Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Material and methods
Results
Discussion
Conclusions
Acknowledgements
Supplementary Material
References
Introduction
Material and methods
Results
Discussion
Conclusions
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(7):2018-2030. doi:10.7150/thno.24382 This issue Cite
Research Paper
A paclitaxel prodrug with bifunctional folate and albumin binding moieties for both passive and active targeted cancer therapy
Lingling Shan1,2, Xin Zhuo1, Fuwu Zhang2, Yunlu Dai2, Guizhi Zhu2, Bryant C. Yung2, Wenpei Fan2, Kefeng Zhai1, Orit Jacobson2, Dale O. Kiesewetter2, Ying Ma2, Guizhen Gao1 ![]() , Xiaoyuan Chen2
, Xiaoyuan Chen2 ![]()
1. Institute of Pharmaceutical Biotechnology, School of Biology and Food Engineering, Suzhou University, Suzhou 234000, China
2. Laboratory of Molecular Imaging and Nanomedicine, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health, Bethesda 20892, USA
Received 2017-12-25; Accepted 2018-2-2; Published 2018-2-16
Citation:
Shan L, Zhuo X, Zhang F, Dai Y, Zhu G, Yung BC, Fan W, Zhai K, Jacobson O, Kiesewetter DO, Ma Y, Gao G, Chen X. A paclitaxel prodrug with bifunctional folate and albumin binding moieties for both passive and active targeted cancer therapy. Theranostics 2018; 8(7):2018-2030. doi:10.7150/thno.24382. https://www.thno.org/v08p2018.htm
Other stylesAbstract

Folate receptor (FR) has proven to be a valuable target for chemotherapy using folic acid (FA) conjugates. However, FA-conjugated chemotherapeutics still have low therapeutic efficacy accompanied with side effects, resulting from complications such as short circulation half-life, limited tumor delivery, as well as high kidney accumulation. Herein, we present a novel FA-conjugated paclitaxel (PTX) prodrug which was additionally conjugated with an Evans blue (EB) derivative for albumin binding. The resulting bifunctional prodrug prolonged blood circulation, enhanced tumor accumulation, and consequently improved tumor therapeutic efficacy.
Methods: Fmoc-Cys(Trt)-OH was coupled onto PTX at the 7'-OH position for further synthesis of ester prodrug FA-PTX-EB. The targeting ability was investigated using confocal microscopy and flow cytometry. The pharmacokinetics of this bifunctional compound was also studied. Meanwhile, cell viability was evaluated in normal cells and three cancer cell lines by MTT assay. In vivo therapeutic effect was tested on FR-α overexpressing MDA-MB-231 tumor model.
Results: Compared with free PTX, the FA-PTX, PTX-EB and FA-PTX-EB prodrugs increased circulation half-life in mice from 2.19 to 3.82, 4.41, and 7.51 h, respectively. Pharmacokinetics studies showed that the FA-PTX-EB delivered more PTX to tumors than FA-PTX and free PTX. In vitro and in vivo studies demonstrated that FA-EB-conjugated PTX induced potent antitumor activity.
Conclusion: FA-PTX-EB showed prolonged blood circulation, enhanced drug accumulation in tumors, higher therapeutic index, and lower side effects than either free PTX or monofunctional FA-PTX and EB-PTX. The results support the potential of using EB for the development of long-acting therapeutics.
Keywords: Evans blue, circulation half-life, paclitaxel prodrug, tumor therapy.
Introduction
Targeted anticancer drug delivery is a promising method to improve the efficacy and safety of therapeutic agents. Among various cell surface targets, folic acid receptor (FR) has been demonstrated as one of the most promising candidates [1-4]. FR is over expressed on the cell membrane of a series of solid tumor cells, including breast, ovarian, and non-small cell lung cancers [5-8]. Conjugation of folic acid, i.e., folate, is a valuable approach to target FR-expressing tissues for chemotherapy.
However, folate-conjugated drugs have poor therapeutic performance and dose limiting side effects, attributing to the poor water solubility, limited blood circulation time, and extremely high kidney accumulation [9-11]. Indeed, poor water solubility has been considered as a causative factor in the slow release of drug molecules from a conjugate and had been previously shown to lead to low therapeutic efficacy [12, 13]. In addition, suboptimal pharmacokinetics or limited blood circulation time likely cause poor therapeutic outcomes [14]. All these considerations highlight the need to understand the pharmacokinetic nature of folate-conjugated small molecule drugs as well as the folic acid receptor-mediated endocytic pathways to enhance folate-conjugated compound drug release[15, 16].
One promising approach to improve drug pharmacokinetics is by hitchhiking albumin to further prolong blood circulation time and enhance drug accumulation in tumors. As one of the most abundant circulating proteins in the blood, albumin is a perfect drug delivery carrier for many endogenous and exogenous compounds [17, 18]. Conjugating drugs to albumin-binding moieties is one of the most successful approaches due to its high efficiency in antitumor drug delivery and low side effects. Among small molecule albumin-binding moieties, Evans blue (EB) exhibits strong binding affinity for serum albumin. EB has been extensively applied in physiologic and clinical investigations, and has been used in routine clinical practice as a way of determining patient plasma volume. By exploiting the high binding affinity of EB to blood albumin in vivo, EB can be used as an effective moiety to improve EB-conjugated drug pharmacokinetic properties [19-21]. Our previous work demonstrated EB conjugates for the realization of long-acting therapeutics and diagnostic molecular probes [22, 23]. In this work, we conjugated truncated EB to FA-PTX to prolong the PTX blood circulation and improve the antitumor therapeutic efficacy.
Pharmacokinetics play a vital role in pharmaceutical research and development [24, 25]. The development of many pharmacologically active compounds has been hindered by poor pharmacokinetics, low solubility, and formulation difficulties. For instance, PTX, one of the most widely used and effective antitumor agents derived from natural sources for a wide spectrum of tumors is highly lipophilic with very poor pharmacokinetics [26, 27]. Abraxane®, a PTX albumin-bound nanoparticle delivery system with particle diameters of ~130 nm, was approved by the US FDA for the treatment of metastatic breast cancer. This nanoparticle formulation has yielded advantages based on decreased toxicity compared to Taxol, a free PTX formulation. However, whether Abraxane® can increase survival and address drug resistance is still unclear [28, 29]. Therefore, substantial challenges for the delivery of PTX remain to be solved.
In this study, PTX derivatives were developed at the 7'-hydroxyl position using Fmoc-Cys(Trt)-OH as a linker. The amino group of the linker was covalently attached to folic acid through an amide bond to synthesize FA-PTX. An EB derivative was further coupled to synthesize FA-PTX-EB prodrug using a maleimide bond. The pharmacokinetic properties of this bifunctional PTX prodrug were significantly enhanced. Consequently, the therapeutic efficacy of the prodrug was augmented in a breast cancer model.
Material and methods
General methods
PTX, folic acid, Fmoc-Cys(Trt)-OH, dicyclohexylcarbodiimide (DCC) and, N-hydroxysuccinimide (NHS) were purchased from Sigma-Aldrich (St. Louis, MO). All other chemicals were purchased from Fisher Scientific (USA). The synthesized compound was purified by using a semi-preparative C18 HPLC column (XTerra Prep RP18, 10 μm, 7.8 × 300 mm, Waters). The purified compound was characterized by a Waters LC-MS system (Milford, MA). The 1H NMR spectra was tested on a Varian NMR spectrophotometer at 500 MHz.
Synthesis of FA-PTX-EB
FA-PTX-EB was prepared in four steps (Scheme S1). 2'-TBSCl-PTX was synthesized as previously reported with slight modifications [30]. Tert-butyldimethylsilyl chloride (TBSCl) (30 mg, 0.234 mmol) and imidazole (82 mg, 1.2 mmol) were dissolved into a dry Dimethylformamide (DMF) (2.0 mL) solution containing PTX (100 mg, 0.117 mmol). The mixture reacted overnight at room temperature under nitrogen. After reaction completion, 15 mL ethyl acetate was added into the mixture and the crude product was washed with 20 mL H2O and 20 mL saturated sodium chloride (NaCl) solution. The resulting compound was collected and dried by Na2SO4 and filtered. After evaporation under reduced pressure, the compound was purified using a silica gel column. The purified compound was collected under reduced pressure to yield a white solid (102.5 mg, yield: 80 %). 100.27 mg 2'-TBSCl-PTX (0.1 mmol, 1 eq.) and 82.23 mg Fmoc-Cys(Trt)-OH (0.14 mmol, 1.2 eq.) were mixed in 10 mL CH2Cl2, and added to 14.27 mg 4-dimethylaminopyridine DMAP (0.117 mmol, 1 eq.). Then 57.51 mg cold EDC was dissolved in 10 mL CH2Cl2 (0.3 mmol) and added dropwise to the mixture over 25 min under room temperature. After 24 h, the reaction mixture was washed by saturated aqueous NaHCO3, and further purified by HPLC (110.5 mg, yield: 82 %). LC-MS: [M+H]+ = 1347.11 calc: 1348.11. 440 mg of folic acid (0.1 mmol) in 5 mL DMF was reacted with DCC and NHS (molar ratio of FA: DDC: NHS is 1: 2 : 2) under room temperature overnight. The residue was washed with water and anhydrous acetone. The extracted FA-NHS was then filtered under reduced pressure (220 mg, yield: 50%). 2'-TBSCl-7'PTX-Cys(Trt)-NH2 (134.81 mg, 0.1 mmol) was mixed in FA-NHS solution (80.7 mg, 0.2 mmol) in dry DMF and reacted under room temperature overnight with monitoring by HPLC (122.5 mg, yield: 56%). FA-2'TBSCl-7'PTX-Cys(Trt) was dissolved in TFA (5 mL) to remove the protection group for 12 h. When HPLC showed complete conversion to the desired compound, it was filtered under reduced pressure. The FA-7'PTX-Cys-SH compound was further purified by HPLC (111.2 mg, yield: 90%). LC-MS (ESI, m/z): 1379.48 ([M + H]+), calc: 1380.48. EB-maleimide (69.37 mg, 0.1 mml) was added into 5 mL DMF FA-PTX-Cys-HS (207.07 mg, 0.15 mmol) solution and reacted for 24 h in the dark at room temperature [22]. The compound was purified using HPLC. The purified FA-PTX-EB was collected as a yellow solid with 75% yield (156.3 mg). LC-MS: [M + H]+ = 2073.13, calc: 2074.13. The chemical structures of 2'-TBSCl-PTX 2'-TBSCl-7'PTX-Cys(Trt)-NH2, FA-2'TBSCl-7'PTX-Cys-SH, and FA-PTX-EB prodrugs were supported by 1H NMR spectroscopy and electrospray ionization mass spectroscopy analysis (Figures S2-S8).
Characterization of FA-PTX-EB
The physicochemical properties of PTX prodrugs were determined, including aqueous solubility and stability. The solubility of FA-PTX, PTX-EB, and FA-PTX-EB were analyzed using a reported method [31]. At room temperature, 100 mg of the three compounds were added into volumetric flasks (10 mL), respectively, and 50 µL of water was injected slowly. When the solution was saturated, the aqueous solubility of FA-PTX, EB-PTX and FA-PTX-EB were measured, respectively. To investigate the release of PTX from different prodrugs in vitro, the three PTX-conjugated prodrugs were incubated in mouse plasma at 37 °C. The sampling times were at 0.5, 1, 2, 4, 8, 12, 24, 48 and 72 h. 50 μL PTX conjugated prodrugs in 10 nM Tris buffer (5 mg/mL) were incubated in 200 μL mouse plasma at 37 °C, respectively. Then, 200 µL of each sample was collected at each designated time point and free PTX was extracted using 2 mL ethyl acetate and analyzed by reversed-phase HPLC with acetonitrile/water gradient.
Cell culture
Human breast cancer (MDA-MB-231), human ovarian cancer (OVCAR3), human primary glioblastoma (U87MG), and human renal epithelial (293T) cell lines were purchased from the American Type Culture Collection (ATCC, Manassas, VA). All cell lines were incubated with different medium at 37 °C under 5% CO2 atmosphere. L-15 (MDA-MB-231), DMEM (OVCAR3, 293T) and MEM (U87MG) medium included 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin, respectively.
Cellular uptake of FA-PTX-EB prodrugs
MDA-MB-231, OVCAR3, U87MG, and 293T were evaluated for FA-α receptor protein expression by Western blot analysis. All cell lines were handled using a reported method [32].
EB-conjugated PTX prodrugs were observed using EB fluorescence for confocal microscopy and flow cytometry experiments. Cell lines were cultured in confocal plates with medium for 4 h, respectively. After 4 h, the cells were fixed on confocal plates directly and observed by confocal laser scanning microscope. PTX-EB and FA-PTX-EB (200 μL, 1 mg/mL) were incubated in 6-well plates with 2 × 105 cells for 4 h, respectively, and then washed with PBS twice. 500 μL suspension in PBS was analyzed by flow cytometry.
Cell viability assay
MDA-MB-231, OVCAR3, U87MG, and 293T cell lines were used to measure the cytotoxicity of FA-PTX-EB, and a standard protocol was followed [33]. The same concentration of PTX, FA-PTX, PTX-EB and FA-PTX-EB from 3 pM to 1.6 nM was added to 5 × 103 cells/well in 96-well plates for 24 h, with six replicates. Then 10 μL MTT solution (5 mg/mL) was added into each well and incubated for 4 h. After changing to 150 μL DMSO, the absorbance of each well was tested by a microplate reader at 490 nm.
Apoptosis study
In vitro effect of FA-PTX-EB prodrugs were measured on three tumor cell lines qualitatively and quantitatively. The change in cell morphology during apoptosis was observed by confocal microscopy. Flow cytometry was used to quantify cell apoptosis [34]. Cells were incubated with 200 μL PTX, FA-PTX, PTX-EB, and FA-PTX-EB (1 mg/mL) for 24 h in confocal plates and washed using PBS (7.4 pH) twice. After adding 500 μL binding buffer, 5 μL propidum iodide and 1 μL Annexin V-FITC were added to the mixture and visualized by confocal microscopy. In the same way, cells were incubated with PTX prodrugs for 24 h in 6-well plates to quantify apoptosis using flow cytometry.
Animal subjects and tumor model
All animals including normal (Kunming) mice and BALB/c athymic nude mice (female) were purchased from Charles River Laboratories (Shanghai, China). All animals were maintained based on the Animal Management Rules of the Ministry of Health of the People's Republic of China (Document NO. 55, 2001) and the guidelines for the Care and Use of Laboratory Animals of Suzhou University.
Following the animal protocol, a folate-deficient rodent diet was fed to the nude mice for 6 days, and MDA-MB-231 tumor cells were inoculated at the right axillary fossa of nude mice [9]. When the tumor volumes reached 80 ~ 150 mm3, the nude mice were used to investigate the targeting ability, pharmacokinetics, and therapy of FA-PTX-EB.
Pharmacokinetics studies
Ten days after MDA-MB-231 tumor cell implantation, mice were randomized into PTX (pure PTX solution), FA-PTX, PTX-EB and FA-PTX-EB groups with an average tumor volume of 98 ± 34 mm3. All mice received 6 mg/kg PTX or 6 mg/kg PTX equivalent of prodrug via a single tail vein injection. After drug injection, three mice per dosing group were sacrificed at 0.12, 0.25, 0.5, 1, 2, 4, 6, 8, 24, and 48 h. Under anesthesia, the blood (500 μL) was collected using terminal cardiac puncture and centrifuged (1500 g, 5 min). Collected tissue samples (liver, kidneys, and tumor) were rinsed with PBS and immediately frozen in liquid nitrogen. All samples were stored at -80 °C before drug analysis.
The 100 μL plasma was extracted with 2 mL of ethyl ether and added to 20 μL of internal standard diazepam (500 ng/mL) with centrifugation at 12,000 rpm for 5 min. The mixture was resolved in 150 μL methanol and analyzed by HPLC. In the same way, the 0.2 g tissues including liver, kidneys, and tumor were homogenized in 1 mL saline solution. Then, the 200 μL homogenate was added to the same volume of acetonitrile and vortexed for 6 min. After the mixture was centrifuged at 12,000 rpm for 10 min, a 20 μL sample was subjected to HPLC analysis (Figure S8). The mobile phase of HPLC contained methanol and water (80 : 20, v : v). PTX was measured at 227 nm. Pharmacokinetic parameters were calculated by DAS 2.0 Pharmacokinetics Software (Chinese Society of Mathematical Pharmacology) with compartmental or non-compartmental modeling approaches. Results were presented as mean ± S.D.
Targeting ability of EB-conjugated FA-PTX in tumor-bearing mice
The targeting ability of PTX-EB and FA-PTX-EB prodrugs was investigated by MDA-MB-231 xenograft tumors (n = 3/group). PTX-EB (0.2 mL, 6 mg/kg PTX equivalent) and FA-PTX-EB (0.2 mL, equal to PTX 6 mg/kg) were administered to mice by tail vein injection, respectively. At 4 h after injection, the tissues (liver, kidneys, tumor) of sacrificed mice were directly visualized by fluorescence microscopy. The tissues were further analyzed using flow cytometry.
Antitumor activity
MDA-MB-231 tumor bearing mice were randomly assigned to five groups (n = 5 per group), respectively. Each group was injected PTX prodrugs by tail vein: (A) PBS buffer (pH 7.4, control), (B) pure PTX solution (6 mg/kg), (C) FA-PTX (6 mg/kg PTX equivalent), (D) PTX-EB (6 mg/kg PTX equivalent), and (E) FA-PTX-EB (6 mg/kg PTX equivalent). Mice experienced three i.v. injections, administered every other day. The PTX prodrug therapeutic efficacies and toxicities on MDA-MB-231 tumor-bearing mice were evaluated by measuring tumor volume and body weight of each mouse every other day. To further evaluate the anti-tumor effect of PTX prodrugs, the liver, kidneys and tumor tissues were excised for pathology and measurement of CD46 protein levels [35].
Statistical analysis
All data are reported as the mean ± SD of n independent measurements. Statistical analysis was performed by using Student's t-test with statistical significance assigned at a p value of < 0.05.
Results
Synthesis and characterization of EB-conjugated PTX targeted prodrugs
The purified FA-PTX-EB and intermediate products (structures shown in Figure 1, synthetic scheme in Scheme S1) were characterized by LC-MS, 1H NMR, and HPLC analysis (Figure S1-S7). As controls, we also synthesized control prodrugs, FA-PTX and PTX-EB, both of which were monofunctional in contrast to bifunctional FA-PTX-EB. To investigate the aqueous solubility of EB-conjugated FA-PTX prodrugs, the direct observations were strictly based on the solubility calculations of PTX prodrugs. The solubility of FA-PTX, PTX-EB, and FA-PTX-EB were 1.121 ± 0.12, 5.03 ± 0.09, and 7.02 ± 0.18 mg/mL, respectively (n = 4). (Figure S8)
In vitro release studies
The release of PTX from these prodrugs (FA-PTX, PTX-EB and FA-PTX-EB) was measured by quantifying the amount of free PTX via HPLC, after incubating the prodrugs in mouse plasma at 37 °C. Figure 1B and Figure S7 showed PTX release from prodrugs as measured by HPLC. The FA-PTX displayed fast release of PTX in vitro, resulting in precipitation of PTX (t1/2 ˂ 4 h). By comparison, the in vitro PTX release from PTX-EB exhibited relatively lower release rates, with t1/2 of 6.82 h. The release of PTX from FA-PTX-EB prodrug had a t1/2 of 9.15 h. Interestingly, EB conjugation enabled more sustainable drug release from prodrugs.
Figure 1
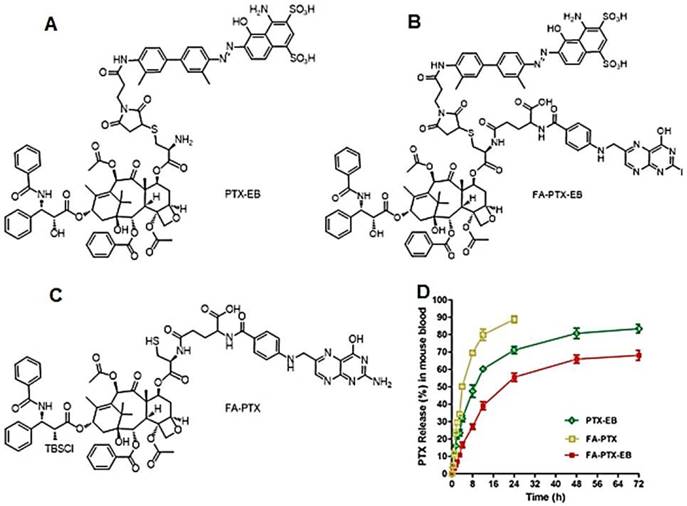
(A-C) Chemical structures of PTX-EB (A), FA-PTX-EB (B), and FA-PTX (C). (D) In vitro release of PTX in mouse plasma. PTX concentration was measured by HPLC. The release was expressed as mean ± standard deviation based on triplicate experiments.
In Vitro Cellular Uptake
To study receptor-mediated cellular uptake of PTX-EB and FA-PTX-EB, the FR-α expression levels on a panel of cells were first evaluated by Western blot. The results showed that the FR-α expression levels were higher in MDA-MB-231 cells compared to OVCAR3 and U87MG tumor cell lines, as shown in Figure 2A, and the FR-α protein expression level was hardly detectable in 293T cells.
The cellular uptake of EB-conjugated prodrugs was studied by fluorescence microscopy in cultured cancer cells at 4 h (Figure 2B). The EB-conjugated PTX prodrug, FA-PTX-EB, displayed increased uptake in FR-α overexpressing MDA-MB-231, OVCAR3, and U87MG tumor cells. Flow cytometry analyses indicated that PTX-EB and FA-PTX-EB were taken up in MDA-MB-231 tumor cells at 35.6% and 65.9%, respectively. 29.1% and 38.1% PTX-EB and FA-PTX-EB were retained in OVCAR3 cells, respectively, and about 23.2% and 32.1% in U87MG cells. In contrast, these two prodrugs were taken up by 293T cells at merely 1.5% and 1.8% (Figure 2C). These findings suggest that FA conjugation increased the targeting selectivity of PTX prodrugs to tumor cells with FR-α expression. The uptake of EB-conjugated prodrugs in the different tumor cells were in agreement with their FR-α expression levels.
In vitro antitumor activity of EB-conjugated PTX prodrugs
The antitumor effect of four cell lines including tumor cell lines MDA-MB-231, OVCAR3, U87MG, and normal cell line 293T were studied in vitro. As shown in Figure 3, for all cell lines, prodrugs displayed dose-dependent antitumor activity. Especially, in all the three different tumor cell lines, FA-PTX-EB prodrug showed inhibition ratios greater than 85%, however, PTX inhibited only a maximum of 63% of tumor cells. In addition, the inhibition ratios of PTX-EB were about 75% in MDA-MB-231, OVCAR3, and U87MG cell lines. The cell inhibition ratio of FA-PTX without EB was only about 68% at the highest dose. The half maximal inhibitory concentration (IC50) of free PTX exhibited higher concentration compared to the EB conjugated FA-PTX. The MDA-MB-231 tumor cells were inhibited to a greater extent over other cancer cell lines owing to the higher expression levels of FR-α receptor. Interestingly, the two EB-conjugated prodrugs exhibited comparatively low cell inhibition ratios relative to normal cells (293T), even at the highest concentration (55%). This result supports the hypothesis that EB-conjugated PTX compound shows lower cytotoxicity to normal cells, and enhances the antitumor ability of free PTX to tumors that overexpress FR.
Figure 2
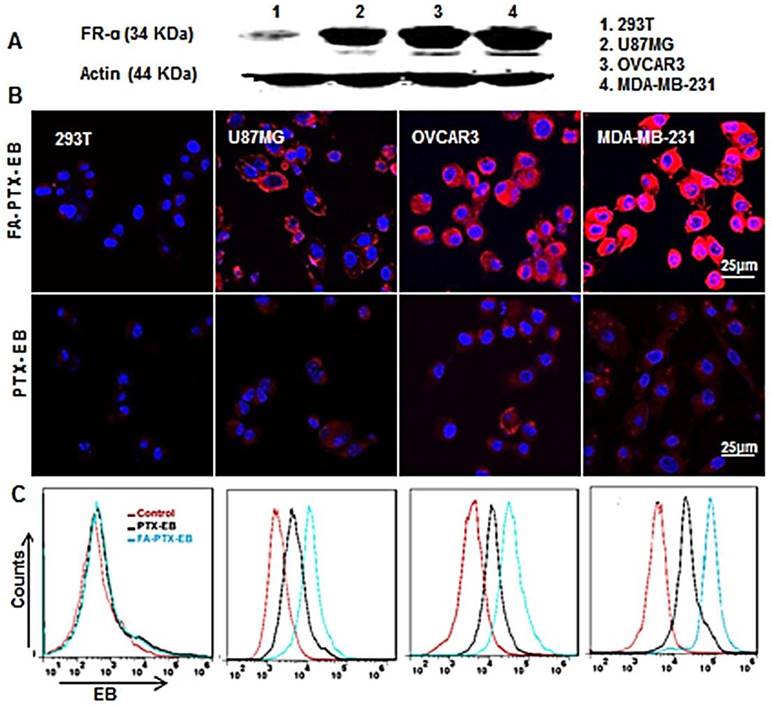
Cellular uptake of FA-PTX-EB in a series of tumor cells with different expression levels of FR-α. (A) Western blot results verify the different expression levels of FR-α on tumor cells and normal cell lines. (B) Confocal microscopy images showing the intracellular uptake ability of FA-PTX-EB and PTX-EB prodrugs in 293T, U87MG, OVCAR3, and MDA-MB-231 cells. The nucleus is stained by DAPI, and the red corresponds with EB fluorescence from EB-conjugated compounds. (C) Flow cytometric analysis of FA-PTX-EB and PTX-EB prodrug in 293T, U87MG, OVCAR3, and MDA-MB-231 cells.
The morphological changes of MDA-MB-231, OVCAR3, and U87MG tumor cells after the incubation of FA-PTX, PTX-EB, and FA-PTX-EB, were observed under confocal microscopy, as shown in Figure 4A. Using annexin V and propidium iodide (PI) staining, we further studied cell apoptosis and necrosis upon treatment with prodrugs [10]. In Figure 4B, the late apoptotic cells (Q4) were evident in different tumor cells treated with PTX, FA-PTX, PTX-EB, and FA-PTX-EB prodrugs. Quantification of flow cytometry results indicated the portions of cells undergoing early apoptosis (Q2) of U87MG, OVCAR3, and MDA-MB-231 were 29.8%, 21.8%, and 33.8% (PTX), 37.1%, 28.8%, and 36% (FA-PTX), 36.8%, 31.5%, and 44.7% (PTX-EB), 47.9%, 50.9%, and 62.3% (FA-PTX-EB), respectively. The percent of Q4 was 3.1%, 2.5%, and 1.3% (PTX), 3.7%, 23.5%, and 34.5% (FA-PTX), 13.2%, 36.5%, and 27.5% (PTX-EB), 12.3%, 22.2%, and 22.7% (FA-PTX-EB), respectively (Figure 4B). The above analyses prove that the prodrugs with EB greatly increased the antitumor effect of low-dose PTX.
Figure 3
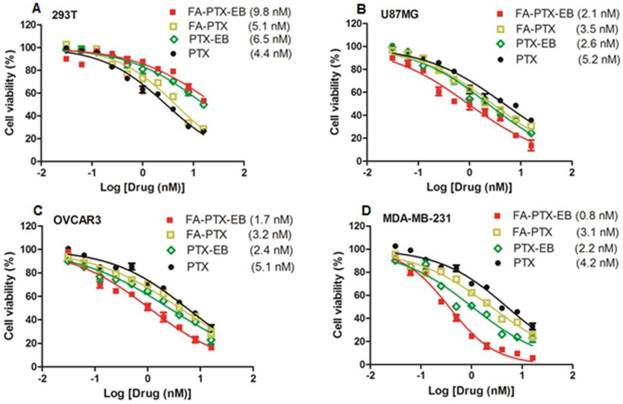
In vitro cytotoxicity of FA-PTX-EB, FA-PTX, PTX-EB, and free PTX against normal cells 293T (A), U87MG (B), OVCAR3 (C), and MDA-MB-231 tumor cells. The numbers in parenthesis are the corresponding IC50 values calculated by GraphPad Prism.
Figure 4
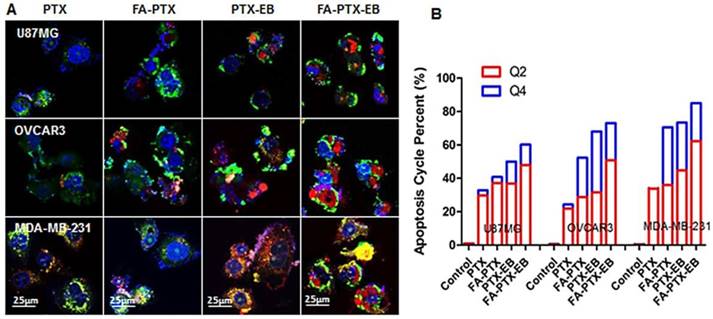
In vitro apoptosis study. (A) The cells were incubated with free PTX, FA-7PTX, 7PTX-EB, or FA-7PTX-EB. One day later, morphology of cell apoptosis was observed by confocal microscopy. Blue: DAPI fluorescence (nucleus stain); Green: Annexin V-FITC (apoptotic cell membrane); and red: Propidium Iodide, PI (apoptotic cell nucleus). (B) The apoptosis cycle percent (%): Q2 + Q4.
Pharmacokinetics and tissue distribution in plasma, liver, kidneys, and tumor
The pharmacokinetics and tissue distribution of PTX and PTX prodrugs were studied by quantifying total amount of PTX in the plasma and different tissue samples from tumor-bearing mice. Figure 5 shows the representative HPLC chromatograms of PTX in mouse plasma and different tissues after i.v. injection of PTX and PTX prodrugs at an equivalent PTX dose of 6 mg/kg. As shown in Figure 5A, following i.v. administration of PTX and PTX prodrugs, the PTX levels in plasma and tissues represented the total concentration of free PTX and PTX released from prodrugs.
Concentration vs. time curves of PTX were generated for plasma, liver, kidneys and tumor from mice receiving treatment of FA-PTX, PTX-EB and FA-PTX-EB, and the pharmacokinetic parameters were then determined (Table 1-4). Cmax levels attained after i.v. injection of prodrugs were 7854 ng/mL (PTX), 8034 ng/mL (FA-PTX), 8396 (PTX-EB), and 9132 ng/mL (FA-PTX-EB), respectively (Table 1). The highest AUC PTX values were observed with FA-PTX-EB and were at least 8.6 times greater than those of FA-PTX. Similarly, in PTX-EB, higher AUC values were observed, at least 4.2 times greater than those of free PTX.
Figure 5
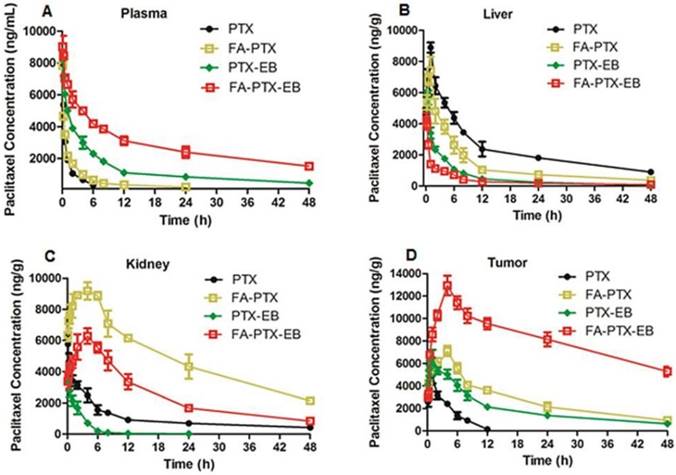
In vivo pharmacokinetic studies. (A) Mean concentration-time profile of PTX in plasma following i.v. administration of a single dose of 6 mg/kg of PTX, FA-PTX, PTX-EB, or FA-PTX-EB in mice. Time courses of PTX levels in tissues of MDA-MB-231 inoculated mice following i.v. administration of a 6 mg/kg dose of PTX or equivalent formulated PTX. (B) Liver. (C) Kidneys. (D) Tumor. Data are shown as the mean concentration ± S.D. (n = 5/group).
Table 1
Pharmacokinetic parameters of PTX prodrugs compared with free PTX. Results represent mean ± S.E. (n = 5)
| Parameter | Units | PTX | FA-PTX | PTX-EB | FA-PTX-EB |
|---|---|---|---|---|---|
| Cmax | ng/mL | 7854 ± 403 | 8034 ± 512 | 8396 ± 422 | 9132 ± 412 |
| AUC | ng.h/mL | 7630 ± 114 | 15657 ± 368 | 59570 ± 428 | 134922 ± 624 |
| t1/2ɑ | h | 0.201 ± 0.01 | 0.129 ± 0. 01 | 0.068 ± 0.001 | 0.033 ± 0.001 |
| t1/2β | h | 2.19 ± 0.32 | 3.82 ± 0.53 | 4.41 ± 0.48 | 7.51 ± 0.68 |
| CL | mL/h/kg | 1.31 ± 0.53 | 1.08 ± 0.46 | 0.58 ± 0.32 | 0.29 ± 0.08 |
Pharmacokinetic parameters were calculated using a two-compartment model. Values are the calculated pharmacokinetic parameter ± the standard error of the estimated single model-dependent calculations
Table 2
Pharmacokinetic parameters of PTX prodrugs compared with free PTX. Results represent mean ± S.E. (n=5)
| Parameter | Units | PTX | FA-PTX | PTX-EB | FA-PTX-EB |
|---|---|---|---|---|---|
| Cmax | ng/mL | 8890 ± 432 | 7412 ± 422 | 6037 ± 511 | 4210 ± 602 |
| AUC | ng.h/mL | 11388 ± 1332 | 61354 ± 1006 | 26453 ± 1315 | 15846 ± 1610 |
| t1/2 | h | 8.455 ± 1.08 | 5.22 ± 0.92 | 1.67 ± 0.48 | 0.395 ± 0.68 |
| CL | mL/h/kg | 37.31 ± 4.03 | 33.84 ± 5.46 | 22.23 ± 1.32 | 14.86 ± 0.98 |
Liver tissue pharmacokinetics were calculated using non-compartmental analysis
Table 3
Pharmacokinetic parameters of PTX prodrugs compared with free PTX. Results represent mean ± S.E. (n = 5)
| Parameter | Units | PTX | FA-PTX | PTX-EB | FA-PTX-EB |
|---|---|---|---|---|---|
| Cmax | ng/mL | 5769 ± 712 | 9181 ± 639 | 3917 ± 233 | 4285 ± 405 |
| AUC | ng.h/mL | 46821 ± 1025 | 234286 ± 2204 | 8659 ± 1062 | 118971 ± 1256 |
| t1/2 | h | 2.15 ± 0.62 | 6.82 ± 0.53 | 1.41 ± 0.48 | 3.29 ± 0.56 |
| CL | mL/min | 28.57 ± 3.03 | 10.21 ± 4.25 | 15.07 ± 2.02 | 7.17 ± 2.09 |
Kidney tissue pharmacokinetics were calculated using non-compartmental analysis
Table 4
Pharmacokinetic parameters of PTX prodrugs compared with free PTX. Results represent mean ± S.E. (n=5)
| Parameter | Units | PTX | FA-PTX | PTX-EB | FA-PTX-EB |
|---|---|---|---|---|---|
| Cmax | ng/mL | 3938 ± 304 | 7108 ± 611 | 6503 ± 455 | 12948 ± 1766 |
| AUC | ng.h/mL | 21511 ± 1244 | 131053 ± 5462 | 31825 ± 1102 | 391016 ± 1232 |
| t1/2 | h | 6.19 ± 0.32 | 44.21 ± 2.53 | 34.41 ± 3.48 | 124.51 ± 4.68 |
| CL | mL/h/kg | 13.11 ± 2.14 | 16.88 ± 5.09 | 22.23 ± 4.32 | 29.86 ± 2.98 |
Tumor tissue pharmacokinetics were calculated using non-compartmental analysis
The mean PTX tissue concentration-time profiles after i.v. injection of all PTX prodrugs are shown in Table 2-4. For free PTX, the highest concentration of drugs was found in the liver. For FA-PTX prodrugs, the highest concentrations were found in the kidneys, and the maximal PTX concentrations of FA-PTX-EB in tumor were 12948 ng/g. In the kidney, the highest concentrations of PTX were 5769, 9181, 3917 and 4285 ng/g for PTX, FA-PTX, PTX-EB, and FA-PTX-EB prodrugs, respectively (Table 3). The highest concentration of PTX displayed in tumor for various prodrugs were as follows: 12948 ng/g (FA-PTX-EB), 7108 ng/g (FA-PTX), and 6503 ng/g (PTX-EB) prodrug. In contrast, the highest PTX concentration of free PTX in tumor was only 3938 ng/g (Table 4).
Targeting ability of FA-PTX-EB in tumor bearing mice
Based on the above pharmacokinetics studies, after injection of PTX-EB and FA-PTX-EB at 4 h, we sacrificed mice bearing MDA-MB-231 tumor xenografts, and excised tissues including liver, kidneys, and tumor for tissue sectioning. Tissue sections were visualized under fluorescence microscopy. In Figure 6A, in comparison with other excretion organs such as the liver and kidneys, the highest fluorescence signal was observed in tumor tissues for FA-PTX-EB. At the same time, tumor tissues from FA-PTX-EB treatment exhibited stronger EB fluorescence signals than PTX-EB, suggesting higher uptake of FA-PTX-EB than PTX-EB.
Figure 6
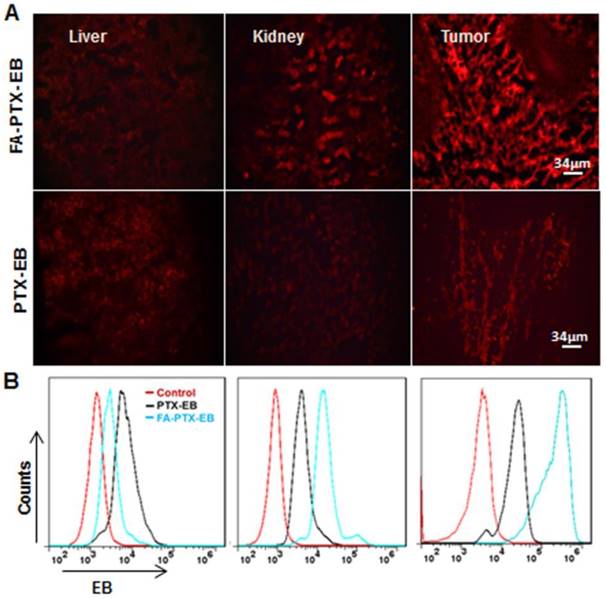
Tissue uptake of FA-PTX-EB and PTX-EB. MDA-MB-231 tumor xenografts at 4 h post-injection of EB-conjugated PTX prodrugs. Tissues (liver, kidneys, and tumor) were immediately excised for sectioning and were visualized by fluorescence microscopy. The red fluorescence from EB-conjugated compounds (A) and corresponding flow cytometry values (B) were observed in liver, kidneys and tumor, respectively. Data represent mean ± standard deviation (SD), n = 5/group.
To further study the tumor targeting ability of the prodrugs, the fluorescence intensity of organ tissues was analyzed by flow cytometry (Figure 6B). The pharmacodynamics of PTX-EB and FA-PTX-EB signals at 4 h in tissues and tumors are described in Figure 6B. At 4 h after injection of FA-PTX-EB, the fluorescence intensity ratio of MDA-MB-231 tumor was 65.98 ± 0.125 %. In contrast, the MDA-MB-231 tumor-bearing mice with administration of PTX-EB prodrugs showed a ratio of 25.12 ± 0.64 % at 4 h post injection.
In vivo therapeutic experiment
Mice bearing MDA-MB-231 tumors were used to evaluate the in vivo anti-tumor efficacy of PTX, FA-PTX, PTX-EB, and FA-PTX-EB. As shown in Figure 7, the change of mice body weight and tumor volume was depicted in different treatment groups. FA-PTX-EB most effectively inhibited tumor growth among all treatment regimens (Figure 7A). The results show that FA-PTX-EB inhibited tumor volume most prominently (74.82%), followed by PTX-EB (50.08%), FA-PTX (45.13%), and PTX (41.04%). Based on tumor weight, the relative inhibition rates were consistent with the results from tumor volume measurements as shown in Figure 7C.
Figure 7
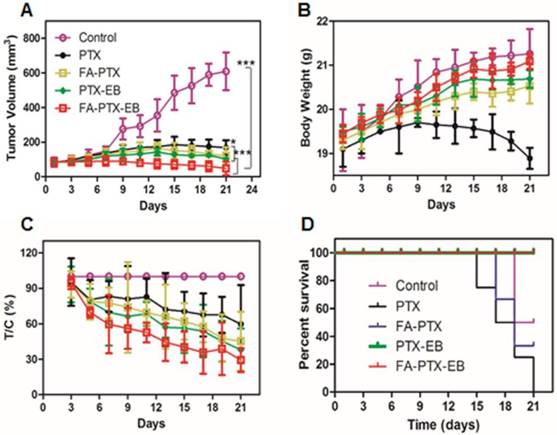
FA-PTX-EB significantly inhibited tumor growth in a breast cancer mouse model. (A) Tumor volumes of MDA-MB-231-bearing mice as a function of time (d) (P values, *P ˂ 0.05, ***P ˂ 0.001, are calculated by t-test). (B) Body weights of MDA-MB-231-bearing mice as a function of time (d). Data were given as mean ± SD (n = 6/group). (C) The relative inhibition rates (%). (D) Kaplan-Meier survival plot.
Figure 8
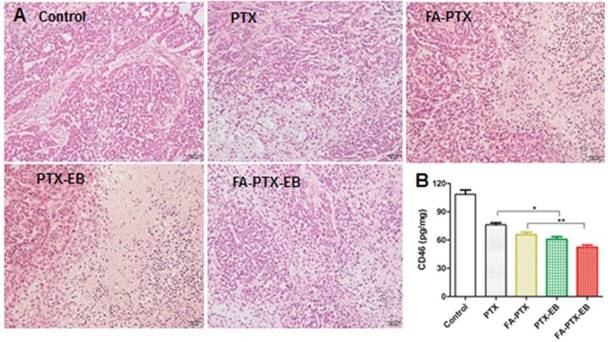
(A) H&E staining of tumor tissues excised from nude mice 21 days after administration of PTX, FA-PTX, PTX-EB and FA-PTX-EB. (B) The level of CD46 protein in tumor tissues was measured by ELISA assay kit (n = 5/group).
The change in body weight was monitored throughout the study period and is presented in Figure 7B. The body weight of mice treated with free PTX showed significant decrease attributed to severe side effects and toxicity of PTX. The body weight loss of the PTX group was 0.65 ± 0.02 g on day 21 post initial treatment, about 3.5% of the initial weight. However, the data showed that the body weight of mice was significantly increased following treatment with FA-PTX, PTX-EB, and FA-PTX-EB groups. Results suggest that these PTX conjugations could significantly reduce the systemic toxicity of PTX.
After treatment with PTX, FA-PTX, PTX-EB, and FA-PTX-EB, liver, kidneys and tumor tissues were excised and further evaluated for the antitumor effect and toxicity by histopathological examination. The representative tumor tissue sections from different groups of treatments were shown in Figure 8. Tumor tissues in the PTX, FA-PTX, and PTX-EB groups exhibited different populations of spherical cells, intercellular blank spots, and necrosis. The FA-PTX-EB group showed larger intercellular empty spaces in tumor tissue that indicate efficacious tumor therapy. Compared with the control group, no distinct pathologic changes were found in the liver and kidneys of the mice injected with PTX, FA-PTX, PTX-EB, and FA-PTX-EB (Figure S9). In addition, we measured the expression levels of membrane cofactor protein CD46 on the tumor tissue. The CD46 values of the FA-PTX-EB treated group were significantly decreased compared with other groups. Overall, these results demonstrated the potent and specific therapeutic effect of FA-PTX-EB.
Discussion
Optimal pharmacokinetics is key to drug development. The limited efficacy of many drugs is often attributed to poor pharmacokinetics that leads to low bioavailability, for example, many folate-conjugated chemotherapeutics failed in clinical trials [36]. Recently, we developed a new class of small molecule albumin binding entities based on the long-circulating EB. EB displays a strong noncovalent binding with both murine and human serum albumin [37-40]. In our previous work, we have extensively studied the albumin binding abilities of EB conjugates with various chemical agents, and have confirmed their strong binding abilities [41, 42]. In this study, we functionalized folate-PTX prodrug with truncated EB as an albumin binding moiety for targeted tumor therapy. The resulting prodrug conjugate, namely FA-PTX-EB, which greatly enhanced aqueous solubility of PTX, dramatically increased blood circulation half-life and tumor uptake of the drug, and decreased the drug accumulation in the kidneys. The fluorescence signal of EB was used to study the dynamic distribution of the prodrugs at the cellular level and the tissue level.
Physicochemical properties of these PTX prodrugs, including FA-PTX, PTX-EB, and FA-PTX-EB, were assayed by HPLC. The water-soluble EB and amino acid cysteine increased the water solubility of PTX prodrug from ∼0.4 μg/mL for PTX to 7.02 mg/mL for the prodrug, and the increase in the water solubility of prodrug with EB was significantly different from prodrug without EB (Figure S8). Interestingly, PTX was sustainably released from PTX-EB and FA-PTX-EB prodrugs, with t1/2 = 6.82 h and 9.15 h, respectively. These results suggest that EB acted as a compatibilizer in PTX prodrugs, resulting in improvements in water solubility, physical stability, and drug release.
The purpose of EB-conjugation is to enhance the blood circulation and tumor tissue retention time of PTX, and to reduce the toxicity of free PTX to normal tissue. Plasma pharmacokinetics and tissue distribution behavior were analyzed, which provided strong evidence of enhanced delivery [43, 44]. In the pharmacokinetics study, pharmacokinetic parameters of PTX from the PTX with EB formulation were significantly better than those from the PTX group without EB (Table 1). The plasma AUC value of PTX from the PTX-EB group was seven times higher compared to the group administered with free PTX, and the AUC value of FA-PTX-EB in plasma was eight times higher than that of FA-PTX. Similarly, the PTX plasma half-life t1/2β value after PTX-EB administration was two times higher than that of free PTX, and the plasma half-life t1/2β value of FA-PTX-EB was two times higher than that of FA-PTX. These results suggested that PTX-conjugated EB prodrugs may circulate for a longer time in blood than PTX prodrug without EB. In addition, EB modification resulted in slower release of PTX from the FA-PTX-EB prodrug over a prolonged period.
Notably, the PTX concentration of EB-PTX prodrugs in the tumor was significantly higher than those of the PTX prodrugs without EB, especially FA-PTX-EB. The primary FR populated tissue (kidneys) comprises the major sites of accumulation for circulating FA conjugates. Therefore, one would expect drug clearance in the FR rich tissues to be significantly greater for FA-PTX conjugated EB than for FA-PTX. The PTX AUC of FA-PTX-EB was lower in the liver and kidneys; and higher in plasma and tumor compared to FA-PTX group. Similar results were observed between the PTX AUC of PTX-EB and PTX groups. Importantly, there were statistically significant formulation-dependent differences observed in the liver and kidneys exposure profiles. This finding may be rationalized by the fact that small molecular albumin binding moieties can extend the blood-circulation time of PTX prodrugs and decrease its accumulation in the liver and kidneys.
The vitamin B9 (folic acid) is one of the well-proven ligands for targeted delivery of imaging or therapeutic agents to tumors. Therefore, in this paper, the cellular uptake ability of FA-PTX conjugated EB was evaluated in vitro and in vivo (Figure 2 and Figure 6). The percentage of cell uptake of FA-PTX-EB (23.09%~65.91%) treated tumor cell groups was far higher than normal cells with low FR-α expression (1.5%, 1.8%, respectively). Compared with prodrug without FA, FA-PTX-EB significantly enhanced targeting ability for tumors via receptor-mediated uptake as evidenced by fluorescence microscopy. In vitro data clearly proved that FA-PTX-EB prodrugs enhanced the cellular uptake and antitumor activity through FR-α targeting. The same results were also displayed in the tissue sections at 4 h post injection of FA-PTX-EB (Figure 6). These results support the notion that FA-PTX-EB improved the targeting ability of PTX to FR-positive tumors. Taken together, this bifunctional drug delivery system enhanced the drug retention time and drug concentration in tumor tissues.
PTX is a potent anticancer agent as a microtubule stabilizer, and it plays a central role in the treatment of breast cancer. Accordingly, PTX has a low IC50 value of 4.5 nM for the human MDA-MB-231 breast cancer cell line (Figure 3B). In comparison to free PTX or prodrug without EB, the IC50 value of PTX-EB or FA-PTX-EB had a lower value of 0.98-2.29 nM, reflecting the enhancement of cytotoxicity via EB conjugation. The IC50 values of FA-PTX-EB as a prodrug were about 3-fold higher, 12 nM in healthy cells, suggesting the specificity of drug cytotoxicity. Notably, FA-PTX-EB or PTX-EB were more effective than FA-PTX or free PTX in vitro. Thus, truncated EB enhanced the intracellular delivery of PTX prodrugs that further led to stronger cytotoxicity in tumor cells. The similar percentage of late apoptotic cells (PI+) in tumor groups was higher for the EB formulations. The percentage of necrotic/dead cells (Q2 + Q4) was 85% for FA-PTX-EB compared with only 34.6% in free PTX treated cells (MDA-MB-231).
The in vivo anticancer efficacy of PTX prodrugs was evaluated in a MDA-MB-231 cancer model after tail vein injection at an equivalent dose of 6 mg/kg PTX (Figure 7). The tumor growth in the FA-PTX-EB group was remarkably inhibited compared to free PTX. Importantly, the tumor growth inhibition correlated highly with significant apoptosis observed in tumor tissues (Figure 8A). No obvious changes of mouse body weights were observed after treatment with prodrugs. In the MDA-MB-231 cancer model, the antitumor effect of FA-PTX-EB was significantly stronger than other PTX prodrug or PTX. The tumor-inhibition of FA-PTX-EB reached 71.6%, which was about 1.2 times higher than FA-PTX and 2 times higher than PTX. These data confirmed that EB and FA cooperatively enhanced the antitumor effect of FA-PTX-EB.
The decrease of mouse weight in the free PTX treatment group demonstrated that free PTX had severe side effects. The mean body weight loss of the PTX group was 0.65 ± 0.02 g, representing a 3.5% loss of the original weight. On the contrary, the increase of body weight of other groups treated with PTX-EB and FA-PTX-EB indicated that the small molecular binding moiety EB improved PTX's water solubility, pharmacokinetic properties, and biodistribution, which collectively contributed to low toxicity. The hematoxylin and eosin (H&E) staining showed no histological signs of toxicity in major organs with all PTX prodrugs (Figure S9). Nevertheless, necrotic areas were clearly shown in the FR-α positive tumors treated with FA-PTX-EB. Similar antitumor effects were observed by CD46 analysis (Figure 8B). The improved therapeutic effect of the FA-PTX-EB prodrug is likely a result of the synergistic function of water soluble cysteine, FA, EPR effect, the “stealth” character of amino acid ester, FA/FR-α mediated endocytosis, and the EB enhanced blood circulation/tumor retention. PTX ester prodrugs are often water-soluble and less toxic, but less active as anticancer agents. However, FA-PTX prodrug with EB prodrug is a unique anticancer prodrug in terms of physical stability, lower systemic toxicity, and higher antitumor efficacy in an MDA-MB-231 xenograft model. Given slower in vitro release and improved physical stability of FA-PTX prodrug with EB vs. FA-PTX without EB prodrug, the EB containing formulation may circulate longer in the bloodstream, increase AUC, reduce the distribution of PTX in non-target tissue, and increase tumor retention time.
Conclusions
Taken together, we have harnessed the advantages of an albumin binding moiety and folate to establish FA-PTX-EB as a bifunctional prodrug for both passive and active targeted delivery of PTX, resulting in long blood circulation, improved pharmacokinetics, high antitumor activity, and low toxicity. Moreover, in comparison with previous PTX formulations, EB-conjugated FA-PTX prodrugs also exhibited higher water solubility, prolonged tumor retention, and potentiated antitumor activity, as well as reduced toxicity. We believe that the strategy of bifunctional targeting will expand the clinical implications of PTX-based cancer therapy and spur the development of similar bifunctional prodrug platforms.
Acknowledgements
The authors are grateful to the Anhui Province Educational Committee Major Project (KJ2016SD62), Key Project of Anhui Educational Committee (KJ2013A262, KJ2015A220, and gxfxZD2016266), Natural Science Foundation Committee of China (NSFC 81000666), Quality Project of AnHui Education Department -The plan of “ChuangKe” Laboratory (2015ckjh109), Research Platform Project of Suzhou University (2016kyf08), and the Intramural Research program, National Institute of Biomedical Imaging and Bioengineering, National Institutes of Health. Lingling Shan was partially funded by the China Scholarship Council (CSC).
Supplementary Material
Supplementary figures.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ono C, Hsyu P-H, Abbas R, Loi C-M, Yamazaki S. Application of physiologically based pharmacokinetic modeling to the understanding of bosutinib pharmacokinetics: prediction of drug-drug and drug-disease interactions. Drug Metab Disposition. 2017;45:390-8
2. Bishai WR. Drug development: locking down metabolism. Nat Chem Biol. 2017;13:925-6
3. Sofias AM, Dunne M, Storm G, Allen C. The battle of “nano” paclitaxel. Adv Drug Del Rev. 2017;122:20-30
4. Nehate C, Jain S, Saneja A, Khare V, Alam N, Dubey RD. et al. Paclitaxel formulations: challenges and novel delivery options. Curr Drug Del. 2014;11:666-86
5. Chen C, Ke J, Zhou XE, Yi W, Brunzelle JS, Li J. et al. Structural basis for molecular recognition of folic acid by folate receptors. Nature. 2013;500:486-9
6. Ledermann J, Canevari S, Thigpen T. Targeting the folate receptor: diagnostic and therapeutic approaches to personalize cancer treatments. Ann Oncol. 2015;26:2034-43
7. Talekar M, Kendall J, Denny W, Garg S. Targeting of nanoparticles in cancer: drug delivery and diagnostics. Anticancer Drugs. 2011;22:949-62
8. Moore KN, Borghaei H, O'Malley DM, Jeong W, Seward SM, Bauer TM. et al. Phase 1 dose-escalation study of mirvetuximab soravtansine (IMGN853), a folate receptor α-targeting antibody-drug conjugate, in patients with solid tumors. Cancer. 2017;123:3080-7
9. Müller C, Struthers H, Winiger C, Zhernosekov K, Schibli R. DOTA conjugate with an albumin-binding entity enables the first folic acid-targeted 177Lu-radionuclide tumor therapy in mice. J Nucl Med. 2013;54:124-31
10. Lee JW, Lu JY, Low P, Fuchs P. Synthesis and evaluation of taxol-folic acid conjugates as targeted antineoplastics. Bioorg Med Chem. 2002;10:2397-414
11. Low PS, Henne WA, Doorneweerd DD. Discovery and development of folic-acid-based receptor targeting for imaging and therapy of cancer and inflammatory diseases. Acc Chem Res. 2007;41:120-9
12. Zhao X, Jia X, Liu L, Zeng J, Tian K, Zhou T. et al. Double-cross-linked hyaluronic acid nanoparticles with pH/reduction dual-responsive triggered release and pH-modulated fluorescence for folate-receptor-mediated targeting visualized chemotherapy. Biomacromolecules. 2016;17:1496-505
13. Villanueva JR, Villanueva LR, Navarro MG. Pharmaceutical technology can turn a traditional drug, dexamethasone into a first-line ocular medicine. A global perspective and future trends. Int J Pharm. 2017;516:342-51
14. Alavijeh MS, Chishty M, Qaiser MZ, Palmer AM. Drug metabolism and pharmacokinetics, the blood-brain barrier, and central nervous system drug discovery. NeuroRx. 2005;2:554-71
15. Vlahov IR, Leamon CP. Engineering folate-drug conjugates to target cancer: from chemistry to clinic. Bioconjug Chem. 2012;23:1357-69
16. Vlahov IR, Santhapuram HKR, You F, Wang Y, Kleindl PJ, Hahn SJ. et al. Carbohydrate-based synthetic approach to control toxicity profiles of folate- drug conjugates. J Org Chem. 2010;75:3685-91
17. Rimac H, Debeljak Ž, Miller L. Displacement of drugs from human serum albumin: from molecular interactions to clinical significance. Curr Med Chem. 2017;24:1930-47
18. Sabbioni G, Turesky RJ. Biomonitoring human albumin adducts: the past, the present, and the future. Chem Res Toxicol. 2016;30:332-66
19. Lin T, Zhao P, Jiang Y, Tang Y, Jin H, Pan Z. et al. Blood-brain-barrier-penetrating albumin nanoparticles for biomimetic drug delivery via albumin-binding protein pathways for antiglioma therapy. ACS Nano. 2016;10:9999-10012
20. Jacobson O, Kiesewetter DO, Chen X. Albumin-binding Evans blue derivatives for diagnostic imaging and production of long-acting therapeutics. Bioconjug Chem. 2016;27:2239-47
21. Liu Y, Wang G, Zhang H, Ma Y, Lang L, Jacobson O. et al. Stable Evans blue derived exendin-4 peptide for type 2 diabetes treatment. Bioconjug Chem. 2015;27:54-8
22. Zhang J, Lang L, Zhu Z, Li F, Niu G, Chen X. Clinical translation of an albumin-binding PET radiotracer 68Ga-NEB. J Nucl Med. 2015;56:1609-14
23. Niu G, Lang L, Kiesewetter DO, Ma Y, Sun Z, Guo N. et al. In vivo labeling of serum albumin for PET. J Nucl Med. 2014;55:1150-6
24. Zhivkova ZD. Quantitative structure-pharmacokinetic relationships for plasma clearance of basic drugs with consideration of the major elimination pathway. J Pharm Pharm Sci. 2017;20:135-47
25. Rizk M, Zou L, Savic R, Dooley K. Importance of drug pharmacokinetics at the site of action. Clin Transl Sci. 2017;10:133-42
26. Kantae V, Krekels EH, Van Esdonk MJ, Lindenburg P, Harms AC, Knibbe CA. et al. Integration of pharmacometabolomics with pharmacokinetics and pharmacodynamics: towards personalized drug therapy. Metabolomics. 2017;13:9
27. Zhang FW, Khan S, Li RC, Smolen JA, Zhang SY, Zhu GZ. et al. Design and development of multifunctional polyphosphoester-based nanoparticles for ultrahigh paclitaxel dual loading. Nanoscale. 2017;9:15773-7
28. Bernabeu E, Cagel M, Lagomarsino E, Moretton M, Chiappetta DA. Paclitaxel: What has been done and the challenges remain ahead. Int J Pharm. 2017;526:474-95
29. Nehate C, Jain S, Saneja A, Khare V, Alam N, Dhar Dubey R. et al. Paclitaxel formulations: challenges and novel delivery options. Curr Drug Deliv. 2014;11:666-86
30. Sugahara S, Kajiki M, Kuriyama H, Kobayashi TR. Paclitaxel delivery systems: the use of amino acid linkers in the conjugation of paclitaxel with carboxymethyldextran to create prodrugs. Biol Pharm Bull. 2002;25:632-41
31. Greenwald RB, Pendri A, Bolikal D. Highly water soluble taxol derivatives: 7-polyethylene glycol carbamates and carbonates. J Org Chem. 1995;60:331-6
32. Yang T, Xu F, Fang D, Chen Y. Targeted proteomics enables simultaneous quantification of folate receptor isoforms and potential isoform-based diagnosis in breast cancer. Sci Rep. 2015;5:16733
33. Shan L, Liu M, Wu C, Zhao L, Li S, Xu L. et al. Multi-small molecule conjugations as new targeted delivery carriers for tumor therapy. Int J Nanomedicine. 2015;10:5571-91
34. Eray M, Mättö M, Kaartinen M, Andersson LC, Pelkonen J. Flow cytometric analysis of apoptotic subpopulations with a combination of Annexin V-FITC, propidium iodide, and SYTO 17. Cytometry. 2001;43:134-42
35. Madjd Z, Durrant LG, Pinder SE, Ellis IO, Ronan J, Lewis S. et al. Do poor-prognosis breast tumours express membrane cofactor proteins (CD46)? Cancer Immunol Immunother. 2005;54:149-56
36. Reddy JA, Westrick E, Vlahov I, Howard SJ, Santhapuram HK, Leamon CP. Folate receptor specific anti-tumor activity of folate-mitomycin conjugates. Cancer Chemother Pharmacol. 2006;58:229-36
37. Chen H, Jacobson O, Niu G, Weiss ID, Kiesewetter DO, Liu Y. et al. Novel “add-on” molecule based on Evans blue confers superior pharmacokinetics and transforms drugs to theranostic agents. J Nucl Med. 2017;58:590-7
38. Chen H, Wang G, Lang L, Jacobson O, Kiesewetter DO, Liu Y. et al. Chemical conjugation of Evans blue derivative: a strategy to develop long-acting therapeutics through albumin binding. Theranostics. 2016;6:243-53
39. Tian R, Jacobson O, Niu G, Kiesewetter DO, Wang Z, Zhu G. et al. Evans blue attachment enhances somatostatin receptor subtype-2 imaging and radiotherapy. Theranostics. 2018;8:735-45
40. Ehlerding EB, Lan X, Cai W. “Albumin hitchhiking” with an Evans blue analog for cancer theranostics. Theranostics. 2018;8:812-4
41. Zhu GZ, Lynn GM, Jacobson O, Chen K, Liu Y, Zhang HM. et al. Albumin/vaccine nanocomplexes that assemble in vivo for combination cancer immunotherapy. Nat Commun. 2017;8:1954
42. Zhang FW, Zhu GZ, Jacobson O, Liu Y, Chen K, Yu GC. et al. Transformative nanomedicine of an amphiphilic camptothecin prodrug for long circulation and high tumor uptake in cancer therapy. ACS Nano. 2017;11:8838-48
43. Chen WY, Zhou XZ, Wu LL, Wu YS, Wang SM, Liu B. et al. A UPLC/MS/MS method for determination of protosappanin B in rat plasma and its application of a pharmacokinetic and bioavailability study. Biomed Chromatogr. 2017;31:e3919
44. Wang F, Shen Y, Xu X, Lv L, Li Y, Liu J. et al. Selective tissue distribution and long circulation endowed by paclitaxel loaded PEGylated poly (ɛ-caprolactone-co-l-lactide) micelles leading to improved anti-tumor effects and low systematic toxicity. Int J Pharm. 2013;456:101-12
Author contact
![]() Corresponding authors: Xiaoyuan Chen, Email: shawn.chengov and Guizhen Gao, Email: szxyggzcom
Corresponding authors: Xiaoyuan Chen, Email: shawn.chengov and Guizhen Gao, Email: szxyggzcom
Citation styles
APA
Shan, L., Zhuo, X., Zhang, F., Dai, Y., Zhu, G., Yung, B.C., Fan, W., Zhai, K., Jacobson, O., Kiesewetter, D.O., Ma, Y., Gao, G., Chen, X. (2018). A paclitaxel prodrug with bifunctional folate and albumin binding moieties for both passive and active targeted cancer therapy. Theranostics, 8(7), 2018-2030. https://doi.org/10.7150/thno.24382.
ACS
Shan, L.; Zhuo, X.; Zhang, F.; Dai, Y.; Zhu, G.; Yung, B.C.; Fan, W.; Zhai, K.; Jacobson, O.; Kiesewetter, D.O.; Ma, Y.; Gao, G.; Chen, X. A paclitaxel prodrug with bifunctional folate and albumin binding moieties for both passive and active targeted cancer therapy. Theranostics 2018, 8 (7), 2018-2030. DOI: 10.7150/thno.24382.
NLM
Shan L, Zhuo X, Zhang F, Dai Y, Zhu G, Yung BC, Fan W, Zhai K, Jacobson O, Kiesewetter DO, Ma Y, Gao G, Chen X. A paclitaxel prodrug with bifunctional folate and albumin binding moieties for both passive and active targeted cancer therapy. Theranostics 2018; 8(7):2018-2030. doi:10.7150/thno.24382. https://www.thno.org/v08p2018.htm
CSE
Shan L, Zhuo X, Zhang F, Dai Y, Zhu G, Yung BC, Fan W, Zhai K, Jacobson O, Kiesewetter DO, Ma Y, Gao G, Chen X. 2018. A paclitaxel prodrug with bifunctional folate and albumin binding moieties for both passive and active targeted cancer therapy. Theranostics. 8(7):2018-2030.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.