Theranostics
13.3
Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(8):2134-2146. doi:10.7150/thno.22641 This issue Cite
Research Paper
Enhancement of Cancer-Specific Protoporphyrin IX Fluorescence by Targeting Oncogenic Ras/MEK Pathway
Ema Yoshioka1*, Vipin Shankar Chelakkot1*, Maria Licursi1, Suzette G Rutihinda1, Jayoti Som1, Leena Derwish1, Justin J King1, Theerawat Pongnopparat2, Karen Mearow1, Mani Larijani1, Ann M Dorward1, Kensuke Hirasawa1 ![]()
1. Division of BioMedical Sciences, Faculty of Medicine, Memorial University of Newfoundland, 300 Prince Philip Dr., St. John's, NL Canada. A1B 3V6 (709) 864-6058
2. Department of Biochemistry, Memorial University of Newfoundland, St. John's, NL Canada
*Authors contributed equally to this work.
Received 2017-9-1; Accepted 2018-2-12; Published 2018-3-8
Citation:
Yoshioka E, Chelakkot VS, Licursi M, Rutihinda SG, Som J, Derwish L, King JJ, Pongnopparat T, Mearow K, Larijani M, Dorward AM, Hirasawa K. Enhancement of Cancer-Specific Protoporphyrin IX Fluorescence by Targeting Oncogenic Ras/MEK Pathway. Theranostics 2018; 8(8):2134-2146. doi:10.7150/thno.22641. https://www.thno.org/v08p2134.htm
Other stylesAbstract

Protoporphyrin IX (PpIX) is an endogenous fluorescent molecule that selectively accumulates in cancer cells treated with the heme precursor 5-aminolevulinic acid (5-ALA). This cancer-specific accumulation of PpIX is used to distinguish tumor from normal tissues in fluorescence-guided surgery (FGS) and to destroy cancer cells by photodynamic therapy (PDT). In this study, we demonstrate that oncogenic Ras/mitogen-activated protein kinase kinase (MEK) pathway can modulate PpIX accumulation in cancer cells.
Methods: To identify Ras downstream elements involved in PpIX accumulation, chemical inhibitors were used. To demonstrate the increase of PpIX accumulation by MEK inhibition, different human normal and cancer cell lines, BALB/c mice bearing mammary 4T1 tumors and athymic nude mice bearing human tumors were used. To identify the mechanisms of PpIX regulation by MEK, biochemical and molecular biological experiments were conducted.
Results: Inhibition of one of the Ras downstream elements, MEK, promoted PpIX accumulation in cancer cells treated with 5-ALA, while inhibitors against other Ras downstream elements did not. Increased PpIX accumulation with MEK inhibition was observed in different types of human cancer cell lines, but not in normal cell lines. We identified two independent cellular mechanisms that underlie this effect in cancer cells. MEK inhibition reduced PpIX efflux from cancer cells by decreasing the expression level of ATP binding cassette subfamily B member 1 (ABCB1) transporter. In addition, the activity of ferrochelatase (FECH), the enzyme responsible for converting PpIX to heme, was reduced by MEK inhibition. Finally, we found that in vivo treatment with MEK inhibitors increased PpIX accumulation (2.2- to 2.4-fold) within mammary 4T1 tumors in BALB/c mice injected with 5-ALA without any change in normal organs. Similar results were also observed in a human tumor xenograft model.
Conclusion: Our study demonstrates that inhibition of oncogenic Ras/MEK significantly enhances PpIX accumulation in vitro and in vivo in a cancer-specific manner. Thus, suppressing the Ras/MEK pathway may be a viable strategy to selectively intensify PpIX fluorescence in cancer cells and improve its clinical applications in FGS.
Keywords: Protoporphyrin IX, fluorescence-guided surgery (FGS), Ras/MEK, ATP binding cassette subfamily B member 1 (ABCB1), ferrochelatase (FECH)
Introduction
Although chemotherapy and radiotherapy have advanced in recent years and are widely used for treating cancer, surgical resection still remains a primary option for most solid tumors [1]. To achieve the ultimate goal of surgical resection and completely remove cancer without damaging healthy tissues, diagnostic imaging is critical. Pre-operative imaging techniques such as magnetic resonance imaging (MRI), computed tomography (CT) and positron emission tomography (PET) provide exquisite structural information of tumors for early detection and surgery planning [2]. However, to accomplish complete removal of tumors with a minimal margin, it is essential to intraoperatively identify tumor borders and small tumor foci. Fluorescence-guided surgery (FGS) is an emerging technique that provides surgeons with intraoperative images to visually identify these boundaries in real-time for optimal tumor resection and healthy tissue preservation [3].
Among the tumor-specific fluorescent probes used for FGS, 5-Aminolevulinic acid (5-ALA) is the front-runner for tumor-normal border detection with a good safety profile and minimal side effects [4-6]. 5-ALA is a small molecule precursor in the heme biosynthesis pathway. It is metabolically converted to protoporphyrin IX (PpIX), an endogenous fluorophore that exhibits red fluorescence (630 nm) under blue light (405 nm) excitation [7]. PpIX can be preferentiallly accumulated in cancer cells compared to normal cells, due to differences in the metabolic rate of the heme biosynthesis pathway [3,7]. Therefore, exogenous 5-ALA administration can be used to increase PpIX accumulation in tumors for use during FGS. In addition, PpIX can also be used in photodynamic therapy (PDT), which takes advantage of the property of PpIX to initiate the generation of reactive oxygen species (ROS) when excited with visible light, leading to cancer cell death [3,7]. The combination of FGS for efficient removal of the tumor mass with immediate PDT for residual disease may be an effective approach to achieve optimal treatment for solid tumors.
5-ALA-FGS has been evaluated in multiple clinical trials with promising results [8,9]. A randomized multicenter phase III trial on malignant gliomas demonstrated that 5-ALA-FGS significantly improved the success rate of complete tumor resection and progression-free survival in malignant glioma patients [10]. Furthermore, 5-ALA-FGS has succeeded as an in situ diagnostic approach for superficial bladder cancer (9,10). In June 2017, the United States Food and Drug Administration (US FDA) approved 5-ALA-FGS to visualize gliomas during surgery [13]. As the pipeline between basic and clinical research is well established, FGS has become an exciting and promising research area where outcomes of basic research directly impact cancer therapy in clinical settings.
Despite its successful use in clinical settings and the FDA approval expanding its applications, 5-ALA-FGS still has room for improvement in certain aspects. For example, the PpIX fluorescence signal is often not strong enough to visualize small satellite tumors around the primary tumor or to clearly delineate the tumor from adjacent normal tissue (tumor ends) [3,14]. Furthermore, the fluorescence signal in tumors is often photobleached with excessive light exposure [15], which is particularly problematic in low grade tumors where the accumulation of PpIX florescence is limited [16]. Thus, to further improve FGS efficacy and widen its applications, a novel approach to intensify PpIX fluorescence is essential.
Ras is a cell signaling molecule that functions as a molecular switch to transduce extracellular signals to the nucleus and subsequently regulates cell proliferation, differentiation and survival [17,18]. These Ras functions are mediated by a broad range of downstream elements, including mitogen-activated protein kinase kinase (MEK), phosphoinositide 3-kinase (PI3K), p38 mitogen-activated protein kinase (p38 MAPK) and c-Jun N-terminal kinase (JNK) [19]. Nearly 30% of all human cancers have activating mutations in Ras, with some variations depending on the cancer type [20]. Furthermore, activating mutations of upstream and downstream elements of Ras are also commonly found in different types of cancer [21,22]. It is believed that the majority of cancer cells have constitutive activation of Ras and its downstream elements. Although oncogenic transformation has been shown to affect 5-ALA-induced PpIX accumulation [23-25], the specific role of oncogenic cell signaling pathways such as Ras in the regulation of cancer-specific accumulation of PpIX is relatively unknown.
In the present study, we sought to determine whether and how active Ras signaling influences PpIX fluorescence accumulation in cancer cells. Our results demonstrate that MEK, one of the Ras downstream elements, regulates the heme biosynthesis pathway and its inhibition increases PpIX fluorescence in a cancer cell-specific manner.
Methods
Cells and reagents
Human cancer and normal cells used in this study were obtained from the American Type Culture Collection (Manassas, VA). Mouse 4T1 mammary tumor cells were obtained from Dr. Jean Marshall (Dalhousie University, Halifax, Canada). All human cancer cell lines used in this study were authenticated by STR DNA analysis (DDC Medical (Fairfield, OH)) or by the Center for Applied Genomics (SickKids, Toronto, Canada). U0126 was purchased from Cell Signaling Technology (Danvers, MA), PD98059, LY294002, SB203580 and SP600125 from Calbiochem (La Jolla, CA), Selumetinib from Selleckchem (Houston, TX) and 5-Aminolevulinic acid from Sigma (Oakville, ON).
Cell culture
All cell lines used in this study were maintained with high glucose Dulbecco's modified Eagle's medium (DMEM) (Invitrogen, Ontario, Canada), supplemented with 10% fetal bovine serum (FBS) and antibiotic-antimycotic mixture (Invitrogen) (100 units/mL penicillin G sodium). For in vitro experiments to determine PpIX formation, cells were plated in 24 well-plates and treated with 5-ALA (5 mM) for 5 h before PpIX measurement.
Animal studies
BALB/c female mice and athymic nude mice obtained from Charles River Laboratories (Montreal, PQ) were housed in a barrier unit within the central animal care facility in the Health Sciences Centre at Memorial University of Newfoundland. Animal care protocols were approved by the Institutional Animal Care Committee, in accordance with the Canadian Council on Animal Care guidelines. At 8 weeks of age, mouse mammary carcinoma 4T1 cells or human colon cancer DLD-1 cells prepared in cell culture media were injected (2×106 cells/100 μL) into the right hind flank of BALB/c female mice and athymic nude mice, respectively. After the development of palpable tumors (5-8 mm in diameter), the mice were randomly assigned to one of the following four groups: 1) U0126 (20 mg/kg body weight (BW) intraperitoneally (i.p.)), 2) i.p. vehicle control (dimethyl sulfoxide (DMSO)/saline), 3) oral Selumetinib (150 mg/kg BW), 4) oral vehicle control (0.5% propyl methyl cellulose in phosphate buffered saline (PBS)). Treatments were performed at 5 and 8 h prior to sacrifice for the drug groups (groups 1 and 3), and at 5 h prior to sacrifice for vehicle controls (groups 2 and 4). In addition, all mice were injected i.p. with 5-ALA (200 mg/kg BW) at 2 h before sacrifice. The tumors and normal organs (brain, colon, kidney, and lung) were then removed and homogenized in radioimmunoprecipitation assay (RIPA) buffer. Tissue homogenates were centrifuged and supernatant was collected for PpIX measurement.
PpIX measurement and high-performance liquid chromatography (HPLC) analysis
The cell lysates, culture supernatant and tissue homogenates were diluted in PBS and PpIX fluorescence was measured using Synergy Mx Fluorescence plate reader (BioTek Instruments Inc. VT) with a 405 nm excitation/630 nm emission filter. PpIX measurements by HPLC were carried out using a previously described method [26]. Briefly, cells (4×106 cells) were pelleted, re-suspended in 50 µL DMEM and then diluted in 200 µL of 60:40 methanol/10 mM potassium monobasic solution. Cell lysate (20 µL) or culture supernatant (20 µL) was injected into a HPLC (Waters 600E/Waters 717 auto-sampler, Waters, Milford, MA) and was separated with a Shimadzu Shim-Pack CLC-ODS (M) 15 cm column (Shimadzu Corporation, Japan) using a buffer consisting of 90% mobile phase A (10 mM potassium phosphate buffer, pH 4.6) and 10% mobile phase B (methanol). PpIX was measured with a fluorescence detector (Shimadzu RF 535 Fluorescence HPLC monitor, Shimadzu, Japan) with excitation set at 400 nm and emission at 620 nm. Measured values were normalized to protein concentration.
Flow cytometry
Cellular PpIX fluorescence was measured on a FACSCalibur (BD Biosciences, (San Jose, CA) at excitation 488 nm and emission 650 nm (FL3). The data was analyzed using FlowJo (FlowJo LLC, OR).
Confocal microscopy analysis
Fluorescence images of 5-ALA-induced PpIX in cells were acquired as described before [27,28]. Briefly, cells were fixed with 4% PFA/PBS and then stained with 4',6-diamidino-2-phenylindole (DAPI) and MitoTracker® Orange (M7510) (Thermo Fisher Scientific). Images were acquired on an Olympus FluoView FV1000 confocal microscope (Olympus) using the virtual channel function with two phases: PpIX (620 nm) and DAPI (488 nm) in phase 1 and MitoTracker Orange (545 nm) in phase 2. The acquired images were analyzed using the FluoView software (Olympus).
Tumor imaging
The tumors were photographed using a Canon 6D camera fitted with 35 mm lens mounted with a yellow 635 nm emission lens filter for fluorescence imaging. White light was used for bright field imaging and a blue light (405 nm) (Storz GmbH, Tuttlingen, Germany) was used for fluorescence imaging. A heat map was generated using the heat map plugin on ImageJ. Image analysis was carried out using ImageJ.
Ferrochelatase (FECH) assay
FECH activity was measured by aerobic enzymatic formation of zinc-protoporphyrin IX (Zn-PpIX) using a modified van Hillegersberg method [29,30]. Cell lysates obtained from DLD-1 cells treated with or without U0126 were incubated with 200 µM PpIX (Sigma Aldrich) in 200 µL assay buffer (0.1 M Tris-HCl, 1 mM palmitic acid (Sigma Aldrich) and 0.3% v/v Tween 20, pH 8.0) and then 50 µL of 2 mM zinc acetate solution was added. The mixture was incubated at 37°C for the indicated period of time. The reaction was terminated by adding 500 µL ice-cold stop buffer (1 mM ethylenediaminetetraacetic acid (EDTA) in 30:70 DMSO/ methanol). The reaction mixture was centrifuged at 14,000 ×g for 10 min and Zn-PpIX in the supernatant was measured using Synergy Mx Fluorescence plate reader (BioTek Instruments Inc. VT) with a 405 nm excitation/590 nm emission filter. Heat-inactivated cell lysates were included as a negative control.
Heme assay
Heme concentration in cell lysates was measured using the Hemin assay kit (Sigma-Aldrich, MO, USA) by following the manufacturer's instructions. Briefly, cells were rapidly homogenized in hemin assay buffer. A master reaction mix consisting of the kit-provided enzyme mix, hemin substrate, hemin assay buffer and hemin probe was prepared and added to the cell lysate and incubated for 60 min at room temperature. The absorbance at 570 nm was measured using a Synergy MX plate reader (BioTek Instruments Inc. VT) against a reagent blank. Heme concentrations were calculated using a standard curve obtained from kit-provided hemin standards.
Quantitative RT-PCR
Cytoplasmic RNA was isolated using TRIzol (Invitrogen, ON, Canada) according to the manufacturer's instructions. RNA (0.5 µg) was reverse transcribed (RT) to cDNA from random hexamers using the first-strand cDNA synthesis kit from Amersham Biosciences (PA, USA). Quantitative RT-PCR (RT-qPCR) was performed using primers shown in Table S1. Primers were validated using a 5-point, 5-fold dilution series. The absence of non-specific amplification was confirmed by observing a single peak in the melt-curve analysis, confirmation of the expected amplicon size by agarose gel analysis and the absence of amplification in the no template control. qPCR was then performed in triplicate on the StepOne Plus (Applied Biosystems CA, USA) using Power SYBR Green PCR Master Mix (Applied Biosystems CA, USA). Cycling conditions were: 95 ºC for 10 min, 40 cycles at 95 ºC for 15 s and 60 ºC for 1 min followed by melt-curve analysis.
Western blot analysis
Protein samples were prepared and western blot was conducted and evaluated as previously described [31]. Anti P-glycoprotein monoclonal antibody (F4) / ATP binding cassette subfamily (ABC) B member 1(ABCB1) was purchased from Thermo Fisher, anti ABCG2/CD338 antibody (3G8) from ORIGENE, and anti ABCB6 and anti GAPDH (6C5) antibodies from Abcam.
Statistical analysis
One-way ANOVA with Tukey's post-hoc test or unpaired t-test was performed using GraphPad Prism 6.07 (GraphPad Software, La Jolla, CA, USA).
Results
Inhibition of Ras/MEK increases PpIX accumulation
To determine whether oncogenic Ras and its downstream elements modulate PpIX accumulation induced by 5-ALA, we used established chemical inhibitors against Ras downstream elements includeing MEK, PI3K, p38 MAPK and JNK (Figure 1). Human colon cancer DLD-1 cells were treated with MEK inhibitor (U0126, PD98059 or Selumetinib), PI3K inhibitor (LY294002), p38 MAPK inhibitor (SB203580), JNK inhibitor (SP600125) or control vehicle (DMSO) for 24 h. 5-ALA was added to all groups for the last 5 h. MEK inhibition by U0126, PD98059 or Selumetinib significantly increased 5-ALA-induced PpIX accumulation in DLD-1 cells (Figure 1A-C), while inhibition of other Ras downstream elements had no effect (Figure 1D-F). The increase in PpIX accumulation by MEK inhibition was confirmed by HPLC analysis (Figure S1). Confocal microscopy analysis also revealed that MEK inhibition increased 5-ALA-induced PpIX fluorescence in DLD-1 cells (Figure 2A). In addition, PpIX fluorescence co-localized with MitoTracker, which suggests that the accumulation occurs within the mitochondria. Flow cytometry analysis further revealed that the MEK inhibitor significantly enhanced PpIX fluorescence in DLD-1 cells in a concentration-dependent manner (Figure 2B). These results clearly demonstrate that an inhibition of the oncogenic Ras/MEK pathway increases PpIX accumulation in human colon cancer cells.
PpIX increase by MEK inhibition is cancer cell specific
To broaden our investigation, we sought to determine whether MEK inhibitors can also increase PpIX accumulation in different types of human cell lines. To test this, human normal colon cells (CCD-18Co), colon cancer cells (DLD-1), normal breast cells (Hs 578Bst) and breast cancer cells (Hs 578T) were treated with U0126 or DMSO for 1, 5, 10 or 20 h (fold changes (Figure 3A) and absolute values (Figure S2)). PpIX accumulation was significantly increased in both cancer cell lines (DLD-1 and Hs 578T cells) after 5, 10 or 20 h treatment with U0126, while there was no significant change in PpIX accumulation in normal colon cells (CCD-18Co) or normal breast cells (Hs 578Bst). To determine whether enhanced PpIX accumulation is a common phenomenon among similar types of human cancer cells, a panel of human breast cell lines was tested (Figure 3B). PpIX accumulation was significantly increased in 4 out of 6 human breast cancer cell lines tested (Hs 578T, MDA-MB-231, T-47D, and MCF7), whereas no significant change was observed in two breast cancer cell lines (MDA-MB-468 and BT-474) and a normal breast cell line (Hs 578Bst). An increase in PpIX accumulation by the MEK inhibitor was similarly observed in other types of cancer cells (lung and prostate) (Figure S3). These results demonstrate that MEK inhibitors increase PpIX accumulation in a cancer cell-specific manner by targeting cellular mechanisms commonly present in a majority of human cancer cells.
Figure 1
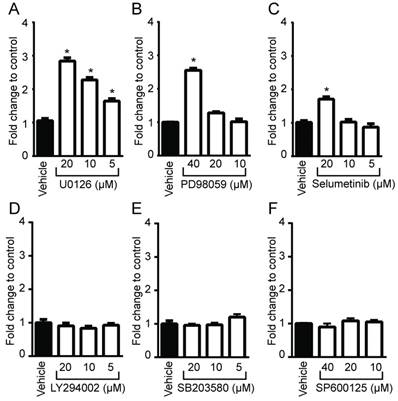
Identification of Ras downstream elements that regulate PpIX accumulation induced by 5-ALA. Human colon cancer DLD-1 cells were treated with control vehicle (Vehicle) or with (A) U0126 (MEK inhibitor), (B) PD98059 (MEK inhibitor), (C) Selumetinib (MEK inhibitor), (D) LY294002 (PI3K inhibitor), (E) SB203580 (p38MAPK inhibitor) or (F) SP600125 (JNK inhibitor) at the indicated concentrations for 20 h and with 5-ALA (5 mM) for the last 5 h. Cellular PpIX in cell lysates was measured using a fluorescence plate reader. Cellular PpIX is presented as fold change compared with vehicle control. Data are expressed as the mean ± SE of 3 independent samples. Statistical analysis was conducted using two-way ANOVA, *p<0.01.
Ras/MEK regulates ABCB1 and FECH activity
Cellular PpIX concentration is a function of the rate of metabolic conversion of 5-ALA and the PpIX retention potential of the cell. Therefore, we examined whether the rate of PpIX efflux from cells was regulated by MEK. Using DLD-1 cells treated with 5-ALA, we found that the MEK inhibitor U0126 increased PpIX within cells but reduced it in cell culture supernatant in a concentration- and time-dependent manner (Figure 4A). This was also confirmed by HPLC analysis (Figure S4). To further examine the role of PpIX efflux in the absence of de novo protein synthesis, cellular PpIX was measured in DLD-1 cells treated with control vehicle or U0126 after cycloheximide treatment (Figure 4B). We found that the cells treated with U0126 retained PpIX for a longer period compared to those treated with vehicle. These results clearly suggest that oncogenic Ras/MEK activity promotes PpIX efflux in cancer cells, and its suppression leads to enhanced PpIX accumulation. This appears to be due to a reduction in the expression of ABCB1, one of the ATP binding cassette transporters (ABCB1, ABCB6 and ABCG2), which are known regulators of PpIX efflux [26,32-36]. The expression of ABCB1, but not ABCB6 or ABCG2, was reduced in DLD-1 cells at the protein and RNA levels when the cells were treated with U0126 (Figure 4C-D).
Another possible mechanism that could increase intracellular PpIX fluorescence is a reduction in its conversion to non-fluorescent product, heme. [30,37]. Thus, we investigated whether PpIX conversion to heme is modulated by Ras/MEK. The chelation of Fe2+ into PpIX generates heme, the end product of the heme biosynthesis pathway, which is catalyzed by the enzyme FECH. To test the activity of the enzyme, cell lysates obtained from DLD-1 cells treated with or without U0126 for 20 h were incubated with exogenous PpIX and zinc acetate. The synthesis of Zn-PpIX, which is catalyzed by endogenous FECH, was measured at various time points. We found that conversion of PpIX into Zn-PpIX was less efficient in U0126-treated cell lysate compared to control cell lysate at 60, 90 and 120 min (Figure 5A). Moreover, when DLD-1 cells were treated with the MEK inhibitor, heme production was decreased in a concentration-dependent manner (Figure 5B). These results indicate that MEK inhibition suppresses FECH-mediated conversion of PpIX to heme, resulting in an intracellular accumulation of PpIX.
Taken together, inhibition of the oncogenic Ras/MEK pathway increases PpIX accumulation in cancer cells by two independent mechanisms, namely the downregulation of ABCB1 expression that mediates PpIX efflux and the reduction of FECH activity that converts PpIX into heme (Figure 5C).
MEK inhibitor treatment enhances PpIX fluorescence in vivo
Next, we asked whether MEK inhibitors can enhance PpIX fluorescence in tumors in vivo using a mouse 4T1 breast cancer model, and human DLD-1 colon cancer model. First, we confirmed the effect of MEK inhibitors on the mouse cancer cells in vitro. In cultured mouse 4T1 breast cancer cells, both U0126 and Selumetinib enhanced 5-ALA-induced PpIX accumulation, similar to the results in human cancer cell lines (Figure 6A). To test the effect of these MEK inhibitors in vivo, BALB/c mice bearing mouse 4T1 breast tumors and athymic nude mice bearing human colon DLD-1 tumor were treated intraperitoneally with U0126 or orally with Selumetinib 5 or 8 h before sacrifice. Control groups received appropriate vehicles via the same route of administration as their experimental counterparts. All 4 groups were then injected intraperitoneally with 5-ALA at 2 h before sacrifice. 5-ALA-induced PpIX accumulation was significantly higher in the 4T1 breast tumors in mice treated with U0126 at 5 h post-injection than in vehicle controls (2.7-fold), which reversed to control levels by 8 h post-U0126 injection (Figure 6B). Selumetinib also significantly increased PpIX accumulation within the 4T1 breast tumors both at 5 h (1.9-fold) and 8 h (2.0-fold) post-treatment compared to vehicle controls (Figure 6C). This accumulation was specific to tumors, as U0126 or Selumetinib treatment did not increase PpIX accumulation in normal organs (Figure 6D).
Figure 2
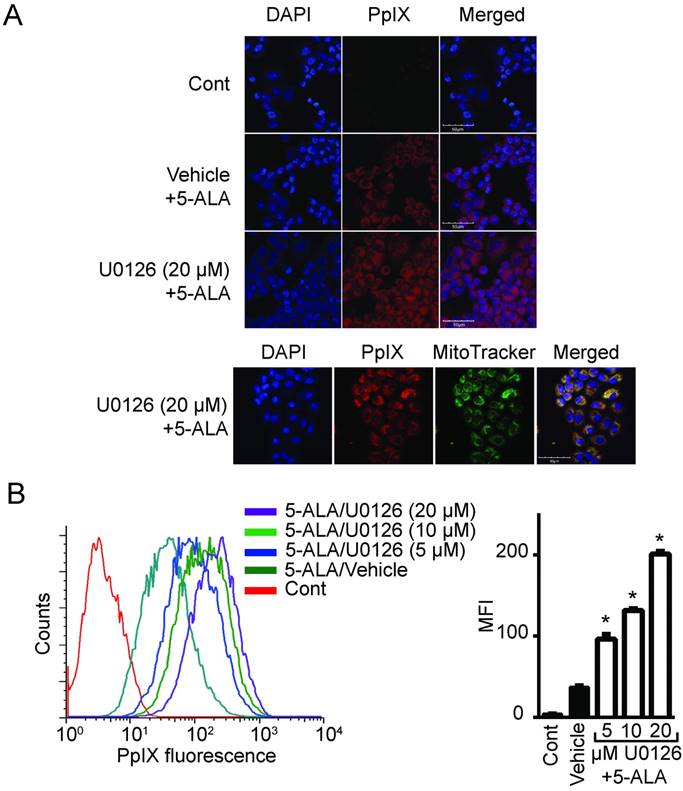
MEK inhibition increases PpIX fluorescence. (A) Fluorescence images of a nuclear marker 4',6-diamidino-2-phenylindole (DAPI) (blue), PpIX fluorescence (red) and mitochondria marker MitoTracker (green) in DLD-1 cells left untreated (Cont), or treated with DMSO (Vehicle) or U0126 for 20 h and with 5-ALA for the last 5 h. (B) Representative histogram (left) and bar graph (right) of PpIX fluorescence measured by flow cytometry in DLD-1 cells treated as described above. Data are expressed as the mean ± SE of mean fluorescence intensity (MFI) of 3 independent samples. Statistical analysis was conducted using two-way ANOVA, *p<0.01.
Figure 3
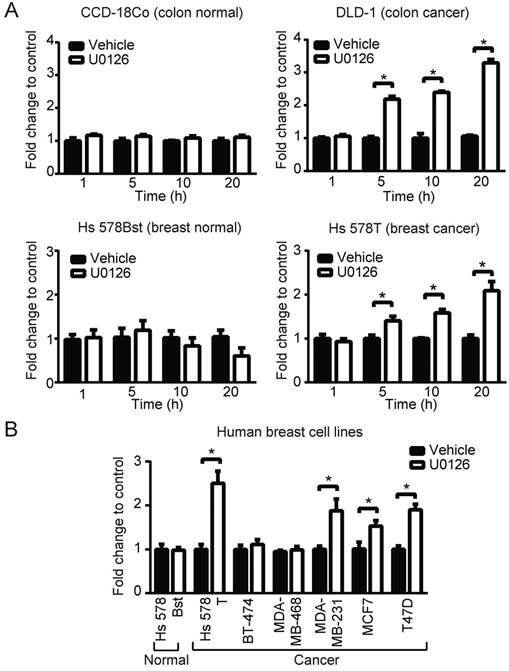
MEK involvement in PpIX accumulation in normal and cancer cell lines. (A) Normal colon cells (CCD-18Co) and colon cancer cells (DLD-1), and normal breast cells (Hs 578Bst) and breast cancer cells (Hs 578T) were treated with or without U0126 (20 µM) and with 5-ALA (5 mM). (B) Normal breast cells (Hs 578Bst), breast cancer cells (Hs 578T, BT-474, MDA-MB-468, MCF7 and T-47D) were treated with or without U0126 (20 µM) for 20 h and with 5-ALA (5 mM) for 5 h. Cellular PpIX was measured using a fluorescence plate reader. Cellular PpIX is presented as fold change compared to vehicle control (CTR). Data are expressed as the mean±SE of 3 independent samples. Statistical analysis was conducted using two-way ANOVA, *p<0.01
To further determine whether MEK inhibition enhances tumor visibility by PpIX fluorescence, the visual fluorescence signal in resected tumors was compared under blue light (405 nm). We found that 5-ALA-induced PpIX fluorescence was stronger in tumors from mice treated with Selumetinib at 8 h post-treatment compared to vehicle controls (Figure 7). The digital quantification of PpIX fluorescence signal confirmed the increase in PpIX fluorescence within tumors by Selumetinib (2.2-fold) (Figure 7C-D), which was similar to the degree of increase measured in tumor homogenates using a fluorescence reader (Figure 6C). Selumetinib treatment also increased 5-ALA-induced PpIX accumulation in human colon DLD-1 tumors in athymic nude mice (Figure 7E). These results demonstrate that MEK inhibitors are capable of efficiently and selectively enhancing PpIX accumulation within tumors in vivo, suggesting that it can intensify the contrast between fluorescence in tumors and the background, which would be advantageous during FGS.
Discussion
It is known that 5-ALA treatment induces PpIX accumulation more efficiently in cancer cells than in normal cells due to their high metabolic activity and activation of the heme biosynthesis pathway [34,38,39]. Since earlier studies have shown that oncogenic transformation increases 5-ALA-induced PpIX accumulation [23-25], we originally speculated that oncogenic Ras activation stimulates the heme biosynthesis pathway, resulting in increased PpIX accumulation in tumors. However, this was not the case as we found an opposite result showing that an inhibition of MEK, one of the Ras downstream elements, promotes PpIX accumulation. While this was unexpected, we were intrigued by the potential of using this property of MEK inhibitors to facilitate PpIX accumulation selectively in cancer cells, which would improve its utility in cancer therapies such as FGS and PDT. Our first critical finding is that MEK inhibition increase 5-ALA-induced PpIX accumulation in a cancer-specific manner, as the same treatment did not increase PpIX in normal cell lines in vitro and in normal organs in vivo. We also discovered that MEK inhibitors increase PpIX accumulation in cancer cells by targeting pathways that effectively reduce the intracellular PpIX concentration by way of export (ABCB1) and metabolic conversion to non-fluorescent heme (FECH). Most importantly, we demonstrated that in vivo treatment of MEK inhibitors can enhance PpIX fluorescence selectively in tumors. This suggests that MEK inhibitors may be valuable in combination with existing PpIX-dependent cancer therapies.
Our study found that MEK inhibition decreases ABCB1 expression, resulting in reduced PpIX efflux from cells. Ras/MEK inhibition is known to suppress ABCB1 expression at its transcriptional [40], posttranslational [41] and functional levels [42]. In agreement with these previous reports, we found that ABCB1 expression is downregulated in cancer cells at both RNA and protein levels by MEK inhibition (Figure 4C-D). In contrast, other primary transporters of PpIX, namely ABCB6 and ABCG2, were not modulated by MEK. Previous studies have also used ABCG2 inhibitors to increase PpIX accumulation in cancer cells and to promote the efficacy of 5-ALA-PDT in vitro [43,44]. In contrast, ABCB6 overexpression increases PpIX accumulation [36]. We found some critical differences between the effects of inhibiting ABCG2 or MEK when tested on a panel of cell lines (Figure S5). We found that the ABCG2 inhibitors increase the PpIX accumulation more rapidly (5 h) and that different types of cell lines were sensitive to the ABCG2 or the MEK inhibitor. More importantly, the effect of ABCG2 inhibitors was not specific to cancer cell lines, as they significantly increased PpIX accumulation in both cancer and normal cell lines, while the MEK inhibition effect was selective to cancer cells. These suggest that ABCB1 expression, which is upregulated by oncogenic Ras/MEK, rather than ABCG2 may underlie the cancer-specific increase of PpIX accumulation induced by MEK inhibitors. To support this, we found that ABCB1 expression was higher on cancer cells than on normal cells (Figure S6A). In addition, when PpIX efflux was compared between normal and cancer cells, MEK inhibition reduced PpIX efflux in cancer cells but not in normal cells (Figure S6B).
Figure 4
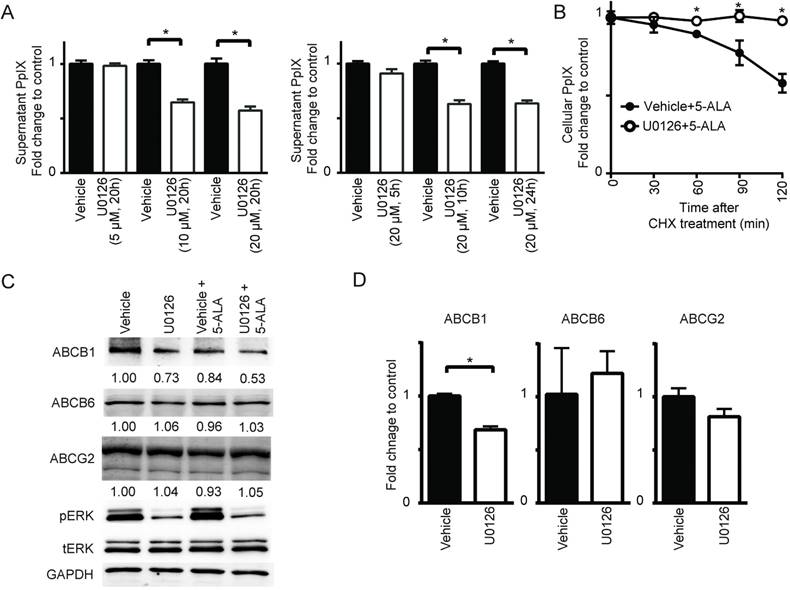
MEK inhibition suppresses PpIX efflux by decreasing ABCB1 expression. (A) DLD-1 cells were treated with DMSO (Vehicle) or U0126 at the indicated concentrations for the indicated time periods and with 5-ALA (5 mM) for the last 5 h. At 1 h before the time points, the cells were washed with PBS and incubated with PBS, and then the PpIX amount in the culture supernatant was measured using a fluorescence plate reader. (B) DLD-1 cells were treated with or without U0126 (20 µM) for 20 h and with 5-ALA (5 mM) for 5 h, and then treated with cycloheximide (CHX) (10 µg/mL, time 0) to block de novo protein synthesis. Cellular PpIX amount was measured at the indicated time points by flow cytometry. (C) Western blot analysis using antibodies against ABCB1, ABCB6, ABCG2, phosphorylated ERK (pERK), total ERK (tERK) and GAPDH on cell lysates obtained from DLD-1 cells treated with DMSO (Vehicle) or U0126 (20 µM) for 20 h and with 5-ALA (5 mM) for 5 h. Numbers below bands are density values relative to GAPDH compared to that of vehicle control. (D) qRT-PCR analysis for ABCB1, ABCB6 and ABCG2 expression in DLD-1 cells treated with DMSO (Vehicle) or U0126 (20 µM) for 20 h. The expression levels were normalized to GAPDH and presented as fold change of vehicle control. Data are expressed as the mean ± SE of 3 independent samples. Statistical analysis was conducted using unpaired t-test, *p<0.01.
Figure 5
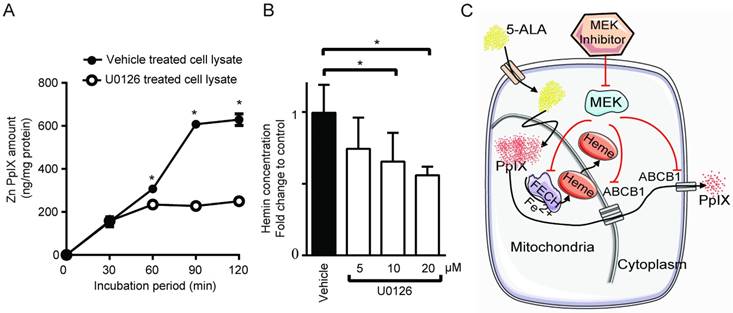
MEK inhibition suppresses FECH activity to convert PpIX into heme. (A) The amount of Zinc-PpIX (Zn-PpIX) was measured at 30, 60, 90 and 120 min after exogenous PpIX was added to the cell lysates obtained from DLD-1 cells treated with or without U0126 (20 µM) for 20 h. (B) The amount of cellular heme was measured in cell lysate obtained from DLD-1 cells treated with or without U0126 (5, 10 or 20 µM) for 20 h. Data are expressed as the mean ± SE of 3 independent samples. Statistical analysis was conducted using two-way ANOVA, *p<0.01, Control vs. U0126. (C) Schematic diagram illustrating the cellular mechanisms for increasing PpIX accumulation by MEK inhibition.
Figure 6
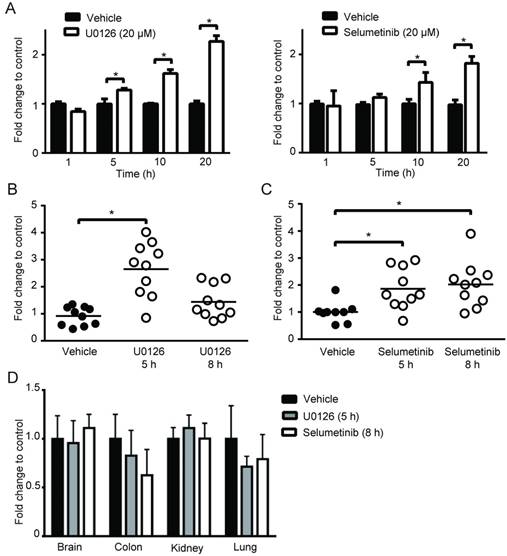
Increase of PpIX fluorescence by MEK inhibitors in vivo. (A) Mouse mammary carcinoma 4T1 cancer cell line was treated with U0126 (20 µM) (left) or with Selumetinib (20 µM) for 1, 5, 10 and 20 h and compared with their respective vehicle controls. 5-ALA (5 mM) was added for the last 5 h. PpIX in tumor homogenates was measured using a fluorescence plate reader. PpIX fluorescence is reported as fold change compared with vehicle control. Data are expressed as the mean ± SE of 3 independent samples. Statistical analysis was conducted using unpaired t-test, *p<0.01. (B, C) Effect of MEK inhibitors on 5-ALA-induced PpIX accumulation in 4T1 tumors in BALB/c mice. Each symbol denotes an individual mouse. Mice were treated with (B) U0126 (20 mg/kg BW i.p.) or (C) Selumetinib (250 mg/kg BW oral) at 5 and 8 h before sacrifice. Controls were treated with their respective vehicle at 5 h before sacrifice. All mice were injected with 5-ALA (200 mg/kg BW) at 2 h prior to sacrifice. Statistical analysis for in vivo studies was conducted using two-way ANOVA, *p<0.05. (D) PpIX fluorescence in various organs from mice treated with U0126 for 5 h, Selumetinib or vehicle for 8 h and 5-ALA for 2 h before sacrifice. Fluorescence in tissue homogenates was measured using a fluorescence plate reader.
Figure 7
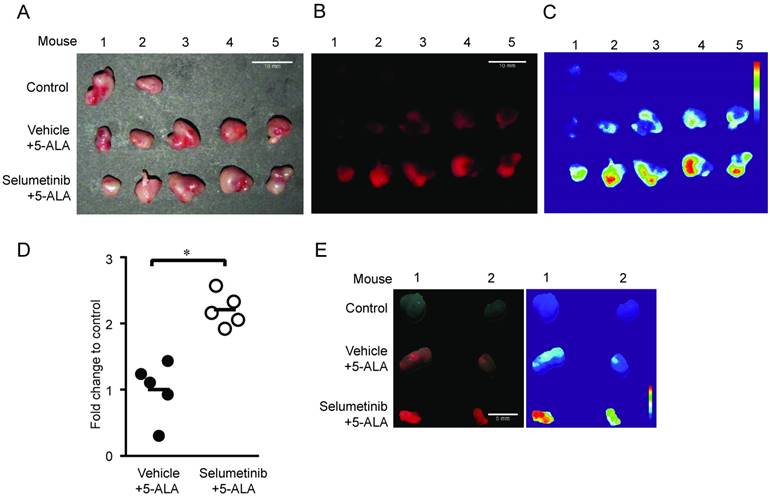
In vivo imaging analysis of PpIX fluorescence. Fluorescence images of 4T1 tumors obtained from BALB/c mice treated with Selumetinib (250 mg/kg BW) or control vehicle at 8 h and with 5-ALA (200 mg/kg BW) at 2 h prior to sacrifice. (A-C) Tumor images under white light (A), visual PpIX fluorescence image under blue light (B) and heat map images of relative fluorescence intensity in the tumors (C). (D) Quantitative analysis of the heat map images. Total fluorescence of each tumor was measured using the metric: integrated density of the tumor - (average background integrated density × area of the tumor). Each circle represents a mouse (5 mice/group) and the bar represents average tumor fluorescence of the group. Statistical analysis was conducted using unpaired t-test, *p<0.05. (E) Fluorescence (left) and heat map (right) images of DLD-1 tumors obtained from athymic nude mice treated with Selumetinib (250 mg/kg BW) or control vehicle for 8 h and 5-ALA (200 mg/kg BW) for 2 h prior to sacrifice.
FECH is another well-established target for increasing PpIX accumulation to promote 5-ALA-PDT efficacy [30,45-47]. FECH is the terminal enzyme in the heme biosynthesis pathway, which inserts a ferrous ion (Fe2+) into PpIX and converts it into heme. Inhibiting FECH by RNAi knockdown or a chemical inhibitor has been shown to increase PpIX fluorescence and 5-ALA-PDT efficacy in human cancer cells [46,47]. Furthermore, vitamin D3 treatment increases PpIX accumulation by suppressing FECH expression [48]. We also identified FECH as another target of MEK. Our results show that MEK inhibition reduces the catalytic activity of FECH and the subsequent formation of heme in cancer cells (Figure 5B). How the Ras/MEK pathway downregulates FECH activity remains to be seen, as, to the best of our knowledge, there is no literature on their interactions. It should be noted that kinase inhibitors can reduce FECH activity in a non-specific manner as FECH is their common off-target [49]. However, this is not the case in our study, as we used three different MEK inhibitors, all of which induced similar increases in PpIX accumulation. Furthermore, kinase inhibitors against other Ras downstream elements (LY294002, SB203580 and SP600125) had no effect on PpIX accumulation. In addition, in silico docking studies demonstrated that the binding affinity of the MEK inhibitors to FECH was significantly lower than that of an established FECH inhibitor (MK2461) and similar to the kinase inhibitors against other Ras downstream elements used in Figure 1 (Figure S7). Thus, our study provides another mechanism by which FECH-mediated conversion of PpIX can be modulated. In summary, MEK inhibitors increase PpIX in cancer cells by targeting two essential limiting factors of PpIX accumulation.
Oncogenic Ras/MEK may modulate other steps in the heme biosynthesis pathway such as 5-ALA uptake and activities of the different heme biosynthesis enzymes, which remain to be determined in future studies. Furthermore, as inhibition of only MEK, but not other Ras downstream elements, increased PpIX accumulation, the Ras/MEK pathway should have unique regulatory mechanisms for ABCB1 and/or FECH, which is essential to be elucidated. It should be also noted that although other Ras downstream elements were not involved in increasing 5-ALA-induced PpIX accumulation, their inhibitors have been reported to improve efficacy of PDT by targeting other mechanisms such as increasing photosensitizer uptake, promoting cancer cell apoptosis and decreasing metastasis [50-54].
Among the breast cancer cell lines tested, the increase in PpIX accumulation by MEK inhibition was not observed in MDA-MB-468 and BT-474 cells even though the MEK inhibitor effectively suppressed the Ras/MEK signaling in these cell lines (data not shown). The breast cancer cells used in this study included triple negative (Hs 578T, MDA-MB-468 and MDA-MD-231), luminal A (ER+, Her2-) (MCF7 and T-47D) and luminal B (ER+, Her2+) (BT-474) subtypes. As MDA-MB-468 and BT-474 cells do not belong to a common subtype, it is unknown at this stage why these cell lines did not respond to the PpIX increase induced by MEK inhibition. As tumor heterogeneity is one of the major problems limiting the efficacy of targeted therapies in clinical settings, it is essential to conduct further studies and identify the resistance mechanisms to the PpIX increase induced by MEK inhibition.
As aberrant activation of the Ras/MEK pathway often found in cancer cells contributes to tumorigenesis, this pathway has been a target of intense research to identify anticancer drug targets, and there are chemical inhibitors targeting the Ras/MEK pathway already being used in clinical settings. For example, two Ras/MEK inhibitors, Trametinib and Cobimetinib, have been approved for treating advanced and metastatic melanoma with a BRAF V600E mutation [55,56]. Furthermore, several second generation Ras/MEK inhibitors including Selumetinib are currently being tested in clinical trials [57]. Our study suggests that some of these Ras/MEK pathway inhibitors may be useful as enhancers of fluorescence signal in 5-ALA-FGS, an innovative technique that can provide surgeons with accurate real time tumor images. One potential application is the surgical removal of gliomas (grades III or IV), a procedure approved by the FDA [13]. For gliomas, however, it is essential for MEK inhibitors to cross the blood brain barrier (BBB) to penetrate into the CNS. To this end, MEK inhibitors (PD0325901 and SL327) [57-59], which can cross the BBB, need to be evaluated in animal models for their efficacy in enhancing 5-ALA-induced PpIX florescence in animal brain tumor models.
In conclusion, the present study provides evidence that oncogenic Ras/MEK can modulate the heme biosynthesis pathway, and its inhibition increases PpIX fluorescence by 2-3-fold in a cancer-specific manner in vitro and in vivo. This is the first study to show involvement of an oncogenic cell signaling pathway in the regulation of PpIX accumulation. Potentiating PpIX fluorescence only in cancer cells would be of advantage in the precise identification of tumor borders and small satellite tumors, which remains a challenge in current 5-ALA-FGS. While further preclinical studies are needed to determine whether the results from animal cancer models could be translated into clinical settings, the combined use of MEK inhibitors with 5-ALA represents a promising strategy for improving PpIX-dependent cancer therapies.
Abbreviations
5-ALA: 5-aminolevulenic acid; ABC: ATP binding cassette; ABCB1: ATP binding cassette sub family B1; ABCB6: ATP binding cassette subfamily B6; ABCG2: ATP binding cassette subfamily G2; ATP: adenosine triphosphate; BW: body weight; CHX: cyclohexamide; Cont: control; CT: computerized tomography; CTR: control; DAPI: 4',6-diamidino-2-phenylindole; DMEM: dulbecco's modified eagle medium; DMSO: dimethyl sulfoxide; EDTA: ethylenediaminetetraacetic acid; FBS: fetal bovine serum; Fe2+: ferrous ion; FECH: ferrochelatase; FGS: fluorescence guided surgery; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; h: hour; HPLC: high performance liquid chromatography; i.p: intraperitoneal; JNK: c-Jun-N-terminal kinases; MEK: /mitogen-activated protein kinase kinase; min: minute; MRI: magnetic resonance imaging; p38 MAPK: p38 mitogen-activated protein kinase; PBS: phosphate buffered saline; PDT: photodynamic therapy; pERK: phosphorylated- extracellular signal-regulated kinase; PET: positron emission tomography; PpIX: protoporphyrin IX; RIPA: radioimmunoprecipitation assay; ROS: reactive oxygen species; RT: reverse transcribed; RT-qPCR: reverse transcribed-quantitative polymerase chain reaction; tERK: total extracellular signal-regulated kinase; US FDA: United States food and drug administration; Zn-PpIX: Zinc-protoporphyrin IX.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by grants (to K.H) from the Canadian Cancer Society (CCSRI) and the Medical Research Fund (MRF) from Memorial University of Newfoundland. V.S.C is a trainee in the Cancer Research Training Program (CRTP) of the Beatrice Hunter Cancer Research Institute (BHCRI) with funds provided by The Terry Fox Foundation, the Prostrate Cancer Fight Foundation (PCFF) and Ride for Dad. The graphical abstract was partly generated using Servier Medical Art, provided by Servier, licensed under a Creative Commons Attribution 3.0 (available at http://www. smart.servier.com).
Competing Interests
The authors have declared that no competing interest exists.
References
1. SEER. SEER Research Data 1973-2014. [cited 2017 Aug 3]. Available from:https://seer.cancer.gov/data/
2. Nguyen QT, Tsien RY. Fluorescence-guided surgery with live molecular navigation — a new cutting edge. Nat Rev Cancer. 2013;13:653-62
3. Guyotat J, Pallud J, Armoiry X, Pavlov V, Metellus P. 5-Aminolevulinic Acid-Protoporphyrin IX Fluorescence-Guided Surgery of High-Grade Gliomas: A Systematic Review. Schramm J, editor. Adv Tech Stand Neurosurg. 2016;68:61-90
4. Agostinis P, Berg K, Cengel K a, Foster TH, Girotti AW, Gollnick SO. et al. Photodynamic therapy of cancer: an update. CA Cancer J Clin. 2011;61:250-81
5. Liu JTC, Meza D, Sanai N. Trends in fluorescence image-guided surgery for gliomas. Neurosurgery. 2014;75:61-71
6. Valdes PA, Bekelis K, Harris BT, Wilson BC, Leblond F, Kim A. et al. 5-Aminolevulinic acid-induced protoporphyrin IX fluorescence in meningioma: qualitative and quantitative measurements in vivo. Neurosurgery. 2014;10(Suppl 1):74-82
7. Ishizuka M, Abe F, Sano Y, Takahashi K, Inoue K, Nakajima M. et al. Novel development of 5-aminolevurinic acid (ALA) in cancer diagnoses and therapy. Int Immunopharmacol. 2011;11:358-65
8. Hussain T, Nguyen QT. Molecular imaging for cancer diagnosis and surgery. Adv Drug Deliv Rev. 2014;66:90-100
9. Cornelius JF, Slotty PJ, Kamp MA, Schneiderhan TM, Steiger HJ, El-Khatib M. Impact of 5-aminolevulinic acid fluorescence-guided surgery on the extent of resection of meningiomas-with special regard to high-grade tumors. Photodiagnosis Photodyn Ther. 2014;11:481-90
10. Stummer W, Pichlmeier U, Meinel T, Wiestler OD, Zanella F, Reulen H-J. et al. Fluorescence-guided surgery with 5-aminolevulinic acid for resection of malignant glioma: a randomised controlled multicentre phase III trial. Lancet Oncol. 2006;7:392-401
11. Kriegmair M, Baumgartner R, Knuechel R, Steinbach P, Ehsan A, Lumper W. et al. Fluorescence photodetection of neoplastic urothelial lesions following intravesical instillation of 5-aminolevulinic acid. Urology. 1994;44:836-41
12. Inoue K, Karashima T, Kamada M, Shuin T, Kurabayashi A, Furihata M. et al. Regulation of 5-aminolevulinic acid-mediated protoporphyrin IX accumulation in human urothelial carcinomas. Pathobiology. 2009;76:303-14
13. Aminolevulinic acid hydrochloride, known as ALA HCl (Gleolan, NX Development Corp.) as an optical imaging agent indicated in patients with gliomas. Center for Drug Evaluation and Research; [cited 2017 Aug 3]. Available from: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm562645.htm
14. Moiyadi A, Syed P, Srivastava S. Fluorescence-guided surgery of malignant gliomas based on 5-aminolevulinic acid: paradigm shifts but not a panacea. Nat Rev Cancer. 2014;14:146
15. Tonn J-C, Stummer W. Fluorescence-guided resection of malignant gliomas using 5-aminolevulinic acid: practical use, risks, and pitfalls. Clin Neurosurg. 2008;55:20-6
16. Floeth FW, Sabel M, Ewelt C, Stummer W, Felsberg J, Reifenberger G. et al. Comparison of (18)F-FET PET and 5-ALA fluorescence in cerebral gliomas. Eur J Nucl Med Mol Imaging. 2011;38:731-41
17. Rocks O, Peyker A, Bastiaens PIH. Spatio-temporal segregation of Ras signals: one ship, three anchors, many harbors. Curr Opin Cell Biol. 2006;18:351-7
18. Shields JM, Pruitt K, McFall A, Shaub A, Der CJ. Understanding Ras: “it ain”t over “til it”s over'. Trends Cell Biol. 2000;10:147-54
19. Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene. 1998;17:1395-413
20. Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7:295-308
21. Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11-22
22. Santarpia L, Lippman SM, El-Naggar AK. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:103-19. Available from: http://www.tandfonline.com/doi/full/10.1517/14728222.2011.645805
23. Yang X, Palasuberniam P, Myers KA, Wang C, Chen B. Her2 oncogene transformation enhances 5-aminolevulinic acid-mediated protoporphyrin IX production and photodynamic therapy response. Oncotarget. 2016;7:57798-810
24. Juzeniene A, Peng Q, Moan J. Milestones in the development of photodynamic therapy and fluorescence diagnosis. Photochem Photobiol Sci. 2007;6:1234-45
25. Li G, Szewczuk MR, Raptis L, Johnson JG, Weagle GE, Pottier RH. et al. Rodent fibroblast model for studies of response of malignant cells to exogenous 5-aminolevulinic acid. Br J Cancer. 1999;80:676-84
26. Zhou S, Zong Y, Ney PA, Nair G, Stewart CF, Sorrentino BP. Increased expression of the Abcg2 transporter during erythroid maturation plays a role in decreasing cellular protoporphyrin IX levels. Blood. 2005;105:2571-6
27. Fan Z, Cui X, Wei D, Liu W, Li B, He H. et al. eEF1A1 binds and enriches protoporphyrin IX in cancer cells in 5-aminolevulinic acid based photodynamic therapy. Sci Rep. 2016;6:25353
28. Kobuchi H, Moriya K, Ogino T, Fujita H, Inoue K, Shuin T. et al. Mitochondrial localization of ABC transporter ABCG2 and its function in 5-aminolevulinic acid-mediated protoporphyrin IX accumulation. PLoS One. 2012;7:e50082
29. Van Hillegersberg R, Van den Berg JW, Kort WJ, Terpstra OT, Wilson JH. Selective accumulation of endogenously produced porphyrins in a liver metastasis model in rats. Gastroenterology. 1992;103:647-51
30. Kemmner W, Wan K, Rüttinger S, Ebert B, Macdonald R, Klamm U. et al. Silencing of human ferrochelatase causes abundant protoporphyrin-IX accumulation in colon cancer. FASEB J. 2008;22:500-9
31. Komatsu Y, Christian SL, Ho N, Pongnopparat T, Licursi M, Hirasawa K. Oncogenic Ras inhibits IRF1 to promote viral oncolysis. Oncogene. 2015;34:3985-93
32. Hagiya Y, Endo Y, Yonemura Y, Takahashi K, Ishizuka M, Abe F. et al. Pivotal roles of peptide transporter PEPT1 and ATP-binding cassette (ABC) transporter ABCG2 in 5-aminolevulinic acid (ALA)-based photocytotoxicity of gastric cancer cells in vitro. Photodiagnosis Photodyn Ther. 2012;9:204-14
33. Kralova J, Kolar M, Kahle M, Truksa J, Lettlova S, Balusikova K. et al. Glycol porphyrin derivatives and temoporfin elicit resistance to photodynamic therapy by different mechanisms. Sci Rep. 2017;7:44497
34. Hadjipanayis CG, Widhalm G, Stummer W. What is the Surgical Benefit of Utilizing 5-Aminolevulinic Acid for Fluorescence-Guided Surgery of Malignant Gliomas? Neurosurgery. 2015;77:663-73
35. Chu ESM, Yow CMN, Shi M, Ho RJY. Effects of photoactivated 5-aminolevulinic acid hexyl ester on MDR1 over-expressing human uterine sarcoma cells. Toxicol Lett. 2008;181:7-12
36. Zhao S-G, Chen X-F, Wang L-G, Yang G, Han D-Y, Teng L. et al. Increased expression of ABCB6 enhances protoporphyrin IX accumulation and photodynamic effect in human glioma. Ann Surg Oncol. 2013;20:4379-88
37. Barman-Aksözen J, Minder EI, Schubiger C, Biolcati G, Schneider-Yin X. In ferrochelatase-deficient protoporphyria patients, ALAS2 expression is enhanced and erythrocytic protoporphyrin concentration correlates with iron availability. Blood Cells Mol Dis. 2015;54:71-7
38. Wyld L, Burn JL, Reed MW, Brown NJ. Factors affecting aminolaevulinic acid-induced generation of protoporphyrin IX. Br J Cancer. 1997;76:705-12
39. Yang X, Palasuberniam P, Kraus D, Chen B. Aminolevulinic Acid-Based Tumor Detection and Therapy: Molecular Mechanisms and Strategies for Enhancement. Int J Mol Sci. 2015;16:25865-80
40. Tsai Y-T, Lozanski G, Lehman A, Sass EJ, Hertlein E, Salunke SB. et al. BRAFV600E induces ABCB1/P-glycoprotein expression and drug resistance in B-cells via AP-1 activation. Leuk Res. 2015;39:1270-7
41. Katayama K, Fujiwara C, Noguchi K, Sugimoto Y. RSK1 protects P-glycoprotein/ABCB1 against ubiquitin-proteasomal degradation by downregulating the ubiquitin-conjugating enzyme E2 R1. Sci Rep. 2016;6:36134
42. Wang Y-J, Zhang Y-K, Zhang G-N, Al Rihani SB, Wei M-N, Gupta P. et al. Regorafenib overcomes chemotherapeutic multidrug resistance mediated by ABCB1 transporter in colorectal cancer: In vitro and in vivo study. Cancer Lett. 2017;396:145-54
43. Nakayama T, Otsuka S, Kobayashi T, Okajima H, Matsumoto K, Hagiya Y. et al. Dormant cancer cells accumulate high protoporphyrin IX levels and are sensitive to 5-aminolevulinic acid-based photodynamic therapy. Sci Rep. 2016;6:36478
44. Zhang J, Lodish HF. Constitutive activation of the MEK/ERK pathway mediates all effects of oncogenic H-ras expression in primary erythroid progenitors. Blood. 2004;104:1679-87
45. Inoue K, Fukuhara H, Kurabayashi A, Furihata M, Tsuda M, Nagakawa K. et al. Photodynamic therapy involves an antiangiogenic mechanism and is enhanced by ferrochelatase inhibitor in urothelial carcinoma. Cancer Sci. 2013;104:765-72
46. Teng L, Nakada M, Zhao S-G, Endo Y, Furuyama N, Nambu E. et al. Silencing of ferrochelatase enhances 5-aminolevulinic acid-based fluorescence and photodynamic therapy efficacy. Br J Cancer. 2011;104:798-807
47. Bhasin G, Kausar H, Athar M. Protoporphyrin-IX accumulation and cutaneous tumor regression in mice using a ferrochelatase inhibitor. Cancer Lett. 2002;187:9-16
48. Anand S, Wilson C, Hasan T, Maytin E V. Vitamin D3 enhances the apoptotic response of epithelial tumors to aminolevulinate-based photodynamic therapy. Cancer Res. 2011;71:6040-50
49. Klaeger S, Gohlke B, Perrin J, Gupta V, Heinzlmeir S, Helm D. et al. Chemical Proteomics Reveals Ferrochelatase as a Common Off-target of Kinase Inhibitors. ACS Chem Biol. 2016;11:1245-54
50. Ge X, Liu J, Shi Z, Jing L, Yu N, Zhang X. et al. Inhibition of MAPK signaling pathways enhances cell death induced by 5-Aminolevulinic acid-photodynamic therapy in skin squamous carcinoma cells. Eur J dermatology. 2016;26:164-72
51. Fateye B, Wan A, Yang X, Myers K, Chen B. Comparison between endothelial and tumor cells in the response to verteporfin-photodynamic therapy and a PI3K pathway inhibitor. Photodiagnosis Photodyn Ther. 2015;12:19-26
52. Fateye B, Li W, Wang C, Chen B. Combination of phosphatidylinositol 3-kinases pathway inhibitor and photodynamic therapy in endothelial and tumor cells. Photochem Photobiol. 2012;88:1265-72
53. Espada J, Galaz S, Sanz-Rodríguez F, Blázquez-Castro A, Stockert JC, Bagazgoitia L. et al. Oncogenic H-Ras and PI3K signaling can inhibit E-cadherin-dependent apoptosis and promote cell survival after photodynamic therapy in mouse keratinocytes. J Cell Physiol. 2009;219:84-93
54. Zhang X, Cai L, He J, Li X, Li L, Chen X. et al. Influence and mechanism of 5-aminolevulinic acid-photodynamic therapy on the metastasis of esophageal carcinoma. Photodiagnosis Photodyn Ther. 2017;20:78-85
55. FDA approves Cotellic as part of combination treatment for advanced melanoma. U.S. Food and Drug Administration; [cited 2017 Aug 4]. Available from: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm471934.htm
56. Bronson J, Black A, Dhar M, Ellsworth B, Robert Merritt J. To Market, To Market—2013. In: Annual Reports in Medicinal Chemistry. 2014;49:437-508
57. El-Hoss J, Kolind M, Jackson MT, Deo N, Mikulec K, McDonald MM. et al. Modulation of endochondral ossification by MEK inhibitors PD0325901 and AZD6244 (Selumetinib). Bone. 2014;59:151-61
58. Ren D-L, Sun A-A, Li Y-J, Chen M, Ge S-C, Hu B. Exogenous melatonin inhibits neutrophil migration through suppression of ERK activation. J Endocrinol. 2015;227:49-60
59. Zhao H, Cui K, Nie F, Wang L, Brandl MB, Jin G. et al. The effect of mTOR inhibition alone or combined with MEK inhibitors on brain metastasis: an in vivo analysis in triple-negative breast cancer models. Breast Cancer Res Treat. 2012;131:425-36
Author contact
![]() Corresponding author: Kensuke Hirasawa, email: kensukeca
Corresponding author: Kensuke Hirasawa, email: kensukeca
Citation styles
APA
Yoshioka, E., Chelakkot, V.S., Licursi, M., Rutihinda, S.G., Som, J., Derwish, L., King, J.J., Pongnopparat, T., Mearow, K., Larijani, M., Dorward, A.M., Hirasawa, K. (2018). Enhancement of Cancer-Specific Protoporphyrin IX Fluorescence by Targeting Oncogenic Ras/MEK Pathway. Theranostics, 8(8), 2134-2146. https://doi.org/10.7150/thno.22641.
ACS
Yoshioka, E.; Chelakkot, V.S.; Licursi, M.; Rutihinda, S.G.; Som, J.; Derwish, L.; King, J.J.; Pongnopparat, T.; Mearow, K.; Larijani, M.; Dorward, A.M.; Hirasawa, K. Enhancement of Cancer-Specific Protoporphyrin IX Fluorescence by Targeting Oncogenic Ras/MEK Pathway. Theranostics 2018, 8 (8), 2134-2146. DOI: 10.7150/thno.22641.
NLM
Yoshioka E, Chelakkot VS, Licursi M, Rutihinda SG, Som J, Derwish L, King JJ, Pongnopparat T, Mearow K, Larijani M, Dorward AM, Hirasawa K. Enhancement of Cancer-Specific Protoporphyrin IX Fluorescence by Targeting Oncogenic Ras/MEK Pathway. Theranostics 2018; 8(8):2134-2146. doi:10.7150/thno.22641. https://www.thno.org/v08p2134.htm
CSE
Yoshioka E, Chelakkot VS, Licursi M, Rutihinda SG, Som J, Derwish L, King JJ, Pongnopparat T, Mearow K, Larijani M, Dorward AM, Hirasawa K. 2018. Enhancement of Cancer-Specific Protoporphyrin IX Fluorescence by Targeting Oncogenic Ras/MEK Pathway. Theranostics. 8(8):2134-2146.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.