Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(12):3078-3089. doi:10.7150/thno.18067 This issue Cite
Research Paper
FKBP3 Promotes Proliferation of Non-Small Cell Lung Cancer Cells through Regulating Sp1/HDAC2/p27
Wenzhuo Zhu1*, Zhao Li1*, Liwen Xiong2, Xiaobo Yu1, Xi Chen1, Qiang Lin1 ![]()
1. Department of Thoracic Surgery, Shanghai General Hospital, Shanghai Jiaotong University School of medicine, Shanghai, China;
2. Department of Thoracic Surgery, Shanghai Chest Hospital, Shanghai Jiaotong University School of Medicine, Shanghai, China.
* Contributed equally.
Received 2016-10-24; Accepted 2017-5-3; Published 2017-7-22
Abstract

FKBP3 is a member of FK506-binding proteins (FKBPs). Little is known about the expression and functional role(s) of FKBP3 in non-small cell lung cancer (NSCLC). In the present study, we demonstrated up-regulation of FKBP3 expression, both at mRNA and protein levels, in NSCLC samples which closely correlated with poor survival in NSCLC patients. In vitro and in vivo experiments revealed that FKBP3 could promote NSCLC cell proliferation. Furthermore, knockdown of FKBP3 significantly decreased histone deacetylase 2 (HDAC2) expression and increased p27 (a cell cycle inhibitor) expression. HDAC2 modulated the acetylation of histone H3K4 by directly binding to the p27 promoter. The proliferation-promoting effect of FKBP3 was dependent on HDAC2 and inhibited by p27. Also, FKBP3 induced HDAC2 promoter activity via inhibiting the ubiquitination of transcription factor Sp1. Additionally, we identified miR-145-5p as a regulator of FKBP3. miR-145-5p overexpression suppressed cell proliferation of NSCLC cells which was abrogated by FKBP3 overexpression. Taken together, our data clearly show that FKBP3/Sp1/HDAC2/p27 control cell proliferation during NSCLC development.
Keywords: FKBP3, miR-145, p27, cell proliferation, NSCLC.
Introduction
Lung cancer is the leading cause of cancer-related deaths [1, 2]. Non-small cell lung cancer (NSCLC) accounts for 80% of lung cancers and is categorized into adenocarcinoma (ADC), squamous cell carcinoma (SCC), large cell carcinoma (LCC) and others [3]. Despite intense research, there are no reliable diagnostic and prognostic markers for NSCLC. Undoubtedly, understanding the molecular mechanisms underlying the formation and development of NSCLC is crucial for the development of more effective approaches for NSCLC treatment.
FKBP3 (also known as FKBP25) is a member of FK506-binding proteins (FKBPs), which express peptidylprolyl cis-trans-isomerase (PPIase) activity and bind immunosuppressant drugs such as FK506 and rapamycin [4]. FKBP3, which is located in the nucleus, is a partner of p53-regulating protein MDM2, regulates p53 and p21 expression [5], and is associated with histone deacetylases (HDAC) activity [6]. Although little is known about the expression and role of FKBP3 in human tumors, the abnormal expression of other FKBPs has been observed in a variety of cancer tissues [7-11]. For instance, FKBP52 is overexpressed in hormone-dependent cancers, such as breast [8] and prostate cancers [11]. FKBP65 has been shown to be associated with colorectal carcinogenesis [10].
In the present study, we analyzed RNA sequencing data on 10 pairs of NSCLC and non-cancerous lung tissues [12] and detected a significant upregulation of FKBP3 in NSCLC tissues. Previously, FKBP3 was reported to be associated with the activity of HDAC1/2 [6], which can regulate the expression of cell cycle-related genes, such as p21 (CDKN1A) and p27 (CDKN1B), by modulating histone acetylation [13, 14]. In this study, we demonstrated that FKBP3 knockdown in NSCLC cells decreased cell proliferation and slowed cell cycle progression by suppressing HDAC2 expression and enhancing the expression of p27. FKBP3 induced HDAC2 promoter activity via transcription factor Sp1.
It is widely known that deregulated expression of miRNAs is involved in a broad spectrum of human malignancies. In lung cancer, extensive alterations of miRNAs expression have been identified which correlated with patients' survival. Consistent with these findings, we observed in the present study that FKBP3 was directly targeted by miR-145-5p which inhibited NSCLC cell proliferation in vitro and in vivo. Thus, miR-145-5p /FKBP3/Sp1/HDAC2/p27 may play a key role in the regulation of cell proliferation during NSCLC development suggesting the clinical potential of FKBP3 in the prognosis of NSCLC.
Materials and Methods
Patients and tissue samples
A total of 135 patients who underwent NSCLC resection surgery (Stage I, II and IIIa) at Shanghai General Hospital between January 2006 and December 2007 were included in our study after giving written informed consents. Patients with a recent history of other cancers, or had recurrent or primary NSCLC and received chemotherapy or radiation therapy before surgical operation were excluded from the study. All participants had complete clinical and pathological follow-up data. The study protocol conformed to the ethical guidelines of the Declaration of Helsinki and was approved by the Institutional Ethical Review Committee of Shanghai General Hospital. Paired NSCLC and adjacent non-tumor tissues obtained from 40 patients were flash-frozen in liquid nitrogen immediately and used for RNA and protein extraction. 135 formalin-fixed paraffin-embedded NSCLC specimens were collected for the detection of FKBP3 protein expression by immunohistochemistry (IHC) analysis.
Bioinformatics analysis
The gene expression data were obtained fromThe Cancer Genome Atlas website (TCGA, https://tcga-data.nci.nih.gov/tcga/) for lung adenocarcinoma (LUAD) projects or from our previous RNA sequencing data deposited in NCBI's Sequence Read Archive database (http://www.ncbi.nlm.nih.gov/sra, AC: SRP075725) [12]. Student's t-test was performed to determine the statistical significance of FKBP3 expression between NSCLC and normal tissues.
Real-time PCR
Total RNA from 40 paired tissue samples or NSCLC cell lines was extracted by the TRIzol reagent (Invitrogen, Carlsbad, CA, USA). After removing genomic DNA contamination with DNase I (Roche, Indianapolis, IN, USA), 2 μg RNA was reverse-transcribed into cDNA with cDNA synthesis kit (Thermo Fisher Scientific, Rockford, IL, USA). Real-time PCR was performed with Maxima SYBR Green qPCR Master Mixes (Thermo Fisher Scientific) on ABI 7300 system (Applied Biosystem, Foster City, CA, USA). GAPDH was served as an internal control. The PCR primers were listed in Table S1. Target gene expression was calculated using the 2-ΔΔCt method.
Stem-loop real-time RT-PCR was carried out to analyze miRNA expression. U6 RNA was used as a miRNA internal control. Briefly, extracted RNAs were converted into cDNAs with stem-loop reverse transcription primers (miR-145-5p, 5'- CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAGGGATTC-3'; U6, 5'- AACGCTTCACGAATTTGCGT -3') with cDNA synthesis kit (Thermo Fisher Scientific). Real-time PCR was then performed using Maxima SYBR Green qPCR Master Mixes (Thermo Fisher Scientific) according to the manufacturer's instructions, with miR-145-5p or U6 -specific forward primer (miR-145-5p, 5'-ACACTCCAGCTGGGGTCCAGTTTTCCCAGGA-3'; U6, 5'- CTCGCTTCGGCAGCACA-3') and a universal reverse primer (5'-TGGTGTCGTGGAGTCG-3').
FKBP3 protein expression by IHC staining
IHC staining of FKBP3 protein expression was performed on 135 NSCLC specimens following standard protocol by using anti-FKBP3 (Ab16654, Abcam, Cambridge, MA, USA; 1:50 dilution). Patients with at least 25% of tumor cells with positive staining, were defined as FKBP3 high expression group and those with less than 25% of tumor cells with positive staining were classified as FKBP3 low expression group.
Cell culture
Human NSCLC cell lines (NCI-H446, NCI-H358, NCI-H1975, NCI-H292 and NCI-A549) were purchased from cell bank of Shanghai Biology Institute, Chinese Academy of Science (Shanghai, China). A549 cells were cultured in DMEM (Life Technologies, Carlsbad, CA, USA), while other cell lines were maintained in RPMI 1640 (Life Technologies). All culture media were supplemented with 10 % fetal bovine serum and 1 % penicillin/streptomycin (Life Technologies).
Small interfering RNAs (siRNAs) and miRNA transfection
Three FKBP3 siRNAs (siRNA1: 5'- GAGGUUCAAUGUUGGAUAU -3', siRNA2: 5'- CUCCCAUGAAAGUUGUUAAU -3' and siRNA3: 5'- CCCACCCAAUGCUAAUAA -3'), HDAC2 siRNA (siHDAC2, 5'-CCCAUAACUUGCUGUUAAA-3'), Sp1 siRNA (siSp1, 5'- CUGGUCAAAUACAGAUCAU-3'), a scramble control siRNA (siNC, 5'-UUCUCCGAACGUGUCACGU-3'), miRNA mimic (miR-145-5p mimic, 5'-GUCCAGUUUUCCCAGGAAUCCCU-3'; miR-30a-5p mimic, 5'-UGUAAACAUCCUCGACUGGAAG-3'; miR-30b-5p mimic, 5'-UGUAAACAUCCUACACUCAGCU-3'; miR-30c-5p mimic, 5'-UGUAAACAUCCUACACUCUCAGC-3'; miR-30d-5p mimic, 5'-UGUAAACAUCCCCGACUGGAAG-3'; and miR-30e-5p mimic, 5'-UGUAAACAUCCUUGACUGGAAG-3') and control mimic (miR-NC, 5'-UUUGUACUACACAAAAGUACUG-3') were synthesized by Genepharm Technologies (Shanghai, China). Transfection was carried out using the Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer's instructions.
FKBP3 overexpression
The full-length human FKBP3 was cloned into the lentiviral expression vector pLVX-puro (Clontech, Palo Alto, CA, USA) by Genewiz Company (Shanghai, China). FKBP3-expressing (FKBP3) and control (Vector) lentivirus were generated as described previously [15].
Western blot analysis
Protein was extracted using RIPA lysis buffer with freshly added protease inhibitor cocktail (Sigma, St. Louis, MO, USA), separated by SDS-PAGE gel, and transferred onto a nitrocellulose membrane (Millipore, Bedford, USA). Membranes were blocked with 5% skim milk, incubated with antibodies against FKBP3, HDAC2, p27, Sp1, (Abcam) and GAPDH (Cell Signaling, Danvers, MA, USA) followed by the HRP-conjugated secondary antibody (Beyotime, Shanghai, China) according to the manufacturers' instructions. Signals were visualized with enhanced chemiluminescence system (Bio-Rad, Richmond, CA, USA).
Cell proliferation assay
Cell proliferation was examined using a Cell Counting Kit (CCK)-8 (Dojindo Lab, Kumamoto, Japan). Briefly, cells were seeded in 96-well plates (2 × 103 cells per well). Cells were transfected with indicated siRNA, siNC, miR-NC or miR-145-5p and/or infected with FKBP3-expressing or Vector lentivirus. After incubation for 0, 24, 48, and 72 h, the CCK-8 reagent was added to each well and cultured for 1 h. Optical density values (OD) of 450 nm were determined using a microplate reader (Bio-Rad). Triplicates were performed at each time point.
Cell cycle profile analysis
At 48 h after siRNA transfection, miRNA transfection, or viral infection, cells were trypsinized and fixed in ice-cold 70 % ethanol. The fixed cells were then pelleted and incubated with ribonuclease A (0.1 mg/mL, Sigma) and PI (0.05 mg/ml, Sigma) at room temperature for 30 min. DNA content was analyzed on a flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Establishment of stable cell lines and Xenograft Study
Lentiviral constructs of FKBP3 knockdown (shFKBP3), miR-145-5p, or control (shNC or miR-NC) were introduced into HEK293 cells to produce lentivirus. After infecting with shFKBP3, shNC, miR-145-5p, or miR-NC lentivirus, stable cell lines were established in NCI-H358 cells by puromycin (Sigma) selection. The transduced cells (2×106) were injected subcutaneously into the flank regions of 4- to 5-week-old female nude mice (Shanghai Laboratory Animal Company, Shanghai, China). Tumors were measured every three days and tumor volumes were calculated using the following formula: volume= 1/2 × (Length × Width2). 27 days after cell implantation, all mice were sacrificed. Tumor xenografts were collected, photographed, weighed and stained with anti-Ki-67 (Abcam). The animal study was performed according to a protocol approved by the Institutional Animal Care and Use Committee of Shanghai Jiao Tong University.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed on NCI-H358 cells transfected with siFKBP3-1 or siNC for 24 h. Cells were treated with 1% formaldehyde at room temperature on a rocking platform to crosslink the chromatin. The chromatin solutions were then incubated with anti-HDAC2 (Abcam), anti-acetyl-histone H3 (K4) (Millipore), anti-Sp1 (Abcam) or control IgG antibody overnight at 4 °C with rotation. Binding was detected by PCR with p27 promoter primers or HDAC2 promoter primers as previously described [16, 17]. The primers are listed in Table S2.
Luciferase reporter assay
The full length HDAC2 promoter [17] and FKBP3 3'-UTR region carrying a putative miR-145-5p binding site was inserted into pGL3 Vector (Promega, Madison, WI). For HDAC2 promoter activity, NCI-H358 cells were transfected with the pGL3-HDAC2 promoter and indicated siRNA, and/or infected with FKBP3-expressing or Vector lentivirus. For FKBP3 luciferase reporter assay, NCI-H358 cells were transfected with miR-145-5p mimics and pGL3-FKBP3WT plasmid or pGL3-FKBP3Mut using Lipofectamine 2000 reagent. Luciferase activity was determined using a Luciferase assay kit (Promega), normalizing to protein concentration and then to a control sample transfected with pGL3.
Immunoprecipitation
Total cell extracts were prepared with RIPA buffer as described above. For immunoprecipitation, the supernatant was incubated with the anti-Sp1 (Abcam) or control IgG antibody at 4 °C for 2 h with rotation. After incubation with protein A agarose beads (Millipore) at 4 °C for 1 h with rotation, the immunoprecipitated complexes were washed with RIPA buffer three times and analyzed by Western blot analysis with anti-ubiquitin (Abcam).
Statistical analysis
Statistical analyses were performed with GraphPad Prism software Version 6.0 (San Diego, CA, USA). Student's t and ANOVA test were carried out to determine the statistical significance between two groups and among more than two groups, respectively. Fisher's exact test was done to assess the relationship between FKBP3 protein expression and clinicopathological features. Overall survival was determined by Kaplan-Meier survival analysis and log-rank test. All in vitro experiments were performed in triplicates and repeated at least three times. The criterion for statistical significance was set at P < 0.05.
Results
Up-regulation of FKBP3 correlates with poor prognosis in NSCLC
Our previous RNA sequencing data on 10 pairs of NSCLC and non-cancerous lung tissues (data were deposited in NCBI's Sequence Read Archive database (http://www.ncbi.nlm.nih.gov/sra, AC: SRP075725) [12] revealed that FKBP3 was significantly up-regulated in NSCLC tissues (Fig. 1A). To further define FKBP3 expression patterns in NSCLC, we examined FKBP3 mRNA levels in 40 pairs of NSCLC and adjacent unaffected tissues. We observed that FKBP3 mRNA was significantly increased in NSCLC tissues compared with adjacent unaffected tissues from the same patient (Fig. 1B). Similar results were obtained by analyzing a TCGA lung adenocarcinoma (LUAD) dataset (Fig. 1C). Western blot analysis showed that FKBP3 was abundant in NSCLC tissues at the translational level (Fig. 1D).
High expression of FKBP3 indicates poor survival of NSCLC. (A) FKBP3 mRNA expression was significantly higher in NSCLC tissues than in paired non-cancerous tissues (n=10) as indicated by RNA-sequencing analysis (P<0.01). (B) mRNA expression of FKBP3 in NSCLC tissues and paired non-cancerous tissues from 40 NSCLC patients by real-time PCR analysis (P<0.0001). (C) FKBP3 expression in lung adenocarcinoma and normal tissues based on TCGA dataset (P<0.0001). (D) Representative FKBP3 protein expression in NSCLC (T1, T2, T3 and T4) and unaffected tissues (N1, N2, N3 ad N4) by Western blots. GAPDH served as a loading control. (E) IHC staining of FKBP3 in NSCLC tissues. Scale bar: 100 μm. (F) Survival analysis of patients' cohort (left panel, P<0.001) and the TCGA dataset (right panel, P<0.01). The survival time in patients with FKBP3 high expression was significantly shorter than that of patients with FKBP3 low expression.
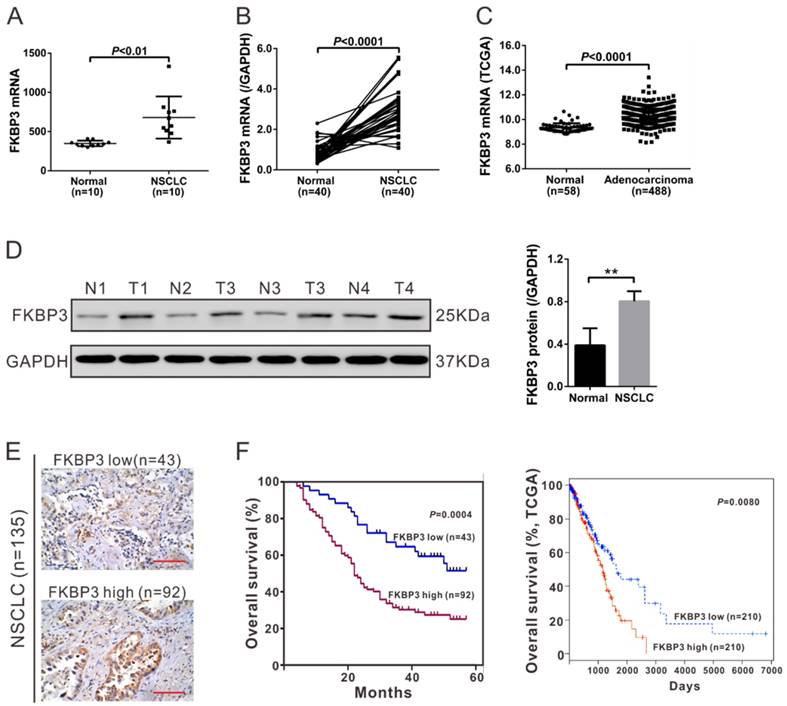
135 NSCLC patients were categorized into two groups by IHC staining (Fig, 1E) comprising an FKBP3 low expression group with less than 25% FKBP3 expression (n=43) and FKBP3 high expression group with more than 25% FKBP3 expression (n=92). To evaluate the correlation between FKBP3 protein expression and clinicopathological characteristics in NSCLC patients, we performed a Fisher's exact test. As shown in Table 1, FKBP3 protein expression was correlated with tumor size and lymph node metastasis. Kaplan-Meier analysis of our patient data and TCGA dataset showed that the overall survival time of patients with FKBP3 low expression was remarkably longer than those with FKBP3 high expression (Fig. 1F). These results suggested that up-regulation of FKBP3 in NSCLC patients contributed to the poor survival.
Correlation of FKBP3 protein expression with patients' features
| Variables | All cases | FKBP3 protein | ||
|---|---|---|---|---|
| Low (n=43) | High (n=92) | P value | ||
| Age at surgery | ||||
| <55 | 53 | 18 | 35 | 0.7078 |
| >=55 | 82 | 25 | 57 | |
| Gender | ||||
| Male | 72 | 22 | 50 | 0.8533 |
| Female | 63 | 21 | 42 | |
| Smoking status | ||||
| Smoker | 46 | 19 | 27 | 0.1188 |
| Non-smoker | 89 | 24 | 65 | |
| Tumor size | ||||
| < 3 cm | 55 | 24 | 31 | 0.0235* |
| >=3 cm | 80 | 19 | 61 | |
| Tumor type | ||||
| SCC | 71 | 24 | 47 | 0.2497 |
| ADC | 46 | 11 | 35 | |
| LCC | 18 | 8 | 10 | |
| Tumor stage | ||||
| I+II | 85 | 32 | 53 | 0.0845 |
| IIIa | 50 | 11 | 39 | |
| Lymph node metastasis | ||||
| Absent | 74 | 30 | 44 | 0.0254* |
| Present | 61 | 13 | 48 | |
Abbreviations: SCC, squamous cell carcinoma; ADC, adenocarcinoma; LCC, large cell carcinoma.
*P<0.05, **P<0.01.
FKBP3 promotes NSCLC proliferation and tumorigenesis
To assess the role of FKBP3 in NSCLC development, we studied the effect of FKBP3 on cell proliferation. First, FKBP3 expression was evaluated in five lung cancer cell lines (Fig. S1). NCI-H358 and NCI-H1975 cells exhibited higher expression of FKBP3 at both mRNA and protein levels, while NCI-H292 had lower expression of FKBP3. We then transfected FKBP3 siRNAs (siRNA1, siRNA2, and siRNA3) or control siRNA (siNC) into NCI-H358 and NCI-H1975 cells. As shown in Fig. S2, siRNA1 reduced FKBP3 expression to less than 10% of the scramble control and was therefore chosen for further experiments.
CCK-8 assay indicated that knockdown of FKBP3 resulted in a decreased cell growth rate at 48 h and 72 h in both NCI-H358 and NCI-H1975 cells. Also, overexpressing FKBP3 in NCI-H292 cells promoted cell growth (Fig. 2B). Next, we explored the mechanisms underlying the proliferation promoting effect of FKBP3 in NSCLC cell lines. Flow cytometry analysis revealed that NCI-H358 and NCI-H1975 cells with FKBP3 knockdown exhibited a significant increase in G0/G1 phase fraction and a notable decrease in the percentages of S and G2/M phase cells. On the contrary, overexpression of FKBP3 in NCI-H292 cells caused an inverse effect (Fig. 2C).
FKBP3 promotes cell proliferation in vitro and in vivo. (A) FKBP3 protein expression in NCI-H358, NCI-H1975, and NCI-H292 cells was analyzed by Western blotting. NCI-H358 and NCI-H1975 cells were transfected with FKBP3 siRNA (siRNA1) or control siRNA (siNC), whereas NCI-H292 cells were infected with FKBP3 expression lentivirus or control vector lentivirus (Vector). (B) Cell proliferation was detected at 0, 24, 48, and 72 h after siRNA transfection or lentiviral infection in NCI-H358, NCI-H1975, and NCI-H292 cells. (C) Cell cycle was analyzed using flow cytometry in NCI-H358 and NCI-H1975 cells with silenced FKBP3, or NCI-H292 cells with FKBP3 overexpression. Data are based on at least 3 independent experiments and shown as mean ± SD. (D) NCI-H358 cells stable transfected with either FKBP3 shRNA (shFKBP3) or control shRNA (shNC) were subcutaneously injected into athymic nude mice (n=6 per group). Tumor volume was evaluated every 3 days for 27 days. (E) At day 27, mice were sacrificed, and tumors were photographed and weighed. (F) Xenograft tumors with Ki-67 immunostaining. **P<0.05, ***P<0.001.
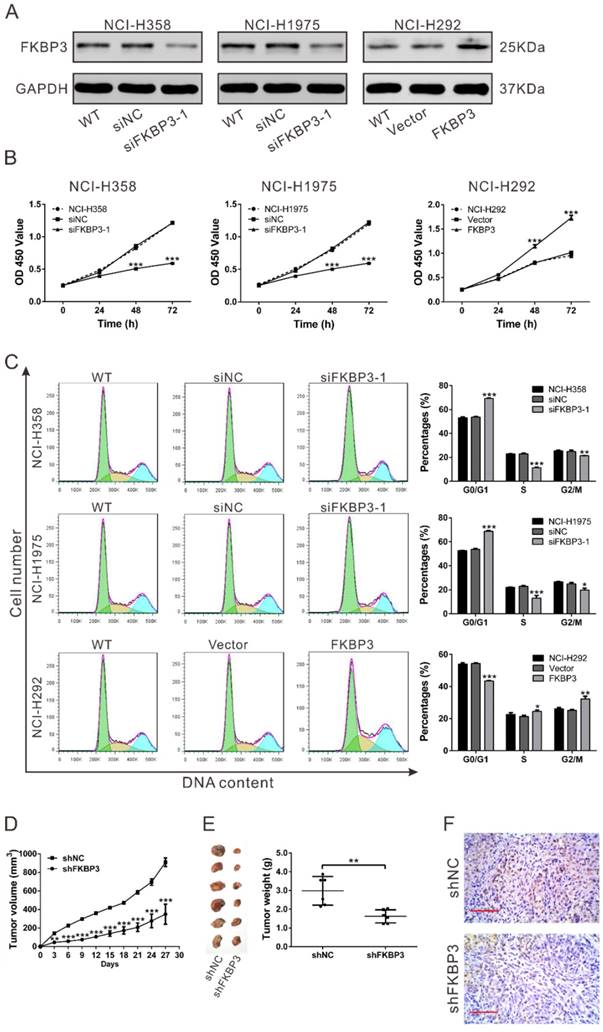
To confirm the effects of FKBP3 on NSCLC cell proliferation in vivo, xenograft tumor assay was performed in nude mice. NCI-H358 cells with FKBP3 knockdown grew slower than the control group (Fig. 2D). Tumors formed from FKBP3 knockdown cells were much smaller than those from the control group at 27 days after cell inoculation (Fig. 2E). Moreover, the number of Ki-67-positive cells was significantly reduced in FKBP3 knockdown group (Fig. 2F). These data indicated that FKBP3 promoted NSCLC cell proliferation, thus playing a critical role in NSCLC development.
FKBP3 knockdown increases p27 level via suppressing HDAC2 expression
We next investigated possible mechanisms by which FKBP3 promoted NSCLC cell proliferation. We analyzed the mRNA levels of proliferation-related genes including p53, CCND1, CDC25A, p16, p21, p27 and MYC in NCI-H358 cells by real-time PCR. As shown in Fig. S3A, p27, an inhibitory regulator of the cell cycle [18], was the most highly increased gene when FKBP3 was knocked down. A previous study reported that FKBP3 was associated with HDAC1/2 activity [6]. By analyzing our RNA-sequencing data [12], we found that HDAC Class I pathway was significantly enriched in tissues with high FKBP3 expression (Fig. S3B). We then analyzed the mRNA levels of HDAC1-9 in NCI-H358 cells by real-time PCR and detected the most significant decrease in HDAC2 when FKBP3 was knocked down (Fig. S3C).
To confirm the effect of FKBP3 on HDAC2 and p27 expression, real-time PCR and Western blot analyses were performed in the FKBP3 knockdown or overexpressing cells. FKBP3 knockdown significantly suppressed HDAC2 expression and enhanced p27 expression at both transcriptional and translational levels, while FKBP3 overexpression exhibited an inverse effect (Figs. 3A and 3B). Further, we performed HDAC2 promoter-luciferase assays and observed that the relative luciferase activity was markedly reduced when HDAC2 promoter was co-transfected with siFKBP3-1 in NCI-H358 cells (Fig. 3C). These data confirmed that FKBP3 knockdown decreased the HDAC2 promoter activity and suppressed HDAC2 transcription.
FKBP3 knockdown increases p27 expression via suppressing HDAC2 expression in NCI-H358 cells. (A, B) Real-time PCR (A) and Western blot analyses (B) of the effects of FKBP3 knockdown or overexpression on p27 and HDAC2 expression. (C) Luciferase Reporter assay was performed to evaluate the activity of the HDAC2 promoter in cells with FKBP3 silencing or overexpression. (D) HDAC2 bound to the p27 promoter as demonstrated by ChIP analysis. Upper, Schematic diagram of primers for ChIP analysis. (E) ChIP analysis of acetyl-histone H3 (K4) on the p27 promoter in NCI-H358 cells transfected with siNC or siFKBP3-1. (F, G) Real-time PCR (F) and Western blot analyses (G) of FKBP3 and HDAC2 expression. (H) Effects of HDAC2 knockdown on cell proliferation of NCI-H358 cells with FKBP3 overexpression. **P<0.01, ***P<0.001.
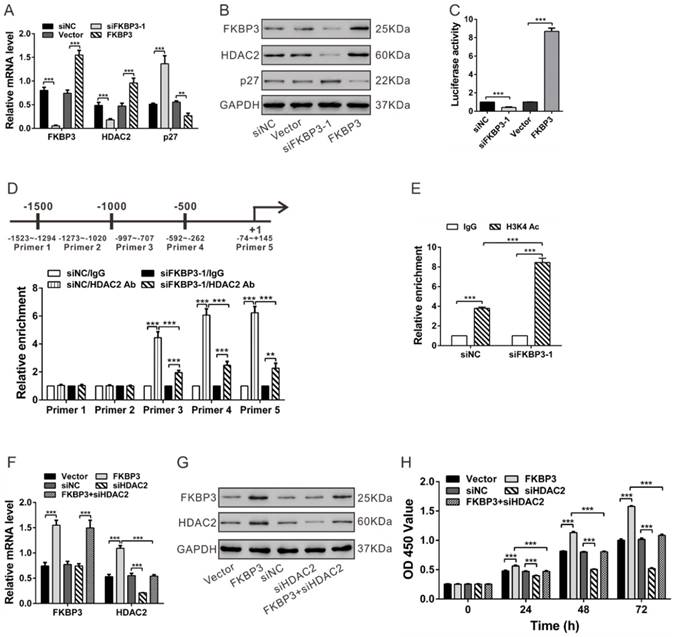
It has been previously reported that the HDAC inhibitor can induce the expression of p27 [13]. We, therefore, performed ChIP assays to analyze whether HDAC2 bound to the p27 promoter in NSCLC cells. In NCI-H358 cells, HDAC2 bound to the p27 promoter regions at -997~-707, -592~-262 and -74~+145, but not to regions at -1523~-1294 and -1273~-1020. The binding levels of HDAC2 at the p27 promoter were decreased following FKBP3 knockdown (Fig. 3D). Acetylation of histone H3 (K4), enriched on the p27 promoter, was significantly reduced when FKBP3 was knocked down (Fig. 3E). These data confirmed that FKBP3 knockdown-induced p27 expression was associated with increased acetylated histone H3 and decreased HDAC2 binding to the p27 promoter.
To test whether the proliferation-promoting effect of FKBP3 was dependent on HDAC2, we knocked down HDAC2 expression in NSCLC cell lines with FKBP3 overexpression. The expression of FKBP3 and HDAC2 was verified by real-time PCR and Western blot analyses (Fig. 3F and 3G). Results of CCK-8 assays showed that HDAC2 knockdown partially abrogated FKBP3-promoted cell proliferation (Fig. 3H). Furthermore, p27 overexpression reduced the proliferation-promoting effects of FKBP2 in NCI-H358 cells (Fig. S4). These data indicated that FKBP3 promoted NSCLC cell proliferation through regulating HDAC2/p27.
FKBP3 regulates HDAC2 transcription via Sp1
We then investigated the mechanism of regulation of HDAC2 promoter activity by FKBP3. It was previously shown that HDAC2 promoter is regulated by Sp1, Sp3 [17], hypoxia-inducible factor 1α (HIF1α) [19], and c-Myc [20]. Luciferase assays showed that HDAC2 promoter was specifically activated by Sp1 overexpression in NCI-H358 cells but not by Sp3, HIF1α or c-Myc overexpression (Fig. S5). We knocked down Sp1 expression in NCI-H358 cells with FKBP3 overexpression and discovered that FKBP3 increased HDAC2 expression in a Sp1-dependent manner at both mRNA and protein levels (Fig. 4A and 4B). Luciferase assays showed that Sp1 knockdown significantly eliminated FKBP3-induced HDAC2 promoter activity (Fig. 4C).
Regulation of FKBP3 on HDAC2 expression is dependent on Sp1. (A, B) Real-time PCR (A) and Western blot analysis (B) of the effect of Sp1 knockdown on HDAC2 expression in NCI-H358 cells with FKBP3 overexpression. (C) Luciferase Reporter assay was performed to evaluate the activity of the HDAC2 promoter. (D) FKBP3 knockdown affected the binding of Sp1 to HDAC2 promoters as demonstrated by ChIP analysis. Upper, Schematic diagram of primers for ChIP analysis. (E, F) Effects of FKBP3 on Sp1 mRNA expression (E) and Sp1 ubiquitination (F). ***P<0.001.
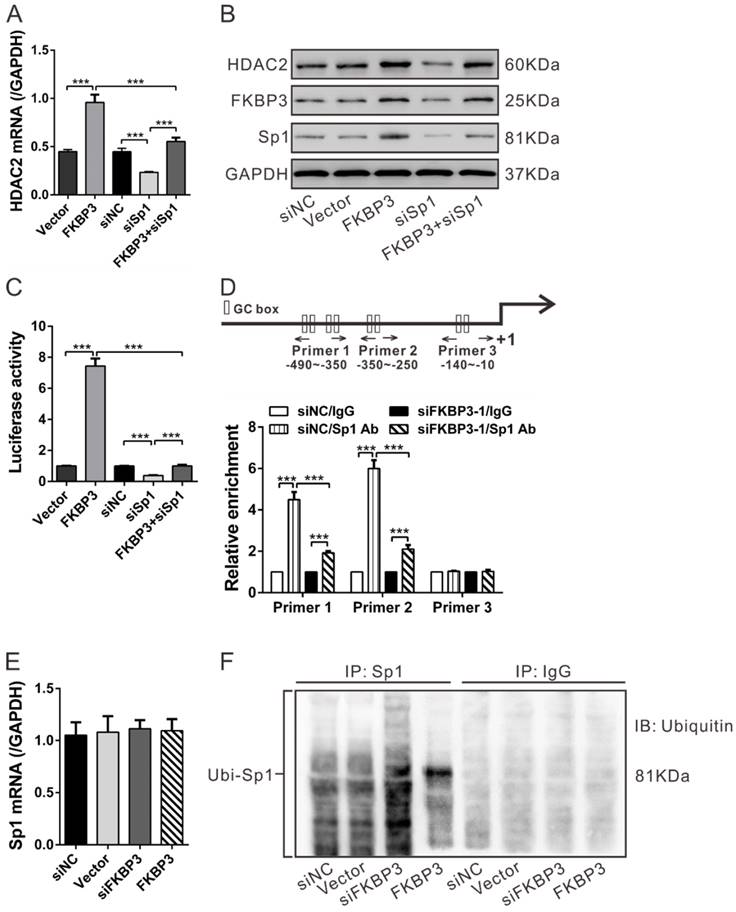
As previously described, Sp1 can bind to the GC boxes of the HDAC2 promoter [17]. We conducted ChIP assays to analyze whether FKBP3 knockdown affected the binding between Sp1 and HDAC2 promoter [17]. In NCI-H358 cells, Sp1 bound to the HDAC2 promoter regions at -490~-350 and -350~-250, but not to regions at -140~-10. The binding of Sp1 at the HDAC2 promoter was decreased following FKBP3 knockdown (Fig. 4D). These data suggest that FKBP3 regulates HDAC2 promoter activity via Sp1.
We also explored the FKBP3 regulation of Sp1 by real-time PCR analysis which showed that FKBP3 overexpression or knockdown in NCI-H358 cells had no effect on Sp1 mRNA expression (Fig. 4E). Considering that FKBP3 overexpression enhanced Sp1 protein expression (Fig. 4B), we speculated that FKBP3 helped to stabilize the Sp1 protein. Previous studies have suggested that Sp1 ubiquitination can regulate its stability in a proteasome-dependent manner [21]. We, therefore, investigated whether FKBP3 affected Sp1 ubiquitination. As shown in Fig. 4F, Sp1 ubiquitination was increased by FKBP3 knockdown but decreased by FKBP3 overexpression. These data suggest that FKBP3 regulates Sp1 expression through its post-translational modification.
FKBP3 is targeted by miR-145-5p
To investigate the upstream regulators of FKBP3, we focused on miRNAs, which are aberrantly expressed in cancer tissues and play key roles in cancer development. To identify potential miRNAs targeting FKBP3, we used three target predicting algorithms (miRnada [http://www.microrna.org/microrna/home.do], PicTar [http://pictar.mdc-berlin.de/], and TargetScan [http://www.targetscan.org/]). Based on these algorithms, we found that miR-145-5p and miR-30 family members targeted FKBP3 mRNA and a potential complimentary binding site was present for miR-145-5p and miR-30 family members within the 3' untranslational region (UTR) of FKBP3 mRNA (Fig. 5A). In real-time PCR analysis of NCI-H358 cells, miR-145-5p significantly repressed FKBP3 expression compared with other miRNAs. We then performed 3'UTR luciferase assay and observed that the relative luciferase activity was markedly reduced when FKBP3 3'UTR (FKBP3WT) was co-transfected with miR-145-5p in NCI-H358 cells. With mutated plasmid (FKBP3Mut), there was no significant difference in luciferase activity between the control (miR-NC) and miR-145-5p transfectants (Fig. 5C). These data suggested that miR-145-5p directly bound to the 3'UTR of the FKBP3 mRNA leading to decreased FKBP3 expression.
FKBP3 is a direct target of miR-145-5p. (A) Complementary miRNA binding sequence in FKBP3 3'UTR. (B) Real-time PCR analysis of the effects of indicated miRNA transfections on FKBP3 mRNA expression in NCI-H358 cells. (C) 3'UTR Luciferase assay (miR-NC and miR-145-5p). WT and Mut indicate the wild-type and mutated plasmid sequences, respectively. (D, E) mRNA and protein levels of FKBP3 in NCI-H358 cells with miR-145-5p and /or FKBP3 overexpression. (F) Effect of enforced overexpression of FKBP3 on cell proliferation of NCI-H358 cells transfected with miR-145-5p. ***P<0.001.
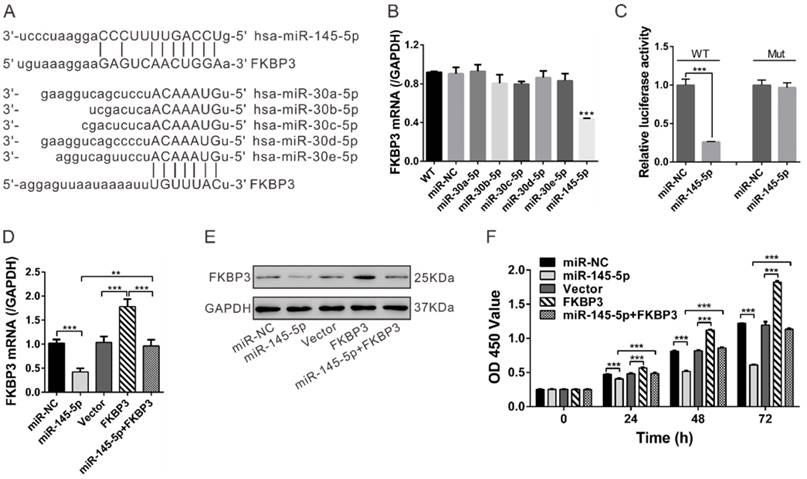
We observed that miR-145-5p inhibited the proliferation of NSCLC cells in vitro and in vivo and decreased HDAC2 mRNA expression but increased p27 mRNA expression (Fig. S6). To test whether restoration of FKBP3 would reverse the inhibitory effect of miR-145 on NSCLC cell proliferation, we overexpressed FKBP3 in miR-145-5p-transfected NSCLC cells. FKBP3 expression was verified by real-time PCR and Western blot analyses (Fig. 5D and 5E). As shown in Figure 5F, FKBP3 overexpression partially abolished miR-145-5p-mediated suppression of cell proliferation.
Correlation analyses in NSCLC tissues
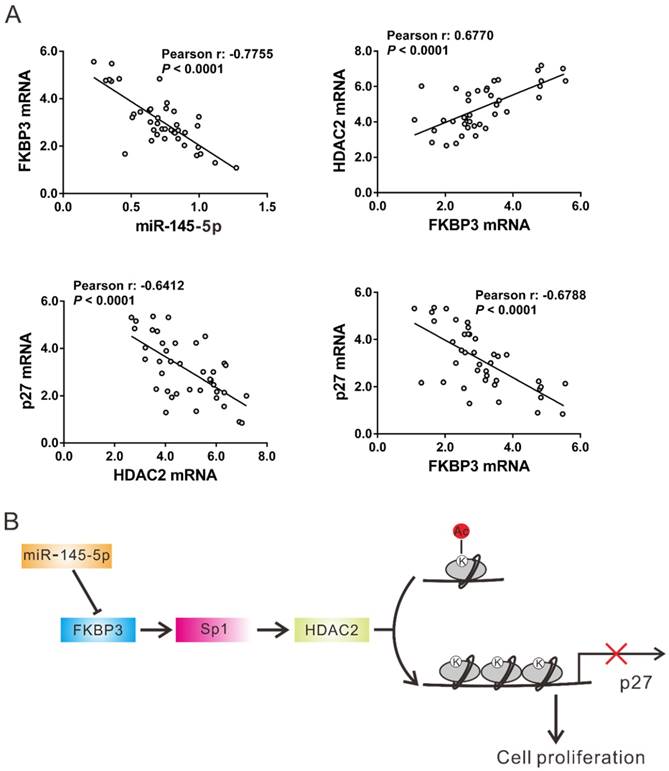
The expression of miR-145-5p, HDAC2, and p27 was assessed in 40 NSCLC tissues by real-time PCR followed by Pearson correlation analysis. FKBP3 mRNA expression was negatively correlated with the expression of miR-145-5p and p27 in NSCLC tissues, whereas there was a positive correlation with HDAC2 expression. A negative correlation was also observed between the expression of HDAC2 and p27 in NSCLC tissues (Fig. 6A). These data further supported the findings in NSCLC cell lines.
Correlation analysis in NSCLC tissues. (A) Pearson correlation scatter plots in NSCLC tissues (n=40). (B) Schematic representation of the regulation of NSCLC proliferation by miR-145-5p /FKBP3/HDAC2/p27.
Discussion
In this study, we identified FKBP3 as an up-regulated gene in NSCLC tissues based on the analysis of RNA sequencing data. Furthermore, we provided evidence for the critical role of FKBP3 in NSCLC progression and proposed a miRNA-mediated mechanism that regulates FKBP3 expression.
FKBPs share similar protein structure and may have a similar biological function. Some members of FKBPs participate in the progression of carcinogenesis as well as chemoresistance and can serve as cancer biomarkers [22]. However, the functional significance of FKBP3, a member of the FKBP family, in cancers has not been investigated. Here, we demonstrated the up-regulation of FKBP3 in NSCLC tissues at both mRNA and protein levels. In vitro and in vivo experiments further demonstrated the growth-promoting role of FKBP3 in NSCLC cells via accelerating cell cycle progression. Our study, which is consistent with previous reports [7-11], revealed the clinical value of FKBPs in cancer development and suggested its prognostic potential.
We also investigated the molecular mechanism by which FKBP3 promotes tumorigenesis. A previous study demonstrated the interaction between FKBP3 and HDAC1/2 [6], which can modulate histone acetylation thus affecting the expression of cell cycle-related genes, such as p21 (CDKN1A) and p27 (CDKN1B) [13, 14]. Our results showed that FKBP3 knockdown significantly reduced HDAC2 expression but induced p27 expression. Given that p27 is an inhibitor of cyclin-dependent kinase 2 (CDK2) [18] and can arrest cells in the G1 phase [23], the increased expression of p27 clearly led to the slower proliferation rate of NSCLC cells. Moreover, ChIP assay demonstrated the direct binding of the HDAC2 protein to the p27 promoter. Knockdown of FKBP3 increased enrichment of histone H3 (K4) acetylation on the p27 promoter ascribing to the decreased expression of HDAC2. HDAC2 knockdown or p27 overexpression partially abrogated FKBP3-promoted NSCLC cell proliferation. In NSCLC tissues, a negative correlation was observed between the expression of FKBP3 and p27 as well as between the expression of HDAC2 and p27 while FKBP3 mRNA expression was positively correlated with HDAC2 expression. Accordingly, we speculated that up-regulated FKBP3 might increase HDAC2 expression which bound to the p27 promoter and reduced acetylated histone H3 (K4), leading to the suppression of p27 expression and the acceleration of cell cycle progression (Fig. 6B).
We also investigated the mechanism of regulation of HDAC2 expression by FKBP3 and found that FKBP3 overexpression induced the activity of the HDAC2 promoter. Transcription factor Sp1, the stability of which is regulated by ubiquitination [21], has been reported to regulate HDAC2 promoter activity in colon cancer cells [17]. Our results showed that FKBP3 increased the mRNA and protein levels of HDAC2 as well as its promoter activity in a Sp1-dependent manner. ChIP assays revealed that the direct binding of Sp1 protein and the HDAC2 promoter was suppressed by the FKBP3 knockdown. FKBP3 overexpression increased Sp1 protein levels by reducing its ubiquitination. Thus, we speculate that up-regulated FKBP3 might induce HDAC2 transcription via Sp1 (Fig. 6B).
It has been well established that miRNAs play a critical role in cancer development [24]. In the present study, we demonstrated that miR-145-5p directly bound to the 3' UTR of FKBP3 thereby decreasing FKBP3 expression at both mRNA and protein levels. miR-145-5p is known to inhibit cell proliferation in NSCLC [25] and liver cancer [26]. A previous study has suggested that miR-145-5p may be a useful prognostic marker for NSCLC as patients with lower levels of miR-145-5p had a shorter time to relapse than those with higher levels [27]. Consistent with these findings, overexpression of miR-145-5p in NSCLC cells repressed cell growth and tumorigenesis. More importantly, FKBP3 overexpression partially abolished miR-145-5p-mediated suppression of cell proliferation. These data suggested that miR-145-5p targeted FKBP3 thus serving as a tumor suppressor in NSCLC.
In conclusion, our study provides evidence that FKBP3 plays an essential role in proliferation and cell cycle progression of NSCLC cells through modulating Sp1/HDAC2/p27. More importantly, our study suggests the clinical value of FKBP3 as a prognostic marker in NSCLC.
Abbreviations
NSCLC: non-small cell type lung cancer; ADC: denocarcinoma; SCC: squamous cell carcinoma; LCC: large cell carcinoma; FKBP: FK506-binding proteins; PPIase: peptidylprolyl cis-trans-isomerase; HDAC: histone deacetylases; miRNA: microRNA; IHC: immunohistochemistry; siRNA: small interfering RNA; CCK-8: Cell Counting Kit-8; OD: optical density; ChIP: chromatin immunoprecipitation; LUAD: lung adenocarcinoma.
Acknowledgements
This study was supported by Natural Science Foundation of China (81372521).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225-49
2. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69-90
3. Koudelakova V, Kneblova M, Trojanec R, Drabek J, Hajduch M. Non-small cell lung cancer-genetic predictors. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2013;157:125-36
4. Jin YJ, Burakoff SJ, Bierer BE. Molecular cloning of a 25-kDa high affinity rapamycin binding protein, FKBP25. J Biol Chem. 1992;267:10942-5
5. Ochocka AM, Kampanis P, Nicol S, Allende-Vega N, Cox M, Marcar L. et al. FKBP25, a novel regulator of the p53 pathway, induces the degradation of MDM2 and activation of p53. FEBS Lett. 2009;583:621-6
6. Yang WM, Yao YL, Seto E. The FK506-binding protein 25 functionally associates with histone deacetylases and with transcription factor YY1. EMBO J. 2001;20:4814-25
7. Lin I-Y, Yen C-H, Liao Y-J, Lin S-E, Ma H-P, Chan Y-J. et al. Identification of FKBP11 as a biomarker for hepatocellular carcinoma. Anticancer research. 2013;33:2763-9
8. Ward BK, Mark PJ, Ingram DM, Minchin RF, Ratajczak T. Expression of the estrogen receptor-associated immunophilins, cyclophilin 40 and FKBP52, in breast cancer. Breast cancer research and treatment. 1999;58:265-78
9. Quinn MC, Wojnarowicz PM, Pickett A, Provencher DM, Mes-Masson A-M, Davis EC. et al. FKBP10/FKBP65 expression in high-grade ovarian serous carcinoma and its association with patient outcome. International journal of oncology. 2013;42:912-20
10. Olesen SH, Christensen LL, Sørensen FB, Cabezón T, Laurberg S, Ørntoft TF. et al. Human FK506 binding protein 65 is associated with colorectal cancer. Molecular & Cellular Proteomics. 2005;4:534-44
11. Lin Jf, Xu J, Tian Hy, Gao X, Chen Qx, Gu Q. et al. Identification of candidate prostate cancer biomarkers in prostate needle biopsy specimens using proteomic analysis. International journal of cancer. 2007;121:2596-605
12. Li Z, Zhu W, Xiong L, Yu X, Chen X, Lin Q. Role of high expression levels of STK39 in the growth, migration and invasion of non-small cell type lung cancer cells. Oncotarget. 2016
13. Krämer O, Knauer S, Zimmermann D, Stauber R, Heinzel T. Histone deacetylase inhibitors and hydroxyurea modulate the cell cycle and cooperatively induce apoptosis. Oncogene. 2008;27:732-40
14. Jung KH, Noh JH, Kim JK, Eun JW, Bae HJ, Xie HJ. et al. HDAC2 overexpression confers oncogenic potential to human lung cancer cells by deregulating expression of apoptosis and cell cycle proteins. J Cell Biochem. 2012;113:2167-77
15. Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G. et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283-98
16. Byun SW, Chang YJ, Chung IS, Moss SF, Kim SS. Helicobacter pylori decreases p27 expression through the delta opioid receptor-mediated inhibition of histone acetylation within the p27 promoter. Cancer Lett. 2012;326:96-104
17. Yang H, Salz T, Zajac-Kaye M, Liao D, Huang S, Qiu Y. Overexpression of histone deacetylases in cancer cells is controlled by interplay of transcription factors and epigenetic modulators. Faseb j. 2014;28:4265-79
18. Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78:67-74
19. Charron CE, Chou P-C, Coutts DJ, Kumar V, To M, Akashi K. et al. Hypoxia-inducible factor 1α induces corticosteroid-insensitive inflammation via reduction of histone deacetylase-2 transcription. Journal of Biological Chemistry. 2009;284:36047-54
20. Zhu P, Martin E, Mengwasser J, Schlag P, Janssen K-P, Göttlicher M. Induction of HDAC2 expression upon loss of APC in colorectal tumorigenesis. Cancer Cell. 2004;5:455-63
21. Chuang J-Y, Wang Y-T, Yeh S-H, Liu Y-W, Chang W-C, Hung J-J. Phosphorylation by c-Jun NH2-terminal kinase 1 regulates the stability of transcription factor Sp1 during mitosis. Molecular biology of the cell. 2008;19:1139-51
22. Solassol J, Mange A, Maudelonde T. FKBP family proteins as promising new biomarkers for cancer. Curr Opin Pharmacol. 2011;11:320-5
23. Kawana H, Tamaru J-i, Tanaka T, Hirai A, Saito Y, Kitagawa M. et al. Role of p27 Kip1 and cyclin-dependent kinase 2 in the proliferation of non-small cell lung cancer. Am J Pathol. 1998;153:505-13
24. Yu S-L, Chen H-Y, Chang G-C, Chen C-Y, Chen H-W, Singh S. et al. MicroRNA signature predicts survival and relapse in lung cancer. Cancer Cell. 2008;13:48-57
25. Chen Z, Zeng H, Guo Y, Liu P, Pan H, Deng A. et al. miRNA-145 inhibits non-small cell lung cancer cell proliferation by targeting c-Myc. J Exp Clin Cancer Res. 2010;29:151
26. Noh JH, Chang YG, Kim MG, Jung KH, Kim JK, Bae HJ. et al. MiR-145 functions as a tumor suppressor by directly targeting histone deacetylase 2 in liver cancer. Cancer Lett. 2013;335:455-62
27. Campayo M, Navarro A, Viñolas N, Diaz T, Tejero R, Gimferrer JM. et al. Low miR-145 and high miR-367 are associated with unfavourable prognosis in resected nonsmall cell lung cancer. European Respiratory Journal. 2013;41:1172-8
Author contact
![]() Corresponding author: Qiang Lin, 100 Haining Rd, Shanghai 200080, China; Tel: +86-021-36123602, Fax: +86-021-63240825. Email: qlin116com.
Corresponding author: Qiang Lin, 100 Haining Rd, Shanghai 200080, China; Tel: +86-021-36123602, Fax: +86-021-63240825. Email: qlin116com.