Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Biogenesis and functions of...
Angiogenesis and...
Pro-angiogenic factors and...
Regulation of miRNAs and lncRNAs...
MiRNAs and neovascularization
miRNA and CNV
miRNA and retinal angiogenesis
The miRNAs that inhibit RNV
The miRNAs that promote RNV
The miRNAs with controversial...
LncRNA and neovascularization
Conclusion
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(12):3155-3167. doi:10.7150/thno.19646 This issue Cite
Review
Decoding Noncoding RNAs: Role of MicroRNAs and Long Noncoding RNAs in Ocular Neovascularization
Yan Zhang1 ![]() , Siwei Cai1, Yurong Jia1, Chen Qi1, Jing Sun1, Hong Zhang1, Fei Wang1, Yunshan Cao2, 3
, Siwei Cai1, Yurong Jia1, Chen Qi1, Jing Sun1, Hong Zhang1, Fei Wang1, Yunshan Cao2, 3 ![]() , Xiaorong Li1
, Xiaorong Li1 ![]()
1. Tianjin Medical University Eye Hospital, Tianjin Medical University Eye Institute, Tianjin Medical University, College of Optometry and Ophthalmology, 300384, China;
2. Department of Heart Failure, Shanghai East Hospital, Tongji University School of Medicine; Research Center for Translational Medicine, Shanghai East Hospital, Tongji University School of Medicine, Shanghai 200120, China;
3. Department of Cardiology, Gansu Provincial Hospital, Lanzhou, Gansu Province, 730000, China.
Received 2017-2-13; Accepted 2017-6-15; Published 2017-7-22
Abstract

Ocular neovascularization is a pathological sequel of multiple eye diseases. Based on the anatomical site into which the abnormal neovessels grow, ocular neovascularization can be categorized into corneal neovascularization, choroidal neovascularization, and retinal neovascularization. Each category is intractable, and may lead to blindness if not appropriately treated. However, the current therapeutic modalities, including laser photocoagulation, vitrectomy surgery, and anti-VEGF drugs, raise concerns due to limited efficacy, damage on retinal parenchyma and vasculature, and the patients' unresponsiveness to the treatments. Therefore, the in-depth study on pathogenesis of and the search for novel therapeutic targets to the ocular neovascularization are needed. During the last 10 years or so, a large number of literatures have emerged indicating a critical role of noncoding RNAs, particularly microRNAs (miRNAs) and long noncoding RNAs (lncRNAs), in the pathogenesis and regulation of the ocular neovascularization. This review summarizes the current understanding of the biosynthesis and functions of the miRNAs and lncRNAs, the regulation of the miRNAs and lncRNAs in neovascular eye diseases, as well as the roles of these noncoding RNAs in the disease models of ocular neovascularization, in the hope that it could provide clues for the pathogenesis of and molecular targets to the ocular neovascularization.
Keywords: eye, neovascularization, noncoding RNAs, microRNAs, long noncoding RNAs.
Introduction
Modern molecular biology has shown that most genes are transcribed into RNAs, yet only 2% of the transcribed RNAs encode proteins, the rest of the transcripts that do not encode proteins are called noncoding RNAs (ncRNAs) [1]. The ncRNAs are categorized into short ncRNAs (< 200 nt) and longer ncRNAs (> 200 nt) [2]. Short ncRNAs include microRNAs (miRNAs), transfer RNAs (tRNAs), piwi-interacting RNAs, small nucleolar RNAs, and small interfering RNAs; whereas longer ncRNAs contain ribosome RNAs and long ncRNAs (lncRNAs) [2].
Biogenesis and functions of miRNAs and lncRNAs
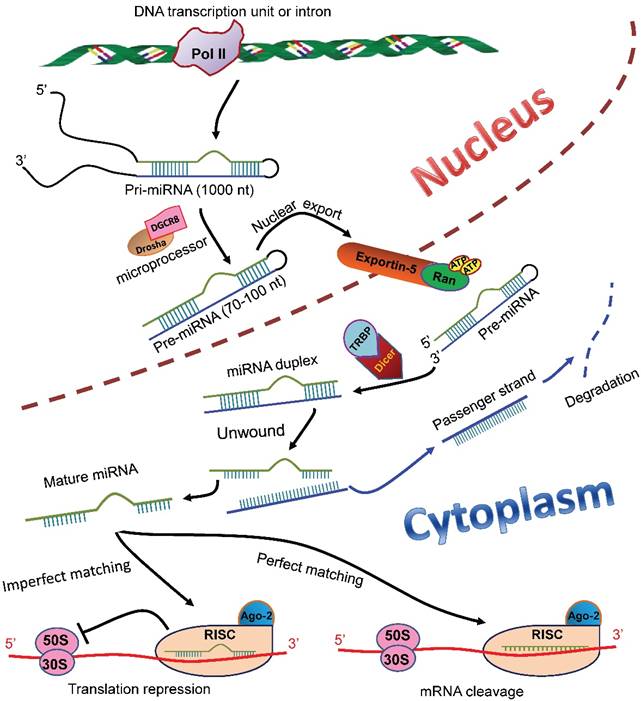
MiRNAs are endogenous ncRNAs that regulate gene expression at post-transcriptional level. In the mammalian cell nucleus, miRNAs are originated from primary miRNAs (pri-miRNAs), the longer transcripts that are transcribed from independent transcription units or introns by RNA polymerase II (Pol II) [3-5]. The pri-miRNAs are then converted into short stem-loop structures (70-100 nt) termed precursor miRNAs (pre-miRNAs) by a microprocessor complex comprising Drosha and DiGeorge syndrome chromosomal region 8 (DGCR8) (Figure 1) [6-10]. A nuclear transport receptor exports the pre-miRNAs to the cytoplasm [11], where an endonuclease Dicer cleaves them and liberates a miRNA-miRNA duplex (Figure 1) [12-14]. The passenger strand of the duplex is degraded; whereas the remaining one becomes a mature miRNA and subsequently is loaded into an RNA-induced silencing complex (RISC) [15-17]. The mature miRNA with the RISC, in most cases, binds to the 3'-untranslated region (3'-UTR) of the target mRNAs to elicit translation inhibition and/or degradation of the target transcripts (Figure 1) [18]. Research has shown that miRNAs play critical roles in a myriad of biological processes, including proliferation [19], differentiation [20, 21], and apoptosis [22] of cells, immune response [23], and angiogenesis [24].
MiRNA biosynthesis and functions in mammalian cells. In the nucleus of a mammalian cell, pri-miRNAs are transcribed from either independent transcription units or introns in the genomic DNA. The pri-miRNAs are then processed by a microprocessor composed of Drosha and DGCR8 to generate pre-miRNAs (70-100 nt), which subsequently are transported to the cytoplasm by a nuclear transporter exportin-5-Ran-GTP. In the cytoplasm, the pre-miRNAs are recognized and bound by TRBP and cleaved into a miRNA duplex by an endonuclease Dicer. The duplex is unwound, and the passenger strand (blue) is degraded. The remaining strand becomes a mature miRNA (green), which is then loaded into the RISC with Ago-2 protein. The miRNA, in most cases, binds to the 3'-UTR of the target mRNAs. If the miRNA completely matches the target sequence, the binding induces degradation of the target mRNA; whereas if the miRNA matches the target imperfectly, the binding induces translational repression of the target mRNA. pri-miRNAs: primary miRNAs; DGCR8: DiGeorge syndrome chromosomal region 8; pre-miRNAs: precursor miRNAs; TRBP: transactivation response RNA-binding protein; RISC: RNA-induced silencing complex; Ago-2: Agronaut-2; 3'-UTR: 3'-untranslated region.
Unlike the miRNAs, lncRNAs are longer ncRNAs located in the nucleus and cytoplasm. The lncRNAs are expressed in a tissue-, developmental stage- and disease state-specific manner, and their biosynthesis involves diverse programs [25]. Most lncRNAs are transcribed by RNA Pol II from genomic loci as the miRNAs [26], nonetheless, they may be 5ʹ‑capped, spliced, and 3'-polyadenylated in unconventional ways. For example, the nascent metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) and nuclear enriched abundant transcript 1 lncRNA transcripts are cleaved by RNase P, simultaneously generating the 3ʹ ends of the mature lncRNAs and the 5' ends of tRNA-like small RNAs [27-29]; some lncRNAs are transcribed from the intronic regions of human genome, processed at the consensus sequences close to 5' splice site and branchpoint, and persist as circular intronic RNAs in the nucleus [30]; In addition, small nucleolar lncRNAs are flanked by small nucleolar RNAs at both 3' and 5' ends, consequently resist degradation and accumulate in the nucleus [31]; and the lncRNAs hosting miRNAs are cleaved to terminate Pol II-mediated transcription by the microprocessor containing Drosha and DGCR8 [32]. Just as their diverse biosynthesis, the lncRNAs regulate gene expression via distinctive mechanisms. For instance, lncRNAs epigenetically repress gene expression by recruiting histone-modifying enzymes to specific domains of a chromosome [33, 34]; alternatively, they may be transcribed from an enhancer of a protein-coding gene by a shared promoter, the resulting lncRNAs may then activate transcription of the protein-coding gene by either recruiting protein partners [35], or directly altering the DNA or RNA target structures. Collectively, lncRNAs can function as signals, decoys, guides, and scaffolds to interact with nucleic acid or protein partners [36, 37], thereby regulating gene expression in a variety of biological processes, including, but not limited to, protein synthesis [38], genetic imprinting [39], cell cycle control [40], alternative splicing [41, 42], and chromatin structure remodeling [43, 44].
Angiogenesis and neovascularization
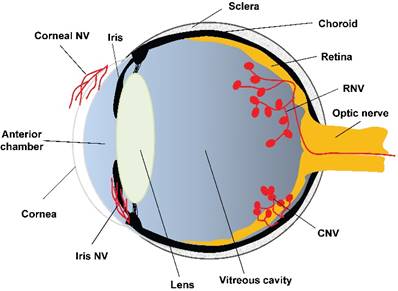
Angiogenesis is a physiological outgrowth of new blood vessels from the pre-existing ones. It involves proliferation, sprouting, migration, and lumen formation of vascular endothelial cells, followed by vascular network remodeling [45]. Under pathological conditions, such as wound healing [46], transplantation [47], and ischemia-induced tissue repair [48, 49], excessive and dysregulated angiogenesis results in neovascularization. According to the literature, neovascularization is associated with more than 30 systemic diseases, such as cancer, rheumatoid arthritis, atherosclerosis, diabetes, and morbid obesity [50]. Whereas ocular diseases, including corneal infection or injury, retinopathy of prematurity (ROP), proliferative diabetic retinopathy (PDR), and wet age-related macular degeneration (wAMD), lead to corneal neovascularization, retinal neovascularization (RNV), and choroidal neovascularization (CNV), respectively (Figure 2). The RNV and CNV manifest as vascular leakage and tissue edema, overexpression of pro-inflammatory factors, and neovessel formation in the retina, retinal pigment epithelium (RPE), and choroid membrane [51, 52].
The anatomical basis of ocular neovascularization. A schematic sagittal section of eye ball illustrates the ocular anatomical structures from anterior to posterior, including cornea, anterior chamber, iris, lens, vitreous cavity, retina, optic nerve, choroid, and sclera. A variety of pathogenic factors can induce neovascularization on cornea (corneal NV), retina (RNV), and choroidal membrane (CNV). In addition, hypoxia, ischemia, or glaucoma may cause neovascularization on iris (iris NV), which is yet beyond the scope of this review. NV: neovascularization; RNV: retinal neovascularization; CNV: choroidal neovascularization.
Pro-angiogenic factors and neovascularization
Vascular endothelial growth factor (VEGF) is a potent endothelial cell-specific mitogen. It has been reported that low expression levels of VEGF result in poor survival rate of mouse embryos and vascular abnormality in adults; conversely, high expression levels of VEGF under hypoxic conditions promote angiogenesis [53-56]. In the eyes, VEGF is closely associated with normal vessel development and disease progression [57]. Peripheral avascular area in retina expresses more VEGF to promote vascular development; whereas abnormally high levels of VEGF under pathological conditions, such as ROP, PDR, and retinal central vein occlusion, cause neovascularization [58]. Therefore, anti-VEGF therapeutic modalities have been used to treat ocular neovascularization and achieved certain efficacy. However, a considerable number of patients do not respond to the anti-VEGFs; moreover, local and systemic adverse events, including subretinal fibrosis, endophthalmitis, intraocular hemorrhage, hypertension, and stroke, may occur following the long-term administration of the anti-VEGF drugs [59, 60]. These phenomena raise concerns about clinical applications of the anti-VEGF drugs, and also prompt the search for other factors besides VEGF, just to name a few, erythropoietin [61, 62], basic fibroblast growth factor [63], platelet-derived growth factor (PDGF) [64], connective tissue growth factor [65], pigment epithelium-derived factor [66], and insulin-like growth factor [67], that may regulate ocular neovascularization by coordinating with VEGF or each other [68]. If this holds true, the anti-neovascular therapy in the eye should target multiple molecules along pro-angiogenic pathways. During the past few years, research has shown that both miRNAs and lncRNAs can bind multiple target genes encoding pro- or anti-angiogenic factors and regulate their expression under pathological conditions, indicating that the miRNAs and lncRNAs may be good molecular targets for intervention of neovascular eye diseases [69-72].
Regulation of miRNAs and lncRNAs in neovascular eye diseases
Indeed, a few studies have been carried out in the last 3 years to measure the circulating or ocular levels of miRNAs in the patients with neovascular eye diseases, such as wAMD and PDR. An Italian group identified that miR-9, miR-23a, miR-27a, miR-34a, miR-126, and miR-146a were upregulated, whereas miR-155 was downregulated in serum of neovascular AMD patients [73]. MiR-146 has also been shown to be upregulated in the aqueous humor of wAMD patients by a Canadian group [74]. Moreover, Cowan and coworkers [75] used a human retinal endothelial culture system and showed that miR-146 was significantly upregulated by activation of NF-κB, a key regulator of inflammatory response; whereas miR-146 targets caspase-recruitment domain-containing protein 10 (CARD10), an essential scaffold/adaptor protein of NF-κB activation pathway. This suggests a negative feedback regulation between miR-146 and NF-κB pathway. Since the inflammatory response is concomitant with the neovascularization in wAMD, the miR-146 upregulation in wAMD patients may implicate a potential of this miRNA in curbing neovascularization by anti-inflammation.
In addition, a Chinese group reported that the expression levels of miR-27a-3p, miR-29b-3p, and miR-195-5p were increased in the serum of wAMD patients, as compared to the healthy controls [76]. By contrast, a German group reported that the levels of miR-301-3p, miR-361-5p, and miR-424-5p were significantly lower in the patients with late-stage neovascular AMD than those in the dry AMD patients and the controls [77].
On the other hand, the levels of miR-126 was significantly decreased in the serum of the patients with PDR, as compared to the control subjects [78], suggesting an association of miR-126 with vascular complications of diabetes, particularly with PDR. In another study, the levels of miR-21, miR-181c, and miR-1179 have been shown to be significantly higher in the serum of the PDR patients than those in the nonproliferative diabetic retinopathy (NPDR) patients and the controls [79].
Therefore, the changes in the circulating and ocular levels of miRNAs in the patients with neovascular eye diseases suggest the clinical relevance of these ncRNAs to the ocular neovascularization.
MiRNAs and neovascularization
MiRNAs and corneal neovascularization
Corneal neovascularization is characterized by excessive blood vessels growing onto the cornea [80]. It is mainly caused by infections with pathogens, such as viruses, bacteria, and fungi, or by physical and chemical injury, or by aberrant expression of endogenous regulators. This ocular surface neovascular disease can result in visual impairments or even blindness. Several lines of research have indicated pathogenic roles of miRNAs in this disease (Table 1).
The effects of miRNAs on ocular neovascularization
| NV/Angiogenesis | ncRNAs | Effects on NV | Molecular targets | References |
|---|---|---|---|---|
| Corneal NV | miR-132 | Promote | Ras-GAP | 81, 82 |
| miR-155 | Promote | SHIP-1, IFN-γRα | 84 | |
| miR-206 | Promote | Cx43 | 85 | |
| miR-21 | Promote | Sprouty2/4 | 86 | |
| miR-184 | Repress | FZD-4, FOG2, PDGF-β, PPAP2B | 87, 88, 89 | |
| miR-204 | Repress | Ang-1 | 88, 90 | |
| CNV | miR-146 | Repress | CARD10 | 73-75 |
| miR-24 | Repress | RCK, PAK4, LIMK2 | 92 | |
| miR-31 | Repress | PDGF-β, HIF-1α | 93 | |
| miR-150 | Repress | CXCR4, DLL4, FZD-4 | 93, 94 | |
| miR-126 | Repress | Spred-1, VEGF, αB-Crystallin | 95 | |
| miR-342-5p | Repress | Endoglin | 96 | |
| miR-29 | Repress | MMP-2 | 97 | |
| miR-23 | Promote | Sprouty2, Sema6A, and Sema6D | 101 | |
| miR-27 | Promote | Sprouty2, Sema6A, and Sema6D | 101 | |
| miR-155 | Promote | SHIP-1 | 102 | |
| miR-132 | Promote | Ras-GAP | 103 | |
| Retinal developmental angiogenesis | miR-126 | Required | Spred-1 | 95 |
| miR-155 | Required | CCN-1 | 105 | |
| miR-132 | Required | Ras-GAP | 82 | |
| miR-342-5p | Repress | Endoglin | 96 | |
| RNV | miR-329 | Repress | CD-146 | 109 |
| miR-31 | Repress | PDGF-β, HIF-1α | 93 | |
| miR-150 | Repress | CXCR4, DLL4, FZD-4 | 93, 94 | |
| miR-184 | Repress | FZD-4 | 93 | |
| miR-200b | Repress | ZEB2, VEGFA | 111 | |
| miR-155 | Promote | SHIP-1 | 102, 105 | |
| miR-132 | Promote | Ras-GAP | 103 | |
| miR-21 | Controversial | PTEN, RhoB | 115, 116 | |
| miR-221/222 | Controversial | c-KIT, CDKN1b, PIK3R1 | 117-121 |
Note: NV stands for neovascularization; CNV choroidal neovascularization; RNV retinal neovascularization.
Infection-induced corneal neovascularization
Herpes simplex virus (HSV) infection on mouse ocular surface elicits corneal neovascularization with a dramatic upregulation of miR-132 expression (10 ~ 20 fold). In addition, the upregulation of miR-132 is preceded by and accompanied with elevated levels of VEGF-A on the cornea; the upregulated expression of miR-132 also requires the functional interaction between IL-17 and its receptor (IL-17R). These results suggest that VEGF-A and IL-17 / IL-17R system may be the up-stream regulators of miR-132 during the HSV-induced corneal infection. Indeed, either blocking VEGF-A with VEGF-trap or depleting VEGF-A-secreting neutrophils significantly reduced miR-132 expression following HSV infection; whereas IL-17R knockout prevented corneal miR-132 upregulation induced by the virus infection [81]. Furthermore, nanoparticle-facilitated delivery of anti-miR-132 ameliorated the HSV-induced corneal neovascularization and stromal keratitis (SK). As for the mechanism, the authors propose that HSV infection in the corneal epithelial cells may induce production of pro-inflammatory cytokines, such as IL-17, which could promote VEGF-A production from the inflammatory cells or vascular endothelial cells. The VEGF-A then acts on the endothelial cells to upregulate miR-132 expression. Whereas miR-132 has been reported to target Ras-GAP [82], which inhibits Ras-GTP through enhancing Ras GTPase activity [83]. Consistent with the report, the authors showed that anti-miR-132 restored the expression levels of Ras-GAP and subdued the angiogenic Ras activity in corneal CD31-enriched cells (possibly vascular endothelial cells). Therefore, the anti-neovascular effects of ani-miR-132 may be ascribed to the reduced Ras activity in corneal vascular endothelial cells during the HSV-induced corneal infection [81]. Moreover, the same group later found that miR-155, another cornea-enriched miRNA that targets SHIP-1 and IFN-γRα, was upregulated to the comparable extent to that of miR-132 in the HSV-infected corneas. The miR-155 upregulation promoted the proliferation of cornea-infiltrating Th1 and Th17 cells as well as their production of IFN-γ and IL17, respectively. This, in turn, led to corneal inflammation after the virus infection. Similarly, the delivery of anti-miR-155 significantly attenuated the HSV-induced SK and corneal neovascularization [84]. Therefore, these studies suggest that the aberrant expression of miRNAs contributes to the pathology following HSV infection, and the miRNAs may be good interventional targets to corneal inflammation and neovascularization induced by HSV infection.
Chemical or physical damage-induced corneal neovascularization
Chemical burn on cornea may cause permanent visual dysfunctions or complete blindness by inducing corneal neovascularization and inflammatory responses. Research has shown that miR-206 and miR-21 were dramatically upregulated in the alkali-burnt mouse corneas as compared to the normal controls; whereas downregulating either miR-206 or miR-21 expression subdued the alkali burn-induced corneal inflammation and neovascularization [85, 86]. Mechanistically, luciferase reporter assay and western blot suggested connexin43 (Cx43) as a direct target of miR-206, and silencing of Cx43 by its siRNA reversed the protective effects of downregulating miR-206 on the alkali-burnt murine corneas [85]. Whereas miR-21 upregulation was positively correlated to VEGF and hypoxia inducible factor-1α (HIF-1α) expression at both transcript and protein levels. Conversely, knocking-down miR-21 expression by its antagomir suppressed the expression of VEGF and HIF-1α. Further analyses indicate an association of the anti-neovascular effects of anti-miR-21 with the Sprouty2/4-mediated reduction in Erk activity [86].
On the other hand, physical stimulus, such as suture, was also used to induce corneal neovascularization. In a suture-induced rat model of corneal neovascularization, corneal expression of miR-184 was significantly downregulated, and that of Frizzled-4 (FZD-4) upregulated, indicating an increased activity of Wnt signaling under such a pathological condition. Moreover, topical applications of miR-184 diminished the area of corneal neovascularization [87]. In the cell culture models, miR-184 mimics inhibited the proliferation and migration of human umbilical vein endothelial cells (HUVECs), and suppressed the VEGF-induced tube formation in simian retinal microvessel endothelial cells; whereas miR-184 inhibitor exerted the opposite effects in these 2 types of cells. An association was suggested in this study between the miR-184's anti-angiogenic effects with the reduced abundance of VEGF and β-catenin, the downstream transcription factor of Wnt signaling pathway [87].
Endogenous regulators of corneal avascularity
MiR-184 and miR-204 are the 2 most abundant miRNAs expressed in corneal endothelia under normal condition. Whereas during the wound healing process in which angiogenesis is turned on to promote recovery, only miR-184 and miR-204, out of 600 miRNAs examined, are significantly downregulated [88]. Therefore, these 2 cornea-enriched miRNAs may serve as endogenous regulators for corneal avascularity. Indeed, a recent study has demonstrated that miR-184 targets the pro-angiogenic factors, including friend of Gata 2 (FOG2), platelet-derived growth factor-β (PDGF-β), and phosphatidic acid phosphatase 2b (PPAP2B) in corneal limbal epithelial keratinocytes. The FOG2 is an upstream regulator of VEGF, whereas PDGF-β and PPAP2B regulate Akt signaling [89]. Therefore, the constitutively high expression of miR-184 in corneal epithelial cells reduces the production of VEGF, PDGF-β, and Akt signaling-mediated metalloproteinases, thereby inhibiting proliferation and migration of the vascular endothelial cells and maintaining avascularity on the cornea [89].
Kather and colleagues [90] recently demonstrated that the miR-204 downregulation was correlated with the upregulation of a pro-angiogenic factor angiopoietin-1 (Ang-1) in the corneas of Kelch-like Ect2-interacting protein (KLEIP)-deficient mice, The KLEIP is essential for corneal structural integrity and transparency. The KLEIP-deficient mice spontaneously develop corneal dystrophy and neovascularization. In addition, bioinformatics and gene expression analyses suggested Ang-1 as a direct target of miR-204 in the HUVECs [90]. These findings indicate a novel, VEGF-independent miR-204/Ang-1 pathway that may lead to corneal dystrophy and neovascularization in KLEIP-/- mouse corneas [90].
miRNA and CNV
CNV refers to the growth of new blood vessels from choroid to RPE and / or subretinal space [91]. The ocular fundus diseases, such as wAMD and pathologic myopia, are the primary causes of CNV and often lead to vision loss. MiRNAs have been reported to function as important regulators of CNV (Table 1).
The miRNAs that inhibit CNV
Multiple miRNAs, including miR-24 [92], miR-31 [93], miR-150 [93, 94], miR-126-3p [95], and miR-342 [96], have demonstrated the ability to inhibit CNV (Table 1). Transfecting most of these miRNA mimics into microvessel endothelial cells suppresses the proliferative, migratory, and tubulogenic functions. Intravitreal or subretinal injections of the miRNAs' mimics or pre-miRNAs significantly reduce the injury area and/or alleviate the vascular leakage in the rodent models of laser-induced CNV.
However, the molecular mechanisms underlying the protective effects vary among individual miRNAs. For instance, miR-24 targets Rho-associated kinase (Rock), P21-activated kinase (Pak), and LIM domain-containing kinase (Limk) in Rho signaling pathway, the pathway crucial for angiogenesis-associated cell division and motility [92]. MiR-31 targets PDGF-β and HIF-1α, which function as pro-angiogenic factors in vascular pericytes and endothelial cells, respectively [93]. Whereas microvessel endothelium-enriched miR-150 is able to target C-X-C chemokine receptor type 4 (CXCR4), Delta like ligand 4 (DLL4), and FZD-4 and downregulate the expression of these genes, suggesting the interference of this miRNA with 3 angiogenic signaling pathways, i.e. SDF-1, Notch, and Wnt pathways. These results also implicate miR-150 as an endothelial cell-intrinsic suppressor of angiogenesis and neovascularization [94]. The cell type-specific function of miRNA is also demonstrated by the action of miR-126-3p. In the HUVECs, where endogenous miR-126-3p is highly expressed and the binding sites on its anti-angiogenic target Sprouty-related EVH1 domain-containing protein 1 (Spred-1) gene are saturated, the cells can only respond to miR-126-3p downregulation, and then the angiogenesis is repressed. By contrast, in the RPE cells, where miR-126-3p is expressed at low level and the binding sites on its targets in this type of cells, the 3'-UTR of VEGF gene and the promoter region of αB-Crystallin gene, are not saturated, the cells respond well to miR-126-3p overexpression, and the production and stability of VEGF protein are reduced, thereby resulting in the profoundly decreased VEGF secretion from the RPE cells and the repressed angiogenesis of the adjacent endothelial cells [95]. Thus the cell type-specific action of miRNA should be cautiously considered before its in vivo application can be performed. The action of miR-342-5p is even more paradoxical. MiR-342-5p downregulates VEGFR2 and VEGFR3 expression, and then represses VEGF-induced Akt signaling in the endothelial cells. Moreover, bioinformatics and gene expression analyses suggest that miR-342-5p targets endoglin, a co-receptor of TGF-β receptor [96]. Therefore, overexpression of miR-342-5p in the HUVECs inhibits TGF-β signaling pathway by decreasing SMAD1/5 phosphorylation [96]. Simultaneously, miR-342-5p promotes endothelial-mesenchymal transition (EMT) by targeting endoglin and interfering with TGF-β signaling [96]. Although EMT may stabilize the vascular structure after developmental sprouting and curb pathological neovascularization, it also elicits fibrosis that may deteriorate photoreceptor and RPE functions under the condition of CNV [91]. Therefore, the pleiotropic effects of miR-342-5p in the context of CNV require further investigations.
The action of miR-29 is different than the above-mentioned CNV-inhibiting miRNAs. The NFκB activation following laser-induced CNV downregulates the expression of miR-29 clusters, including miR-29a, b, and c. The miR-29 cluster target matrix metalloproteinase-2 (MMP-2) [97]. The MMP-2 has been shown to contribute to CNV [98-100], partly due to its ability to degrade basement membranes and extracellular matrix proteins and to promote migration of neovascular endothelial cells. Therefore, despite lacking direct evidence, these results indicate an inhibitory role of miR-29 cluster in CNV through targeting MMP-2.
The miRNAs that promote CNV
It has been reported that a few miRNAs, such as miR-23 [101], miR-27 [101], miR-155 [102], and miR-132 [103], can promote CNV (Table 1). These miRNAs are significantly upregulated in the choroidal tissue of the rodent models following laser injury [101, 102]. In the cell culture models, knocking down miR-23 and 27 with locked nucleic acid-modified anti-miRNAs inhibits tubulogenesis of the HUVECs and cell outgrowth in an ex vivo aortic ring assay [101]; whereas downregulation of miR-155 expression in human retinal microvessel endothelial cells by lentivirus suppresses the VEGF-induced proliferation, migration, and tube formation [102]. In the animal models, more than 90% reduction in miR-23 and 27 abundance results in greater than 50% shrinkage in the volume of the laser-induced CNV [101]; anti-miR-155 and anti-miR-132 also cause comparable reduction in the CNV lesion volume [102, 103]. At the molecular level, miR-23 and 27 target the negative regulators of MAPK- and VEGFR2-mediated pro-angiogenic pathways, including Sprouty2, Sema6A, and Sema6D [101]; miR-155 targets an SH2 domain-containing inositol 5'-phosphatase (SHIP-1), a negative regulator of PI3K/Akt pathway [102]; whereas miR-132, as mentioned above, targets Ras-GAP, an inhibitor of Ras signaling activity [82, 103]. So far, the mechanisms of action of the CNV-promoting miRNAs have been straightforward. They are upregulated at the site of CNV, and target the negative arms of pro-angiogenic pathways.
miRNA and retinal angiogenesis
The miRNAs essential for normal developmental angiogenesis
Normal developmental angiogenesis in retina is a physiological process that has been extensively studied due to easy access of the vasculature, definitive readout of the assays, and orchestrated regulatory programs during the development [60, 104].
One approach to identify the roles of miRNA in developmental angiogenesis is the loss of function study. For example, in the miR-126 deficient mice, the superficial vascular plexus in retina covers 30% less area as compared to the wild type (WT) counterpart from postnatal 6 (P6) to P8. Moreover, very few retinal vessels in miR-126-/- mice are found in the intermediate and deep plexus at P10 when the normal vessels sprout into the deeper plexus [95]. Similarly, knocking out miR-155 leads to mild, but quantifiably significant defects during retinal vessel development, such as decreased junctional density and increased vascular length, compared to the normal retinal vessels [105]. In addition, the defects in the miR-126 or miR-155-deficient mice were rescued or even reversed by knocking out their respective target genes, Spred-1 and CCN family member 1 (CCN-1) [95, 105], indicating that these miRNAs modulate retinal vascular development through regulating their target expression. If knockout animals are not available, then knocking down the expression of the miRNA of interest is an alternative approach to achieve the loss of function. It has been reported that an intraocular injection of anti-miR-132 leads to 50% reduction in the deeper retinal plexus covering area, without affecting the coverage of superficial plexus and perivascular cells [82].
The other approach to reveal the miRNAs' role in developmental retinal angiogenesis is the gain of function study. An intravitreal injection of miR-155 mimics to WT pups generates the retinal vascular phenotype opposite to that in miR-155-/- mice, such as delayed migration of superficial plexus along retina radius, hyperproliferative endothelial cells, and the increased anastomosis at the expanding vascular front [105]. By contrast, lentivirus-mediated intravitreal delivery of miR-342-5p agomir to WT pups results in diminished vascularized area and less proliferative endothelial cells in developing retinas [96]. Therefore, these studies suggest a crucial role of miRNA in developmental retinal angiogenesis.
The miRNAs that regulate pathological neovascularization
RNV is a severe and blinding pathology incurred by common but intractable retinal diseases, such as ROP, PDR, and central retinal vein occlusion [106]. The animal model mainly used in the RNV research is a mouse model of oxygen-induced retinopathy (OIR). In the OIR model, P7 pups are exposed to 75% oxygen for 5 days, and then raised in normal air for another 5 days. When exposed to hyperoxia, the developing vasculature halts extension, regresses, and forms a large avascular area in the central retina, and this is the vaso-obliteration phase of the OIR model; whereas returning of the pups to normal air subjects the retinal vasculature to a relative hypoxia, upregulates pro-angiogenic factor expression, and generates neovascular tufts at the junction between vascularized and avascularized regions, and this is the neovascularization phase of the OIR model [94, 107, 108]. The pathology of RNV in the OIR model is similar to that in the ROP and PDR patients. In the past 5 years, a large number of literatures have emerged demonstrating associations of miRNAs with pathological RNV. However, the roles of individual miRNAs in the RNV remain case-dependent, and even controversial. Therefore, the miRNAs are categorized into 3 groups based on their reported effects on the RNV, i.e. the miRNAs inhibiting RNV, the miRNAs promoting RNV, and those with controversial effects (Table 1).
The miRNAs that inhibit RNV
In the OIR model, miR-329, miR-31, miR-150, and miR-184 have been shown to exert the anti-neovascular effects (Table 1).
MiR-329 targets CD146, an endothelial marker functioning as a co-receptor for VEGFR-2 to promote neovascularization. In the OIR retinas, CD146 was significantly upregulated, while miR-329 downregulated during the neovascularization phase. An intravitreal injection of miR-329 into the OIR pups suppressed the CD-146 overexpression and reduced the RNV by 40%, although it did not ameliorate the vaso-obliteration in the OIR retinas [109]. Mechanistic study suggests that miR-329 may hamper the endothelial response to VEGF-induced SRC kinase/p38 MAPK/NF-κB activation via negatively regulating the CD146 expression [109].
A microarray analysis in an earlier study revealed the significant downregulation of miR-31, -150, and -184 in the OIR mouse retinas, as compared to the normal controls. An intravitreal injection of pre-miR-31, -150, or -184 significantly reduced the ischemia-induced RNV [93]. In a more recent study, miR-150 was identified as an endothelial cell-intrinsic suppressor of pathological neovascularization, and exhibited inhibitory effects on both CNV and RNV [94]. Additionally, as discussed above, another recent study found that miR-184 was able to antagonize the suture-induced corneal neovascularization [87]. These studies suggest that the RNV-inhibiting miRNAs are usually downregulated in the retinas with neovascularization, and the intravitreal injection of miRNA mimics or overexpression of pre-miRNAs is a potential therapeutic strategy to the RNV.
On the other hand, an Australian group generated a murine model with selective disruption of Müller cells by tamoxifen-induced Müller cell-specific expression of a Diphtheria toxin gene. In this model, the disruption of Müller cell leads to blood-retina barrier breakdown, deep retinal neovascularization, and photoreceptor degeneration [110]. More interestingly, a miRNA array identified 9 differentially expressed miRNAs in the Müller cell-deficient retinas. Among these 9 miRNAs, miR-200b was significantly downregulated; the expression of its potential targets, zinc finger E-box binding homeobox 2 (ZEB2) and VEGFA genes, was significantly upregulated. Furthermore, intravitreal injections of miR-220b mimics attenuated the vascular leakage, whereas injections of miR-200b inhibitor exacerbated the leakage in the retinas with the mild vasculopathy [111]. However, neither the mimics nor the inhibitor of miR-200b had significant effects on the retinas with severe vasculopathy, which might be related to the dosage applied to the retinal vasculopathy at this stage.
The miRNAs that promote RNV
The miR-155 and miR-132 have been shown to promote RNV in the OIR model (Table 1). The miR-155 was significantly upregulated in the OIR retinas, in comparison to the normal counterparts. Either downregulating miR-155 expression by anti-miR-155 [102] or knocking out miR-155 [105] resulted in the reduced avascular areas and neovascular tufts in the OIR retinas.
On the other hand, the pathological endothelium-enriched miR-132 appears to be a ubiquitous promoter for neovessel formation, including the HSV-induced corneal neovascularization [81], the developmental retinal angiogenesis [82], and the laser-induced CNV [103], as discussed in the previous sections. Moreover, an intravitreal delivery of anti-miR-132 not only prevented the formation of neovascular tufts in the OIR retinas, but also reduced the number of angiomas in the retinas of Vldlr knockout mice [103]. The Vldlr-/- mouse serves as a model for idiopathic juxtafoveal macular telangiectasia and retinal angiomatous proliferation, a rare retinal disease characterized by intraretinal and subretinal neovascularization [112]. Further, the anti-miR-132 caused the simultaneous downregulation of multiple pro-angiogenic and pro-inflammatory factors, while the conventional suppression of VEGF signaling pathway using anti-VEGFs may result in a compensatory upregulation of the pro-angiogenic and pro-inflammatory factors [103], which may contribute to the subsequent reduced efficacy of the anti-VEGF agents. Therefore, anti-miR-132 could be one of the most promising therapeutic modalities for neovascular eye diseases.
The miRNAs with controversial effects on RNV
MiR-21 is one of the important miRNAs associated with tumor growth, angiogenesis, and metastasis. Overexpression of miR-21 in prostate cancer cells induces tumor angiogenesis through targeting phosphatase and tensin homolog deleted on chromosome 10 (PTEN), activating AKT and ERK1/2 pathways, and upregulating HIF-1α and VEGF expression [113, 114]. In the eyes, miR-21 is the most highly expressed miRNA in the primary bovine retinal microvascular endothelial cells, and inhibition of miR-21 significantly reduced proliferation, migration, and tube formation of these cells [115]. These two lines of research indicate miR-21 as a pro-angiogenic molecule. However, the results of another study demonstrated that overexpression of miR-21 in HUVECs significantly reduced cell proliferation, migration, and tube formation; whereas inhibition of endogenous miR-21 expression leads to the opposite effects. Further mechanistic investigation indicates that miR-21 may target RhoB to interfere with actin filament-mediated stress fiber formation, thereby antagonizing angiogenesis via inhibiting migration and tubulogenesis of endothelial cells [116]. The discrepancies of these results could be due to the distinct cell culture and assay systems employed in the studies, but also indicate a species-specific effect of this miRNA.
Moreover, miR-221 and miR-222 belong to the same family and their genes are located in close proximity on Xp11.3 chromosome. They inhibit endothelial cell angiogenesis by targeting stem cell factor receptor c-kit and indirectly regulating the expression of endothelial nitric oxide synthase [117, 118]. However, in vascular smooth muscle cells, miR-221/222 have pro-proliferative, pro-migratory, and anti-apoptotic effects. These results suggest that the biological functions of miR-221/222 in vascular walls are cell type-specific [119, 120]. In addition, the latest research finds that miR-221 is essential for angiogenesis in zebrafish, miR-221 knockdown phenocopied the defects accompanied by loss of Flt4 receptor on the tip cells at the vascular endothelial front [121]. Furthermore, miR-221 can promote tip cell proliferation and migration through repressing cyclin-dependent kinase inhibitor 1b (CDKN1b), and phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1) [121]. These results identify miR-221 as a regulator of tip cell migration and proliferation during angiogenesis. The opposite effects of miR-221/222 in different in vitro and in vivo models highlight the complicated regulatory mechanisms of this miRNA in angiogenesis, and warrant further study on the functions of miR-221/222 in retinal vessel development and RNV.
LncRNA and neovascularization
During the last 2 years, the function of lncRNAs in ocular neovascularization is becoming a hotspot in the field. Xu et al. [122] studied the expression profiling of lncRNAs in the OIR model by a microarray analysis. The results showed that the expression levels of 326 or 51 lncRNAs were significantly altered in the vaso-obliteration or neovascularization phase of this model, respectively [122]. Moreover, lncRNAs/mRNAs co-expression networks were constructed based on the Pearson correlation analysis. Gene ontology enrichment analysis of the mRNAs co-expressed with the lncRNAs indicated that the biological modules during the vaso-obliteration phase were correlated with chromosome organization, extracellular region, and guanylate cyclase activator activity; whereas the modules in the neovascularization phase correlated with cell proliferation, extracellular region, and guanylate cyclase regulator activity. KEGG pathway analysis implicated MAPK signaling pathway as the most significantly enriched pathway in both phases. Importantly, Vax2os1 and Vax2os2 were dynamically expressed in both phases of the OIR model, and their expression levels were significantly altered in aqueous humor of wAMD patients, indicating a potential role of the lncRNAs in the pathogenesis of RNV and CNV [122].
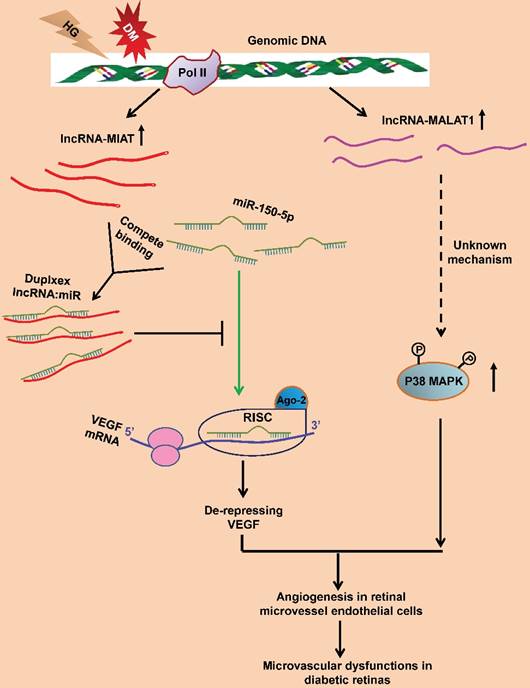
LncRNA-myocardial infarction-associated transcript (lncRNA-MIAT) is enriched in endothelial cells and regulates migration and sprouting of these cells [123]. Hyperglycemia upregulates lncRNA-MIAT expression in the endothelial cells and the retinas of diabetic animals. In addition, lncRNA-MIAT knockdown alleviates retinal pericyte loss, capillary degeneration, vascular leakage, and inflammation in diabetic rats; and significantly reduces proliferation, migration, and tubulogenesis of endothelial cells [124]. Further study identifies that lncRNA-MIAT functions as a competing endogenous RNA (ceRNA) to specifically bind and sequester miR-150-5p, thereby de-repressing the target of miR-150-5p, VEGF, in the retinal endothelial cells (Figure 2). Hence the ceRNA regulatory network, composed of lncRNA-MIAT, miR-150-5p, and VEGF, provides a novel insight into the pathogenic mechanism for ocular neovascularization [124].
LncRNA-MALAT1 is another endothelial cell-enriched lncRNA. Under the physiological condition, downregulating lncRNA-MALAT1 with either siRNA or locked nucleic acid GapmeRs switches the HUVECs from a proliferative state to a pro-migratory and pro-sprouting state [125]. Furthermore, knockout lncRNA-MALAT1 also results in similar phenotypes in the developing retinal vasculature, including delayed vessel extension, reduced density at the vascular front, and decreased proliferating cells [125]. Microarray and quantitative PCR analyses further suggest that the pro-migratory and anti-proliferative effects of silencing MALAT1 may be due to its downregulating the S-phase cell cycle cyclins and upregulating the cell cycle inhibitory genes [125]. These results implicate that during normal development, the high expression of lncRNA-MALAT1 in endothelial cells may help maintain these cells in a proliferative state.
On the other hand, lncRNA-MALAT1 dysregulation is also implicated in the pathogenesis of diabetes-related microvascular disorders. LncRNA-MALAT1 expression is upregulated in the retinas of Streptozotocin-induced diabetic rats and db/db mice. LncRNA-MALAT1 knockdown ameliorates the diabetes-induced microvascular dysfunctions, including pericyte loss, acellular capillary structure, vascular leakage, and neuroinflammation in retina [126]. Moreover, lncRNA-MALAT1 knockdown inhibits proliferation, migration, and tube formation of retinal microvessel endothelial cells. These protective effects were mediated by the crosstalk between lncRNA-MALAT1 and p38 MAPK signaling pathway [126] (Figure 3). The results indicate lncRNA-MALAT1 upregulation as a critical pathogenic factor for the diabetes-induced microvascular dysfunctions.
Pathogenic mechanisms of lncRNA-MIAT and lncRNA-MALAT1 in RNV. In the HG-treated retinal microvessel endothelial cells and in Type I and Type II DM animals, the expression of lncRNA-MIAT (red lines) and lncRNA-MALAT1 (purple lines) is upregulated. LncRNA-MIAT, with the increased amplitude, can competitively bind to miR-150-5p (green), resulting in an inhibition of miR-150-5p's loading into the RISC and binding to the target, VEGF mRNA (green arrow). The expression of VEGF is, therefore, de-repressed. The elevated levels of VEGF then lead to a pro-angiogenic response in retinal microvessel endothelial cells and microvascular dysfunctions in retinas of diabetic animals. On the other hand, the upregulated lncRNA-MALAT1 expression enhances p38 MAPK activity through an unknown mechanism (dashed arrow). The enhanced p38 MAPK activity causes angiogenesis in retinal endothelial cells and microvascular dysfunctions in diabetic retinas. RNV: retinal neovascularization; HG: high glucose; DM: diabetes mellitus.
Conclusion
In general, miRNAs can be classified into 3 categories based on their effects on ocular neovascularization. One is the miRNAs that promote neovascularization, for example, miR-206 and miR-21 boost corneal neovascularization, miR-23 and miR-27 enhance CNV, whereas miR-132 and miR-155 promote all types of neovascularization discussed; Another category is the neovascularization-repressing miRNAs, which include miR-204 and miR-184 that inhibit corneal neovascularization, miR-24, miR-126, miR-342-5p, and miR-29 that inhibit CNV, miR-329 and miR-200b that suppress RNV, as well as miR-31 and miR-150 that suppress both CNV and RNV. Finally, there are some miRNAs, such as miR-21 and miR-221/222, with controversial functions towards neovascularization (Table 1). On the other hand, a few lncRNAs, including lncRNA-MIAT and lncRNA-MALAT1, have been identified within the last 3 years as the new regulators of retinal angiogenesis or neovascularization (Figure 2), although the regulatory mechanisms underlying individual lncRNAs may vary and to a large extent, remain to be investigated.
Therefore, research results suggest that miRNAs or lncRNAs might be good molecular targets for intervention of neovascular eye diseases. Yet, one major problem is that each miRNA or lncRNA usually has several targets, and targeting a single miR or lncRNA could have non-intended effects. Further, each protein-coding mRNA might be a target for several miRs or lncRNAs, and targeting a single miR or lncRNA might not be effective enough to halt neovascularization. Notwithstanding, it would still be interesting to test the efficacy of combination of these miRNAs or lncRNAs in pre-clinical studies, facilitating translation of the research results on the ncRNAs into novel therapeutic modalities to ocular neovascularization.
Abbreviations
miRNAs: microRNAs; lncRNAs: long noncoding RNAs; ncRNAs: noncoding RNAs; tRNAs: transfer RNAs; pri-miRNAs: primary miRNAs; Pol II: RNA polymerase II; pre-miRNAs: precursor miRNAs; DGCR8: DiGeorge syndrome chromosomal region 8; RISC: RNA-induced silencing complex; 3'-UTR: 3'-untranslated region; MALAT1: metastasis-associated lung adenocarcinoma transcript 1; ROP: retinopathy of prematurity; PDR: proliferative diabetic retinopathy; wAMD: wet age-related macular degeneration; RNV: retinal neovascularization; CNV: choroidal neovascularization; RPE: retinal pigment epithelium; VEGF: vascular endothelial growth factor; PDGF: platelet-derived growth factor; CARD10: caspase-recruitment domain-containing protein 10; NPDR: nonproliferative diabetic retinopathy; HSV: herpes simplex virus; IL-17 receptor: IL-17R; SK: stromal keratitis; Cx43: connexin43; HIF-1α: hypoxia inducible factor-1α; FZD-4: Frizzled-4; HUVECs: human umbilical vein endothelial cells; FOG2: friend of Gata 2; PDGF-β: platelet-derived growth factor-β; PPAP2B: phosphatidic acid phosphatase 2b; Ang-1: angiopoietin-1; KLEIP: Kelch-like Ect2-interacting protein; Rock: Rho-associated kinase; Pak: P21-activated kinase; Limk: LIM domain-containing kinase; CXCR4: C-X-C chemokine receptor type 4; DLL4: Delta like ligand 4; Spred-1: Sprouty-related EVH1 domain-containing protein 1; EMT: endothelial-mesenchymal transition; MMP2: Matrix metalloproteinase 2; EMT: endothelial-mesenchymal transition; SHIP-1: SH2 domain-containing inositol 5'-phosphatase; WT: wild type; OIR: oxygen-induced retinopathy; ZEB2: zinc finger E-box binding homeobox 2; PTEN: phosphatase and tensin homolog deleted on chromosome 10; CDKN1b: cyclin-dependent kinase inhibitor 1b; PIK3R1: phosphoinositide-3-kinase regulatory subunit 1; TGF-β: transforming growth factor-β; SRC: sarcoma; NF-κB: nuclear factor-κB; RhoB: Ras homolog gene family member B; Erk: extracellular signal-regulated kinase; MAPK: mitogen-activated protein kinase; KEGG: Kyoto Encyclopedia of Genes and Genomes; lncRNA-MIAT: lncRNA-myocardial infarction-associated transcript; ceRNA: competing endogenous RNA.
Acknowledgements
This research work was supported by the grants from the 45th Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry to Y.Z., the key projects of Tianjin Municipal Science and Technology Commission (17JCZDJC35600 to Y.Z., 14JCZDJC36200 to C.Q.), and the National Natural Science Foundation of China (81460072) and CAS "Light of West China" Program to Y.C.
Author contributions
Y.Z. conceived, extensively revised, formatted, and submitted the manuscript. S.C. and Y.J. wrote the first draft of the manuscript. C.Q., J.S., H.Z., and F.W. searched and archived the literature, and discussed on contents of the manuscript. Y.C. and X.L. organized and supervised the composition, and critically reviewed the manuscript prior to submissions.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Human Genome Sequencing Consortium I. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931-45
2. Michalik KM, You X, Manavski Y. et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114:1389-97
3. Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA. 2004;10:1957-66
4. Lee Y, Kim M, Han J. et al. MicroRNA genes are transcribed by RNA polymerase II. EMBO J. 2004;23:4051-60
5. Ozsolak F, Poling LL, Wang Z. et al. Chromatin structure analyses identify miRNA promoters. Genes Dev. 2008;22:3172-83
6. Denli AM, Tops BB, Plasterk RH. et al. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231-5
7. Gregory RI, Yan KP, Amuthan G. et al. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235-40
8. Han J, Lee Y, Yeom KH. et al. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016-27
9. Han J, Lee Y, Yeom KH. et al. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell. 2006;125:887-901
10. Lee Y, Ahn C, Han J. et al. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415-9
11. Yi R, Qin Y, Macara IG. et al. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003;17:3011-6
12. Fukunaga R, Han BW, Hung JH. et al. Dicer partner proteins tune the length of mature miRNAs in flies and mammals. Cell. 2012;151:533-46
13. Jiang F, Ye X, Liu X. et al. Dicer-1 and R3D1-L catalyze microRNA maturation in Drosophila. Genes Dev. 2005;19:1674-9
14. Saito K, Ishizuka A, Siomi H. et al. Processing of pre-microRNAs by the Dicer-1-Loquacious complex in Drosophila cells. PLoS Biol. 2005;3:e235
15. Chendrimada TP, Gregory RI, Kumaraswamy E. et al. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740-4
16. Matranga C, Tomari Y, Shin C. et al. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell. 2005;123:607-20
17. Rand TA, Petersen S, Du F. et al. Argonaute2 cleaves the anti-guide strand of siRNA during RISC activation. Cell. 2005;123:621-9
18. Baek D, Villen J, Shin C. et al. The impact of microRNAs on protein output. Nature. 2008;455:64-71
19. Li C, Li X, Gao S. et al. microRNA-133a Inhibits Proliferation of Gastric Cancer Cells By Downregulating ERBB2 Expression. Oncol Res. 2017 doi: 10.3727/096504017X14847395834985
20. Malpeli G, Barbi S, Zupo S. et al. Identification of microRNAs implicated in the late differentiation stages of normal B cells suggests a central role for miRNA targets ZEB1 and TP53. Oncotarget. 2017;8:11809-26
21. Yue B, Wu J, Wang Y. et al. Expression Profiles Analysis and Functional Characterization of microRNA-660 in Skeletal Muscle Differentiation. J Cell Biochem. 2017 doi: 10.1002/jcb.25901
22. Takai T, Yoshikawa Y, Inamoto T. et al. A Novel Combination RNAi toward Warburg Effect by Replacement with miR-145 and Silencing of PTBP1 Induces Apoptotic Cell Death in Bladder Cancer Cells. Int J Mol Sci. 2017;18:E179
23. Zhi L, Yu Y, Li X. et al. Molecular Control of Innate Immune Response to Pseudomonas aeruginosa Infection by Intestinal let-7 in Caenorhabditis elegans. PLoS Pathog. 2017;13:e1006152
24. Chistiakov DA, Orekhov AN, Bobryshev YV. The role of miR-126 in embryonic angiogenesis, adult vascular homeostasis, and vascular repair and its alterations in atherosclerotic disease. J Mol Cell Cardiol. 2016;97:47-55
25. Thum T, Fiedler J. LINCing MALAT1 and angiogenesis. Circ Res. 2014;114:1366-8
26. Kornienko AE, Guenzl PM, Barlow DP. et al. Gene regulation by the act of long non-coding RNA transcription. BMC Biol. 2013;11:59
27. Hutchinson JN, Ensminger AW, Clemson CM. et al. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics. 2007;8:39
28. Ulitsky I, Shkumatava A, Jan CH. et al. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011;147:1537-50
29. Wilusz JE, Freier SM, Spector DL. 3' end processing of a long nuclear-retained noncoding RNA yields a tRNA-like cytoplasmic RNA. Cell. 2008;135:919-32
30. Zhang Y, Zhang XO, Chen T. et al. Circular intronic long noncoding RNAs. Mol Cell. 2013;51:792-806
31. Yin QF, Yang L, Zhang Y. et al. Long noncoding RNAs with snoRNA ends. Mol Cell. 2012;48:219-30
32. Dhir A, Dhir S, Proudfoot NJ. et al. Microprocessor mediates transcriptional termination of long noncoding RNA transcripts hosting microRNAs. Nat Struct Mol Biol. 2015;22:319-27
33. Nagano T, Mitchell JA, Sanz LA. et al. The Air noncoding RNA epigenetically silences transcription by targeting G9a to chromatin. Science. 2008;322:1717-20
34. Pandey RR, Mondal T, Mohammad F. et al. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell. 2008;32:232-46
35. Orom UA, Shiekhattar R. Noncoding RNAs and enhancers: complications of a long-distance relationship. Trends Genet. 2011;27:433-9
36. Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81:145-66
37. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011;43:904-14
38. Jang S, Shin H, Lee J. et al. Regulation of BC200 RNA-mediated translation inhibition by hnRNP E1 and E2. FEBS Lett. 2017;591:393-405
39. Liu H, Shang X, Zhu H. LncRNA/DNA binding analysis reveals losses and gains and lineage specificity of genomic imprinting in mammals. Bioinformatics. 2017;33:1431-6
40. Qiao Q, Li H. LncRNA FER1L4 suppresses cancer cell proliferation and cycle by regulating PTEN expression in endometrial carcinoma. Biochem Biophys Res Commun. 2016;478:507-12
41. El BG, Patel RS, Carter G. et al. MALAT1 in human adipose stem cells modulates survival and alternative splicing of PKCdeltaII in HT22 cells. Endocrinology. 2017;158:183-95
42. Tripathi V, Ellis JD, Shen Z. et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol Cell. 2010;39:925-38
43. Bohmdorfer G, Wierzbicki AT. Control of Chromatin Structure by Long Noncoding RNA. Trends Cell Biol. 2015;25:623-32
44. Han P, Chang CP. Long non-coding RNA and chromatin remodeling. RNA Biol. 2015;12:1094-8
45. Saint-Geniez M, D'Amore PA. Development and pathology of the hyaloid, choroidal and retinal vasculature. Int J Dev Biol. 2004;48:1045-58
46. Ridiandries A, Bursill C, Tan J. Broad-Spectrum Inhibition of the CC-Chemokine Class Improves Wound Healing and Wound Angiogenesis. Int J Mol Sci. 2017;18:E155
47. Inomata T, Mashaghi A, Di Zazzo A. et al. Kinetics of Angiogenic Responses in Corneal Transplantation. Cornea. 2017;36:491-6
48. Renault MA, Vandierdonck S, Chapouly C. et al. Gli3 regulation of myogenesis is necessary for ischemia-induced angiogenesis. Circ Res. 2013;113:1148-58
49. Wei Y, Gong J, Xu Z. et al. Nrf2 in ischemic neurons promotes retinal vascular regeneration through regulation of semaphorin 6A. Proc Natl Acad Sci U S A. 2015;112:E6927-36
50. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873-87
51. Campochiaro PA. Retinal and choroidal neovascularization. J Cell Physiol. 2000;184:301-10
52. Grossniklaus HE, Kang SJ, Berglin L. Animal models of choroidal and retinal neovascularization. Prog Retin Eye Res. 2010;29:500-19
53. Carmeliet P, Ferreira V, Breier G. et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435-9
54. Ferrara N, Carver-Moore K, Chen H. et al. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439-42
55. Lee S, Chen TT, Barber CL. et al. Autocrine VEGF signaling is required for vascular homeostasis. Cell. 2007;130:691-703
56. Uemura A, Kusuhara S, Katsuta H. et al. Angiogenesis in the mouse retina: a model system for experimental manipulation. Exp Cell Res. 2006;312:676-83
57. Hoeben A, Landuyt B, Highley MS. et al. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev. 2004;56:549-80
58. Yamakawa M, Liu LX, Date T. et al. Hypoxia-inducible factor-1 mediates activation of cultured vascular endothelial cells by inducing multiple angiogenic factors. Circ Res. 2003;93:664-73
59. Falavarjani KG, Nguyen QD. Adverse events and complications associated with intravitreal injection of anti-VEGF agents: a review of literature. Eye (Lond). 2013;27:787-94
60. Gariano RF, Gardner TW. Retinal angiogenesis in development and disease. Nature. 2005;438:960-6
61. Xu H, Zhang L, Gu L. et al. Subretinal delivery of AAV2-mediated human erythropoietin gene is protective and safe in experimental diabetic retinopathy. Invest Ophthalmol Vis Sci. 2014;55:1519-30
62. Zhang J, Wu Y, Jin Y. et al. Intravitreal injection of erythropoietin protects both retinal vascular and neuronal cells in early diabetes. Invest Ophthalmol Vis Sci. 2008;49:732-42
63. Li D, Xie K, Zhang L. et al. Dual blockade of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (FGF-2) exhibits potent anti-angiogenic effects. Cancer Lett. 2016;377:164-73
64. Jo N, Mailhos C, Ju M. et al. Inhibition of platelet-derived growth factor B signaling enhances the efficacy of anti-vascular endothelial growth factor therapy in multiple models of ocular neovascularization. Am J Pathol. 2006;168:2036-53
65. Hu B, Zhang Y, Zeng Q. et al. Intravitreal injection of ranibizumab and CTGF shRNA improves retinal gene expression and microvessel ultrastructure in a rodent model of diabetes. Int J Mol Sci. 2014;15:1606-24
66. Haurigot V, Villacampa P, Ribera A. et al. Long-term retinal PEDF overexpression prevents neovascularization in a murine adult model of retinopathy. PLoS One. 2012;7:e41511
67. Dong L, Nian H, Shao Y. et al. PTB-associated splicing factor inhibits IGF-1-induced VEGF upregulation in a mouse model of oxygen-induced retinopathy. Cell Tissue Res. 2015;360:233-43
68. Qazi Y, Maddula S, Ambati BK. Mediators of ocular angiogenesis. J Genet. 2009;88:495-515
69. Caporali A, Emanueli C. MicroRNA regulation in angiogenesis. Vascul Pharmacol. 2011;55:79-86
70. Hartmann D, Thum T. MicroRNAs and vascular (dys)function. Vascul Pharmacol. 2011;55:92-105
71. Landskroner-Eiger S, Moneke I, Sessa WC. miRNAs as modulators of angiogenesis. Cold Spring Harb Perspect Med. 2013;3:a6643
72. Wang S, Koster KM, He Y. et al. miRNAs as potential therapeutic targets for age-related macular degeneration. Future Med Chem. 2012;4:277-87
73. Romano GL, Platania C, Drago F. et al. Retinal and Circulating miRNAs in Age-Related Macular Degeneration: An In vivo Animal and Human Study. Front Pharmacol. 2017;8:168
74. Menard C, Rezende FA, Miloudi K. et al. MicroRNA signatures in vitreous humour and plasma of patients with exudative AMD. Oncotarget. 2016;7:19171-84
75. Cowan C, Muraleedharan CK, O'Donnell JR. et al. MicroRNA-146 inhibits thrombin-induced NF-kappaB activation and subsequent inflammatory responses in human retinal endothelial cells. Invest Ophthalmol Vis Sci. 2014;55:4944-51
76. Ren C, Liu Q, Wei Q. et al. Circulating miRNAs as Potential Biomarkers of Age-Related Macular Degeneration. Cell Physiol Biochem. 2017;41:1413-23
77. Grassmann F, Schoenberger PG, Brandl C. et al. A circulating microrna profile is associated with late-stage neovascular age-related macular degeneration. PLoS One. 2014;9:e107461
78. Barutta F, Bruno G, Matullo G. et al. MicroRNA-126 and micro-/macrovascular complications of type 1 diabetes in the EURODIAB Prospective Complications Study. Acta Diabetol. 2017;54:133-9
79. Qing S, Yuan S, Yun C. et al. Serum miRNA biomarkers serve as a fingerprint for proliferative diabetic retinopathy. Cell Physiol Biochem. 2014;34:1733-40
80. Hsu CC, Chang HM, Lin TC. et al. Corneal neovascularization and contemporary antiangiogenic therapeutics. J Chin Med Assoc. 2015;78:323-30
81. Mulik S, Xu J, Reddy PB. et al. Role of miR-132 in angiogenesis after ocular infection with herpes simplex virus. Am J Pathol. 2012;181:525-34
82. Anand S, Majeti BK, Acevedo LM. et al. MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat Med. 2010;16:909-14
83. McCormick F. ras GTPase activating protein: signal transmitter and signal terminator. Cell. 1989;56:5-8
84. Bhela S, Mulik S, Gimenez F. et al. Role of miR-155 in the pathogenesis of herpetic stromal keratitis. Am J Pathol. 2015;185:1073-84
85. Li X, Zhou H, Tang W. et al. Transient downregulation of microRNA-206 protects alkali burn injury in mouse cornea by regulating connexin 43. Int J Clin Exp Pathol. 2015;8:2719-27
86. Zhang Y, Zhang T, Ma X. et al. Subconjunctival injection of antagomir-21 alleviates corneal neovascularization in a mouse model of alkali-burned cornea. Oncotarget. 2017;8:11797-808
87. Zong R, Zhou T, Lin Z. et al. Down-Regulation of MicroRNA-184 Is Associated With Corneal Neovascularization. Invest Ophthalmol Vis Sci. 2016;57:1398-407
88. An J, Chen X, Chen W. et al. MicroRNA Expression Profile and the Role of miR-204 in Corneal Wound Healing. Invest Ophthalmol Vis Sci. 2015;56:3673-83
89. Park JK, Peng H, Yang W. et al. miR-184 exhibits angiostatic properties via regulation of Akt and VEGF signaling pathways. FASEB J. 2017;31:256-65
90. Kather JN, Friedrich J, Woik N. et al. Angiopoietin-1 is regulated by miR-204 and contributes to corneal neovascularization in KLEIP-deficient mice. Invest Ophthalmol Vis Sci. 2014;55:4295-303
91. Jian L, Panpan Y, Wen X. Current choroidal neovascularization treatment. Ophthalmologica. 2013;230:55-61
92. Zhou Q, Anderson C, Zhang H. et al. Repression of choroidal neovascularization through actin cytoskeleton pathways by microRNA-24. Mol Ther. 2014;22:378-89
93. Shen J, Yang X, Xie B. et al. MicroRNAs regulate ocular neovascularization. Mol Ther. 2008;16:1208-16
94. Liu CH, Sun Y, Li J. et al. Endothelial microRNA-150 is an intrinsic suppressor of pathologic ocular neovascularization. Proc Natl Acad Sci U S A. 2015;112:12163-8
95. Zhou Q, Anderson C, Hanus J. et al. Strand and Cell Type-specific Function of microRNA-126 in Angiogenesis. Mol Ther. 2016;24:1823-35
96. Yan XC, Cao J, Liang L. et al. miR-342-5p Is a Notch Downstream Molecule and Regulates Multiple Angiogenic Pathways Including Notch, Vascular Endothelial Growth Factor and Transforming Growth Factor beta Signaling. J Am Heart Assoc. 2016:5
97. Cai J, Yin G, Lin B. et al. Roles of NFkappaB-miR-29s-MMP-2 circuitry in experimental choroidal neovascularization. J Neuroinflammation. 2014;11:88
98. Berglin L, Sarman S, van der Ploeg I. et al. Reduced choroidal neovascular membrane formation in matrix metalloproteinase-2-deficient mice. Invest Ophthalmol Vis Sci. 2003;44:403-8
99. Lambert V, Wielockx B, Munaut C. et al. MMP-2 and MMP-9 synergize in promoting choroidal neovascularization. FASEB J. 2003;17:2290-2
100. Ohno-Matsui K, Uetama T, Yoshida T. et al. Reduced retinal angiogenesis in MMP-2-deficient mice. Invest Ophthalmol Vis Sci. 2003;44:5370-5
101. Zhou Q, Gallagher R, Ufret-Vincenty R. et al. Regulation of angiogenesis and choroidal neovascularization by members of microRNA-23~27~24 clusters. Proc Natl Acad Sci U S A. 2011;108:8287-92
102. Zhuang Z, Xiao-qin, Hu H. et al. Down-regulation of microRNA-155 attenuates retinal neovascularization via the PI3K/Akt pathway. Mol Vis. 2015;21:1173-84
103. Westenskow PD, Kurihara T, Aguilar E. et al. Ras pathway inhibition prevents neovascularization by repressing endothelial cell sprouting. J Clin Invest. 2013;123:4900-8
104. Stahl A, Connor KM, Sapieha P. et al. The mouse retina as an angiogenesis model. Invest Ophthalmol Vis Sci. 2010;51:2813-26
105. Yan L, Lee S, Lazzaro DR. et al. Single and Compound Knock-outs of MicroRNA (miRNA)-155 and Its Angiogenic Gene Target CCN1 in Mice Alter Vascular and Neovascular Growth in the Retina via Resident Microglia. J Biol Chem. 2015;290:23264-81
106. Campochiaro PA. Molecular pathogenesis of retinal and choroidal vascular diseases. Prog Retin Eye Res. 2015;49:67-81
107. Lange C, Ehlken C, Stahl A. et al. Kinetics of retinal vaso-obliteration and neovascularisation in the oxygen-induced retinopathy (OIR) mouse model. Graefes Arch Clin Exp Ophthalmol. 2009;247:1205-11
108. Sun Y, Liu CH, SanGiovanni JP. et al. Nuclear receptor RORalpha regulates pathologic retinal angiogenesis by modulating SOCS3-dependent inflammation. Proc Natl Acad Sci U S A. 2015;112:10401-6
109. Wang P, Luo Y, Duan H. et al. MicroRNA 329 suppresses angiogenesis by targeting CD146. Mol Cell Biol. 2013;33:3689-99
110. Shen W, Fruttiger M, Zhu L. et al. Conditional Mullercell ablation causes independent neuronal and vascular pathologies in a novel transgenic model. J Neurosci. 2012;32:15715-27
111. Chung SH, Gillies M, Yam M. et al. Differential expression of microRNAs in retinal vasculopathy caused by selective Muller cell disruption. Sci Rep. 2016;6:28993
112. Dorrell MI, Aguilar E, Jacobson R. et al. Antioxidant or neurotrophic factor treatment preserves function in a mouse model of neovascularization-associated oxidative stress. J Clin Invest. 2009;119:611-23
113. Liu LZ, Li C, Chen Q. et al. MiR-21 induced angiogenesis through AKT and ERK activation and HIF-1alpha expression. PLoS One. 2011;6:e19139
114. Weber M, Baker MB, Moore JP. et al. MiR-21 is induced in endothelial cells by shear stress and modulates apoptosis and eNOS activity. Biochem Biophys Res Commun. 2010;393:643-8
115. Guduric-Fuchs J, O'Connor A, Cullen A. et al. Deep sequencing reveals predominant expression of miR-21 amongst the small non-coding RNAs in retinal microvascular endothelial cells. J Cell Biochem. 2012;113:2098-111
116. Sabatel C, Malvaux L, Bovy N. et al. MicroRNA-21 exhibits antiangiogenic function by targeting RhoB expression in endothelial cells. PLoS One. 2011;6:e16979
117. Poliseno L, Tuccoli A, Mariani L. et al. MicroRNAs modulate the angiogenic properties of HUVECs. Blood. 2006;108:3068-71
118. Urbich C, Kuehbacher A, Dimmeler S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc Res. 2008;79:581-8
119. Dentelli P, Rosso A, Orso F. et al. microRNA-222 controls neovascularization by regulating signal transducer and activator of transcription 5A expression. Arterioscler Thromb Vasc Biol. 2010;30:1562-8
120. Liu X, Cheng Y, Yang J. et al. Cell-specific effects of miR-221/222 in vessels: molecular mechanism and therapeutic application. J Mol Cell Cardiol. 2012;52:245-55
121. Nicoli S, Knyphausen CP, Zhu LJ. et al. miR-221 is required for endothelial tip cell behaviors during vascular development. Dev Cell. 2012;22:418-29
122. Xu XD, Li KR, Li XM. et al. Long non-coding RNAs: new players in ocular neovascularization. Mol Biol Rep. 2014;41:4493-505
123. Faghihi MA, Wahlestedt C. Regulatory roles of natural antisense transcripts. Nat Rev Mol Cell Biol. 2009;10:637-43
124. Yan B, Yao J, Liu JY. et al. lncRNA-MIAT regulates microvascular dysfunction by functioning as a competing endogenous RNA. Circ Res. 2015;116:1143-56
125. Michalik KM, You X, Manavski Y. et al. Long noncoding RNA MALAT1 regulates endothelial cell function and vessel growth. Circ Res. 2014;114:1389-97
126. Liu JY, Yao J, Li XM. et al. Pathogenic role of lncRNA-MALAT1 in endothelial cell dysfunction in diabetes mellitus. Cell Death Dis. 2014;5:e1506
Author contact
![]() Corresponding authors: Yan Zhang, Ph.D., Tianjin Medical University Eye Hospital, Tianjin Medical University Eye Institute, College of Optometry and Ophthalmology, Tianjin Medical University, Tianjin, 300384, China. Fax: 86-22-86428777; Telephone: 86-22-86428863; Email: yanzhang04edu.cn Yunshan Cao, MD., Ph.D., Department of Heart Failure, Shanghai East Hospital, Tongji University School of Medicine; Research Center for Translational Medicine, Shanghai East Hospital, Tongji University School of Medicine, Shanghai 200120, China; Department of Cardiology, Gansu Provincial Hospital, Lanzhou, Gansu Province, 730000, China. Fax: 86-931-8281921; Telephone: 86-931-8281795; Email: yunshancaocom Xiaorong Li, MD, Tianjin Medical University Eye Hospital, Tianjin Medical University Eye Institute, College of Optometry and Ophthalmology, Tianjin Medical University, Tianjin, 300384, China. Fax: 86-22-86428777; Telephone: 86-22-86428808; Email: xliedu.cn
Corresponding authors: Yan Zhang, Ph.D., Tianjin Medical University Eye Hospital, Tianjin Medical University Eye Institute, College of Optometry and Ophthalmology, Tianjin Medical University, Tianjin, 300384, China. Fax: 86-22-86428777; Telephone: 86-22-86428863; Email: yanzhang04edu.cn Yunshan Cao, MD., Ph.D., Department of Heart Failure, Shanghai East Hospital, Tongji University School of Medicine; Research Center for Translational Medicine, Shanghai East Hospital, Tongji University School of Medicine, Shanghai 200120, China; Department of Cardiology, Gansu Provincial Hospital, Lanzhou, Gansu Province, 730000, China. Fax: 86-931-8281921; Telephone: 86-931-8281795; Email: yunshancaocom Xiaorong Li, MD, Tianjin Medical University Eye Hospital, Tianjin Medical University Eye Institute, College of Optometry and Ophthalmology, Tianjin Medical University, Tianjin, 300384, China. Fax: 86-22-86428777; Telephone: 86-22-86428808; Email: xliedu.cn