Impact Factor
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results and Discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2018; 8(2):518-532. doi:10.7150/thno.21194 This issue Cite
Research Paper
A Transferrin-Conjugated Hollow Nanoplatform for Redox-Controlled and Targeted Chemotherapy of Tumor with Reduced Inflammatory Reactions
Jun Zhou1*, Menghuan Li1*, Wei Qi Lim2*, Zhong Luo1,3 ![]() , Soo Zeng Fiona Phua2, Runlan Huo1, Liqi Li4, Ke Li3, Liangliang Dai3, Junjie Liu3, Kaiyong Cai3, Yanli Zhao2,5
, Soo Zeng Fiona Phua2, Runlan Huo1, Liqi Li4, Ke Li3, Liangliang Dai3, Junjie Liu3, Kaiyong Cai3, Yanli Zhao2,5 ![]()
1. School of Life Science, Chongqing University, Chongqing 400044, P. R. China;
2. Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371;
3. Key Laboratory of Biorheological Science and Technology, Ministry of Education, Chongqing University, Chongqing 400044, P. R. China;
4. Department of General Surgery, Xinqiao Hospital, Third Military Medical University, Chongqing 400037, China;
5. School of Materials Science and Engineering, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798.
* These authors contributed equally to this work.
Received 2017-5-26; Accepted 2017-9-17; Published 2018-1-1
Abstract

Purpose: In this study, we report the design, development and evaluation of a hollow drug delivery nanoplatform for cancer therapy in vitro and in vivo. This composite nanosystem was prepared by modifying hollow mesoporous silica nanoparticles (HMSNs) with transferrin (Tf) targeting moieties via redox-liable linkage, and was capable of delivering therapeutic cargos (doxorubicin) specifically to the tumor site and subsequently releasing them in an on-demand manner. Moreover, the Tf corona could simultaneously reduce the inflammatory response after intravenous administration in vivo.
Methods: Nanostructural morphology of the drug delivery system was observed by scanning electron microscope and transmission electron microscope. The preparation process was monitored primarily using Fourier-transform infrared spectroscopy, dynamic light scattering, nitrogen adsorption/desorption isotherm, and thermogravimetric analysis. The release profile in solution was monitored by fluorescence spectroscopy. In vitro drug delivery efficacy was evaluated on MDA-MB-231 breast cancer cell line using confocal laser scanning microscopy, MTT assay and flow cytometry. In vitro inflammatory response was evaluated on RAW264.7 macrophage cells. In vivo therapeutic experiments were carried out using in situ mouse breast cancer models.
Results: The experimental results evidently demonstrate that the developed nanocarrier could effectively deliver anticancer drugs to the tumor site in a targeted manner and release them in response to the elevated glutathione level inside tumor cells, resulting in improved anticancer efficacy both in vitro and in vivo. Moreover, the Tf conjugation significantly ameliorated the inflammatory reaction triggered by the administration of the nanocarrier.
Conclusions: This manuscript demonstrated that the Tf-conjugated HMSNs could enhance the delivery efficiency of anticancer drugs, while simultaneously alleviating the adverse side effects. The current study presents a promising integrated delivery system toward effective and safe cancer treatment.
Keywords: attenuation of inflammation, hollow mesoporous silica nanoparticles, redox-responsive release, targeted chemotherapy, transferrin conjugation.
Introduction
The advent of nanotechnology has greatly impacted various aspects of anticancer research, as it offers innovative and unprecedented technological solutions to difficult clinical problems in oncology.[1-5] Remarkably, one area of particular interest in nano-oncology is the development of nanoparticle-based drug reservoirs to efficiently deliver therapeutic agents to the cancer cells in a targeted manner, which may potentially enhance therapeutic efficiency of drugs while reducing their adverse side effects.[6-8] Some of the most prominent examples in this regard are the nanoparticle carriers conjugated with cancer cell targeting moieties on their surface, which allow the drug-loaded nanoparticles to be concentrated at the tumor region while limiting their uptake by healthy cells/tissues.[9, 10] For instance, folic acid, a natural occurring compound that could be massively produced, has been used as the targeting ligand for a variety of cancer indications, especially ovarian cancer.[11-13] Similarly, several types of antibodies that could specifically bind to cancer biomarkers have also been evaluated in preclinical trials.[14-16] Among these frequently used targeting moieties, much attention has been focused on transferrin (Tf) and the associated receptor-mediated endocytic and transcytic pathways.[17] Tf is a blood plasma glycoprotein in the human body that is primarily responsible for iron transport, and Tf receptor (Tf-R) has been found to be overexpressed in many types of cancer cells.[18, 19] It should also be noted that the expression level of Tf-R in normal cells is low and almost negligible,[20] therefore, enhancing the application potential of Tf as the targeting moiety for the cancer-specific drug delivery.[21, 22]
An emerging trend in Tf-dependent tumor-specific drug delivery is to conjugate Tf onto the surface of mesoporous silica nanoparticles (MSNs).[19, 23, 24] MSNs are silica nanoparticles with a regular array of mesopores, featuring large surface area and high pore volume. It has been consistently demonstrated in many reports that MSNs could be loaded with large amounts of small-molecule anticancer drugs and deliver these therapeutic agents to their intracellular destinations in a Trojan horse manner.[25, 26] The benefit of modifying MSNs with Tf is two-fold: The coupled Tf could improve the delivery efficiency of the loaded drugs specifically to the cancer cells, as well as blocking the pore openings to drug leakage during the transportation process.[27, 28] Therefore, developing Tf-conjugated MSN nanoplatforms would enable the targeted and controlled delivery of the anticancer drugs with enhanced biocompatibility and minimize adverse side effects in the cancer treatment.
With the rapid expansion of knowledge on tumor biology and genetics, conventional approaches for the design and implementation of nanoparticle carriers have been challenged by a series of new questions, which are largely associated with their clinical safety in both short and long term administration.[29, 30] A demanding criterion among these emerging issues is the pro-inflammatory property of the drug delivery nanoplatforms, which describes the inflammatory response of the physiological environment provoked by exogenous compounds.[31-33] For example, Wolinsky et al reported in a recent review that many acid-degradable synthetic nanomaterials may accumulate at their site of biological action and cause inflammation in the peripheral tissue.[34] Naahidi et al demonstrated in greater detail that biodegradable poly(lactic-co-glycolic acid) (PLGA) nanoparticles could inflict strong inflammation when they are introduced into loosely connective tissue surrounding nerves, and thus are unsuitable for drug delivery to these locations.[35] Rigorous investigation has been carried out on elucidating the pro-inflammatory mechanism of nanoparticles and mitigating, or even eliminating the inflammation responses. Among these emerging approaches, modulating the protein corona of nanoparticles has drawn extensive attention. Specifically, Wan et al demonstrated that by deglycosylating protein-capsulated silica nanoparticles, the nanoparticle-protein complex shows enhanced adhesion to the cell membrane of two types of THP-1 differentiated macrophages, resulting in increasing pro-inflammatory responses compared to their glycosylated form.[36] Walkey et al meticulously investigated the connection between the physicochemical properties of nanobiomaterials and their interactions with physiological systems, and concluded that the surface absorption of serum proteins plays critical roles in the macrophage uptake of the nanoparticles, thus further impacting their delivery efficiency and potential toxicity.[37] These studies collectively demonstrated the importance of protein corona properties for the regulation of the nanoparticle-cell interactions, of which the underlying principles and mechanisms could facilitate the rational design and application of clinically translatable drug delivery nanoplatforms.
With the information above, a novel tumor-specific redox-sensitive drug delivery nanoplatform was developed to enhance the chemotherapeutic efficacy of small-molecule anticancer drugs. Specifically, Tf was exploited as the targeting moiety and conjugated onto the surface of doxorubicin (DOX)-loaded hollow mesoporous nanoparticles (HMSNs). The HMSN is a more advanced form of MSNs that has greater drug loading capacity and improved biosafety.[38, 39] Due to the relative size of Tf to the pore width of HMSN, the Tf moiety in close proximity to the silica surface could efficiently block the pore openings so as to prevent the premature drug release.[23] The conjugation of Tf was achieved through a biocompatible one-pot click reaction, which is a well-established approach for the non-invasive incorporation of various biomacromolecules including proteins and nucleotides.[40, 41] Moreover, a disulfide bond has been integrated into the molecular linkage between Tf and HMSN, which could be specifically cleaved by the elevated intracellular redox potential in the tumor cells.[42] As Tf is a human protein that exists in almost all types of biological fluids, we also hypothesized that the Tf-conjugated hollow nanoplatform could have reduced pro-inflammatory effect in vivo, which would be favorable for clinical translation.
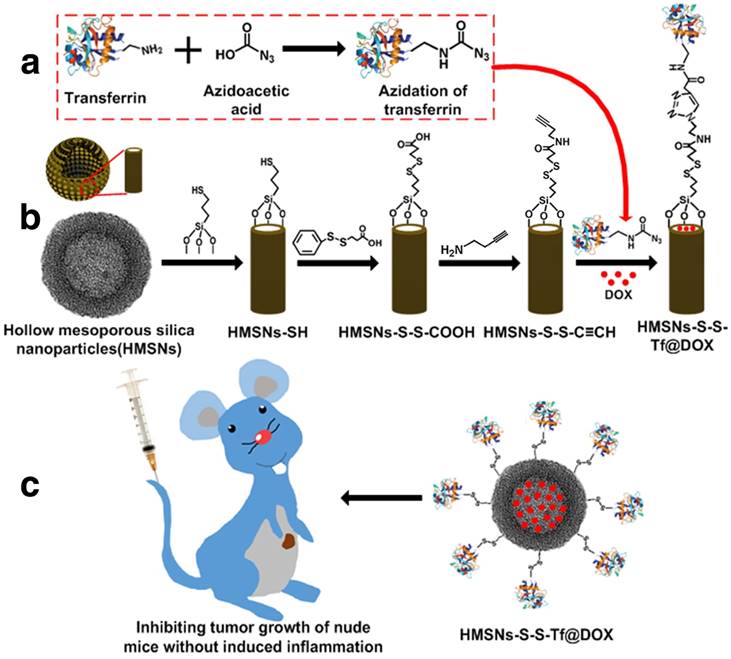
Scheme 1 presents a detailed illustration of the therapeutic process in vivo with the Tf-conjugated redox-sensitive hollow nanoplatform. The DOX-loaded nanocarriers were administered intravenously and concentrated at the tumor site through the enhanced permeability and retention effect, and eventually internalized by the cancer cells via the receptor mediated endocytosis. The Tf moieties would then be removed from the particle surface in the intracellular redox condition, as a result of which the anticancer drugs would be released from the nanocarrier and confer sustained damage to the cancer cells.
(a,b) Fabrication of redox-trigged HMSNs by using a disulfide bond as the intermediate linker. (c) Illustration of the intracellular redox-trigged HMSNs for targeted tumor therapy in vitro and in vivo.
Results and Discussion
Preparation and characterization of the hollow nanoplatform
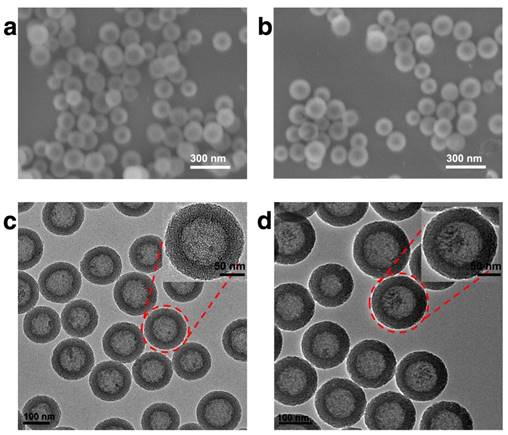
In this study, HMSN was chosen as the scaffold for the subsequent biofunctionalization, on account of its high drug loading capacity, biological inertness and versatile surface chemistry. The synthesis of the hollow mesoporous silica nanoparticle was achieved via a well-established template method with essential modifications. Typically, dense silica nanoparticles were firstly prepared via Stöber method and then coated with a layer of mesoporous silica. The dense silica core could be removed by etching with sodium carbonate while the mesoporous silica shell remained intact (Fig. S1). As demonstrated by the SEM result (Fig. 1a), the as-prepared HMSNs are spherical with homogeneous morphology. It could be further observed in the TEM image (Fig. 1c) that the HMSNs are highly uniform and monodisperse with distinct nanochannels. By using a particle size analyzer, it was found that the average diameter of HMSNs was 158 nm. After the drug loading and Tf conjugation, the average diameter of HMSN-S-S-Tf@DOX slightly increased to 167 nm, while the monodispersity was maintained (Table S1 and Fig. S2). For dynamic light scattering (DLS) characterizations, the nanoparticle samples were suspended in solutions and used fresh to improve their monodispersity. It should be noted that the size distribution features of the nanoparticle samples were still maintained after incubating for more than 4 days in biomimicking buffer solutions, indicating their good stability in a bio-relevant environment (Fig. S3). Compared to the unmodified form, the boundary and hierarchical mesoporous network of HMSN-S-S-Tf@DOX became more blurry after the conjugation of Tf moieties (Fig. 1b,d), indicating the successful attachment of the organic components. It is also worth noting that the monodispersity of HMSN series was well retained after the drug loading and Tf modification, and no noticeable changes in their size were detected. Using a method similar to a previous report,(41) the amount of Tf proteins on a single HMSN was calculated to be ~800 units. Specifically for the calculation process, the mass of a single dense silica nanoparticle was first obtained based on the hydrodynamic diameter of the dense silica nanoparticles (measured using DLS) and density of SiO2, which was found to be 1.7 × 10-15 g. As dense silica nanoparticles were used as the template for the synthesis of HMSNs, it is reasonable to assume that the number of HMSNs is roughly equal to the number of dense silica nanoparticles added into the reaction. By correlating to the amount of HMSNs-S-S-Tf collected after anhydration, the mass of a single HMSN-S-S-Tf could be calculated to be 1.18 × 10-15 g. Based on the TGA characterizations, the mass fraction of Tf in HMSN-S-S-Tf was determined to be ~9%. Combining the mass of HMSN-S-S-Tf calculated above and that of a single Tf protein (~80 kDa), it could be concluded that the average number of Tf proteins on a single HMSN was ~800.
Representative morphologies of unmodified HMSNs and HMSNs-S-S-Tf@DOX. SEM images of (a) free HMSNs and (b) HMSNs-S-S-Tf@DOX. Scale bar: 300 nm. TEM images of (c) free HMSNs and (d) HMSNs-S-S-Tf@DOX. Scale bar: 100 nm.
The N2 absorption-desorption isotherms of unmodified HMSNs, HMSNs-SH, HMSNs-S-S-C≡C, HMSNs-S-S-Tf and the drug-loaded HMSNs-S-S-Tf@DOX were then performed to study their porosity and surface area (Fig. S4). Raw HMSNs showed a typical type IV curve with an apparent hysteresis loop. A quantitative analysis of the results further revealed that the HMSNs have a BET surface area of 1297.544 m2/g and a pore volume of 1.1859 cm3/g, as well as a narrow pore size distribution centered at 2.012 nm. The BET surface area drops drastically as the modification continues, and eventually becomes 396.378 m2/g for the HMSN-S-S-Tf sample. In contrast, only a slight decrease was observed for the BJH pore size, from 2.012 nm for HMSNs to 1.959 nm for HMSNs-S-S-Tf@DOX (Table S2). Combined with the TEM observations, these results are an immediate sign that the HMSNs have been successfully conjugated with Tf moieties and loaded with the anticancer drug DOX, of which the entrapment efficiency was calculated to be 13.86%. Furthermore, Fourier transform infrared spectroscopy (FTIR) was employed to characterize the stepwise surface functionalization of HMSNs, and the results are illustrated in Fig. S5. All these evidences consistently prove the successful multi-step surface engineering of HMSNs. Additionally, TGA was used to quantify the DOX encapsulation (Fig. S6) and the results revealed a DOX loading amount of ~20 wt%, again confirming the superior drug loading capability of HMSNs compared with various types of conventional MCM-41 type MSNs.[38, 39, 43]
Release profiles in solution
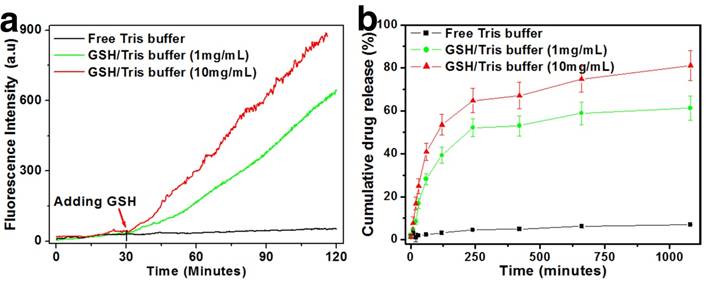
An initial assessment of the drug release behavior and redox sensitivity of the nanoplatform was conducted in Tris buffer of neutral pH (Fig. 2). GSH was added into the solution at graded concentrations to mimic the different intracellular redox environments. Due to the intrinsic fluorescence property of DOX, it could be used as an indicator for the monitoring of drug release.[44] In the real-time release tests, the drug-loaded HMSNs-S-S-Tf@DOX were first dispersed in neutral Tris buffer and only a small increase in DOX fluorescence (<30 a.u.) was observed after 2 h of incubation, which is possibly due to the disassociation of the surface-absorbed DOX molecules. The negligible amount of DOX release immediately suggests the excellent cargo encapsulation stability of the nanoplatform. However, for the HMSN-S-S-Tf@DOX sample that was treated with 1 mM of GSH for the same length of time, the fluorescence intensity of released DOX increased to >630 a.u. Furthermore, an evident positive correlation was found between the DOX releasing rate and the concentration of GSH. When the GSH concentration increased to 10 mM, the accumulated fluorescence intensity of DOX increased to almost 900 a.u. The DOX release profile is theoretically consistent with our proposed release mechanism, where the disulfide linkage would be readily cleaved by GSH through reduction to remove the capping moieties and release the entrapped DOX. To quantitatively study the encapsulation stability and release kinetics of the nanoplatform across a larger time scale, the incubation period was extended to 24 h. It is demonstrated in Fig. 2b that even after 24 h of continual incubation, the drug leakage in normal physiological conditions is still <5%. While for the two sample groups with added 1 and 10 mg/mL of GSH, the accumulative DOX release percentage was ~60% and ~81%, respectively. Together, the results of these release tests consistently demonstrated the excellent encapsulation efficiency of the nanoplatform in normal physiological environment as well as its sensitivity to redox stimulus.
Redox-sensitive DOX release from Tf-capped HMSNs in vitro. (a) Real-time release profiles over 2 h under the reductive GSH stimulus. (b) Long-term release profiles over 26 h under the reductive GSH stimulus.
Cellular interaction of the engineered nanoplatform
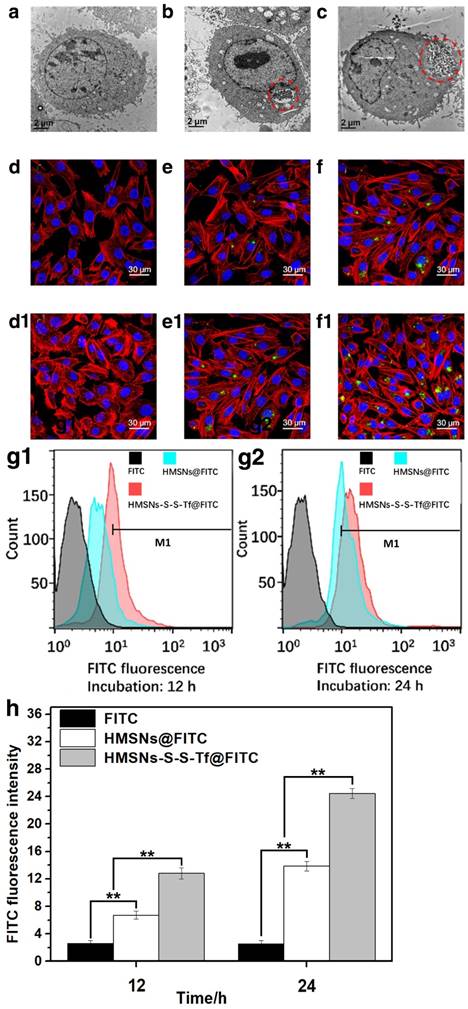
The uptake, allocation and biological impact of the nanocarrier at cellular level were firstly investigated using transmission electron microscopy (Fig. 3a-c) on MDA-MB-231 cells, since MDA-MB-231 cell line is one of the major breast cancer cell lines currently used in oncological research. MDA-MB-231 cell line was originally established from the pleural effusion of metastatic breast cancer, and one of its key features is the elevated expression level of Tf receptors.[45-47] Herein, the MDA-MB-231 cells were incubated with HMSNs or HMSNs-S-S-Tf and then cut into ultra-thin sections for TEM analysis, of which the results were compared to MDA-MB-231 cells incubated on tissue culture polystyrene plates (TCPS). The formation of endosomes was observed in both HMSN and HMSN-S-S-Tf groups, and the endocytosed amount of the Tf-conjugated nanoparticles was much greater than the unmodified ones, immediately suggesting the enhanced cellular uptake due to receptor-mediated endocytosis (an enlarged depiction of the internalized nanoparticles can be found in Fig. S7). Additionally, by comparing those cell samples that have been incubated with nanoparticles (HMSNs and HMSNs-S-S-Tf) to the control group, no noticeable changes were detected in cell morphology and membrane integrity, which implies the good biocompatibility of the nanocarriers.
Based on the above findings, the amount of internalized nanoparticles in cancer cells was investigated using confocal laser scanning microscope (CLSM) (Fig. 3d,e,f,d1,e1,f1). Fluorescein isothiocyanate (FITC), a widely-used bio-probe with strong fluorescence, was loaded into the HMSNs and HMSNs-S-S-Tf as the model drug for the subsequent optical study (denoted as HMSNs@FITC and HMSNs-S-S-Tf@FITC for the convenience of readers). Briefly, MDA-MB-231 cells were incubated with HMSNs@FITC and HMSNs-S-S-Tf@FITC for 12 h or 24 h at biologically relevant doses, followed by fixation and staining for the CLSM characterization. For the CLSM images, the yellowish green color is the FITC fluorescence, while the red and blue colors represent the dye-stained cytoskeleton and cell nuclei, respectively. Consistent with the previous TEM results, it could be firstly observed that all MDA-MB-231 cell samples incubated with nanoparticles retained the normal spindle shape, and their cell morphology/cytoskeleton structure/nuclei shape are almost identical to those cells in the control group that were incubated with equivalent concentration of free FITC. By comparing panel e to f and e1 to f1, an apparent positive correlation was observed between the amount of endocytosed nanoparticles and the length of incubation period in both experimental groups, which could be explained by the well-established endocytic mechanism of nanoparticles.[48] Additionally, the FITC fluorescence in the images were quantitatively analyzed using ImageJ, in which the cumulative FITC fluorescence (intensity × area) in the selected areas were integrated. The results showed that the integral fluorescence intensity ratios of e to e1 and f to f1 were 0.55:1 and 0.5:1, respectively.
Due to the limited number of cells included in the fluorescence analysis, we also performed flow cytometry and inductively coupled plasma (ICP) analysis with large cell populations to eliminate bias and improve the cogency of the results, and the results are shown separately in Fig. 3g1/g2/h, Fig. S8 and Fig. S9. As shown by the flow cytometry data, the ratio of internalized HMSNs to HMSNs-S-S-Tf was 0.42:1 after 12 h and 0.60:1 after 24 h. The percentage ratios of MDA-MB-231 cells with HMSN uptake to that of HMSN-S-S-Tf were 0.355:1 after 12 h and 0.656:1 after 24 h, both of which are also consistent to the results calculated by ICP (0.36:1 after 12 h and 0.55:1 after 24 h) as well as CLSM data. The comparative analysis demonstrated that the Tf-conjugated HMSNs always have superior cellular uptake efficiency than those unmodified ones regardless of the incubation time, again implying the targeting effect of the Tf moiety to cancer cells.
Anticancer activity of the nanoplatform on MDA-MB-231 cells: an in vitro study
As demonstrated by the results above, the Tf-conjugated nanoplatform could be readily internalized by cancer cells without eliciting noticeable cellular damage. In this section, the anticancer efficacy of HMSN-S-S-Tf@DOX was studied on MDA-MB-231 cells. CLSM was firstly exploited to investigate the biological impact of various samples on cancer cells (Fig. 4). The imaging results revealed that except for the control and HMSN groups, varied extents of nucleus deformation and disintegration were observed for those cancer cells incubated with DOX, HMSN@DOX and HMSN-S-S-Tf@DOX, which are apparent signs of apoptosis. For all three DOX-containing sample groups, the nuclei of the cancer cells showed a purplish pink color, which was a blend of the red DOX fluorescence and the original blue staining. The enrichment of DOX fluorescence in the cancer cell nuclei could be explained by its mechanism of action; the DOX molecules would selectively interact with DNA by intercalation and prevent the replication process [49]. It should also be noted that the remaining red fluorescence in the cytoplasm was almost negligible for the HMSN-S-S-Tf@DOX group, which suggests that the DOX release from the nanoplatform is complete.
Representative TEM images showing a MDA-MB-231 cell grown on (a, control group) TCPS and those treated with (b) free HMSNs or (c) HMSNs-S-S-Tf after 12 h. Endocytosed nanoparticles were marked with red circles. Scale bar: 2 μm. Representative CLSM images showing the distributions of (d and d1) free FITC, (e and e1) HMSNs@FITC, and (f and f1) HMSNs-S-S-Tf@FITC within MDA-MB-231 cells after incubations of 12 h and 24 h, respectively. Scale bar: 50 μm. Red: cytoskeleton, blue: cell nuclei, green: FITC-loaded nanoparticles. Flow cytometric evaluation of the nanoparticle intake with or without Tf conjugation in panel (g1, 12h) and (g2, 24h), respectively. The results were compared quantitatively in panel (h). (n = 6, **p < 0.01)
Representative CLSM images of MDA-MB-231 cells after incubation on (a and a1, control group) TCPS or with (b and b1) HMSNs, (c and c1) DOX, (d and d1) HMSNs@DOX and (e and e1) HMSNs-S-S-Tf@DOX for 24 h, respectively. Scale bar: 20 μm. The merged results of H33258 and DOX channels are displayed in panel a-e, while panel a1-e1 are the visualization of cells under DOX channel. Red: DOX, blue: cell nuclei.

Subsequently, the anticancer efficacy of the nanoplatform on live breast cancer cells was studied quantitatively with methylthiazoly tetrazolium (MTT) assays. From the cell viability data in Fig. S10 and S11, HMSN and HMSN-S-S-Tf both demonstrate excellent biocompatibility as their inhibitory effects on the cancer cells were minimal. It was also verified that the conjugation of Tf could further reduce the cytotoxicity of the HMSN substrate. More details were revealed from the comparative analysis of cytotoxicity data in Fig. S12. For the two sample groups of HMSN@DOX and HMSN-S-S-Tf@DOX, the incubation with the drug-loaded nanoparticles resulted in a strong inhibitory effect on the growth of breast cancer cells in a time and dose-dependent fashion. Under 24 h of incubation the HMSN-S-S-Tf always showed lower mean cell viability compared to the HMSN group, although no significant difference was detected between the two sample groups for any dosing condition. However, when the incubation time was extended to 48 h, the disparities in cell growth and viability at all nanoparticle concentrations increased, which is associated with the different endocytic pathway and release mechanism for each type of nanoparticles as well as the presence of molecular drug pumps on the cancer cell membrane. On one hand, the Tf conjugation is capable of improving the particle internalization by receptor-mediated endocytosis, essentially increasing the DOX uptake by the cancer cells. On the other hand, efflux pumps are usually overexpressed on the cancer cell membrane as a defense mechanism against chemotherapies, which can readily remove the DOX molecule in the cytoplasm [50]. For those unmodified HMSNs, the DOX molecules were released in a one-time burst and created a transient high cytoplasmic concertation of DOX, only to be effused by the transmembrane protein pumps later and result in less effective chemotherapy. In contrast, the surface modification of the HMSNs could regulate the release rate of drug molecules from the nanochannels and maintain the intracellular DOX concentration at a relatively stable level while not impairing its biological effectiveness, therefore providing enhanced drug advantage in comparison to raw HMSNs. The advantage of HMSNs-S-S-Tf@DOX was further validated in a comparative cell viability test, in which breast cancer cells were incubated with HMSNs-S-S-Tf@DOX and free DOX of equivalent concentration (Fig. S12). For the incubation period of 24 h, both the free DOX and HMSN-S-S-Tf@DOX samples displayed dose-dependent cytotoxicity to cancer cells. When the equivalent DOX dosage was below 4 μg/mL, free DOX showed greater cytotoxicity. As the DOX concentration increased above this value, the cytotoxicity of HMSNs-S-S-Tf@DOX began to increase rapidly and eventually surpassed that of the free DOX. A similar trend was also observed when the incubation was prolonged to 48 h. As a small-molecule anticancer drug, the cellular intake of free DOX is achieved primarily through rapid passive diffusion, resulting in relatively greater toxicity to cancer cells at the initial stage. For HMSNs-S-S-Tf@DOX, their entry into cancer cells is realized through relatively slower endocytic pathway, and the cytotoxicity of the released DOX is further influenced by the release kinetics of the HMSN carrier. As the equivalent DOX dose increases to a clinically relevant level, the cancer cell killing efficiency of HMSNs-S-S-Tf@DOX quickly surpasses free DOX, again confirming superior therapeutic efficiency and alleviated side effects of HMSNs-S-S-Tf@DOX in comparison with free DOX.
In addition to the CLSM and MTT results, the pro-apoptotic effect of the nanoplatform on MDA-MB-231 breast cancer cell line was quantitatively measured via flow cytometry to further clarify the underlying therapeutic mechanisms. The fluorescence of FITC and propidium iodide (PI) was used as the indicator for the evaluation of cell apoptosis after treatment with various samples. PI has frequently been used in combination with FITC to provide better understanding of the cell apoptosis from the early stage to the late stage. As revealed in Fig. S13 and Table S3, the incubation with unmodified HMSNs only has negligible impact on the cell activity, with a minimal increase of 2.4% in the relative amount of apoptotic cells (6.6%) compared to the TCPS control (4.2%). The treatment of free DOX resulted in a potent apoptotic effect of 34.6%, which was slightly higher than that of the HMSN@DOX group. This could be explained by the rapid passive diffusion of DOX molecules into the MDA-MB-231 cells and resultant acute cytotoxicity. The highest pro-apoptotic efficiency (50.6%) was found in HMSN-S-S-Tf@DOX group, owing to more advantageous drug release kinetics. Collectively, these in vitro investigations consistently demonstrated that the HMSN-S-S-Tf nanocarrier could improve the intracellular delivery efficiency of DOX and enhance its therapeutic effect in vitro.
Immunotoxicity in vitro: inflammation responses in macrophages
To study the immunotoxicity of the nanoplatform, the inflammatory macrophage response of RAW264.7 macrophage cells to HMSNs@FITC and HMSNs-S-S-Tf@FITC was thoroughly evaluated (Fig. 5). For the macrophage cells in the control group, they all displayed round shapes with a large cell nucleus in the center. However, for the cells incubated with HMSNs@FITC, they acquired a highly dendritic morphology with emerging filopodia and actin filaments close to the membrane surface. These observations immediately suggest the pro-inflammatory activation of the RAW264.7 cells after the incubation with the unmodified HMSNs.[51] In comparison, macrophage cells incubated with the HMSNs-S-S-Tf@FITC retained the circular morphology with minimal filopodia and actin excretion, which indicates that most of the macrophage cells remained inactivated. Similar to the characterization of the endocytosis efficiency by MDA-MB-231 cells in the previous sections, we also used flow cytometry and ICP to quantify the impact of Tf functionalization on the macrophage phagocytic efficiency of HMSNs, and the ratio of internalized HMSNs to HMSNs-S-S-Tf was 1.46:1 (Fig. 5d-e) by flow cytometric study and 1.57:1 (Fig. S14) by ICP test. The observations collectively demonstrated that the Tf conjugation could reduce the pro-inflammatory effect of the HMSN substrate. It is also evident that the cell sample incubated with HMSNs-S-S-Tf@FITC has higher cell density compared to the HMSN@FITC sample group, which is almost at a comparable level to the TCPS group. The observation was further validated quantitatively using MTT assays, revealing that the amount of RAW264.7 cells after incubating with HMSN-S-S-Tf@FITC for 24 h was statistically higher than the HMSN@FITC group (Fig. S11). The greater cell viability is another proof of the biocompatibility of the HMSN-S-S-Tf nanoplatform.
In vivo anticancer activity
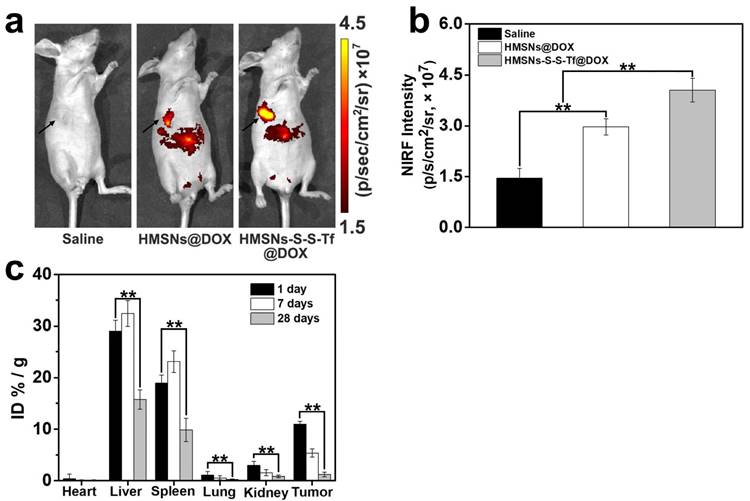
The therapeutic effect of the nanoplatform was further investigated in vivo by monitoring the MDA-MB-231 tumor growth in nude mouse models. The impact of the Tf-conjugation on the biodistribution of nanoparticles was firstly investigated. From the in vivo imaging results shown in Fig. 6 (panel a and b), it could be observed that the DOX fluorescence of the HMSN-S-S-Tf@DOX group at the tumor site was significantly greater than that of HMSN@DOX, indicating that HMSNs-S-S-Tf has an excellent tumor-targeting capability in vivo. Consistently, larger amounts of modified nanoparticles were successfully internalized at the tumor site after 1 day of administration in comparison with unmodified HMSNs (Fig. S15). Moreover, the HMSN-S-S-Tf nanoparticles could be cleared from the tumor tissues and steadily removed from the body after the drug release (Fig. 6c). Fig. 8a shows representative images of the tumors on the 21st day after various treatments. It was observed that the administration of saline and HMSNs resulted in rapid and uncontrolled tumor growth, while tumor growth inhibition effects to different extents were found in the three groups of DOX (III), HMSNs@DOX (IV), and HMSNs-S-S-Tf@DOX (V). The survival time of mice medicated with HMSNs-S-S-Tf@DOX was prolonged as compared to others, giving the highest survival rate (>60%) among all groups after 2 months of incubation (Fig. 7).
More details were revealed by the quantitative analysis of the tumor volumes across the experimental period (Fig. 8b). It was found that the volumes of the MDA-MB-231 tumors all increased in the four groups of saline, HMSNs, DOX and HMSNs@DOX, while a steadily downward trend was observed for mice administered with HMSNs-S-S-Tf@DOX. Specifically, the HMSN@DOX group showed higher growth inhibitory effect compared to that of the free DOX group, which is likely attributed to the enhanced DOX accumulation at the tumor site due to the EPR effect as well as the sustained kinetics of DOX release from the nanocarrier. Furthermore, the HMSNs-S-S-Tf@DOX group exhibited the greatest growth inhibitory efficiency as compared to other therapeutic treatments, as a result of the combined function of the targeting effect and redox-dependent drug release. These findings were also consistently verified by the changes in the relative tumor weight on the 21st day after various treatment (Fig. 8c). These results demonstrate that the HMSNs-S-S-Tf could greatly facilitate the intracellular delivery of DOX and improve the eventual therapeutic outcome.
In addition to the quantification of the tumor growth, haemotoxylin and eosin (H&E) staining was performed to analyze the changes in tumor tissues (Fig. 8d-h). Under microscopic view, no apparent cell/tissue damage was observed in the stained cross sections of tumor samples from the saline and HMSN groups, again confirming the biocompatibility of the nanosubstrates. For the free DOX group, cell death and scattered intercellular gaps were spotted in the tumor tissue with relatively low occurrence, while more severe tumor cell death was found in the tissue samples from the HMSN@DOX group. Nevertheless, the most severe and widespread cell death among all the experimental groups was observed in those tumors treated with HMSNs-S-S-Tf@DOX. The H&E staining result is another immediate evidence that by loading DOX into the HMSN-S-S-Tf nanocarrier, the anticancer effect of DOX could be further improved for better therapeutic outcome.
Representative CLSM images showing RAW 264.7 macrophage cells treated with (a) free FITC, (b) HMSNs@FITC, and (c) HMSNs-S-S-Tf@FITC after 24 h. Scale bar: 25 μm. Red: cytoskeleton, blue: cell nuclei, green: FITC-loaded nanoparticles. (d) Nanoparticle intake as measured by flow cytometry. (e) Quantitative analysis of the flow cytometry data. (n = 6, **p < 0.01)
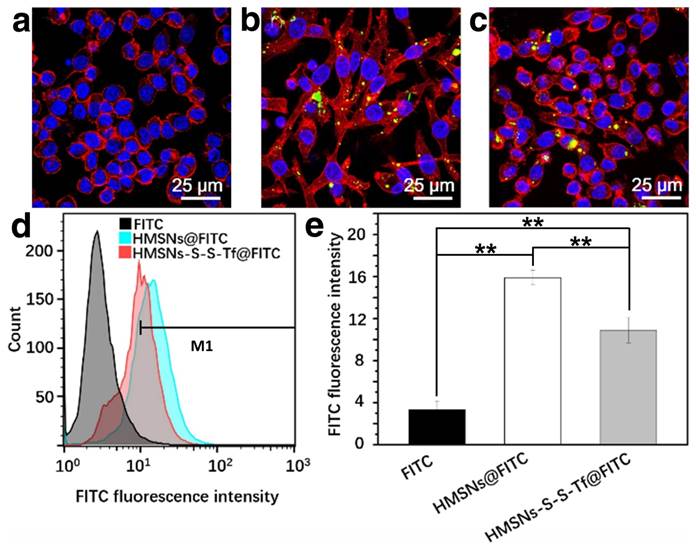
(a) In vivo NIRF images and (b) NIRF intensities of the mice bearing MDA-MB-231 tumor injected with saline, HMSNs@DOX, and HMSNs-S-S-Tf@DOX. (c) Biodistribution of HMSNs-S-S-Tf in major organs and tumor tissue of nude mice at different time of intervals. (n = 6, **p < 0.01)
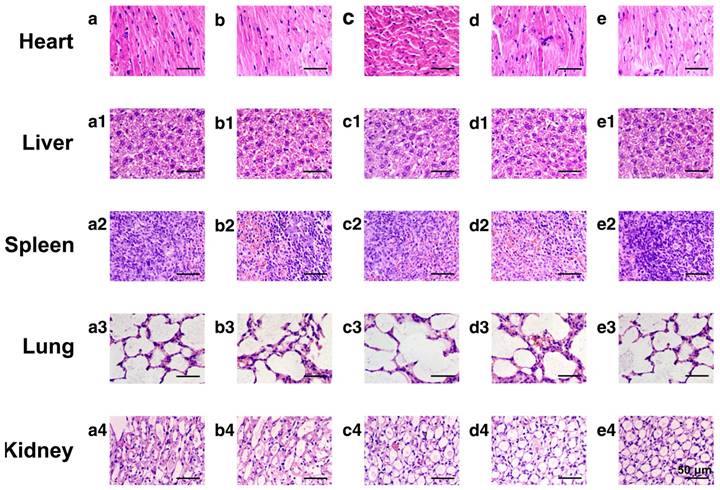
To evaluate the potential systemic toxicity of the nanoplatform, the body weight of the mice after different treatments were monitored throughout the experimental period (21 days, Fig. S16). The initial values of the average body weight for all groups were maintained at the same level of ~18.4 g. However, after 21 days of incubation, the average body weight of mice administered with DOX was still as low as ~19.6 g, while the average body weights of mice treated with saline, HMSNs, HMSNs@DOX and HMSNs-S-S-Tf@DOX increased to 23.1 g, 23.0 g, 21.2 g and 21.6 g, respectively. Additionally, major organs from the tumor-bearing mice in each group were also harvested on the 21st day and stained with H&E for histological analysis. It is well known that DOX molecules could bind to myocardial cardiolipin with high affinity and damage the myocardial cells.[52-54] As shown in Fig. 9, compared with the control groups, the administration of free DOX resulted in severe myocardial damage, while no significant histological changes were observed in the cross-sections of organs from the other experimental groups. All these results suggest that the nanocarrier itself is highly biocompatible and capable of mitigating the toxic side effects of DOX, effectively improving the safety of the nanoparticle-mediated targeted chemotherapy.
Macrophage phagocytosis and inflammation response to the nanosystem in vivo
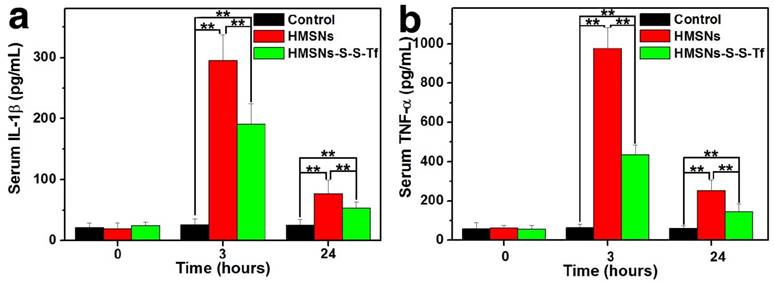
For the in vivo evaluation of the inflammation response to the Tf-conjugated hollow nanoplatform, we firstly examined the plasma concentration of nanoplatform after administration. The analysis was carried out on an ICP system and the results are shown in Fig. S17. It was found that compared to mice administered with HMSN, the administration of HMSN-S-S-Tf resulted in higher nanoparticle concentration in plasma throughout the experimental period of the study (up to 24 h). Specifically, the blood circulation half-life of HMSNs-S-S-Tf was found to be 5.12 ± 1.34 h, in comparison to 2.21 ± 0.89 h for HMSNs. Consistent with the in vitro macrophage phagocytosis evaluations where the fluorescence intensity of FITC-labelled nanoparticles and Si content in macrophage cells were measured and compared, the results indicate that the conjugation of Tf on the HMSN surface could lower its chance of being captured by the macrophage cells in vivo. Additionally, the expression levels of two representative cytokines interleukin-1β (IL-1β) and tumor necrosis factor α (TNF-α) were quantitatively measured and comparatively analyzed. These two cytokines are secreted by macrophage cells and have been used as clinically relevant markers for inflammation responses.[55, 56] Herein, the concentrations of IL-1β and TNF-α in human blood after the incubation with different nanoparticles for varied durations were quantified by enzyme linked immunosorbent assay (ELISA). As displayed in Fig. 10, the administration of unmodified HMSNs resulted in a massive increase in the blood concentration of IL-1β and TNF-α after 3 h of incubation, which were almost 15 times and 19 times that of the TCPS group, respectively. In contrast, the extent of the concentration increase of the two cytokines was much less for those macrophages incubated with HMSNs-S-S-Tf for 3 h, which were around 9 times and 8.7 times that of IL-1β and TNF-α concentrations for TCPS. Similar trends were also observed for the IL-1β and TNF-α concentrations when the incubation period was extended to 24 h. From these results it is immediately evident that unmodified HMSNs would inflict severe side effects in vivo including inflammation and other immune reactions, and the Tf modification could reduce the nanoparticle-induced inflammatory response associated with the administration of the nanoparticles.
Survival rate of tumor-bearing mice after the treatment with saline (control group), HMSNs, DOX, HMSNs@DOX, and HMSNs-S-S-Tf@DOX for 60 days, respectively.
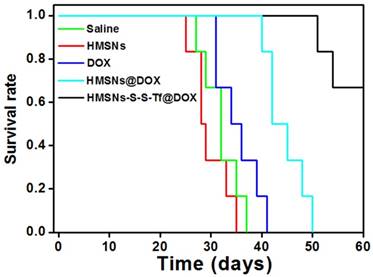
(a) Representative morphologies of tumor tissues after being treated with saline (I, control group), HMSNs (II), DOX (III), HMSNs@DOX (IV), or HMSNs-S-S-Tf@DOX (V) for 21 days. (b) Continual measurements of tumor sizes by digital vernier calliper in vivo after the treatments. (c) Final weights of tumor tissues after treatment for 21 days. Histological observation of in situ apoptosis in tumor tissues by using the H&E staining method, after treatment with (d) saline, (e) HMSNs, (f) DOX, (g) HMSNs@DOX, or (h) HMSNs-S-S-Tf@DOX for 21 days, respectively. Blue: cell nuclei. (n = 6, **p < 0.01)
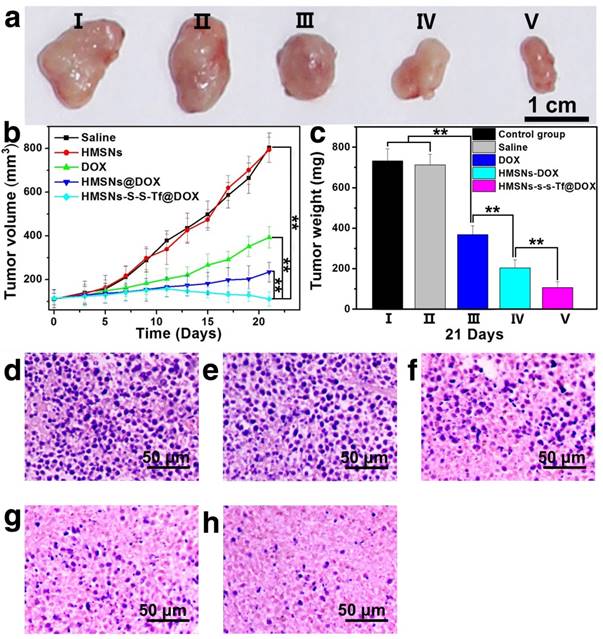
Representative morphologies of vital organs after treatment with (a-a4) saline as a control group, (b-b4) HMSNs, (c-c4) DOX, (d-d4) HMSNs@DOX, or (e-e4) HMSNs-S-S-Tf@DOX for 21 days, respectively. Panel a-e: heart; Panel a1-e1: liver; Panel a2-e2: spleen; Panel a3-e3: lung; Panel a4-e4: kidney.
Concentrations of the inflammatory factors in mice after treatment with saline (control group), HMSNs, and HMSNs-S-S-Tf@DOX for 3 h and 24 h respectively, based on cytokines (a) interleukin-1β (IL-1β) and (b) tumor necrosis factor α (TNF-α). (n = 6, **p < 0.01).
Conclusions
In summary, we have successfully developed a tumor-targeted drug delivery nanoplatform with high DOX loading capacity, redox sensitivity and immune tolerance by conjugating Tf onto the surface of HMSNs via disulfide linkage. It has been consistently validated by experimental results in vitro and in vivo that the HMSN-S-S-Tf carriers could be selectively enriched at the tumor site and efficiently internalized by the cancer cells, as the result of the combined effort of the EPR effect and receptor-mediated endocytosis. The disulfide linkage could be readily cleaved in the intracellular redox environment, eventually resulting in the sustained and effective tumor growth suppression. Moreover, the Tf corona could enhance the stability of the nanoplatform in blood circulation and simultaneously ameliorate the inflammatory reactions provoked by the intravenous administration, which are critical for the clinical translation of the nanoformulation. Therefore, this nanoplatform may provide new avenues for the development of anticancer nanomedicine with enhanced clinical relevance.
Abbreviations
BET: Brunauer-Emmett-Teller; CLSM: confocal laser scanning microscope; DLS: dynamic light scattering; DOX: doxorubicin; ELISA: enzyme linked immunosorbent assay; FITC: fluorescein isothiocyanate; FTIR: Fourier transform infrared spectroscopy; GSH: glutathione; H&E: haemotoxylin and eosin; HMSN: hollow mesoporous silica nanoparticles; ICP: inductively coupled plasma mass spectrometry; IL-1β: interleukin-1β; MTT: methylthiazoly tetrazolium; SEM: scanning electron microscope; TCPS: tissue culture polystyrene plates; TEM: transmission electron microscope; Tf: Transferrin; Tf-R: transferrin receptors; TGA: thermogravimetric analysis; TNF-α: tumor necrosis factor α.
Acknowledgements
This work was financially supported by the Natural Science Foundation of China (51602034, 51603024, and 21628401), Fundamental Research Funds for the Central Universities (106112017CDJXFLX0014 and 106112017CDJZRGJ0002), and SingHealth-NTU Research Collaborative Grant (SHS-NTU/009/2016). The authors sincerely thank the Department of General Surgery in Third Military Medical University for providing murine tumor models.
Author Contributions
J.Z., Z.L. and Y.Z conceived the idea and M.L., W.Q.L. and S.Z.F.P. helped in designing the experiments. J.Z., M.L., W.Q.L., S.Z.F.P., L.L., K.L., L.D., and J.L. performed the experiments. J.Z., M.L., W.Q.L., S.Z.F.P., L.L., K.L., L.D., and J.L. analyzed the data. R.H. helped with the revision. J.Z. and M.L. wrote the manuscript with contributions from Z.L., K.C. and Y.Z. All authors approved the final version.
Supplementary Material
Detailed synthesis and characterization procedures, SEM images, size distribution, FTIR spectra, TGA, TEM images, ICP data, cytotoxic effects, quantitative flow cytometry analysis, and weight changes of nude mice. Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zeng S, Baillargeat D, Ho H-P, Yong K-T. Nanomaterials enhanced surface plasmon resonance for biological and chemical sensing applications. Chem Soc Rev. 2014;43:3426-52
2. Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5:161-71
3. Lammers T, Aime S, Hennink WE, Storm G, Kiessling F. Theranostic nanomedicine. Acc Chem Res. 2011;44:1029-38
4. Sun TM, Zhang YS, Pang B, Hyun DC, Yang MX, Xia YN. Engineered nanoparticles for drug delivery in cancer therapy. Angew Chem-Int Edit. 2014;53:12320-64
5. Mura S, Couvreur P. Nanotheranostics for personalized medicine. Adv Drug Del Rev. 2012;64:1394-416
6. Kwon IK, Lee SC, Han B, Park K. Analysis on the current status of targeted drug delivery to tumors. J Controlled Release. 2012;164:108
7. Godin B, Tasciotti E, Liu XW, Serda RE, Ferrari M. Multistage nanovectors: From concept to novel imaging contrast agents and therapeutics. Acc Chem Res. 2011;44:979-89
8. Brannon-Peppas L, Blanchette JO. Nanoparticle and targeted systems for cancer therapy. Adv Drug Del Rev. 2012;64:206-12
9. Couvreur P. Nanoparticles in drug delivery: Past, present and future. Adv Drug Del Rev. 2013;65:21-3
10. Wicki A, Witzigmann D, Balasubramanian V, Huwyler J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J Controlled Release. 2015;200:138-57
11. van Dam GM, Themelis G, Crane LMA, Harlaar NJ, Pleijhuis RG, Kelder W. et al. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-[alpha] targeting: first in-human results. Nat Med. 2011;17:1315-9
12. Vlashi E, Kelderhouse LE, Sturgis JE, Low PS. Effect of folate-targeted nanoparticle size on their rates of penetration into solid tumors. ACS Nano. 2013;7:8573-82
13. Mackowiak SA, Schmidt A, Weiss V, Argyo C, von Schirnding C, Bein T. et al. Targeted drug delivery in cancer cells with red-light photoactivated mesoporous silica nanoparticles. Nano Lett. 2013;13:2576-83
14. Fernandes E, Ferreira JA, Andreia P, Luís L, Barroso S, Sarmento B. et al. New trends in guided nanotherapies for digestive cancers: A systematic review. J Controlled Release. 2015;209:288-307
15. Dawidczyk CM, Kim C, Park JH, Russell LM, Lee KH, Pomper MG. et al. State-of-the-art in design rules for drug delivery platforms: Lessons learned from FDA-approved nanomedicines. J Controlled Release. 2014;187:133-44
16. Steichen SD, Caldorera-Moore M, Peppas NA. A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur J Pharm Sci. 2013;48:416-27
17. Yameen B, Choi WI, Vilos C, Swami A, Shi J, Farokhzad OC. Insight into nanoparticle cellular uptake and intracellular targeting. J Controlled Release. 2014;190:485-99
18. Muthu MS, Kutty RV, Luo Z, Xie J, Feng S-S. Theranostic vitamin E TPGS micelles of transferrin conjugation for targeted co-delivery of docetaxel and ultra bright gold nanoclusters. Biomaterials. 2015;39:234-48
19. Li J, Qu X, Payne GF, Zhang C, Zhang Y, Li J. et al. Biospecific self-assembly of a nanoparticle coating for targeted and stimuli-responsive drug delivery. Adv Funct Mater. 2015;25:1404-17
20. Luck AN, Mason AB. Structure and dynamics of drug carriers and their interaction with cellular receptors: Focus on serum transferrin. Adv Drug Del Rev. 2013;65:1012-9
21. Spillmann CM, Naciri J, Algar WR, Medintz IL, Delehanty JB. Multifunctional liquid crystal nanoparticles for intracellular fluorescent imaging and drug delivery. ACS Nano. 2014;8:6986-97
22. Rosen CB, Kodal AL, Nielsen JS, Schaffert DH, Scavenius C, Okholm AH. et al. Template-directed covalent conjugation of DNA to native antibodies, transferrin and other metal-binding proteins. Nat Chem. 2014;6:804-9
23. Ferris DP, Lu J, Gothard C, Yanes R, Thomas CR, Olsen J-C. et al. Synthesis of biomolecule-modified mesoporous silica nanoparticles for targeted hydrophobic drug delivery to cancer cells. Small. 2011;7:1816-26
24. Hwang AA, Lu J, Tamanoi F, Zink JI. Functional nanovalves on protein-coated nanoparticles for in vitro and in vivo controlled drug delivery. Small. 2015;11:319-28
25. Argyo C, Weiss V, Bräuchle C, Bein T. Multifunctional mesoporous silica nanoparticles as a universal platform for drug delivery. Chem Mater. 2014;26:435-51
26. Chinen AB, Guan CM, Ferrer JR, Barnaby SN, Merkel TJ, Mirkin CA. Nanoparticle probes for the detection of cancer biomarkers, cells, and tissues by fluorescence. Chem Rev. 2015;115:10530-74
27. Yang J, Fan L, Wang F, Luo Y, Sui X, Li W. et al. Rapid-releasing of HI-6 via brain-targeted mesoporous silica nanoparticles for nerve agent detoxification. Nanoscale. 2016;8:9537-47
28. Tian Y, Guo R, Jiao Y, Sun Y, Shen S, Wang Y. et al. Redox stimuli-responsive hollow mesoporous silica nanocarriers for targeted drug delivery in cancer therapy. Nanoscale Horiz. 2016;1:480-7
29. Sharifi S, Behzadi S, Laurent S, Laird Forrest M, Stroeve P, Mahmoudi M. Toxicity of nanomaterials. Chem Soc Rev. 2012;41:2323-43
30. Ding CQ, Zhu AW, Tian Y. Functional surface engineering of C-dots for fluorescent biosensing and in vivo bioimaging. Acc Chem Res. 2014;47:20-30
31. Fadeel B, Garcia-Bennett AE. Better safe than sorry: Understanding the toxicological properties of inorganic nanoparticles manufactured for biomedical applications. Adv Drug Del Rev. 2010;62:362-74
32. Barua S, Mitragotri S. Challenges associated with penetration of nanoparticles across cell and tissue barriers: A review of current status and future prospects. Nano Today. 2014;9:223-43
33. Park K. Controlled drug delivery systems: Past forward and future back. J Controlled Release. 2014;190:3-8
34. Wolinsky JB, Colson YL, Grinstaff MW. Local drug delivery strategies for cancer treatment: Gels, nanoparticles, polymeric films, rods, and wafers. J Controlled Release. 2012;159:14-26
35. Naahidi S, Jafari M, Edalat F, Raymond K, Khademhosseini A, Chen P. Biocompatibility of engineered nanoparticles for drug delivery. J Controlled Release. 2013;166:182-94
36. Wan S, Kelly PM, Mahon E, Stöckmann H, Rudd PM, Caruso F. et al. The “sweet” side of the protein corona: Effects of glycosylation on nanoparticle-cell interactions. ACS Nano. 2015;9:2157-66
37. Walkey CD, Olsen JB, Guo H, Emili A, Chan WCW. Nanoparticle size and surface chemistry determine serum protein adsorption and macrophage uptake. J Am Chem Soc. 2012;134:2139-47
38. Luo Z, Ding X, Hu Y, Wu S, Xiang Y, Zeng Y. et al. Engineering a hollow nanocontainer platform with multifunctional molecular machines for tumor-targeted therapy in vitro and in vivo. ACS Nano. 2013;7:10271-84
39. Tang F, Li L, Chen D. Mesoporous silica nanoparticles: Synthesis, biocompatibility and drug delivery. Adv Mater. 2012;24:1504-34
40. Lang K, Chin JW. Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem Rev. 2014;114:4764-806
41. Boutureira O, Bernardes GJL. Advances in chemical protein modification. Chem Rev. 2015;115:2174-95
42. Ma X, Teh C, Zhang Q, Borah P, Choong C, Korzh V. et al. Redox-responsive mesoporous silica nanoparticles: a physiologically sensitive codelivery vehicle for siRNA and doxorubicin. Antioxid Redox Signal. 2014;21:707-22
43. Slowing II, Vivero-Escoto JL Wu CW, Lin VSY. Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv Drug Del Rev. 2008;60:1278-88
44. Luo Z, Hu Y, Cai K, Ding X, Zhang Q, Li M. et al. Intracellular redox-activated anticancer drug delivery by functionalized hollow mesoporous silica nanoreservoirs with tumor specificity. Biomaterials. 2014;35:7951-62
45. Yang DC, Jiang XP, Elliott RL, Head JF. Inhibition of growth of human breast carcinoma cells by an antisense oligonucleotide targeted to the transferrin receptor gene. Anticancer Res. 2001;21:1777-87
46. Daniels TR, Delgado T, Helguera G, Penichet ML. The transferrin receptor part II: targeted delivery of therapeutic agents into cancer cells. Clin Immunol. 2006;121:159-76
47. Kawamoto M, Horibe T, Kohno M, Kawakami K. A novel transferrin receptor-targeted hybrid peptide disintegrates cancer cell membrane to induce rapid killing of cancer cells. BMC Cancer. 2011;11:359
48. Oh N, Park JH. Endocytosis and exocytosis of nanoparticles in mammalian cells. Int J Nanomedicine. 2014;9:51-63
49. Momparler RL, Karon M, Siegel SE, Avila F. Effect of adriamycin on DNA, RNA, and protein synthesis in cell-free systems and intact cells. Cancer Res. 1976;36:2891-5
50. Cooray HC, Shahi S, Cahn AP, van Veen HW, Hladky SB, Barrand MA. Modulation of p-glycoprotein and breast cancer resistance protein by some prescribed corticosteroids. Eur J Pharmacol. 2006;531:25-33
51. Luo Z, Hu Y, Xin R, Zhang B, Li J, Ding X. et al. Surface functionalized mesoporous silica nanoparticles with natural proteins for reduced immunotoxicity. J Biomed Mater Res A. 2014;102:3781-94
52. Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. New Engl J Med. 2009;339:900-5
53. Pai VB, Nahata MC. Cardiotoxicity of chemotherapeutic agents: incidence, treatment and prevention. Drug Saf. 2000;22:263-302
54. Takemura G, Fujiwara H. Doxorubicin-induced cardiomyopathy from the cardiotoxic mechanisms to management. Prog Cardiovasc Dis. 2007;49:330-52
55. Choy EHS, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. New Engl J Med. 2001;344:907-16
56. Gabay C, Kushner I. Acute-phase proteins and other systemic responses to inflammation. New Engl J Med. 1999;340:448-54
Author contact
![]() Corresponding authors: E-mail address: zhaoyanliedu.sg (Y. Zhao); luozhong918edu.cn (Z. Luo)
Corresponding authors: E-mail address: zhaoyanliedu.sg (Y. Zhao); luozhong918edu.cn (Z. Luo)