Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results and Discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Materials and Methods
Results and Discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(11):2924-2939. doi:10.7150/thno.20094 This issue Cite
Research Paper
Al18F-Labeling Of Heat-Sensitive Biomolecules for Positron Emission Tomography Imaging
Frederik Cleeren1, Joan Lecina1, Muneer Ahamed1, Geert Raes2, 3, Nick Devoogdt4, Vicky Caveliers4, Paul McQuade5, Daniel J Rubins5, Wenping Li5, Alfons Verbruggen1, Catarina Xavier4, Guy Bormans1 ![]()
1. Laboratory for Radiopharmaceutical research, Department of Pharmacy and Pharmacology, University of Leuven, Leuven, Belgium;
2. Laboratory of Cellular and Molecular Immunology, Vrije Universiteit Brussel, Brussels, Belgium;
3. VIB Laboratory of Myeloid Cell Immunology, Vrije Universiteit Brussel, Brussels, Belgium;
4. In Vivo Cellular and Molecular Imaging Center, Vrije Universiteit Brussel, Brussels, Belgium;
5. Translational Biomarkers, Merck Research Laboratories, Merck & Co., 770 Sumneytown Pike, West Point, Pennsylvania 19486, United States.
Received 2017-3-15; Accepted 2017-6-2; Published 2017-7-14
Citation:
Cleeren F, Lecina J, Ahamed M, Raes G, Devoogdt N, Caveliers V, McQuade P, Rubins DJ, Li W, Verbruggen A, Xavier C, Bormans G. Al18F-Labeling Of Heat-Sensitive Biomolecules for Positron Emission Tomography Imaging. Theranostics 2017; 7(11):2924-2939. doi:10.7150/thno.20094. https://www.thno.org/v07p2924.htm
Other stylesAbstract

Positron emission tomography (PET) using radiolabeled biomolecules is a translational molecular imaging technology that is increasingly used in support of drug development. Current methods for radiolabeling biomolecules with fluorine-18 are laborious and require multistep procedures with moderate labeling yields. The Al18F-labeling strategy involves chelation in aqueous medium of aluminum mono[18F]fluoride ({Al18F}2+) by a suitable chelator conjugated to a biomolecule. However, the need for elevated temperatures (100-120 °C) required for the chelation reaction limits its widespread use. Therefore, we designed a new restrained complexing agent (RESCA) for application of the AlF strategy at room temperature.
Methods. The new chelator RESCA was conjugated to three relevant biologicals and the constructs were labeled with {Al18F}2+ to evaluate the generic applicability of the one-step Al18F-RESCA-method.
Results. We successfully labeled human serum albumin with excellent radiochemical yields in less than 30 minutes and confirmed in vivo stability of the Al18F-labeled protein in rats. In addition, we efficiently labeled nanobodies targeting the Kupffer cell marker CRIg, and performed µPET studies in healthy and CRIg deficient mice to demonstrate that the proposed radiolabeling method does not affect the functional integrity of the protein. Finally, an affibody targeting HER2 (PEP04314) was labeled site-specifically, and the distribution profile of (±)-[18F]AlF(RESCA)-PEP04314 in a rhesus monkey was compared with that of [18F]AlF(NOTA)-PEP04314 using whole-body PET/CT.
Conclusion. This generic radiolabeling method has the potential to be a kit-based fluorine-18 labeling strategy, and could have a large impact on PET radiochemical space, potentially enabling the development of many new fluorine-18 labeled protein-based radiotracers.
Keywords: Al18F-labeling, PET, human serum albumin, nanobody, affibody.
Introduction
Positron emission tomography (PET) provides non-invasive, sensitive and specific imaging of molecular interactions in vivo, and can be used to study the function of cells, receptors, neurotransmitters, genes and drug pharmacokinetics [1]. Among positron-emitting radioisotopes, the halogen fluorine-18 is the most commonly used radionuclide due to its optimal chemical properties and favorable nuclear decay characteristics [2]. The low maximum positron energy (0.635 MeV) of fluorine-18 enables the acquisition of high resolution PET images as a result of the short range of tissue penetration prior to annihilation [3, 4]. Fluorine-18 has a half-life of 109.8 min, which is short enough to avoid prolonged irradiation of subjects, yet still long enough to allow extended in vivo investigations and distribution to remote imaging centres without an on-site cyclotron [2, 5]. The success and medical impact of PET using 2-[18F]fluoro-2-deoxy-D-glucose ([18F]FDG), led to a fast worldwide proliferation of PET camera's and fluorine-18 producing cyclotron centres [6]. Consequently, fluorine-18 is now readily available in numerous hospitals and research centres, and the infrastructure is in place to expand its application far beyond [18F]FDG [7].
In addition to using small organic molecules, researchers in the molecular imaging community are increasingly interested in using peptides and high molecular weight biomolecules for use as PET-radiopharmaceuticals, in line with the growth of the share of biologic- based drugs in the global pharmaceutical market [8]. The molecular imaging technique, immuno-PET, combines the high in vivo target affinity and selectivity of antibodies or antibody fragments and the high resolution and sensitivity of PET for imaging ligand-target interactions. An immuno-PET tracer should exhibit a high target-to-background ratio, high target specificity, high stability, sufficient solubility and low immunogenicity [9]. The advantage of using mAbs (150 kDa) as vector molecules in immuno-PET is their (sub)nanomolar affinity, excellent specificity and high accumulation at the target site [10]. Moreover, the availability of a large number of FDA-approved mAbs for imaging purposes can stimulate clinical translation [9]. However, as a result of the long residence time in blood of full-length antibodies, imaging with sufficient contrast between target and non-target tissues can only be performed several days post-injection, with peak contrast generally obtained after 2-4 days. Hence, due to the slow kinetics, radiolabeling with long-lived radionuclides such as zirconium-89 (89Zr, t1/2: 3.3 days) or iodine-124 (124I, t1/2: 4.2 days) is required, causing a high radiation burden for the patient (20-40 mSv per scan). The use of long-lived radionuclides is impractical for routine clinical use and, moreover, has a severe social impact on patients as only limited visiting time of family is advised for radioprotection reasons.
Advancements in biotechnology have led to the bioengineering of many vector molecules with shorter biological half-lives, and compatible with a short-lived PET radionuclide such as fluorine-18, that can be used as an alternative to mAbs [11]. Examples of suitable vector molecules are 80-kDa minibodies [12], 55-kDa antigen-binding fragments (Fab) [13], 28-kDa single-chain variable fragment (scFv) [14], 18-kDa designed ankyrin repeat proteins (DARPins) [15], 15-kDa antibody fragments derived from heavy-chain only antibodies (VHH or nanobodies) [16], 7-kDa affibody molecules [17], 7-kDa albumin-binding domain derived affinity proteins (ADAPTs) [18] and many others [11]. These radiolabeled vector molecules are expected to play an increasingly important role in cancer diagnosis, treatment selection, and monitoring of molecularly targeted therapeutics. However, the incorporation of fluorine-18 into heat-sensitive and complex biomolecules creates substantial challenges for radiochemists, and so only a limited number of methods are currently available.
Conventional radiofluorination strategies involve carbon-fluorine bond formation in anhydrous aprotic solvents using aliphatic or aromatic nucleophilic substitution reactions, generally at high temperatures (Figure 1A). A solution to overcome the complications caused by the low nucleophilicity of [18F]F- in aqueous media is the use of prosthetic groups (Figure 1B). Fluorine-18 labeled prosthetic groups (or secondary labeling agents) are small and simple fluorine-18 containing molecules that facilitate the radiolabeling process of complex molecules [19]. Some well-known prosthetic groups are N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB) [20], 6-[18F]fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester ([18F]F-Py-TFP) [21] and N-[2-(4-[18F]fluorobenzamido)ethyl]maleimide ([18F]FBEM) [22]. This approach avoids exposing the biomolecule to harsh conditions, but intrinsically requires time-consuming synthetic procedures that often include several purification steps (e.g. HPLC). Hence, there is a need for innovative, efficient radiolabeling methods that will boost the use of fluorine-18 labeled biomolecules in PET research and clinical studies.
Several non-carbon elements favour strong bonds (i.e. high bond enthalpy) with fluorine, with preservation of high kinetic stability of the fluorine containing compounds formed. Moreover, lower activation energy is required for formation of these bonds in comparison to carbon-fluorine bonds [8]. This has been the basis for a new research field in 18F-radiochemistry. Currently, boron, silicon and aluminum are being investigated as elements allowing fluorination in an aqueous environment. An excellent review by Bernard-Gauthier et al. deals with the current status of both the 18F-SiFA and 18F-BF3 technologies for fluorine-18 labeling, and their use in preclinical PET imaging [23]. The strength of the inorganic approach is that it can entail a single, potentially very fast, radiochemical reaction step in, at least partially, aqueous media [8], that can be easily automated for routine production.
Figure 1
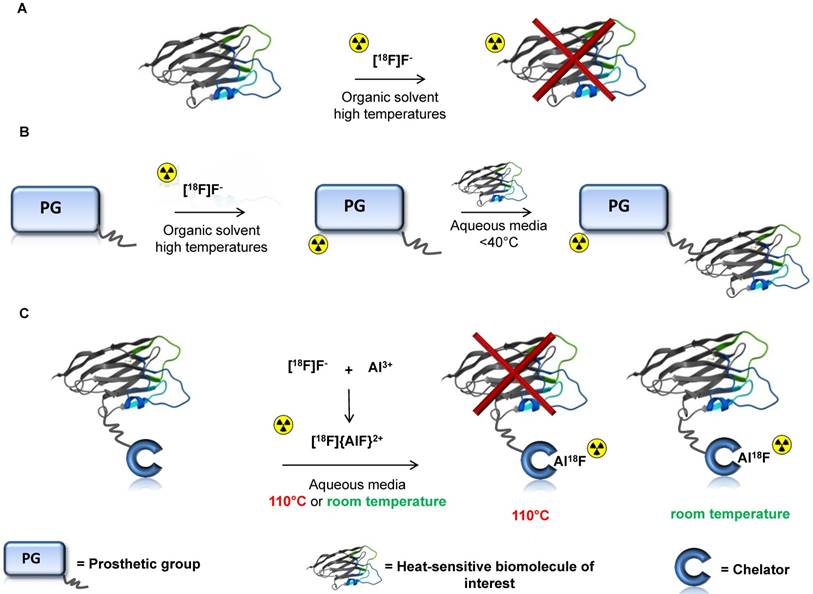
Overview of fluorine-18 radiolabeling methods for heat-sensitive biomolecules A: Conventional radiofluorination strategies in anhydrous aprotic solvents using aliphatic or aromatic nucleophilic substitution reactions at high temperatures are not compatible with heat-sensitive biomolecules B: Fluorine-18 labeled prosthetic groups facilitate the radiolabeling process of complex molecules. This approach avoids exposing the biomolecule to harsh conditions but intrinsically requires a time consuming synthetic procedure to obtain the radiolabeled prosthetic group that often includes several purification steps C: One-pot radiolabeling of biomolecules with fluorine-18 via the Al18F-method at 110 °C using macrocyclic Al18F-chelators (not compatible with heat-sensitive biomolecules) or at room temperature using a restrained complexing agent (RESCA) as Al18F-chelator.
Al18F-chelation is a relatively new strategy that allows one-pot radiofluorination of biomolecules in aqueous media [24]. In this method, fluorine is firmly bound to Al3+ (>670 KJ/mol), forming aluminum mono[18F]fluoride ([18F]{AlF}2+), and this moiety is subsequently chelated by a suitable chelator conjugated to a biomolecule (Figure 1C) [25].
A wide range of peptides have been labeled using the Al18F-method with macrocyclic chelators such as the pentadentate (N3O2-donor) ligand 1,4,7-triazacyclononane-1,4-diacetate (NODA) and the hexadentate (N3O3-donor) ligand 1,4,7-triazacyclononane-1,4,7-triacetate (NOTA) [26]. This has allowed radiolabeling with [18F]{AlF}2+ of relatively small peptides such as αvβ3 integrin-binding peptides [27], and also larger peptides, such as exendin-4 [28] and affibodies [29]. The first clinical study using peptides labeled via the Al18F-method was published in 2013 [30]. Although the macrocyclic chelators used in these methods have shown a lot of potential, the approach has limited applicability due to the high temperature required for the complexation reaction (≥ 100 °C) [31]. These harsh reaction conditions are clearly not compatible with heat-sensitive biomolecules (Figure 1C).
Recently, we developed a new restrained complexing agent, ((±)-H3RESCA), that allows efficient chelation of [18F]{AlF}2+ using mild labeling conditions. (±)-H3RESCA is an acyclic pentadentate ligand with an N2O3 coordinative set of donor atoms. As a result, (±)-H3RESCA showed excellent labeling properties at room temperature and its Al18F-complex demonstrated high in vitro and in vivo stability. Indeed, the development of this new chelator makes it possible to radiolabel heat-sensitive biomolecules with fluorine-18 in one radiolabeling step (Figure 1C). The synthesis of this new chelator, in addition to the synthesis of bifunctional derivatives, in depth characterization of the corresponding Al18F-complex, and evaluation of in vitro and in vivo stability of the Al18F-complex, is described in a patent [32] and will be reported elsewhere. Here we demonstrate the generic potential of the Al18F-RESCA-method by labeling human serum albumin, a nanobody targeting the Kupffer cell marker CRIg, and an affibody targeting HER2 (PEP04314).
Materials and Methods
General
All reagents and solvents were purchased from Sigma-Aldrich (Bornem, Belgium), Fluka (Bornem, Belgium), Fisher (Doornik, Belgium) or Acros Organics (Geel, Belgium). The synthesis of (±)-H3RESCA and synthesis of bifunctional derivatives is described in patent WO/2016/065435 [32]. All buffers used for derivatization and labeling of proteins were chelexed (chelex, 100 sodium form (Sigma Aldrich), 2 g/l, 30 min stirring at rt and filtration with a 0.45 µm polyamide filter (Sartorius Stedim Biotech, Göttingen, Germany)). Fluorine-18 was produced on site using a cyclotron (IBA Cyclone 18/9, IBA, Louvain-la-Neuve, Belgium) by irradiation of H218O with 18-MeV protons. Radioactivity was measured using an ionization chamber-based activity meter (Capintec Radioisotope Calibrator CRC-721, Ramsey, NJ, USA).
A Dionex Ultimate 3000 UPLC System (Thermo Fisher Scientific, Sunnyvale, USA) coupled in series to a UV detector, a 3-inch NaI(Tl) radioactivity detector, and an ultra-high resolution time-of-flight mass spectrometer with electron spray ionization (ESI) (MaXis Impact, Bruker, Bremen, Germany) was used for analysis of human serum albumin and nanobody constructs. For HSA, a massPREP on-line desalting cartridge (2.1 mm x 10 mm, Waters, Milford, USA) was applied using the following method. Solvent A (H2O, 0.1% HCOOH) and solvent B (acetonitrile, 0.1% HCOOH), flow rate 0.4 ml/min. The elution gradient was: 0-1 min: 95% A; 1-3 min: from 95% A to 20% A; 3-5 min: 95% A. For nanobody constructs, a Zorbax SB-C3 column (1.8 µm, 3.0 mm x 100 mm, Agilent) was used with following gradient for (radio)-LC-HRMS: Solvent A (H2O, 0.1% HCOOH) and solvent B (acetonitrile, 0.1% HCOOH), flow rate 0.3 ml/min. The elution gradient was: 0-1 min: 99% A; 1-7 min: from 99% A to 1% A; 7-10 min: 1% A; 10.1-12 min: 99% A. UV monitoring of the eluate was performed at 280 nm. For deconvolution analysis of the raw mass spectral data, the software program DataAnalysis (Bruker Daltonik, Bremen, Germany) was used. Calculated average neutral molecular ion mass values were obtained using Compas Isotope Pattern (version 3.2, Bruker) software.
Autoradiography was performed using phosphor storage screens (super-resolution screen, Perkin Elmer, Waltham, USA). Screens were read in a Cyclone Plus system (Perkin Elmer), and images were analyzed using Optiquant software (Perkin Elmer). Size-exclusion chromatography (SEC) was carried out with a system consisting of a Merck-Hitachi L-6200 pump and a Merck Hitachi L-4200 UV-VIS detector coupled in series to a 3-inch NaI(Tl) radioactivity detector on a Superdex 200 10/300 GL column (for HSA constructs), or on a Superdex 75 10/300 GL column (for nanobody constructs) (GE Healthcare Bio-Science AB, Uppsala, Sweden) using sodium phosphate buffer (0.01 M, pH 7.4, 0.14 M NaCl), as eluent at a flow rate of 0.5 mL/min. CRIg-deficient mice (C57Bl/6 background) were generously provided by Genentech. V4m119 nanobodies were generated and produced as described previously [33].
Synthesis of (±)-H3RESCA-HSA
A solution of (±)-H3RESCA-tetrafluorophenyl ester ((±)-H3RESCA-TFP, 80 µL, 5.64 x 10-6 moles) in DMSO was added in a 15-molar excess to a solution of human serum albumin (1.5 ml, 3.7609 x 10-7 moles, CAF-DCF, Brussels, Belgium) in 0.05 M sodium bicarbonate pH 8.6, and the mixture was incubated for 2 h at rt. The conjugate was purified by gel filtration in sodium acetate buffer (0.1 M, pH 4.5, PD-10 column, GE Healthcare Bio-Science AB), and the concentration of (±)-H3RESCA-HSA was determined spectrophotometrically at 280 nm (Nanodrop 2000, Thermo Fisher scientific). The purified product was analyzed by SEC. UV detection of the eluate was performed at 280 nm. The chelator-to-protein ratio was estimated by ESI-TOF-HRMS analysis.
Radiosynthesis and purification of (±)-[18F]AlF(RESCA)-HSA
After production in a cyclotron, [18F]F- was separated from [18O]H2O by trapping on a SepPak Light Accel plus QMA anion exchange cartridge (Cl- form; Waters). The cartridge was washed with water (3 mL, HPCE grade, Sigma-Aldrich), [18F]F- (3.9 GBq) was eluted from the cartridge with an aqueous solution of sodium chloride 0.9% (0.3 mL, Mini-Plasco, solution for injection, B. Braun, Diegem, Belgium), and the eluate was diluted with water (0.45 mL, HPCE grade, Sigma-Aldrich). This [18F]F- solution (250 µl, 1.25 GBq) was added to 25 µL of 2 mM aluminum chloride (AlCl3) in sodium acetate buffer (0.1 M, pH 4.5). The solution was incubated at rt for 5 min to form {Al18F}2+. A solution of (±)-H3RESCA-HSA (0.5 mL, 75 nmoles) in sodium acetate buffer (0.1 M, pH 4.5) was added to the freshly prepared {Al18F}2+ solution, and was incubated at room temperature (20-22 °C) for 12 min. The pH of the final reaction mixture was checked, and was found to be pH 4.6. After incubation, the mixture was purified by gel filtration with phosphate buffered saline (PBS, 0.1 M pH 7.4, PD-10 column, GE Healthcare Bio-Science AB). Radiochemical yield was calculated based on the amount of radioactivity eluted from the column relative to the starting activity of [18F]F-. Radiochemical identity and purity were assessed by SEC. The identity of the main compound was confirmed using non-radioactive HSA as reference material.
Synthesis of (±)-H3RESCA-NbV4m119
A solution of (±)-H3RESCA-TFP (23 µl, 1.4294 x 10-6 moles) in DMSO was added in 12-molar excess to a solution of NbV4m119 (2.5 ml, 1.1912 x 10-7 moles,) in 0.05 M sodium bicarbonate pH 8.6, and the mixture was incubated for 3 h at room temperature. The conjugate was purified by gel filtration with a sodium acetate buffer (0.1 M, pH 4.5, PD-10 column, GE Healthcare Bio-Science AB), and the concentration of (±)-H3RESCA-NbV4m119 was determined spectrophotometrically at 280 nm (Nanodrop 2000, Thermo Fisher scientific) using the corrected extinction coefficient (ε = 35,425 M-1cm-1). Analysis of the purified compound was performed by SEC. The chelator-to-protein ratio was estimated by ESI-TOF-HRMS analysis.
Radiosynthesis and semi-automated purification of (±)-[18F]AlF(RESCA)-NbV4m119
[18F]F- was separated from [18O]H2O by trapping on a SepPak Light Accel plus QMA anion exchange cartridge (Cl- form; Waters). The cartridge was washed with water (3 mL, HPCE grade, Sigma-Aldrich), [18F]F- was eluted from the cartridge with an aqueous solution of sodium chloride 0.9% (0.3 mL, Mini-Plasco, solution for injection, B. Braun), and the eluate was diluted with water (2.7 ml, HPCE grade, Sigma-Aldrich). The [18F]F- solution (0.5 ml) was added to 12.5 µL of 2 mM aluminum chloride (AlCl3) in sodium acetate buffer (0.1 M, pH 4.5). The solution was incubated at rt for 5 min to form {Al18F}2+. A solution of (±)-H3RESCA-NbV4m119 (1.5 mL, 0.55 mg/ml) in sodium acetate buffer (0.1 M, pH 4.5) was added to the freshly prepared {Al18F}2+ solution and the mixture was incubated at ambient temperature (20-22 °C) for 12 min. After incubation, the mixture was purified using four Hitrap desalting columns in series (GE Healthcare Bio-Science AB) using sodium phosphate buffer (0.01 M, pH 7.4, 0.14 M NaCl) as eluent at a flow rate of 1 mL/min. A UV detector and 3-inch NaI(Tl) radioactivity detector were coupled in series to be able to collect the purified product and to calculate the radiochemical yield. The collected fraction was filtered through a 0.22-µm filter (Millex-GV, 13 mm, Merck KGaA, Darmstadt, Germany), and diluted with sodium phosphate buffer (0.01 M, pH 7.4, 0.14 M NaCl). Radiochemical identity and purity were assessed by SEC and by radio-LC-HRMS. The identity of the main compound was confirmed using unlabeled NbV4m119 as reference material.
In vitro stability studies
A volume of 100 µL of purified (±)-[18F]AlF(RESCA)-HSA (22 MBq) or (±)-[18F]AlF(RESCA)-NbV4m119 (24 MBq) was added to freshly isolated rat plasma (1 mL). The mixtures were incubated at 37 °C and analysed at 1, 2, 3, and 4 h after the start of incubation by SEC.
Biodistribution and microPET studies with (±)-[18F]AlF(RESCA)-HSA and (±)-[18F]AlF(RESCA)-NbV4m119
Quantification of radioactivity for biodistribution studies was performed using an automated gamma counter equipped with a 3-inch NaI(Tl) well crystal coupled to a multichannel analyser, mounted in a sample changer (Perkin Elmer 1480 Wizard 3q). Counts were corrected for background radiation, physical decay and counter dead time. Animals were housed in individually ventilated cages in a thermo-regulated (~22 °C), humidity-controlled facility under a 12h-12h light-dark cycle, with access to food and water ad libitum. All animal experiments were conducted according to the Belgian code of practice for the care and the use of animals, after approval from the university animal ethics committee.
The biodistribution study of (±)-[18F]AlF(RESCA)-HSA was carried out in healthy female Wistar rats (body weight: 187-225 g) at 1 h, 3 h, and 6 h post injection (p.i.; n = 4/time point). Rats were injected with (±)-[18F]AlF(RESCA)-HSA (2-7.5 MBq, 17-62 µg) via a tail vein under anesthesia (2.5% isoflurane in O2 at 1 L/min flow rate), and sacrificed by decapitation at above specified time points. The biodistribution study of (±)-[18F]AlF(RESCA)-NbV4m119 was carried out in healthy male C57BL/6 mice (body weight: 22-26 g) at 1 h and 3 h p.i. (n = 3/time point) or in CRIg-deficient mice (CRIg-/-, C57BL/6 background, body weight: 20-23 g) at 3 h p.i. (n = 3/time point). Mice were injected with freshly filtered (0.22 µm, Millipore) (±)-[18F]AlF(RESCA)-NbV4m119 (7-11 MBq, 8-13 µg ) via a tail vein under anesthesia (2.5% isoflurane in O2 at 1 L/min flow rate), and sacrificed by decapitation at above specified time points. Blood and major organs were collected in tared tubes and weighed. The radioactivity in blood, organs, and other body parts was counted using an automated γ-counter. Results are presented as standardized uptake values (SUV; tissue activity (MBq/g)/[injected dose (MBq)/body weight (g)]).
Small animal whole-body PET imaging was performed using a FOCUSTM 220 microPET scanner (Concorde Microsystems, Knoxville, US), and PMOD software (v 3.2, PMOD Technologies, Zürich, Switzerland) was used to analyze the images. The animals were kept under gas anaesthesia during the entire procedure (2.5% isoflurane in O2 at 1 L/min flow rate). A whole-body PET image of a healthy rat was obtained 180 min after intravenous injection of (±)-[18F]AlF(RESCA)-HSA (96 MBq, 798 µg). A 60-min dynamic microPET scan was performed on a healthy male C57BL/6 mouse (body weight: 24 g) and CRIg-deficient mouse (CRIg-/-, C57BL/6 background, body weight: 23 g) simultaneously. The scan was started immediately after intravenous injection of sterile filtered (0.22 µm, Millipore) (±)-[18F]AlF(RESCA)-NbV4m119 (11 MBq, 13 µg).
Synthesis of (±)-H3RESCA-PEP04314 and H2NOTA-PEP04314
Five mg of lyophilized PEP04314 (0.71 µmol, Affibody AB, Solna, Sweden) was added to 0.5 mL of 0.2 M ammonium acetate buffer (pH 7.5), and the solution was added to a reaction vial containing 0.4 mg (0.72 µmol) of (±)-H3RESCA-maleimide or maleimide-mono-amide-NOTA (Macrocyclics, Plano, Tx, USA). The reaction mixture was kept at room temperature for 90 min before being transferred to an ultracel 3K centrifugal filter (Merck) containing 3 mL of 0.1 M ammonium acetate buffer (pH 4) and centrifuged at 4000 rpm for 90 min. The flow-through was discarded, and 4 mL of fresh 0.1 M ammonium acetate buffer (pH 4) was added. The filter was then centrifuged again for 90 min, and the flow-through was discarded. Purified (±)-H3RESCA-PEP04314 or H2NOTA-PEP04314 was collected via reverse spin in 1 mL of 0.1 M ammonium acetate buffer (pH 4.5), and stored at -70 °C in 100 µL aliquots prior to use. Purity of the final product was determined via a Waters Acquity LC/MS system equipped with a Waters Xselect CSH C18 column (250 mm ×10 mm, 130 Å) at a flow rate of 5 mL/min using a gradient of EtOH (10-40% over 15 min) and 0.1% formic acid.
Radiosynthesis (±)-[18F]AlF(RESCA)-PEP04314 and [18F]AlF(NOTA)-PEP04314
A SepPak Light Accel plus QMA anion exchange cartridge (HCO3- form; Waters) containing [18F]-fluoride was first washed with 1.5 mL of ultrapure water (Water Ultra Trace Elemental Analysis Grade, Fisher Chemical, Pittsburgh, USA), and then [18F]-fluoride was eluted with 1.0 mL of 0.4 M KHCO3. An aliquot of 100 μL of the eluted [18F]-fluoride solution was added to a reaction vial containing 10 μL acetic acid, 50 μL AlCl3 solution (2 mM in 0.1 M NaOAc buffer, pH 4), and 125 μL 0.1 M NaOAc pH 4. The solution was incubated for 2 min at room temperature prior to addition of 0.5 mg (±)-H3RESCA-PEP04314 or H2NOTA-PEP04314 in 400 µL of a 1:1 solution of ethanol and 0.1 M NaOAc pH 4. The reaction mixture was heated for 15 min at 37 °C for (±)-H3RESCA-PEP04314 and at 100 °C for H2NOTA-PEP04314. After heating, the reaction mixture was transferred to a vial containing 0.7 mL of 0.1% formic acid, and mixed and purified by HPLC on a Waters Xselect CSH C18 column (250 mm ×10 mm, 130 Å) at a flow rate of 5 mL/min using a gradient of EtOH (10-40% over 15 min) and 0.1% formic acid. The peak corresponding to (±)-[18F]AlF(RESCA)-PEP04314 or [18F]AlF(NOTA)-PEP04314 was collected and transferred to a sterile vial using normal saline as a rinse to give (±)-[18F]AlF(RESCA)-PEP04314 or [18F]AlF(NOTA)-PEP04314. Specific activity and radiochemical purity were determined via a Waters Acquity LC/MS system on a Waters Xselect CSH C18 column (250 mm ×10 mm, 130 Å) at a flow rate of 5 mL/min using a gradient of EtOH (10-40% over 15 min) and 0.1% formic acid and a β-RAM Model 4 radio-HPLC detector (IN/US Systems, Brandon, FL, USA).
PET/CT studies with (±)-[18F]AlF(RESCA)-PEP04314 and [18F]AlF(NOTA)-PEP04314 in non-human primates
PET studies were conducted in healthy adult rhesus monkeys (n=4, rhesus monkey A-D, weight 7.6 ± 1.8 kg). All procedures were conducted in accordance with the guidelines of the Institutional Animal Care and Use Committee of Merck (West Point, PA) and guidelines for the care and use of mammals in neuroscience and behavioral research (National Research Council 2003). Animals were housed in temperature- and humidity-controlled rooms in fully AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care) accredited facilities and fed a commercially prepared high protein monkey diet (Lab Diet no. 5045, PMI Nutrition International, Brentwood, MO); water was offered ad libitum. Fresh fruits and vegetables were provided daily and animal housing rooms were maintained on a twelve hour light/dark cycle.
Animals were fasted and initially anesthetized with ketamine (10 mg/kg IM), and then maintained under anesthesia using IV propofol (5 mg/kg induction, 0.6 mg/kg/min maintenance). Following the initial induction with propofol, the animal was intubated and ventilated with medical grade compressed air at ~10 mL/breath/kg and 23 respirations per minute. Monkeys were instrumented with a temperature probe, a pulse oximeter and an end tidal CO2 monitor. End tidal CO2 was maintained at 40 ± 2 mm Hg, with body temperature maintained between 37-38 °C using a T-pump warming pump (Gaymar Industrial, Orchard Park, NY) with K-module heating pads (Harvard Apparatus, Holliston, MA)
PET data were acquired on a Siemens Biograph 64 TruePoint PET/CT scanner (Siemens Medical Solutions, Malvern, PA).The PET scanner was cross-calibrated with the dose calibrator using an 18F cylinder according to the manufacturer's standard operating procedures. Following a whole body CT scan used to correct for photon attenuation and scatter, 29-149 MBq ( 9-48 µg) of (±)-[18F]AlF(RESCA)-PEP04314 (n=3; n=1 in rhesus monkey A, n=1 in rhesus monkey B, n=1 in rhesus monkey C) or [18F]AlF(NOTA)-PEP04314 (n=6; n=2 in rhesus monkey A, n=2 in rhesus monkey B, n=2 in rhesus monkey D) was injected as a 2-min bolus IV. A whole body dynamic emission scan was acquired for 180 minutes, encompassing five fields of view (24 static frames). Venous samples to measure blood levels of radiotracer were obtained throughout the scan. Following PET data acquisition, a whole body CT with contrast (~3 ml Omnipaque/kg) was obtained.
PET data were reconstructed using the vendor-suppled iterative method with 4 iterations and 8 subsets. CT-based attenuation and scatter correction were carried out as implemented by the camera manufacturer. Regions of Interest (ROIs) were drawn for liver, kidney cortex, heart blood pool, salivary glands, bladder, lung, and muscle using both the PET and CT images to guide ROI placement, and time activity curves (TACs) were calculated. Bone ROIs were drawn on the lumbar spine. Results are presented as standardized uptake values (SUV; tissue activity (MBq/cm3)/[injected dose (MBq)/body weight (g)]). Additional ROIs were drawn to encompass all of the bladder and kidney, and these were compared to the total radioactivity measured in the image to calculate the percentage injected dose (%ID) for each organ. Radiotracer levels in venous blood samples were fit to a bi-exponential clearance curve, and alpha and beta clearance half-lives in blood were estimated.
Statistical analysis
Quantitative data are expressed as mean ± standard deviation (STDEV) unless stated otherwise. Means were compared using the unpaired two-tailed Student t-test. Values were considered statistically significant for P < 0.05.
Results and Discussion
(±)-[18F]AlF(RESCA)-Human serum albumin
The best method to assess the in vivo stability of a chelate is to attach it to a suitable vector that is slowly cleared from plasma and thus remains in circulation for an extended period of time. Human serum albumin (HSA, 66.5 kDa) is a heat-sensitive globular protein (melting temperature, Tm, of 71°C at 0.5 mg/ml) [34] with a primary sequence made up of 585 amino acid residues, and is the most abundant protein in human plasma (3.5-5 g/dL) [35]. It accounts for about 70% of the plasma colloid osmotic pressure. Its high solubility, stability and plasma half-life of approximately 16-18 h make HSA the ideal vector to evaluate the stability of the Al18F-chelate in vivo. Moreover, the Al18F-labeled construct can potentially be used for PET blood pool imaging applications [36].
HSA possesses 59 lysine residues that can be used for the introduction of chelators via reaction of ɛ-amino groups with an excess of (±)-H3RESCA-TFP (Figure S1). (±)-H3RESCA-TFP ester was efficiently conjugated to HSA providing (±)-H3RESCA-HSA with a chelator-to-protein ratio of 3, estimated by ESI-TOF-HRMS analysis, and no aggregates or degradation products were detected (Figure S2). (±)-H3RESCA-HSA was labeled directly with [18F]{AlF}2+ and a good radiochemical yield (RCY 52-63%) was achieved using mild reaction conditions (rt, 12 min, pH 4.6) compatible with the structural and functional integrity of proteins. (±)-[18F]AlF(RESCA)-HSA was purified using a disposable size-exclusion cartridge, providing the final product in high radiochemical purity, as assessed by size-exclusion chromatography (Figure 2).
This method provided higher radiochemical yields in considerably shorter overall synthesis time than other radiofluorination strategies such as the prosthetic group approach using [18F]F-Py-TFP described by Basuli et al. [36] and the Cu(I)mediated 1,3-dipolar [3+2]cycloaddition reaction reported by Ramenda et al. [37] (Table 1). (±)-[18F]AlF(RESCA)-HSA showed excellent stability in vitro. After 4 h incubation in rat plasma at 37 °C, 92% of the Al18F-tracer was still intact as assessed by size exclusion chromatography (Figure S3).
Figure 2
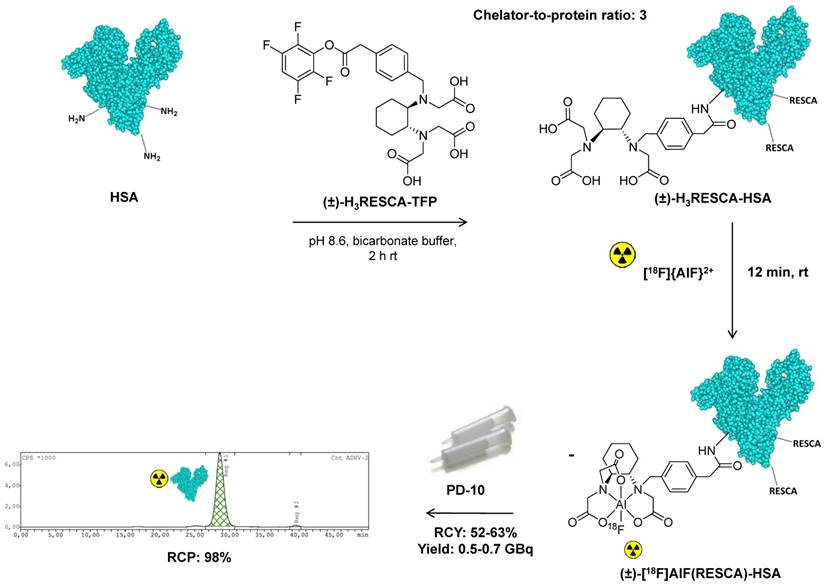
Synthesis and radiolabeling of (±)-H3RESCA-HSA with [18F]{AlF}2+. (±)-H3RESCA-TFP ester was conjugated to HSA providing (±)-H3RESCA-HSA. (±)-[18F]AlF(RESCA)-HSA was prepared in high RCY and RCP, as assessed by size-exclusion chromatography (SEC), in less than 30 min (starting after elution of fluorine-18 from the anion exchange QMA cartridge). SEC-radio-chromatogram of purified (±)-[18F]AlF(RESCA)-HSA with a retention time of 28.5 min. HSA = human serum albumin (PDB ID:1E7H), rt = room temperature (20-25°C), RCY = radiochemical yield (calculated based on the amount of radioactivity eluted from disposable desalting column relative to the starting activity of [18F]F-), RCP = radiochemical purity.
Table 1
Comparison of different radiofluorination methods for labeling the heat-sensitive protein HSA.
| Method | (±)-[18F]AlF(RESCA)-HSA | [18F]F-Py-HSA [36] | Cu(I)mediated 1,3-dipolar [3+2]cycloaddition [37] |
|---|---|---|---|
| RCY* | 52-63% (n=3) | 18-35% (n=30) | 15-20% |
| RCP | > 98% | > 98% | > 95% |
| Synthesis time** | < 30 min | 90 min | 120 min |
| Tb | 8.6 h (in rats) | 4.8 h# (in rats) | 31 min (in mice) |
RCY: radiochemical yield; RCP: radiochemical purity; Tb: blood biological half-life
*Calculated based on the amount of radioactivity eluted from disposable desalting column relative to the starting activity of [18F]F-**Starting from [18F]F- # Data for [18F]F-Py-RSA (rat serum albumin)
Biodistribution of (±)-[18F]AlF(RESCA)-HSA in rats showed high retention in blood with SUV: 11.8 ± 0.6, 10.1 ± 0.7, and 8.0 ± 0.1 at 1 h, 3 h, and 6 h, respectively (Figure 3A, Table S1). Blood data points were fitted with a one- component exponential equation (y = 12.75e(-0.08x); R² = 0.9998), from which the blood biological half-life (Tb) was calculated to be 8.7 h (Figure S4). This indicates that the introduction of RESCA residues into HSA and subsequent radiolabeling using the Al18F-method did not alter the structural and functional integrity of HSA. In contrast, the radiolabeling method using a Cu(I)mediated 1,3-dipolar [3+2]cycloaddition significantly affected functional integrity of HSA, resulting in a Tb of only 31 minutes [37]. Fluorine-18 in the form of fluoride and also [18F]{AlF}2+ are known to accumulate in bone [38, 39]. Bone is a highly vascular tissue [40], so this might explain the observed bone uptake at 1 h p.i. However, only minor increase in bone uptake was observed over time, indicating high in vivo stability of the Al18F-labeled protein conjugate. Moreover, (±)-[18F]AlF(RESCA)-HSA showed favorable properties for PET blood pool imaging applications. A whole body PET image of a healthy rat 180 min after intravenous injection of (±)-[18F]AlF(RESCA)-HSA is shown in Figure 3B. The ventricles of the heart and the peripheral vasculature are well visualized. Other organs can be observed but retain lower concentrations of fluorine-18 than the central vasculature. The tissue concentrations in the microPET study are consistent with the ex vivo biodistribution results at comparable time points.
(±)-[18F]AlF(RESCA)-Nanobody
Nanobodies (Nb, VHH) are heat-sensitive antigen-binding fragments derived from heavy-chain-only antibodies occurring naturally in Camelid species and can be considered as magic bullets for immuno-PET imaging [41]. Nanobodies bind their antigens very fast and specifically with high affinity in vivo, whereas unbound nanobody is rapidly cleared from blood by the kidneys [42]. Using nanobodies labeled with a positron emitting radionuclide, high contrast PET images can be acquired as early as 1 hour after tracer administration [43]. This early imaging allows the use of short-lived PET radionuclides such as fluorine-18. Xavier et al. recently reported successful 18F-labeling of HER2-targeting nanobodies using the prosthetic group [18F]SFB [44]. The same method was used by Blykers et al. and Bala et al. for radiofluorination of nanobodies targeting the macrophage mannose receptor (MMR) [45] and the vascular cell adhesion molecule (VCAM1) [46], respectively.
However, this multistep synthesis is laborious, and only limited yields of purified radiolabeled product were obtained with moderate apparent molar radioactivity (Table 2). We have evaluated the Al18F-method for radiolabeling of the nanobody NbV4m119 [47], targeting the complement receptor of the Ig superfamily (CRIg) [48]. CRIg has a pronounced expression on Kupffer cells, which are resident liver macrophages. Moreover, it has been shown that CRIg is more abundant in rheumatoid arthritis (RA) synovial tissue than in normal synovium [33]. Consequently, CRIg is a promising marker to monitor changes in Kupffer cell dynamics [49] and for specific molecular imaging of RA, allowing non-invasive quantification of joint inflammation [33].
Characterization of a radiolabeled nanobody is important in order to confirm that the derivatization and radiolabeling procedure does not affect the nanobody integrity. We have therefore explored radio-liquid chromatography in combination with high-resolution mass spectrometry (radio-LC-HRMS) to analyze NbV4m119 before and after derivatization with (±)-H3RESCA-TFP ester. The theoretical average neutral molecular mass ([M]) of unlabeled NbV4m119 is 14577.85 Da [M] (calculated for C643H950N188O196S4), and the observed molecular mass after deconvolution was 14577.61 ± 0.02 Da [M] (Figure S5). We also observed the presence of a deamination product with an observed molecular mass of 14560.76 ± 0.02 Da (calculated for C643H947N187O196S4 [M]: 14560.82). This deamination product was expected and can be formed spontaneously during manipulation and storage of the protein.
Figure 3
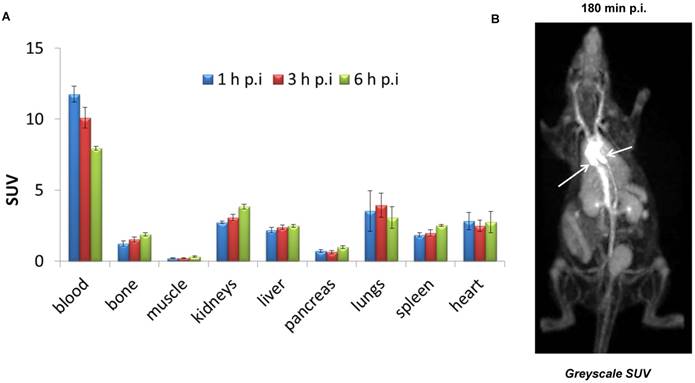
Biodistribution and PET imaging of rats after intravenous injection of (±)-[18F]AlF(RESCA)-HSA. Biodistribution (A) of (±)-[18F]AlF(RESCA)-HSA in rats at 1 h, 3 h, and 6 h p.i. (n=4/group) and whole-body PET image (B) of a healthy rat 180 min after intravenous injection of (±)-[18F]AlF(RESCA)-HSA (greyscale SUV (g/ml): max, white; min, black) White arrows indicate ventricles of the heart. Results of biodistribution are presented as standardized uptake values (SUV; tissue activity (MBq/g)/[injected dose (MBq)/body weight (g)]). Blood data points were fitted with a one- component exponential equation: y = 12.75e(-0.08x); R² = 0.9998, blood biological half-life (Tb) = 8.66 h.
Table 2
Comparison of two radiofluorination methods for labeling of nanobodies
| Method | Al18F-RESCA | [18F]SFB [44] | [18F]SFB [45] | [18F]SFB [46] |
|---|---|---|---|---|
| Target | CRIg | HER2 | MMR | VCAM-1 |
| Nanobody | V4m119 | 2Rs15d | 3.49 sdAb | cAbVCAM-1-5 |
| RCY | 35-53%* (n=3) | 5-15%** (n=3) | 5-15%** | / |
| RCP | > 98% | > 98% | > 97% | > 99% |
| Synthesis time# | < 35 min | 180 min | 180 min | 180 min |
| GBq/µmol ## | 80-85 | 17-33 | 10-30 | / |
| Overall batch activity | 2-2.2 GBq | 0.2- 0.5 GBq | / | / |
RCY: radiochemical yield; RCP: radiochemical purity
*Calculated based on the preparative chromatogram and relative to radioactivity of [18F]F-/[18F]{AlF}2+ **Calculated based on the amount of radioactivity eluted from disposable desalting column relative to the starting activity of [18F]F- #Starting from [18F]F- ##Apparent molar radioactivity at the end of purification
NbV4m119 possesses five lysine residues in its amino acid sequence, therefore several lysine groups can react with the (±)-H3RESCA-TFP active ester via the free amino functionalities (Figure S6). (±)-H3RESCA-TFP ester was efficiently conjugated to NbV4m119 providing (±)-H3RESCA-NbV4m119 with a chelator-to-protein ratio of 1.5, estimated by ESI-TOF-HRMS analysis, and no aggregates or degradation products were observed (Figure S7). The theoretical average neutral molecular masses for single and dual conjugation are 14979.26 Da [M] (calculated for C664H973N189O203S4) and 15397.70 Da [M] (calculated for C685H999N191O210S4), respectively. The observed molecular masses for (±)-H3RESCA-NbV4m119 after deconvolution were 14979.35 ± 0.02 Da and 15397.69 ± 0.02 Da [M], respectively.
(±)-H3RESCA-NbV4m119 was labeled with [18F]{AlF}2+, achieving good radiochemical yields using mild conditions and the crude reaction mixture was purified using four Hitrap desalting columns in series yielding the purified batch (±)-[18F]AlF(RESCA)-NbV4m119 (Table 2 and Figure 4). (±)-[18F]AlF(RESCA)-NbV4m119 was analyzed with radio-RPLC-HRMS and SEC, eluting at a retention time of 5.6 min and 26.9 min, respectively (Figure S8 and Figure 4) to check the radiochemical purity of the radiolabeled nanobody. No aggregation or degradation products were observed. The radiofluorination method provided higher radiochemical yields, higher overall batch activity and higher apparent molar radioactivity in considerably shorter overall synthesis time than the prosthetic group approach using [18F]SFB (Table 2). Analysis with SEC revealed that 96% of the Al18F-tracer was still intact after 6 h incubation in the storage buffer (Figure S9), and 91.5% of the Al18F-tracer was still intact after 3 h incubation in rat plasma at 37 °C, indicating excellent in vitro stability of (±)-[18F]AlF(RESCA)-NbV4m119 (Figure S10).
Since the molecular size of nanobodies is below the threshold for renal glomerular filtration (<60 kDa), nanobodies are generally cleared quickly from blood, primarily via renal clearance. Only 0.20 ± 0.02 and 0.13 ± 0.02 SUV of the radioactivity was observed in blood after 1 h and 3 h p.i. respectively, indicating fast blood clearance (Table S2). It is known that nanobodies are efficiently reabsorbed in the renal proximal tubuli, and Gainkam et al. showed elegantly that the megalin receptor plays an important role in this mechanism using megalin-knockout mice [50]. Kidney retention is also significantly affected by the presence of a hexahistidine tag, which is genetically inserted in the structure of NbV4m119 for the purpose of immobilized metal ion affinity chromatography purification [50].
As expected, high uptake in kidneys (SUV: 37.6 ± 4.5) was observed at 1 h p.i. Figure S11 shows an ex vivo autoradiogram of a renal tissue slice of a naive WT mouse injected with (±)-[18F]AlF(RESCA)-NbV4m119 and sacrificed at 1 h p.i. The radioactivity is mainly concentrated in the renal cortex indicating that the kidney retention may be caused by tubular reabsorption and subsequent internalization. Remarkably, at 3 h p.i. kidney uptake was decreased drastically (SUV: 8.9 ± 2.3) with a corresponding fraction being excreted with the urine, strongly indicating that resulting (radio)catabolites are non-residualizing. Urine analysis shows the presence of mainly radiometabolites of (±)-[18F]AlF(RESCA)-NbV4m119, confirming the hypothesis of renal metabolism followed by elimination of the catabolites via the urine (Figure S12-S13). This high rate of in vivo degradation was also reported previously for 99mTc-labeled nanobodies targeting the epidermal growth factor receptor (EGFR). In this study the authors found that only 10% of 99mTc-labeled nanobody in urine was intact at 1 h p.i. indicating that this degradation is specific for nanobodies rather than dependent on the radiolabeling method [50].
Zheng et al. investigated CRIg mRNA levels in collagen-induced arthritis (CIA) mice to determine whether this marker is expressed homogeneously in the body or specifically in certain tissues during inflammation [33]. High CRIg mRNA levels were found in liver (the CRIg receptor has a pronounced expression on CD11bintermediate F4/801 Kupffer cells in the liver) and low or undetectable quantities in bone marrow, lymph nodes, spleen and lungs. In accordance with these results (±)-[18F]AlF(RESCA)-NbV4m119 showed high uptake in liver (SUV: 2.3 ± 0.2, liver-to-muscle ratio of 33 ± 2.1) at 3 h p.i. in healthy mice, but low uptake in other organs (Figure 5A). In CRIg-/- mice no specific uptake of (±)-[18F]AlF(RESCA)-NbV4m119 could be observed in the liver (SUV: 0.27 ± 0.1, P < 0.001) at 3 h p.i., indicating excellent in vivo CRIg specificity of the tracer. This demonstrates that the Al18F-RESCA labeling method does not affect the functional integrity of nanobodies.
Higher kidney uptake in the CRIg-/- mice at 3 h p.i compared with the kidney uptake in the WT mice at 3 h p.i. (P < 0.005) may be explained by the lower liver uptake in CRIg-/- mice, resulting in higher availability of [18F]AlF-RESCA-NbV4m119 towards the renal elimination pathway. It is important to minimize residence time of (±)-[18F]AlF(RESCA)-NbV4m119 in plastic syringes to avoid aggregation, which might result in increased lung uptake. Probably, aggregation over time in these syringes explains the relative high variation in lung uptake seen in the biodistribution study. Aggregation of proteins is a common problem and might be related to this specific nanobody or labeling method. To overcome this issue, different agents can be added to the final formulation buffer that promote protein solubility (e.g. 0.1% w/v polysorbate 80) [51].
Although we showed excellent in vitro stability of (±)-[18F]AlF(RESCA)-NbV4m119 in rat plasma, the biodistribution study showed an increase of bone uptake over time, indicating minor levels of demetallation and/or defluorination of the compound (SUV: 0.6 ± 0.1 and 1.0 ± 0.3 at 1 h and 3 h p.i., respectively). One should consider the harsh environment in kidney lysosomes as a possible explanation for the observed elevated bone uptake. Indeed, after glomerular filtration, nanobodies are internalized and transported to lysosomes [50]. Here, (±)-[18F]AlF(RESCA) may be degraded, and as a result [18F]{AlF}2+ and/or [18F]F- could be recycled back into circulation, resulting in increased bone uptake values. Non-specific bone accumulation might compromise the utility of this tracer for noninvasive quantification of joint inflammation in vivo.
Figure 4
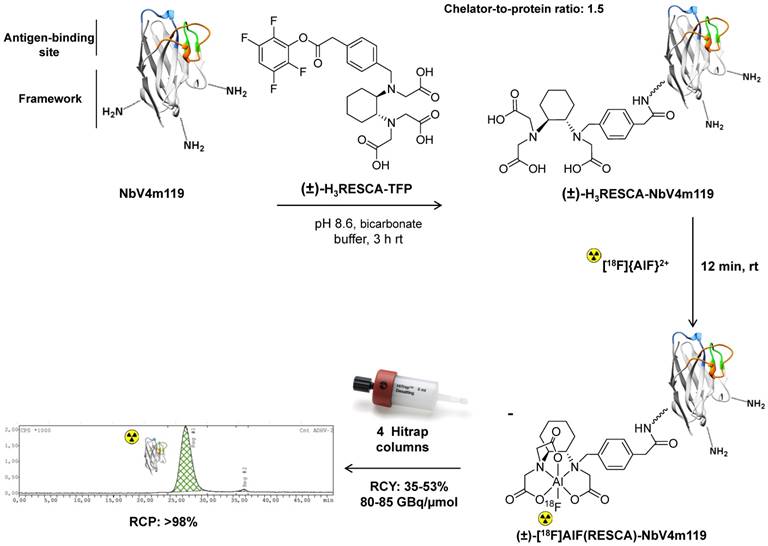
Synthesis and radiolabeling of (±)-H3RESCA-NbV4m119 with [18F]{AlF}2+. (±)-H3RESCA-TFP ester was conjugated to NbV4m119 providing (±)-H3RESCA-NbV4m119. (±)-[18F]AlF(RESCA)-NbV4m119 was prepared in high radiochemical yield. Purification was accomplished with SEC using 4 Hitrap desalting columns in series, affording highly pure (±)-[18F]AlF(RESCA)-NbV4m119 in less than 35 min (starting after elution of fluorine-18 from the anion exchange QMA cartridge). SEC Radio-chromatogram of purified (±)-[18F]AlF(RESCA)-NbV4m119 eluting with a retention time of 26.9 min. rt = room temperature (20-25 °C), RCY = radiochemical yield (calculated based on the preparative chromatogram and relative to radioactivity of [18F]F-/[18F]{AlF}2+), RCP = radiochemical purity.
Figure 5
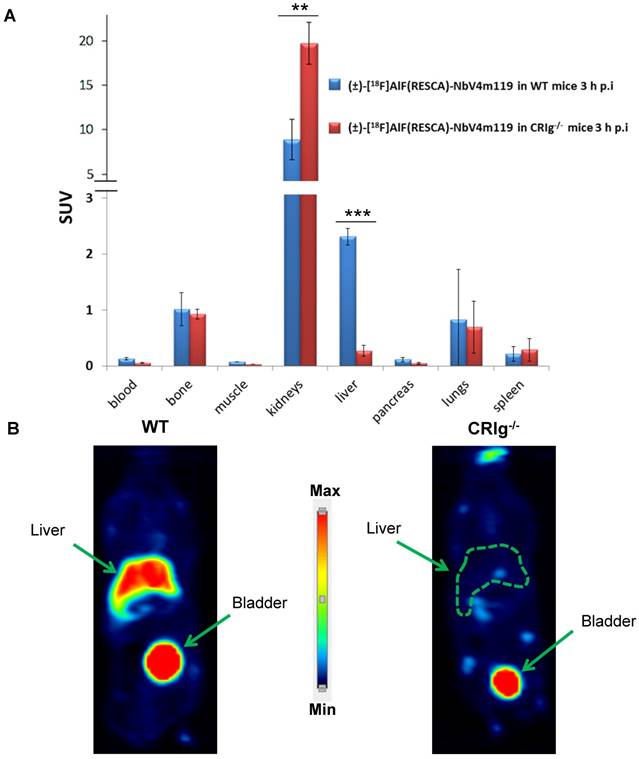
Biodistribution and PET imaging of WT and CRIg-/- mice after intravenous injection of (±)-[18F]AlF(RESCA)-NbV4m119 A: Biodistribution of (±)-[18F]AlF(RESCA)-NbV4m119 in wild-type mice (WT) and CRIg-/- mice (CRIg-/-) at 3 h p.i (n=3/group). Biodistribution results are presented as standardized uptake value (SUV; tissue activity (MBq/g)/[injected dose (MBq)/body weight (g)]). **P < 0.005***P < 0.001 B: Coronal microPET images. Summed images (30-60 min p.i., time weighted average) performed on naive WT and CRIg-/- mice.
Nevertheless, the calculated bone uptake is limited and might be species-dependent. Whole-body PET imaging clearly showed specific uptake of (±)-[18F]AlF(RESCA)-NbV4m119 in the liver, whereas no liver uptake was observed in the CRIg knockout mice, confirming high specificity for CRIg (Figure 5B). In vivo PET imaging did not show major bone uptake after 1 h and high liver-to background images were obtained, indicating favorable in vivo imaging properties of (±)-[18F]AlF(RESCA)-NbV4m119.
[18F]AlF-RESCA-Affibody
Affibody molecules are non-immunoglobulin-based scaffold proteins characterized by high target specificity and binding affinity (Kd in the low-nanomolar to picomolar range) [52, 53]. These small proteins have been generated against different cancer-associated molecular targets and have been labeled with different radionuclides (68Ga, 18F, 64Cu, 99mTc, and 111In) suitable for immuno-PET and SPECT [54-59]. As for nanobodies, the half-life of fluorine-18 perfectly matches the fast pharmacokinetics of affibody molecules [60]. Indeed, the small size of fluorine-18 labeled affibody molecules is responsible for fast clearance, providing high contrast images within one to three hours after injection. Furthermore, introduction of an unpaired cysteine residue at the c-terminus of the protein allows site-specific conjugation of chelators or radiolabeled groups via maleimide-thiol reactions, avoiding heterogeneous tracer populations.
To show the feasibility of radiolabeling affibody molecules using the Al18F-RESCA method, and to further investigate the in vivo stability of the (±)-[18F]AlF(RESCA)-complex, we radiolabeled a generic affibody and conducted PET/CT studies in healthy non-human primates. In general, affibodies are considered to be heat-stable. However, Da Pieve et al. labeled a NOTA-derivatized affibody targeting HER3 with [18F]AlF using standard one-pot-reaction conditions (pH 4, 100 °C for 15 min) [29]. The reported radiochemical conversion and the apparent molar radioactivity of the radioconjugate were 38.8 ± 5.8% (non-decay corrected) and 6.0-11.9 GBq/μmol, respectively. Unfortunately, thermal degradation of the radioconjugate was observed, compromising the final yield of the reaction. As shown by Heskamp et al., good to excellent yields can also be obtained at 90°C for labeling of affibodies that are more sensitive [61]. Nevertheless, direct radiolabeling at low temperature (<40°C) using the Al18F-RESCA method could provide an interesting alternative to avoid possible thermal degradation.
As a proof of concept, we successfully derivatized affibody molecules (PEP04314, also known as ZHER2:2891) targeting HER2, exploiting site-specific maleimide-thiol reactions with (±)-H3RESCA-maleimide to afford (±)-H3RESCA-PEP04314. As a control, we also derivatized affibody molecules site-specifically with maleimide-mono-amide-NOTA to afford H2NOTA-PEP04314. The purity of the final products was determined with LC/MS (Figure S14). (±)-H3RESCA-PEP04314 and H2NOTA-PEP04314 were labeled with [18F]{AlF}2+ in radiochemical yields of respectively 20 ± 7% (n=4) and 8 ± 6% (n=4). Labeling conditions for both compounds were identical (15 min, pH 4, 50% v/v ethanol), except the reaction temperature was 37 °C for (±)-H3RESCA-PEP04314 and 100 °C for H2NOTA-PEP04314. For this particular affibody (PEP04314), no thermal degradation products were observed, even after 15 minutes incubation at 100 °C. The crude reaction mixtures were purified using preparative RP-HPLC to give (±)-[18F]AlF(RESCA)-PEP04314 or [18F]AlF(NOTA)-PEP04314 in high radiochemical purity (> 98% for both compounds) and with apparent molar radioactivity of respectively 23.2 ± 3.4 GBq/µmol and 21.6 ± 5.4 GBq/µmol at the end of purification. Total synthesis time, starting from [18F]F-, was less than 30 minutes for both compounds. PET/CT studies in healthy male non-human primates were performed to evaluate the distribution profile and in vivo stability of both (±)-[18F]AlF(RESCA)-PEP04314 (n=3) and [18F]AlF(NOTA)-PEP04314 (n=6).
Preclinical studies have shown that the nature of the chelator might have a significant effect on the biodistribution of affibody molecules. However this effect is difficult to predict [54]. The attachment of the macrocyclic chelator, NOTA, to affibody molecules involves one of the carboxylic groups of the chelator. As a result, only two carboxyl groups remain available for chelation of [18F]{AlF}2+, forming a neutral complex. In contrast, the RESCA ligand conjugated to affibody molecules has three carboxylic groups available for chelation of [18F]{AlF}2+, resulting in a complex with a negative net charge. This net charge might shift elimination more to renal clearance and affect kidney retention. On the other hand, the more lipophilic nature of RESCA derivative (due to the presence of a trans-cyclohexyl building block and an additional phenyl ring) in comparison with NOTA could shift the elimination of the radiolabeled affibody to hepatic clearance.
PET/CT images of (±)-[18F]AlF(RESCA)- PEP04314 and [18F]AlF(NOTA)-PEP04314 in a healthy rhesus monkey, with the corresponding time-activity curves and chemical structures, are shown in Figure 6. Tissue uptake values (SUV120-180 min) of both compounds are shown in Table S3. Similar distribution profiles were observed for both tracers with slightly slower blood clearance for (±)-[18F]AlF(RESCA)-PEP04314. Radiotracer levels in venous samples were fit to a bi-exponential clearance curve, and alpha and beta clearance half-lives in blood were calculated to be respectively 0.08 ± 0.05 h and 1.09 ± 0.23 h for [18F]AlF(NOTA)-PEP04314 and 0.04 ± 0.01 h and 2.70 ± 0.43 h for (±)-[18F]AlF(RESCA)-PEP04314. The sum of percentage injected dose (%ID) in kidneys and urinary bladder (average 120-180 min p.i) was 28.8 ± 2.4 %ID for (±)-[18F]AlF(RESCA)-PEP04314 and 29.9 ± 3.7 %ID for [18F]AlF(NOTA)-PEP04314. This indicates that the difference in net charge and lipophilicity of the chelator seem to compensate each other, resulting in a similar renal clearance pattern for both compounds.
Higher liver uptake was observed for (±)-[18F]AlF(RESCA)-PEP04314 at early time points, probably due to the slower blood clearance (Figure 6). However, significantly lower retention in kidney cortex was observed for (±)-[18F]AlF(RESCA)-PEP04314 (standardized uptake value 120-180 min after tracer administration (SUV120-180 min) 32.5 ± 8.2) than [18F]AlF(NOTA)-PEP04314 (SUV 120-180 min: 56.8 ± 11.0), and higher accumulation of (±)-[18F]AlF(RESCA)-PEP04314 in the bladder was also observed in the PET/CT images. The %ID120-180 min in bladder was 10.6 ± 0.8 %ID for (±)-[18F]AlF(RESCA)-PEP04314 and only 3.8 ± 1.0 %ID for [18F]AlF(NOTA)-PEP04314. The use of residualizing radiometals, including [18F]AlF(NOTA) complexes [25], results in extended retention of radioactivity in kidneys because these stable radiocomplexes are not able to diffuse out of the proximal tubuli cells. This may cause high radiation burden to the kidneys, hinder diagnostic accuracy, and limit therapeutic applications. Compared to [18F]AlF(NOTA)-PEP04314, (±)-[18F]AlF(RESCA)-PEP04314 shows less activity accumulating in the kidneys due to faster clearance to the bladder. Nonetheless, retention in kidneys is still high compared to non-residualizing 18F-labeling strategies for affibody molecules such as the prosthetic group approach using [18F]FBEM [62]. The residualizing effect is not only important regarding kidney retention, but might also occur in target cells of interest (e.g. cancer cells) if there is a high rate of internalization. In tumor cells the residualizing effect is beneficial, as tumor uptake will be higher and last longer.
Importantly, only minor bone uptake was observed for both tracers, with slightly higher values for (±)-[18F]AlF(RESCA)-PEP04314 (SUV120-180 min 2.3 ± 0.7) than [18F]AlF(NOTA)-PEP04314 (SUV120-180 min 1.0 ± 0.3), indicating high in vivo stability of both tracers in rhesus monkeys. The increasing bone uptake observed for (±)-[18F]AlF(RESCA)-PEP04314 over time indicates that the Al18F-complex formed with the macrocyclic chelator NOTA shows somewhat higher in vivo stability than the Al18F-complex formed with the acyclic chelator RESCA, confirming the trade-off between reactivity of the chelators towards [18F]{AlF}2+ and the in vivo stability of the resulting complexes.
Figure 6
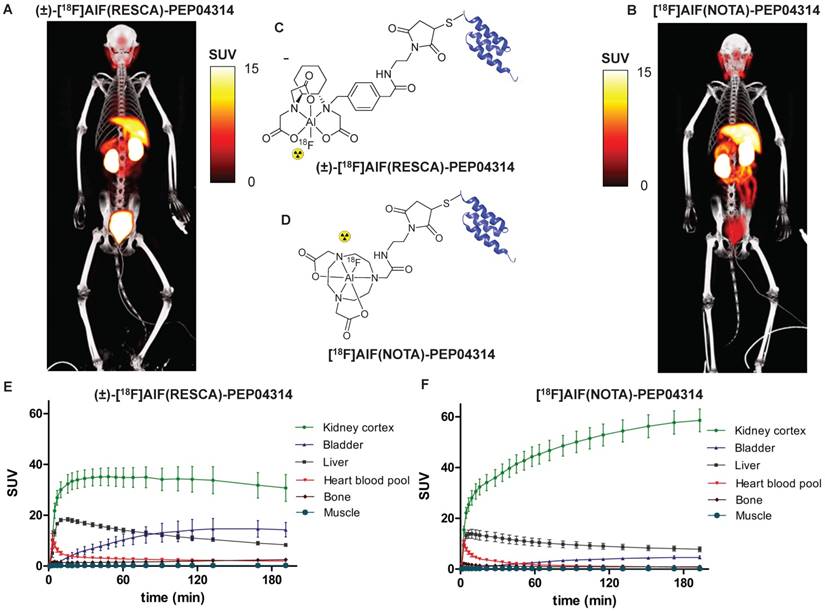
PET/CT imaging of rhesus monkey after intravenous injection of (A) (±)-[18F]AlF(RESCA)-PEP04314 and comparison with (B) [18F]AlF(NOTA)-PEP04314. Summed PET/CT images (120-180 min p.i., time weighted average) performed on healthy rhesus monkey, same animal for both compounds (rhesus monkey A). Structures of (C) (±)-[18F]AlF(RESCA)-PEP04314 and (D) [18F]AlF(NOTA)-PEP04314. Time activity curves (TAC) of different organs after intravenous injection of (E) (±)-[18F]AlF(RESCA)-PEP04314 (n=3) and (F) [18F]AlF(NOTA)-PEP04314 (n=6), data are presented as average standardized uptake values (SUV; tissue activity (MBq/cm3)/[injected dose (MBq)/body weight (g)]) ± SEM.
Conclusions
Fluorine-18 is currently the radionuclide of choice for PET because of its favorable nuclear decay characteristics and high production capacity. In this study, we show that the Al18F-method using the recently developed chelator RESCA provides an efficient one-step approach to radiolabel heat-sensitive biomolecules at low temperature (< 37°C). We successfully labeled human serum albumin with excellent radiochemical yields in less than 30 minutes and confirmed in vivo stability of the Al18F-labeled protein in rats. Moreover, we efficiently labeled nanobodies targeting the Kupffer cell marker CRIg and performed µPET studies in healthy and CRIg deficient mice to demonstrate that the proposed radiolabeling method does not affect the functional integrity of the protein. Finally, an affibody targeting HER2 (PEP04314) was labeled site-specifically, and the distribution profile of (±)-[18F]AlF(RESCA)-PEP04314 in a rhesus monkey was compared with that of [18F]AlF(NOTA)-PEP04314 using whole-body PET/CT. The total synthesis time (starting from [18F]F-) for all compounds was less than 35 minutes, which is considerably shorter than for other radiofluorination methods. Additionally, in vivo PET imaging in rats, mice, and non-human primates showed excellent imaging properties for all tracers. This new optimized radiolabeling method can be considered a breakthrough in fluorine-18 radiochemistry. It is easy to implement and can be generically applied for the development of numerous new fluorine-18 labeled heat-sensitive biomolecules.
Abbreviations
{Al18F}2+: aluminum mono[18F]fluoride; RESCA: Restrained Complexing Agent; PET: Positron emission tomography; [18F]FDG: 2-[18F]fluoro-2-deoxy-D-glucose; [18F]SFB: N-succinimidyl 4-[18F]fluorobenzoate; [18F]F-Py-TFP: 6-[18F]fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester; [18F]FBEM: N-[2-(4-[18F]fluorobenzamido)ethyl]maleimide; NODA: 1,4,7-triazacyclononane-1,4-diacetate; NOTA: 1,4,7-triazacyclononane-1,4,7-triacetate; HSA: Human serum albumin; RCY: radiochemical yield; RCP: radiochemical purity; Nb: nanobody; CRIg: complement receptor of the Ig superfamily; RA: rheumatoid arthritis; SUV: standardized uptake value.
Acknowledgements
The authors thank Julie Cornelis, Ivan Sannen, Pieter Haspeslagh and Jana Hemelaers from the Laboratory for Radiopharmaceutical Research, and Ann Van Santvoort, Tine Buelens, Michel Koole, Andrey Postnov from the Department of Nuclear Medicine. This research received support from IWT Flanders (SBO 130065 MIRIAD) and FWO (G0D8817N).
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Weissleder R. Scaling down imaging: molecular mapping of cancer in mice. Nat Rev Cancer. 2002;2:1-8
2. Serdons K, Verbruggen A, Bormans GM. Developing new molecular imaging probes for PET. Methods. 2009;48:104-111
3. Le Bars D. Fluorine-18 and medical imaging: Radiopharmaceuticals for positron emission tomography. J Fluor Chem. 2006;127:1488-1493
4. Naumova AV, Modo M, Moore A. et al. Clinical imaging in regenerative medicine. Nat Biotechnol. 2014;32:804-818
5. Huyvetter MD, Xavier C, Caveliers V. et al. Radiolabeled nanobodies as theranostic tools in targeted radionuclide therapy of cancer. Expert Opin Drug Deliv. 2014;11:1939-1954
6. Boellaard R, Delgado-Bolton R Oyen W. et al. FDG PET/CT : EANM procedure guidelines for tumour imaging : version 2. Eur J Nucl Med Mol Imaging. 2015;42:328-354
7. Wadsak W, Mitterhauser M. Basics and principles of radiopharmaceuticals for PET / CT. Eur J Radiol. 2010;73:461-469
8. Smith GE, Sladen HL, Biagini SCG. et al. Inorganic approaches for radiolabelling biomolecules with fluorine-18 for imaging with positron emission tomography. Dalton Trans. 2011;40:6196-6205
9. Vaneycken I, D'huyvetter M, Hernot S. et al. Immuno-imaging using nanobodies. Curr Opin Biotechnol. 2011;22:877-881
10. De Meyer T, Muyldermans S, Depicker A. Nanobody-based products as research and diagnostic tools. Trends Biotechnol. 2014;32:263-270
11. Wu AM. Engineered antibodies for molecular imaging of cancer. Methods. 2014;65:139-147
12. Pandit-Taskar N, O'Donoghue JA, Ruan S. et al. First-in-Human Imaging with 89Zr-Df-IAB2M Anti-PSMA Minibody in Patients with Metastatic Prostate Cancer: Pharmacokinetics, Biodistribution, Dosimetry, and Lesion Uptake. J Nucl Med. 2016;57:1858-1864
13. Chakravarty R, Goel S, Valdovinos HF. et al. Matching the Decay Half-Life with the Biological Half-Life: ImmunoPET Imaging with 44Sc-Labeled Cetuximab Fab Fragment. Bioconjug Chem. 2014;25:2197-2204
14. Sharma SK, Wuest M, Way JD. et al. Synthesis and pre-clinical evaluation of an 18F-labeled single-chain antibody fragment for PET imaging of epithelial ovarian cancer. Am J Nucl Med Mol Imaging. 2016;6:185-198
15. Goldstein R, Sosabowski J, Livanos M. et al. Development of the designed ankyrin repeat protein ( DARPin ) G3 for HER2 molecular imaging. Eur J Nucl Med Mol Imaging. 2015;42:288-301
16. Keyaerts M, Xavier C, Heemskerk J. et al. Assessment of HER2 Expression in Breast Carcinoma. J Nucl Med. 2016;57:27-33
17. Sandström M, Lindskog K, Velikyan I. et al. Biodistribution and Radiation Dosimetry of the Anti-HER2 Affibody Molecule 68Ga-ABY-025 in Breast Cancer Patients. J Nucl Med. 2016;57:867-871
18. Garousi J, Lindbo S, Honarvar H. et al. Influence of the N-terminal composition on targeting properties of radiometal-labeled anti- HER2 scaffold protein ADAPT6. Bioconjug Chem. 2016;27:2678-2688
19. Richter S, Wuest M, Bergman CN. et al. Rerouting the metabolic pathway of (18)F-labeled peptides: the influence of prosthetic groups. Bioconjug Chem. 2015;26:201-212
20. Thonon D, Goblet D, Goukens E. et al. Fully Automated Preparation and Conjugation of N-Succinimidyl 4-[18F]Fluorobenzoate ([18F]SFB) with RGD Peptide Using a GEFASTlabTM Synthesizer. Mol Imaging and Biol. 2011;13:1088-1095
21. Olberg DE, Arukwe JM, Grace D. et al. One step radiosynthesis of 6-[(18)F]fluoronicotinic acid 2,3,5,6-tetrafluorophenyl ester ([(18)F]F-Py-TFP): a new prosthetic group for efficient labeling of biomolecules with fluorine-18. J Med Chem. 2010;53:1732-1740
22. Kiesewetter DO, Jacobson O, Lang L. et al. Automated radiochemical synthesis of [18F]FBEM: A thiol reactive synthon for radiofluorination of peptides and proteins. Appl Radiat Isot. 2011;69:410-414
23. Bernard-Gauthier V, Bailey JJ, Liu Z. et al. From Unorthodox to Established: The Current Status of 18F-Trifluoroborate- and 18F-SiFA-Based Radiopharmaceuticals in PET Nuclear Imaging. Bioconjug Chem. 2015;27:267-279
24. McBride WJ, D'Souza CA, Sharkey RM. et al. The radiolabeling of proteins by the [18F]AlF method. Appl Radiat Isot. 2012;70:200-204
25. McBride WJ, Sharkey RM, Goldenberg DM. Radiofluorination using aluminum-fluoride (Al18F). Eur J Nucl Med Mol Imaging Research. 2013;3:36-47
26. Laverman P, McBride WJ, Sharkey RM. et al. Al(18)F labeling of peptides and proteins. J Labelled Comp Radiopharm. 2014;57:219-223
27. Wang W, Liu Z, Li Z. One-Step 18F Labeling of Non-Peptidic Bivalent Integrin α. Bioconjug Chem. 2015;26:24-28
28. Kiesewetter DO, Guo N, Guo J. et al. Evaluation of an [(18)F]AlF-NOTA Analog of Exendin-4 for Imaging of GLP-1 Receptor in Insulinoma. Theranostics. 2012;2:999-1009
29. Da Pieve C, Allott L, Martins CD. et al. Efficient [18F]AlF Radiolabeling of ZHER3:8698 Affibody Molecule for Imaging of HER3 Positive Tumors. Bioconjug Chem. 2016;27:1839-1849
30. Wan W, Guo N, Pan D. et al. First Experience of 18F-Alfatide in Lung Cancer Patients Using a New Lyophilized Kit for Rapid Radiofluorination. J Nucl Med. 2013;54:691-698
31. Cleeren F, Lecina J, Billaud EMF. et al. New chelators for low temperature Al18F-labeling of biomolecules. Bioconjug Chem. 2016;27:790-798
32. Bormans GM, Cleeren F, Lecina J. et al. Methods for low temperature fluorine-18 radiolabelling of biomolecules (WO/2016/065435).
33. Zheng F, Put S, Bouwens L. et al. Nanobodies for Early and Preclinical Diagnosis in a Mouse Model of Rheumatoid Arthritis. J Nucl Med. 2014;55:824-830
34. Wetzel R, Becker M, Behlke J. et al. Temperature behaviour of human serum albumin. Eur J Biochem. 1980;104:469-478
35. Bernardi M, Ricci CS, Zaccherini G. Role of human albumin in the management of complications of liver cirrhosis. J Clin Exp Hepatol. 2014;4:302-311
36. Basuli F, Li C, Xu B. et al. Synthesis of fluorine-18 radio-labeled serum albumins for PET blood pool imaging. Nucl Med Biol. 2015;42:219-225
37. Ramenda T, Kniess T, Bergmann R. et al. Radiolabelling of proteins with fluorine-18 via click chemistry. Chem Commun. 2009;48:7521-7523
38. Jadvar H, Desai B, Conti PS. Sodium 18F-fluoride PET/CT of bone, joint, and other disorders. Semin Nucl Med. 2015;45:58-65
39. Chatalic KLS, Franssen GM, Van Weerden WM. et al. Preclinical comparison of Al18F- and 68Ga-labeled gastrin-releasing peptide receptor antagonists for PET imaging of prostate cancer. J Nucl Med. 2014;55:2050-2056
40. Lafage-Proust MH, Roche B, Langer M. et al. Assessment of bone vascularization and its role in bone remodeling. BoneKEy Rep. 2015;4:1-8
41. De Vos J, Devoogdt N, Lahoutte T. et al. Camelid single-domain antibody-fragment engineering for (pre)clinical in vivo molecular imaging applications: adjusting the bullet to its target. Expert Opin Biol Ther. 2013;13:1149-1160
42. D'Huyvetter M, Vincke C, Xavier C. et al. Targeted radionuclide therapy with A 177Lu-labeled anti-HER2 nanobody. Theranostics. 2014;4:708-720
43. Xavier C, Vaneycken I, D'huyvetter M. et al. Synthesis, Preclinical Validation, Dosimetry, and Toxicity of 68Ga-NOTA-Anti-HER2 Nanobodies for iPET Imaging of HER2 Receptor Expression in Cancer. J Nucl Med. 2013;54:776-784
44. Xavier C, Blykers A, Vaneycken I. et al. 18F-nanobody for PET imaging of HER2 overexpressing tumors. Nucl Med Biol. 2016;43:247-252
45. Blykers A, Schoonooghe S, Xavier C. et al. PET imaging of MMR-expressing macrophages in tumor stroma using 18F-radiolabeled camelid single-domain antibody fragments. J Nucl Med. 2015;56:1265-1271
46. Bala G, Blykers A, Xavier C. et al. Targeting of vascular cell adhesion molecule-1 by 18F-labelled nanobodies for PET/CT imaging of inflamed atherosclerotic plaques. Eur Heart J Cardiovasc Imaging. 2016;17:1001-1008
47. Wen Y, Ouyang Z, Schoonooghe S. et al. Structural evaluation of a nanobody targeting complement receptor Vsig4 and its cross reactivity. Immunobiology. in press
48. Helmy KY, Katschke KJ, Gorgani NN. et al. CRIg: A macrophage complement receptor required for phagocytosis of circulating pathogens. Cell. 2006;124:915-927
49. Zheng F, Sparkes A, De Baetselier P. et al. Molecular Imaging with Kupffer Cell-Targeting Nanobodies for Diagnosis and Prognosis in Mouse Models of Liver Pathogenesis. Mol Imaging Biol. 2017;19:49-58
50. Gainkam LOT, Caveliers V, Devoogdt N. et al. Localization, mechanism and reduction of renal retention of technetium-99m labeled epidermal growth factor receptor-specific nanobody in mice. Contrast Media Mol Imaging. 2011;6:85-92
51. Bondos SE, Bicknell A. Detection and prevention of protein aggregation before, during, and after purification. Analytical biochemistry. 2003;316:223-231
52. Nygren PA. Alternative binding proteins: Affibody binding proteins developed from a small three-helix bundle scaffold. FEBS J. 2008;275:2668-2676
53. Strand J, Nordeman P, Honarvar H. et al. Site-Specific Radioiodination of HER2-Targeting Affibody Molecules using 4-Iodophenethylmaleimide Decreases Renal Uptake of Radioactivity. ChemistryOpen. 2015;4:174-182
54. Westerlund K, Honarvar H, Norrström E. et al. Increasing the Net Negative Charge by Replacement of DOTA Chelator with DOTAGA Improves the Biodistribution of Radiolabeled Second-Generation Synthetic Affibody Molecules. Mol Pharm. 2016;13:1668-1678
55. Krämer-Marek G, Shenoy N, Seidel J. et al. 68Ga-DOTA-Affibody Molecule for In Vivo Assessment of HER2/ neu Expression with PET. Eur J Nucl Med Mol Imaging. 2011;38:1967-1976
56. Kiesewetter DO, Krämer-Marek G, Ma Y. et al. Radiolabeling of HER2 specific Affibody molecule with F-18. J Fluor Chem. 2008;129:799-805
57. Cheng Z, De Jesus OP, Kramer DJ. et al. 64Cu-Labeled Affibody Molecules for Imaging of HER2 Expressing Tumors. Mol Imaging Biol. 2010;12:316-324
58. Ahlgren S, Wallberg H, Tran TA. et al. Targeting of HER2-Expressing Tumors with a C-Terminally Engineered Cysteine. J Nucl Med. 2009;50:781-789
59. Perols A, Honarvar H, Strand J. et al. Influence of DOTA Chelator Position on Biodistribution and Targeting Properties of 111In-Labeled Synthetic Anti-HER2 Affibody Molecules. Bioconjug Chem. 2012;23:1661-1670
60. Price EW, Orvig C. Matching chelators to radiometals for radiopharmaceuticals. Chem Soc Rev. 2014;43:260-290
61. Heskamp S, Laverman P, Rosik D. et al. Imaging of Human Epidermal Growth Factor Receptor Type 2 Expression with 18F-Labeled Affibody Molecule ZHER2:2395 in a Mouse Model for Ovarian Cancer. J Nucl Med. 2012;53:146-153
62. Kramer-Marek G, Kiesewetter DO, Martiniova L. et al. [18F]FBEM-ZHER2:342-Affibody molecule—a new molecular tracer for in vivo monitoring of HER2 expression by positron emission tomography. Eur J Nucl Med Mol Imaging. 2008;35:1008-1018
Author contact
![]() Corresponding author.
Corresponding author.
Citation styles
APA
Cleeren, F., Lecina, J., Ahamed, M., Raes, G., Devoogdt, N., Caveliers, V., McQuade, P., Rubins, D.J., Li, W., Verbruggen, A., Xavier, C., Bormans, G. (2017). Al18F-Labeling Of Heat-Sensitive Biomolecules for Positron Emission Tomography Imaging. Theranostics, 7(11), 2924-2939. https://doi.org/10.7150/thno.20094.
ACS
Cleeren, F.; Lecina, J.; Ahamed, M.; Raes, G.; Devoogdt, N.; Caveliers, V.; McQuade, P.; Rubins, D.J.; Li, W.; Verbruggen, A.; Xavier, C.; Bormans, G. Al18F-Labeling Of Heat-Sensitive Biomolecules for Positron Emission Tomography Imaging. Theranostics 2017, 7 (11), 2924-2939. DOI: 10.7150/thno.20094.
NLM
Cleeren F, Lecina J, Ahamed M, Raes G, Devoogdt N, Caveliers V, McQuade P, Rubins DJ, Li W, Verbruggen A, Xavier C, Bormans G. Al18F-Labeling Of Heat-Sensitive Biomolecules for Positron Emission Tomography Imaging. Theranostics 2017; 7(11):2924-2939. doi:10.7150/thno.20094. https://www.thno.org/v07p2924.htm
CSE
Cleeren F, Lecina J, Ahamed M, Raes G, Devoogdt N, Caveliers V, McQuade P, Rubins DJ, Li W, Verbruggen A, Xavier C, Bormans G. 2017. Al18F-Labeling Of Heat-Sensitive Biomolecules for Positron Emission Tomography Imaging. Theranostics. 7(11):2924-2939.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.