Stoorvogel W, Wauben MHM. Activated T cells recruit exosomes secreted by dendritic cells via LFA-1. Blood. 2009;113:1977-81
7. Mittelbrunn M, Gutierrez-Vazquez C, Villarroya-Beltri C, Gonzalez S, Sanchez-Cabo F, Gonzalez MA. et al. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat Commun. 2011:2
8. Clayton A, Harris CL, Court J, Mason MD, Morgan BP. Antigen-presenting cell exosomes are protected from complement-mediated lysis by expression of CD55 and CD59. Eur J Immunol. 2003;33:522-31
9. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. 2011;29:341-5
10. Sun D, Zhuang X, Xiang X, Liu Y, Zhang S, Liu C. et al. A novel nanoparticle drug delivery system: the anti-inflammatory activity of curcumin is enhanced when encapsulated in exosomes. Mol Ther. 2010;18:1606-14
11. Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ. et al. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35:2383-90
12. Morse MA, Garst J, Osada T, Khan S, Hobeika A, Clay TM. et al. A phase I study of dexosome immunotherapy in patients with advanced non-small cell lung cancer. J Transl Med. 2005;3:9
13. Admyre C, Bohle B, Johansson SM, Focke-Tejkl M, Valenta R, Scheynius A. et al. B cell-derived exosomes can present allergen peptides and activate allergen-specific T cells to proliferate and produce T(H)2-like cytokines. J Allergy Clin Immun. 2007;120:1418-24
14. Fuhrmann G, Neuer AL, Herrmann IK. Extracellular vesicles - A promising avenue for the detection and treatment of infectious diseases? European Journal of Pharmaceutics and Biopharmaceutics. 2017;118:56-61
15. Tamai K, Tanaka N, Nakano T, Kakazu E, Kondo Y, Inoue J. et al. Exosome secretion of dendritic cells is regulated by Hrs, an ESCRT-0 protein. Biochem Bioph Res Co. 2010;399:384-90
16. Pant S, Hilton H, Burczynski ME. The multifaceted exosome: biogenesis, role in normal and aberrant cellular function, and frontiers for pharmacological and biomarker opportunities. Biochem Pharmacol. 2012;83:1484-94
17. Arenaccio C, Anticoli S, Manfredi F, Chiozzini C, Olivetta E, Federico M. Latent HIV-1 is activated by exosomes from cells infected with either replication-competent or defective HIV-1. Retrovirology. 2015:12
18. Athman JJ, Wang Y, McDonald DJ, Boom WH, Harding CV, Wearsch PA. Bacterial Membrane Vesicles Mediate the Release of Mycobacterium tuberculosis Lipoglycans and Lipoproteins from Infected Macrophages. J Immunol. 2015;195:1044-53
19. van Niel G, Porto-Carreiro I, Simoes S, Raposo G. Exosomes: a common pathway for a specialized function. J Biochem. 2006;140:13-21
20. Thery C, Boussac M, Veron P, Ricciardi-Castagnoli P, Raposo G, Garin J. et al. Proteomic Analysis of Dendritic Cell-Derived Exosomes: A Secreted Subcellular Compartment Distinct from Apoptotic Vesicles. The Journal of Immunology. 2001;166:7309-18
21. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2:569-79
22. Montecalvo A, Larregina AT, Shufesky WJ, Stolz DB, Sullivan MLG, Karlsson JM. et al. Mechanism of transfer of functional microRNAs between mouse dendritic cells via exosomes. Blood. 2012;119:756-66
23. Matsuo H, Chevallier J, Mayran N, Le Blanc I, Ferguson C, Faure J. et al. Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science. 2004;303:531-4
24. Lee Y, El Andaloussi S, Wood MJ. Exosomes and microvesicles: extracellular vesicles for genetic information transfer and gene therapy. Hum Mol Genet. 2012;21:R125-34
25. Montecalvo A, Shufesky WJ, Stolz DB, Sullivan MG, Wang ZL, Divito SJ. et al. Exosomes as a short-range mechanism to spread alloantigen between dendiritic cells during T cell allorecognition. J Immunol. 2008;180:3081-90
26. Qazi KR, Gehrmann U, Jordo ED, Karlsson MCI, Gabrielsson S. Antigen-loaded exosomes alone induce Th1-type memory through a B cell-dependent mechanism. Blood. 2009;113:2673-83
27. Segura E, Amigorena S, Thery C. Mature dendritic cells secrete exosomes with strong ability to induce antigen-specific effector immune responses. Blood Cell Mol Dis. 2005;35:89-93
28. Thery C, Duban L, Segura E, Veron P, Lantz O, Amigorena S. Indirect activation of naive CD4(+) T cells by dendritic cell-derived exosomes. Nat Immunol. 2002;3:1156-62
29. Andre F, Chaput N, Schartz NEC, Flament C, Aubert N, Bernard J. et al. Exosomes as potent cell-free peptide-based vaccine. I. Dendritic cell-derived exosomes transfer functional MHC class I/peptide complexes to dendritic cells. J Immunol. 2004;172:2126-36
30. Chaput N, Schartz NEC, Andre F, Taieb J, Novault S, Bonnaventure P. et al. Exosomes as potent cell-free peptide-based vaccine. II. Exosomes in CpG adjuvants efficiently prime naive Tc1 lymphocytes leading to tumor rejection. J Immunol. 2004;172:2137-46
31. Luketic L, Delanghe J, Sobol PT, Yang PC, Frotten E, Mossman KL. et al. Antigen presentation by exosomes released from peptide-pulsed dendritic cells is not suppressed by the presence of active CTL. J Immunol. 2007;179:5024-32
32. Admyre C, Johansson SM, Paulie S, Gabrielsson S. Direct exosome stimulation of peripheral human T cells detected by ELISPOT. European Journal of Immunology. 2006;36:1772-81
33. Utsugi-Kobukai S, Fujimaki H, Hotta C, Nakazawa M, Minami M. MHC class I-mediated exogenous antigen presentation by exosomes secreted from immature and mature bone marrow derived dendritic cells. Immunol Lett. 2003;89:125-31
34. Muntasell A, Berger AC, Roche PA. T cell-induced secretion of MHC class II-peptide complexes on B cell exosomes. Embo J. 2007;26:4263-72
35. Schorey JS, Harding CV. Extracellular vesicles and infectious diseases: new complexity to an old story. J Clin Invest. 2016;126:1181-9
36. Pawliczek T, Crump CM. Herpes simplex virus type 1 production requires a functional ESCRT-III complex but is independent of TSG101 and ALIX expression. J Virol. 2009;83:11254-64
37. Sadeghipour S, Mathias RA. Herpesviruses hijack host exosomes for viral pathogenesis. Semin Cell Dev Biol. 2017;67:91-100
38. Garcia E, Pion M, Pelchen-Matthews A, Collinson L, Arrighi JF, Blot G. et al. HIV-1 trafficking to the dendritic cell-T-cell infectious synapse uses a pathway of tetraspanin sorting to the immunological synapse. Traffic. 2005;6:488-501
39. Alenquer M, Amorim MJ. Exosome Biogenesis, Regulation, and Function in Viral Infection. Viruses. 2015;7:5066-83
40. Gould SJ, Booth AM, Hildreth JEK. The Trojan exosome hypothesis. P Natl Acad Sci USA. 2003;100:10592-7
41. Watanabe T, Sorensen EM, Naito A, Schott M, Kim S, Ahlquist P. Involvement of host cellular multivesicular body functions in hepatitis B virus budding. Proc Natl Acad Sci U S A. 2007;104:10205-10
42. Tamai K, Shiina M, Tanaka N, Nakano T, Yamamoto A, Kondo Y. et al. Regulation of hepatitis C virus secretion by the Hrs-dependent exosomal pathway. Virology. 2012;422:377-85
43. Bukong TN, Momen-Heravi F, Kodys K, Bala S, Szabo G. Exosomes from Hepatitis C Infected Patients Transmit HCV Infection and Contain Replication Competent Viral RNA in Complex with Ago2-miR122-HSP90. Plos Pathog. 2014:10
44. Feng Z, Hensley L, McKnight KL, Hu F, Madden V, Ping L. et al. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature. 2013;496:367-71
45. McKnight KL, Xie L, Gonzalez-Lopez O, Rivera-Serrano EE, Chen X, Lemon SM. Protein composition of the hepatitis A virus quasi-envelope. Proc Natl Acad Sci U S A. 2017;114:6587-92
46. Nagashima S, Jirintai S, Takahashi M, Kobayashi T. et al. Hepatitis E virus egress depends on the exosomal pathway, with secretory exosomes derived from multivesicular bodies. J Gen Virol. 2014;95:2166-75
47. Dreux M, Garaigorta U, Boyd B, Decembre E, Chung J, Whitten-Bauer C. et al. Short-Range Exosomal Transfer of Viral RNA from Infected Cells to Plasmacytoid Dendritic Cells Triggers Innate Immunity. Cell Host & Microbe. 2012;12:558-70
48. Feng ZD, Hensley L, McKnight KL, Hu FY, Madden V, Ping LF. et al. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature. 2013;496:367 -+
49. Ramakrishnaiah V, Thumann C, Fofana I, Habersetzer F, Pan QW, de Ruiter PE. et al. Exosome-mediated transmission of hepatitis C virus between human hepatoma Huh7.5 cells. P Natl Acad Sci USA. 2013;110:13109-13
50. Rozmyslowicz T, Majka M, Kijowski J, Murphy SL, Conover DO, Poncz M. et al. Platelet- and megakaryocyte-derived microparticles transfer CXCR4 receptor to CXCR4-null cells and make them susceptible to infection by X4-HIV. Aids. 2003;17:33-42
51. Mack M, Kleinschmidt A, Bruhl H, Klier C, Nelson PJ, Cihak J. et al. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: A mechanism for cellular human immunodeficiency virus 1 infection. Nat Med. 2000;6:769 -+
52. Pileri P, Uematsu Y, Campagnoli S, Galli G, Falugi F, Petracca R. et al. Binding of hepatitis C virus to CD81. Science. 1998;282:938-41
53. Chang CC, Hsu HJ, Yen JH, Lo SY, Liou JW. A Sequence in the loop domain of hepatitis C virus E2 protein identified in silico as crucial for the selective binding to human CD81. Plos One. 2017:12
54. Zhang J, Randall G, Higginbottom A, Monk P, Rice CM, McKeating JA. CD81 is required for hepatitis C virus glycoprotein-mediated viral infection. J Virol. 2004;78:1448-55
55. Raymond AD, Campbell-Sims TC, Khan M, Lang M, Huang MB, Bond VC. et al. HIV Type 1 Nef Is Released from Infected Cells in CD45(+) Microvesicles and Is Present in the Plasma of HIV-Infected Individuals. Aids Res Hum Retrov. 2011;27:167-78
56. Ali SA, Huang MB, Campbell PE, Roth WW, Campbell T, Khan M. et al. Genetic Characterization of HIV Type 1 Nef-Induced Vesicle Secretion. Aids Res Hum Retrov. 2010;26:173-92
57. Arenaccio C, Chiozzini C, Columba-Cabezas S, Manfredi F, Affabris E, Baur A. et al. Exosomes from Human Immunodeficiency Virus Type 1 (HIV-1)-Infected Cells License Quiescent CD4(+) T Lymphocytes To Replicate HIV-1 through a Nef- and ADAM17-Dependent Mechanism. J Virol. 2014;88:11529-39
58. Aqil M, Naqvi AR, Bano AS, Jameel S. The HIV-1 Nef protein binds argonaute-2 and functions as a viral suppressor of RNA interference. PLoS One. 2013;8:e74472
59. Gray LR, Gabuzda D, Cowley D, Ellett A, Chiavaroli L, Wesselingh SL. et al. CD4 and MHC class 1 down-modulation activities of nef alleles from brain- and lymphoid tissue-derived primary HIV-1 isolates. J Neurovirol. 2011;17:82-91
60. Lenassi M, Cagney G, Liao M, Vaupotic T, Bartholomeeusen K, Cheng Y. et al. HIV Nef is secreted in exosomes and triggers apoptosis in bystander CD4+ T cells. Traffic. 2010;11:110-22
61. Flanagan J, Middeldorp J, Sculley T. Localization of the Epstein-Barr virus protein LMP 1 to exosomes. Journal of General Virology. 2003;84:1871-9
62. Meckes DG Jr, Gunawardena HP, Dekroon RM, Heaton PR, Edwards RH, Ozgur S. et al. Modulation of B-cell exosome proteins by gamma herpesvirus infection. Proc Natl Acad Sci U S A. 2013;110:E2925-33
63. Verweij FJ, van Eijndhoven MAJ, Hopmans ES, Vendrig T, Wurdinger T, Cahir-McFarland E. et al. LMP1 association with CD63 in endosomes and secretion via exosomes limits constitutive NF-kappa B activation. Embo J. 2011;30:2115-29
64. Hurwitz SN, Nkosi D, Conlon MM, York SB, Liu X, Tremblay DC. et al. CD63 Regulates Epstein-Barr Virus LMP1 Exosomal Packaging, Enhancement of Vesicle Production, and Noncanonical NF-kappa B Signaling. J Virol. 2017:91
65. Meckes DG Jr, Shair KH, Marquitz AR, Kung CP, Edwards RH, Raab-Traub N. Human tumor virus utilizes exosomes for intercellular communication. Proc Natl Acad Sci U S A. 2010;107:20370-5
66. Ceccarelli S, Visco V, Raffa S, Wakisaka N, Pagano JS, Torrisi MR. Epstein-Barr virus latent membrane protein 1 promotes concentration in multivesicular bodies of fibroblast growth factor 2 and its release through exosomes. Int J Cancer. 2007;121:1494-506
67. Schaefer MR, Wonderlich ER, Roeth JF, Leonard JA, Collins KL. HIV-1 Nef targets MHC-I and CD4 for degradation via a final common beta-COP-dependent pathway in T cells. Plos Pathog. 2008:4
68. Lenassi M, Cagney G, Liao MF, Vaupotic T, Bartholomeeusen K, Cheng YF. et al. HIV Nef is Secreted in Exosomes and Triggers Apoptosis in Bystander CD4(+) T Cells. Traffic. 2010;11:110-22
69. Nguyen DG, Booth A, Gould SJ, Hildreth JEK. Evidence that HIV budding in primary macrophages occurs through the exosome release pathway. J Biol Chem. 2003;278:52347-54
70. Gutzeit C, Nagy N, Gentile M, Lyberg K, Gumz J, Vallhov H. et al. Exosomes Derived from Burkitt's Lymphoma Cell Lines Induce Proliferation, Differentiation, and Class-Switch Recombination in B Cells. J Immunol. 2014;192:5852-62
71. Pfeffer S, Zavolan M, Grasser FA, Chien MC, Russo JJ, Ju JY. et al. Identification of virus-encoded microRNAs. Science. 2004;304:734-6
72. Yang HJ, Huang TJ, Yang CF, Peng LX, Liu RY, Yang GD. et al. Comprehensive profiling of Epstein-Barr virus-encoded miRNA species associated with specific latency types in tumor cells. Virol J. 2013:10
73. Pegtel DM, Cosmopoulos K, Thorley-Lawson DA, van Eijndhoven MAJ, Hopmans ES, Lindenberg JL. et al. Functional delivery of viral miRNAs via exosomes. P Natl Acad Sci USA. 2010;107:6328-33
74. Klibi J, Niki T, Riedel A, Pioche-Durieu C, Souquere S, Rubinstein E. et al. Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein-Barr virus-infected nasopharyngeal carcinoma cells. Blood. 2009;113:1957-66
75. Wiley RD, Gummuluru S. Immature dendritic cell-derived exosomes can mediate HIV-1 trans infection. P Natl Acad Sci USA. 2006;103:738-43
76. Izquierdo-Useros N, Naranjo-Gomez M, Erkizia I, Puertas MC, Borras FE, Blanco J. et al. HIV and Mature Dendritic Cells: Trojan Exosomes Riding the Trojan Horse?. Plos Pathog. 2010:6
77. Narayanan A, Iordanskiy S, Das R, Van Duyne R, Santos S, Jaworski E. et al. Exosomes Derived from HIV-1-infected Cells Contain Trans-activation Response Element RNA. J Biol Chem. 2013;288:20014-33
78. Beatty WL, Russell DG. Identification of mycobacterial surface proteins released into subcellular compartments of infected macrophages. Infect Immun. 2000;68:6997-7002
79. Beatty WL, Ullrich HJ, Russell DG. Mycobacterial surface moieties are released from infected macrophages by a constitutive exocytic event. Eur J Cell Biol. 2001;80:31-40
80. Giri PK, Kruh NA, Dobos KM, Schorey JS. Proteomic analysis identifies highly antigenic proteins in exosomes from M. tuberculosis-infected and culture filtrate protein-treated macrophages. Proteomics. 2010;10:3190-202
81. Singh PP, Smith VL, Karakousis PC, Schorey JS. Exosomes Isolated from Mycobacteria-Infected Mice or Cultured Macrophages Can Recruit and Activate Immune Cells In Vitro and In Vivo. J Immunol. 2012;189:777-85
82. Bhatnagar S, Shinagawa K, Castellino FJ, Schorey JRS. Exosomes released from macrophages infected with intracellular pathogens stimulate a proinflammatory response in vitro and in vivo. Blood. 2007;110:3234-44
83. Bhatnagar S, Schorey JS. Exosomes released from infected macrophages contain mycobacterium avium glycopeptidolipids and are proinflammatory. J Biol Chem. 2007;282:25779-89
84. Anand PK, Anand E, Bleck CKE, Anes E, Griffiths G. Exosomal Hsp70 Induces a Pro-Inflammatory Response to Foreign Particles Including Mycobacteria. Plos One. 2010:5
85. Schorey JS, Bhatnagar S. Exosome function: From tumor immunology to pathogen biology. Traffic. 2008;9:871-81
86. Yang CJ, Ruffner MA, Kim SH, Robbins PD. Plasma-derived MHC class II+ exosomes from tumor-bearing mice suppress tumor antigen-specific immune responses. European Journal of Immunology. 2012;42:1778-84
87. Mahon RN, Sande OJ, Rojas RE, Levine AD, Harding CV, Boom WH. Mycobacterium tuberculosis ManLAM inhibits T-cell-receptor signaling by interference with ZAP-70, Lck and LAT phosphorylation. Cell Immunol. 2012;275:98-105
88. Harding CV, Boom WH. Regulation of antigen presentation by Mycobacterium tuberculosis: a role for Toll-like receptors. Nat Rev Microbiol. 2010;8:296-307
89. Sheridan C. Exosome cancer diagnostic reaches market. Nat Biotechnol. 2016;34:359-60
90. Hu G, Gong AY, Roth AL, Huang BQ, Ward HD, Zhu G. et al. Release of Luminal Exosomes Contributes to TLR4-Mediated Epithelial Antimicrobial Defense. Plos Pathog. 2013:9
91. WHO: Geneva, Switzerland. High-priority target product profiles for new tuberculosis diagnostics: Report of a consensus meeting. 28-29 April 2014. http://www.who.int/tb/publications/tpp_report/en/
92. Kruh-Garcia NA, Wolfe LM, Chaisson LH, Worodria WO, Nahid P, Schorey JS. et al. Detection of Mycobacterium tuberculosis peptides in the exosomes of patients with active and latent M. tuberculosis infection using MRM-MS. PLoS One. 2014;9:e103811
93. Singh PP, Li L, Schorey JS. Exosomal RNA from Mycobacterium tuberculosis-Infected Cells Is Functional in Recipient Macrophages. Traffic. 2015;16:555-71
94. Vogel R, Willmott G, Kozak D, Roberts GS, Anderson W, Groenewegen L. et al. Quantitative sizing of nano/microparticles with a tunable elastomeric pore sensor. Anal Chem. 2011;83:3499-506
95. Dragovic RA, Gardiner C, Brooks AS, Tannetta DS, Ferguson DJP, Hole P. et al. Sizing and phenotyping of cellular vesicles using Nanoparticle Tracking Analysis. Nanomed-Nanotechnol. 2011;7:780-8
96. Kozak D, Anderson W, Vogel R, Chen S, Antaw F, Trau M. Simultaneous size and zeta-potential measurements of individual nanoparticles in dispersion using size-tunable pore sensors. ACS Nano. 2012;6:6990-7
97. Ibsen SD, Wright J, Lewis JM, Kim S, Ko SY, Ong J. et al. Rapid Isolation and Detection of Exosomes and Associated Biomarkers from Plasma. Acs Nano. 2017;11:6641-51
98. van der Vlist EJ, Nolte-'t Hoen EN, Stoorvogel W, Arkesteijn GJ, Wauben MH. Fluorescent labeling of nano-sized vesicles released by cells and subsequent quantitative and qualitative analysis by high-resolution flow cytometry. Nat Protoc. 2012;7:1311-26
99. Aras O, Shet A, Bach RR, Hysjulien JL, Slungaard A, Hebbel RP. et al. Induction of microparticle- and cell-associated intravascular tissue factor in human endotoxemia. Blood. 2004;103:4545-53
100. Koliha N, Wiencek Y, Heider U, Jungst C, Kladt N, Krauthauser S. et al. A novel multiplex bead-based platform highlights the diversity of extracellular vesicles. J Extracell Vesicles. 2016;5:29975
101. Im H, Shao HL, Park YI, Peterson VM, Castro CM, Weissleder R. et al. Label-free detection and molecular profiling of exosomes with a nano-plasmonic sensor. Nat Biotechnol. 2014;32:490-U219
102. Shao HL, Chung J, Lee K, Balaj L, Min C, Carter BS. et al. Chip-based analysis of exosomal mRNA mediating drug resistance in glioblastoma. Nat Commun. 2015:6
103. Liang K, Liu F, Fan J, Sun D, Liu C, Lyon CJ. et al. Nanoplasmonic quantification of tumour-derived extracellular vesicles in plasma microsamples for diagnosis and treatment monitoring. Nature Biomedical Engineering. 2017;1:0021
104. Madison MN, Okeoma CM. Exosomes: Implications in HIV-1 Pathogenesis. Viruses. 2015;7:4093-118
105. Ohno S, Drummen GPC, Kuroda M. Focus on Extracellular Vesicles: Development of Extracellular Vesicle-Based Therapeutic Systems. Int J Mol Sci. 2016:17
106. Gyorgy B, Hung ME, Breakefield XO, Leonard JN. Therapeutic Applications of Extracellular Vesicles: Clinical Promise and Open Questions. Annu Rev Pharmacol. 2015;55:439-64
107. Armstrong JPK, Holme MN, Stevens MM. Re-Engineering Extracellular Vesicles as Smart Nanoscale Therapeutics. Acs Nano. 2017;11:69-83
108. Ferguson SW, Nguyen J. Exosomes as therapeutics: The implications of molecular composition and exosomal heterogeneity. J Control Release. 2016;228:179-90
109. Cheng Y, Schorey JS. Exosomes carrying mycobacterial antigens can protect mice against Mycobacterium tuberculosis infection. European Journal of Immunology. 2013;43:3279-90
110. Qazi KR, Gehrmann U, Domange Jordo E, Karlsson MC, Gabrielsson S. Antigen-loaded exosomes alone induce Th1-type memory through a B-cell-dependent mechanism. Blood. 2009;113:2673-83
111. Kalra H, Drummen GPC, Mathivanan S. Focus on Extracellular Vesicles: Introducing the Next Small Big Thing. Int J Mol Sci. 2016:17
112. Fuhrmann G, Herrmann IK, Stevens MM. Cell-derived vesicles for drug therapy and diagnostics: Opportunities and challenges. Nano Today. 2015;10:397-409
113. Lai RC, Yeo RWY, Tan KH, Lim SK. Exosomes for drug delivery - a novel application for the mesenchymal stem cell. Biotechnol Adv. 2013;31:543-51
114. Wiklander OPB, Nordin JZ, O'Loughlin A, Gustafsson Y, Corso G, Mager I. et al. Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. Journal of Extracellular Vesicles. 2015:4
115. De Jong WH, Hagens WI, Krystek P, Burger MC, Sips AJAM, Geertsma RE. Particle size-dependent organ distribution of gold nanoparticles after intravenous administration. Biomaterials. 2008;29:1912-9
116. Zhang YJ, Liu DQ, Chen X, Li J, Li LM, Bian Z. et al. Secreted Monocytic miR-150 Enhances Targeted Endothelial Cell Migration. Mol Cell. 2010;39:133-44
117. Mizrak A, Bolukbasi MF, Ozdener GB, Brenner GJ, Madlener S, Erkan EP. et al. Genetically Engineered Microvesicles Carrying Suicide mRNA/Protein Inhibit Schwannoma Tumor Growth. Mol Ther. 2013;21:101-8
Author contact
![]() Corresponding author: wanmhedu.cn and tyhuedu
Corresponding author: wanmhedu.cn and tyhuedu