Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(13):3228-3242. doi:10.7150/thno.19893 This issue Cite
Research Paper
Combination of AAV-TRAIL with miR-221-Zip Therapeutic Strategy Overcomes the Resistance to TRAIL Induced Apoptosis in Liver Cancer
Sisi Ma1, Jiazeng Sun1, Yabin Guo2, Peng Zhang3, Yanxin Liu1, Dexian Zheng1, Juan Shi1 ![]()
1. National Laboratory of Medical Molecular Biology, Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100005, China;
2. Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Medical Research Center, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, China;
3. Department of Oncology, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan 430030, China.
Received 2017-3-2; Accepted 2017-5-29; Published 2017-7-22
Citation:
Ma S, Sun J, Guo Y, Zhang P, Liu Y, Zheng D, Shi J. Combination of AAV-TRAIL with miR-221-Zip Therapeutic Strategy Overcomes the Resistance to TRAIL Induced Apoptosis in Liver Cancer. Theranostics 2017; 7(13):3228-3242. doi:10.7150/thno.19893. https://www.thno.org/v07p3228.htm
Other stylesAbstract

TNF-related apoptosis-inducing ligand (TRAIL) possesses the capacity to induce apoptosis in a wide variety of tumor cells without affecting most normal cells. However, it has now emerged that many primary cancer cells are resistant to TRAIL monotherapy. Overcoming the intrinsic or acquired TRAIL resistance is desirable for TRAIL-mediated cancer therapy. In this study, we found that the miR-221/222 cluster was up-regulated in TRAIL-resistant liver cancer cells. Specific inhibitors of miR-221 and/or miR-222, called sponge, TuD and miR-Zip were constructed, and their ability to overcome TRAIL resistance was compared. Among them, AAV-mediated gene therapy using co-expression of TRAIL with miR-221-Zip showed the most synergistic activity in the induction of apoptosis in vitro. In vivo treatment of nude mice bearing human TRAIL-resistant liver cancer xenografts with AAV-TRAIL-miR-221-Zip also led to growth inhibition. This sensitizing effect of miR-221-Zip was associated with increased expression of PTEN, the miR-221 target, as well as with decreasing levels of Survivin. Moreover, miR-221 expression was concomitant with promotion of Survivin expression and suppression of PTEN expression. TRAIL sensitivity of cancer cells isolated from liver cancer tissues or from patients was significantly correlated with miR-221 expression. And miR-221 blood expression levels in liver cancer patients were correlated with TRAIL sensitivity, thus it had the potential to be a predictor of TRAIL sensitivity in liver cancer. These data suggested the potential of combining AAV-TRAIL with miR-221-Zip as a therapeutic intervention for liver cancer.
Keywords: AAV, gene therapy, TRAIL, miR-221, TRAIL resistance.
Introduction
Liver cancer is the sixth most frequent cancer globally, and the second leading cause of cancer death. Liver cancer is particularly insensitive to chemotherapy. Surgery is standard for liver cancer caught early, but only a third of cases are identified at this stage. These problems emphasize the urgency of identifying liver cancer patients early on and establishing new therapeutic targets for successful intervention.
The Apo2L/tumor necrosis factor (TNF)-α-related apoptosis-inducing ligand (TRAIL) was identified as a member of the TNF superfamily that induces apoptosis [1, 2]. A number of studies have shown that TRAIL can induce apoptosis in a variety of tumor cell lines without affecting most normal cells. Currently, different TRAIL-R agonists, including TRAIL itself, and various agonistic monoclonal antibodies against the two human TRAIL death receptors are now in clinical trials as novel cancer therapeutics [3, 4]. Recent some breakthroughs related to strategies to natural Sensitizers, miRNAs and nanotechnologically delivered TRAIL to the target site to promote TRAIL-mediated cell death [5, 6]. Given promising preclinical results, the unique feature of selectivity for cancer cells has drawn considerable attention to TRAIL as a potential therapeutic agent against human cancers, a significant proportion of human cancer cells, especially in liver cancer, are resistant to TRAIL-induced apoptosis. Furthermore, repeated application of TRAIL to TRAIL-susceptible cancer cells results in the selection and expansion of TRAIL-resistant cells [7]. A variety of mechanisms involved in the escape from TRAIL-induced cytotoxicity in some cancer cells are reported [8]. And rebalancing pro-apoptotic and anti-apoptotic proteins would restore apoptosis in hepatocellular carcinoma cells to TRAIL [9]. Several means to overcome TRAIL resistance of cancer cells have been carried out, including combination with chemotherapeutic drugs, radiation or with targeted small molecules such as BH3 mimetics, Smac mimetics or inhibitors of kinases or the proteasome [10]. But the side effects from both chemotherapy and radiation itself are extensive. And the small molecules are always targeting a single pathway which is unlikely to be fully successful to overcome the resistance. But it is imminent to establish the addition of efficient with cancer-cell-selective TRAIL-sensitizing strategy to overcome TRAIL resistance. Furthermore, pre-clinical studies suggest that it is also necessary to identify suitable biomarkers for pre-selection of patients responsive to the combination therapy.
The European Commission approved in 2012 the use of uniQure's drug Glybera, an rAAV1-based vector for intramuscular injection [11, 12]. This is the first viral vector that has achieved regulatory approval in the West, indicating that AAV is a potentially useful tool in the field of gene therapy. We previously showed that AAV-mediated TRAIL gene therapy suppressed the growth of tumor cells transplanted in the liver of mice [13]. However, in our in-depth preclinical study, we found that a majority of Liver cancers were resistant to TRAIL therapy. To identify novel mechanisms implicated in TRAIL resistance, Garofalo et al. performed genome-wide expression profiling of miRNAs in four different Non-small cell lung cancer (NSCLC) cell lines [14]. They showed that high expression levels of miR-221 and miR-222 are needed to maintain the intrinsic TRAIL-resistant phenotype in NSCLC and liver cancer [14]. MiR-148a was also reported to be down-regulated in NSCLC cells with acquired TRAIL-resistance [15]. Additionally, miR-25 targets DR4 and promotes apoptosis resistance in cholangiocarcinoma [16]. However, no systematic analysis of miRNA expression in acquired TRAIL-resistance liver cancer was performed.
In this study, to improve the therapeutic effect of AAV-TRAIL and to identify novel mechanisms implicated in acquired TRAIL resistance in liver cancer, a microRNA expression microarray was used to screen for miRNAs that were aberrantly expressed in acquired TRAIL-resistance liver cancer cells. We found that up-regulated miR-221/222 are associated with TRAIL-derived resistance, and the combination of miR-221-Zip with TRAIL using AAV vector delivery promotes the therapeutic effect of TRAIL in TRAIL-resistant liver tumors by simultaneously targeting extrinsic and intrinsic pathways. What counts was TRAIL sensitivity of primary liver cancer cells from patient tumor samples was negatively correlated with miR-221 expression. And serum miR-221 also showed negative correlation with TRAIL sensitivity of primary liver cancer cells, suggesting that serum miR-221 could be used as a potential predictor of TRAIL sensitivity in liver cancer, thereby providing the possibility of developing an individualized treatment based on the TRAIL-induced apoptosis pathway.
Methods
Bioinformatics analysis
The miRNA microarray was carried out using the Exiqon miRCURY LNA microRNA array, 7th generation (miRBase v18) from the KangChen Bio-tech company (Shanghai, China). The threshold value used to screen differentially expressed miRNAs was a fold change of ≥2.0 or ≤0.5, a P-value of less than 0.01, and a normalized signal value, indicating the relative abundance to the transcript, of ≥2.0.
CCK-8 assay
The apoptosis rate of cells was measured using the CCK-8 Celling Counting Kit (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer's instructions.
Serum total RNA extraction with spiked-in control microRNA mimic
To normalize the serum RNA extraction efficiency, a synthesized Caenorhabditis elegans microRNA mimic (Shanghai GeneChem, Shanghai, China), cel-miR-39, was used as the spike-in control. Cel-miR-39 (5 fmol) was added to each 200 μl of serum sample. Total RNA was isolated from serum using the mirVana PARIS Kit (Ambion, Austin, TX, USA) following the manufacturer's protocol for the total RNA isolation procedure. Briefly, 200 μl of serum was mixed with an equal volume of 2× denaturing solution followed by organic extraction using acid-phenol and chloroform. The aqueous phase was mixed with 1.25 volumes of room temperature 100% ethanol. After washing three times, RNA was finally eluted using 100 μl 95 °C elution solution or nuclease-free water.
Flow cytometry
HepG2 cells were harvested after transfection with combination therapy constructs and washed twice using cold PBS containing 0.5% BSA. They were then resuspended in 1×Binding Buffer at a concentration of 1×106 cells/ml, and 100μl of the solution (1×105 total cells) were transferred to a 5 ml culture tube. Next, 5μl of FITC Annexin V and 5μl PI were added, and the cells were gently vortexed and then incubated for 15 min at RT (25°C) in the dark. A total of 400μl of 1×Binding Buffer was added to each tube, and the samples were analyzed by flow cytometry within 1 hr using a C6 FlowCytometer® Instrument (BD Biosciences, San Jose, CA, USA).
Patients and specimens
All patients in this study met the following inclusion criteria: Resected nodules were identified as liver cancer by pathological examination; no anticancer treatments were given before surgery; complete resection of all tumor nodules was verified by the cut surface being free of cancer by pathological examination. Venous blood (5 mL) was collected from each patient prior to surgery. Blood samples were centrifuged at 2500 rpm and 4°C for 10 min. Supernatants were recovered and stored at -80°C until further analysis. Informed consents for the use of samples were obtained from all patients and approval was obtained from the hospital.
Cell culture, treatment and transfection
Human liver cancer cell lines of Bel-7402, HuH7 were obtained from Type Culture Collection of the Chinese Academy of Sciences, Shanghai, China. Human liver cancer cell lines of SMMC-771, human primary embryonic liver cells CCC-HEL-1 were obtained from Cell Culture Center, Chinese Academy of Medical Sciences (Beijing, China). Human liver cancer cell lines of HepG2 and human embryonic kidney HEK 293T was from ATCC (Manassas, VA). All cell lines were identified by morphology and growth characteristics. Bel-7402 cells were cultured in RPMI 1640 supplemented with 10% FBS (Hyclone, Logan, UT, USA), 100 U/mL penicillin and 100μg/mL streptomycin at 37℃ in 5% CO2. Huh7, SMMC-7721, CCC-HEL-1 and 293T cells were cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin and 100μg/mL streptomycin at 37℃ in 5% CO2. HepG2 cells were cultured in MEM-EBSS supplemented with NEAA, 10% FBS, 100 U/mL penicillin and 100μg/mL streptomycin at 37℃ in 5% CO2. MiR-221/222 mimics (dsRNA oligonucleotides) and inhibitors (single-strand chemically modified oligonucleotides) from Invitrogen (Carlsbad, CA, USA) were used for the overexpression and inhibition of the mature miRNAs. These mimics and inhibitors were transfected at a final concentration of 10 nM into cells using LipofectamineTM 2000 reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. The combination therapy constructs were transfected at a final concentration of 10 nM using LipofectamineTM 2000 reagent for 48 hrs. Negative controls were transfected to serve as matched controls.
Quantitative RT-PCR analysis
Total RNA was extracted from tissues using the miRNeasy Mini kit (Qiagen, Hilden, Germany) following the manufacturer instructions. Total RNA containing small RNA was extracted from 500 μL plasma using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) and the miRNeasy Mini kit according to the manufacturer instructions. cDNA was synthesized via M-MLV reverse transcriptase (Promega, Madison, WI, USA). Oligo (dT)15 was used as the RT primers for reverse transcription of mRNAs. MiR-221/222 and U6 snRNA were reverse transcribed using specific RT primers. Quantitative RT-PCR was performed in an Step-One real-time PCR System (Applied Biosystems, Foster City, CA, USA) using the SYBR Green PCR Mix (Takara, Dalian, China) at 95 °C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min according to the manufacturer's instructions. The comparative Ct method was used to quantify target genes relative to the endogenous control. For the mRNAs, data were normalized to the endogenous GAPDH or beta-actin control. For the miRNAs, U6 snRNA or cel-miR-39 was used as the endogenous control. All PCR reactions were performed in triplicate. The primers used for PCR are listed in Table S1.
Western blot analysis
Protein extracts were resolved through 12% SDS-PAGE, transferred to PVDF membranes, and probed with antibodies against human Caspase-9, Caspase-8, Caspase-3 (Cell Signaling Technology, Danvers, MA, USA), PTEN (Cell Signaling Technology, Danvers, MA, USA), Survivin (Cell Signaling Technology, Danvers, MA, USA), PARP (Cell Signaling Technology, Danvers, MA, USA), MDR1 (Cell Signaling Technology, Danvers, MA, USA) and P27 (Cell Signaling Technology, Danvers, MA, USA), and normalized to the endogenous GAPDH (Kangcheng, China) or tubulin (Cell Signaling Technology, Danvers, MA, USA) control. Peroxidase-conjugated anti-mouse or rabbit IgG (Cell Signaling Technology, Danvers, MA, USA) was used as secondary antibody, and the antigen-antibody reaction was visualized by enhanced chemiluminescence assay (ECL, ThermoFisher Scientific, Cleveland, OH, USA).
Isolation and culture of liver cancer hepatocytes
Hepatocyte isolation was performed based on previous isolation protocol with modifications [17]. Tumor liver specimens were obtained from the primary tumor and transferred in ice-cold DMEM-F12 supplemented with 10% FBS. Liver specimens were treated with pre-warmed 0.1% Collagenase Type IV (Sigma Chemicals, Germany) and filtered through a 70 μm cell strainer (BD, USA). The pellet containing the liver cells was resuspended in ice cold DMEM-F12 with 20% FBS and triple centrifugation at 81 g for 12 min at 4°C was performed in order to separate the purified hepatocyte population (pellet) from the non-parenchymal cells (supernatant). Next, hepatocytes remained in culture for few hours in 60-mm dishes without collagen I in order to achieve maximum hepatocyte purity, by removing possible remaining nonparenchymal cells. Primary liver cancer cells from 8 liver cancer patients were isolated successfully from total 20 liver cancer patients.
Constructs and AAV package
The U6 promoter was PCR amplified and cloned into a T vector (TAKARA, Japan).
The sequences of miR-221-sponge, miR-222-sponge, miR-221-222-sponge, miR-221-TuD, miR-222-TuD, miR-221-Zip, miR-222-Zip and miR-221-222-Zip were designed and individually inserted into the T vector after the U6 promoter. The poly-A sequence was synthesized and inserted into the pAM/CAG-TRAIL vector after the TRAIL sequence. Then, the U6 promoter and several inhibitors were cut from the T vector and inserted into the pAM/CAG-TRAIL vector after the poly-A sequence. The rAAV virus was generated by standard production and purification protocols [18]. The rAAV titer was quantified by real-time PCR analysis, and the CAG primer used for qPCR is listed in Table S1.
Animal experiments
Female 5-week-old BALB/c nude mice were kept under specific pathogen-free conditions. Animal experiments proceeded in accordance with the guidelines of the National Institutes of Health. HepG2 cells were injected into the mice. The 1ⅹ1010 Gps AAV was transported into the tumor and the tumor size was monitored every 3 or 4 days until tumor volume reached 100 mm3, at which point the animals were sacrificed. Tumor volume (V) was calculated using the formula V = (ab2)/2, in which a is the longest and b the shortest diameter of the tumor.
Statistical analysis
Results of quantitative data in this study are expressed as the mean ±SD. Significant differences between groups were compared using two-tailed ANOVA via t test. A P value of less than .05 was considered significant (* P-values < 0.05, ** P-values <0.01, *** P-values < 0.001).
Results
MiR-221 and miR-222 were up-regulated both in primary and acquired TRAIL resistant liver cancer cells
The IC50 of a panel of liver cancer cells to TRAIL were determined by CCK-8 assay. We defined a liver cancer cell line as TRAIL resistant if greater than 50% of the cells were viable in response to a TRAIL concentration of 1000ng/ml for 24 hours treatment. As listed in Figure S1 and Table S2, HepG2 and Huh7 cells are intrinsic TRAIL-resistant liver cancer cell lines. HepG2 showed the highest IC50 at 5806 ng/ml and Bel-7402 showed the lowest IC50 to TRAIL.
To investigate mechanisms involved in TRAIL resistance in liver cancer, we generated TRAIL-resistant Bel-7402R cells by exposing parental TRAIL-sensitive Bel-7402 cells (Bel-7402S) to a stepwise increase in TRAIL concentration (1-1000 ng/ml) over a period of 2 months. As shown in Figure S2, Bel-7402R was more resistant to TRAIL compared to the parental Bel-7402S cells.
We explored the differential miRNA expression profiles between Bel-7402R and Bel-7402S by microarray technology. Microarray data have been deposited in the NCBI Gene Expression Omnibus and are accessible through the GEO Series accession number GSE74130. The threshold value used to screen differentially expressed miRNAs was a fold change of ≥2.0 or ≤0.5, a P-value of less than 0.01, and a normalized signal value, indicating the relative abundance to the transcript, of ≥2.0. As shown in Figure 1A, 11 miRNAs were up-regulated and 2 were down-regulated in Bel-7402R cells. Six of the altered miRNAs were randomly selected and validated by real time quantitative RT-PCR (qRT-PCR) (Figure S3). Among these miRNAs, miR-221 was markedly up-regulated with the highest relative abundance in acquired TRAIL-resistant Bel-7402 cells.
MiR-221 and miR-222 are two highly homologous miRNAs encoded in tandem on the same chromosome. Moreover, they have an identical seed sequence. MiR-222 was also detected upregulated in Bel-7402R cells in the microarray assays (the fold change is 1.832). Therefore, we simultaneously detected the expression of miR-221/222 in liver cancer cell lines treated with TRAIL. We observed a significant increase of miR-221/222 in the expression of both in Bel-7402, SMMC-7721, Huh7, and HepG2 cells treated with TRAIL for 24 hrs (Figure 1B).
To investigate the miR-221/222 expression in liver cancer after TRAIL treatment in vivo, the TRAIL-sensitive Bel-7402 liver cancer cells were injected into the right armpit of BALB/c nude mice. Then AAV-EGFP or AAV-TRAIL was introduced into the mice by intratumoral injection, and the tumor volumes were measured for about one month. The tumors in the AAV-TRAIL group were significantly smaller than those in the AAV-EGFP group (Figure S4). We then collected the residual tumor and cultured the cells in vitro, and stimulated them with TRAIL or PBS alone. The result indicated that cells from residual tumors isolated from the AAV-TRAIL group (Bel-7402-post) were more resistant to TRAIL compared to the control Bel-7402 cells (Bel-7402-pre) (Figure S5), which indicated that the TRAIL-sensitive liver cancer cells acquired TRAIL-resistance during AAV-TRAIL therapy. As is shown in Figure 1C, AAV-TRAIL induced up-regulation of miR-221/222 in acquired TRAIL-resistant liver cancer cell Bel-7402-post. Next, AAV-TRAIL was injected into subcutaneous HepG2 tumors in nude mice. One month later, tumors were collected to detect the expression of miR-221/222. As is shown in Figure 1D, AAV-TRAIL induced up-regulation of miR-221/222 in intrinsically TRAIL-resistant HepG2 tumors.
The expression of miR-221/222 in Bel-7402, SMMC-7721, Huh7, and HepG2 were also detected. As shown in Figure 1E, the expression of miR-221/222 in these liver cancer cells were was significantly correlated with TRAIL sensitivity. These results indicated that MiR-221 and miR-222 were up-regulated both in primary and acquired TRAIL resistant liver cancer cells.
MiR-221/222 knockdown increases TRAIL sensitivity in liver cancer
We next examined the effects of miR-221/222 on cell survival and TRAIL resistance in liver cancer. To test whether miR-221/222 in TRAIL-resistant HepG2 could change the response to TRAIL, miR-221 or miR-222 mimics or inhibitors were transfected into HepG2 cells, and then the cells were stimulated with 100 ng/ml TRAIL. The results showed that when transfected with miR-221 or miR-222 mimics, the viability of TRAIL-stimulated cells was increased compared to those treated with the control mimics (Figure S6). When HepG2 cells were treated with a combination of TRAIL and miR-221/222 inhibitor, significant synergy in the potentiation of cytotoxicity was observed. This phenomenon was also observed in TRAIL-resistant Bel-7402R cells (Figure S6). Furthermore, an Annexin V-FITC assay revealed an increase in TRAIL sensitivity after miR-221 or miR-222 inhibitor was transfected into TRAIL-resistant HepG2 cells (Figure S7A). The activation of Caspases 9 and 8 was easily identified in HepG2 cells exposed to the combination of TRAIL and miR-221 inhibitor, as evidenced by the reduced expression of the preform of these Caspases and the appearance of cleavage products (Figure S7B), indicating that miR-221 inhibitor overcome the resistance to TRAIL through the intrinsic and extrinsic apoptotic pathway simultaneously. Cleaved PARP and activation of Caspase 3 also implied that combination of TRAIL and miR-221/222 inhibitor showed significant synergy in the potentiation of cytotoxicity in TRAIL-resistance liver cancer cells.
The combination AAV constructs of TRAIL and specific miR-221/222 inhibitors efficiently reduce TRAIL-resistance
Because inhibition of miR-221/222 could enhance the sensitivity of cells to TRAIL, we wanted to combine TRAIL and miR-221/222 inhibitors to increase the therapeutic effect of TRAIL on liver cancer in vivo. Various methods have been used to date to inhibit miRNAs [19-22]. MicroRNA sponges, 'tough decoys' (TuDs) and miRZip have been reported to be newer and more efficient specific inhibitors of miRNA [23-25]. Therefore, we designed eight kinds of miRNA inhibitors, including miR-221-Sponge, miR-222-Sponge, miR-221-222-Sponge, miR-221-TuD, miR-222-TuD, miR-221-Zip, miR-222-Zip and miR-221-222-Zip, which were cloned into pAM-CAG, an AAV expression vector, and co-expressed with TRAIL (Figure 2A). We generated a dual promoter vector based on pAM-CAG with dual gene expression cassettes. TRAIL expression was under the control of CAG promoter, while the expression of these inhibitors was driven by the U6 promoter, a Pol III promoter commonly used to express small RNAs (Figure 2B).
Figure 1
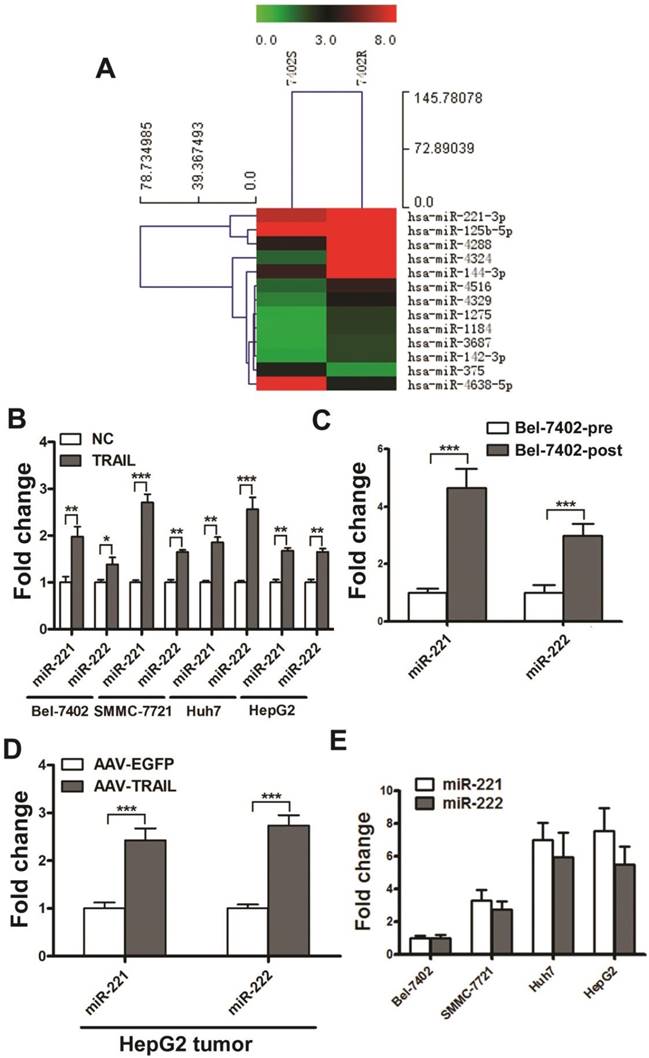
MiR-221 and miR-222 were up-regulated both in primary and acquired TRAIL resistant liver cancer cells. (A) Heat map representation of changes in expression of 13 miRNAs from Bel-7402S and Bel-7402R cells. (B) qRT-PCR analysis of the expression of miR-221 and miR-222 in Bel-7402, SMMC-7721, Huh7 and HepG2 cells during 50 ng/ml TRAIL stimulation. (C) qRT-PCR analysis of the expression of miR-221 and miR-222 in Bel-7402-pre and Bel-7402-post cells. (D) qRT-PCR analysis of the expression of miR-221 and miR-222 in HepG2 tumors after AAV-EGFP and AAV-TRAIL injection. (E) qRT-PCR analysis of the expression of miR-221 and miR-222 in Bel-7402, SMMC-7721, Huh7 and HepG2 cells. *** p<0.001; ** p<0.01; * p<0.05.
Figure 2
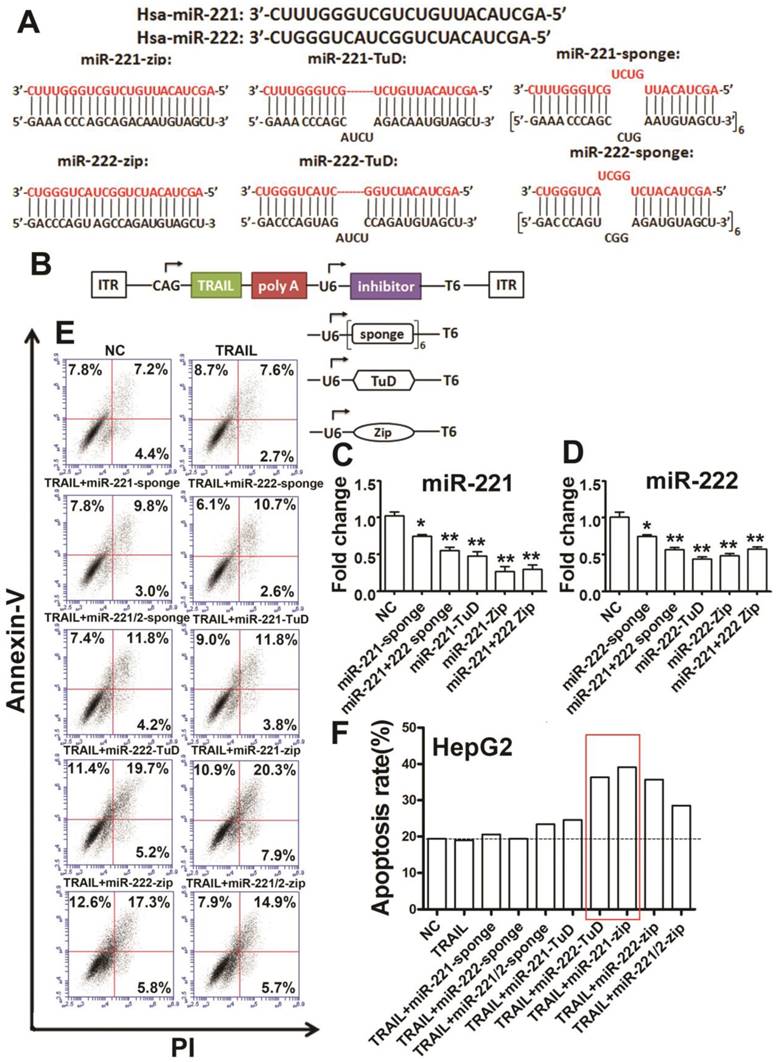
The combination constructs of TRAIL and specific miR-221/222 inhibitors reduce TRAIL-resistance. (A) Design of the miR-221/222 sponges, TuDs and miR-Zips. (B) Design of the two-promoter TRAIL expression system with specific miR-221/222 inhibitors. (C) qRT-PCR analysis of the relative expression of miR-221 in HEK 293T cells after transfection with the indicated constructs. (D) qRT-PCR analysis of the relative expression of miR-222 in HEK 293T cells after transfection with the indicated constructs. (E, F) Flow cytometry analysis of the apoptosis rate of HepG2 cells after transfection with the indicated combination constructs. ***p<0.001; ** p<0.01; * p<0.05.
We transfected the co-expression constructs into HEK293T cells to detect the function of the inhibitors. These results showed that miR-221-sponge, miR-221-222-sponge, miR-221-TuD, miR-221-Zip and miR-221-222-Zip could efficiently inhibit miR-221 (Figure 2C). Additionally, miR-222-sponge, miR-221-222-sponge, miR-222-TuD, miR-222-Zip and miR-221-222-Zip could efficiently inhibit miR-222 (Figure 2D). Next, we transfected the same vectors into HepG2 cells to detect apoptosis. We found that the apoptosis rate increased in the co-expression group more than in the single TRAIL group (Figure 2E, F), indicating that the combination therapy of TRAIL and specific miR-221/222 inhibitors could efficiently reduce TRAIL-resistance and enhance TRAIL sensitivity, especially in the TRAIL/miR-222-TuD co-expression and TRAIL/miR-221-Zip co-expression group.
AAV-TRAIL-miR-221-Zip increased TRAIL-induced apoptosis in liver cancer in vivo
Because all eight specific miR-221/222 inhibitors could promote TRAIL-induced apoptosis in vitro, we selected the two inhibitors/TRAIL co-expression constructs with the highest TRAIL sensitivity promoting activity (miR-222-TuD/TRAIL and miR-221-Zip/TRAIL) to package into AAV virus. qRT-PCR was used to detect the expression of TRAIL, miR-221 and miR-222 in the HEK 293T cells infected by the recombinant virus. The results showed that TRAIL was significantly overexpressed after virus infection (Figure S8A). Additionally, miR-221 was significantly inhibited in the AAV-TRAIL-miR-221-Zip group (Figure S8B) and the expression level of miR-222 was decreased in the AAV-TRAIL-miR-222-TuD group (Figure S8C).
To detect the combined therapeutic effect in liver cancer, we further established s.c. HepG2 tumor xenografts in BALB/c nude mice. When the tumors were grown to approximately 100 mm3, the mice were randomly divided into four groups: AAV-EGFP, AAV-TRAIL, AAV-TRAIL-miR-222-TuD and AAV-TRAIL-miR-221-Zip. The appropriate virus administered to the mice by intratumoral injection. It was noteworthy that the dose of virus administrated was much lower than our previous study [13, 26]. Results of this experiment showed that AAV-TRAIL could not inhibit the growth of HepG2 tumors, consistent with our results in vitro. Furthermore, there was no obvious difference in the AAV-TRAIL and AAV-TRAIL-miR-222-TuD groups when compared to the negative control group, while the tumors in the AAV-TRAIL-miR-221-Zip group were smaller than those in the other three groups (Figure 3A-C). To explore the mechanism of tumor growth suppression mediated by administration of AAV-TRAIL-miR-221-Zip, TUNEL analysis, cleaved Caspase-3 and cleaved PARP staining of subcutaneous tumor sections was performed. As shown in Figure 3D, there was no significant difference in the level of apoptosis in tumor between the control group and AAV-TRAIL, AAV-TRAIL-miR-222-TuD. However, a significantly higher level of apoptotic cell death was detectable in the tumor treated with AAV-TRAIL-miR-221-Zip. qRT-PCR was then used to detect the expression of miR-222 and miR-221 in tumors. The expression level of miR-221 decreased in the AAV-TRAIL-miR-221-Zip group (Figure 3E). Meanwhile, the expression level of miR-222 in AAV-TRAIL-miR-222-TuD group was somewhat decreased compared to the AAV-EGFP group (Figure 3F). These data indicated that the combination therapy of TRAIL and miR-221-Zip could reverse TRAIL resistance in liver cancer cells.
MiR-221-Zip promotes TRAIL-sensitivity through the molecules in apoptotic pathway in liver cancer
Because AAV-TRAIL-miR-221-Zip treatment group showed the greatest liver cancer growth inhibiting activity in vitro and in vivo, we focus the study on miR-221 inhibition synergized with TRAIL. Previous reports indicated that PTEN and p27 were the target genes of miR-221/222, leading to regulation of TRAIL resistance [14]. Therefore, we detected the expression of PTEN and p27 in HepG2 tumors injected with AAV-TRAIL-miR-221-Zip. As shown in Figure 3G, PTEN and p27 were increased in the AAV-TRAIL-miR-221-Zip group, which meant that miR-221-Zip could inhibit the function of miR-221 in vivo. Meanwhile, it was also shown that PTEN and p27 were decreased in the AAV-TRAIL group which indicated that PTEN and p27 downregulation might account for TRAIL resistant.
We next detected the expression of PTEN and p27 in liver cancer cell lines. We found that the expression level of PTEN and p27 were positively correlated to TRAIL sensitivity (Figure 4A). Other critical proteins related to TRAIL resistance, including Survivin, FLIP, XIAP, Mcl-1, were also detected. As shown in Figure 4A, Survivin negatively correlated to TRAIL sensitivity in liver cancer cells, while FLIP, XIAP, Mcl-1 did not show any relationship with TRAIL sensitivity. MDR1 expression in cancer cells confers drug resistance to many anticancer drugs [27], so we detect the MDR1 expression in liver cancer cell lines. As shown in Figure 4A, MDR1 did not show any relationship with TRAIL sensitivity. Furthermore, Western blot showed the promotion of PTEN, p27 expression in cells transfected with miR-221 inhibitors (Figure 4B). HepG2 cells transfected with miR-221 inhibitors also lead to significant inhibition of Survivin (Figure 4B). It was reported that the lack of surface DR4/DR5 is sufficient to render cancers resistant to TRAIL-induced apoptosis. However, we found there was no obvious difference of DR4 and DR5 expression in cells transfected with miR-221 inhibitors. And MDR1 expression also did not changed in miR-221 inhibitors transfecting cells (Figure S9). These results indicated that miR-221 inhibitors enhanced TRAIL induced apoptosis in TRAIL-resistant liver cancer cells by regulating molecules in apoptosis pathways.
Figure 3
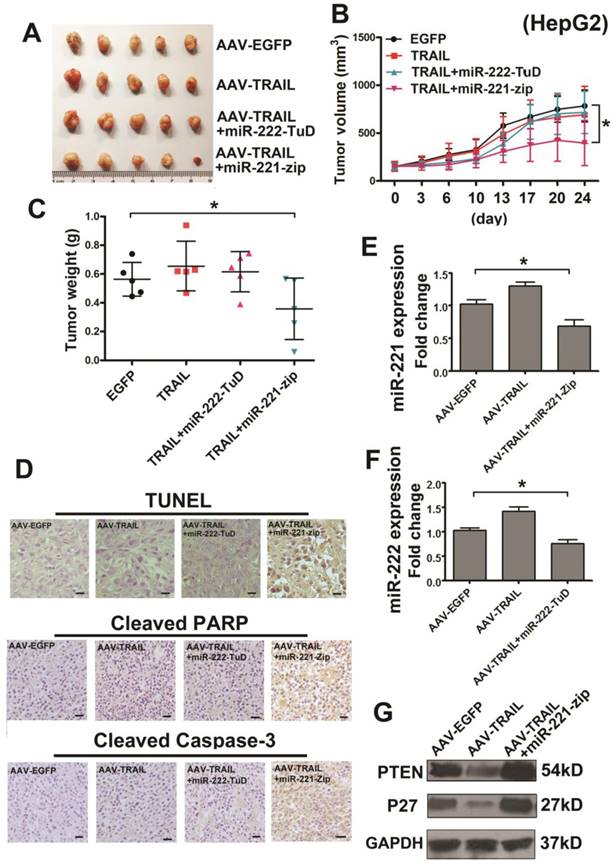
The AAV-TRAIL-miR-221-Zip increases TRAIL-induced apoptosis in liver cancer in vivo. Approximately 5×106 HepG2 cells in 100 μl PBS were s.c. injected into 4-6-week-old BALB/c female nude mice. After 14 days, the tumors had grown to approximately 100 mm3 and the mice were randomly divided into four groups for intratumor AAV delivery. Tumor length and width were measured with a caliper every 3 or 4 days for 24 days. (A) Images of tumors at the experimental end point. (B) Growth curves of the xenograft tumors. (C) Tumor weight was measured at the experimental end point. (D) TUNEL assay of cellular apoptosis, cleaved PARP and Caspase-3 staining in the tumor tissues of four groups. (E) qRT-PCR analysis of the relative expression of miR-221 in the tumors of indicated groups. (F) qRT-PCR analysis of the relative expression of miR-222 in the tumors of indicated groups. (G) Western blot analysis of PTEN and p27 in the tumors of indicated groups. * p<0.05.
Figure 4
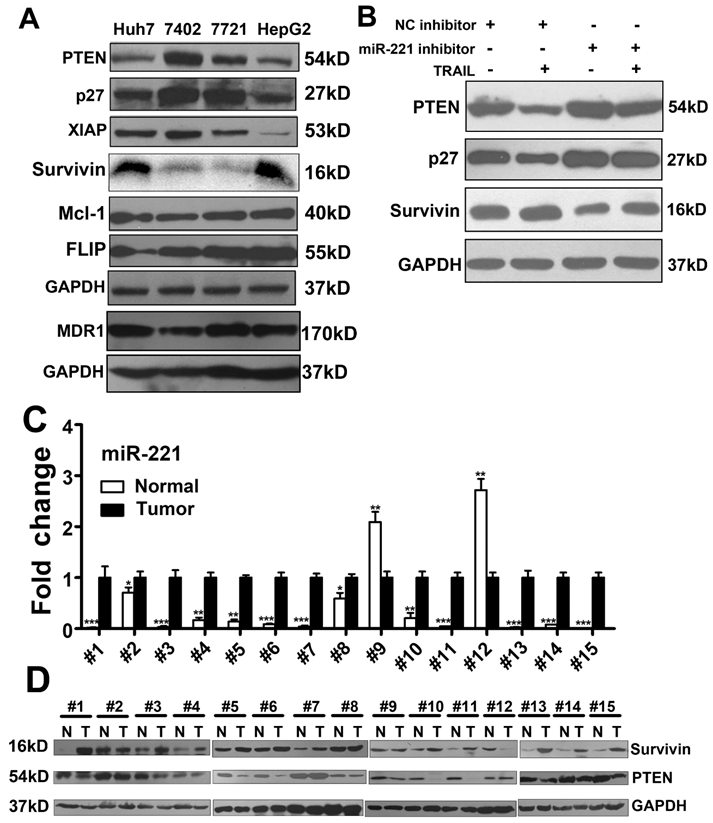
MiR-221-Zip promotes TRAIL-sensitivity through the molecules in apoptotic pathway in liver cancer. (A) Western blot analysis of the expression of PTEN, p27, XIAP, FLIP, Mcl-1, MDR1 and Survivin in Huh7, Bel-7402, SMMC-7721 and HepG cells. (B) Western blot analysis of the expression of PTEN, p27 and Survivin in HepG2 cells after TRAIL stimulation and/or transfection with a miR-221 inhibitor. (C) qRT-PCR analysis the expression of miR-221 in liver cancer tissues from patients. (D) Western blot analysis of the expression of PTEN and Survivin in liver cancer tissues from patients. ***p<0.001; ** p<0.01; * p<0.05.
We further analyzed expression of miR-221 and p27, PTEN and Survivin in liver cancer patients. Expression of each was recapitulated from a large cohort of liver cancer patients available from the NCBI Gene expression Omnibus (GEO) database (accession number GSE22058), and data were analyzed as scatter plots. As shown in Figure S10, miR-221and Survivin were significantly overexpressed in liver cancer, while P27 and PTEN were significantly down-regulated. Furthermore, we observed that miR-221 expression was concomitant with promotion of Survivin mRNA expression in this cohort of liver cancer patients (Figure S11).
As the results in Figure 4A, we found that PTEN and Survivin expression showed highest correlation with TRAIL sensitivity in liver cancer cell lines. We then analyzed the expression of miR-221, Survivin and PTEN in 15 pairs of liver cancer and corresponding non-tumor tissues. As seen in Figure 4C, miR-221 was highly expressed in liver cancer tissues relative to non-tumor tissues. Survivin and PTEN expression in these tumor sets was then checked by Western blot (Figure 4D). Interestingly, most tumor samples showed high expression of Survivin and low expression of PTEN. And miR-221 expression showed significant positive correlation with Survivin expression (Figure S12A), but no obvious correlation between miR-221 expression and PTEN expression was observed. The expression of miR-221 in the serum of these patients was also detected. As shown in Figure S12B, serum miR-221 was also positively correlated with Survivin expression and no obvious correlation between serum miR-221 expression and PTEN expression was observed.
Primary liver cancer cells from eight patients' tumor samples were isolated and cultured. The TRAIL IC50 of these cells was detected (Table S3). MiR-221, Survivin, and PTEN expression was analyzed by qRT-PCR or Western blot (Figure 5A, B). These results indicated that miR-221 expression in this primary liver cancer cells positively correlated with Survivin expression, negatively correlated with PTEN expression and also TRAIL sensitivity of primary liver cancer cells (Figure 5C-E). The expression of miR-221 in the serum of these patients was also detected. As shown in Figure 5F and 5G, serum miR-221 also showed positive correlation with Survivin expression and negative correlation with TRAIL sensitivity of primary liver cancer cells. Together, these results indicated that PTEN and Survivin might play a crucial role in TRAIL resistance in liver cancer and suggested that serum miR-221 could be used as a potential predictor of TRAIL sensitivity in liver cancer.
Figure 5
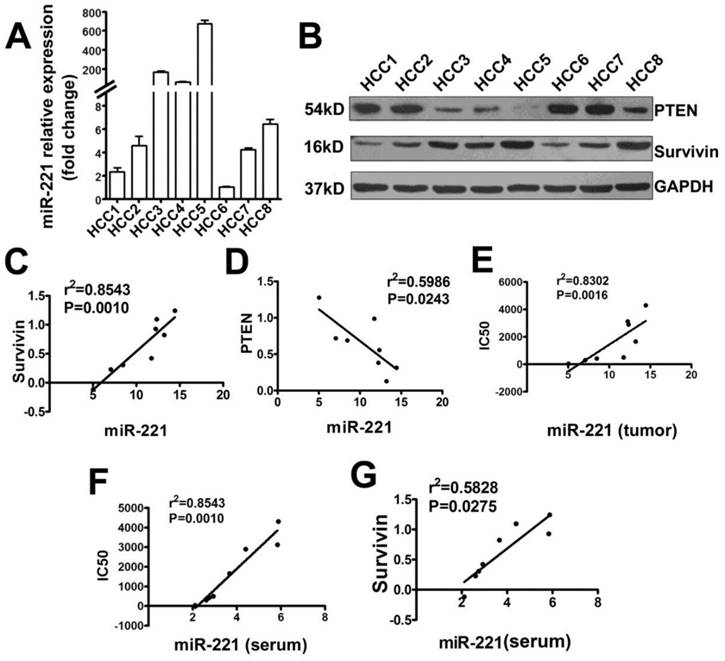
The correlation analysis of miR-221 expression with Survivin, PTEN expression or IC50 of liver cancer cells to TRAIL. (A) qRT-PCR analysis of the expression of miR-221 in primary liver cancer (Hepatocellular Carcinoma; HCC) cells from eight patients' tumor samples. (B) Western blot analysis of the expression of PTEN and Survivin in eight primary HCC cells. (C) The correlation analysis of miR-221 and Survivin expression in primary liver cancer cells. (D) The correlation analysis of miR-221 and PTEN expression in primary liver cancer cells. (E) The correlation analysis of miR-221 expression and IC50 of the primary liver cancer cells to TRAIL. (F) The correlation analysis of miR-221 expression in serum of liver cancer patients and IC50 of the primary liver cancer cells to TRAIL. (G) The correlation analysis of miR-221 expression in serum of liver cancer patients with Survivin expression in the primary liver cancer cells.
PTEN down-regulates Survivin expression in liver cancer cells
Survivin is considered a critical protein related to apoptosis resistance. Here, we found that Survivin expression correlated with miR-221 expression and TRAIL sensitivity in liver cancer cell lines and patients. Moreover, Survivin was clearly down-regulated in the presence of a miR-221 inhibitor, indicating that Survivin was important in TRAIL resistance. Therefore, we introduced Survivin siRNA into Bel-7402 cells and HepG2 cells and then treated the cells with TRAIL. CCK-8 results from this assay showed that TRAIL induced more apoptosis in cells transfected with Survivin siRNA (Figure 6A, B).
Next, we investigated the reason for Survivin down-regulation induced by inhibition of miR-221. It has been suggested that miRNAs can increase gene expression by binding to promoter regions [28] or the 5'UTR [29]. We analyzed the 4000 bp upstream of the TSS along with the whole sequence of Survivin mRNA including the 5'UTR and also 3'UTR. However, no possible miR-221 target site was found. So we hypothesized that the gene targeted by miR-221might be negative regulator of Survivin. Because AKT is regulated by PI3K signaling and has been shown to be hyper activated through the loss of PTEN, and because it has also been reported that Survivin expression is up-regulated by PI3K/Akt, we analyzed the activity of this pathway in liver cancer cells. As shown in Figure 6C, the expression level of PI3K, phosphorylation AKT and Survivin was elevated in PTEN siRNA transfected HepG2 and Bel-7402 cells. Importantly, the decreased apoptosis seen in PTEN-knockdown liver cancer cells was rescued by knocking down Survivin expression (Figure 6D). Therefore, the results indicated that Survivin upregulation induced by miR-221 overexpression might be related to PTEN downregulation.
Figure 6
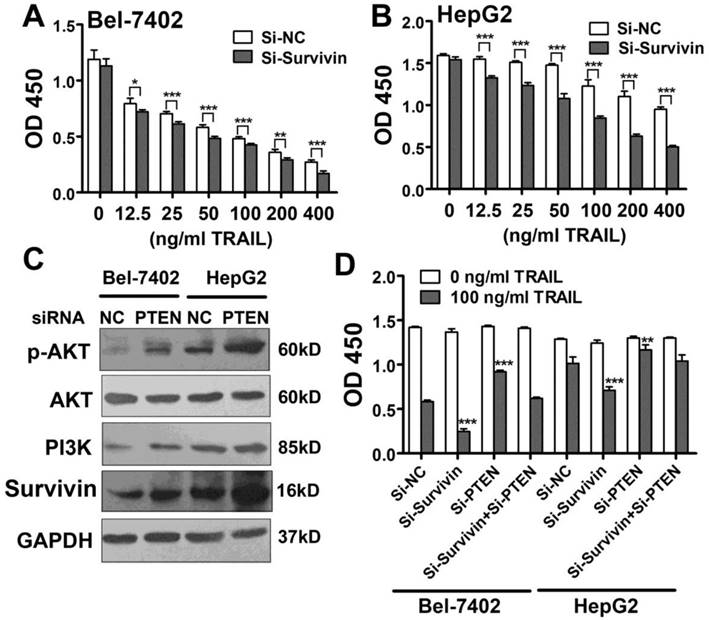
PTEN down-regulates Survivin expression in liver cancer cells, and the miR-221-Zip functions through Zip endogenous miR-221. (A, B) CCK-8 analysis of apoptosis in Bel-7402 and HepG2 cells treated with indicated TRAIL concentrations after transfection with Survivin siRNA. (C) Western blot analysis of the expression of p-AKT, PI3K, Survivin, AKT levels in Bel-7402 and HepG2 cells after transfection with PTEN siRNA. (D) CCK-8 analysis of apoptosis in Bel-7402 and HepG2 cells treated with indicated TRAIL concentrations after transfection with Survivin siRNA and/or PTEN siRNA. ***p<0.001; ** p<0.01; * p<0.05.
Toxicity of strategy of TRAIL combined with miR-221 inhibitors in vitro
Our results suggested that AAV-TRAIL-miR-221-Zip was a potential agent for use against TRAIL-resistant liver cancer. Therefore, it was important to investigate the toxicity of AAV-TRAIL-miR-221-Zip to normal cells. There was no obvious apoptosis induced by TRAIL in human primary embryonic liver cells CCC-HEL-1 cell line (Figure S13A). Next, we transfected the cells with miR-221/222 mimics or inhibitors followed by stimulation with TRAIL to detect the toxicity of the combined treatment (Figure S13B). Furthermore, AAV-TRAIL-miR-221-Zip infected CCC-HEL-1 to detect the cytotoxicity (Figure S13C). These experiments showed no significant toxicity on cell growth, which indicated the safety of AAV-TRAIL-miR-221-Zip.
Discussion
Although many studies have elucidated TRAIL signaling and its crosstalk with other signaling pathways [30-36], it seems we have only reached the tip of the iceberg in understanding the mechanism of TRAIL resistance. Conventional chemotherapeutic drugs, as well as irradiation, can sensitize TRAIL-resistant cells to undergo apoptosis. Thus, these agents enhance the therapeutic potential of TRAIL in TRAIL-sensitive cells and sensitize TRAIL-resistant cells [37]. However, the toxicity of chemotherapy drugs is widely known, so target-specific drug with low side effect to overcome TRAIL resistance needs to be developed. Experimental evidence demonstrates that modulation of specific miRNA alterations in cancer cells using miRNA replacement or anti-miRNA technologies can restore miRNA activities and repair gene regulatory networks. Numerous animal studies for miRNA-based therapy offer the hope of targeting miRNAs as an alternative cancer treatment. Here we provide a possible strategy to reduce TRAIL resistance by combining AAV-TRAIL with miR-221-Zip with no obvious toxicity to normal hepatocyte.
MiR-221/222 has been considered to act as oncogenes in many cancers including liver cancer. It is well known that liver cancer express high levels of miR-221[38, 39]. In these tumors, miR-221 increases cell proliferation [40] and tumor progression [41-43] and inhibits apoptosis [39]. Our results indicated that the high level expression of miR-221/222 in liver cancer might be the reason of TRAIL resistance in majority liver cancer cells. What's more important is, we found although miR-221 upregulated in liver cancer, the expression level of miR-221 in serum was correlated with TRAIL sensitivity. It is the first time to suggest that serum miR-221 could be used as a potential predictor of TRAIL sensitivity in liver cancer.
Recent studies reported on modulation of miR-221/222 as therapeutic strategies to counteract their tumor-promoting effects. A miRNA sponge for miR-221 has been developed and showed strong suppression of this miRNA together with upregulation of its targets within HCC cells [44]. Intravenous treatment of transgenic mice with 2ʹ-O-methyl modified oligonucleotides targeting miR-221 led to the suppression of liver and circulating miR-221, inducing tumor growth inhibition. No relevant systemic toxicity has been reported [45]. Pineau et al. introduced LNA-modified antimiR-221 and antimiR-222 into liver cancer cells to reduce the growth of liver cancer cells [46]. Recently, pharmacokinetics and pharmacodynamics study of a 13-mer LNA-inhibitor-miR-221 in mice and non-human primates indicate the suitability of LNA-i-miR-221 for clinical use and provide pilot data for safety analysis [47]. Here we provide a possible strategy by AAV mediated TRAIL plus miR-221-Zip expression to reduce TRAIL resistance. Our study also suggested the efficiency and safety of AAV mediated miRNA expression as a strategy in cancer therapy.
In our previous study, high dose of AAV-TRAIL suppressed s.c. or metastatic liver tumors [13, 26]. However, the dose of the AAV virus administrated into tumor in this study was much lower than our previous study [13, 26]. We showed that AAV-mediated TRAIL combined with miR-221-Zip gene therapy significantly suppressed the growth of human liver tumor cells transplanted in mice even be administrated with much lower dose virus. Large scale production of AAV is costly and time-consuming, so it will be advantageous to reduce the therapy dose of the virus. Meanwhile, reducing dose might be helpful for prevent adverse side effects.
Although derived from the same precursor and carrying identical seed sequences, miR-221 and miR-222 showed different “preferences” in their function. MiR-221-Zip showed more significant enhancement of the antitumor efficacy of AAV-TRAIL in liver cancer than miR-222-TuD. However, in our study on lung cancer, miR-222-TuD showed more significant enhancement of AAV-TRAIL-induced apoptosis than miR-221-Zip (unpublished data). Therefore, we wondered whether miR-221 and miR-222 could function through the combination of sequences other than the seed sequence. It is well known that some miRNAs, such as RNAa, play their roles independent of seed sequence [48] so this represents an interesting hypothesis to explain the functional differences between miR-221 and miR-222 needs further study.
Although we demonstrated PTEN was a critical target of miR-221 related to TRAIL resistant, miR-221 expression in tumor tissues and serum of 15 liver cancer patients showed no obvious correlation with PTEN expression in tumor tissues. This inconsistent might be due to tumor tissues, far from being homogenous lumps of cells, consist of different cell types. It also might be due to the limited number of tumor samples. It would be helpful to collect more clinical samples to clarify this problem. But the expression level of miR-221 in serum was demonstrated correlated with TRAIL sensitivity exactly suggested that serum miR-221 could be used as a potential predictor of TRAIL sensitivity in liver cancer.
Abbreviations
TRAIL: tumor necrosis factor-α-related apoptosis-inducing ligand; AAV: adeno associated virus; HCC: Hepatocellular carcinoma; FITC: fluorescein isothiocyanate; IC50: 50% inhibitive concentration; LNA: locked nucleic acid; TuD: tough decoys; qRT-PCR: quantitative real-time polymerase chain reaction; TSS: transcription start site; UTR: untranslated regions.
Acknowledgements
This work was partially supported by the Program for the Innovation of New Drugs (2013ZX09103003-007), the National Natural Science Foundation of China (Grant No.81372200, 81572755), Beijing Natural Science Foundation (Grant No. 5164039) and CAMS Initiative for Innovative Medicine (CAMS-I2M Grant No. 2016-I2M-1-001)
Author contributions
J. S. contributed to the conception and design of the research. S. M., J. S. and P. Z. performed the experiments, analyzed data. S. M. wrote the manuscript and prepared the figures. Y. G. analyzed the GEO data. J.S., Y. L. and D. Z. edited and revised the manuscript.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK. et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673-82
2. Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. 1996;271:12687-90
3. Wajant H, Gerspach J, Pfizenmaier K. Tumor therapeutics by design: targeting and activation of death receptors. Cytokine Growth Factor Rev. 2005;16:55-75
4. Koschny R, Walczak H, Ganten TM. The promise of TRAIL-potential and risks of a novel anti-cancer therapy. J Mol Med (Berl). 2007;85:923-35
5. Farooqi AA, Gadaleta CD, Ranieri G, Fayyaz S, Marech I. New Frontiers in Promoting TRAIL-Mediated Cell Death: Focus on Natural Sensitizers, miRNAs, and Nanotechnological Advancements. Cell Biochem Biophys. 2016;74:3-10
6. Farooqi AA, De Rosa G. TRAIL and microRNAs in the treatment of prostate cancer: therapeutic potential and role of nanotechnology. Appl Microbiol Biotechnol. 2013;97:8849-57
7. Zhu H, Zhang L, Huang X, Davis JJ, Jacob DA, Teraishi F. et al. Overcoming acquired resistance to TRAIL by chemotherapeutic agents and calpain inhibitor I through distinct mechanisms. Mol Ther. 2004;9:666-73
8. Trivedi R, Mishra DP. Trailing TRAIL Resistance: Novel Targets for TRAIL Sensitization in Cancer Cells. Front Oncol. 2015;5:69
9. Fayyaz S, Yaylim I, Turan S, Kanwal S, Farooqi AA. Hepatocellular carcinoma: targeting of oncogenic signaling networks in TRAIL resistant cancer cells. Mol Biol Rep. 2014;41:6909-17
10. Lemke J, von Karstedt S, Zinngrebe J, Walczak H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014;21:1350-64
11. Ylä-Herttuala S. Endgame: glybera finally recommended for approval as the first gene therapy drug in the European union. Mol Ther. 2012;20:1831-2
12. Miller N. Glybera and the future of gene therapy in the European Union. Nat Rev Drug Discov. 2012;11:419
13. Ma H, Liu Y, Liu S, Kung HF, Sun X, Zheng D. et al. Recombinant adeno-associated virus-mediated TRAIL gene therapy suppresses liver metastatic tumors. Int J Cancer. 2005;116:314-21
14. Garofalo M, Di Leva G, Romano G, Nuovo G, Suh SS, Ngankeu A. et al. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell. 2009;16:498-509
15. Joshi P, Jeon YJ, Laganà A, Middleton J, Secchiero P, Garofalo M. et al. MicroRNA-148a reduces tumorigenesis and increases TRAIL-induced apoptosis in NSCLC. Proc Natl Acad Sci U S A. 2015;112:8650-5
16. Razumilava N, Bronk SF, Smoot RL, Fingas CD, Werneburg NW, Roberts LR. et al. miR-25 targets TNF-related apoptosis inducing ligand (TRAIL) death receptor-4 and promotes apoptosis resistance in cholangiocarcinoma. Hepatology. 2012;55:465-75
17. Doumba PP, Nikolopoulou M, Gomatos IP, Konstadoulakis MM, Koskinas J. Co-culture of primary human tumor hepatocytes from patients with hepatocellular carcinoma with autologous peripheral blood mononuclear cells: study of their in vitro immunological interactions. BMC Gastroenterol. 2013;13:17
18. Shi J, Zheng D, Liu Y, Sham MH, Tam P, Farzaneh F. et al. Overexpression of soluble TRAIL induces apoptosis in human lung adenocarcinoma and inhibits growth of tumor xenografts in nude mice. Cancer Res. 2005;65:1687-92
19. Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4:721-6
20. Hutvágner G, Simard MJ, Mello CC, Zamore PD. Sequence-specific inhibition of small RNA function. PLoS Biol. 2004;2:E98
21. Meister G, Landthaler M, Dorsett Y, Tuschl T. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10:544-50
22. Ørom UA, Kauppinen S, Lund AH. LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene. 2006;372:137-141
23. Xie J, Ameres SL, Friedline R, Hung JH, Zhang Y, Xie Q. et al. Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat Methods. 2012;9:403-9
24. Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M. et al. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-γ. Nat Immunol. 2011;12:861-9
25. Haraguchi T, Ozaki Y, Iba H. Vectors expressing efficient RNA decoys achieve the long-term suppression of specific microRNA activity in mammalian cells. Nucleic Acids Res. 2009;37:e43
26. Zhang Y, Ma H, Zhang J, Liu S, Liu Y, Zheng D. AAV-mediated TRAIL gene expression driven by hTERT promoter suppressed human hepatocellular carcinoma growth in mice. Life Sci. 2008;82:1154-61
27. Santos SA, Paulo A. Small molecule inhibitors of multidrug resistance gene (MDR1) expression: preclinical evaluation and mechanisms of action. Curr Cancer Drug Targets. 2013;13:814-28
28. Suzuki K, Ahlenstiel C, Marks K, Kelleher AD. Promoter Targeting RNAs: unexpected contributors to the control of HIV-1 transcription. Mol Ther Nucleic Acids. 2015;4:e222
29. Guo ZW, Xie C, Yang JR, Li JH, Yang JH, Zheng L. MtiBase: a database for decoding microRNA target sites located within CDS and 5'UTR regions from CLIP-Seq and expression profile datasets. Database (Oxford). 2015 pii: bav102
30. So J, Pasculescu A, Dai AY, Williton K, James A, Nguyen V. et al. Integrative analysis of kinase networks in TRAIL-induced apoptosis provides a source of potential targets for combination therapy. Sci Signal. 2015;8:rs3
31. Cha HY, Lee BS, Chang JW, Park JK, Han JH, Kim YS, Shin YS, Byeon HK, Kim CH. Downregulation of Nrf2 by the combination of TRAIL and Valproic acid induces apoptotic cell death of TRAIL-resistant papillary thyroid cancer cells via suppression of Bcl-xL. Cancer Lett. 2016;372:65-74
32. Fayyaz S, Yaylim I, Turan S, Kanwal S, Farooqi AA. Hepatocellular carcinoma: targeting of oncogenic signaling networks in TRAIL resistant cancer cells. Mol Biol Rep. 2014;41:6909-17
33. Farooqi AA, Yaylim I, Ozkan NE, Zaman F, Halim TA, Chang H. Restoring TRAIL mediated signaling in ovarian cancer cells. Arch Immunol Ther Exp (Warsz). 2014;62:459-74
34. Fulda S. Tumor-necrosis-factor-related apoptosis-inducing ligand (TRAIL). Adv Exp Med Biol. 2014;818:167-80
35. Refaat A, Abd-Rabou A, Reda A. TRAIL combinations: The new 'trail' for cancer therapy. Oncol Lett. 2014;7:1327-32
36. Du J, Wang Y, Chen D, Ji G, Ma Q, Liao S. et al. BAY61-3606 potentiates the anti-tumor effects of TRAIL against colon cancer through up-regulating DR4 and down-regulating NF-κB. Cancer Lett. 2016;383:145-153
37. Shankar S, Srivastava RK. Enhancement of therapeutic potential of TRAIL by cancer chemotherapy and irradiation: mechanisms and clinical implications. Drug Resist Updat. 2004;7:139-56
38. Garofalo M, Condorelli GL, Croce CM, Condorelli G. MicroRNAs as regulators of death receptors signaling. Cell Death Differ. 2010;17:200-8
39. Garofalo M, Condorelli G, Croce CM. MicroRNAs in diseases and drug response. Curr Opin Pharmacol. 2008;8:661-7
40. Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F. et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257-61
41. Zhang C, Zhang J, Zhang A, Wang Y, Han L, You Y. et al. PUMA is a novel target of miR-221/222 in human epithelial cancers. Int J Oncol. 2010;37:1621-6
42. Galardi S, Mercatelli N, Giorda E, Massalini S, Frajese GV. et al. miR-221 and miR-222 expression affects the proliferation potential of human prostate carcinoma cell lines by targeting p27Kip1. J Biol Chem. 2007;282:23716-24
43. le Sage C, Nagel R, Egan DA, Schrier M, Mesman E, Mangiola A. et al. Regulation of the p27(Kip1) tumor suppressor by miR-221 and miR-222 promotes cancer cell proliferation. EMBO J. 2007;26:3699-708
44. Moshiri F, Callegari E, D'Abundo L, Corrà F, Lupini L, Sabbioni S. et al. Inhibiting the oncogenic mir-221 by microRNA sponge: toward microRNA-based therapeutics for hepatocellular carcinoma. Gastroenterol Hepatol Bed Bench. 2014;7:43-54
45. Callegari E, Elamin BK, Giannone F, Milazzo M, Altavilla G, Fornari F. et al. Liver tumorigenicity promoted by microRNA-221 in a mouse transgenic model. Hepatology. 2012;56:1025-33
46. Pineau P, Volinia S, McJunkin K, Marchio A, Battiston C, Terris B. et al. miR-221 overexpression contributes to liver tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:264-9
47. Gallo Cantafio ME, Nielsen BS, Mignogna C, Arbitrio M, Botta C, Frandsen NM. et al. Pharmacokinetics and Pharmacodynamics of a 13-mer LNA-inhibitor-miR-221 in Mice and Non-human Primates. Mol Ther Nucleic Acids. 2016;5:e326
48. Guo D, Barry L, Lin SS, Huang V, Li LC. RNAa in action: from the exception to the norm. RNA Biol. 2014;11:1221-5
Author contact
![]() Corresponding author: Juan Shi (shijuanttcom, juanshipumc.edu.cn), Institute of Basic Medical Sciences, 5 Dong Dan San Tiao, Beijing 100005, China. Tel: +86 10-69156430, Fax: +86 10 6510 5102
Corresponding author: Juan Shi (shijuanttcom, juanshipumc.edu.cn), Institute of Basic Medical Sciences, 5 Dong Dan San Tiao, Beijing 100005, China. Tel: +86 10-69156430, Fax: +86 10 6510 5102
Citation styles
APA
Ma, S., Sun, J., Guo, Y., Zhang, P., Liu, Y., Zheng, D., Shi, J. (2017). Combination of AAV-TRAIL with miR-221-Zip Therapeutic Strategy Overcomes the Resistance to TRAIL Induced Apoptosis in Liver Cancer. Theranostics, 7(13), 3228-3242. https://doi.org/10.7150/thno.19893.
ACS
Ma, S.; Sun, J.; Guo, Y.; Zhang, P.; Liu, Y.; Zheng, D.; Shi, J. Combination of AAV-TRAIL with miR-221-Zip Therapeutic Strategy Overcomes the Resistance to TRAIL Induced Apoptosis in Liver Cancer. Theranostics 2017, 7 (13), 3228-3242. DOI: 10.7150/thno.19893.
NLM
Ma S, Sun J, Guo Y, Zhang P, Liu Y, Zheng D, Shi J. Combination of AAV-TRAIL with miR-221-Zip Therapeutic Strategy Overcomes the Resistance to TRAIL Induced Apoptosis in Liver Cancer. Theranostics 2017; 7(13):3228-3242. doi:10.7150/thno.19893. https://www.thno.org/v07p3228.htm
CSE
Ma S, Sun J, Guo Y, Zhang P, Liu Y, Zheng D, Shi J. 2017. Combination of AAV-TRAIL with miR-221-Zip Therapeutic Strategy Overcomes the Resistance to TRAIL Induced Apoptosis in Liver Cancer. Theranostics. 7(13):3228-3242.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.