Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(7):1766-1781. doi:10.7150/thno.22788 This issue Cite
Research Paper
Young Bone Marrow Sca-1 Cells Rejuvenate the Aged Heart by Promoting Epithelial-to-Mesenchymal Transition
Jiao Li1,2,3, Shu-Hong Li2, Jun Wu2, Richard D. Weisel2,3, Alina Yao2, William L. Stanford4, Shi-Ming Liu1 ![]() , Ren-Ke Li2,3
, Ren-Ke Li2,3 ![]()
1. Department of Cardiology, Second Affiliated Hospital of Guangzhou Medical University, Guangzhou, China
2. Toronto General Research Institute, Division of Cardiovascular Surgery, University Health Network, Toronto, Canada
3. Division of Cardiac Surgery, Department of Surgery, University of Toronto; Toronto, Canada
4. Regenerative Medicine Program, Ottawa Hospital Research Institute, Department of Cellular and Molecular Medicine, University of Ottawa
Received 2017-9-11; Accepted 2018-1-13; Published 2018-2-12
Citation:
Li J, Li SH, Wu J, Weisel RD, Yao A, Stanford WL, Liu SM, Li RK. Young Bone Marrow Sca-1 Cells Rejuvenate the Aged Heart by Promoting Epithelial-to-Mesenchymal Transition. Theranostics 2018; 8(7):1766-1781. doi:10.7150/thno.22788. https://www.thno.org/v08p1766.htm
Other stylesAbstract

Background: To improve the regenerative capacity of aged individuals, we reconstituted bone marrow (BM) of aged mice with young Sca-1 cells, which repopulated cardiac progenitors and prevented cardiac dysfunction after a myocardial infarction (MI). However, the mechanisms involved were incompletely elucidated. This study aimed to investigate whether young, highly regenerative BM Sca-1 cells exert their cardio-protective effects on the aged heart through reactivation of the epithelial-to-mesenchymal transition (EMT) process.
Methods: In vitro, BM Sca-1 cells were co-cultured with epicardial-derived cells (EPDCs) under hypoxia condition; mRNA and protein levels of EMT genes were measured along with cellular proliferation and migration. In vivo, BM Sca-1+ or Sca-1- cells from young mice (2-3 months) were transplanted into lethally-irradiated old mice (20-22 months) to generate chimeras. In addition, Sca-1 knockout (KO) mice were reconstituted with wild type (WT) BM Sca-1+ cells. The effects of BM Sca-1 cell on EMT reactivation and improvement of cardiac function after MI were evaluated.
Results: In vitro, BM Sca-1+ cells increased EPDC proliferation, migration, and EMT relative to Sca-1- cells and these effects were inhibited by a TGF-β blocker. In vivo, more young BM Sca-1+ than Sca-1- cells homed to the epicardium and induced greater host EPDC proliferation, migration, and EMT after MI. Furthermore, reconstitution of Sca-1 KO mice with WT Sca-1+ cells was associated with the reactivation of EMT and improved cardiac function after MI.
Conclusions: Young BM Sca-1+ cells improved cardiac regeneration through promoting EPDC proliferation, migration and reactivation of EMT via the TGF-β signaling pathway.
Keywords: stem cells, aging, rejuvenation, heart
Introduction
In heart development, the fetal epicardium secretes factors that promote growth of the myocardium [1]. A subpopulation of epicardial cells also undergo epithelial-to-mesenchymal transition (EMT) to form epicardium-derived cells (EPDCs) that migrate to the underlying myocardium where they give rise to coronary vascular smooth muscle cells, adventitial, and interstitial fibroblasts [2]. EPDCs have been reported to differentiate into cardiomyocytes (CMs) and endothelial cells during normal heart development, although this process remains controversial [3]. The adult epicardium is quiescent under normal physiological conditions, but can be reactivated and produces EPDCs after injury [4]. Following myocardial infarction (MI) in the adult mouse heart, a rapid and robust epicardial response has been well studied [5]. The epicardial response is characterized by an initial expression of an embryonic gene program. Using lineage analyses in the mouse, van Wijk et al. demonstrated the formation of a WT1 (wilms tumor 1)-positive sub-epicardial mesenchyme, from which predominantly fibroblasts were derived, followed by coronary endothelium and, at later stages, a modest number of CMs [4]. Furthermore, by complementary paracrine actions, EPDCs and cardiac progenitor cells can synergistically improve cardiac function after MI [6]. Therefore, activated EPDCs can contribute to new vasculature and CMs to repair the injured heart.
In mammals, the transcription factors Tbx18 and WT1 are expressed in the proepicardium and embryonic epicardium during development [7, 8] and are down regulated in normal adult epicardium [9]. Previous studies showed that in fetal heart, cardiac WT1 expression was largely restricted to the epicardium [3, 9, 10], and in postnatal mouse and human heart, WT1 was also detected in epicardial cells, although at a reduced frequency compared with fetal epicardium [9]. The expression of these molecules is upregulated after injury [9, 11, 12]. Zhou et al. showed that fetal epicardial genes Wt1, Tbx18, and Raldh2 were dynamically upregulated after MI, peaking between 1 and 5 days and declining to near-baseline levels by 4 weeks, and that the fraction of WT1+ cells increased from approximately 25% in normal heart to approximately 75% after MI [9]. Therefore, WT1 and TBX18 were considered as the first sign of the reactivation of epicardial genes after MI.
During EMT in many tissues, epithelial cells lose their junctions and apical-basal polarity, reorganize their cytoskeleton, downregulate their epithelial gene expression signature and activate genes that help to define the mesenchymal phenotype—increased cell protrusions and motility—and, in many cases, enable the development of an invasive phenotype. Thus, the reactivation of EMT after MI can be confirmed stepwise. First, at the transcription level, the Snail proteins (encoded by Snai1 and Snai2) are the key factors that drive EMT progression. Snail1 and Snail2 (also known as Slug) are activated by EMT-inducing stimuli such as TGF-β and Notch [13, 14, 15]. Snail binds to conserved E-box elements within the promoters of adhesion protein genes such as E-cadherin, directly repressing their transcription. Loss of E-cadherin is considered to be a fundamental event in EMT. Therefore, Snail1 and Slug are usually defined as the re-activation of EMT at the transcriptional level. The second step during EMT is the downregulation of the expression of epithelial proteins. Two epithelial markers that are consistently down-regulated upon EMT induction are Krt14 [16, 17] and BVES [18]. Therefore, Krt14 and BVES are generally examined to confirm the down-regulation of the epithelial genes. The third step of EMT is the deconstruction of cell junctions. The dissolution of tight junctions during EMT is accompanied by decreased claudin and occluding expression and the diffusion of zonula occludens 1 (ZO-1) from cell-cell contacts [19]. The fourth step during EMT is the induction of mesenchymal genes, including Calponin [20], Snail [21], Sox9 [22], Cdh6 [23], Col7a1[24], MMP10 [25], and OPG [26, 27, 28]. Therefore, the diffusion of ZO-1 and the induction of Calponin, Cdh6 and Collage7a1 are considered as the sign of mesenchymal transition. Lastly, EMT is characterized by increased cell contractility and actin stress fiber formation, and increased cell protrusions and motility through SMA expression [9, 29].
In the aged heart, diminished stem cell numbers decrease tissue regeneration capacity after injury. Previously, we showed that young bone marrow (BM) cells can restore aged recipient progenitors in both the BM and, most important, the myocardium, by BM reconstitution [30]. Subsequently, we found that stem cell antigen 1+ (Sca-1+) cells are a key BM cell type involved in the rejuvenation of the aged heart [31]. However, the underlying mechanisms remain unknown. Considering the reported functional role of epicardial cells and EMT in the repair of the infarcted myocardium, we postulated that BM Sca-1 cells may exert their cardio-protective effects through activation of epicardial cells, thereby stimulating the EMT process.
The present study investigated the possible cross-talk between BM Sca-1 cells and the cardiac EMT process using in vitro and in vivo experimental models, including reconstitution of Sca-1 knockout (KO) mice with wild type (WT) BM Sca-1+ cells to study the effects of Sca-1 cell on EMT and the molecular mechanisms responsible for Sca-1 cell-mediated EMT activation after MI.
Methods
Animal procedures: The Animal Care Committee of the University Health Network approved all experimental procedures, which were carried out according to the Guide for the Care and Use of Laboratory Animals (NIH, revised 2011).
Sca-1+ and Sca-1- BM cells from young (Y) GFP transgenic mice (C57BL/6-Tg-GFP mice, The Jackson Laboratory) were used to reconstitute irradiated old (O, C57BL/6) wild type recipient mice, generating Y(Sca1+)-O and Y(Sca1-)-O chimeras as described previously (Figure S1) [31]. Male Sca-1 knockout (KO) mice (C57BL/6 background) and their wild type (WT) littermates aged 2-3 months were kindly provided by Dr William L. Stanford [32].
Myocardial infarction and cardiac function measurement: Twelve weeks after BM reconstitution, coronary occlusion was performed in Y(Sca-1+)-O and Y(Sca-1-)-O chimeras as well as Sca-1 KO and (Sca-1+)-KO mice as previously reported [30]. In brief, mice were anesthetized with 2% isoflurane and given buprenorphine (0.05 mg/kg) for analgesia. Mice were intubated and ventilated with 2% isoflurane. Through a thoracotomy, the pericardium was dissected and the left anterior descending (LAD) coronary artery was ligated. Cell proliferation was measured 3 and 7 days post-MI. The EMT process of epicardial cells was evaluated 3 days post-MI (Figure S1B). Cardiac function was measured with echocardiography before and at 7, 14, 21 and 28 days after MI [33, 34, 35]. Briefly, mice were sedated with a 2% isoflurane (Pharmaceutical Partners of Canada) nosecone. Echocardiographic examinations were performed using a GE Vivid 7 ultrasound system (GE Healthcare Canada) with an i13L transducer. Depth and frequency were set at 1 cm and 14 MHz, respectively. Short-axis views were obtained from the parasternal approach. LV dimensions (left ventricular end-diastolic internal diameter (LVIDd) and end-systolic internal diameter (LVIDs)) were measured in M-mode. Ejection fraction was calculated as follows: (LVIDd3 - LVIDs3) / LVIDd3 × 100. Fractional shortening was calculated as follows: (LVIDd - LVIDs) / LVIDd × 100. Twenty-eight days after MI, the hearts were arrested and fixed at physiologic pressures. Hearts were then cut into 1 mm sections and photographed for morphometry and processed for histological staining. The infarct area fraction was calculated by computerized planimetry (Image-Pro Plus, Media Cybernetics) of digital images of three Masson's trichrome-stained serial LV sections taken at 1.0 mm intervals along the longitudinal axis. To assess the effect of BM cells on myocardial regeneration, the area occupied by myocytes in the infarct zone was measured and expressed as a percentage of the total infarct area [36]. The infarct area was defined as the entire area of LV that contained scar in myocardial sections stained with Masson's trichrome. The scar thickness was measured by computerized planimetry and presented as an average of wall thickness measurements taken at the middle and at each edge of the scar area at its thinnest point. All morphometric analyses were performed by investigators who were blind to the treatment allocation.
Statistical analysis: All values are expressed as mean ± SD. Analyses were performed using GraphPad InStat software (La Jolla, California, USA). Student's t-test was used for two-group comparisons. Comparisons of parameters among three or more groups were analyzed using one-way analysis of variance (ANOVA) followed by Tukey or two-way ANOVA with repeated measures over time followed by Bonferroni post-hoc tests for multiple comparisons. Differences were considered statistically significant at P<0.05.
Results
BM Sca-1+ cells homed to the epicardium and increased proliferation of host epicardial cells after MI
Sca-1+ and Sca-1- BM cells from young (Y) GFP transgenic mice were separated by immunomagnetic activated cell sorting and were extensively characterized with established markers for hematopoietic, mesenchymal and angiogenic progenitors using flow cytometry. The BM Sca-1+ cells were identified as multipotent progenitors with greater hematopoietic and angiogenic potentials (Figure S2). The sorted Sca-1+ and Sca-1- BM cells were used to reconstitute irradiated old (O) wild type recipient mice, generating Y(Sca1+)-O and Y(Sca1-)-O chimeras, respectively. MI was induced 12 weeks after BM reconstitution. First, the BM-derived cells, identified as GFP+, were quantified at baseline and 3 and 7 days post-MI in the infarcted area of the chimeric hearts (Figure 1A). The total number of GFP+ cells in Y(Sca1+)-O chimeras was higher than in Y(Sca1-)-O chimeras at 3 and 7 days post-MI. In comparison to the number of GFP+ cells homed to 3 layers of chimeric hearts, the number of GFP+ cells in epicardium was significantly higher than those in the myocardium and endocardium at 3 and 7 days post-MI (Figure 1B). In the epicardium, the number of GFP+ cells in Y(Sca1+)-O chimeras was significantly higher than in Y(Sca1-)-O chimeras.
Next, the epicardial cells, identified as WT1+ (a specific marker for epicardial cells), were quantified at 3 and 7 days post-MI in the infarcted area of the chimeric hearts. We found the total number of WT1+ cells (from all three layers of heart in the infarct zone) in Y(Sca1+)-O chimeras was significantly higher than that in Y(Sca1-)-O chimeras at 3 days post-MI (Figure S3). In both chimeric hearts, the number of WT1+ cells in the epicardium was significantly higher than the number of cells in the myocardium and endocardium at 3 days post-MI (Figure 1C). The number of WT1+ cells in the epicardium of Y(Sca1+)-O chimeras was significantly higher than in Y(Sca1-)-O chimeras at 3 days post-MI. However, the number of WT1+ cells in the epicardium at 7 days post-MI declined significantly compared with that at 3 days post-MI and there were more WT1+ cells in the myocardium and endocardium in Y(Sca1+)-O chimeric hearts, suggesting possible migration of epicardial cells (Figure 1C). To determine the origin of the epicardial cells, the number of donor-derived epicardial cells (WT1+GFP+) or host-derived epicardial cells (WT1+GFP-) in the infarcted region of the chimeric hearts was quantified (Figure 1D). Although the WT1+GFP- cells outnumbered the WT1+GFP+ cells in both groups, the magnitude of the increase in WT1+GFP- cells was much higher in the Y(Sca1+)-O than the Y(Sca1-)-O group, especially at 3 days post-MI, suggesting the possible activation of host epicardial cells after MI by BM Sca-1+ cells primarily homed to the epicardium.
To obtain further evidence for the activation of epicardial cells by BM Sca-1 cells, BrdU was used to label proliferating cells in Y(Sca1+)-O and Y(Sca1-)-O chimeras. Immunofluoresencent labeling was used to identify donor (GFP+)- or host (GFP-)-derived proliferating (BrdU+) epicardial (WT1+) cells in the infarcted region at 3 and 7 days post-MI (Figure 1E). The number of proliferating epicardial cells (WT1+BrdU+) peaked at 3 days post-MI in both chimeric hearts. However, the number of proliferating epicardial cells in Y(Sca1+)-O chimeric hearts was significantly higher than in Y(Sca1-)-O chimeric hearts at both 3 and 7 days post-MI (Figure 1F). Analysis of the origin of the proliferating epicardial cells revealed that there were more host-derived proliferating epicardial cells (WT1+BrdU+GFP-) than donor-derived proliferating epicardial cells (WT1+BrdU+GFP+) in Y(Sca1+)-O chimeric hearts compared to Y(Sca1-)-O chimeric hearts at 3 days post-MI (Figure 1G). This finding indicates that BM Sca-1+ cells primarily increased the proliferation of host epicardial cells after MI. The same trend was observed at 7 days post-MI (Figure 1H).
For the first time, we showed the cardiac resident BM Sca-1+ cells in the epicardium contributed to the stimulation of the host epicardial cell proliferation. This novel observation led us to build the hypothesis that young BM Sca-1+ cells may contribute to cardiac regeneration by promoting EPDC proliferation, migration and activation of EMT.
Homed Sca-1+ cells activated the EMT process after MI
To confirm that homed Sca-1+ cells not only stimulated epicardial cell proliferation but also activated the EMT process of epicardial cells after MI, the expression of EMT embryonic epicardial genes (WT1, Tbx18), transcriptional genes (Slug, Snail), mesenchymal genes (Calponin, Cdh6, Col7a1) and epithelial genes (BVES, Krt14) were quantified by RT-qPCR in Y(Sca1+)-O and Y(Sca1-)-O chimeric hearts at baseline and in the whole infarcted area at 3 days post-MI. The expression of EMT embryonic epicardial genes (WT1, Tbx18; Figure 2A), transcriptional genes (Slug, Snail; Figure 2B), and mesenchymal genes (Calponin, Cdh6, Col7a1; Figure 2C) was significantly higher in the infarcted zone of Y(Sca1+)-O than Y(Sca1-)-O chimeric hearts at 3 days post-MI. Conversely, the expression of EMT epithelial genes (BVES, Krt14) was significantly lower in the infarcted zone of Y(Sca1+)-O than Y(Sca1-)-O chimeric hearts at 3 days post-MI (Figure 2D). The increased expression of mesenchymal genes and the corresponding decreased expression of epicardial genes after MI in the Y(Sca1+)-O chimeras suggest the activation of the EMT process by BM Sca-1+ cells. Indeed, immunolabeling of mesenchymal proteins, SMA (Figure 2E), vimentin (Figure 2F), and calponin (Figure 2G) in the infarcted region showed greater mesenchymal protein expression in Y(Sca1+)-O than Y(Sca1-)-O chimeras (Figure S4). These results were confirmed by Western blot showing significantly greater SMA, vimentin and calponin protein levels in the infarcted area of Y(Sca1+)-O than Y(Sca1-)-O chimeras at 3 days post-MI (Figure 2H).
Co-culture of BM Sca-1+ cells with EPDCs under hypoxia conditions increased proliferation and migration and activated the EMT process in EPDCs
To confirm the in vivo finding that BM Sca-1+ cells stimulated EPDC proliferation, we isolated epicardial cells and co-cultured them with BM Sca-1+ or Sca-1- cells under normoxia and hypoxia conditions. The purity of EPDCs was confirmed by immunofluorescent staining of WT1 (Figure 3A). BM Sca-1+ cells stimulated more EPDC proliferation than BM Sca-1- cells under hypoxia conditions (Figure 3B). This result was confirmed with BrdU labeling of EPDCs (Figure 3C-D). Cell migration ability was detected by transwell and wound-scratch assays (Figure 3E-H), and was significantly greater in EPDCs co-cultured with BM Sca-1+ cells than BM Sca-1- cells under hypoxia conditions.
Figure 1
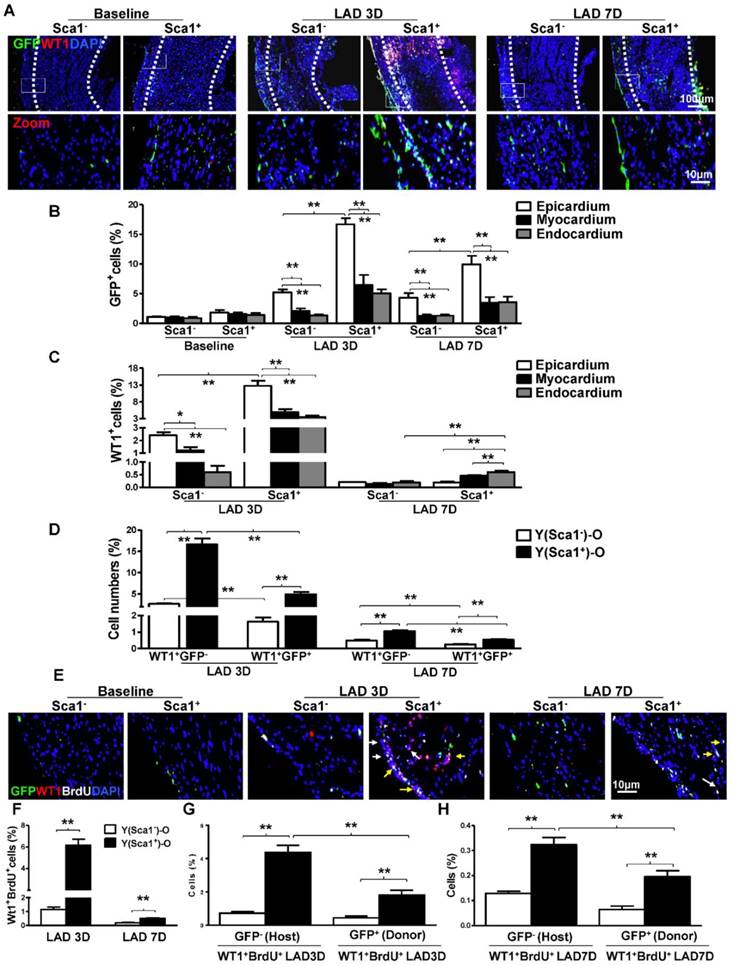
BM Sca-1+ cells homed to the epicardium and increased proliferation of host epicardial cells after MI. Sca-1+ and Sca-1- bone marrow (BM, 2×106) cells from young (Y) GFP (green fluorescent protein) transgenic mice were used to reconstitute irradiated old (O, 9.5 Gy) wild type mice, generating Y(Sca1+)-O and Y(Sca1-)-O chimeras, respectively. Twelve weeks after BM reconstitution, coronary occlusion was performed to induce myocardial infarction (MI). To detect cell proliferation after MI, BrdU (50 mg/kg) was administered to mice by intraperitoneal injection for 3 consecutive days. Coronary artery ligation was performed 1 day later. (A) Immunofluorescent staining of GFP and WT1 in chimeric hearts at baseline, 3 and 7 days post-MI. (B) Quantification of GFP+ in the epicardium, myocardium, and endocardium of chimeric hearts at baseline and in the infarcted region at 3 and 7 days post-MI. (C) Quantification of WT1+ cells in the epicardium, myocardium, and endocardium of the infarct area in the chimeric hearts. (D) Quantification of WT1+GFP- or WT1+GFP+ cells in the infarcted region of the chimeric hearts. (E) Immunofluorescent staining of GFP, WT1, and BrdU in chimeric hearts at baseline, 3 and 7 days post-MI. Yellow arrows indicate GFP, WT1, and BrdU triple-positive cells. White arrows indicate WT1 and BrdU double-positive cells. (F) Quantification of WT1+BrdU+ (proliferating epicardial cells) cells in the infarcted region of the chimeric hearts at 3 and 7 days post-MI. Quantification of WT1+BrdU+GFP- (host-derived proliferating epicardial cells) and WT1+BrdU+GFP+ cells (donor-derived proliferating epicardial cells) in the infarcted region of the chimeric hearts at 3 (G) and 7 (F) days post-MI. n= 6/group, mean ± SD; *P<0.05, **P<0.01. BrdU: bromodeoxyuridine; WT1: wilms tumor 1.
Figure 2
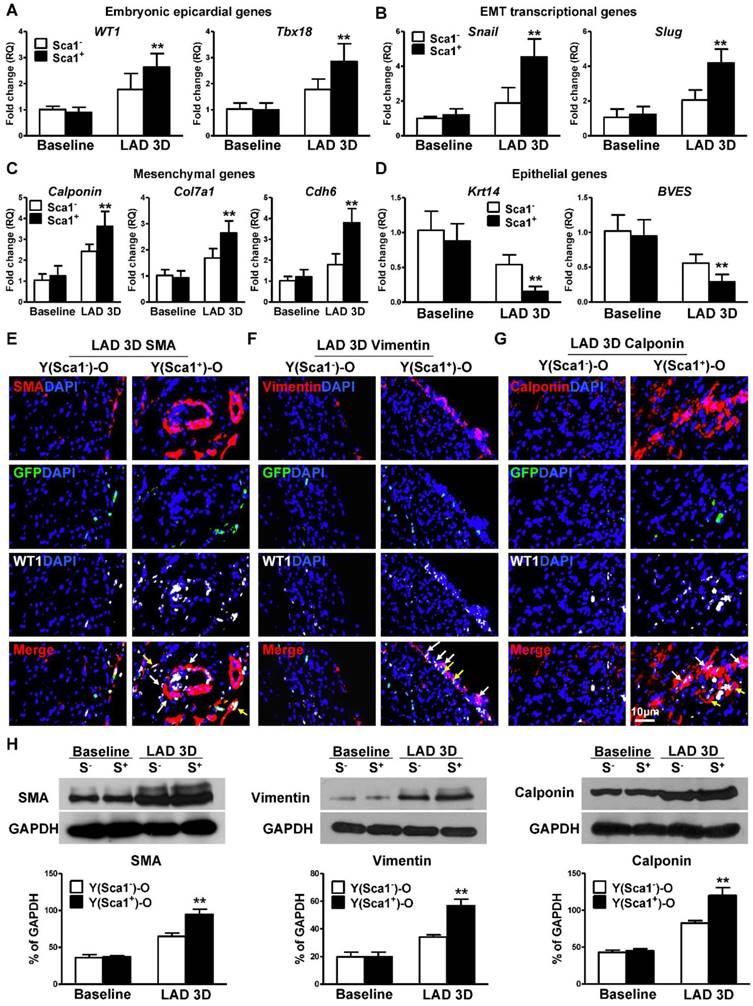
BM Sca-1+ cells activated EMT of epicardial cells after MI. Bone marrow (BM; 2×106) Sca-1+ and Sca-1- cells from young (Y) GFP (green fluorescent protein) transgenic mice were used to reconstitute irradiated old (O, 9.5 Gy) wild type mice, generating Y(Sca1+)-O and Y (Sca1-)-O chimeras, respectively. Twelve weeks after BM reconstitution, coronary occlusion was performed to induce myocardial infarction (MI). Quantification of EMT embryonic epicardial genes (A), transcriptional genes (B), mesenchymal genes (C), and epithelial genes (D) at baseline and in the infarcted region at 3 days post-MI by RT-qPCR. (E) Immunofluorescent staining of smooth muscle actin (SMA) at 3 days post-MI. Yellow arrows indicate GFP, WT1, and SMA triple-positive cells (donor-derived epicardial cell obtained the mesenchymal phenotype). White arrows indicate WT1 and SMA double-positive cells (host-derived epicardial cell obtained the mesenchymal phenotype). (F) Immunofluorescent staining of vimentin at 3 days post-MI. (G) Immunofluorescent staining of calponin at 3 days post-MI. (H) Protein levels of SMA, vimentin, and calponin (mesenchymal phenotype) at baseline and in the infarcted region of the chimeric hearts were determined by Western blot and normalized by GAPDH. n=6/group, mean ± SD; **P<0.01.
Figure 3
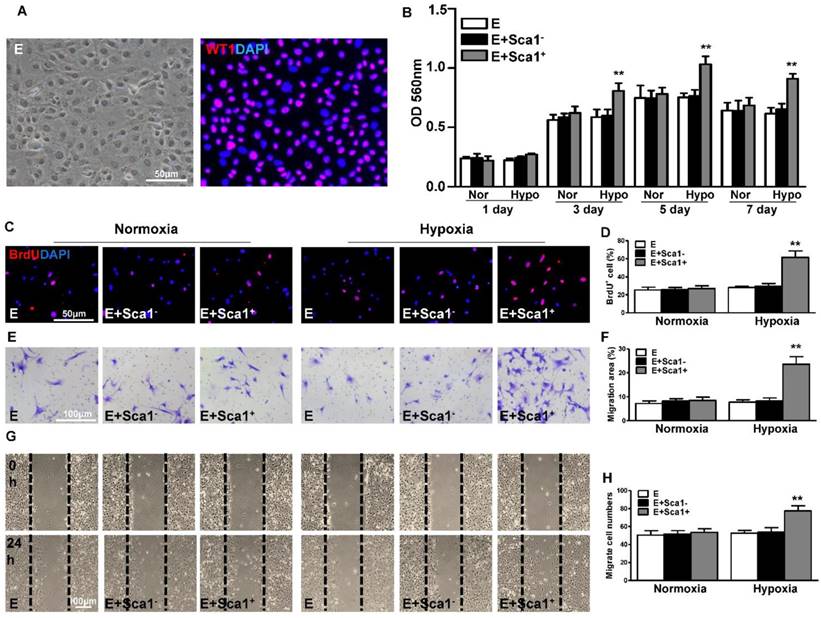
Co-culture of BM Sca-1+ cells with EPDCs under hypoxia conditions increased proliferation and migration of EPDCs. Epicardial-derived cells (EPDCs, abbreviated as E) were isolated and co-cultured with bone marrow (BM) Sca-1+ cells or Sca-1- cells under normoxia and hypoxia (0.1% O2) conditions. (A) Representative images of EPDCs and immunofluorescent labeling of WT1. (B) Cell proliferation was determined by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. (C, D) Immunofluorescent staining of BrdU and quantification of BrdU+ (proliferating epicardial cells) cells. EPDCs, co-cultured with BM Sca-1+ cells or Sca-1- cells under normoxia (Nor) and hypoxia (Hypo) conditions for 72 h, were pulse-chased with BrdU (10 µM) for labeling of proliferative cells. Cell migration was evaluated by the transwell (E, F) and wound-scratch (G, H) assays after co-culture for 24 h. n=6/group, mean ± SD; **P<0.01. BrdU: bromodeoxyuridine.
Next, the expression of EMT transcriptional genes (Slug, Snail; Figure 4A), epithelial genes (BVES, Krt14; Figure 4B), and mesenchymal genes (Calponin, Cdh6, Col7a1; Figure 4C) was evaluated by RT-qPCR. The expression of EMT transcriptional and mesenchymal genes was significantly higher in the Sca-1+ cell co-cultured group compared with both the control (EPDC alone) and Sca-1- cell co-cultured groups. Conversely, the expression of EMT epithelial genes (BVES, Krt14) was significantly lower in the Sca-1+ cell co-cultured group. Accordingly, immunofluoresent labeling revealed that BM Sca-1+ cells stimulated the expression of mesenchymal proteins, such as SMA, vimentin, and calponin in EPDCs under hypoxia conditions (Figure 4D). Conversely, EPDCs were reorganized and lost the cell-cell contacts marked by ZO-1 expression (tight junction protein for EPDCs) by co-culture with BM Sca-1+ cells under hypoxia conditions (Figure 4D). The upregulation of mesenchymal proteins, vimentin and calponin, in the BM Sca-1+ cells stimulated EPDCs was confirmed by Western blot (Figure 4E). All of these data illustrated the ability of BM Sca-1+ cells to activate the EMT process in EPDCs under hypoxia conditions.
BM Sca-1+ cells activated the EMT process in EPDCs through TGF-β1 signaling
To identify the possible mediators by which BM Sca-1+ cells activate the EMT process in EPDCs, BM Sca-1+ and Sca-1- cells were isolated and subjected to hypoxia conditions. RT-qPCR was performed to screen for possible targets. Under hypoxia conditions, BM Sca-1+ cells had significantly higher expression levels of TGF-β1 (Figure 5A), PDGFB, and FGF-2 (Figure S5) than BM Sca-1- cells.
Figure 4
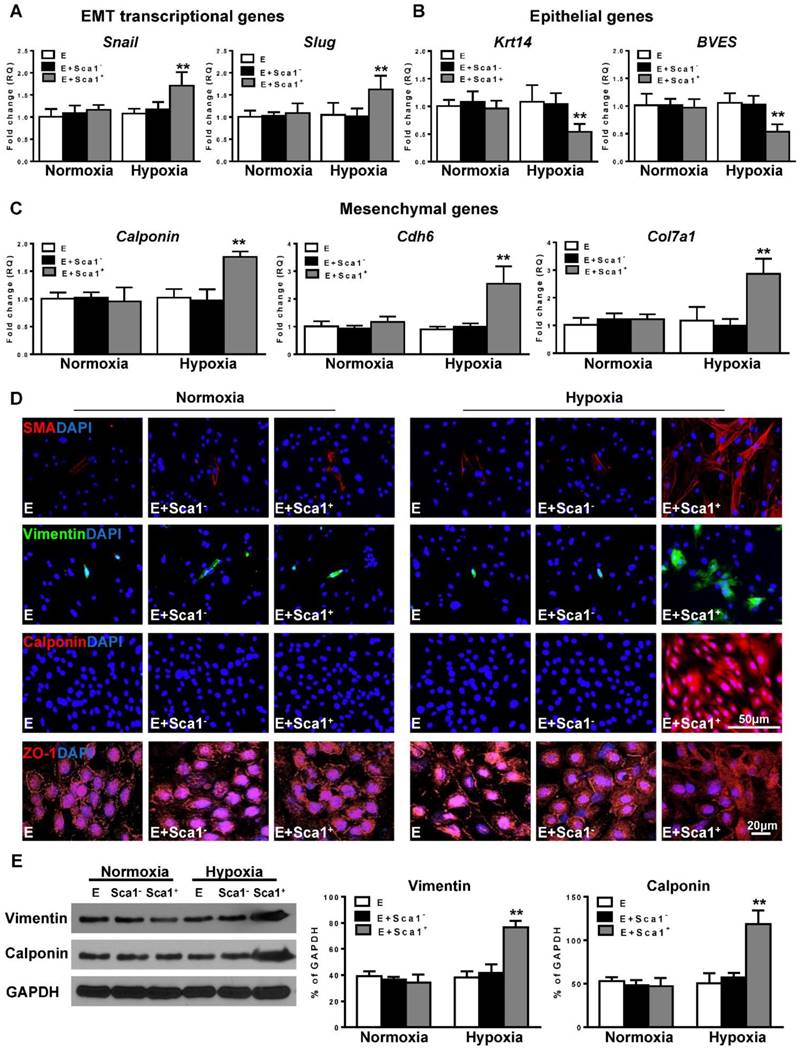
Co-culture of BM Sca-1+ cells with EPDCs under hypoxia conditions activated EMT of EPDCs. Epicardial-derived cells (EPDCs, abbreviated as E) were separately co-cultured with bone marrow (BM) Sca-1- cells or Sca-1+ cells under normoxia and hypoxia (0.1% O2) conditions for 72 h. Young Sca1+ or Sca1- BM cells (1×105/cm2) in serum-free DMEM medium were plated in the transwell cell culture inserts (1 μm diameter pores). EPDCs (2×104/cm2) were cultured in the lower compartment. Quantification of the expression of EMT transcriptional genes (A), epithelial genes (B) and mesenchymal genes (C) in EPDCs. (D) Immunofluorescent labeling of mesenchymal markers (smooth muscle actin (SMA), vimentin, calponin) and epithelial tight junction protein (ZO-1) in EPDCs. (E) Protein levels of vimentin and calponin in EPDCs were determined by Western blot and normalized by GAPDH. n=6/group, mean ± SD; **P<0.01.
An ELISA assay confirmed that BM Sca-1+ cells secreted significantly more TGF-β1 homodimer than BM Sca-1- cells (Figure 5B). TGF-β1 was also measured in the supernatant and cell lysate of EPDCs with or without hypoxia for 72 h. It showed that EPDC secreted TGF-β1 but at much lower amounts than BM Sca-1+ cells (Figure 5A-B). The level of TGF-β1 mRNA was also significantly higher in Y(Sca1+)-O than Y(Sca1-)-O chimeric hearts 3 days post-MI (Figure 5C). To confirm that TGF-β1 was the mediator involved in the BM Sca-1 cell-activation of EMT in EPDCs, EPDCs were separately co-cultured with BM Sca-1- cells, Sca-1+ cells, TGF-β1 or Sca-1+ cells with a TGF-β1 blocking antibody under hypoxia conditions. As shown in Figure 5D, BM Sca-1+ cells increased cell proliferation in EPDCs to the same extent as TGF-β1. However, this effect of BM Sca-1+ cells was completely blocked when a TGF-β1 blocking antibody was used. The same observation was found with BrdU pulse-chasing, and showed that the effect of BM Sca-1+ cells on the proliferation of EPDCs was blocked by a TGF-β1 blocking antibody (Figure 5E). Moreover, the TGF-β1 blocking antibody inhibited BM Sca-1+ cell-stimulated EPDC migration when measured by both the transwell (Figure 5F) and wound-scratch (Figure 5G) assays. On the other hand, the insufficiency of Sca1- cells to stimulate EPDC migration was rescued by the addition of TGF-β1 and EPDC migration was restored to a level comparable with the Sca-1+ cell- or TGF-β1-treated groups (Figure 5F-G).
Figure 5
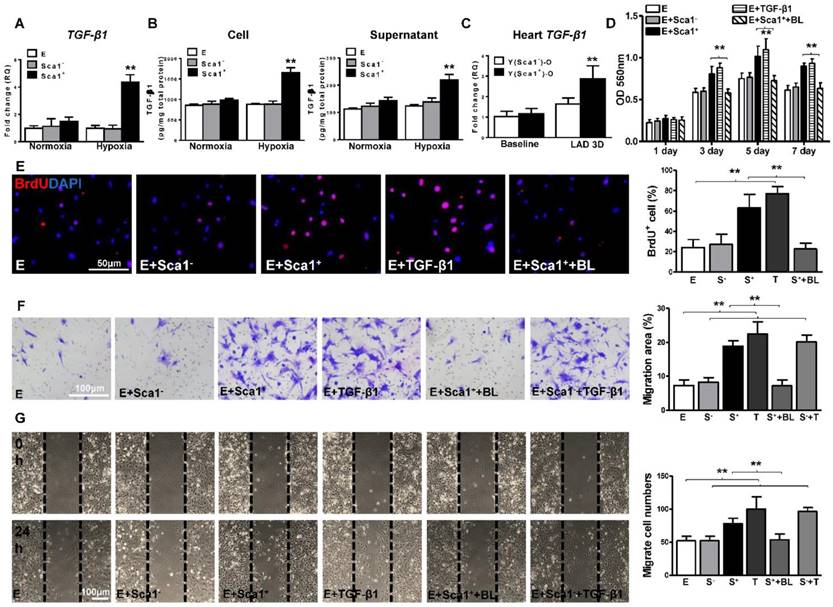
BM Sca-1+ cells increased proliferation and migration of EPDCs through TGF-β1 signaling. Epicardial-derived cells (EPDCs, abbreviated as E), bone marrow (BM) Sca-1+ and Sca-1- cells were isolated and subjected to normoxia and hypoxia (0.1% O2) conditions. (A) Higher expression levels of TGF-β1 mRNA were found in BM Sca-1+ cells than in EPDCs and Sca-1- cells under hypoxia conditions for 72 h. (B) TGF-β1 homodimer secretion in EPDCs, BM Sca-1+ and Sca-1- cell lysate and culture medium under normoxia and hypoxia conditions for 72 h was quantified by ELISA. (C) TGF-β1 mRNA expression was measured in the Y(sca-1+)-O and Y(sca-1-)-O chimeric hearts at baseline and in the infarcted area at 3 days post-MI. Epicardial-derived cells (EPDCs, abbreviated as E) were co-cultured with BM Sca-1- cells (S-), Sca-1+ cells (S+), TGF-β1 (T, 5 ng/mL), Sca-1+ cells with TGF-β1 blocking antibody (S++BL, 1 µg/mL) or Sca-1- cells with TGF-β1 (S-+T) under hypoxia conditions. The following assays were conducted in EPDCs: (D) MTT assay measured cell proliferation; (E) EPDCs, co-cultured with BM Sca-1+ cells or Sca-1- cells under normoxia and hypoxia conditions for 72 h, were pulse-chased with BrdU (10 µM) for labeling of proliferative cells; (F) transwell and (G) wound-scratch assays measured migration areas and number of EPDCs after co-culture for 24 h, respectively. Insufficiency of Sca1- cells on EPDC migration can be rescued by TGF-β1, and EPDC migration was restored to a level comparable with that of the Sca-1+ cell- or TGF-β1-treated groups. n=6/group, mean ± SD; **P<0.01. BrdU: bromodeoxyuridine; MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
To further confirm that TGF-β1 not only mediated BM Sca-1+ cell-stimulated EPDC proliferation and migration but was also involved in the activation of the EMT process by BM Sca-1+ cells, an RT-qPCR array was conducted to determine the expression levels of the genes related to EMT in EPDCs. Indeed, the EMT transcriptional genes, Snail and Slug, were upregulated by BM Sca-1+ cells to the same extent as TGF-β1 (Figure 6A) and this upregulation was lost when a TGF-β1 blocking antibody was presented (Figure 6A). Conversely, the expression of EMT epithelial genes was decreased in EPDCs co-cultured with BM Sca-1+ cells, but this effect was reversed by a TGF-β1 blocking antibody (Figure 6B). An inverse correlation was found for mesenchymal gene (calponin, Col7a1, Cdh6) expression in EPDCs where expression was increased by both BM Sca-1+ cells and TGF-β1. However, the upregulation of mesenchymal genes by BM Sca-1+ cells was blocked by adding a TGF-β1 blocking antibody (Figure 6C). In agreement with the gene expression profile, BM Sca-1+ cells increased mesenchymal protein (SMA, vimentin, and calponin) expression and reorganized epithelial tight junction protein (ZO-1) expression to a level comparable to the TGF-β1-treated EPDCs. However, the effects of BM Sca-1+ cells on the EMT protein expression of EPDCs were also blocked by the TGF-β1 blocking antibody (Figure 6D). Quantification of the mesenchymal proteins (vimentin, calponin) by Western blot showed the same trend with a TGF-β1 blocking antibody, which blocked the upregulation of mesenchymal proteins in EPDCs by BM Sca-1+ cells (Figure 6E).
Reconstitution of Sca-1 KO mice with WT BM Sca-1+ cells restored EMT response and cardiac function after MI
To further seek definitive evidence, Sca-1 KO mice were utilized and reconstituted with WT BM Sca-1+ cells for 3 months, then underwent MI. Cardiac function was evaluated by echocardiography at baseline (before MI) and at 7, 14, 21, and 28 days after MI in WT, Sca-1 KO, and Sca-1 KO reconstituted with WT BM Sca-1+ cells mice ((Sca-1+)-KO; Figure 7A). After MI, there was a significant decrease in fractional shortening (FS; Figure 7B) and ejection fraction (EF; Figure 7C) and an increase in left ventricular internal end systolic dimension (LVIDS; Figure 7D) and left ventricular internal end-diastolic dimension (LVIDD; Figure 7E) in Sca-1 KO mice compared with WT mice. However, there was an improvement in all of these parameters in the Sca-1 KO mice that received BM Sca-1+ reconstitution when compared with both WT and Sca-1 KO mice (Figure 7A-E). To assess the effect of BM cells on myocardial regeneration, the area occupied by myocytes in the infarcted zone was measured and expressed as a percentage of the total infarcted area. The fraction of viable myocardium in the total infarcted area at 28 days post-MI was significantly less in Sca-1 KO mice than in WT mice (Figure 7F-G). However, there was more viable myocardium in the total infarcted area in the Sca-1 KO mice that received BM Sca-1+ reconstitution when compared with both WT and Sca-1 KO mice, suggesting greater myocardial regeneration stimulated by BM Sca-1+ cells (Figure 7F-G). Similarly, Scar thickness was significantly lower in Sca-1 KO mice than in WT mice (Figure 7H). However, the scar thickness was even greater in Sca-1 KO mice that received BM Sca-1+ reconstitution when compared with both the WT and Sca-1 KO mice (Figure 7F-H).
To confirm that WT BM Sca-1+ cells indeed activated the EMT process. The expression of EMT embryonic epicardial genes (WT1, Tbx18), transcriptional genes (Slug, Snail), mesenchymal genes (Calponin, Cdh6, Col7a1) and epithelial genes (BVES, Krt14) were quantified by RT-qPCR in the infarcted region of WT, Sca-1 KO, and (Sca-1+)-KO mouse hearts at 3 days post-MI. The expression of EMT embryonic epicardial genes (Figure 8A), transcriptional genes (Figure 8B), and mesenchymal genes (Figure 8C) was significantly lower in Sca-1 KO than WT mice hearts at 3 days post-MI. Conversely, the expression of EMT epithelial genes was significantly higher in Sca-1 KO mice hearts compared with that of the WT mice (Figure 8D). However, after WT BM Sca-1+ cell reconstitution, the expression of EMT embryonic epicardial genes, transcriptional genes, and mesenchymal genes was significantly higher in (Sca-1+)-KO mouse hearts compared with the other two groups. And, the expression of EMT epithelial genes was significantly lower in (Sca-1+)-KO hearts compared with the other two groups (Figure 8A-D). Indeed, immunolabeling of mesenchymal proteins, SMA, vimentin, and calponin (Figure 8E), showed greater mesenchymal protein expression in the infarcted region of (Sca-1+)-KO mouse hearts compared with the other two groups at 3 days post-MI (Figure S6). These results were confirmed by Western blot showing significantly greater SMA, vimentin and calponin protein levels in the infarcted region of (Sca-1+)-KO mouse hearts compared with the other two groups (Figure 8F). All these results clearly established that reconstitution of Sca-1 KO mice with WT BM Sca-1+ cells restored the EMT response and cardiac function after MI.
Figure 6
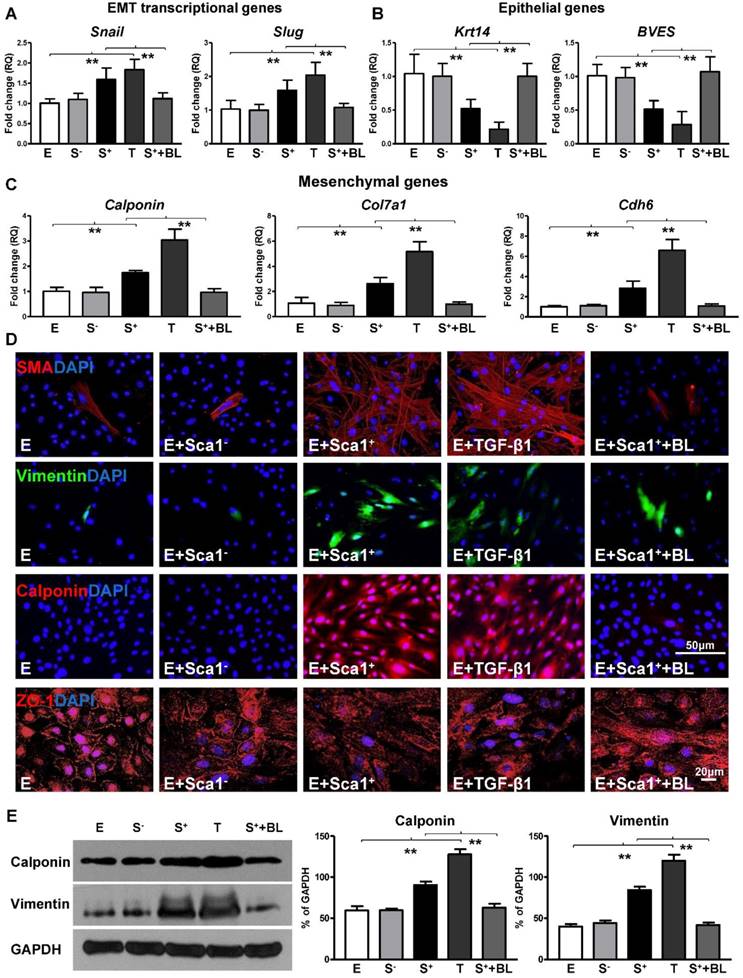
BM Sca-1+ cells activated EMT of EPDCs through TGF-β1 signaling. Epicardial-derived cells (EPDCs, abbreviated as E) were separately co-cultured with Sca-1- cells (S-), Sca-1+ cells (S+), TGF-β1 (T, 5 ng/mL) or Sca-1+ cells with a TGF-β1 blocking antibody (S++BL, 1 µg/mL) under hypoxia (0.1% O2) conditions for 72 h. Young Sca1+ or Sca1- BM cells (1×105/cm2) in serum-free DMEM medium were plated in the transwell cell culture inserts (1 μm diameter pores). EPDCs (2×104/cm2) were cultured in the lower compartment. EMT transcriptional genes (A), epithelial genes (B) and mesenchymal genes (C) in EPDCs were quantified by RT-qPCR. (D) Immunofluorescent staining of mesenchymal markers (smooth muscle actin, vimentin, calponin) and epithelial tight junction protein (ZO-1) in EPDCs. (E) The protein levels of vimentin and calponin in EPDCs were determined by Western blot and normalized by GAPDH. n=6/group, mean ± SD; **P<0.01.
Figure 7
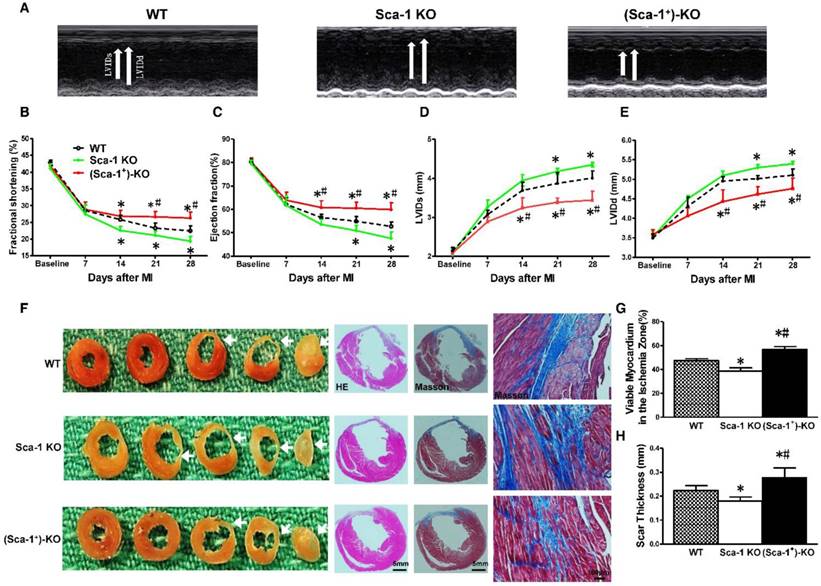
Reconstitution of Sca-1 KO mice with WT BM Sca-1+ cells restored cardiac function after MI. Sca-1 knock-out (KO) mice were reconstituted with wild type (WT) bone marrow (BM) Sca-1+ cells (2×106, (Sca-1+)-KO) for 3 months, then underwent myocardial infarction (MI). Cardiac function was measured by echocardiography at baseline (before MI), 7, 14, 21, and 28 days after MI in WT, Sca-1 KO and (Sca-1+)-KO groups. (A) Representative M-mode echocardiographic images. (B) Fractional shortening. (C) Left ventricular internal end systolic dimension (LVIDs). (D) Left ventricular internal end-diastolic dimension (LVIDd). (E) Eject fraction (EF). (F) Representative images of whole sectioned hearts; H&E and Masson's Trichrome staining in WT, Sca-1 KO and (Sca-1+)-KO groups at 28 days after MI. (G) Viable myocardium (identified as red with Trichrome's staining) in the ischemia zone was quantified and expressed as a percentage of the total infarcted area. (H) Scar size thickness is presented as an average of wall thickness measurements taken at the middle and at each edge of the scar area at its thinnest point. (B-E), n=5/group; (G-H), n=6/group; mean ± SD; * vs. WT, P<0.01; # vs. KO, P<0.05.
Discussion
The present study demonstrated that reconstitution of aged BM with young Sca-1 cells promoted the activation of the cardiac EMT process, thereby restoring cardiac function post-MI. BM Sca-1+ cells primarily homed to the epicardium and activated host epicardial cells after MI. In vitro, co-culture assays showed that BM Sca-1+ cells stimulated more proliferation and migration of epicardial cells than did BM Sca-1- cells. Subsequently, BM Sca-1+ cells activated epicardial cell EMT and increased mesenchymal protein expression under hypoxia conditions. Furthermore, we found that BM Sca-1+ cells activated the EMT process of EPDCs through TGF-β1, and blocking the TGF-β1 signaling pathway inhibited the activation of EMT by BM Sca-1+ cells. Our in vivo study also demonstrated that cardiac function after MI in Sca-1 KO mice was depressed compared with WT mice. The EMT response after MI in Sca-1 KO mice hearts was lower compared with WT mice. However, after BM reconstitution of Sca-1 KO mice with WT BM Sca-1+ cells, cardiac function was restored in the reconstituted Sca-1 KO mice and this effect was likely mediated through the activation of EMT. The present study provides further insight into the cellular and molecular mechanisms underlying the ability of BM Sca-1+ cells to improve cardiac repair post-MI. Identification of these mechanisms may provide targets for new therapies to rejuvenate the aged heart and restore heart function after a MI.
Extensive research has been done to investigate the Sca-1-related signaling pathway. It has been shown that Sca-1 may serve as a negative regulator of Src family kinase signaling [37]. Sca-1 antisense expression resulted in premature activation of Fyn, sustained proliferation, and reduced differentiation in C2C12 myoblasts [38]. Sca-1 was also shown to be involved in ERK and PI3K signaling pathway regulation. Research has revealed that the MAPK/ERK pathway plays a synergistic role with the Wnt/β-Catenin pathway to promote proliferation of Sca-1+ hepatic progenitors [39]. Sca-1 expression activated the ERK pathway and promoted the proliferation of myeloid cells during bacteremia [40]. The proliferation of BM Sca-1+ cells can be activated by FGF-2 and FGF-4 through the ERK1/2 and PI3K-Akt signaling pathways [41]. In addition, Sca-1+ cells can survive under oxygen and glucose deprivation conditions by PI3K/Akt-dependent caspase-3 down regulation [42]. In the present study, we compared differential growth factor expression between BM Sca-1+ cells and Sca-1- cells under normoxia and hypoxia conditions and found that BM Sca-1+ cells secreted more TGF-β1 homodimer than BM Sca-1- cells under hypoxia conditions, which is one of the key factors modulating the EMT process of EPDCs.
Figure 8
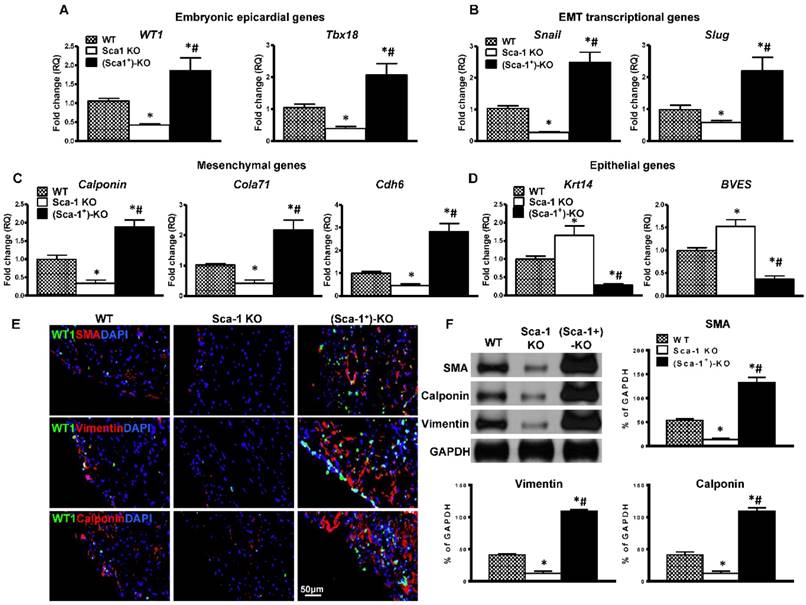
Reconstitution of Sca-1 KO mice with WT BM Sca-1+ cells restored EMT response after MI. Sca-1 knock-out (KO) mice were reconstituted with wild type (WT) bone marrow (BM) Sca-1+ cells (2×106, (Sca-1+)-KO) for 3 months, then underwent myocardial infarction (MI). Quantification of EMT embryonic epicardial genes (A), transcriptional genes (B), mesenchymal genes (C), and epithelial genes (D) at the infarcted region of WT, Sca-1 KO and (Sca-1+)-KO hearts at 3 days post-MI by RT-qPCR. (E) Immunofluorescent staining of smooth muscle actin (SMA), vimentin, and calponin at the infarcted region of WT, Sca-1 KO and (Sca-1+)-KO hearts at 3 days post-MI. (F) Protein levels of SMA, vimentin, and calponin at the infarcted region of WT, Sca-1 KO and (Sca-1+)-KO hearts were determined by Western blot and normalized by GAPDH. (A-D), n=5/group; (F), n=6/group; mean ± SD; * vs. WT, P<0.01; # vs. KO, P<0.05.
EMT is regulated through several pathways that are triggered by TGF-β, HGF, EGF, FGF, VEGF, Wnt, SHH, IL6, HIF1α, and other proteins. Among these, the TGF-β family signaling plays a predominant role. In development, the TGF-β superfamily appears to promote both epicardial and endocardial EMT. Molin et al [43], using in situ hybridization, showed that by embryonic day 12.5 the epicardium expresses all three TGF-β genes. Studies [44, 45] using cardiac explants either from 4 days in ovo embryonic chick heart or from embryonic day (E) 11.5 mouse embryos grown on collagen matrices, showed that TGF-β1 and TGF-β2 induce loss of epithelial morphology, cytokeratin, and membrane-associated ZO-1 and increase the smooth muscle markers calponin and caldesmon. Inhibition of activin receptor-like kinase (ALK) 5 blocks these effects. Sridurongrit et al. [46] using GATA5-Cre-mediated Alk5 inactivation confirmed the requirements of ALK5 and, thus, TGF-β signaling in epicardium in mouse embryos, where cGATA5-Cre-mediated Alk5 inactivation abrogated EMT in vitro and caused abnormal epicardial attachment, impaired myocardial growth, and coronary vessel abnormalities in vivo. Both Goumans [47] and Barnett [48] and colleagues, using in vitro models for human adult epicardial cells, showed that TGF-β induced loss of epithelial characteristics and initiated the onset of mesenchymal differentiation in human adult epicardial cells. All this evidence pointed to the role of TGF-β as an important EMT regulator of epicardial cells during development.
TGF-β also plays an important regulatory role under the pathological condition of MI. Following MI, TGF-β isoforms are markedly induced and rapidly expressed in the infarcted myocardium [49]. Members of the TGF-β family, via their potent effects on the epicardium, are capable of playing a central role in infarcted healing, cardiac repair and left ventricular remodeling. However, the diversity of their effects elicits multiple, and often opposing cellular responses. Experimental studies suggest that TGF-β signaling is a central mediator involved in the inflammatory and fibrotic phase of the infarcted healing [49]. Regarding its role in the inflammatory response, TGF-β is generally considered as pro-inflammatory in this setting [49]. However, its effects on macrophages are primarily deactivating, suppressing chemokine and pro-inflammatory cytokine synthesis. In addition, Frangogiannis et al. showed that TGF-β inhibits chemokine synthesis in cytokine-stimulated endothelial cells [50]. Ikeuchi et al. found that inhibition of TGF-β signaling in the limb skeletal muscles resulted in enhanced neutrophil infiltration and increased pro-inflammatory cytokine and chemokine gene expression when applied early following coronary occlusion [51]. These findings suggested an important role for TGF-β signaling in resolution of inflammation and repression of cytokine and chemokine gene synthesis. In addition, mice with disruption of TSP-1, a critical activator of TGF-β [52] or TSP-1 null mice exhibit enhanced and prolonged inflammation following MI. For the fibrotic response, TGF-β1 is mostly pro-fibrotic. TGF-β1 expression post-MI increases fibroblasts and promotes matrix deposition by inducing extracellular matrix protein expression by fibroblasts and by inhibiting matrix degradation through upregulation of TIMPs and PAI-1 [49]. TGF-β is critically involved in phenotypic modulation of fibroblasts into myofibroblasts and induces acquisition of the myofibroblastic phenotype in isolated cardiac fibroblasts [53]. Both Tsutsui [51] and Fujiwara [54] and colleagues showed that TGF-β inhibition after the inflammatory phase of infarcted healing resulted in decreased fibrous tissue deposition in the infarcted area.
Regarding therapeutic interventions of TGF-β, experimental studies suggested that the effects of anti-TGFβ treatment are depended on the timing of the intervention [54]. Anti-TGF therapy within 24 h following infarction enhanced cytokine and chemokine synthesis and increased neutrophil infiltration, resulting in exacerbated left ventricular dysfunction and increased mortality [51]. In contrast, late TGF-β inhibition decreased interstitial fibrosis in the remodeling heart, reducing left ventricular dilatation and dysfunction. Thus TGF-β inhibition during the inflammatory phase of infarcted healing is detrimental. On the other hand, late TGF-β inhibition has beneficial actions through attenuation of fibrotic and hypertrophic remodeling.
To our knowledge, the present study is the first one to identify TGF-β1 as a key modulator of BM Sca-1+ cell-mediated re-activation of EMT of EPDCs. We used an RT-qPCR array to screen for possible targets and found significantly higher expression levels of TGF-β1, PDGFB, and FGF-2 in hypoxia-treated BM Sca-1+ cells. Most importantly, by using a TGF-β1-blocking antibody, we excluded other factors and confirmed that BM Sca-1+ cells, through TGF-β1, increased EPDE migration, decreased the expression of EMT epithelial genes, induced the EMT transcriptional genes and the mesenchymal genes. Furthermore, we found that BM Sca-1+ cells, through TGF-β1, increased mesenchymal protein expression. All this evidence supported the notion that TGF-β1 was the mediator involved in BM Sca-1 cell-mediated re-activation of EMT in EPDCs. On the other hand, as mentioned above, TGF-β1 is pro-fibrotic and the epicardium, particularly in the ischemic setting, is a rich source of fibroblasts. Therefore, it is critical to understand the complexity of TGF-β signaling and modify it to therapeutically shift the balance of EPDC contribution towards EMT activation and away from fibrosis.
Reactivation of the embryonic program of epicardial cells after an MI is an essential component of the process of intrinsic cardiac repair. Recent studies in zebrafish found that, after injury, epicardial tissue can generate a new epithelial cover for the exposed myocardium: the epicardial cells undergo EMT, and the cells migrate to the wound, providing new vasculature for muscle regeneration [55]. In mammals, the fetal epicardium can also undergo EMT to form CMs, coronary ECs, smooth muscle cells, and interstitial fibroblast lineages [56]. Although the adult epicardium is quiescent under normal physiological conditions, it can be reactivated after injury and produce EPDCs [4]. However, data on the fate of EPDCs using lineage tracing in the mouse are controversial. Van Wijk et al. found that the formation of a WT1+ sub-epicardial mesenchyme, from which mostly fibroblasts were derived, resulted in a small contribution of coronary endothelium and, at later stages, CMs [4]. However, Zhou et al. found no epicardial CM or endothelial cell contribution post-MI from WT1+ EPDCs, but did find secretion of pro-angiogenic paracrine signals by WT1+ EPDCs. They postulated that EPDCs secreted growth factors that promoted angiogenesis, reduced infarct size, and improved heart function [9]. Consistent with these findings, we found reactivation of the EMT process after MI, which was characterized by active proliferation of epicardial cells and subsequent transition to the mesenchymal lineage. Most important, we demonstrated that young BM Sca-1+ cells primarily homed to the epicardium of the aged recipient and augmented the EMT process. Homed BM Sca-1+ cells stimulated proliferation and subsequent activation of EMT in host epicardial cells after MI through TGF-β1 signaling and these effects may significantly contribute to improved cardiac function.
Conclusions
We demonstrated the effects of BM Sca-1+ cells on promoting the EMT process of epicardial cells using in vivo BM reconstitution and in vitro co-culture assays. Furthermore, we identified TGF-β1 as a key modulator of BM Sca-1+ cell-mediated activation of the EMT of EPDCs. Future studies should be conducted using transgenic mice to better elucidate the cellular and molecular mechanisms responsible for BM Sca-1+ cell-mediated cardiac regeneration through reactivation of the EMT process.
Abbreviations
α-SMA: smooth muscle actin; BM: bone marrow; BrdU: bromodeoxyuridine; EF: eject fraction; EMT: epithelial-to-mesenchymal transition; EPDCs: epicardial-derived cells; LAD: ligation of the left anterior descending artery; LVIDs: left ventricular internal end-systolic dimension; LVIDd: left ventricular internal end-diastolic dimension; MI: myocardial infarction; Sca-1: stem cell antigen 1; WT1: wilms tumor 1.
Acknowledgements
We thank Dr. Leigh Botly for help with manuscript preparation and editing.
Source of Funding
This work was supported by a grant from the Canadian Institutes of Health Research (332652 to R-K.L.). R-K.L. holds a Tier 1 Canada Research Chair in Cardiac Regeneration. J.L. is a recipient of a Chinese Government Scholarship supported by the National Natural Science Foundation of China (81401156) as well as the grant from the Science and Technology Planning Project of Guangdong Province, China (2016A020215166, 2017A020215084).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Olivey HE, Svensson EC. Epicardial-myocardial signaling directing coronary vasculogenesis. Circulation research. 2010;106:818-32
2. Reese DE, Mikawa T, Bader DM. Development of the coronary vessel system. Circulation research. 2002;91:761-8
3. Zhou B, Ma Q, Rajagopal S, Wu SM, Domian I, Rivera-Feliciano J. et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature. 2008;454:109-13
4. van Wijk B, Gunst QD, Moorman AF, van den Hoff MJ. Cardiac regeneration from activated epicardium. PloS one. 2012;7:e44692
5. Smart N, Bollini S, Dube KN, Vieira JM, Zhou B, Davidson S. et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature. 2011;474:640-4
6. Winter EM, van Oorschot AA, Hogers B, van der Graaf LM, Doevendans PA, Poelmann RE. et al. A new direction for cardiac regeneration therapy: application of synergistically acting epicardium-derived cells and cardiomyocyte progenitor cells. Circulation Heart failure. 2009;2:643-53
7. Kraus F, Haenig B, Kispert A. Cloning and expression analysis of the mouse T-box gene Tbx18. Mechanisms of development. 2001;100:83-6
8. Moore AW, Schedl A, McInnes L, Doyle M, Hecksher-Sorensen J, Hastie ND. YAC transgenic analysis reveals Wilms' tumour 1 gene activity in the proliferating coelomic epithelium, developing diaphragm and limb. Mechanisms of development. 1998;79:169-84
9. Zhou B, Honor LB, He H, Ma Q, Oh JH, Butterfield C. et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. The Journal of clinical investigation. 2011;121:1894-904
10. Wilm B, Ipenberg A, Hastie ND, Burch JB, Bader DM. The serosal mesothelium is a major source of smooth muscle cells of the gut vasculature. Development. 2005;132:5317-28
11. Limana F, Bertolami C, Mangoni A, Di Carlo A, Avitabile D, Mocini D. et al. Myocardial infarction induces embryonic reprogramming of epicardial c-kit(+) cells: role of the pericardial fluid. Journal of molecular and cellular cardiology. 2010;48:609-18
12. Bollini S, Vieira JM, Howard S, Dube KN, Balmer GM, Smart N. et al. Re-activated adult epicardial progenitor cells are a heterogeneous population molecularly distinct from their embryonic counterparts. Stem cells and development. 2014;23:1719-30
13. Medici D, Hay ED, Olsen BR. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Molecular biology of the cell. 2008;19:4875-87
14. Barrallo-Gimeno A, Nieto MA. The Snail genes as inducers of cell movement and survival: implications in development and cancer. Development. 2005;132:3151-61
15. Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nature reviews Cancer. 2007;7:415-28
16. Chamulitrat W, Schmidt R, Chunglok W, Kohl A, Tomakidi P. Epithelium and fibroblast-like phenotypes derived from HPV16 E6/E7-immortalized human gingival keratinocytes following chronic ethanol treatment. European journal of cell biology. 2003;82:313-22
17. Ke XS, Qu Y, Goldfinger N, Rostad K, Hovland R, Akslen LA. et al. Epithelial to mesenchymal transition of a primary prostate cell line with switches of cell adhesion modules but without malignant transformation. PloS one. 2008;3:e3368
18. Wada AM, Reese DE, Bader DM. Bves: prototype of a new class of cell adhesion molecules expressed during coronary artery development. Development. 2001;128:2085-93
19. Huang RY, Guilford P, Thiery JP. Early events in cell adhesion and polarity during epithelial-mesenchymal transition. Journal of cell science. 2012;125:4417-22
20. Hill CR, Sanchez NS, Love JD, Arrieta JA, Hong CC, Brown CB. et al. BMP2 signals loss of epithelial character in epicardial cells but requires the Type III TGFbeta receptor to promote invasion. Cellular signalling. 2012;24:1012-22
21. Murray SA, Carver EA, Gridley T. Generation of a Snail1 (Snai1) conditional null allele. Genesis. 2006;44:7-11
22. Sakai D, Suzuki T, Osumi N, Wakamatsu Y. Cooperative action of Sox9, Snail2 and PKA signaling in early neural crest development. Development. 2006;133:1323-33
23. Inoue T, Inoue YU, Asami J, Izumi H, Nakamura S, Krumlauf R. Analysis of mouse Cdh6 gene regulation by transgenesis of modified bacterial artificial chromosomes. Developmental biology. 2008;315:506-20
24. Vindevoghel L, Lechleider RJ, Kon A, de Caestecker MP, Uitto J, Roberts AB. et al. SMAD3/4-dependent transcriptional activation of the human type VII collagen gene (COL7A1) promoter by transforming growth factor beta. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:14769-74
25. Wilkins-Port CE, Higgins PJ. Regulation of extracellular matrix remodeling following transforming growth factor-beta1/epidermal growth factor-stimulated epithelial-mesenchymal transition in human premalignant keratinocytes. Cells, tissues, organs. 2007;185:116-22
26. Sakata M, Shiba H, Komatsuzawa H, Fujita T, Ohta K, Sugai M. et al. Expression of osteoprotegerin (osteoclastogenesis inhibitory factor) in cultures of human dental mesenchymal cells and epithelial cells. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 1999;14:1486-92
27. Corallini F, Gonelli A, D'Aurizio F, di Iasio MG, Vaccarezza M. Mesenchymal stem cells-derived vascular smooth muscle cells release abundant levels of osteoprotegerin. European journal of histochemistry: EJH. 2009;53:19-24
28. Vidal NO, Brandstrom H, Jonsson KB, Ohlsson C. Osteoprotegerin mRNA is expressed in primary human osteoblast-like cells: down-regulation by glucocorticoids. The Journal of endocrinology. 1998;159:191-5
29. Smith CL, Baek ST, Sung CY, Tallquist MD. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circulation research. 2011;108:e15-26
30. Li SH, Sun Z, Brunt KR, Shi X, Chen MS, Weisel RD. et al. Reconstitution of aged bone marrow with young cells repopulates cardiac-resident bone marrow-derived progenitor cells and prevents cardiac dysfunction after a myocardial infarction. European heart journal. 2013;34:1157-67
31. Li SH, Sun L, Yang L, Li J, Shao Z, Du GQ. et al. Young Bone-Marrow Sca-1(+) Stem Cells Rejuvenate the Aged Heart and Improve Function after Injury through PDGFRbeta-Akt pathway. Scientific reports. 2017;7:41756
32. Ito CY, Li CY, Bernstein A, Dick JE, Stanford WL. Hematopoietic stem cell and progenitor defects in Sca-1/Ly-6A-null mice. Blood. 2003;101:517-23
33. Fazel S, Chen L, Weisel RD, Angoulvant D, Seneviratne C, Fazel A. et al. Cell transplantation preserves cardiac function after infarction by infarct stabilization: augmentation by stem cell factor. The Journal of thoracic and cardiovascular surgery. 2005;130:1310
34. Fazel S, Cimini M, Chen L, Li S, Angoulvant D, Fedak P. et al. Cardioprotective c-kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. The Journal of clinical investigation. 2006;116:1865-77
35. Fujii H, Tomita S, Nakatani T, Fukuhara S, Hanatani A, Ohtsu Y. et al. A novel application of myocardial contrast echocardiography to evaluate angiogenesis by autologous bone marrow cell transplantation in chronic ischemic pig model. Journal of the American College of Cardiology. 2004;43:1299-305
36. Dawn B, Tiwari S, Kucia MJ, Zuba-Surma EK, Guo Y, Sanganalmath SK. et al. Transplantation of bone marrow-derived very small embryonic-like stem cells attenuates left ventricular dysfunction and remodeling after myocardial infarction. Stem cells. 2008;26:1646-55
37. Holmes C, Stanford WL. Concise review: stem cell antigen-1: expression, function, and enigma. Stem cells. 2007;25:1339-47
38. Epting CL, Lopez JE, Shen X, Liu L, Bristow J, Bernstein HS. Stem cell antigen-1 is necessary for cell-cycle withdrawal and myoblast differentiation in C2C12 cells. Journal of cell science. 2004;117:6185-95
39. Jin C, Samuelson L, Cui CB, Sun Y, Gerber DA. MAPK/ERK and Wnt/beta-Catenin pathways are synergistically involved in proliferation of Sca-1 positive hepatic progenitor cells. Biochemical and biophysical research communications. 2011;409:803-7
40. Melvan JN, Siggins RW, Stanford WL, Porretta C, Nelson S, Bagby GJ. et al. Alcohol impairs the myeloid proliferative response to bacteremia in mice by inhibiting the stem cell antigen-1/ERK pathway. Journal of immunology. 2012;188:1961-9
41. Choi SC, Kim SJ, Choi JH, Park CY, Shim WJ, Lim DS. Fibroblast growth factor-2 and -4 promote the proliferation of bone marrow mesenchymal stem cells by the activation of the PI3K-Akt and ERK1/2 signaling pathways. Stem cells and development. 2008;17:725-36
42. Lu G, Haider HK, Jiang S, Ashraf M. Sca-1+ stem cell survival and engraftment in the infarcted heart: dual role for preconditioning-induced connexin-43. Circulation. 2009;119:2587-96
43. Molin DG, Bartram U, Van der Heiden K, Van Iperen L, Speer CP, Hierck BP. et al. Expression patterns of Tgfbeta1-3 associate with myocardialisation of the outflow tract and the development of the epicardium and the fibrous heart skeleton. Developmental dynamics: an official publication of the American Association of Anatomists. 2003;227:431-44
44. Compton LA, Potash DA, Mundell NA, Barnett JV. Transforming growth factor-beta induces loss of epithelial character and smooth muscle cell differentiation in epicardial cells. Developmental dynamics: an official publication of the American Association of Anatomists. 2006;235:82-93
45. Austin AF, Compton LA, Love JD, Brown CB, Barnett JV. Primary and immortalized mouse epicardial cells undergo differentiation in response to TGFbeta. Developmental dynamics: an official publication of the American Association of Anatomists. 2008;237:366-76
46. Sridurongrit S, Larsson J, Schwartz R, Ruiz-Lozano P, Kaartinen V. Signaling via the Tgf-beta type I receptor Alk5 in heart development. Developmental biology. 2008;322:208-18
47. Bax NA, van Oorschot AA, Maas S, Braun J, van Tuyn J, de Vries AA. et al. In vitro epithelial-to-mesenchymal transformation in human adult epicardial cells is regulated by TGFbeta-signaling and WT1. Basic research in cardiology. 2011;106:829-47
48. Olivey HE, Mundell NA, Austin AF, Barnett JV. Transforming growth factor-beta stimulates epithelial-mesenchymal transformation in the proepicardium. Developmental dynamics: an official publication of the American Association of Anatomists. 2006;235:50-9
49. Bujak M, Frangogiannis NG. The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovascular research. 2007;74:184-95
50. Frangogiannis NG, Mendoza LH, Lewallen M, Michael LH, Smith CW, Entman ML. Induction and suppression of interferon-inducible protein 10 in reperfused myocardial infarcts may regulate angiogenesis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2001;15:1428-30
51. Ikeuchi M, Tsutsui H, Shiomi T, Matsusaka H, Matsushima S, Wen J. et al. Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovascular research. 2004;64:526-35
52. Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A. et al. Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111:2935-42
53. Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S. et al. NAD(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circulation research. 2005;97:900-7
54. Okada H, Takemura G, Kosai K, Li Y, Takahashi T, Esaki M. et al. Postinfarction gene therapy against transforming growth factor-beta signal modulates infarct tissue dynamics and attenuates left ventricular remodeling and heart failure. Circulation. 2005;111:2430-7
55. Lepilina A, Coon AN, Kikuchi K, Holdway JE, Roberts RW, Burns CG. et al. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell. 2006;127:607-19
56. Cai CL, Martin JC, Sun Y, Cui L, Wang L, Ouyang K. et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature. 2008;454:104-8
Author contact
![]() Corresponding author: Shi-Ming Liu, MD, Department of Cardiology, Second Affiliated Hospital of Guangzhou Medical University, Guangzhou 510260, China. Tel: 86-020-34153522; Fax: 86-20-3415-3709; gzliushimingcom and Ren-Ke Li, MD, PhD, Toronto Medical Discovery Tower, Room 3-702, 101 College Street, Toronto, Ontario, Canada M5G 1L7. Tel: 1-416-581-7492; Fax: 1-416-581-7493; renkelica
Corresponding author: Shi-Ming Liu, MD, Department of Cardiology, Second Affiliated Hospital of Guangzhou Medical University, Guangzhou 510260, China. Tel: 86-020-34153522; Fax: 86-20-3415-3709; gzliushimingcom and Ren-Ke Li, MD, PhD, Toronto Medical Discovery Tower, Room 3-702, 101 College Street, Toronto, Ontario, Canada M5G 1L7. Tel: 1-416-581-7492; Fax: 1-416-581-7493; renkelica
Citation styles
APA
Li, J., Li, S.H., Wu, J., Weisel, R.D., Yao, A., Stanford, W.L., Liu, S.M., Li, R.K. (2018). Young Bone Marrow Sca-1 Cells Rejuvenate the Aged Heart by Promoting Epithelial-to-Mesenchymal Transition. Theranostics, 8(7), 1766-1781. https://doi.org/10.7150/thno.22788.
ACS
Li, J.; Li, S.H.; Wu, J.; Weisel, R.D.; Yao, A.; Stanford, W.L.; Liu, S.M.; Li, R.K. Young Bone Marrow Sca-1 Cells Rejuvenate the Aged Heart by Promoting Epithelial-to-Mesenchymal Transition. Theranostics 2018, 8 (7), 1766-1781. DOI: 10.7150/thno.22788.
NLM
Li J, Li SH, Wu J, Weisel RD, Yao A, Stanford WL, Liu SM, Li RK. Young Bone Marrow Sca-1 Cells Rejuvenate the Aged Heart by Promoting Epithelial-to-Mesenchymal Transition. Theranostics 2018; 8(7):1766-1781. doi:10.7150/thno.22788. https://www.thno.org/v08p1766.htm
CSE
Li J, Li SH, Wu J, Weisel RD, Yao A, Stanford WL, Liu SM, Li RK. 2018. Young Bone Marrow Sca-1 Cells Rejuvenate the Aged Heart by Promoting Epithelial-to-Mesenchymal Transition. Theranostics. 8(7):1766-1781.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.