Theranostics
13.3
Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(8):2094-2106. doi:10.7150/thno.22069 This issue Cite
Research Paper
Development of a novel albumin-based and maleimidopropionic acid-conjugated peptide with prolonged half-life and increased in vivo anti-tumor efficacy
Junnan Feng1, Chuanke Zhao1 ![]() , Lixin Wang1, Like Qu1, Hua Zhu2, Zhi Yang2, Guo An3, Huifang Tian4, Chengchao Shou1
, Lixin Wang1, Like Qu1, Hua Zhu2, Zhi Yang2, Guo An3, Huifang Tian4, Chengchao Shou1 ![]()
1. Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Department of Biochemistry and Molecular Biology, Peking University Cancer Hospital & Institute, Beijing 100142, China
2. Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Department of Nuclear Medicine, Peking University Cancer Hospital & Institute, Beijing 100142, China
3. Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Department of Laboratory Animal, Peking University Cancer Hospital & Institute, Beijing 100142, China.
4. Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education/Beijing), Central Laboratory, Peking University Cancer Hospital & Institute, Beijing 100142, China.
Received 2017-7-25; Accepted 2018-2-6; Published 2018-3-7
Citation:
Feng J, Zhao C, Wang L, Qu L, Zhu H, Yang Z, An G, Tian H, Shou C. Development of a novel albumin-based and maleimidopropionic acid-conjugated peptide with prolonged half-life and increased in vivo anti-tumor efficacy. Theranostics 2018; 8(8):2094-2106. doi:10.7150/thno.22069. https://www.thno.org/v08p2094.htm
Other stylesAbstract

Angiogenesis plays a critical role in tumor aggressiveness, and a lot of anti-angiogenic agents have been used in clinical therapy. The therapeutic efficacy of peptides are generally restricted by the short in vivo life-time, thus, we were interested in developing a novel albumin-based and maleimidopropionic acid-conjugated peptide to prolong the half-life and improve the anti-tumor effect.
Methods: We developed a peptide F56 with a maleimidopropionic acid (MPA) at the C-terminal (denoted as F56-CM), which allows immediate and irreversible conjugation with serum albumin. Biological property and anti-tumor activity of F56-CM were evaluated in vitro and in vivo.
Results: We showed that F56-CM reduced migration and tube formation of endothelial cells in vitro and inhibited the generation of subintestinal vessels (SIV) in zebrafish embryos in vivo. F56-CM inhibited vascular endothelial growth factor (VEGF) induced phosphorylation of VEGFR1 and activation of the PI3K-AKT axis. Furthermore, F56-CM rapidly conjugated with albumin upon intravenous injection and extended the biological half-life of F56 from 0.4249 h to 6.967 h in rats. Compared with F56, F56-CM exhibited stronger anti-tumor activity on both BGC-823 gastric cancer and HT-29 colon cancer xenografts in nude mice, and the statistical difference was remarkable. More significantly, the efficacy of F56-CM inhibiting lung metastasis of BGC-823 cells was also better than that of F56. The inhibition rates were 62.1% and 78.9% for F56 and F56-CM respectively when administrated every day, and 43.8% and 63.1% when administrated every four days at equal dose.
Conclusions: Taken together, our results demonstrated that F56-CM has considerable potential for cancer therapy.
Keywords: F56, MPA, albumin conjugate, anti-tumor, VEGFR1
Introduction
Angiogenesis plays an important role in the growth, invasion and metastasis of malignant tumor [1, 2]. Among numerous anti-angiogenesis strategies, the VEGF signaling pathway has been considered a predominant target in controlling cancer progression [3]. After the approval by the US Food and Drug Administration (FDA) of Avastin (bevacizumab), a recombinant humanized monoclonal IgG1 antibody against VEGF-A, for the treatment of metastatic colorectal cancer (mCRC) combined with chemotherapy [4], several other anti-angiogenic drugs targeting VEGF/VEGFR have been developed [5, 6]. However, “off-target” effects and/or resistance may reduce the therapeutic efficacy of anti-angiogenesis drugs [7, 8]; thus, devising new anti-angiogenic drugs with selected anti-tumor targets is necessary.
In our previous study, we screened out a VEGFR1-specific peptide F56 (WHSDMEWWYLLG) that could act as an effective competitor against VEGF binding to VEGFR1 [9]. We showed that F56 inhibited blood vessel formation in the chicken chorioallantoic membrane (CAM) system and zebrafish embryos [10]. In addition, F56 inhibited tumor growth in BALB/c nude mice and lung metastasis of B16 melanoma cells in C57BL/6 mice [10]. Consistent with these observations, F56 inhibited VEGF-induced phosphorylation of VEGFR1 in human umbilical vein endothelial cells (HUVEC), as well as phosphorylation of the PI3K-AKT axis [8]. Recently, we also demonstrated that 125I-F56 [11] and 64Cu-DOTA-F56 [12] could specifically target VEGFR1 for noninvasive molecular imaging of gastric tumors. Moreover, some groups designed F56-conjugated nanoparticles for neovasculature imaging [13] or utilized the specificity of F56 to enhance the therapeutic effect of cytotoxic drugs [14, 15]. Thus, F56 offers potent benefits for cancer therapy and diagnosis. Despite these advantages, F56 is still facing a grand challenge in clinical application due to its short in vivo half-life.
Albumin (MW 66 kDa), the most abundant plasma protein in blood, presents at 35-50 mg/mL and has a half-life of 19 days in humans [16]. This long half-life is thought to be mainly due to neonatal Fc receptor (FcRn)-mediated recycling and Megalin/Cubilin complex rescue from renal clearance [17, 18]. In addition, albumin has good biocompatibility and biodegradability, and thus serves as a versatile carrier for drug delivery [19]. Some studies showed that proliferating tumors are inclined to actively accumulate abundant albumin for nutrition supply [20] and this promising ability of albumin to target and accumulate selectively in tumor tissues can be attributed to the enhanced permeability and retention (EPR) effect [21, 22]. Thus far, three main strategies are generally used for binding drugs to albumin. The first strategy is by fusing the gene of peptides or proteins of interest to the albumin gene [23-25]. The second strategy is to utilize the fatty acid binding sites of albumin by linking drugs with long-chain fatty acids [26]. The third involves the covalent and irreversible conjugation of drugs to albumin by biofunctional spacers. For instance, Chen et al. conjugated peptides to a small molecular weight albumin-binding moiety, truncated Evans blue, to improve their intrinsic pharmacokinetics [27]. In addition, albumin-bound paclitaxel nanoparticles (Abraxane®) [28], pegylated liposomal doxorubicin (Doxil®/Caelyx®) [29], and paclitaxel-containing polymeric micelles (Genexol-PM) [30], have been translated into clinical applications.
Multiple species such as rodents, dogs, rabbits, monkeys, and humans all possess a conserved cysteine residue of albumin (Cys34 in human), which is the only free thiol group in this protein [31, 32]. Furthermore, cysteine-34 is located in the hydrophobic crevice of albumin, suggesting that its reactivity is limited [33]. This nucleophilic thiol group has good chemical reactivity for further structural modification, leading to novel bioactive constructs that will adopt an extended half-life [34].
Maleimidopropionic acid (MPA) modification offers an attractive platform for targeted drug delivery, which allows a specific and stable covalent attachment to cysteine 34 of albumin to form a thiosuccinimide bond [35]. Such conjugation reacted with serum albumin can be performed in vivo by administering the compound directly into the human patient. Several successful combinations have confirmed the advantages of combination macromolecular therapy. Xie et al. chemically modified a peptide of HIV fusion inhibitor with MPA to achieve a remarkably extended in vivo half-life [31]. Simon et al. coupled two biorthogonal chemistries (one of them maleimide-cysteine thiol coupling) to assemble stable conjugates with serum albumin for half-life extension [36].
In this study, we developed a peptide F56 with MPA at the C-terminal (F56-CM) and investigated its specific albumin-conjugating characteristics, pharmacokinetic performance, and anti-tumor activity.
Materials and Methods
Reagents and antibodies
Recombinant human VEGF165 (239-VE-010) was obtained from R&D (Minneapolis, MN, USA). Recombinant human serum albumin (HSA) was purchased from Sigma-Aldrich (St Louis, MO, USA). Mouse serum albumin (MSA) was from Equitech-Bio (Kerrville, TX, USA). Cyclophosphamide (CTX) was from Jiangsu Hengrui Medicine (Lianyungang, China). Avastin was from Roche (Basel, Switzerland). The Growth Factor Reduced Matrigel (356230) was obtained from BD Biosciences (Franklin Lakes, NJ, USA). Anti-VEGFR2 (#2479), anti-phospho-VEGFR2 (Y1175) (#2478), anti-PI3K (#4292), anti-Phospho-PI3 Kinase p85 (Y458)/p55 (Y199) (#4228), anti-AKT (#4691), anti-phospho-AKT (S473) (#4060) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-VEGFR1 mAb (TA303515) was from OriGene (Rockville, MD, USA). Anti-phospho-VEGFR1 (Y1213) rabbit antibody (AF4170) was purchased from R&D. Anti-albumin (#46293) was purchased from Santa Cruz (Santa Cruz, CA, USA). Anti-F56/F56-CM monoclonal antibody 9G10 and 9G10-HRP were generated in our lab [10].
Peptide synthesis
The peptides were synthesized by Frontier Biotechnologies Inc. (Nanjing, China). The MPA unit was chemically synthesized to the N-terminal of F56 peptide to generate F56-AM. F56-BM and F56-CM were constructed by insertion of a lysine into the F56 peptide and then conjugation with MPA through the -NH2 group. The linker connecting the MPA unit to the peptides is a synthetic chain termed AEEA for [2-(2-aminoethoxy)-ethoxy] acetic acid. F56 has a MW of 1622.8, and those of F56-AM, F56-BM, F56-CM were 1919.0, 1860.7 and 2046.7, respectively. The purity of the peptides was above 99% as confirmed by high-performance liquid chromatography (HPLC).
Preparation and characterization of peptide-albumin conjugates
Peptides were solubilized in dimethyl sulfoxide solution at a concentration of 10 mM and then diluted to 1 mM into a solution of 25% albumin (in 100 mM NaH2PO4/Na2HPO4, pH 7.4). The samples were incubated at 37 °C for 3 h. The conjugation reactions were found to be very rapid, and there was no maleimide hydrolysis observed. The resulting mixtures of peptides and albumin were at a 1:3 molar ratio. Albumin conjugates were identified using a matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF) system. α-cyano-4- hydroxycinnamic acid (CCA) was used as the matrix for detecting peptide, while 3, 5-Dimethoxy-4- hydroxycinnamic acid (SA) was used as the matrix for detecting protein.
Cell culture and transfection
HUVEC were isolated and cultured as previously described [10]. Human tumor cell lines of HT-29 (colon carcinoma), BGC-823 (gastric carcinoma) and mouse melanoma cell line B16 were cultured in RPMI-1640 with 10% FBS in 5% CO2 at 37 °C. For the small interference RNA (siRNA) transfection experiment, cells were transfected with specific siRNAs or a negative control (NC) using siRNA Mate (GenePharma, Suzhou, China) according to the manufacturer's protocol. The siRNA sequences were as follows: VEGFR1 #1 5'- GCCGGAAGUUGUAUGGU ATT-3', VEGFR1 #2 5'-CGUGGCUACUCGUUAAUUATT-3', VEGFR1 #3 5'-GCUGUACCUACUUCAAAGA TT-3'. The NC siRNA sequence was 5'-UUCUCCG AACGUGUCACGUTT-3'. All siRNAs were synthesized by GenePharma. Efficiency of knock-down was validated by Western blot.
Western blot analysis
Cells were homogenized in lysis buffer containing 50 mM Tris-HCl (pH 7.0), 150 mM NaCl, 2 mM EDTA, 1% SDS, 2 mM Dithiothreitol and 1× protease inhibitor cocktail (Roche). Protein concentration was determined by a BCA Protein Assay Kit (Thermo Fisher Scientific, Tewksbury, MA, USA). Lysates (50 μg per sample) were subjected to Western blot with indicated antibodies and GAPDH was used as loading control. For quantitation, the optical densities of protein bands were analyzed by Image Pro Plus 6.0 software.
Tube formation assay
A total of 50 μL growth factor reduced Matrigel was layered in a 96-well plate and allowed to polymerize by incubating at 37 °C for 30 min. HUVECs were trypsinized and resuspended in serum-free EBM-2 culture medium containing 0.1% BSA. Before exposure to VEGF (10 ng/mL), cells (1.5×104 cells/well) were pre-incubated with peptides for 30 min and then plated onto the Matrigel layer. After 6 h of incubation, the tubes were observed and photographed under a microscope. For quantitation, the images were converted to 8-bit type and processed by HUVEC phase contrast analysis to produce tube area, number of tube branches and total tube length parameters by using the Image J software with the plug-in for angiogenesis analysis. The total tube length was chosen for assessing the tube formation function in this study and the analyses were repeated for at least three independent assays.
Cell migration assay
HUVECs were washed twice with serum-free culture medium, trypsinized mildly, and resuspended in the serum-free EBM-2 culture medium containing 0.1% BSA. Cell suspensions were pre-incubated with different peptides, as indicated, at 37 °C in 5% CO2 for 30 min prior to migration assay. Then, cells (5×104 cells in a final volume of 150 μL) were plated in the upper chamber of transwell inserts with 8 μm pores. The lower chambers contained 700 μL culture medium plus 0.1% BSA with or without peptides and VEGF (20 ng/mL). The cells were allowed to migrate for 8 h at 37 °C. The wells were fixed in methanol and stained with 0.1% crystal violet solution for 30 min at room temperature. Cells that remained in the upper chamber were removed with a cotton wool. The polycarbonate membrane was then removed and sealed on the slide by resin, and the cells that penetrated to the lower side of the membrane were counted in nine randomly selected fields under a microscope.
Zebrafish angiogenesis model
Transgenic zebrafish assessment was performed by Hunter Biotechnology (Hangzhou, China). Peptides were injected into the yolk sac of 2 dpf zebrafish by the microinjection method. There were 30 zebrafish treated in each experimental group. After being incubated for another 24 h, 10 zebrafish of each group were randomly selected, and fluorescence of the subintestinal vessels (SIVs) was detected and photographed using a multi-purpose zoom microscope system (AZ 100, Nikon). Image analysis was performed by calculating the SIV area using the Nikon NIS-Elements D 3.10 Advanced image processing software.
Immunofluorescence assay
HUVECs were seeded on glass coverslips. The next day, cells were treated with peptides for 30 min. Then, cells were fixed with 4% paraformaldehyde for 20 min and permeabilized with 0.1% Triton X-100 in PBS for 5 min. After blocking with 5% BSA/PBS for 1 h, cells were incubated with anti-VEGFR1 or 9G10 antibody at 4 °C overnight and subsequently with FITC- or TRITC-labeled secondary antibody at room temperature for 45 min. Nuclei were counterstained with DAPI (5 µg/mL) and images were captured by the Leica TCS SP5 laser confocal microscope.
Ethics
The animal study was approved by the Biomedical Ethical Committee of Peking University Cancer Hospital & Institute (Permit Number: Animal-2012-01) and performed along established institutional animal welfare guidelines concordant with the US guidelines (NIH Publication #85-23, revised in 1985).
Pharmacokinetics of peptides in vivo
Male BALB/c mice (Vital River Laboratories, Beijing, China) were administered F56-CM (5 mg/mL in PBS, injection volume, 10 mL/kg) through the angular vein. Blood samples were taken at the indicated time points, centrifuged (800 rcf for 10 min at 4 °C), and the plasma samples were aliquoted and stored frozen. The diluted (1: 50) plasma samples were subjected to Western blot.
Because albumin has a half-life of 1.9 days in rats and 1.0 day in mice [37], we selected old female Sprague-Dawley rats (7-8 weeks, weights ranging 322-347 g) from Vital River Laboratories for the single-dose pharmacokinetic study. 64Cu-DOTA-F56 or 64Cu-DOTA-F56-CM was administered to rats (7.4 mBq, i.v.). Each treatment group consisted of 2 rats. Blood samples (20 µL) were collected before injection and up to 96 h after dosing and determined by gamma counting. These results were then used to calculate the half-life of peptides in blood. The blood pharmacokinetic parameters of 64Cu-DOTA-F56 and 64Cu-DOTA-F56-CM were analyzed using a two-compartmental model and Graph Pad Prism 5.01, referring to relevant literature [38, 39].
Xenograft and pneumonic metastasis studies
In the tumor growth assay, female BALB/c nude mice (Vital River Laboratories) aged 4-5 weeks were used and housed under specific pathogen-free conditions. HT-29 or BGC-823 cells were injected s.c. into the right flank of each mouse. When tumor sizes reached 100 mm3, mice were randomly divided into 7 groups and administrated different drugs: (A) PBS (control), (B) CTX (80 mg/kg/7d, as the positive group), (C) Avastin (5 mg/kg/7d), (D) F56 (5 mg/kg/d, as Q1D), (E) F56 (5 mg/kg/4d, as Q4D), (F) F56-CM (5 mg/kg/d, as Q1D), (G) F56-CM (5 mg/kg/4d, as Q4D). The body weights and tumor volumes of the mice were monitored over time. After drug withdrawal, mice were observed for a week and then sacrificed. The weight and volume of tumors were then analyzed.
For the tumor metastasis assay, B16 melanoma cells were inoculated into C57BL/6 mice (Vital River Laboratories) intravenously and drugs were delivered by vein injection every day in the following treatment groups: (A) PBS (control), (B) Avastin (5 mg/kg), (C) F56 (1 mg/kg), (D) F56 (5 mg/kg), (E) F56-CM (1 mg/kg), (F) F56-CM (5 mg/kg). After continuous administration for 14 days, mice were sacrificed, lungs were dissected and the number of metastatic foci per lung was counted carefully by the naked eye. Alternatively, BGC-823 cells (1.0 × 106 cells in 0.2 mL medium) were injected into the tail vein of BALB/c NOD SCID mice (female, Vital River Laboratories) and drugs were delivered as indicated. The mice were sacrificed at the 21st experimental day. The lung organs were fixed with Bouin's solution and the numbers of peripheral tumor nodules were calculated. The metastatic nodules were confirmed by H&E staining.
Statistical analysis
Values represent the mean ± SD of at least two independent experiments. P values were calculated by two-tailed Student's t-test or one-way Analysis of variance (ANOVA) using Graph Pad Prism 5.01. P values less than 0.05 were considered statistically significant.
Results
Synthesis and characteristics of MPA-modified F56 peptides
Three F56-derived peptides, F56-AM, F56-BM, and F56-CM, were synthesized with a single MPA at different residual positions (Figure 1A). After co-incubating with HSA in vitro, the products were subjected to mass spectrometric analysis to investigate their conjugating activities with HSA. The insets of Figure 1B show that the single peak of HSA (66412 Da) is close to its expected mass (66437 Da). Besides the same peak as HSA (66412 Da), F56-AM-HSA has another peak of 68233 Da, which is 1821 Da larger than that of HSA due to the conjugation of F56-AM. Similarly, both F56-BM-HSA and F56-CM-HSA show another ~1800 Da larger mass than that of HSA, confirming that the reaction ratio was 1:1. A negative control of HSA incubated with F56 peptide did not show a larger peak besides the position of HSA. Peptides were then subjected to CAM assay (Figure 1C) and tube formation assay (Figure 1D) to determine the anti-angiogenic activities, while the commercialized anti-angiogenic drug Avastin was used as a positive control. It was found that F56-CM, compared to F56-AM and F56-BM, showed an enhanced efficacy to counteract VEGF-induced angiogenesis (Figure 1C-D). Based on these results, F56-CM was selected for further studies.
In order to confirm the precise reaction ratio, we set up a series of ratios of F56-CM: HSA from 3:1 to 1:5 (mol/mol). Figure S1A displays the MALDI chromatographic curves of F56-CM, HSA and F56-CM-HSA at different ratios. The prominent peaks of F56-CM and HSA alone were 2046 Da and 66441 Da, respectively. When F56-CM was superfluous (F56-CM: HSA, 3:1), the conjugate peak occurred at 68510 Da and 70611 Da, while the F56-CM peak still existed. When the reaction was at an equal molar ratio (F56-CM: HSA, 1:1), the conjugate peak was unambiguous and single at 68324 Da, but the F56-CM peak had disappeared, indicating that the conjugate was formed completely. We also attempted to increase the ratio to 1:3 and 1:5. However, the peaks of HSA (mass: 66380) and F56-CM-HSA (mass: 68468) were unaltered, indicating that the grafting ratio of F56-CM to HSA was not increased. We concluded that the optimal covalent attachment of F56-CM to HSA occurs at an equal molar ratio, as suggested by another study [36]. Immunoblotting analysis showed that the apparent molecular weight of HSA was ~66 kDa, as detected with an albumin-specific antibody (Figure S1B, lower panel). With the F56-specific antibody 9G10, a band was found at ~66 kDa of F6-CM after incubating with HSA (Figure S1B, upper panel), suggesting the existence of a conjugate of F56-CM and HSA. Meanwhile, no band was found of F56 plus HSA (Figure S1B, upper panel), which was consistent with the mass spectrometric analysis (Figure 1B), suggesting the absence of conjugation.
F56-CM associates with HUVEC cells in a VEGFR1-dependent pattern and blocks phosphorylation of VEGFR1, PI3K and Akt in HUVEC cells
Consistent with F56's adhesion with endothelial cells [10], F56-CM also interacted with HUVEC cells, as examined by cell ELISA assay (Figure 2A). The HUVEC-binding abilities of F56 and F56-CM were similar, while that of F56-CM-HSA was weaker (Figure 2A), but still significantly increased compared with the control group. By immunofluorescence staining, F56-CM was shown to co-localize with VEGFR1 in HUVEC cells (Figure 2B). When VEGFR1 was ablated by transfection with specific siRNA (Figure 2C), F56-CM's association with HUVEC cells was decreased (Figure 2B), indicating that VEGFR1 facilitates the adhesion of F56-CM to HUVEC cells. To clarify the signaling events associated with binding of F56-CM and F56-CM-HSA with HUVEC cells, we investigated the effect of peptides on the phosphorylation of VEGFR1, VEGFR2, PI3K and AKT. The phosphorylation of VEGFR1 induced by VEGF was markedly down-regulated by F56, F56-CM, and F56-CM-HSA treatment, and the phosphorylation of PI3K and AKT were also reduced, but VEGF-induced phosphorylation of VEGFR2 was unchanged (Figure 2D-E). These results further support the specificity of F56-CM in counteracting VEGFR1 signaling, and the covalent conjugation with HSA did not affect its bioactivities.
F56-CM inhibits tube formation and migration of HUVECs and the angiogenesis of zebrafish embryo SIVs
To examine the biological effects of F56-CM, we firstly performed a proliferation assay. VEGF treatment, as expected, enhanced the proliferation of HUVEC cells, but this effect was not affected by F56 or F56-CM (Figure 3A). Next, we examined the effect of peptides on the tube formation ability of HUVEC cells on Matrigel. F56, F56-CM, and F56-CM-HSA inhibited the tube formation of HUVEC cells induced by VEGF, but HSA alone had no inhibitory effect (Figure 3B). In the migration assay, to minimize the influence of VEGF-promoted cell growth, the migrating time was set as 8 h after seeding cells in the top chamber and the drugs had little influence on cell viability at indicated concentrations. Compared with the vehicle control, VEGF-promoted HUVEC migration was inhibited by F56, F56-CM, and F56-CM-HSA, and differences in the inhibition rates of all three peptides had no statistical significance (Figure 3C). These data demonstrated that F56-CM retained the anti-angiogenesis ability in vitro and this ability was not compromised after conjugating to albumin. We further validated the ability of F56-CM to conjugate with MSA (Figure S2A-B) and its ability to inhibit tube formation (Figure S2C) and migration (Figure S2D) of HUVEC after conjugating with MSA.
Figure 1
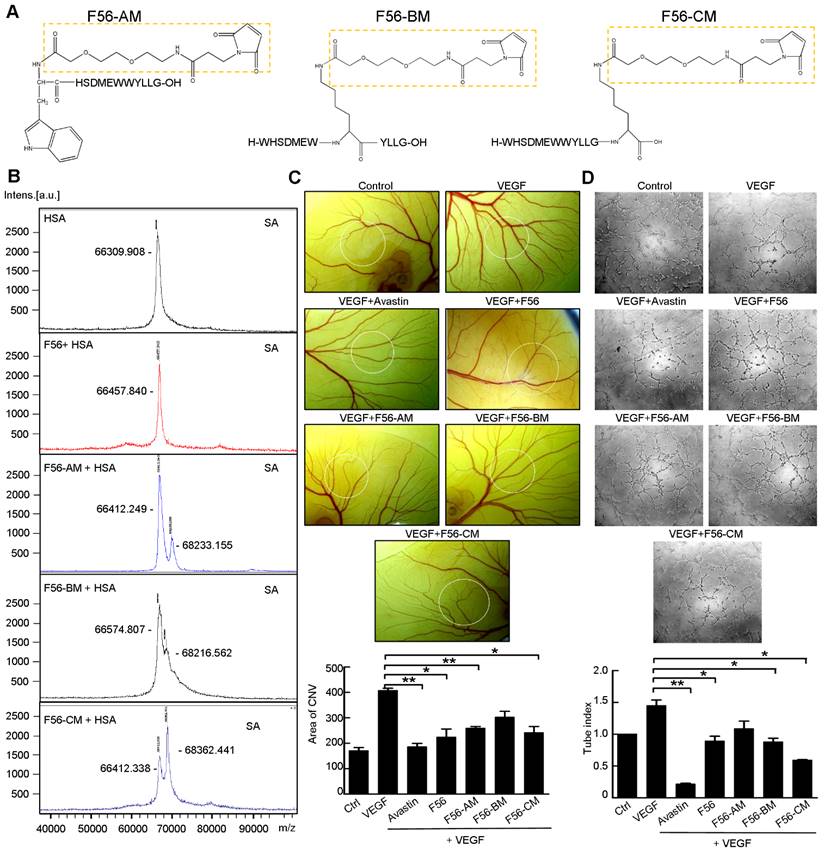
Identification of peptides reacted with HSA and their anti-angiogenic efficacy in vitro. (A) Structures of F56-AM, F56-BM, F56-CM, prepared by solid-phase synthesis. The dotted boxes represent the AEEA-MPA unit. (B) MALDI mass spectra of HSA or peptides incubated with HSA. The measured molecular weights are marked next to the peak. (C) CAM assay. After 48 h of incubation, vessel formation was observed under a stereomicroscope (upper panel). The formation of vessels was quantified by comparing the vascular area to the CAM area (lower panel). n = 5 per group. (D) Tube formation assay. HUVECs were pre-incubated with peptides for 30 min before exposure to VEG and then seeded on Matrigel. After 6 h of incubation, tube-like structures were photographed (upper panel) and quantified (lower panel). Data indicate mean ± SD of three independent experiments. *P < 0.05; **P < 0.01.
Figure 2
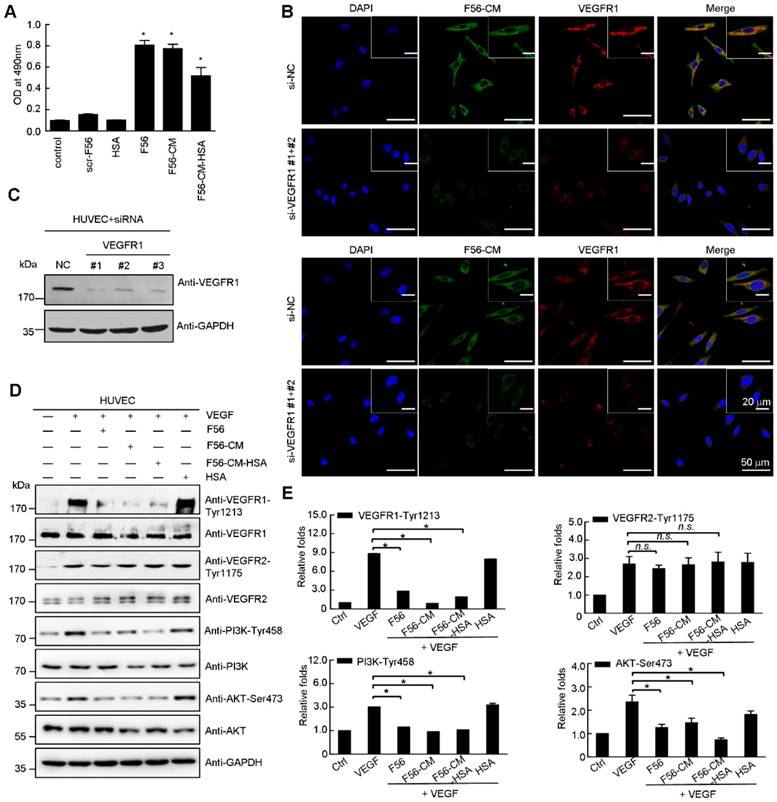
F56-CM associates with HUVEC cells in a VEGFR1-dependent fashion and reduces phosphorylation of VEGFR1, PI3K and AKT in HUVEC cells. (A) Cell ELISA assay of binding of F56 and F56-CM to HUVEC. The vertical axis represents the absorbance at 490 nm. (B) Immunofluorescence analysis of binding of F56 and F56-CM to HUVEC cells. HUVEC cells seeded on coverslips were transfected with indicated siRNAs for 48 h, treated with F56 or F56-CM (green) for 30 min, followed by immunostainings of VEGFR1 (red) and F56 (green). DNA was counterstained with DAPI (blue). Co-localization is shown by merged signals (yellow). (C) Knockdown efficiencies of VEGFR1. HUVEC cells were transfected with 50 nM siRNAs against VEGFR1 for 48 h. Lysates were analyzed by Western blot with indicated antibodies. (D) HUVEC cells were starved overnight. After treatment with F56, F56-CM, F56-CM-HSA or HSA at a concentration of 50 μM for 30 min,10 ng/mL VEGF-A was added to the medium for 10 min stimulation, and the expressions of indicated proteins were assayed by Western blot. (E) Quantification of the relative gray value of (D), which was calculated by normalizing to that of the loading control, and the value of untreated sample was set as 100%. Data indicate mean ± SD of three independent experiments. *P < 0.05; n.s., no significance.
To assess the anti-angiogenic ability of peptides in vivo, we utilized the endothelial cell fluorescently labeled transgenic zebrafish model. The data showed that Avastin, as a positive control, inhibited the angiogenesis of SIV plexus in zebrafish embryos. F56 and F56-CM reduced the angiogenesis of zebrafish embryos in a dose-dependent way, and the inhibitory effects were comparable to Avastin at a dosage of 10 pmol. In addition, at a dosage of 3 pmol, the inhibition rate of F56-CM was better than that of 3 pmol F56 and had statistical significance (Figure 3D), indicating that F56-CM maintained its endogenous anti-angiogenic activity and exhibited higher activity than F56 at a concentration of 3 pmol.
Figure 3
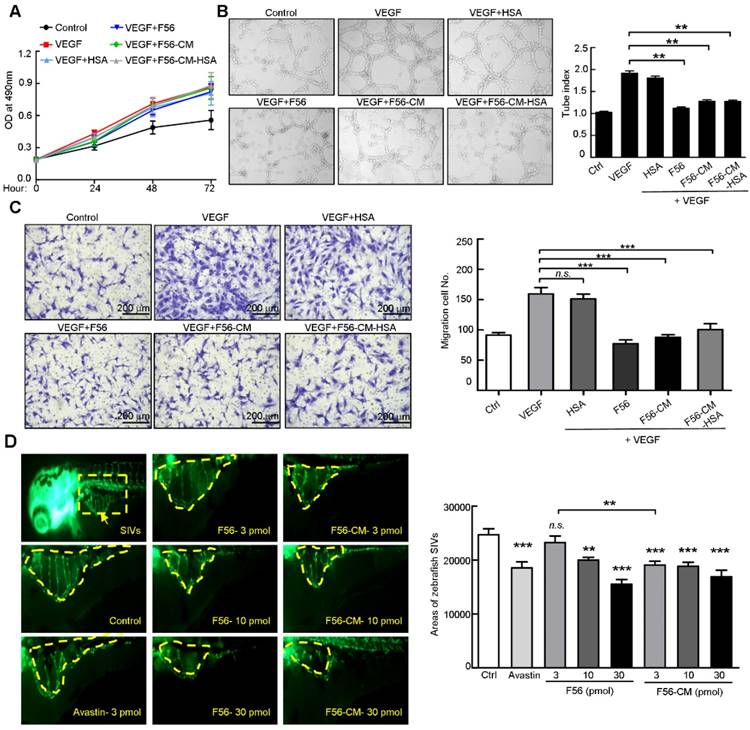
F56-CM inhibits tube formation and migration of HUVEC and the angiogenesis of zebrafish embryo SIVs. (A) Proliferation assay. HUVEC cells were treated with VEGF (10 ng/mL) alone or in combination with HSA or peptides; the proliferation was measured by MTT assay. (B) Tube formation assay. HUVEC cells were pre-incubated with peptides for 30 min before exposure to VEGF (10 ng/mL) and then seeded on Matrigel. After incubation for 6 h, formation of tube-like structures was photographed from randomly chosen fields (left) and the total pipe length of the tube-like structure was quantified (right). (C) Migration assay. HUVEC cells were starved for 24 h and then seeded into transwell inserts in the presence of indicated doses of peptides for 8 h. Cells reaching the bottom side of the transwell membrane were stained and representative images are displayed (left; scale bar, 200 μm) and the numbers of migrated HUVEC cells were quantified (right). (D) Inhibitory effects of F56 and F56-CM on angiogenesis of the zebrafish embryo. Embryos with suitable developmental stages were selected, then peptides were injected into the yolk sac of 2 dpf (2 days after fertilization) zebrafish. Avastin was used as the positive control. When incubated to 3 dpf, fluorescence of the SIVs (subintestinal vessels) were detected and photographed (left) and the SIVs areas were quantified (right). Data represent mean ± SD of three to four independent experiments. **P < 0.01; ***P < 0.001; n.s., no significance.
Pharmacokinetic profile and long-circulating feature of F56-CM in vivo
To verify the in vivo conjugating effect of F56-CM to albumin, BALB/c mice were administered F56-CM and blood samples were taken at the indicated time points. Immunoblotting analysis showed that the albumin band was clearly visible at all of the time points at ~66 kDa, as detected with an albumin-specific antibody (Figure 4A, lower panel). With the F56-specific antibody 9G10, no band at ~66 kDa was found in the pre-injection sample. However, the bands became visible at 5 min and persisted in all samples taken beyond this time point. In fact, the intensity was decreased at 12 h after injection (Figure 4A, upper panel). This is a strong evidence showing that F56-CM was rapidly attached to albumin in plasma after administration and the maintenance time was significantly prolonged. A very faint band was observed above F56-CM-albumin conjugate (Figure 4A, upper panel), corresponding to albumin dimer, which is often observed when analyzing albumin with gel electrophoresis. No other bands were found in the gel, demonstrating that other kinds of binding forms were negligible compared with the covalent binding of F56-CM with albumin.
The radio-labeled conjugates, 64Cu-DOTA-F56/F56-CM, were synthesized by the reaction of DOTA-F56/F56-CM peptide with 64Cu2+ at 60 °C for 30 min incubation (Figure 4B). There were more than 85% intact 64Cu-DOTA-F56 and 64Cu-DOTA-F56-CM after 24 h of incubation in 3 conditions: 0.01 M PBS at pH 7.4, 0.1 M NaAc at pH 5.5, and 5% HSA (Figure S3A), showing that 64Cu-DOTA-F56 and 64Cu-DOTA-F56-CM have excellent stabilities. HUVEC, BGC-823, and HT-29 cells showed good uptake stabilities of 64Cu-DOTA-F56 and 64Cu-DOTA-F56-CM at 10, 30, 60, and 120 min (Figure S3B). The uptake of both peptides could be dramatically reduced by co-incubating with excess unlabeled F56 (Figure S3B).
Positron emission tomography (PET) imaging of nude mice bearing BGC-823 and HT-29 xenografts was performed on a micro-PET scanner. It was shown that accumulation of 64Cu-DOTA-F56-CM in tumor sites was much stronger than that of 64Cu-DOTA-F56 at 4 h after injection (Figure 4C-D). This result was further confirmed by the biodistribution assay. Liver and kidney of mice bearing BGC-823 (Figure 4E) and HT-29 (Figure 4F) tumor had relatively high uptake of both peptides. Importantly, 64Cu-DOTA-F56-CM (BGC-823: 1.27 %IA/g, HT-29: 4.69 %IA/g) accumulated more in tumor than 64Cu-DOTA-F56 (BGC-823: 0.55 %IA/g, HT-29: 3.55 % IA/g) did at 4 h after injection. Meanwhile, the radioactivity of 64Cu-DOTA-F56 in the blood (BGC-823: 2.55 %, HT-29: 2.11 %IA/g) was also lower than that of 64Cu-DOTA-F56-CM (BGC-823: 6.50 %IA/g, HT-29: 3.53 %IA/g) (Figure 4E-F).
The pharmacokinetic profile of 64Cu-DOTA-F56 and 64Cu-DOTA-F56-CM were compared by intravenous administration in SD rats at an equivalent radioactivity of 7.4 mBq. The blood concentration vs. time curves were investigated at indicated time points. As shown in Figure 4G, 64Cu-DOTA-F56 was quickly removed from the circulating system after administration. In contrast, 64Cu-DOTA-F56-CM exhibited a delayed blood clearance. The pharmacokinetic parameters determined through the statistical moments analysis showed that the half-life of 64Cu-DOTA-F56-CM was 6.967 h, while that of 64Cu-DOTA-F56 was 0.4249 h, indicating that a majority of the administered 64Cu-DOTA-F56-CM was stored in drug-albumin form to avoid being quickly degraded. Hence, the bond linking MPA to albumin is highly stable in vivo, and F56-CM peptide is rendered more stable against rapid renal clearance and/or peptidase degradation. These results indicate that F56-CM exhibits an improved pharmacokinetic profile compared with F56 in vivo.
F56-CM inhibits tumor growth and metastasis in vivo
Tumor-bearing mice models were employed to evaluate whether the improvement in pharmacokinetic behavior of F56-CM could confer enhanced therapeutic efficacy and allow less frequent dosing. CTX and Avastin were used as positive controls. We evaluated the activities of peptides administrated every day (Q1D) and every four days (Q4D) in the xenograft models of BGC-823 and HT-29 cells in nude mice. The inhibition rates on BGC-823 xenografts were 79.54% (Avastin), 65.9% (CTX), 50% (F56-Q1D), 38.63% (F56-Q4D), 68.18% (F56-CM-Q1D) and 56.81% (F56-CM-Q4D), respectively (Figure 5B). The difference in inhibition rates between F56 (Q1D) and F56-CM (Q1D) has statistical significance (p = 0.0325). And, the inhibition rates on HT-29 xenograft were 77.91% (Avastin), 78.02% (CTX), 75.58% (F56-Q1D), 43.02% (F56-Q4D), 76.98% (F56-CM-Q1D) and 67.44% (F56-CM-Q4D), respectively (Figure 5C). The difference in inhibition rate between F56 (Q4D) and F56-CM (Q4D) also has statistical significance (p = 0.0256). Contrary to F56, there was excellent conservation of anti-tumor activity for F56-CM with infrequent dosing and with initiation of dosing delayed until 4 days after injection. Additionally, no significant changes in body weight were observed in all the mice (data not shown). These results demonstrated that F56-CM has potent anti-tumor growth ability and possesses a better therapeutic index than F56.
To investigate the effect of F56-CM on tumor metastasis, we first injected mouse B16 melanoma cells in C57 mice via caudal veins, and then measured the metastatic foci on the lungs (Figure 6A). At a dose of 1 or 5 mg/kg, both F56 and F56-CM significantly inhibited lung metastasis of B16 cells (Figure 6B). Because injection of HT-29 cells failed to establish a pulmonary metastasis model (data not shown), we then chose BGC-823 cells to assess the ability of F56-CM to inhibit tumor metastasis. After being fixed and dyed, the white metastatic nodules on lungs were observed clearly (Figure 6C). The lung metastatic nodules were confirmed by H&E staining (Figure 6D). The inhibition rates of drugs were 62.7% (Avastin), 82.8% (CTX), 62.1% (F56-Q1D), 43.8% (F56-Q4D), 78.9% (F56-CM-Q1D) and 63.1% (F56-CM-Q4D), respectively (Figure 6E). The difference in inhibition rates between F56 (Q1D) and F56-CM (Q1D) has statistical significance (p = 0.0426), and also the difference in inhibition rates between F56 (Q4D) and F56-CM (Q4D) has statistical significance (p = 0.0328).
Figure 4
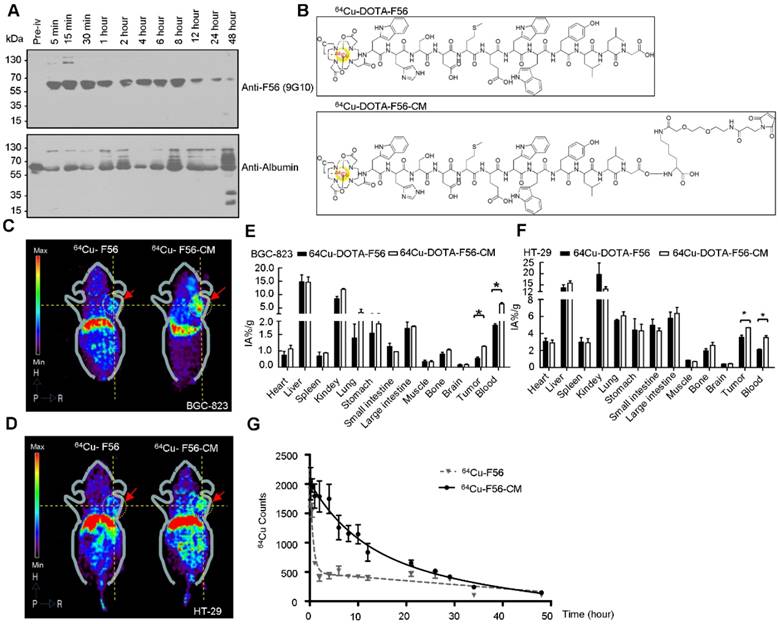
Pharmacokinetic profile, micro-PET imaging and biodistribution analysis in vivo. (A) Identification of F56-CM conjugated to albumin, before and after intravenous injection of 5 mg/kg F56-CM in BALB/c mice. Blood samples were taken at the indicated time points, diluted 1:50, and subjected to Western blot using the F56-specific antibody and the albumin-specific antibody. (B) The structures of radiolabeled 64Cu-DOTA-F56 and 64Cu-DOTA-F56-CM. (C-D) Micro-PET imaging of nu/nu mice bearing BCG-823 (C) and HT-29 (D) tumor cells. Images were acquired at 4 h after injection of 64Cu-DOTA-F56 and 64Cu-DOTA-F56-CM. Dotted circles indicate positions of tumors. (E-F) Biodistribution at 8 h after i.v. injection of 64Cu-DOTA-F56 and 64Cu-DOTA-F56-CM in nude mice xenografted with BGC-823 and HT-29 cells. Data were obtained using the radioactivity count method, plotted as % injected activity per gram of tissue (%IA/g). (G) Female Sprague-Dawley rats were given a single intravenous injection of 64Cu-DOTA-F56 or 64Cu-DOTA-F56-CM. Blood samples (20 µL) were collected before injection and up to 96 h after dosing and determined by gamma counting. Data represent mean ± SD of three independent experiments. *P < 0.05.
Discussion
The efficacies of therapeutic proteins and peptides are generally limited by their rapid clearance from the body [40]. The in vivo half-life of protein-based drugs is often a few hours and for peptides it is generally less than two hours, mostly due to renal filtration or proteolysis [41]. In this study, we engineered a long-lasting peptide named F56-CM with the strategy of MPA modification. Our results demonstrated that F56-CM rapidly reacted with albumin upon injection and prolonged the biological half-life of F56 from 0.4249 h to 6.967 h in rats. Tube formation and transgenic zebrafish assays confirmed that F56-CM has an anti-angiogenic effect similar to F56 in vitro and in vivo. Additionally, F56-CM exhibited better anti-tumor growth and anti-metastasis activity than F56 at an equal dose.
Some other groups have sought to achieve high specificity and affinity with F56 by engineering F56 conjugates [42, 43]. Attempts to target tumors with F56-bearing liposomes have failed, probably due to the hydrophobicity of F56, which promoted aggregation and caused leakage of free DOX from the liposomal carrying F56 [42]. Brunel et al. modified F56 with four arginines to improve its solubility and then conjugated it to viral nano-particles (VNP); the product was able to target VEGFR-1 on endothelial cell lines and VEGFR-1-expressing tumor xenografts in mice [43]. The peptide F56-CM we designed in this study is superior for several reasons: the high selectivity and affinity towards thiol groups, the lack of byproducts, and the reaction can be done in aqueous solution without catalysts or heating [35].
Figure 5
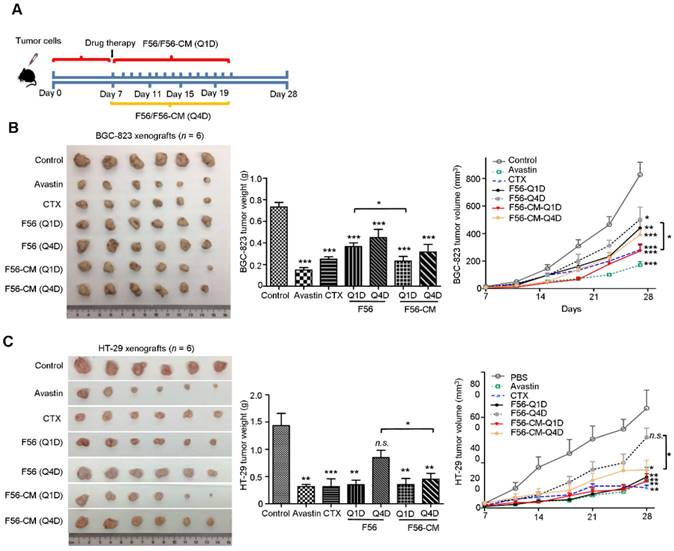
F56-CM inhibits tumor growth in xenograft mice models. (A) Schema for subcutaneous implantation of BGC-823 or HT-29 cells into the nude mice on day 0 and therapeutic injection of indicated drugs on day 7. The mice were sacrificed on day 28. Tumor nodules, tumor weights and tumor growth curves of BALB/c nu/nu mice bearing BGC-823 gastric cancer cells (B) and HT-29 colon cancer cells (C) are shown. Data represent mean ± SD (n = 6). *P < 0.05; **P < 0.01; *** P < 0.001; n.s., no significance.
The results reported here highlighted the potential utility of F56-CM in several aspects. First, the reaction between F56-CM and albumin is rapid and stable. Although F56-CM's binding activity with HUVEC, its effects on inhibiting VEGFR1 phosphorylation and suppressing tube formation and migration of HUVEC were slightly weaker than those of F56, results from the in vivo assays showed that F56-CM has better tumor-targeting, a longer half-life, and stronger anti-tumor and anti-metastasis activities than F56. Therefore, the disadvantage of restrained binding of F56-CM to its target, VEGFR1 receptor on the endothelial cells, could be markedly overridden by prolonged retention in the body. Moreover, our data showed that F56-CM extended the half-life of F56 from 0.4249 h to 6.967 h in rats. However, the half-life of albumin in different species is generally based upon body weight. For example, it has been estimated as 1, 1.9, 5.55 and 19 days in mice, rats, rabbits and humans, respectively [37]. Drug clearance in humans is known to be much slower than in rodents [44] and, thus, the anti-tumor duration of F56-CM may be substantially longer in humans than the ~6.9 h in rats calculated in the present study. However, the effects need to be further validated in controlled clinical trials. Second, when injected into tumor-bearing mice, the accumulation of F56-CM in the tumor was interestingly superior to that of F56. Some studies have reported that proliferating tumors are inclined to actively accumulate abundant albumin for nutrition supply [20]. Hence, it is also possible that albumin serves in an active participatory role rather than merely acting as a protein carrier. Third, it should also be mentioned that F56-CM is obviously a foreign entity in humans. However, HSA, as an endogenous protein, diminishes immunogenicity and thereby significantly reduces the toxic side effects of anticancer agents [45]. In addition, compared to F56, F56-CM improved tumor-specific biodistribution, which to a certain degree could decrease off-target effects. Furthermore, the reduced injection frequency of drugs could decrease the total accumulated dose, which is related to intractable drug resistance [46]. Finally, unlike demanding genetic engineering, MPA modification is flexible to synthesize [47], and such a drug candidate could significantly reduce the cost of treatment and improve treatment compliance and patients' quality of life.
Figure 6
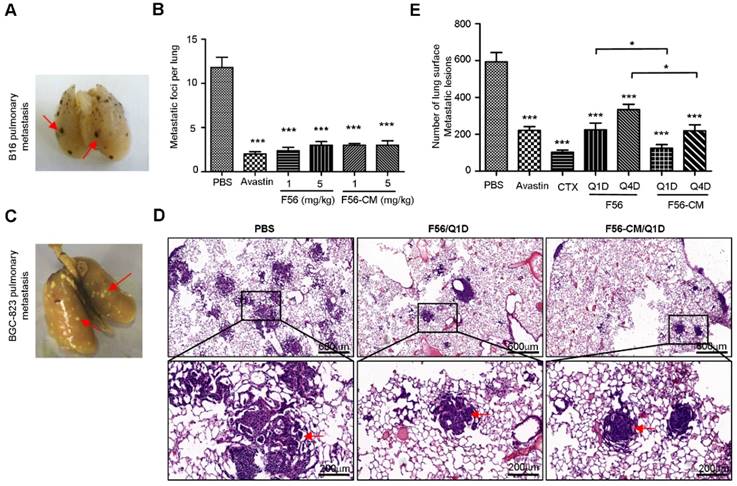
F56-CM inhibits tumor pulmonary metastasis in a xenograft mouse model. (A) C57BL/6 mice were injected with 4 × 106 B16 melanoma cells via the tail vein. After continuous administration of drugs for 14 days, lungs were dissected and the number of metastatic foci per lung was counted carefully by the naked eye. A representative image is shown and the red arrows indicate B16 pulmonary metastatic nodules. (B) Quantification of the metastatic foci per lung from (A). (C) Representative image of a lung of a BALB/c NOD SCID mouse injected with BGC-823 cells. The red arrows indicate pulmonary metastatic nodules. (D) Images of representative metastatic nodules in the lungs of SCID mice (H&E staining). (E) Quantification of peripheral BGC-823 tumor nodules on the lungs. Data represent mean ± SD (n = 6). *P < 0.05; **P < 0.01.
An overall scheme of this study is depicted in the Graphical Abstract. In summary, our results indicate that F56-CM exhibited a more robust potency to enable anti-angiogenic ability and improved antitumor activity compared to F56. This enhanced in vivo performance of F56-CM is mostly due to its albumin-conjugating property. Based on these results, we believe that F56-CM has considerable pharmaceutical potential for practical application in cancer therapy.
Abbreviations
VEGF: vascular endothelial growth factor; SIV: subintestinal vessels; MPA: maleimidopropionic acid; FDA: Food and Drug Administration; mCRC: metastatic colorectal cancer; CAM: chicken chorioallantoic membrane; HUVEC: human umbilical vein endothelial cells; FcRn: neonatal Fc receptor; EPR: enhanced permeability and retention; siRNA: small interference RNA; NC: negative control; VNP: viral nano-particles; PET: positron emission tomography.
Acknowledgements
This work was supported by National Basic Research Program of China (2015CB553906).
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249-57
2. Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17:1359-70
3. Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8:210-21
4. Hurwitz H, Fehrenbacher L, Novotny W. et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335-42
5. Choueiri TK, Schutz FA, Je Y. et al. Risk of arterial thromboembolic events with sunitinib and sorafenib: a systematic review and meta-analysis of clinical trials. J Clin Oncol. 2010;28:2280-5
6. Pan D, Schmieder AH, Wang K. et al. Anti-angiogenesis therapy in the Vx2 rabbit cancer model with a lipase-cleavable Sn2 taxane phospholipid prodrug using α(v)β3-targeted theranostic nanoparticles. Theranostics. 2014;4:565-78
7. Hall AP, Mitchard T, Rolf MG. et al. Femoral Head Growth Plate Dysplasia and Fracture in Juvenile Rabbits Induced by Off-target Antiangiogenic Treatment. Toxicol Pathol. 2016;44:866-73
8. Bottsford-Miller JN, Coleman RL, Sood AK. Resistance and escape from antiangiogenesis therapy: clinical implications and future strategies. J Clin Oncol. 2012;30:4026-34
9. An P, Lei H, Zhang J. et al. Suppression of tumor growth and metastasis by a VEGFR-1 antagonizing peptide identified from a phage display library. Int J Cancer. 2004;111:165-73
10. Zhou Z, Zhao C, Wang L. et al. A VEGFR1 antagonistic peptide inhibits tumor growth and metastasis through VEGFR1-PI3K-AKT signaling pathway inhibition. Am J Cancer Res. 2015;5:3149-61
11. Zhu H, Zhao C, Liu F. et al. 125I-F56 Peptide as Radioanalysis Agent Targeting VEGFR1 in Mice Xenografted with Human Gastric Tumor. ACS Med Chem Lett. 2017;8:266-9
12. Zhu H, Zhao C, Liu F. et al. Radiolabeling and evaluation of 64Cu-DOTA-F56 peptide targeting vascular endothelial growth factor receptor 1 in the molecular imaging of gastric cancer. Am J Cancer Res. 2015;5:3301-10
13. Xu QH, Shi JY, Zhang J. et al. Comparison of tumor neovasculature-targeted paramagnetic nanoliposomes for MRI in mice xenograft models. Clin Transl Oncol. 2014;16:395-401
14. Shamay Y, Golan M, Tyomkin D. et al. Assessing the therapeutic efficacy of VEGFR-1-targeted polymer drug conjugates in mouse tumor models. J Control Release. 2016;229:192-9
15. Luan X, Guan YY, Lovell JF. et al. Tumor priming using metronomic chemotherapy with neovasculature-targeted, nanoparticulate paclitaxel. Biomaterials. 2016;95:60-73
16. Merlot AM, Kalinowski DS, Richardson DR. Unraveling the mysteries of serum albumin-more than just a serum protein. Front Physiol. 2014;5:299
17. Larsen MT, Kuhlmann M, Hvam ML. et al. Albumin-based drug delivery: harnessing nature to cure disease. Mol Cell Ther. 2016;4:3
18. Cui S, Verroust PJ, Moestrup SK. et al. Megalin/gp330 mediates uptake of albumin in renal proximal tubule. Am J Physiol. 1996;27:900-7
19. Liu Z, Chen X. Simple bioconjugate chemistry serves great clinical advances: albumin as a versatile platform for diagnosis and precision therapy. Chem Soc Rev. 2016;45:1432-56
20. Fanali G, di Masi A, Trezza V. et al. Human serum albumin: from bench to bedside. Mol Aspects Med. 2012;33:209-90
21. Kim B, Lee C, Lee ES. et al. Paclitaxel and curcumin co-bound albumin nanoparticles having antitumor potential to pancreatic cancer. Asian J Pharm Sci. 2016;11:708-14
22. Tang B, Fang G, Gao Y. et al. Lipid-albumin nanoassemblies co-loaded with borneol and paclitaxel for intracellular drug delivery to C6 glioma cells with P-gp inhibition and its tumor targeting. Asian J Pharm Sci. 2015;10:363-71
23. Herzog E, Harris S, Henson C. et al. Biodistribution of the recombinant fusion protein linking coagulation factor IX with albumin (rIX-FP) in rats. Thromb Res. 2014;133:900-7
24. Khunti K, Damci T, Meneghini L. et al. Study of Once Daily Levemir (SOLVE™): insights into the timing of insulin initiation in people with poorly controlled type 2 diabetes in routine clinical practice. Diabetes Obes Metab. 2012;14:654-61
25. Buysschaert M, D'Hooge D, Preumont V. et al. ROOTS: A multicenter study in Belgium to evaluate the effectiveness and safety of liraglutide (Victoza®) in type 2 diabetic patients. Diabetes Metab Syndr. 2015;9:139-42
26. Krenzel ES, Chen Z, Hamilton JA. Correspondence of fatty acid and drug binding sites on human serum albumin: a two-dimensional nuclear magnetic resonance study. Biochemistry. 2013;52:1559-67
27. Chen H, Wang G, Lang L. et al. Chemical Conjugation of Evans Blue Derivative: A Strategy to Develop Long-Acting Therapeutics through Albumin Binding. Theranostics. 2016;6:243-53
28. Ma P, Mumper RJ. Paclitaxel Nano-Delivery Systems: A Comprehensive Review. J Nanomed Nanotechnol. 2013;4:1000164
29. Stirland DL, Nichols JW, Miura S. et al. Mind the gap: a survey of how cancer drug carriers are susceptible to the gap between research and practice. J Control Release. 2013;172:1045-64
30. Lammers T, Kiessling F, Hennink WE. et al. Drug targeting to tumors: principles, pitfalls and (pre-) clinical progress. J Control Release. 2012;161:175-87
31. Xie D, Yao C, Wang L. et al. An albumin-conjugated peptide exhibits potent anti-HIV activity and long in vivo half-life. Antimicrob Agents Chemother. 2010;54:191-6
32. Zhao D, Zhang H, Tao W. A rapid albumin-binding 5-fluorouracil prodrug with a prolonged circulation time and enhanced antitumor activity. Biomater Sci. 2017;5:502-10
33. Kratz F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J Control Release. 2008;132:171-83
34. Zeng J, Davies MJ. Evidence for the formation of adducts and S-(carboxymethyl) cysteine on reaction of alpha-dicarbonyl compounds with thiol groups on amino acids, peptides, and proteins. Chem Res Toxicol. 2005;18:1232-41
35. Gunnoo SB, Madder A. Chemical Protein Modification through Cysteine. Chembiochem. 2016;17:529-53
36. Simon M, Frey R, Zangemeister-Wittke U. et al. Orthogonal assembly of a designed ankyrin repeat protein-cytotoxin conjugate with a clickable serum albumin module for half-life extension. Bioconjugate Chem. 2013;24:1955-66
37. Nguyen A, Reyes AE 2nd, Zhang M. et al. The pharmacokinetics of an albumin-binding Fab (AB.Fab) can be modulated as a function of affinity for albumin. Protein Eng Des Sel. 2006;19:291-7
38. Yang Z, Zheng S, Harrison WJ. et al. Long-Circulating Near-Infrared Fluorescence Core-Cross-Linked Polymeric Micelles: Synthesis, Characterization, and Dual Nuclear/Optical Imaging. Biomacromolecules. 2007;8:3422-8
39. Wen X, Wu QP, Ke S. et al. Improved radiolabeling of PEGylated protein: PEGylated annexin V for noninvasive imaging of tumor apoptosis. Cancer Biother Radiopharm. 2003;18:819-27
40. Caliceti P, Veronese FM. Pharmacokinetic and biodistribution properties of poly (ethylene glycol)-protein conjugates. Adv Drug Deliv Rev. 2003;55:1261-77
41. Shechter Y, Mironchik M, Rubinraut S. et al. Albumin-insulin conjugate releasing insulin slowly under physiological conditions: a new concept for long-acting insulin. Bioconjug Chem. 2005;16:913-20
42. Herringson TP, Altin JG. Effective tumor targeting and enhanced anti-tumor effect of liposomes engrafted with peptides specific for tumor lymphatics and vasculature. Int J Pharm. 2011;411:206-14
43. Brunel FM, Lewis JD, Destito G. et al. A hydrazone ligation strategy to assemble multifunctional viral nanoparticles for cell imaging and tumor targeting. Nano Lett. 2010;10:1093-7
44. Kreyling WG, Semmler-Behnke M, Takenaka S. et al. Differences in the biokinetics of inhaled nano- versus micrometer-sized particles. Acc Chem Res. 2013;46:714-22
45. Han JH, Oh YK, Kim DS. et al. Enhanced hepatocyte uptake and liver targeting of methotrexate using galactosylated albumin as a carrier. Int J Pharm. 1999;188:39-47
46. Q Zhang. Comparison of Efficacy and Drug Resistance between Consecutive and Intermittent Injection of Azithromycin Treating Mycoplasma Pneumonia. Gut. 2001;49:214-9
47. Jeger S, Zimmermann K, Blanc A. et al. Site-specific and stoichiometric modification of antibodies by bacterial transglutaminase. Angew Chem Int Ed Engl. 2010;49:9995-7
Author contact
![]() Corresponding authors: Chengchao Shou, Peking University Cancer Hospital & Institute, 52 Fucheng Road, Beijing 100142, China. Tel: 0086-10-88196766, Fax: 0086-10-88122437, E-mail: cshousina.com. and Chuanke Zhao, Peking University Cancer Hospital & Institute, 52 Fucheng Road, Beijing 100142, China. Tel: 0086-10-88196769, Fax: 0086-10-88122437, E-mail: zhaochkedu.cn.
Corresponding authors: Chengchao Shou, Peking University Cancer Hospital & Institute, 52 Fucheng Road, Beijing 100142, China. Tel: 0086-10-88196766, Fax: 0086-10-88122437, E-mail: cshousina.com. and Chuanke Zhao, Peking University Cancer Hospital & Institute, 52 Fucheng Road, Beijing 100142, China. Tel: 0086-10-88196769, Fax: 0086-10-88122437, E-mail: zhaochkedu.cn.
Citation styles
APA
Feng, J., Zhao, C., Wang, L., Qu, L., Zhu, H., Yang, Z., An, G., Tian, H., Shou, C. (2018). Development of a novel albumin-based and maleimidopropionic acid-conjugated peptide with prolonged half-life and increased in vivo anti-tumor efficacy. Theranostics, 8(8), 2094-2106. https://doi.org/10.7150/thno.22069.
ACS
Feng, J.; Zhao, C.; Wang, L.; Qu, L.; Zhu, H.; Yang, Z.; An, G.; Tian, H.; Shou, C. Development of a novel albumin-based and maleimidopropionic acid-conjugated peptide with prolonged half-life and increased in vivo anti-tumor efficacy. Theranostics 2018, 8 (8), 2094-2106. DOI: 10.7150/thno.22069.
NLM
Feng J, Zhao C, Wang L, Qu L, Zhu H, Yang Z, An G, Tian H, Shou C. Development of a novel albumin-based and maleimidopropionic acid-conjugated peptide with prolonged half-life and increased in vivo anti-tumor efficacy. Theranostics 2018; 8(8):2094-2106. doi:10.7150/thno.22069. https://www.thno.org/v08p2094.htm
CSE
Feng J, Zhao C, Wang L, Qu L, Zhu H, Yang Z, An G, Tian H, Shou C. 2018. Development of a novel albumin-based and maleimidopropionic acid-conjugated peptide with prolonged half-life and increased in vivo anti-tumor efficacy. Theranostics. 8(8):2094-2106.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.