Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(3):627-643. doi:10.7150/thno.22177 This issue Cite
Research Paper
Cardiomyocyte-Restricted Low Density Lipoprotein Receptor-Related Protein 6 (LRP6) Deletion Leads to Lethal Dilated Cardiomyopathy Partly Through Drp1 Signaling
Zhidan Chen1#, Yang Li1#, Ying Wang1#, Juying Qian1, Hong Ma1, Xiang Wang1, Guoliang Jiang1, Ming Liu1, Yanpeng An2, Leilei Ma1, Le Kang1, Jianguo Jia1, Chunjie Yang1, Guoping Zhang1, Ying Chen3, Wei Gao1, Mingqiang Fu1, Zheyong Huang1, Huiru Tang2, Yichun Zhu3, Junbo Ge1, Hui Gong1 ![]() , Yunzeng Zou1
, Yunzeng Zou1 ![]()
1. Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital, and Institutes of Biomedical Sciences, Fudan University, Shanghai 200032, China.
2. State Key Laboratory of Genetic Engineering, Zhongshan Hospital and School of Life Sciences, Fudan University. International Centre for Molecular Phenomics, Collaborative Innovation Center for Genetics and Development, Shanghai 200438, China.
3. Department of Physiology and Pathophysiology, Shanghai Medical College, Fudan University, Shanghai 200032, China.
# These authors contributed equally to this work.
Received 2017-7-31; Accepted 2017-10-14; Published 2018-1-1
Abstract

Low density lipoprotein receptor-related protein 6 (LRP6), a wnt co-receptor, regulates multiple functions in various organs. However, the roles of LRP6 in the adult heart are not well understood.
Methods: We observed LRP6 expression in heart with end-stage dilated cardiomyopathy (DCM) by western blot. Tamoxifen-inducible cardiac-specific LRP6 knockout mouse was constructed. Hemodynamic and echocardiographic analyses were performed to these mice.
Results: Cardiac LRP6 expression was dramatically decreased in patients with end-stage dilated cardiomyopathy (DCM) compared to control group. Tamoxifen-inducible cardiac-specific LRP6 knockout mice developed acute heart failure and mitochondrial dysfunction with reduced survival. Proteomic analysis suggests the fatty acid metabolism disorder involving peroxisome proliferator-activated receptors (PPARs) signaling in the LRP6 deficient heart. Accumulation of mitochondrial targeting to autophagosomes and lipid droplet were observed in LRP6 deletion hearts. Further analysis revealed cardiac LRP6 deletion suppressed autophagic degradation and fatty acid utilization, coinciding with activation of dynamin-related protein 1 (Drp1) and downregulation of nuclear TFEB (Transcription factor EB). Injection of Mdivi-1, a Drp1 inhibitor, not only promoted nuclear translocation of TFEB, but also partially rescued autophagic degradation, improved PPARs signaling, and attenuated cardiac dysfunction induced by cardiac specific LRP6 deletion.
Conclusions: Cardiac LRP6 deficiency greatly suppressed autophagic degradation and fatty acid utilization, and subsequently leads to lethal dilated cardiomyopathy and cardiac dysfunction through activation of Drp1 signaling. It suggests that heart failure progression may be attenuated by therapeutic modulation of LRP6 expression.
Keywords: LRP6, Autophagy, Heart failure, mTOR, TFEB
Introduction
Cardiomyopathy, such as hypertrophic cardiomyopathy or dilated cardiomyopathy, is one of the most common causes of heart failure that is characterized by cardiac remodeling and contractile dysfunction. Genetic mutations and altered expression of many signaling molecules and structural proteins have been indicated in the development of cardiomyopathy [1, 2]. However, the molecular mechanisms underlying the progression from adaptive cardiac remodeling to decompensated heart failure remain largely unknown. Strategies targeting new molecules or factors are urgently needed to prevent this transition under pathological condition. In this study, we describe low density lipoprotein receptor-related protein 6 (LRP6) as a novel regulator in the progression of cardiac dysfunction.
LRP6, belonging to the low density lipoprotein (LDL) receptor-related family, is a transmembrane cell surface protein and a wnt coreceptor, inducing the wnt canonical signaling pathway involved in embryonic development[3], body fat and glucose homeostasis[4], and bone cell metabolism[5] etc. It has been widely studied in the development of several organs including cardiac specification and morphogenesis [6]. In addition to organ development, a potential function of LRP6 in coronary heart disease owing to high blood lipid has been reported [7, 8]. The mutation Y418H in LRP6 weakens the wnt3a signaling pathway and impairs endothelial cell functions, which contributes to normolipidemic familial coronary artery disease (CAD) [9]. Moreover, the C allele of rs11054731 within LRP6 was an independent risk factor for CAD in Chinese [10]. In our recent study, we found the activation of LRP6 was associated with the proliferation of cardiac side population cells during pressure overload in vitro and in vivo [11]. Despite recent reports that LRP6 acts as a scaffold protein regulating cardiac gap junction assembly [12], surprisingly little is known about the role of LRP6 in the adult heart, especially in adult cardiomyocyte or the development of heart failure.
Here, we will explore: 1) the expression of LRP6 in human heart tissue with dilated cardiomyopathy; 2) the role of LRP6 in the development of heart failure by inducible cardiac-specific LRP6 knockout mice; and 3) the potential molecular mechanism. This study allowed us to identify a novel role of LRP6 in heart failure, indicating that modulation of LRP6 may be a new strategy to prevent heart failure.
Methods
Detailed methods are described in the Supplementary Material.
Human heart samples
Left ventricular samples from patients with dilated cardiomyopathy were obtained from explanted hearts undergoing heart transplantation at Zhongshan hospital, Fudan University. Non-failing hearts without obvious cardiac dysfunction were obtained from organ donor networks or cardiac donors whose hearts were rejected for transplant. For western blot analysis and immunostaining experiments, 3 rejected healthy donor hearts and 3 end-stage heart failure patients with dilated cardiomyopathy were studied.
Mouse models
LRP6 flox homo (fl/fl) mice (LRP6fl/fl) were kindly gifted by Prof. Bart O. Williams (Skeletal Disease and Tumor Microenvironment and Center for Cancer and Cell Biology, Van Andel Research Institute, Michigan USA). Cardiac-specific conditional LRP6 knockout mice were generated by crossing LRP6fl/fl and α-myosin heavy chain (α-MHC) Mer-Cre-Mer Tg mice (α MHC-MCM, termed MCM), and LRP6 expression was knocked out in cardiomyocytes by tamoxifen injection (30 mg/kg i.p.) for 3 consecutive days. Cardiac-specific heterozygous LRP6 knockout (MCM/LRP6fl/+) mice were generated by crossing LRP6 flox hetero (LRP6fl/+) mice and MCM mice. Mdivi-1 (50 mg/kg, i.p.) was injected to MCM/LRP6fl/fl mice at 12 h after tamoxifen or diluent injection, and dimethyl sulphoxide (DMSO) treatment was used as control.
Mitochondrial function analysis
Cardiac mitochondria were isolated using a mitochondrial isolation kit (Beyotime). ATP (adenosine triphosphate) levels in heart tissue were determined using an ATP assay kit (Beyotime). Mitochondrial membrane potential was detected using a mitochondrial membrane potential assay kit (Beyotime). Mitochondrial complex activities were examined by MitoCheck Complex I, II-III (Cayman Chemical Company, USA) and IV (Sigma USA). All the details are described in supplementary methods.
Echocardiography and hemodynamic analysis
Echocardiography was performed at the determined time. Mice were anaesthetized with inhalation of isoflurane and M-mode images were obtained with a RMV 707 scan head on the Vevo 770 (VisualSonics Inc., Toronto, Canada). Left ventricular cavity diastolic dimension (LVID;d) and wall thickness in left ventricular diastolic anterior wall (LVAW;d), left ventricular diastolic posterior wall (LVPW;d) and ejection fraction (EF) were assessed. Heart rate (HR) was maintained at more than 450 bpm. Hemodynamic assessment was performed by a 1.4 F pressure catheter (SPR 671, Millar Instruments) inserted into the aorta and left ventricle through the right common carotid artery. Left ventricular systolic pressure (LVSP), left ventricular end diastolic pressure (LVEDP), left ventricular developed pressure (LVDP), +dp/dt and -dp/dt were recorded by Powerlab system (AD Instruments, Castle Hill, Australia) through the transducer.
Mass spectrometry-based proteomics
The detailed methods are described in the Supplementary Methods.
GC-FID/MS analysis of fatty acid composition in heart tissue
The detailed methods are described in the Supplementary Methods.
Immunofluorescence staining
Frozen left ventricular tissue slides were incubated with LRP6 antibody (1:50; Cell signaling) and Alexa Fluor® Plus 488 conjugated goat anti-rabbit secondary antibody (1:1000 Life Technology). Cardiomyocytes were isolated from adult mice as in a previous study [13]. Acute isolated adult cardiomyocytes were incubated in MitoTracker (1:5000, invitrogen) for 5 min. The cells were fixed with 4% paraformaldehyde followed by blocking with 2% BSA in PBS, and then incubated with LRP6 antibody as mentioned above. To observe cardiomyocyte hypertrophy, the left ventricular tissue slides were incubated with wheat germ agglutinin (WGA) Alexa Fluor 568 conjugate (1:100; Invitrogen). Fluorescence images were obtained using a confocal microscope (Zeiss, Germany).
Western blot analysis
The heart tissue or cardiac mitochondria were lysed for western blot analysis in a standard routine with specific antibodies (Table S1). Quantitative analysis was performed by LAS-3000 imaging system (FUJIFILM Inc, Tokyo, Japan).
Morphological analysis
Hematoxylin and eosin (HE) staining was performed to examine cardiac structure as in a previous study [14]. The ultrastructure of heart tissue was analyzed by transmission electron microscopy. The accumulation of lipid droplets in heart tissue was analyzed by Oil red O staining. All detailed methods are described in Supplementary Methods.
Statistical analysis
Data are expressed as mean ± SEM. Student's t-test was applied to two group comparisons. One-way ANOVA with Bonferroni post-hoc test was used for multiple group comparisons. The difference in mean values between tamoxifen or diluent injected MCM or MCM-LRP6fl/fl was evaluated by two-way ANOVA, followed by Bonferroni post-tests. Statistical analyses were carried out using Prism 5 (GraphPad). Values of p < 0.05 were considered statistically significant.
Study approval
All animal experiment protocols were approved by the Animal Care and Use Committee of Fudan University and conformed to the Guidelines for the Care and Use of Laboratory Animals published by the NIH. All human heart tissue samples were obtained in accordance with ethics committee approval (Approval No: B2014-084) at Zhongshan hospital, Fudan University. Consent for research use of explanted heart tissues or the donor heart tissues was obtained in all cases. A detailed protocol and informed consent documents were reviewed by Fudan University, Zhongshan Hospital Review Board.
Results
LRP6 is downregulated in heart tissue of patients with dilated cardiomyopathy
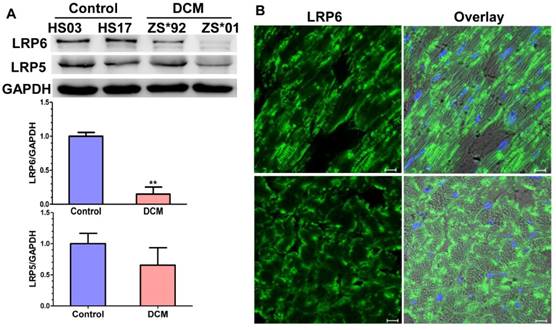
We first quantified LRP6 and LRP5 protein levels in heart samples from patients with end-stage dilated cardiomyopathy (DCM) or subjects without obvious cardiovascular diseases (Table S3), and observed that LRP6 was sharply downregulated in heart tissues with DCM (Figure 1A, Figure S1). Although LRP5 showed reduced expression in heart samples with DCM, there was no statistical significance between the two groups. Immunofluorescence staining of heart tissue from C57BL/6 mice revealed LRP6 was predominately expressed in membranes of cardiomyocytes (Figure 1B). These data suggest that downregulation of LRP6 in cardiomyocytes may be involved in the development of cardiac dysfunction in patients with DCM.
LRP6 expression in heart tissues. (A) Western blot analysis of LRP6 and LRP5 expression in human heart tissues. 3 rejected healthy donor hearts (control) and 3 end-stage heart failure patients with dilated cardiomyopathy (DCM) were studied. ** p<0.01 vs. control group. (B) Immunofluorescence staining of LRP6 expression in left ventricular tissue from C57BL/6 mice. Green: LRP6; blue: nucleus. Scale bars, 10 μm.
Cardiomyocyte-specific deletion of LRP6 causes dilated cardiomyopathy
To investigate whether knockdown of LRP6 in cardiomyocytes contributes to maladaptive cardiac dysfunction in DCM heart, we employed the tamoxifen-inducible Mer-Cre-Mer system to delete LRP6 specifically in cardiomyocytes. Mice with a “floxed” LRP6 gene were bred to the tamoxifen-inducible α-myosin heavy chain (α-MHC) Mer-Cre-Mer Tg mice (termed MCM) to generate α-MHC Mer-Cre-Mer/LRP6fl/fl mice (termed MCM/LRP6fl/fl). MCM or MCM/LRP6fl/fl male mice aged 8-12-week were used in the present study. Echocardiographic analysis revealed no difference in basal cardiac function between MCM and MCM-LRP6fl/fl mice before tamoxifen injection and deletion of LRP6 (Table S4). After intraperitoneal injection of tamoxifen for 3 consecutive days, MCM/LRP6fl/fl allowed for inducible deletion of LRP6 specifically in cardiomyocytes. Experimental strategy in vivo is shown in Figure 2A. LRP6 level declined ~90% in myocardial tissue (Figure 2B) while the levels were unaffected in the kidney (Figure S2A) and other tissues (data not shown) from tamoxifen-injected-MCM/LRP6fl/fl. LRP5 expression didn't show alteration in these mice (Figure S2C). Diluent-injected littermate MCM-LRP6fl/fl and tamoxifen-injected littermate MCM or LRP6fl/fl served as control groups. Tamoxifen injection didn't induce the decrease in cardiac LRP6 expression in MCM mice (Figure S2B). Surprisingly, we observed that ~35% mice died and most of the surviving mice developed overt cardiac dysfunction within 7 days after tamoxifen injection in MCM/LRP6fl/fl, whereas none of tamoxifen-injected MCM died during the experimental process after tamoxifen injection (Figure 2C).
Cardiomyocyte-specific deletion of LRP6 causes cardiac dysfunction. (A) Schematic presentation of experimental strategy in vivo. (B) Western blot analysis of LRP6 expression in heart tissue from MCM/LRP6fl/fl mice after treatment with diluent or tamoxifen for 3 days. ** p<0.001 vs. diluent group (n=3/group). (C) Survival proportion was analyzed in MCM mice and MCM/LRP6fl/fl mice for 60 days following three-day-treatment with tamoxifen. n=14/MCM/LRP6fl/fl group, n=6/MCM group. (D) Echocardiographic analysis of fraction shortening (FS) in MCM or MCM/LRP6fl/fl injected with tamoxifen at different time-points during the experimental process. n=7-14/MCM/LRP6fl/fl group, n=6/MCM group. (E) Representative M-Mode echocardiographs. (F-G) Fraction shortening (FS) and left ventricular cavity diastolic dimension (LVID.d) were analyzed by echocardiography. n=6-8/group. (H-K) dp/dt, -dp/dt, left ventricular systolic pressure (LVSP) and left ventricular end diastolic pressure (LVEDP) were determined by hemodynamic analysis. n=8/group. (L) Representative photographs of gross hearts. (M) Hematoxylin-eosin (HE) stains of longitudinal heart sections. Scale bars, 1 mm. n=3-8/group. (N) Histological analysis of heart tissues. Upper lane: WGA staining, scale bars, 20 μm; middle lane: HE staining, scale bars, 10 μm; lower lane: oil red O staining, scale bars, 50 μm. n=3-10/group. (O) Heart/body ratio (HW/BW) and (P) Cross section area (CSA) of cardiomyocyte were quantitatively analyzed. HW/BW: n=6-12/group; CSA: n=3/group. MCM or MCM/LRP6fl/fl mice were injected with diluent or tamoxifen respectively for 3 consecutive days. These parameters were analyzed from day 2 to day 4 post-injection of tamoxifen or diluent. *p<0.05; **p<0.01; **p<0.001 vs. diluent group.
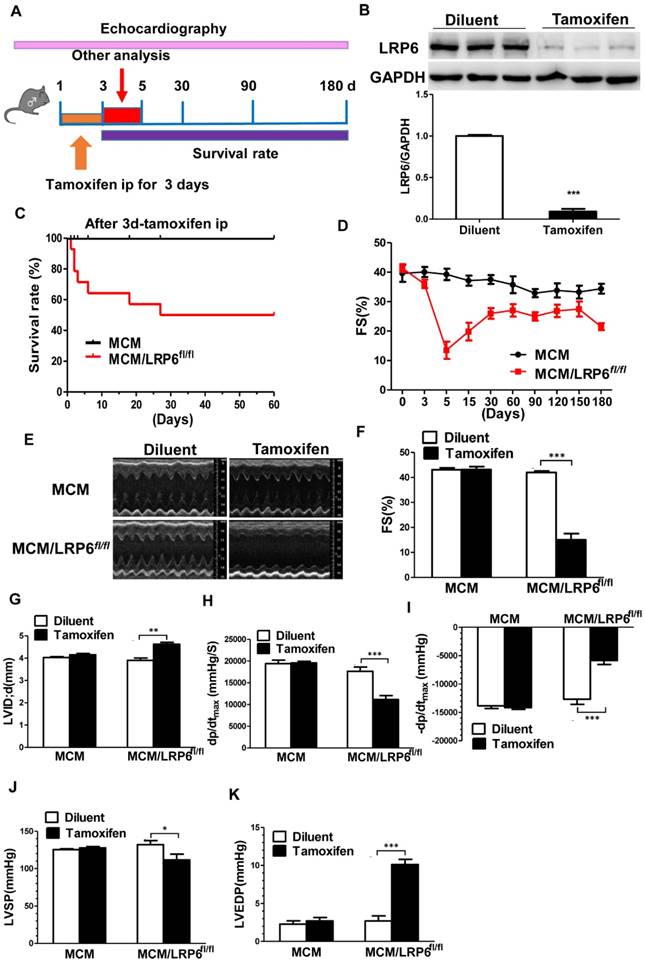
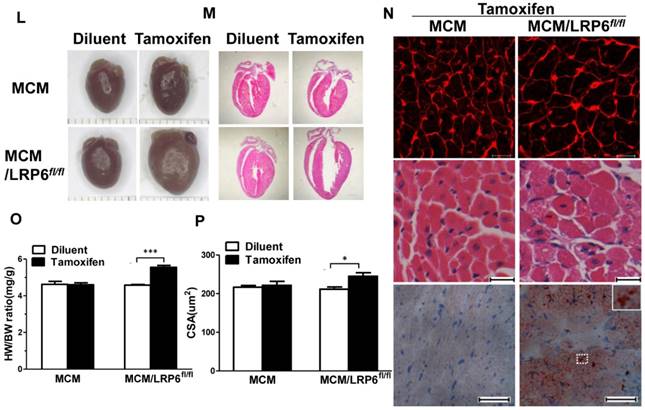
Fractional shortening (FS) showed a sharp decline in MCM/LRP6fl/fl starting at day 2 post-tamoxifen injection; it recovered in some mice that survived but remained much lower than that in control mice through the whole duration of the study (Figure 2D-F). Ejection fraction (EF) showed similar results (Figure S3A). Ventricular M-mode imaging showed left ventricular cavity diastolic dimension (LVID;d) was greater, but left ventricular anterior diastolic walls (LVAW;d) and posterior diastolic walls (LVPW;d) were thinner in tamoxifen-injected MCM-LRP6fl/fl than in diluent-injected mice (Figure 2G, Figure S3B,C). Cardiomyocyte-specific deletion of LRP6 also resulted in significant decreased left ventricle systolic pressure (LVSP), increased left ventricular diastolic pressure (LVEDP), and impaired peak rates of ventricular contraction and relaxation (±dp/dtmax) (Figure 2H-K). Moreover, tamoxifen-injected MCM-LRP6fl/fl showed significantly enlarged heart size and increased heart weight/body weight ratio (HW/BW) or lung weight/body weight (LW/BW) ratio compared with control mice (Figure 2O, Figure S3G). Histological analysis of H&E-stained longitudinal sections of hearts confirmed dilated ventricular chamber with evidence of cardiomyocyte hypertrophy in tamoxifen-injected MCM-LRP6fl/fl compared to control mice (Figure 2L-P). In addition, H&E staining revealed vacuolated myocytes and oil red O staining showed positive red staining in tamoxifen-injected MCM/LRP6fl/fl hearts, indicating intracellular accumulation of lipid (Figure 2N). In MCM mice, tamoxifen injection didn't induce these mentioned effects. But tamoxifen-injected MCM/LRP6fl/fl showed little apoptosis or fibrosis in left ventricular tissue as control group by TUNEL staining or Masson's trichrome staining (data not shown).
In heterozygous mice (termed MCM/LRP6fl/+), LRP6 level declined ~50%, but EF or FS didn't alter after tamoxifen injection (Figure S4A,B). None of these mice died or exhibited any signs of cardiac dysfunction during 30 days observation after tamoxifen injection. However, within 2 weeks (adaptive stage of pressure overload) after transverse aorta constriction (TAC), tamoxifen-injected-MCM/LRP6fl/+ were susceptible to sudden cardiac death (43%). Both EF and FS significantly decreased in tamoxifen-injected-MCM/LRP6fl/+ compared to sham group, while they showed little difference in diluent-injected-MCM between TAC and sham group (Figure S4C). This suggests that downregulation of LRP6 in cardiomyocytes predisposes mice to cardiac dysfunction or heart failure in response to pressure overload.
Taken together, cardiomyocyte-specific deletion of LRP6 causes lethal dilated cardiomyopathy and cardiac dysfunction in adult mice.
Cardiomyocyte-specific deletion of LRP6 greatly impairs architecture of intercalated disk, and induces the accumulation of autophagosome and lipid droplet
Electron microscopy revealed abnormal intercalated disks in the LRP6 deletion heart. In normal myocardium, the long and step-like indercalated disk (ID) could be frequently observed (21 intercalated disks from 7 mice), but in the LRP6 deletion heart, 13 IDs (21 intercalated disks from 7 mice were observed) displayed the obvious wide gap junction or the wavy or bent Z disk between the adjacent cardiomyocytes, and 2 IDs were destroyed and difficult to detect (Figure S5A). In addition to abnormal IDs, we observed obvious mitochondrial targeting to autophagosomes and lipid accumulation in left ventricular sections from the LRP6 deletion heart (Figure 3A). In heterozygous mice, electron microscopy analysis revealed some mitochondrial targeting to autophagosomes but no obvious lipid droplets or abnormal ID (Figure S5B).
Cardiomyocyte-specific deletion of LRP6 impairs mitochondrial function. (A) Representative electron microscopy images of hearts from MCM/LRP6fl/fl or MCM at day 2 after 3-day consecutive injection with tamoxifen. Scale bars: 5 μm, n=3/group. (B) Relative cardiac ATP production in MCM or MCM/LRP6fl/fl at day 2 after 3-day consecutive injection with tamoxifen or diluent. *p<0.05 vs. diluent-injected-MCM/LRP6fl/fl. n=3-6/group. (C) Mitochondrial membrane potential assessment with JC-1 staining. Mitochondria were freshly isolated from heart tissue of mice as in B. *p<0.05; vs. diluent-injected-MCM/LRP6fl/fl. n=4-8/group. (D) Mitochondrial swelling induced by Ca2+ (50 μM). Left panel: each data curve represents the average of 3 individual measurements. Right panel: the decrease in optical density at 520 nm at 10 min induced by Ca2+. *p<0.05 vs. MCM group. n=3/group. (E-G) Mitochondrial respiratory chain complex I, II/III, and IV activity, respectively. The activity in mitochondria from diluent-injected MCM hearts or MCM/LRP6fl/fl ones is expressed as 1. *p<0.05, **p<0.01 vs. diluent-injected mice. n=3/group. (H-J) GC-FID/MS analysis of fatty acid composition in heart tissue from tamoxifen-injected MCM or MCM/LRP6fl/fl mice. Quantitative analysis of C16:0, C18:0, C18:1n9, C18:2n6 (H) UFA, PUFA, SFA, MUFA (I) and PUFA/UFA, PUFA/MUFA, MUFA/UFA, PUFA/SFA (J) in heart tissue. PUFA: polyunsaturated fatty acids; UFA: unsaturated fatty acids; MUFA: monounsaturated fatty acids; SFA: saturated fatty acids. *p<0.05; **p<0.01 vs. MCM mice. n=10/group.
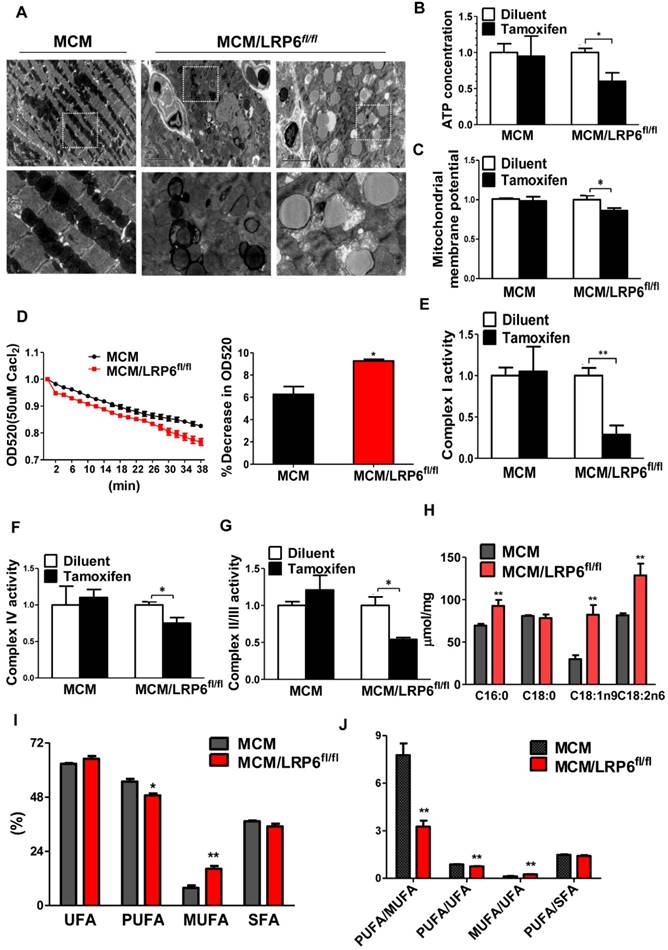
Cardiac ID contains a complex protein network that is critical for electrical continuity and chemical communication between adjacent cardiomyocytes [15]. We then performed electrocardiogram measurement to examine whether the wide gap junction induced by LRP6 deletion would lead to arrhythmias. No obvious arrhythmia events were observed in cardiac LRP6 deletion mice (data not shown). Cardiac mitophagy or lipid accumulation is accompanied by mitochondrial dysfunction [16]. In parallel, cardiac ATP level was significantly lower in tamoxifen-injected MCM/LRP6fl/fl than in diluent-injected ones (Figure 3B). Mitochondrial membrane potential was evaluated with JC-1 assay. Tamoxifen-injected MCM/LRP6fl/fl showed a decrease of red to green fluorescence ratio, indicating depolarization of mitochondrial membrane potential in myocardium (Figure 3C). Furthermore, mitochondrial swelling assay revealed that decrease in absorbance at 520 nm (an indicator of mPTP opening) was significantly greater in isolated cardiac mitochondria from tamoxifen-injected MCM/LRP6fl/fl than that from tamoxifen-injected MCM, suggesting that mPTP opening is accelerated by cardiomyocyte-specific LRP6 deletion (Figure 3D).
Mitochondrial complexes I, II/III, and IV activities were also significantly attenuated in MCM/LRP6fl/fl hearts after tamoxifen injection compared to those in control mouse hearts (Figure 3E-G). In MCM mice, tamoxifen injection showed little difference in these above-mentioned effects. Thus, cardiac LRP6 depletion resulted in mitochondrial dysfunction, which impaired the production of ATP, contributing to severe heart failure. Immunostaining and western blot analysis showed LRP6 is expressed on the membrane of cardiomyocytes but not in mitochondria (Figure S6A, B). The expressions of β-catenin in cardiac nuclei or cytosol showed little difference between diluents- and tamoxifen- injected MCM/LRP6fl/fl (Figure S7A, Figure 5). There was no difference in active β-catenin expression in heart tissue between LRP6 deletion mice and control ones (Figure S7B). The mRNA of Axin2, Lef1 Tcf7l2, and β-catenin target genes revealed little alteration in LRP6 deletion hearts compared to control ones (Figure S7C). This suggests that LRP6 has little possibility of directly regulating mitochondrial function in cardiomyocytes, and the deletion-induced mitochondrial dysfunction may be independent of canonical wnt signaling.
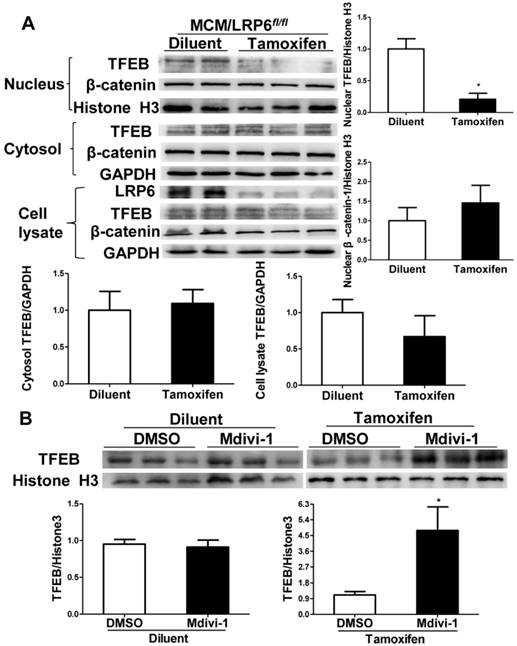
Cardiomyocytes-specific deletion of LRP6 inhibits TFEB nuclear translocation. (A) Western blot analysis of TFEB and β-catenin expression in nucleus, cytosol and tissue lysates of hearts from diluent-injected or tamoxifen-injected MCM/LRP6fl/fl. *p<0.05 vs. diluent-injected MCM/LRP6fl/fl. n=4/group. (B) Western blot analysis of nuclear TFEB expression in hearts from tamoxifen-injected MCM/LRP6fl/fl with pre-injection of DMSO or Midvi-1. *p<0.05 vs. DMSO group; n=6/group. Analysis of hearts from MCM/LRP6fl/fl at day 1 after 3-day consecutive injection with tamoxifen or diluent.
Cardiomyocyte-specific deletion of LRP6 greatly impairs autophagic degradation and fatty acid utilization
To identify the molecular function of LRP6 in the heart, protein expression profiling was analyzed by proteomics in heart tissue from MCM/LRP6fl/fl at day 1 after three-day treatment with tamoxifen or diluent. 64 proteins were upregulated and 15 proteins were downregulated in MCM/LRP6fl/fl mice treated with tamoxifen compared to those treated with diluent (Figure S8A). Function analysis revealed differentially expressed proteins were involved in multiple functions such as cell-cell signaling and interaction, inflammatory response, metabolic disease, lipid metabolism and molecular transport, etc. (Figure S8B, Table S5). In addition to involvement in cell-cell signaling and interaction, most of the proteins that were altered by LRP6 deletion were mapped onto mitochondrial pathways and fatty acid metabolism such as peroxisome proliferator-activated receptors (PPARs) activation, ERK/MAPK signaling and caveolar-mediated signaling etc (Table S6). Fatty acid analysis by GC-FID/MS indicated LRP6 deletion induced the higher levels of C16:0, C18:1n9 and C18:2n6 than in control hearts. The LRP6 deletion heart also has higher MUFA level and MUFA-to-UFA ratio with lower PUFA level, PUFA-to-MUFA and PUFA-to-UFA ratios than controls (Figure 3H-J). Further western analysis confirmed that PPARα and PPARδ, the prominent regulators for maintaining basal cardiac fatty acid oxidation, decreased sharply, while PPARγ coactivator-1α (PGC-1α), caveolar 3 (muscle-specific caveolin family member), and lipoprotein ligase showed little alteration in LRP6 deletion hearts compared to control hearts (Figure 4A, Figure S9A). The activation of ERK1/2 showed little difference between the two groups (Figure S9B). Quantitative PCR analysis revealed little alteration in mt-DNA-encoded genes: mt-Cytb (encoding cytochrome b), mt-Co1 (encoding the cytochrome c oxidase subunit 1) and mt-Nd1(encoding NADH dehydrogenase-1) in the LRP6 deficient heart (Figure S9C). We also detected mRNAs of those genes related to lipogenesis. These included acetyl CoA carboxylase 1 (Acc1), stearoyl-CoA desaturase (Scd) and fatty acid synthase (Fas). There were no differences in Acc1, Scd1 and Fas mRNAs levels between LRP6 deletion heart and control heart (Figure S9D). These data suggest that cardiac LRP6 deletion has little effect on mitochondrial biogenesis or fatty acid synthesis, but it may impair fatty acid utilization by decreasing the expression of PPARs which leads to lipid accumulation in the heart.
Cardiomyocyte-specific deletion of LRP6 inhibits autophagic degradation. (A) Western blot analysis of LC3-II, PPARα, PPARδ, PGC1α and LC3b (LC3-I: the upper band; LC3-II: the down band) in heart tissue from diluent or tamoxifen-injected MCM or MCM/LRP6fl/fl mice (B) Western blot analysis of LRP6, p-Drp1, Drp1, p-mTOR, mTOR, p-AMPK, AMPK, PINK1 in heart tissue from the same mice as in (A). (C) Western blot analysis of p-Drp1, Drp1, PINK1, VDAC1, Found1, Parkin, Tom22, expression in cardiac mitochondria in tamoxifen-injected MCM or MCM/LRP6fl/fl mice. *p<0.05, **p<0.01vs. diluent-injected MCM/LRP6fl/fl (n=3-4/group). Analysis of hearts or cardiac mitochondria from MCM/LRP6fl/fl or MCM at day 1 after 3-day consecutive injection with tamoxifen or diluent.
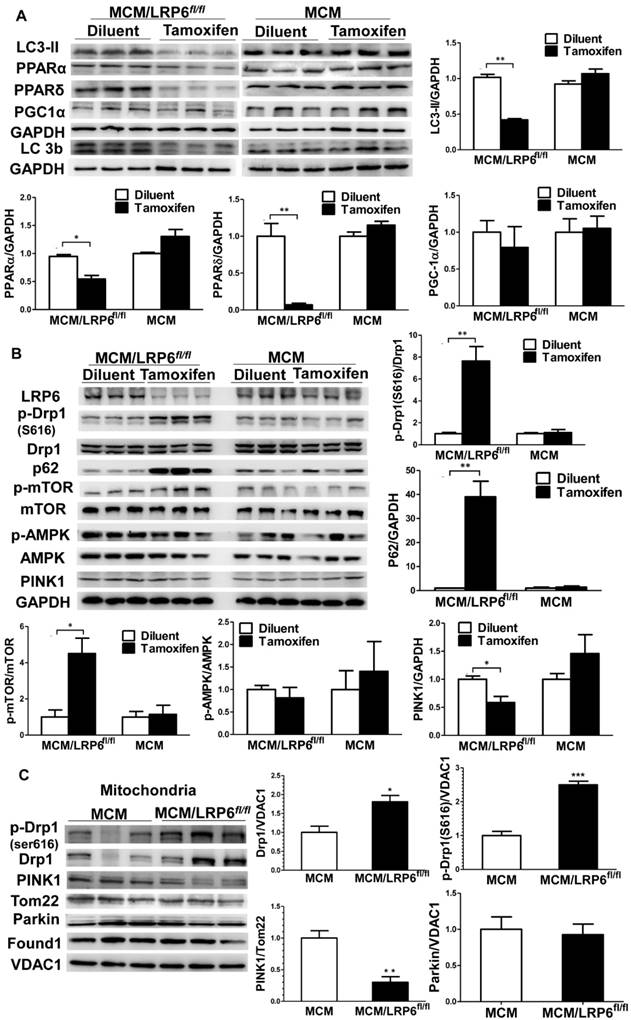
A recent study suggests there is crosstalk between autophagy and lipid metabolism [17]. Given that obvious mitochondrial targeting to autophagosomes was observed in LRP6 deletion hearts, we next investigated the role of LRP6 in mediating autophagy in myocardium in vivo. There was significantly less LC3-II, an indicator of autophagic flux, and significantly more p62, a protein degraded by autophagy, in hearts from tamoxifen-injected MCM/LRP6fl/fl than those from diluent-injected ones (Figure 4A,B). These data suggest cardiac LRP6 deletion may result in the inhibition of autophagic or mitophagic degradation. Selective mitophagic elimination usually requires Parkin, a cytosolic ubiquitin ligase, to be translocated to damaged mitochondria from cytosol in PTEN-induced putative kinase 1 (PINK1, the mitochondrial kinase)-dependent manner [18, 19]. We observed that cardiac PINK1 expression and its translocation to mitochondria were both suppressed, and mitochondrial Parkin expression didn't change significantly in tamoxifen-injected-MCM/LRP6fl/fl hearts compared to those in control hearts (Figure 4B,C). Dynamin-related protein 1 (Drp1), a cytoplasmic GTPase, which can be recruited to mitochondria, plays an important role in autophagy or mitophagy[20]. LRP6 deletion promotes cardiac Drp1 phosphorylation at ser616 and its translocation to mitochondria (Figure 4B,C). In myocardial sections from DCM patients, p-Drp1 (ser616) was higher than in control group (Figure S10). The activation mammalian target of rapamycin (mTOR) by stress or hormones usually suppresses proteolysis by proteasomes or autophagy [21]. Ingenuity Pathway Analysis (IPA) showed that mTOR was one of the proteins which could interact with Drp1 (Table S7). The present data revealed that p-mTOR level was greatly increased in tamoxifen-injected MCM/LRP6fl/fl hearts compared to that in diluent-injected ones (Figure 4B). 5'-adenosine monophosphate-activated protein kinase (AMPK), a key player of metabolism pathways, mediates autophagy or mitophagy through inhibition of mTOR [14, 22]. Although p-AMPK level was decreased in tamoxifen-injected MCM/LRP6fl/fl hearts compared to that in diluent-injected ones, there was no statistical difference in p-AMPK/AMPK between the two groups (Figure 4B). In MCM mice, tamoxifen injection didn't influence these proteins' expression.
Inhibition of autophagy increases lipid storage and impairs its utilization, which then further suppresses autophagy [17]. These data suggest that LRP6 deletion inhibits autophagy, which may perturb fatty acid oxidation, and vice versa, leading to myocardial lipid accumulation and cardiac dysfunction. The activation of Drp1 may be involved in the process.
Cardiomyocyte-specific deletion of LRP6 leads to a decrease in TFEB in the nucleus, associated with activation of Drp1.
Transcription factor EB (TFEB) has been reported to link the autophagic pathway to cellular lipid metabolism [23]. Dephosphorylated TFEB transloncates from cytosol to the nucleus to promote autophagy, mitochondrial biogenesis and β-oxidation [24]. We examined TFEB expression in whole cell lysate, nuclei and cytosol from heart tissue respectively. There was no obvious difference in TFEB expression in whole cell lysate or cytosol from heart tissue between tamoxifen-injected MCM/LRP6fl/fl mice and diluent-injected ones. However, nuclear TFEB expression was significantly lower in tamoxifen-injected MCM/LRP6fl/fl hearts than in dilution-injected ones (Figure 5A), while it showed little alteration between tamoxifen-injected MCM hearts and diluent-injected ones (Figure S11). TFEB can be phosphorylated by ERK2 [23] or mTOR [24, 25], which inhibits its translocation to the nucleus. Cardiac LRP6 deletion greatly promoted mTOR phosphorylation (Figure 4B), but it showed little effect on the phosphorylation of ERK1/2 (Figure S9B). We then examined the effect of Drp1 activation on mTOR phosphorylation and TFEB translocation. Further analysis revealed that mdivi-1 suppressed the phosphorylation of mTOR and attenuated the decrease in TFEB nuclear expression in cardiac-specific LRP6 deletion mice, while in control mice, mdivi-1 showed little effect on nuclear TFEB expression (Figure 5B). This suggests that the inhibition of TFEB nuclear translocation induced by LRP6 specific deletion in cardiomyocytes may be dependent on the activation of Drp1/mTOR.
Drp1 inhibitor greatly improves autophagic inhibition and reduces the size of lipid droplets in LRP6 deletion hearts
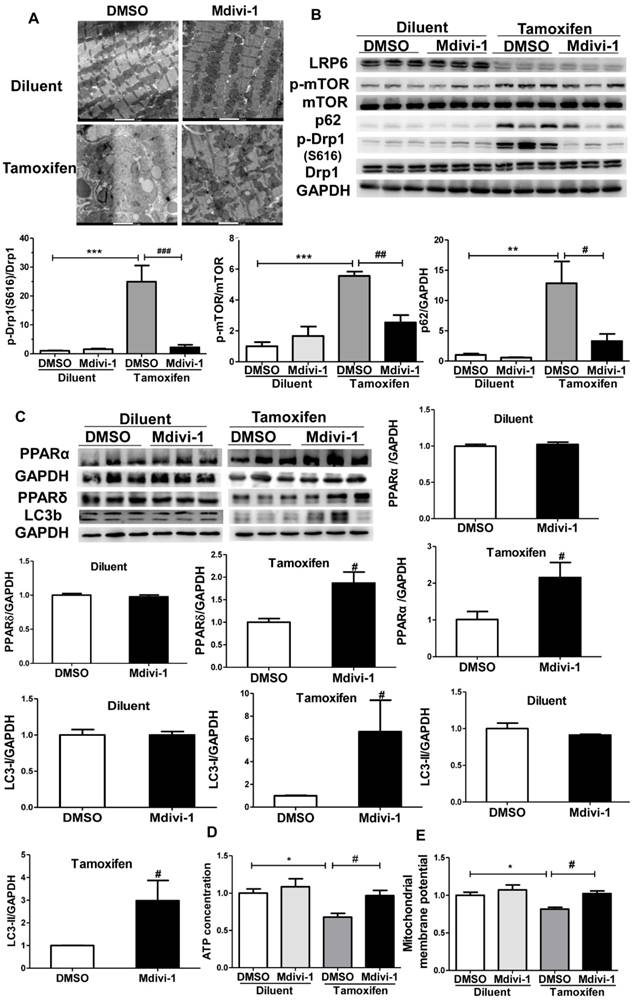
To explore the effects of Drp1 activation on autophagic inhibition, we treated MCM/LRP6fl/fl with mdivi-1 by intraperitoneal injection [26] after diluent- or tamoxifen- injection. Electron microscopy analysis showed that mdivi-1 treatment improved the accumulation of autophagosome and reduced the size of lipid droplets in cardiac tissue sections from tamoxifen-injected MCM/LRP6fl/fl (Figure 6A), while it showed little effects in diluent-injected MCM/LRP6fl/fl mice. In addition, mdivi-1 treatment attenuated the increase in the expression of p62, p-mTOR, p-Drp1 and the decrease in the expression of LC3-II in tamoxifen-injected MCM/LRP6fl/fl (Figure 6B,C). It also improved the expression of PPARα or PPARδ in tamoxifen-injected MCM/LRP6fl/fl (Figure 6C). In diluent-injected MCM/LRP6fl/fl, mdivi-1 treatment didn't alter the expression of these proteins. In addition, the decreased ATP production and mitochondrial membrane potential induced by LRP6 deletion were significantly improved by mdivi-1 injection (Figure 6D,E). These data indicate Drp1 inhibitor greatly improves autophagic inhibition and lipid accumulation, which subsequently attenuates mitochondrial dysfunction induced by LRP6-specific deletion in cardiomyocytes.
Drp1 inhibitor attenuates the autophagic degradation and mitochondrial dysfunction induced by LRP6 deletion in cardiomyocytes. (A) Electron microscopy images (scale bars, 1 μm), of hearts from tamoxifen-injected MCM/LRP6fl/fl treated with mdivi-1 or the same volume of DMSO. n=3/group. (B) Western blot analysis of LRP6, p-Drp1, Drp1, p-mTOR, mTOR, and P62 in heart tissue from diluent or tamoxifen-injected MCM/LRP6fl/fl mice treated with mdivi-1 or the same volume of DMSO. (C) Western blot analysis of PPARα, PPARδ and LC3b in heart tissue from diluent or tamoxifen-injected MCM/LRP6fl/fl mice treated with mdivi-1 or the same volume of DMSO. n=3-6/group. (D-E) Cardiac ATP production and mitochondrial membrane potential were analyzed in mice as in (B). n=4/group. *p<0.05; **p<0.01; ***p<0.001 vs. DMSO group; #p<0.05, ##p<0.01, ###p<0.001 vs. tamoxifen+DMSO group.
Drp1 inhibitor greatly improves cardiac dysfunction induced by LRP6 deletion
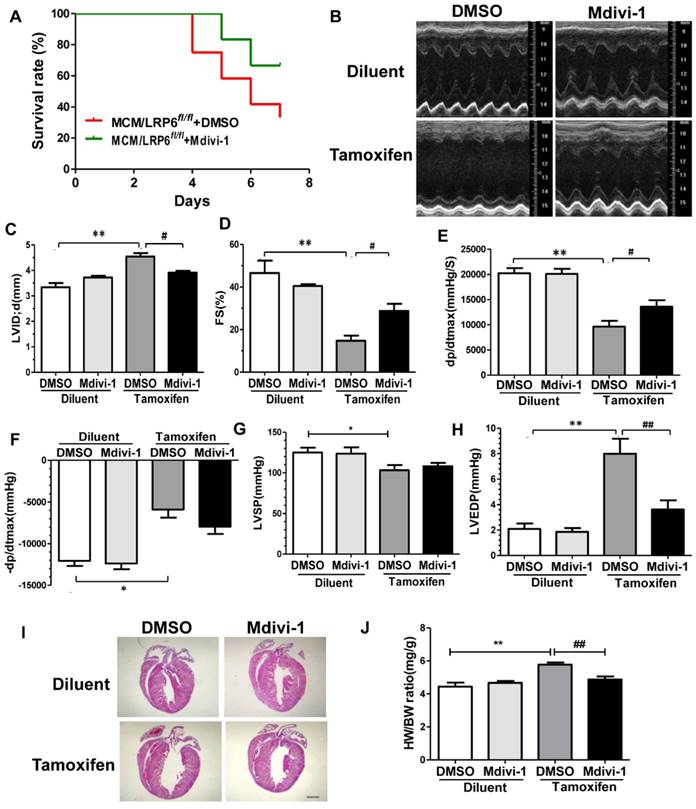
We then examined the effect of Drp1 inhibitor on cardiac function in MCM/LRP6fl/fl mice injected with tamoxifen or diluent. Mdivi-1 significantly increased survival rate of MCM/LRP6fl/fl mice within 7 days after tamoxifen injection (Figure 7A). Echocardiography analysis showed that mdivi-1 greatly attenuated the decrease in FS, EF and LVPW;d, and the increase in LVID induced by cardiomyocyte-specific LRP6 deletion (Figure 7B-D, Figure S12A,B). In parallel, mdivi-1 partly weakened the decrease in dp/dtmax and the increase in LVEDP induced by cardiomyocyte-specific LRP6 deletion (Figure 7E,H). Although -dp/dtmax, LVSP and LVAW;d were higher in mdivi-1-treated-LRP6 deletion mice compared to those in DMSO-treated ones, there was no statistical difference between the two groups (Figure 7F,G, Figure S12C). Histological analysis of H&E-stained longitudinal sections of hearts indicated the dilated ventricular chamber was greatly attenuated by mdivi-1 treatment compared to DMSO treatment in tamoxifen-injected MCM-LRP6fl/fl (Figure 7I). Mdivi-1 injection also greatly attenuated the increased HW/BW ratio induced by cardiac-specific LRP6 deletion (Figure 7J). These data revealed that Drp1 inhibitor greatly improved cardiac dysfunction induced by Drp1-specific deletion in cardiomyocytes. This may be related to upregulation of nuclear TFEB induced by mdivi-1 in cardiac-specific LRP6 knockout mice.
Drp1 inhibitor improves cardiac dysfunction induced by LRP6 deletion in cardiomyocyte. (A) Survival proportion was analyzed in MCM/LRP6fl/fl mice treated with mdivi-1 or DMSO within 7 days after tamoxifen injection. n=12/group. (B) Representative M-Mode echocardiographs from diluent or tamoxifen-injected MCM/LRP6fl/fl mice treated with DMSO or mdivi-1. (C-D) Echocardiographic analysis of LVID.d and FS in mice from (A). n=3-6/group. (E-H) Hemodynamic analysis of dp/dtmax, -dp/dtmax, LVSP and LVEDP in mice from (A). n=6-8/group. (I) HE staining of longitudinal heart sections from mice from (A). n=3-6/group. (J) HW/BW was analyzed in mice from (A). n=3-6/group *p<0.05; **p<0.01 vs. DMSO group. #p<0.05, ##p<0.01 vs. tamoxifen+DMSO group.
Discussion
In this study, we identified the downregulation of LRP6 in human heart tissue with end-stage dilated cardiomyopathy. Cardiomyocyte-specific lack of LRP6 led to acute heart failure resulting from impaired myocardial autophagy and lipid accumulation (Figure 8). Inhibition of Drp1 partly attenuated cardiac dysfunction observed in cardiomyocyte-LRP6-null mice by regulating the activation of mTOR and TFEB nuclear localization. The present study demonstrates novel roles of LRP6 in establishing physiological cardiac function and maintaining mitochondrial homeostasis in adult mice with implications for human heart disease.
A proposed model for cardiac-specific deletion of LRP6 induced cardiac dysfunction. Tamoxifen-inducible cardiac-specific LRP6 knockout promotes Drp1 phosphorylation at ser616 and its translocation to mitochondria, which leads to mitochondrial fragmentation and autophagosome formation. Meanwhile, LRP6 deletion-induced phosphorylation of Drp1 also enhances p-mTOR level, which inhibits TFEB nuclear translocation by decreasing TFEB phosphorylation. TFEB-induced autophagy genes as well as fatty acids metabolism genes PPARs are suppressed, resulting in the inhibition of mitochondrial degradation and fatty acids oxidation, which contribute to cardiomyocyte death and cardiac dysfunction.
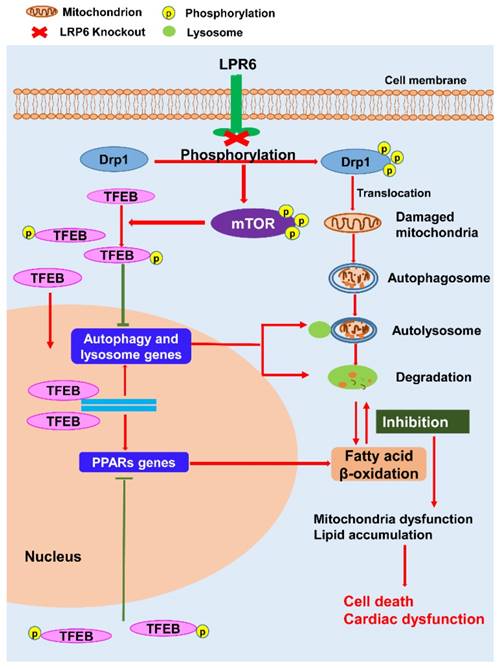
Recently, Li J et al. reported cardiac deletion of LRP6 predisposed mice to lethal arrhythmias by reducing connexin43 (Cx43) gap junction plaques [12], but they reported no obvious symptoms of heart failure in cardiac LRP6 deletion mice in the absence of stress. This might be explained by the higher LRP6 knockdown efficacy in our study than in theirs. In their study, cardiac LRP6 expression declined ~50% in cardiac-specific LRP6 knockout mice, while in our study, the expression decreased to ~10% compared to control mice. In the cardiac-specific heterozygous LRP6 deletion mice, cardiac LRP6 expression decreased to 49%, and no cardiac dysfunction was observed in the present study; but, the mice were predisposed to cardiac dysfunction or sudden death during pressure overload. The difference in LRP6 knockdown efficacy between our study and theirs may be owing to several factors such as tamoxifen dose, cre activity or age of mice. However, the present study revealed the impaired structure of ID in the LRP6 deletion heart, which may partly explain lethal arrhythmias of these mice in response to stress in the other study [12].
In the presence of Wnts or Frizzled proteins, LRP6 is activated, leading to the inhibition of cytosolic β-catenin degradation. The accumulated β-catenin enters into the nucleus and binds to T cell factor-lymphoid enhancer factor (TCF-LEF) family, the DNA binding transcription factors, to promote the expression of target genes and regulate various cellular activities [27]. However, in this study, cardiac-specific LRP6 deletion didn't alter β-catenin expression and its cellular distribution. This suggests that cardiac dysfunction induced by LRP6 deletion is independent of Wnt/β-catenin signaling.
More than 95% of cardiac ATP generated comes from oxidative phosphorylation in the mitochondria under physiological conditions [28, 29]. We observed the decreased ATP production and mitochondrial dysfunction were accompanied by obvious accumulation of mitochondrial targeting to autophagosome and lipid droplet in LRP6 deletion hearts, indicating the critical role of mitochondrial autophagy and fatty acid metabolism disorder in the process. Proteomics analysis revealed that PPARs signaling involved in the cardiac dysfunction induced by LRP6 deletion. PPARα and PPARδ play a prominent role in the regulation of cardiac fatty acid metabolism by transcriptional activation of genes encoding critical enzymes in fatty acid metabolism [30]. Cardiac deactivation of the PPARα or PPARδ deficiency depresses myocardial fatty acid oxidation and develops lipotoxic cardiomyopathy [31-33]. The expression of PPARα or PPARδ was sharply decreased in LRP6 deficient hearts. But, the expression of PPARγ coactivator-1α (PGC-1α), the transcriptional coactivator of the PPARs and stimulator of mitochondrial biogenesis [34], showed little alteration in LRP6 deficient hearts, and mitochondrial DNA copy number revealed similar results, suggesting cardiac LRP6 deletion has little effect on mitochondrial biogenesis or content. However, the higher level of medium- and long-chain fatty acids suggests the inhibition of fatty acid oxidation in LRP6 deletion hearts. The excess free fatty acids can cause lipotoxicity-induced cardiac dysfunction [35]. Moreover, the significant LRP6 deletion-induced decreases in PUFA-to-UFA and PUFA-to-MUFA ratios in heart tissue observed here is indicative of enhanced peroxidation of polyunsaturated fatty acids and subsequent oxidative stress, which contribute to lipid accumulation or excess reactive oxygen species [36]. This is supported by our observation of LRP6 deletion-induced heart failure. These data suggest the reduced expression of PPARα and PPARδ may contribute to the ATP production collapse by inhibition of fatty acid oxidation.
Fatty acid β-oxidation is coupled with induction of autophagy to deliver fatty acid to mitochondria in liver [37] [17]. Recently, similar mechanisms have been reported in heart tissue. An M et al. demonstrated that the activation of autophagy can inhibit lipid storage and subsequently improve cardiac dysfunction in obese hearts [38]. Selective autophagic mitochondria removal, mitophagy, plays a key role in maintaining the oxidative phosphorylation ability of mitochondria [39]. Parkin, an E3 ubiquitin ligase, translocates to mitochondria from cytosol to promote autophagic removal of damaged mitochondria via activation by PINK1 [40]. Elevated LC3b-II expression and decreased p62 levels indicate the activation of autophagy or degradation of autophagosomes [20, 21]. PINK1 expression was decreased and Parkin expression showed no alteration in mitochondria from LRP6 deletion hearts, and cardiac LRP6 deletion led to less LC3b-II or more p62 expression than control hearts, indicating the inhibition of autophagic/mitophagic degradation in LRP6 deletion hearts. AMPK, a critical regulator of energy balance, promotes myocardial autophagy through suppression of mTOR [14, 41]. Cardiac LRP6 deletion showed little effect on the activation of AMPK level but it enhanced the p-mTOR level. This suggests that LRP6 deletion mediates other signaling to activate mTOR, which induces the inhibition of autophagic degradation.
Cardiac LRP6 deletion also promoted the phosphorylation of Drp1 at ser616 and its mitochondrial translocation. Drp1 inhibitor greatly attenuated the phosphorylation of Drp1 and mTOR in LRP6 deletion hearts. It also greatly improved the inhibition of autophagic degradation, lipid accumulation and cardiac dysfunction induced by LRP6 deletion. Drp1, a small GTPase, mediates mitochondrial fission by translocating to mitochondria and mitochondrial fusion by phosphorylation at S637, to maintain balance between fission and fusion in mammalian cells. Drp1 activity is rapidly increased by phosphorylation of serine 616[28]. Endogenous Drp1 mediates mitochondrial autophagy including autophagic removal of mitochondria in cardiomyocyte [20, 42, 43]. There are multiple posttranscriptional modifications of Drp1, such as phosphorylation, S-nitrosylation or O-GlcNAcylation [29]. C452F mutation on Drp1 increases Drp1 GTPase activity, but it impairs autophagic clearance of mitochondria and ATP production by failure of Drp1 disassembly, which results in heart failure and DCM [44]; whether Drp1 phosphorylation at ser616 is involved in these effects is largely unknown. Excessive mitochondrial fission results in mitochondrial fragmentation, depolarization and mPTP opening, which lead to initiation of mitophagy [45]. In LRP6 deletion hearts, Drp1 activation induced not only mitochondrial fragmentation but also the phosphorylation of mTOR. Chronic activation of mTOR suppresses autophagy including autophagic degradation, which is mechanistically involved in the development of heart failure or Alzheimer's disease [46, 47]. A previous study provided evidence that activation of myocardial autophagic degradation is associated with a better prognosis in patients with DCM [42]. Rapamycin, a kind of mTOR inhibitor, could improve left ventricular function of early-stage DCM by upregulating autophagy [43]. Inhibition of autophagic degradation leads to the accumulation of depolarized mitochondria, impairs lipid metabolism and increases lipid storage [17]. We speculate LRP6 deletion-induced phosphorylation of Drp1 at ser616 initiates the formation of autophagosome but it also promotes the activation of mTOR, which inhibits autophagic degradation. This could explain the accumulation of mitochondrial targeting to autophagosome and lipid droplet in the LRP6 deficient hearts. We then explored the downstream signals responsible for the inhibition of the autophagic degradation in LRP6 deletion hearts.
TFEB, the basic helix-loop-helix leucine zipper transcription factor, is a master regulator for the autophagy-lysosome pathway, by promoting the expression of autophagy and lysosomal genes [48]. It also regulates mitochondrial biogenesis and lipid catabolism by driving expression of relative genes such as PPARα and PGC-1α [23, 48]. In physiological condition, TFEB is mainly localized in cytosol, but it can rapidly translocate to the nucleus to activate the autophagy-lysosomal pathway in response to stress such as starvation [23]. Cellular localization of TFEB is mainly regulated by post-translational modification such as phosphorylation of TFEB at multiple sites by ERK2, mTOR and protein kinase Cβ [48, 49]. Phosphorylation of TFEB greatly limits nuclear localization and then inhibits autophagic process. The present data revealed TFEB expression was decreased in the nucleus but showed no obvious alteration in the cytosol in LRP6 deletion hearts. Although activation of Rag GTPases promotes the phosphorylation of mTORC1, which inhibits the nuclear localization of TFEB [49], few studies reported the relationship between Drp1 phosphorylation and the cellular localization of TFEB. Drp1 selective inhibitor greatly suppressed the activation of mTOR and increased nucleus TFEB expression in LRP6 deletion hearts. It also improved the decrease in the expression of PPARα, PPARδ and LC3b. These data suggested that LRP6 deletion impaired autophagic degradation and fatty acid oxidation by Drp1/mTOR/TFEB pathway.
LRP6 negatively regulates GSK3β activity [50] and GSK3β-mediated-Drp1-phosphorylation induces mitochondrial fragmentation in Alzheimer's disease (AD) [51]. LRP6-deletion-dependent Drp1 phosphorylation may be regulated by GSK3β, which promotes autophagic inhibition and cell death. Put into an even wider perspective, other potential factors or pathways regulated by LRP6 deletion are also likely to contribute to myocardial dysfunction.
In summary, we identified cardiac LRP6 was downregulated in patients with DCM, and LRP6 deletion in adult cardiomyocytes induced acute heart failure from impaired mitochondrial autophagy and lipid accumulation through Drp1/mTOR/TFEB signaling. Taken together, these findings could provide novel insights into the critical roles of LRP6 in the regulation of cardiac function. This may have important implications for development of new therapeutic strategies to prevent the progression of heart failure.
Abbreviations
LRP6: lipoprotein receptor-related protein 6; DCM: dilated cardiomyopathy; PPARs: proliferator-activated receptors; Drp1: dynamin-related protein 1; TFEB: Transcription factor EB; LDL: low density lipoprotein; CAD: coronary artery disease; α-MHC: α-myosin heavy chain; MCM: Mer-Cre-Mer Tg; ATP: adenosine triphosphate; DMSO: dimethyl Sulphoxide; LVID;d: left ventricular cavity diastolic dimension; LVAW;d: left ventricular diastolic anterior wall; LVPW;d: left ventricular diastolic posterior wall; EF: ejection fraction; HR: Heart rate; LVSP: Left ventricular systolic pressure; LVEDP: left ventricular end diastolic pressure; LVDP: left ventricular developed pressure; WGA: wheat germ agglutinin; HW/BW: heart weight/body weight ratio, LW/BW: lung weight/body weight ratio; TAC: transverse aorta constriction; ID: indercalated disk; PUFA: polyunsaturated fatty acids; UFA: unsaturated fatty acids; MUFA: monounsaturated fatty acids; SFA: saturated fatty acids. PGC-1α: PPARγ coactivator-1α; ACC1:acetyl CoA carboxylase 1, SCD: stearoyl-CoA desaturase; FAS: fatty acid synthase, PINK1: PTEN-induced putative kinase 1; Drp1: Dynamin-related protein 1; IPA: ingenuity pathway analysis; mTOR: mammalian target of rapamycin; AMPK: 5'-adenosine monophosphate-activated protein kinase; ERK: extracellular regulated protein kinases; MAPK: mitogen-activated protein kinase; Acc1: acetyl CoA carboxylase 1; Scd: stearoyl-CoA desaturase; Fas: fatty acid synthase, Cx43: connexin43; GSK3β: glycogen synthase kinase-3β.
Supplementary Material
Supplementary methods, figures and tables.
Acknowledgements
We thank Prof. Bart O. Williams (Van Andel Research Institute, Michigan USA) for providing LRP6fl/fl mice, Bingyu Li, Hongyang Gao, Zhang Yu, Guoquan Yan and Lei Zhang for excellent technical assistance. This work was supported by National Natural Science Foundation of China (31430039, 81570353; 81220108003), National Program on Key Basic Research Project of China (2014CBA02003), China Postdoctoral Science Foundation (2015M571493) and Foundation for Key Researcher by Zhongshan Hospital, Fudan University (2015ZSYXGG06).
Author contribution
HG conceived the study, analyzed the data, drafted and revised the manuscript. ZC performed the majority of the experiments and analyzed data. YL conducted in vivo studies including echocardiographic and hemodynamic analysis. YW performed part of western blot analysis and Real-time PCR analysis. JQ received approval and provided human heart tissues. HM, XW, GJ, LM and JJ performed morphological analysis. YA and HT performed GC-FID/MS Analysis. CY constructed lentivirus. GZ performed proteomics analysis. YC conducted confocal analysis. LM and LK aided with echocardiographic analysis. WG contributed to Oil red O staining. MF, ZH, YZhu and JG provided materials. YZou revised the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hill JA, Olson EN. Cardiac plasticity. New Engl J Med. 2008;358:1370-80
2. Xin M, Olson EN, Bassel-Duby R. Mending broken hearts: cardiac development as a basis for adult heart regeneration and repair. Nat Rev Mol Bio. 2013;14:529-41
3. Kokubu C, Heinzmann U, Kokubu T, Sakai N, Kubota T, Kawai M. et al. Skeletal defects in ringelschwanz mutant mice reveal that Lrp6 is required for proper somitogenesis and osteogenesis. Development. 2004;131:5469-80
4. Liu W, Singh R, Choi CS, Lee HY, Keramati AR, Samuel VT. et al. Low density lipoprotein (LDL) receptor-related protein 6 (LRP6) regulates body fat and glucose homeostasis by modulating nutrient sensing pathways and mitochondrial energy expenditure. J Biol Chem. 2012;287:7213-23
5. Williams BO, Insogna KL. Where Wnts went: the exploding field of Lrp5 and Lrp6 signaling in bone. J Bone Miner Res. 2009;24:171-8
6. Song L, Li Y, Wang K, Zhou CJ. Cardiac neural crest and outflow tract defects in Lrp6 mutant mice. Dev Dynam. 2010;239:200-10
7. Ye ZJ, Go GW, Singh R, Liu W, Keramati AR, Mani A. LRP6 protein regulates low density lipoprotein (LDL) receptor-mediated LDL uptake. J Biol Chem. 2012;287:1335-44
8. Mani A, Radhakrishnan J, Wang H, Mani A, Mani MA, Nelson-Williams C. et al. LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science. 2007;315:1278-82
9. Guo J, Li Y, Ren YH, Sun Z, Dong J, Yan H. et al. Mutant LRP6 Impairs Endothelial Cell Functions Associated with Familial Normolipidemic Coronary Artery Disease. Int J Mol Sci. 2016;17:1173
10. Wang H, Liu QJ, Chen MZ, Li L, Zhang K, Cheng GH. et al. Association of common polymorphisms in the LRP6 gene with sporadic coronary artery disease in a Chinese population. Chin Med J (Engl). 2012;125:444-9
11. Chen Z, Xu J, Ye Y, Li Y, Gong H, Zhang G. et al. Urotensin II inhibited the proliferation of cardiac side population cells in mice during pressure overload by JNK-LRP6 signalling. J Cell Mol Med. 2014;18:852-62
12. Li J, Li C, Liang D, Lv F, Yuan T, The E. et al. LRP6 acts as a scaffold protein in cardiac gap junction assembly. Nat commun. 2016;7:11775
13. Gong H, Wang YX, Zhu YZ, Wang WW, Wang MJ, Yao T. et al. Cellular distribution of GPR14 and the positive inotropic role of urotensin II in the myocardium in adult rat. J Appl Physiol (1985). 2004;97:2228-35
14. Xu X, Hua Y, Nair S, Bucala R, Ren J. Macrophage migration inhibitory factor deletion exacerbates pressure overload-induced cardiac hypertrophy through mitigating autophagy. Hypertension. 2014;63:490-9
15. Noorman M, van der Heyden MA, van Veen TA, Cox MG, Hauer RN, de Bakker JM. et al. Cardiac cell-cell junctions in health and disease: Electrical versus mechanical coupling. J Mol Cell Cardiol. 2009;47:23-31
16. Ren J, Pulakat L, Whaley-Connell A, Sowers JR. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J Mol Med (Berl). 2010;88:993-1001
17. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M. et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131-5
18. Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J. et al. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010;8:e1000298
19. Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795-803
20. Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J. et al. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264-78
21. Zhao J, Goldberg AL. Coordinate regulation of autophagy and the ubiquitin proteasome system by MTOR. Autophagy. 2016;12:1967-70
22. Hardie DG. AMPK and autophagy get connected. EMBO J. 2011;30:634-5
23. Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O. et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15:647-58
24. Ding WX. Drinking coffee burns hepatic fat by inducing lipophagy coupled with mitochondrial beta-oxidation. Hepatology. 2014;59:1235-8
25. Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903-14
26. Park SW, Kim KY, Lindsey JD, Dai Y, Heo H, Nguyen DH. et al. A selective inhibitor of drp1, mdivi-1, increases retinal ganglion cell survival in acute ischemic mouse retina. Invest Ophth Vis Sci. 2011;52:2837-43
27. He X, Semenov M, Tamai K, Zeng X. LDL receptor-related proteins 5 and 6 in Wnt/beta-catenin signaling: arrows point the way. Development. 2004;131:1663-77
28. Archer SL. Mitochondrial dynamics-mitochondrial fission and fusion in human diseases. New Engl J Med. 2013;369:2236-51
29. Hall AR, Burke N, Dongworth RK, Hausenloy DJ. Mitochondrial fusion and fission proteins: novel therapeutic targets for combating cardiovascular disease. Brit J Pharmacol. 2014;171:1890-906
30. Gilde AJ, van der Lee KA, Willemsen PH, Chinetti G, van der Leij FR, van der Vusse GJ. et al. Peroxisome proliferator-activated receptor (PPAR) alpha and PPARbeta/delta, but not PPARgamma, modulate the expression of genes involved in cardiac lipid metabolism. Circ Res. 2003;92:518-24
31. Barger PM, Kelly DP. PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc Med. 2000;10:238-45
32. Wang P, Liu J, Li Y, Wu S, Luo J, Yang H. et al. Peroxisome proliferator-activated receptor {delta} is an essential transcriptional regulator for mitochondrial protection and biogenesis in adult heart. Circ Res. 2010;106:911-9
33. Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N. et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. 2004;10:1245-50
34. Finck BN. The PPAR regulatory system in cardiac physiology and disease. Cardiovasc Res. 2007;73:269-77
35. Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P. et al. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813-22
36. Matsuzawa-Nagata N, Takamura T, Ando H, Nakamura S, Kurita S, Misu H. et al. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metabolism. 2008;57:1071-7
37. Sinha RA, You SH, Zhou J, Siddique MM, Bay BH, Zhu X. et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J Clin Invest. 2012;122:2428-38
38. An M, Ryu DR, Won Park J, Ha Choi J, Park EM, Eun Lee K. et al. ULK1 prevents cardiac dysfunction in obesity through autophagy-meditated regulation of lipid metabolism. Cardiovasc Res. 2017;113:1137-1147
39. Wang K, Klionsky DJ. Mitochondria removal by autophagy. Autophagy. 2011;7:297-300
40. Poole AC, Thomas RE, Andrews LA, McBride HM, Whitworth AJ, Pallanck LJ. The PINK1/Parkin pathway regulates mitochondrial morphology. P Natl Acad Sci USA. 2008;105:1638-43
41. Guo R, Ren J. Deficiency in AMPK attenuates ethanol-induced cardiac contractile dysfunction through inhibition of autophagosome formation. Cardiovasc Res. 2012;94:480-91
42. Saito T, Asai K, Sato S, Hayashi M, Adachi A, Sasaki Y. et al. Autophagic vacuoles in cardiomyocytes of dilated cardiomyopathy with initially decompensated heart failure predict improved prognosis. Autophagy. 2016;12:579-87
43. Xie K, Jin B, Li Y, Luo X, Zhu J, Ma D. et al. Modulating autophagy improves cardiac function in a rat model of early-stage dilated cardiomyopathy. Cardiology. 2013;125:60-8
44. Cahill TJ, Leo V, Kelly M, Stockenhuber A, Kennedy NW, Bao L. et al. Resistance of Dynamin-related Protein 1 Oligomers to Disassembly Impairs Mitophagy, Resulting in Myocardial Inflammation and Heart Failure. J Biol Chem. 2015;290:25907-19
45. Tatsuta T, Langer T. Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J. 2008;27:306-14
46. Sciarretta S, Volpe M, Sadoshima J. Mammalian target of rapamycin signaling in cardiac physiology and disease. Circ Res. 2014;114:549-64
47. Kim DI, Lee KH, Gabr AA, Choi GE, Kim JS, Ko SH. et al. Abeta-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim Biophys Acta. 2016;1863:2820-34
48. Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S. et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429-33
49. Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S. et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012;31:1095-108
50. Beagle B, Mi K, Johnson GV. Phosphorylation of PPP(S/T)P motif of the free LRP6 intracellular domain is not required to activate the Wnt/beta-catenin pathway and attenuate GSK3beta activity. J Cell Biochem. 2009;108:886-95
51. Yan J, Liu XH, Han MZ, Wang YM, Sun XL, Yu N. et al. Blockage of GSK3beta-mediated Drp1 phosphorylation provides neuroprotection in neuronal and mouse models of Alzheimer's disease. Neurobiol Aging. 2015;36:211-27
Author contact
![]() Corresponding authors: Hui Gong, PhD or Yunzeng Zou, MD, PhD; Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital, Fudan University, 180 Feng Lin Road, Shanghai 200032, China. Email: gonghui2005edu.cn or zou.yunzengsh.cn
Corresponding authors: Hui Gong, PhD or Yunzeng Zou, MD, PhD; Shanghai Institute of Cardiovascular Diseases, Zhongshan Hospital, Fudan University, 180 Feng Lin Road, Shanghai 200032, China. Email: gonghui2005edu.cn or zou.yunzengsh.cn