Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Material and Methods
Results
Discussion
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(11):2914-2923. doi:10.7150/thno.20355 This issue Cite
Research Paper
Mutational Landscapes of Smoking-Related Cancers in Caucasians and African Americans: Precision Oncology Perspectives at Wake Forest Baptist Comprehensive Cancer Center
Ville Kytola1, 2, 10*, Umit Topaloglu1, 2*, Lance D. Miller1, 2*, Rhonda L. Bitting1, 3, Michael M. Goodman1, 3, Ralph B. D`Agostino Jr1, 4, Rodwige J. Desnoyers1, 3, Carol Albright1, 3, George Yacoub1, 3, Shadi A. Qasem1, 5, Barry DeYoung1, 5, Vesteinn Thorsson11, Ilya Shmulevich11, Meng Yang1, 2, 12, Anastasia Shcherban1, 2, 10, Matthew Pagni1, 7, Liang Liu1, Matti Nykter10, Kexin Chen12, Gregory A. Hawkins1, 15, Stefan C. Grant1, 3, W. Jeffrey Petty1, 3, Angela Tatiana Alistar1, 3, Edward A. Levine1, 6, Edgar D. Staren1, 6, Carl D. Langefeld4, Vincent Miller13, Gaurav Singal13, Robin M. Petro1, 3, Mac Robinson1, William Blackstock1, 8, Bayard L. Powell1, 3, Lynne I. Wagner1, 9, Kristie L. Foley1, 9, Edward Abraham14, Boris Pasche1, 2, 3 ![]() , Wei Zhang1, 2, 15
, Wei Zhang1, 2, 15 ![]()
1. Wake Forest Baptist Comprehensive Cancer Center, Wake Forest Baptist Medical Center, Winston Salem, NC, USA 27157;
2. Department of Cancer Biology, Wake Forest School of Medicine, Winston Salem, NC, USA 27157;
3. Department of Internal Medicine-Section of Hematology and Oncology, Wake Forest School of Medicine, Winston Salem, NC, USA 27157;
4. Department of Biostatistical Sciences, Wake Forest School of Medicine, Winston Salem, NC, USA 27157;
5. Department of Laboratory Medicine and Pathology, Wake Forest School of Medicine, Winston Salem, NC, USA 27157;
6. Department of General Surgery-Section of Surgical Oncology, Wake Forest School of Medicine, Winston Salem, NC, USA 27157;
7. Department of Radiology, Wake Forest School of Medicine, Winston Salem, NC, USA 27157;
8. Department of Radiation Oncology, Wake Forest School of Medicine, Winston Salem, NC, USA 27157;
9. Department of Social Sciences and Health Policy, Wake Forest School of Medicine, Winston Salem, NC, USA 27157;
10. Institute for Biosciences and Medical Technology, University of Tampere, Tampere, Finland 33520;
11. Institute for Systems Biology, Seattle, WA, USA 98109;
12. Department of Epidemiology and Biostatistics, Tianjin Medical University Cancer Institute and Hospital, Tianjin, China 300060;
13. Foundation Medicine, Cambridge, MA, USA 02141;
14. Wake Forest School of Medicine, Winston Salem, NC, USA 27157;
15. Center for Genomics and Personalized Medicine Research, Wake Forest School of Medicine, Winston Salem, NC, USA 27157.
* Equal contributions
Received 2017-3-31; Accepted 2017-4-21; Published 2017-7-12
Abstract

Background: Cancers related to tobacco use and African-American ancestry are under-characterized by genomics. This gap in precision oncology research represents a major challenge in the health disparities in the United States.
Methods: The Precision Oncology trial at the Wake Forest Baptist Comprehensive Cancer Center enrolled 431 cancer patients from March 2015 to May 2016. The composition of these patients consists of a high representation of tobacco-related cancers (e.g., lung, colorectal, and bladder) and African-American ancestry (13.5%). Tumors were sequenced to identify mutations to gain insight into genetic alterations associated with smoking and/or African-American ancestry.
Results: Tobacco-related cancers exhibit a high mutational load. These tumors are characterized by high-frequency mutations in TP53, DNA damage repair genes (BRCA2 and ATM), and chromatin remodeling genes (the lysine methyltransferases KMT2D or MLL2, and KMT2C or MLL3). These tobacco-related cancers also exhibit augmented tumor heterogeneities. Smoking related genetic mutations were validated by The Cancer Genome Atlas dataset that includes 2,821 cases with known smoking status. The Wake Forest and The Cancer Genome Atlas cohorts (431 and 7,991 cases, respectively) revealed a significantly increased mutation rate in the TP53 gene in the African-American subgroup studied. Both cohorts also revealed 5 genes (e.g. CDK8) significantly amplified in the African-American population.
Conclusions: These results provide strong evidence that tobacco is a major cause of genomic instability and heterogeneity in cancer. TP53 mutations and key oncogene amplifications emerge as key factors contributing to cancer outcome disparities among different racial/ethnic groups.
Introduction
Advances in genomics and informatics have validated the importance of individuality in cancer diagnosis and treatment. Evidence illustrates that cancer is a disease of genetic and epigenetic causality, profoundly affected by environment and lifestyle [1]. An increasing number of genetic alterations have been characterized that drive the pathogenesis of cancer and convey therapeutic actionability [2]. These driver mutations often are not restricted to a specific cancer type, histology or patient demographic. This unprecedented molecular understanding of individual cancers has ushered in a new era of health care coined precision medicine [3].
Precision medicine has begun a reprogramming of clinical oncology practice [4, 5]. Specialization in organ-oriented disease is being supplemented with molecular target assessment and targeted treatment across cancer types [6-9]. New clinical trial models (e.g., BASKET trial, NCI-MATCH) emphasize treatment decisions based on druggability of gene mutations rather than tumor histology [10, 11]. Precision medicine consortia have formed to test this new mode of cancer management [12]. The Precision Medicine Exchange Consortium (PMEC) is one such consortium, consisting of eight major cancer centers in the US, including the Wake Forest Baptist Comprehensive Cancer Center (WFBCCC).
To investigate the relationship between precision medicine-derived cancer genomic correlates and patient demographics at WFBCCC, 431 cancer patients were enrolled into the Wake Forest Precision Oncology Initiative trial. This patient cohort reflects the patient population in the WFBCCC catchment area with a high representation of tobacco-related cancers (e.g., lung, colorectal, and bladder) and African American (AA) ancestry (13.5%). In the WFBCCC catchment area, (22% of adults are current tobacco users versus 19% nationally). Cigarette smoke is a known carcinogen, causing defined mutational signatures [13, 14]. However, smoking-related genetic changes in cancer are not well-characterized. Even more unclear is whether the mutational events differ between AA and Caucasian cancer patients, despite AA cancer patients having a poorer prognosis, including cancer-related and higher overall mortality rates [15].
Here, we report the characterization of the mutational landscapes of our unique cohort of cancer patients with findings validated in The Cancer Genome Atlas (TCGA) dataset. We also provide examples of mutation directed treatment in these patients to demonstrate the clinical impact of precision oncology initiatives.
Material and Methods
Patient Cohort
Four hundred thirty-one cancer patients from the catchment area of the WFBCCC participated in the IRB-approved Wake Forest Precision Oncology Initiative (POI) from March 1, 2015 to May 30, 2016. African American status is based on self-reported ancestry. Each patient was consented for research analysis of sequencing results. Tumor specimens were evaluated by two board-certified pathologists to confirm diagnosis and classification. Tumor biopsies and surgical specimens were formalin-fixed and paraffin-embedded immediately following acquisition, according to standard clinical protocol. Tumor blocks of sufficient cellularity (>20%) and limited necrosis were selected and submitted to Foundation Medicine for FoundationOne® testing. The clinical management process is shown in Figure S1 and supplementary methods. ClinicalTrials.gov Identifier: NCT02566421
Genomic Profiling
Tumor tissue was subjected to Next Generation Sequencing (NGS) to identify mutations, rearrangements and copy number alterations spanning 415 cancer-related genes that make up the FoundationOne ® (F1) test (Foundation Medicine, Cambridge, MA) (Supplementary Methods) [16].
Statistical and Bioinformatic Analysis
Nonsynonymous somatic mutation calls were quantified. Patients were assigned to low or high mutation load groups based on the cohort mean mutation number. Fisher's exact test and Benjamini-Hochberg multiple testing adjustments were used to determine associations between mutation load and DNA damage genes and chromatin remodeling genes. Smoking status was defined by self-reported smoking history obtained from Cancer Registry and/or Epic Electronic Medical Record. Never smokers were defined as respondents who smoked less than 100 cigarettes in their lifetime. Based on evidence that smoking cessation reduces cancer risk by half at five years, active smokers at the time of clinical data collection and those who had quit smoking within the previous five years were considered current/recent smokers [17, 18]. Those having quit more than five years prior to data collection were defined as former smokers. Only white (Caucasian) and black (AA patients) were included in disparities analyses, as these are the two main ethnic groups of the WFBCCC catchment area. Other racial/ethnic populations were underrepresented in the sample (less than 5%). Analyses for discovery of smoking-related mutations focused on genes with functional roles in DNA Damage Repair and Chromatin Remodeling. Each set of analyses used the Cochran-Mantel- Haenszel test to uncover associations between smoking status (defined as an ordinal variable - Never, Former, Recent) and gene mutation. Fisher's exact test was used to assess significance (p < 0.05) of gene mutation frequencies that differed with respect to low and high mutation load and racial status (Caucasians versus AA). The Hochberg (1988) approach was used to adjust for multiple testing [19]. MutSig algorithm, MutSigCV, was used to evaluate the significance of mutated genes. All analyses were performed with R statistical computing software version 3.3.0 [20]. Mutagenic processes and tumor clonality were analyzed with R packages somaticSignatures and SciClone, respectively (Supplementary Methods) [21].
Results
Mutational Analysis
We analyzed 431 cancer patients from the catchment area of the WFBCCC that participated in the IRB-approved Wake Forest Precision Oncology Initiative (POI). Patient demographics are summarized in Table 1, Table S1 and Figure 1. In our patients, the most frequently mutated genes were tumor suppressor genes TP53, APC, FAT1, RB1, BRCA2, and NF1; Wnt signaling pathway genes LRP1B and APC; oncogenes KRAS, PIK3A, DNA damage repair (DDR) genes (ATM, BRCA2); chromosomal integrity genes (TERT), and chromatin remodeling (CR) genes (KMT2D or MLL2, KMT2C or MLL3, ARID1A, ARID1B, EP300) (Figure 2A).
Demographics of patients (N = 431) in the Precision Oncology Initiative
| Characteristic | No. (%) |
|---|---|
| SMOKING HISTORY | |
| Current | 127 (29.5) |
| Former | 145 (33.6) |
| Never | 159 (36.9) |
| RACE | |
| White or Caucasian | 356 (82.6) |
| Black or African American | 58 (13.5) |
| American Indian or Alaska Native | 2 (0.5) |
| Asian | 5 (1.2) |
| Native Hawaiian or Pacific Islander | 1 (0.2) |
| Other | 9 (2.1) |
| ETHNICITY | |
| Hispanic or Latino | 8 (1.9) |
| Not Hispanic or Latino | 423 (98.1) |
| CANCER STATUS | |
| Metastasis | 185 (42.9) |
| Primary | 220 (51.0) |
| Unknown | 26 (6.0) |
| DISEASE STAGE | |
| Stage 0 | 2 (0.5) |
| Stage 1 | 31 (7.2) |
| Stage 2 | 36 (8.4) |
| Stage 3 | 85 (19.7) |
| Stage 4 | 231 (53.6) |
| Undetermined/Unknown | 46 (10.7) |
| TUMOR TYPE | |
| Lung | 90 (20.9) |
| Colorectal | 56 (13.0) |
| Other | 43 (10.0) |
| Cup | 41 (9.5) |
| Brain | 31 (7.2) |
| Sarcoma | 30 (7.0) |
| Head/Neck | 26 (6.0) |
| Other GI | 21 (4.9) |
| Breast | 18 (4.2) |
| Bladder | 16 (3.7) |
| Pancreas | 16 (3.7) |
| Ovary/Uterus | 14 (3.2) |
| Appendix | 10 (2.3) |
| Kidney | 10 (2.3) |
| Prostate | 9 (2.1) |
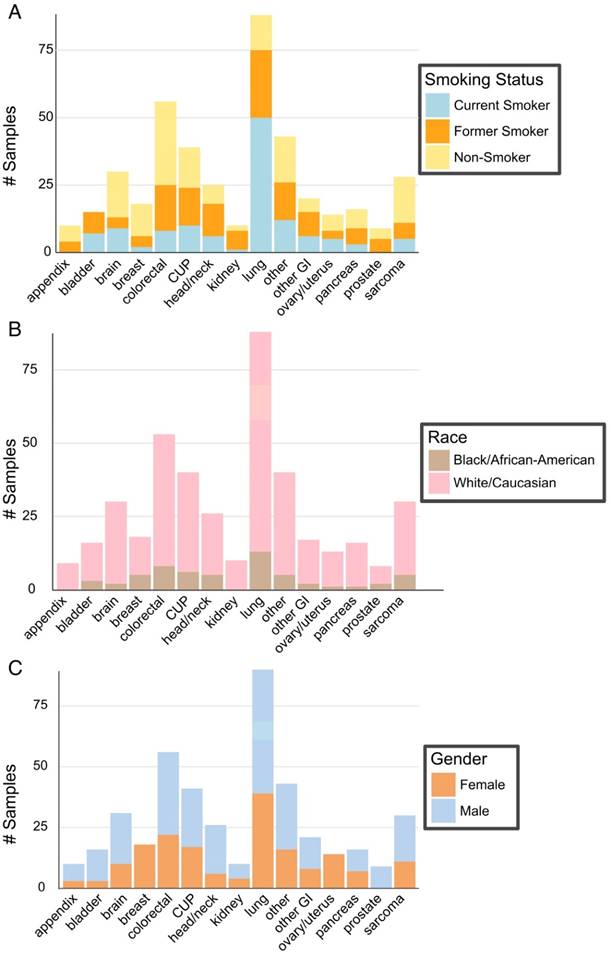
Patient Demographics. (A) Patient smoking status within the context of type of cancer. (B) Patient race as described within cancer type. (C). Gender of patients within each cancer type.
Global Landscape of Somatic Mutations. (A) Global somatic mutational landscape of all patients for the top 30 genes having the largest fraction of mutations. Top and left bar charts show the number of mutations and percent of mutated samples, respectively. The lower part of panel A summarizes clinical information from each patient. (B) Somatic mutational landscape for major cancer groups for the same 30 genes seen in (A). Cancer group-wise mutational patterns show large similarities (TP53) but also striking differences (KRAS, APC, TERT) between cancer groups.
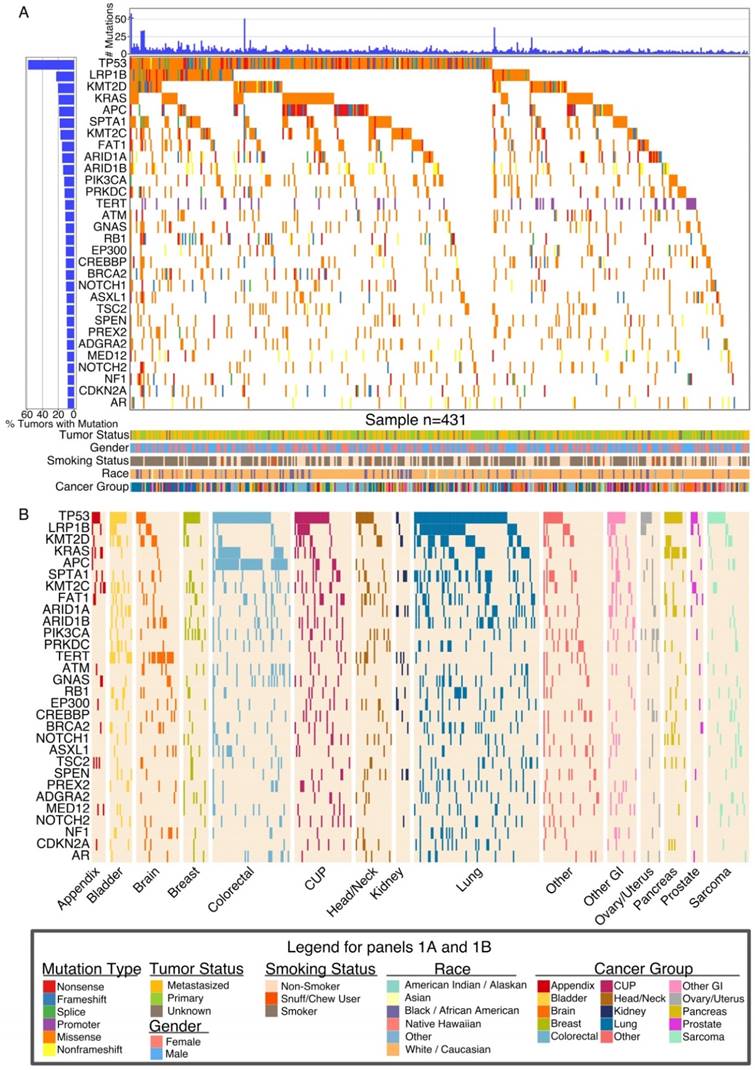
Some of the observed gene mutations were expected. For example, TP53 showed a uniformly high frequency of mutation across all cancer types while APC was predominantly mutated in colorectal cancer. KRAS was mutated at high frequency in pancreatic, colorectal, and lung cancer. LRP1B was frequently mutated only in lung cancer (43 of 90, 47.8%). Another gene highly mutated in lung cancer was SPTA1 (33 of 90, 37%) that has unknown oncogenic functions. Analysis of The Cancer Genome Atlas (TCGA) lung cancer cohort (adenocarcinomas and squamous) validated the frequent mutation of SPTA1 gene (Figure S2). EPHA3 and EPHA5 were also frequently mutated in both our and TCGA cohorts (Figure S2). TERT, which codes for telomerase and is involved in the longevity of tumor cells, was found to be frequently mutated at a promoter hot spot (-124C > T) in brain tumors (16 of 31, 53%), bladder cancers (9 of 16, 56%), and head/neck cancers (6 of 26, 23%), consistent with recent reports [22, 23]. In contrast, the TERT promoter is rarely mutated in colorectal, lung or pancreatic cancer, or soft tissue sarcoma (Figure 2B).
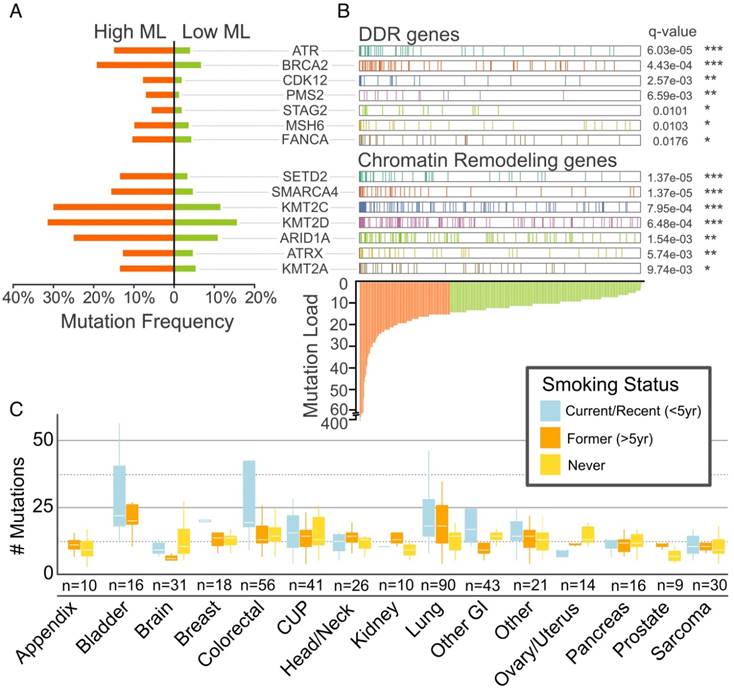
A striking observation was the remarkably high mutation rates of DDR and CR genes in our cohort and their association with high-mutational load (Figure 3A, B, C), underscoring the highly unstable genome associated with smoking-related cancers that dominate our cohort.
Associations between DDR/CR Gene Mutation Frequency, Tumor Mutational Load and Smoking Status. DDR and CR genes are mutated at higher frequency (A) and in tumors with higher mutation load (B). High and low mutational load (ML) designations are based on above-mean (orange) and below-mean (light green) mutation count, respectively. Nonsynonymous protein-altering mutations (SNVs, in/dels, rearrangements) and copy number deletions were included. **** (p < 0.0001); *** (p < 0.001); ** (p < 0.01); * (p < 0.05), Fisher's exact test and Benjamini-Hochberg adjusting. (C) Mutational load across cancer groups as a function of smoking status. Boxes mark the interquartile range (25th-75th percentile) of the distribution while the whiskers demarcate the 5th and 95th percentiles. The white line marks the median of the distribution.
Large numbers of gene mutations (hypermutation phenotype) and copy number alterations (chromosomal instability or CIN) represent two different types of genomic instability [24]. We observed that the CIN phenotype exhibited variable patterns in different cancer types, with extensive overall changes in lung and colorectal cancers (Figure S3A). Despite high mutation rates, two smoking-related cancer types, bladder and head/neck cancers, did not show extensive copy number alterations (Figure 3C, Figure S3A). Among the most extensively amplified genes were oncogenes, including ERRB2, MYC, MET, CDK6, and EGFR (e Figure 3B). Two cases exhibited amplification of immunosuppressing genes PD-L1 (CD274) and PD-L2 (PDCD1LG2), suggesting a role for anti-PD-1 or anti-PD-L1 therapy. Genes frequently deleted in our advanced cancer cohort are CDKN2A/B and PTEN (Figure S3B).
Gene Mutations Associated with Smoking
In our cohort, proportions of smokers were similar in AAs and Caucasian-Americans (38 of 58, 216 of 356, respectively; Fisher exact test p-value, 0.56). Current/recent smokers exhibited a significantly higher mutational load (mean = 20.5, median = 14.0) than former smokers (mean = 13.0, median = 11.5; p = 0.017, 2-sided t-test) and never smokers (mean = 12.3, median = 11.0; p = 0.029, 2-sided t-test). Analysis of total mutations per cancer showed a heterogeneous pattern (Figure S4) with lung, bladder, and colorectal cancer exhibiting high tumor mutational load. Appendiceal, brain, and prostate cancers exhibited the lowest mutational load.
Analysis of the mutational signatures characterized by nucleotide changes in the context of neighboring nucleotides identified three major signatures (Figure 4A). Current smokers, former smokers and never smokers exhibited distinct mutational signatures (Figure 4B). Many DDR and CR genes exhibited associations with smoking status (Figure 4C), with a greater frequency of mutation in current/recent or former smokers as compared to never smokers. After adjusting for multiple testing, mutations in two DDR genes - CDK12 and BRCA2 met the criteria for statistical significance (p = 0.0069 and 0.016, respectively). Similarly, the CR gene KMT2D met the criteria for statistical significance (p = 0.0087), while two others (KDM6A and SMARCA4) were nominally significant (p = 0.026 and 0.032, respectively) (Figure 4C, Table S2).
Association of Gene Alterations and Smoking. (A) Three somatic mutational signatures present in the cohort. X-axis consists of adjacent nucleotides to the mutated base indicated on top of each column. Y-axis describes strength of contribution for a given triplet formed of altered base and adjacent nucleotides. (B) Contribution of each signature to smoking categories. (C) Significant smoking related alterations in DNA damage repair (DDR) and chromatin remodeling (CR) genes defined by the Cochran-Mantel-Haenzel test. (D) Validation for the smoking associated mutations in TCGA data. The validation dataset consists of 675 current smokers, 1351 former smokers and 795 never smokers. (E) Significant association between high tumor clonality and smoking. * (p < 0.05), ** (p < 0.01)
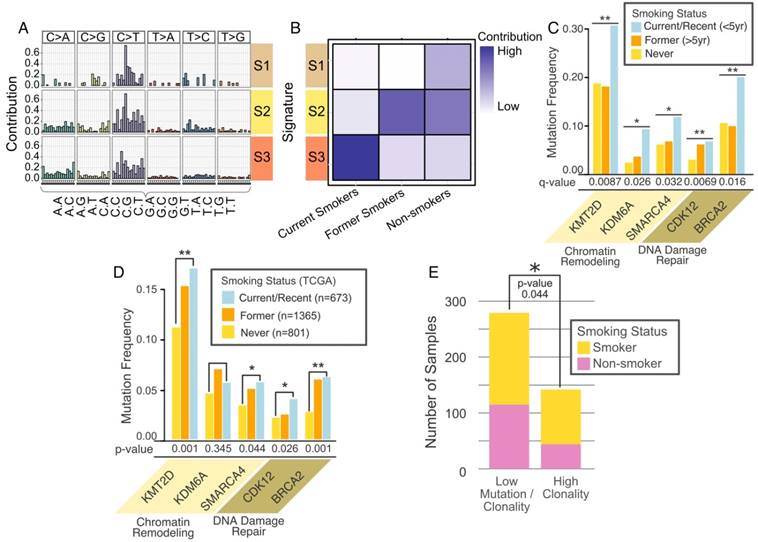
To begin validation in the TCGA cohort we found that among the solid tumors, mutation and smoking status data was available in 2,821 cases. As shown in Figure 4D, current and former smokers have similar mutation frequencies of these genes. This analysis showed that most smoking-related gene mutations found in our cohort (e.g., KMT2D, BRCA2) were validated in the TCGA cohort.
Intratumoral clonal heterogeneity poses serious challenges to precision oncology treatment [25]. Tumors comprised of multiple clones with different mutational events may require multiple targeting strategies; in combination or in sequence. We quantified tumor clonal heterogeneity based on clustering of variant allele fractions (Figure S5A, B, see Methods). Mutation rates for 48% of patients were relatively low with no clonal diversity. For the others, 19, 23, and 10% of cases exhibited 1, 2 or more than 2 clones, respectively, based on clonality analysis (Figure S5C). Higher clonality was associated with smoking (Figure 4E).
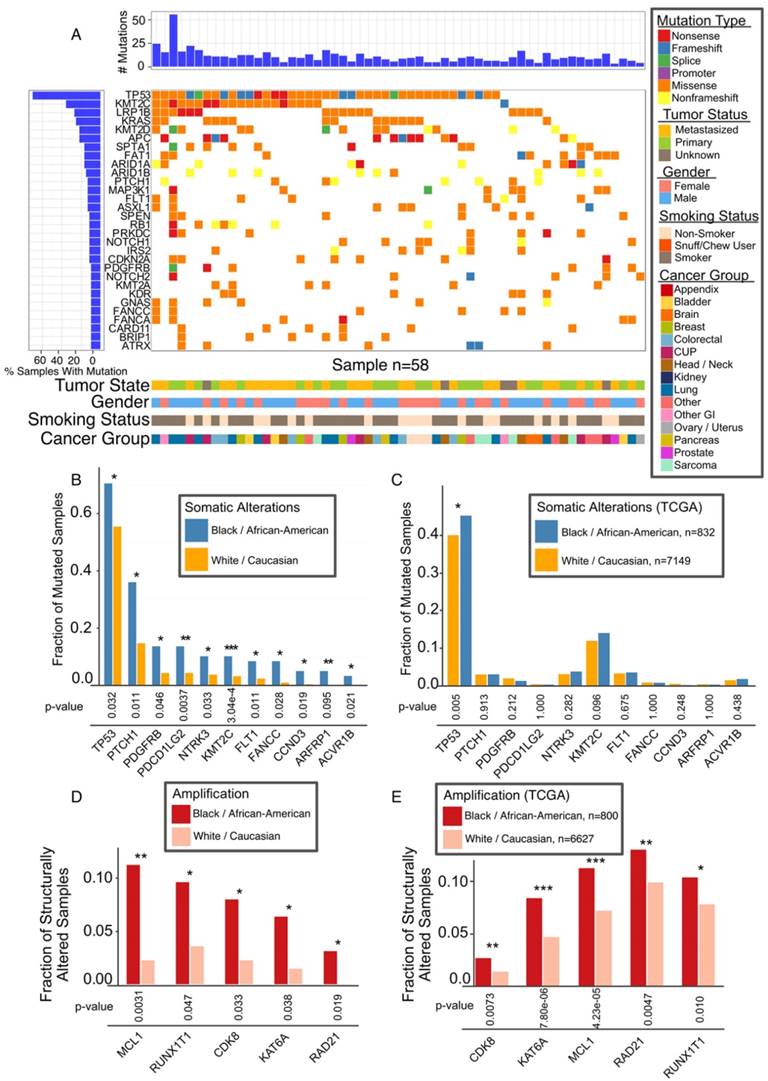
Gene Mutations Associated with Race
The overall mutational landscape of AA patients is similar to that of the whole WFBCCC cohort, the majority of which are Caucasian patients (Figure 5A and Figure 2A). However, our analysis revealed differential mutation rates in the key genes, TP53 and KMT2C (Figure 5B). In the TCGA cohort, there are 842 AA and 7,149 Caucasian cases with mutation data and 892 AA and 7,679 Caucasians with gene amplification data. TP53 (p = 0.027), and to a lesser extent, KMT2C (p = 0.093), were more frequently mutated in AA patients in the TCGA cohort (Figure 5C). Gene copy number analysis revealed marked differences in five oncogenes in our cohort (Figure 5D); all of them were found to be more significantly amplified in AA in the TCGA cohort (Figure 5E).
Association of Mutations with AA Population. (A) Mutational landscape in AA cancers, 30 most frequently mutated genes. (B) Mutation frequencies among Caucasian and AA for significant race-associated genes. (C) Validation of TP53 mutation in AA in TCGA data. (D) Most common copy number alterations in AA. Genes with significant difference after adjustment are marked with asterisk. (E) Genes with significant connection between race and alteration status validated TCGA data are marked with asterisk * (p < 0.05), ** (p < 0.01), *** (p <0.001).
Precision Oncology Case Reports
The essence of precision oncology is to match mutational information with drugs that have shown therapeutic efficacy in targeting the mutated protein. Oncologists at WFBCCC have designed clinical treatment regimens based on genomics testing in our Precision Oncology Trial and patients have shown remarkable responses. Key examples are described in Supplementary Material.
Discussion
Smokers and AAs are more prevalent among our disproportionately rural, Appalachian/Piedmont catchment area population. Thus, we are able to uniquely interrogate mutations associated with these two understudied populations. This undertaking has provided a number of insights. Among the most interesting discoveries are the revelations that DDR and CR genes are highly mutated in current/former smokers, and smoking is associated with augmented clonal evolution (clonality) and tumor heterogeneities. This is consistent with recent genomic characterization of smoking related cancers [26]. These results provide strong evidence that genomic instability is a fundamental hallmark of cancer and the events underlying the regulation of genome stability are centered on interactions with environmental factors and lifestyle.
AA cancer patients have a more dismal prognosis, which represents a key health disparity challenge in the US. Our genomics analysis revealed a number of genes mutated at different frequencies in AA and Caucasian cancer patients. After further analysis of the larger independent TCGA cohort, mutations of the tumor suppressor gene TP53 still emerged as a more common event in AA cancer patients. Notably, in our cohort of lung cancer, mutation rates for a number of genes including TP53 are higher than that observed in the TCGA cohort, consistent with the predominance of advanced and smoking-related cancers in our cohort. However, our analysis showed that the enriched mutation in TP53 in AA is not driven by lung cancer in the cohort because TP53 mutation rates are similar in AA and Caucasian Americans (p = 0.5). TP53 has long been recognized as a critical control gene for genome stability [27]. Numerous studies have shown that mutations of TP53 are associated with poor prognosis in cancer [28]. Therefore, genomic stability regulated by TP53 may be a key factor that contributes to cancer outcome disparities among different racial groups. The limitation of this study is the size of cohort enrolled in precision oncology initiatives due to the enrollment criteria and cost associated with the clinical sequencing tests. Therefore, future data sharing effort will enable pooled analysis of all the major precision oncology programs in the country to determine whether genetic events such as increased TP53 mutation rates are observed in all major cancer types and their relationship with smoking. Interestingly, during the review of our study, a recent paper focusing on lung cancer reported overall similar mutation frequencies between AA and Caucasian American, however they also observed more prevalent TP53 mutation in AA subgroup than Caucasian group [29].
In addition to gaining insight into the knowledge of genetic/molecular mechanisms of cancer development/progression, a key benchmark for precision oncology initiatives is the translatability of genomics information to more accurately targeted and beneficial treatments in patients [5, 30-33]. Several successful examples at WFBCCC are reported here and described in supplemental document.
There is no doubt that increasing numbers of cancer patients will benefit from the precision oncology design. There are, however, a number of important challenges and limitations [34].
First, the current precision oncology initiatives focus more on advanced metastatic cancer patients. Many of these patients die within 3-4 months of the genomic testing, before treatment decisions can be rendered. Thus, genomic testing should extend to patients with newly diagnosed metastatic disease, with the hypothesis that a patient with longer expected survival will benefit more from precision treatment. Retesting of tumors from recurrent patients will identify treatment-associated mutations to revise therapeutic strategies. Secondly, drug availability is a major problem [35-37]. Many FDA approved but off-label drugs, are not covered by insurance. Getting access to these off-label drugs on a compassionate basis invariably requires the resources and extra time of physicians. There clearly is a need for a streamlined process of drug acquisition for precision oncology to reach its full potential. Thirdly, genomic testing reveals many gene mutations without information about whether these mutations are deleterious (driver mutations). Thus, there is a clear need for efficient high throughput laboratory assays to identify functional mutations [38]. Fourthly, intratumoral heterogeneity poses a significant obstacle for sustained treatment response to a single agent therapy [25, 39]. Our clonality analyses showed that different clones exist in a fraction of tumors with different potential driver mutations. Therefore, precision oncology requires an understanding of tumor clonality to inform the design of combination or sequential therapy with different drugs. Finally, from the patients' perspective, these complexities are compounded by the psychosocial and ethical considerations inherent to the genomic profiling process [40, 41]. In this newly evolving paradigm, patients and providers need to navigate care from a patient-centered framework. In the decision making process, smoking status and ethnicity should clearly be considered because of the association with differential mutation rates.
Acknowledgements
We thank the physicians and nurses who participated in the POI. We thank the patients who provided consent to participate in the POI knowing they may not directly derive benefit.
Funding
The work is partially supported by the Cancer Center Support Grant from the National Cancer Institute to the Comprehensive Cancer Center of Wake Forest Baptist Medical Center (P30 CA012197) and funding for the Genome Data Analysis Centers from the National Institutes of Health (U24 CA143835 to I.S. and W.Z.). W.Z. is supported by Hanes and Willis Professorship in Cancer and a Fellowship by the National Foundation for Cancer Research. B.P. is supported by Charles L. Spurr Professorship of Medicine.
Supplementary Material
Supplementary figures and tables.
Competing Interests
V.M. and G.S. are employees of and stockholders in Foundation Medicine. No other authors declared Conflicts of Interest.
References
1. Kolonel LN, Altshuler D, Henderson BE. The multiethnic cohort study: exploring genes, lifestyle and cancer risk. Nat Rev Cancer. 2004;4:519-27
2. Carr TH, McEwen R, Dougherty B. et al. Defining actionable mutations for oncology therapeutic development. Nat Rev Cancer. 2016;16:319-29
3. Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793-5
4. Morris LG, Chandramohan R, West L. et al. The Molecular Landscape of Recurrent and Metastatic Head and Neck Cancers: Insights From a Precision Oncology Sequencing Platform. JAMA Oncol. 2016;3:244-55
5. Kris MG, Johnson BE, Berry LD. et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA. 2014;311:1998-2006
6. Sonnenblick A, Ponde N, Piccart M. Metastatic breast cancer: The Odyssey of personalization. Mol Oncol. 2016;10:1147-59
7. Tran NH, Cavalcante LL, Lubner SJ. et al. Precision medicine in colorectal cancer: the molecular profile alters treatment strategies. Ther Adv Med Oncol. 2015;7:252-62
8. Pasche B, Grant SC. Non-small cell lung cancer and precision medicine: a model for the incorporation of genomic features into clinical trial design. JAMA. 2014;311:1975-6
9. Jurgensmeier JM, Eder JP, Herbst RS. New strategies in personalized medicine for solid tumors: molecular markers and clinical trial designs. Clin Cancer Res. 2014;20:4425-35
10. Ornes S. Core Concept: Basket trial approach capitalizes on the molecular mechanisms of tumors. Proc Natl Acad Sci U S A. 2016;113:7007-8
11. ECOG-ACRIN Cancer Research Group. NCI-MATCH / EAY131 Interim Analysis.; 2016. http://ecog-acrin.org/nci-match-eay131/interim-analysis
12. Harris MH, DuBois SG, Glade Bender JL. et al. Multicenter Feasibility Study of Tumor Molecular Profiling to Inform Therapeutic Decisions in Advanced Pediatric Solid Tumors: The Individualized Cancer Therapy (iCat) Study. JAMA Oncol. 2016;2:608-15
13. Pfeifer GP, Denissenko MF, Olivier M. et al. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene. 2002;21:7435-51
14. Blackford A, Parmigiani G, Kensler TW. et al. Genetic mutations associated with cigarette smoking in pancreatic cancer. Cancer Res. 2009;69:3681-8
15. DeSantis CE, Siegel RL, Sauer AG. et al. Cancer statistics for African Americans, 2016: Progress and opportunities in reducing racial disparities. CA Cancer J Clin. 2016;66:290-308
16. Frampton GM, Fichtenholtz A, Otto GA. et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023-31
17. La Vecchia C, Franceschi S, Bosetti C. et al. Time since stopping smoking and the risk of oral and pharyngeal cancers. J Natl Cancer Inst. 1999;91:726-8
18. U.S. Department of Health and Human Services. US Surgeon General's Report, 2010: U.S. Department of Health and Human Services. A Report of the Surgeon General. How Tobacco Smoke Causes Disease: What It Means to You.. Atlanta: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2010 [accessed 2015 Oct 19]. https://www.cdc.gov/tobacco/data_statistics/sgr/2010/consumer_booklet/
19. Hochberg Y. A sharper Bonferroni procedure for multiple tests of significance. Biometrika. 1988;75:800-2
20. R Core Team. A language and environment for statistical computing. 3.3.0 ed. Vienna, Austria: R Foundation for Statistical Computing. 2016 https://www.R-project.org
21. Miller CA, White BS, Dees ND. et al. SciClone: inferring clonal architecture and tracking the spatial and temporal patterns of tumor evolution. PLoS Comput Biol. 2014;10:e1003665
22. Ceccarelli M, Barthel FP, Malta TM. et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell. 2016;164:550-63
23. Vinagre J, Almeida A, Populo H. et al. Frequency of TERT promoter mutations in human cancers. Nat Commun. 2013;4:2185
24. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330-7
25. Chen K, Yang D, Li X. et al. Mutational landscape of gastric adenocarcinoma in Chinese: implications for prognosis and therapy. Proc Natl Acad Sci U S A. 2015;112:1107-12
26. Alexandrov LB, Ju YS, Haase K. et al. Mutational signatures associated with tobacco smoking in human cancer. Science. 2016;354:618-22
27. Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15-6
28. Robles AI, Harris CC. Clinical outcomes and correlates of TP53 mutations and cancer. Cold Spring Harb Perspect Biol. 2010;2:a001016
29. Campbell JD, Lathan C, Sholl L. et al. Comparison of Prevalence and Types of Mutations in Lung Cancers Among Black and White Populations. JAMA Oncol. 2017 doi: 10.1001/jamaoncol.2016.6108
30. MacConaill LE, Lindeman NI, Rollins BJ. Brave-ish New World-What's Needed to Make Precision Oncology a Practical Reality. JAMA Oncol. 2015;1:879-80
31. Meric-Bernstam F, Johnson A, Holla V. et al. A decision support framework for genomically informed investigational cancer therapy. J Natl Cancer Inst. 2015;107:doi 10.1093/jnci/djv098
32. Wheler J, Lee JJ, Kurzrock R. Unique molecular landscapes in cancer: implications for individualized, curated drug combinations. Cancer Res. 2014;74:7181-4
33. Mody RJ, Wu YM, Lonigro RJ. et al. Integrative Clinical Sequencing in the Management of Refractory or Relapsed Cancer in Youth. JAMA. 2015;314:913-25
34. Jameson JL, Longo DL. Precision medicine-personalized, problematic, and promising. N Engl J Med. 2015;372:2229-34
35. Miller FA, Hayeems RZ, Bytautas JP. et al. Testing personalized medicine: patient and physician expectations of next-generation genomic sequencing in late-stage cancer care. Eur J Hum Genet. 2014;22:391-5
36. Sohal DP, Rini BI, Khorana AA. et al. Prospective Clinical Study of Precision Oncology in Solid Tumors. J Natl Cancer Inst. 2016;108:doi 10.1093/jnci/djv332
37. Darrow JJ, Sarpatwari A, Avorn J. et al. Practical, legal, and ethical issues in expanded access to investigational drugs. N Engl J Med. 2015;372:279-86
38. Carter H, Chen S, Isik L. et al. Cancer-specific high-throughput annotation of somatic mutations: computational prediction of driver missense mutations. Cancer Res. 2009;69:6660-7
39. Linnekamp JF, Wang X, Medema JP. et al. Colorectal cancer heterogeneity and targeted therapy: a case for molecular disease subtypes. Cancer Res. 2015;75:245-9
40. Lolkema MP, Gadellaa-van Hooijdonk CG, Bredenoord AL. et al. Ethical, legal, and counseling challenges surrounding the return of genetic results in oncology. J Clin Oncol. 2013;31:1842-8
41. Biankin AV, Piantadosi S, Hollingsworth SJ. Patient-centric trials for therapeutic development in precision oncology. Nature. 2015;526:361-70
Author contact
![]() Corresponding authors: Wei Zhang or Boris Pasche, Department of Cancer Biology, Wake Forest Baptist Comprehensive Cancer Center, 1 Medical School Blvd., Winston-Salem, NC 27157. wezhangedu or bpascheedu
Corresponding authors: Wei Zhang or Boris Pasche, Department of Cancer Biology, Wake Forest Baptist Comprehensive Cancer Center, 1 Medical School Blvd., Winston-Salem, NC 27157. wezhangedu or bpascheedu