Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(11):2822-2836. doi:10.7150/thno.19068 This issue Cite
Research Paper
C-X-C Motif Chemokine 10 Impairs Autophagy and Autolysosome Formation in Non-alcoholic Steatohepatitis
Xiang Zhang1, William KK Wu2, Weiqi Xu1, Kwan Man3, Xiaojuan Wang1, Juqiang Han1, 4, Wing Yan Leung1, Ruonan Wu1, Ken Liu1, 5, Jun Yu1 ![]()
1. Institute of Digestive Disease and The Department of Medicine and Therapeutics, State Key Laboratory of Digestive Disease, Li Ka Shing Institute of Health Sciences, CUHK Shenzhen Research Institute, The Chinese University of Hong Kong, Hong Kong;
2. Department of Anesthesia and Intensive Care, The Chinese University of Hong Kong, Hong Kong;
3. Department of Surgery, LKS Faculty of Medicine, The University of Hong Kong, Hong Kong;
4. Institute of liver disease, Beijing Military General Hospital, Beijing, China;
5. Faculty of Medicine, University of Sydney, Sydney, Australia.
Received 2017-1-5; Accepted 2017-5-9; Published 2017-7-8
Citation:
Zhang X, Wu WKK, Xu W, Man K, Wang X, Han J, Leung WY, Wu R, Liu K, Yu J. C-X-C Motif Chemokine 10 Impairs Autophagy and Autolysosome Formation in Non-alcoholic Steatohepatitis. Theranostics 2017; 7(11):2822-2836. doi:10.7150/thno.19068. https://www.thno.org/v07p2822.htm
Other stylesAbstract

C-X-C motif chemokine 10 (CXCL10) is a crucial pro-inflammatory factor in chronic hepatitis. Autophagy dysregulation is known to contribute to hepatic inflammatory injury. Hence, we investigated the regulatory effect of CXCL10 on the autophagosome-lysosome system during non-alcoholic fatty liver disease (NAFLD) development. The effect of CXCL10 ablation by neutralizing monoclonal antibody (mAb) or genetic knockout on autophagic flux was evaluated in cultured hepatocytes and animal models of NAFLD. Results demonstrated that CXCL10 ablation protected against hepatocyte injury in vitro and steatohepatitis development in mice. Autophagic flux impairment was rectified by CXCL10 inhibition using anti-CXCL10 mAb in AML-12 and HepG2 liver cell lines and primary hepatocytes as evidenced by the attenuated accumulation of p62/SQSTM1 and LC3-II proteins and increased autophagic protein degradation. Impaired autophagic flux was significantly restored by CXCL10 knockout or anti-CXCL10 mAb in mice. Bafilomycin A1, an inhibitor of autolysosome formation, abolished the rectifying effect of anti-CXCL10 mAb or CXCL10 knockdown in AML-12 and primary hepatocytes, indicating CXCL10 impaired late-stage autophagy in NAFLD. Anti-CXCL10 mAb treatment also increased the fusion of LC3-positive autophagosomes with lysosomes in HepG2 cells challenged with palmitic acid, suggesting that CXCL10 ablation restored autolysosome formation. Consistently, the number of autolysosomes was significantly increased by CXCL10 knockout in mice as shown by electron microscopy. In conclusion, upregulated CXCL10 in steatohepatitis impairs autophagic flux by reducing autolysosome formation, thereby inhibiting autophagic protein degradation and the accumulation of ubiquitinated proteins, leading to the development of steatohepatitis.
Keywords: Cytokine, non-alcoholic fatty liver disease, autophagy, lysosome, autolysosome.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is currently the most common cause of chronic liver disease worldwide. The prevalence of NAFLD in Hong Kong has increased to over 27% in the general population (1). The pathological spectrum of NAFLD ranges from simple steatosis to nonalcoholic steatohepatitis (NASH), the latter of which can lead to progressive liver fibrosis, cirrhosis and eventually hepatocellular carcinoma (HCC). However, the molecular mechanisms driving NAFLD progression remain largely elusive and thus treatment options for this disease remain limited. There is a compelling need for understanding the molecular pathogenesis of NASH to provide new insights into mechanism-based therapy.
Autophagy is a critical catabolic pathway that targets cellular components for lysosomal degradation (2). During this process, the cytoplasm is engulfed by double-membrane vesicles. The resulting autophagosomes ultimately fuse with lysosomes in which the engulfed contents are degraded (3). Autophagy has been implicated in pathophysiological processes including lipid metabolism (4), insulin resistance (5, 6), inflammation (7), oxidative stress (8) and endoplasmic reticulum (ER) stress (6). In the liver, decreased autophagic function is associated with the progression of steatosis to steatohepatitis (9). Patients with NASH also display impaired autophagic function (9). However, the molecular mechanisms underlying autophagic dysfunction in the pathogenesis of NASH are still largely unclear.
Chemokines play important roles in the regulation of autophagy (10). A crosstalk between autophagy and chemokine production has been indicated (10), in which inhibition of autophagy enhances pro-inflammatory cytokine expression (7). In influenza virus infection, C-X-C motif chemokine 10 (CXCL10), a pro-inflammatory mediator, is induced by the autophagic response (11). CXCL10 is also implicated in mediating inflammation in various types of hepatitis, including hepatitis C virus infection, hepatitis B virus infection and steatohepatitis (12). However, it is uncertain whether CXCL10 could regulate autophagy in NAFLD development. In this study, we aim to elucidate the role and molecular consequences of CXCL10 in regulating autophagy during the development of steatohepatitis using cultured hepatocytes and CXCL10 knockout (CXCL10-/-) mouse model. Here, we show for the first time that CXCL10 impairs the autophagosome-lysosome system in the development of steatohepatitis. We also provide evidence that elimination of CXCL10 by genetic ablation or antibody-mediated neutralization restores autolysosome formation in NAFLD, thus promoting degradation of endogenous substrates, including ubiquitinated proteins, p62/SQSTM1 and damaged mitochondria. These findings suggest that CXCL10 contributes to defective autolysosome formation that impairs the autophagic pathway in the evolution of steatohepatitis.
Materials and Methods
Cell culture
The human hepatoma cell line HepG2 and immortalized hepatocyte cell line LO2 were cultured in Dulbecco's Modified Eagle's Medium (DMEM) medium (Thermo Fisher Scientific, Waltham, MA). The immortalized mouse hepatocyte cell line AML-12 was cultured in DMEM/F-12 medium (Thermo Fisher Scientific). Primary hepatocytes were isolated from C57BL/6 mice and cultured in William's E medium (Thermo Fisher Scientific). AML-12 and LO2 cells were subsequently exposed to control medium or DMEM/F12 medium deficient in methionine and choline (MCD) for 24h. HepG2 cells and primary hepatocytes were treated with 200μM palmitic acid or bovine serum albumin (BSA) control for 24h. For CXCL10 inhibition, cells were treated with 1μg/mL anti-CXCL10 mAb or isotype-matched control IgG2A antibody for 24h. Primary hepatocytes and AML-12 cells were also treated with 0.3nM or 10nM bafilomycin A1 (Sigma-Aldrich, St. Louis, MO) to determine autophagic flux. For the gain of function of CXCL10, cells were administrated with 10ng/mL CXCL10 recombinant protein for 24h (PeproTech, Rocky Hill, NJ). To evaluate the fusion of autophagosomes with lysosomes, HepG2 cells were transfected with RFP-GFP-LC3 plasmid and the immunofluorescence signals were detected by confocal microscopy.
Animals and treatments
CXCL10-/- and C57BL/6 wildtype (WT) mice were randomly fed with MCD diet to induce NASH or the corresponding control diet supplemented with choline bitartrate (2g/kg) and DL-methionine (3g/kg) (ICN Biomedicals, Costa Mesa, CA) for 4 weeks. CXCL10-/- and WT mice were also fed with high-fat high-cholesterol diet (HFHC) (Specialty feeds, Glen Forrest, WA, Australia) or normal chow for 8 weeks. In another experiment, WT mice were administered with anti-CXCL10 monoclonal antibodies (mAb) (MAB466, R&D systems, Minneapolis, MN) or control mAb (MAB006, R&D systems) by intraperitoneal injection (50 μg per mouse) for 10 days after induction of steatohepatitis by feeding mice with the MCD diet for 3 weeks. Animals were housed in a temperature-controlled room under a 12h light /12h dark cycle and had free access to food and water. All animal studies were performed in accordance with guidelines approved by the Animal Experimentation Ethics Committee of the Chinese University of Hong Kong.
Human subjects
The relationship between CXCL10 and p62/SQSTM1 levels was investigated in human serum samples from 129 patients with biopsy-proven NAFLD and 66 healthy subjects. Two experienced pathologists scored the histological grading and staging of NAFLD. Control subjects had normal liver histology or less than 5% of hepatic triglyceride content measured by proton-magnetic resonance spectroscopy with no history of diabetes or hypertension, while NAFLD patients were defined by the presence of steatosis (12). All subjects had given written informed consent and the study protocol was approved by the Clinical Research Ethics Committee of the Chinese University of Hong Kong.
Autophagic protein degradation assay
Click-iT AHA Alexa Fluor 488 Protein Synthesis HCS Assay kit (Thermo Fisher Scientific) was used to determine the long-lived protein degradation. Isolated primary hepatocytes and HepG2 cells were treated with palmitic acid challenged with 1μg/mL anti-CXCL10 mAb or isotype-matched control IgG2A antibody, or 10ng/mL CXCL10 recombinant protein for 24h. Cells were first cultured in methionine-free medium to deplete the intracellular L-methionine. Click-iT L-azidohomoalanine (AHA) reagent was added for 18h and the cultured cells were incubated with complete medium for 2h. The cells were then subject to a click reaction to tag the AHA-containing proteins with a fluorescently-tagged alkyne probe. Lastly, the fluorescence intensity of the cells was analyzed by flow cytometry.
Western blot analysis
Total proteins were extracted using CytoBuster Protein Extraction Reagent containing a protease inhibitor cocktail (Roche, Indianapolis, MN) and Phosphatase Inhibitor Cocktail (Roche). Western blot was performed as previously described with the following primary antibodies: LC3B (NB100-2220, Novus Biologicals, Littleton, CO), p62/SQSTM1 (H00008878, Novus Biologicals), cathepsin D (sc-10725, Santa Cruz), Lysosomal-associated membrane protein (LAMP2) (NBP1-71692, Novus Biologicals), Rubicon (8465, Cell Signaling Technology, Danvers, MA), phospho-AMPKα (2535, Cell Signaling), phospho-mTOR (2971, Cell Signaling), phosphor-p70 ribosomal protein S6 kinase (p70 S6 Kinase) (9205, Cell Signaling), phosphor-4E-BP1 (2855, Cell Signaling), ubiquitin (3936S, Cell Signaling), GRP78 (sc-13968, Santa Cruz Biotechnology, Santa Cruz, CA), phosphorylated inositol-requiring enzyme 1α (p-IRE1α) (NB100-2323, Novus Biologicals), IRE1α (3294, Cell Signaling), C/EBP homologous protein (CHOP) (2895, Cell Signaling), X-box binding protein 1 (XBP-1) (sc-7160, Santa Cruz), β-Actin (sc-4778, Santa Cruz), and GAPDH (sc-25778, Santa Cruz) .
Transmission electron microscopy in liver tissues
Liver sections with a size less than 1mm3 were fixed in 2.5% glutaraldehyde in cacodylate buffer at 4°C. The tissues were then post-fixed in 1% osmium tetroxide for 1h at room temperature, dehydrated and infiltrated with epoxy resin/propylene oxide and epoxy resin. The tissues were sliced into 60-80nm sections. The sections were then observed under a Philips CM100 transmission electron microscope with a TENGRA 2.3K X 2.3K TEM camera.
Statistical analysis
Differences between two groups were compared by non-parametric Mann-Whitney U test or Student's t test. Multiple group comparisons were made by one-way ANOVA after Bonferroni's correction or Kruskal-Wallis test (GraphPad Software, San Diego, CA). The co-expression pattern was determined by Spearman's rank-sum correlation analysis. Data were expressed as mean ± SEM and considered significant at P < 0.05.
Additional experimental methods are in Supplementary Materials and Methods.
Results
CXCL10 is associated with impaired autophagy and the clearance of autophagosomes in hepatocytes
To elucidate the role of CXCL10 in autophagic response in the development of steatohepatitis, autophagic flux was determined by measuring the protein levels of LC3 and p62/SQSTM1 in two cell line models of steatohepatitis (i.e. HepG2 cells treated with palmitic acid and AML-12 cells exposed to MCD medium) treated with or without anti-CXCL10 mAb. After exposing HepG2 cells to palmitic acid for 24h, CXCL10 levels were significantly increased in the culture medium (Fig. S1A), concomitant with the increased lipid accumulation and lipid peroxide formation in hepatocytes (Fig. S1B-C). The protein levels of LC3-II and p62/SQSTM1 were significantly increased in HepG2 cells supplemented with palmitic acid for 24h (Fig. 1A), suggesting that the clearance of autophagosomes is compromised and autophagic flux is impaired in experimental steatohepatitis (13). However, when palmitic acid-challenged HepG2 cells were treated with anti-CXCL10 mAb, the protein expression of LC3-II and p62/SQSTM1 were decreased (Fig. 1A) in parallel with decreased steatosis and oxidative injury (Fig. S1B-C). Similar phenomena were observed in MCD medium-challenged AML-12 cells (Fig. 1A) (12). The inhibition of CXCL10 release by anti-CXCL10 mAb in AML-12 cells was confirmed by macrophage transwell chemotaxis assay (Fig. S2).
Figure 1
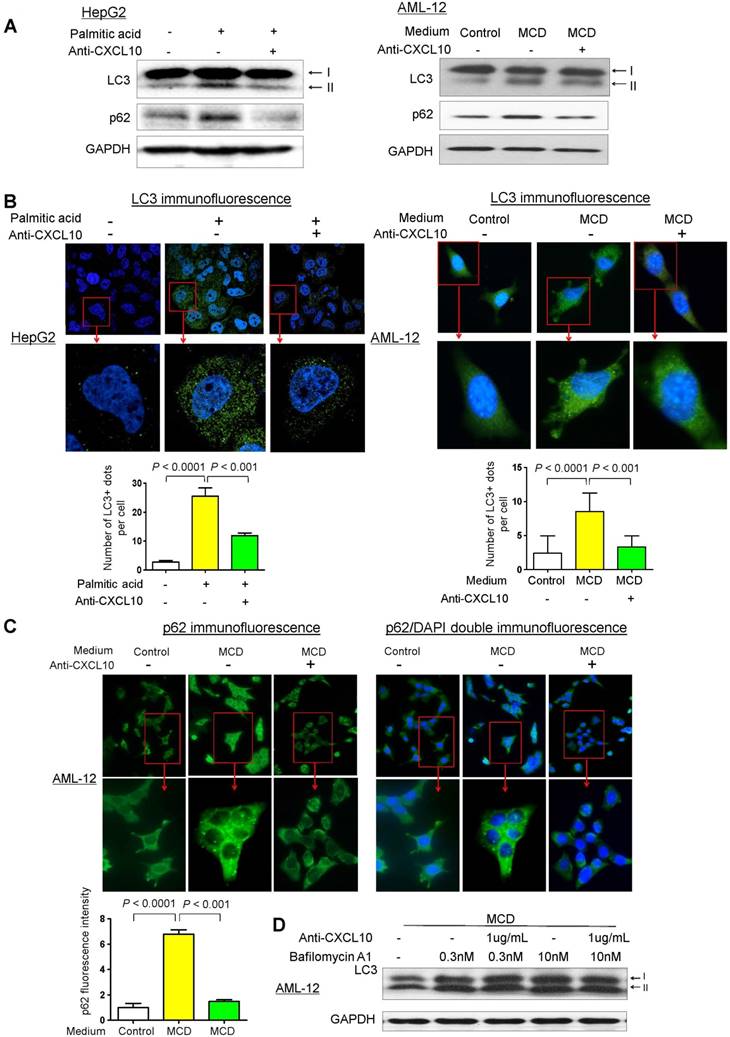
Restoration of autophagy by CXCL10 neutralization in hepatocytes. (A) LC3 and p62/SQSTM1 protein levels in MCD medium cultured mouse AML-12 hepatocytes and palmitic acid treated human HepG2 hepatocytes administrated with control or anti-CXCL10 mAb (1μg/mL); (B) Decreased LC3 positive autophagic vacuoles (LC3+ dots) was demonstrated by immunofluorescence in MCD-cultured AML-12 and palmitic acid treated HepG2 hepatocytes administrated with 1μg/mL anti-CXCL10 mAb compared to hepatocytes treated with control mAb, autophagosomes were quantified by counting the number of LC3 positive (LC3+) dots or vacuoles per cells (a minimum of 50 cells per preparation in two independent experiments); (C) Decreased positive staining of p62/SQSTM1 was determined by immunofluorescence in MCD medium cultured AML-12 hepatocytes administrated with 1μg/mL anti-CXCL10 mAb than control mAb, the intensities were quantified by Image J software. (D) Anti-CXCL10 mAb could not reduce the expression of LC3-II in the presence of bafilomycin A1 in AML-12 hepatocytes.
To confirm the inhibitory effect of CXCL10 on autophagic flux, we analyzed the accumulation of LC3-positive autophagic vacuoles in HepG2 and AML-12 cells by immunofluorescent staining with the LC3 antibody. Under confocal microscopy, LC3-positive dots were more frequently observed in both palmitic acid-challenged HepG2 cells and MCD medium-challenged AML-12 compared to the control cells (Fig. 1B). In contrast, CXCL10 neutralization by anti-CXCL10 mAb blunted the increase of LC3-positive dots in HepG2 under palmitic acid (P < 0.001) and AML-12 under MCD medium (P < 0.001) (Fig. 1B). Likewise, immunofluorescence analysis of p62/SQSTM1 demonstrated decreased accumulation of p62/SQSTM1 in anti-CXCL10 mAb-treated AML-12 cells in MCD medium (Fig. 1C), suggesting that CXCL10 neutralization by its antibody could restore autophagic flux in hepatocytes with steatohepatitis changes.
To confirm the effect by which CXCL10 inhibition restored autophagic flux, we treated AML-12 cells with bafilomycin A1, which inhibits lysosomal enzyme activity and the fusion of autophagosomes with lysosomes. We examined the amount of newly formed autophagosomes through LC3-II immunoblotting. AML-12 hepatocytes exposed in MCD medium showed increased LC3-II level after bafilomycin A1 application, whereas CXCL10 inhibition by anti-CXCL10 mAb did not decrease LC3-II in the presence of bafilomycin A1 (Fig. 1D), suggesting the decreased autophagosome degradation, rather than increased formation, is the cause of autophagosome accumulation by CXCL10.
We also confirmed the direct inhibitory effect of CXCL10 on autophagy by treating AML-12 hepatocytes with CXCL10 recombinant protein. p62/SQSTM1 and LC3-II protein levels were increased by recombinant CXCL10 in AML-12 hepatocytes, accompanied by increased intracellular lipid accumulation and lipid peroxidation (Fig. S3A-B). Collectively, these results indicate that CXCL10 is associated with impaired autophagy in hepatocytes in steatohepatitis.
CXCL10 inhibits autophagic protein degradation in hepatocytes
To further evaluate the detrimental role of CXCL10 in autophagic function, we quantified the degradation of long-lived proteins, which is a classic and gold standard method of measuring autophagic flux (14), in hepatocytes treated with anti-CXCL10 mAb or challenged with recombinant CXCL10 protein. Autophagic protein degradation was measured by the Click-iT AHA, which is a surrogate for L-methionine with an azide group that is incorporated into proteins during protein synthesis. Inhibition of autophagy or inhibition of lysosome proton pump activity has been shown to increase the signal from fluorescent long-lived proteins (15). Herein we found that treatment with palmitic acid in both primary hepatocytes and HepG2 cell lines increased the amount of fluorescently-labelled long-lived proteins compared to control, suggesting the impairment of autophagic flux by palmitic acid (Fig. 2A-B). Conversely, CXCL10 neutralization by anti-CXCL10 mAb blunted the increases in fluorescent signals in primary hepatocytes (P < 0.01) and HepG2 under palmitic acid (P < 0.0001) (Fig. 2A-B). Consistently, fluorescently-labelled long-lived proteins were increased by recombinant CXCL10 protein treatment compared to control in both primary hepatocytes and HepG2 cells (P < 0.05) (Fig. 2A-B), indicating that CXCL10 inhibits autophagic proteolysis of long-lived proteins in hepatocytes. Taken together, these results further confirmed that CXCL10 impairs autophagic flux in hepatocytes.
CXCL10 knockout restores autophagy in mice steatohepatitis
To elucidate the role of CXCL10 in autophagy in the development of steatohepatitis in vivo, autophagic flux was determined by measuring hepatic protein levels of LC3 and p62/SQSTM1 in CXCL10-/- and C57BL/6 WT mice. WT mice fed with MCD diet developed more pronounced steatohepatitis compared to CXCL10-/- mice fed with the same diet (Fig. S4A). Increased protein levels of p62/SQSTM1 and LC3-II were observed in MCD-fed WT mice with severe steatohepatitis compared with control diet-fed WT mice with normal liver histology (Fig. 3A). Hepatic accumulation of p62/SQSTM1 and LC3-II expression was significantly decreased in CXCL10-/- mice compared to WT mice fed with MCD diet, indicating that restoration of autophagic flux was due to CXCL10 deficiency (Fig. 3A). However, p62 mRNA levels did not change in CXCL10-/- and WT mice (Fig. S4B), indicating that the decreased p62 levels in CXCL10-/- mice were caused by autophagic degradation but not reduced oxidative stress and inflammation. To further confirm the effect of CXCL10 in inhibiting autophagic flux in steatohepatitis, we detected LC3-positive autophagosomes by LC3 immunofluorescence staining in liver tissue sections of CXCL10-/- mice and WT mice. A reduced accumulation of autophagosomes was found in CXCL10-/- mice compared to WT mice fed with MCD diet (Fig. 3B). Likewise, reduced accumulation of p62/SQSTM1 protein by immunofluorescence staining was observed in CXCL10-/- mice as compared with WT mice fed with MCD (P < 0.05, Fig. 3B). These findings were further corroborated by a second dietary steatohepatitis model, which showed that LC3-II and p62/SQSTM1 protein levels, as well as lipid peroxidation, were decreased in primary hepatocytes isolated from HFHC-fed CXCL10-/- mice compared to those from HFHC-fed WT mice (Fig. S5A-B). Notably, these effects could be abolished by bafilomycin A1 (Fig. S5C). Collectively, these results suggest that CXCL10 could be associated with the accumulation of autophagosomes as a result of impaired autophagic flux in steatohepatitis.
Figure 2
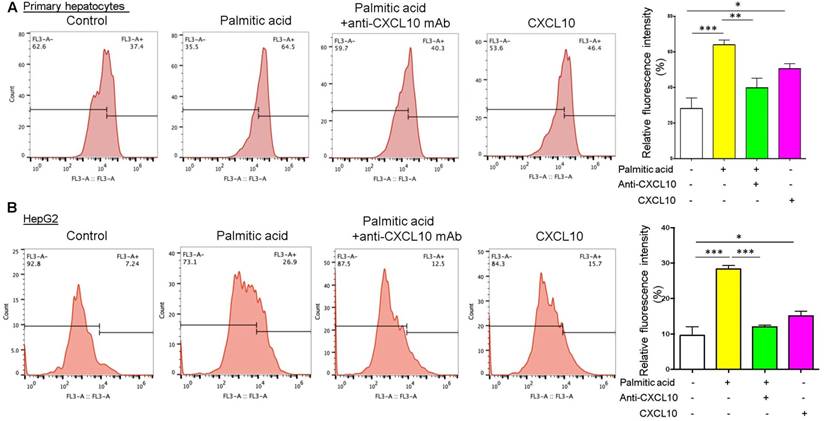
CXCL10 inhibits autophagic proteolysis of long-lived proteins in hepatocytes. Fluorescently-labelled long-lived proteins were decreased by anti-CXCL10 mAb treatment and increased by recombinant CXCL10 protein treatment compared to control in both (A) primary hepatocytes and (B) HepG2 cells. AHA-positive fluorescent was quantified by flow cytometry as described in the materials and methods. *P < 0.05, **P < 0.01, ***P < 0.0001.
CXCL10 neutralization restores autophagy in mice with steatohepatitis
To determine whether anti-CXCL10 mAb treatment could restore impaired autophagy in mice, we assessed the status of the autophagosome-lysosome system in MCD-fed WT mice with or without the administration of anti-CXCL10 mAb. As shown in Fig. 3C, anti-CXCL10 mAb treatment reduced LC3-II and p62/SQSTM1 levels in MCD-fed C57BL/6 mice compared to untreated MCD-fed mice by Western blot analysis, suggesting CXCL10 neutralization by its antibody increased autophagic flux. Moreover, immunofluorescence of LC3 and p62/SQSTM1 showed decreased accumulation of autophagosomes and p62/SQSTM1 in mice administered with anti-CXCL10 mAb (Fig. 3D-E), indicating that CXCL10 neutralization restores autophagy activity by preventing autophagosome accumulation in steatohepatitis.
CXCL10 impairs autophagy at the late stage by blocking the fusion of autophagic vesicles with lysosomes
Autophagy is comprised of early and late stages. During the early stage, there is a formation of double-membrane bound vacuoles (autophagosomes), while the late stage is characterized by the production of autolysosomes (through the fusion of autophagosomes with lysosomes) and lysosome-dependent degradation. The accumulation of autophagosomes, LC3-II and p62/SQTM1, found in WT mice with steatohepatitis and their restoration in CXCL10-/- mice suggests that CXCL10 might mediate late-stage autophagic impairment in the pathogenesis of steatohepatitis. To confirm this postulation, we stained anti-CXCL10 mAb-treated cells with anti-LC3 and anti-LAMP1 antibodies to visualize autophagosomes and lysosomes, respectively. HepG2 cells treated with palmitic acid showed a substantial dissociation between LC3 and LAMP1, which respectively correspond to autophagosomes and late lysosomes (Fig. 4A). In contrast, anti-CXCL10 mAb treatment increased fusion of LC3-positive autophagosomes with LAMP1-positive lysosomes in HepG2 cells under palmitic acid condition as indicated by increased LAMP1/LC3 co-localization (Fig. 4A).
Figure 3
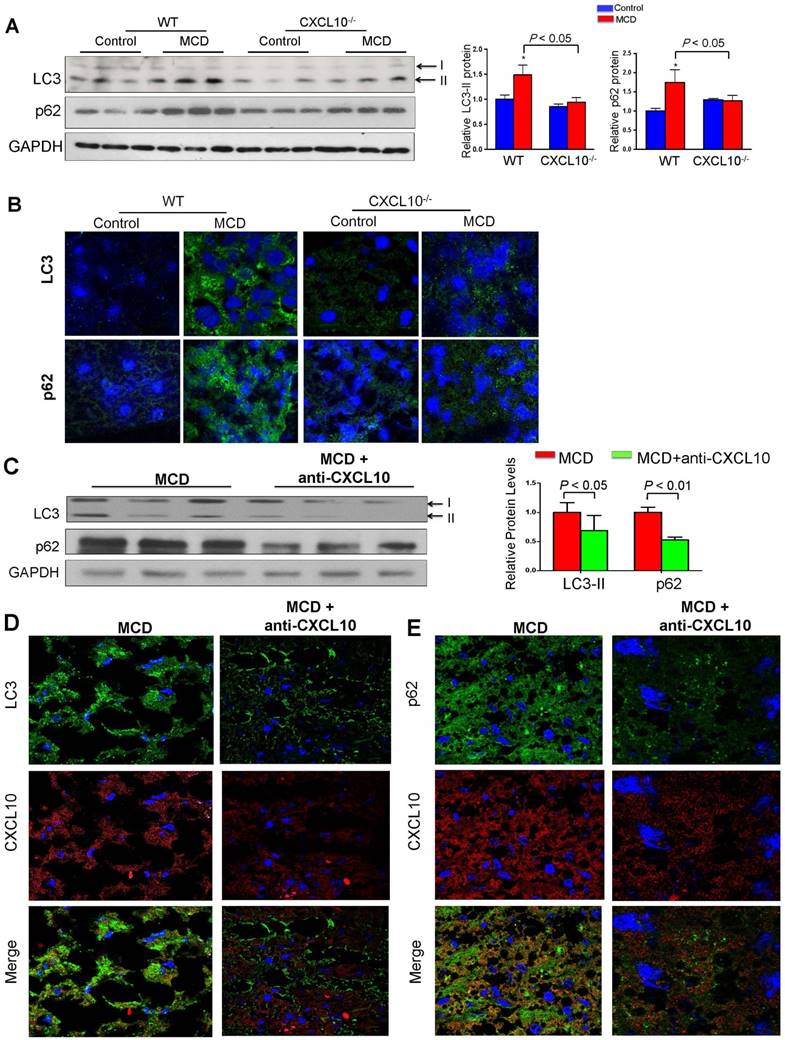
CXCL10 ablation by genetic knockout or antibody neutralization prevents hepatic autophagic impairment induced by experimental steatohepatitis in mice. (A) Hepatic LC3 and p62/SQSTM1 protein levels in the liver sections of the WT and CXCL10-/- mice fed with control or methionine and choline deficient (MCD) diet for 4 weeks. GAPDH was detected as loading control; (B) Representative immunofluorescent staining of LC3 and p62/SQSTM1 in the livers of WT and CXCL10-/- mice fed with control or MCD diet for 4 weeks; (C) LC3-II and p62/SQSTM1 protein expression were downregulated in MCD-fed WT mice administrated with anti-CXCL10 mAb (50 μg per mouse) than mice administrated with control mAb by Western blot; (D) Representative immunofluorescent staining of LC3 and CXCL10 in MCD-fed WT mice administrated with anti-CXCL10 mAb or control mAb; (E) Representative immunofluorescent staining of p62/SQSTM1 and CXCL10 in MCD-fed WT mice administrated with anti-CXCL10 mAb or control mAb.
Figure 4
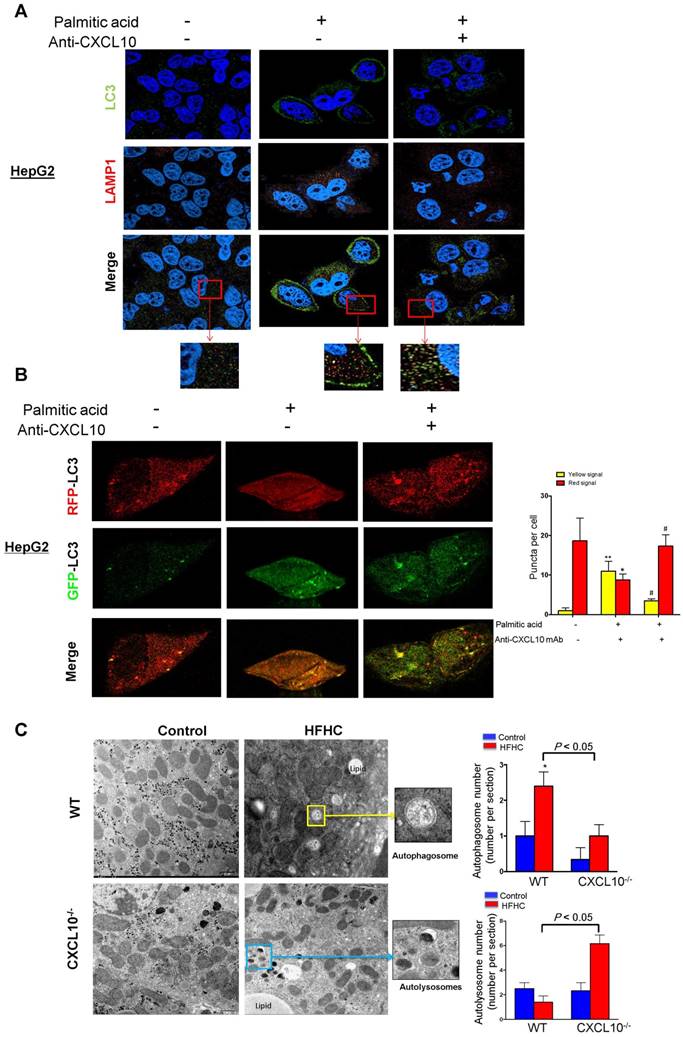
CXCL10 impairs autophagosome-lysosome fusion. (A) LC3 and LAMP1 co-localization was increased by anti-CXCL10 mAb treatment in palmitic acid-treated HepG2 hepatocytes compared to cells treated with control mAb by co-immunofluorescent staining. (B) RFP- and GFP-labeled LC3 was transfected into HepG2 cells. The overlapped signals (yellow) of the RFP-LC3 (red) and GFP-LC3 (green) fluorescence were increased in palmitic acid-treated HepG2 cells compared with control cells by confocal microscope; whereas the overlapped signals (yellow) in HepG2 cells were reduced by anti-CXCL10 mAb treatment. *P < 0.05 vs control, **P < 0.01 vs control, #P < 0.05 vs palmitic acid; (C) Electron microscope images showed the accumulation of autophagosomes (yellow arrows) were mainly observed in WT mice with HFHC, while autolysosomes (blue arrows) were frequently identified in CXCL10-/- mice compared with WT mice, autophagosomes and autolysosomes were counted from electron microscopy.
Moreover, we transfected RFP/GFP dual-labeled LC3 into HepG2 cells treated with palmitic acid in the presence or absence of anti-CXCL10 mAb to evaluate autophagosomes and autolysosomes. As shown in Fig. 4B, we found a clear increase in the overlapped signals (yellow) of RFP-LC3 (red) and GFP-LC3 (green) fluorescence in palmitic acid-treated HepG2 cells compared with control cells (Fig. 4B), indicating the accumulation of unfused autophagosomes. In the presence of anti-CXCL10 mAb, the overlapped signals (yellow) in HepG2 cells were dramatically reduced, suggesting fewer autophagosomes but increased autolysosomes (Fig. 4B). These results indicate that the impaired late-stage autophagy by CXCL10 is mediated by blocking the fusion of autophagic vesicles with lysosomes in steatohepatitis.
We subsequently performed electron microscopy to determine the histological changes of autophagic structures including autophagosomes and autolysosomes in liver tissue from CXCL10-/- and WT mice. Autophagosomes, known as initial autophagic vacuoles, have a double membrane that encloses cytosol and organelles with normal morphology. Autolysosomes, known as late autophagic vacuoles, have one limiting membrane enclosing cytoplasmic material and organelles at various stages of degradation. As shown in Fig. 4C, the accumulation of autophagosomes was mainly observed in WT mice, but not in CXCL10-/- mice fed with HFHC diet, while autolysosomes were more frequently identified in CXCL10-/- compared with WT mice fed with HFHC diet (Fig. 4C). These results further suggest that CXCL10 inhibits autophagic flux by impairing the fusion of autophagosomes with lysosomes in the development of nutritional steatohepatitis.
CXCL10 weakens lysosomal acidification in steatohepatitis
The potential mechanisms underlying impaired autophagy by CXCL10 was evaluated by characterizing lysosomal alterations in steatohepatitis. Acidification of autolysosomes is crucial for activating cathepsins and effecting proteolysis of substrates, thus contributing to the maintenance of autophagic flux. Procathepsin D is a short-lived inactive precursor of lysosomal aspartyl protease which is cleaved to mature cathepsin D upon acidification in lysosomes. Therefore, reduced procathepsin D/cathepsin D protein expression ratio indicates the induction of lysosomal acidification. We found that the procathepsin D/cathepsin D ratio was significantly decreased by anti-CXCL10 mAb treatment in MCD-fed C57/BL6 WT mice (P < 0.05, Fig. 5A). In addition, measurement of average lysosomal pH with LysoSensor Yellow/Blue DND-160-dextran indicated that lysosomal acidification was restored by anti-CXCL10 mAb treatment in MCD medium-cultured hepatocytes (Fig. 5B). These data suggest that CXCL10 impairs lysosomal acidification in steatohepatitis.
In addition, LAMP2 protein level was upregulated when AML-12 cells were cultured in MCD medium compared to control medium and this upregulation was blunted by anti-CXCL10 mAb (Fig. 5C). In keeping with this in vitro result, decreased LAMP2 protein expression was observed in MCD-fed mice administered with anti-CXCL10 mAb compared with MCD-fed mice (Fig. 5C). In addition, the co-localization of p62/SQSTM1 and LAMP2, which indicates the impaired autolysosome recycling (16), was clearly diminished in MCD-fed mice administered with CXCL10 mAb compared with MCD-fed mice as determined by co-immunofluorescent staining of p62/SQSTM1 and LAMP2 (Fig. S6A). The decreased p62/SQSTM1 in lysosomes by anti-CXCL10 mAb treatment was corroborated by decreased co-localization of p62/SQSTM1 and LAMP2 in CXCL10-/- mice compared to WT mice fed with MCD diet (Fig. S6B). Collectively, these results indicate that CXCL10 neutralization by its antibody restores autophagosome-lysosome fusion and autolysosome formation in steatohepatitis.
CXCL10 impairs autophagosome-lysosome fusion through regulating Rubicon
To explore the mechanism underlying CXCL10-mediated suppression of autophagosome-lysosome fusion, we evaluated Rubicon expression in hepatocytes treated with or without anti-CXCL10 mAb. Rubicon is a Beclin 1-interacting negative regulator of autophagosome-lysosome fusion and plays a pathogenic role in NASH (17). Our results showed that incubation of the HepG2 cell line with palmitic acid for 24h increased Rubicon protein levels. Conversely, anti-CXCL10 mAb added to HepG2 cells with palmitic acid significantly reduced Rubicon levels (Fig. 5D). Consistently, recombinant CXCL10 protein increased Rubicon protein levels in AML-12 cell line (Fig. S7). Therefore, CXCL10 may suppress autophagosome-lysosome fusion through the regulation of Rubicon.
AMPK/mTOR signaling is a well-known pathway involved in the regulation of autophagy. Therefore, we examined the effect of CXCL10 on this pathway. After 24h treatment with palmitic acid, phospho-AMPKα levels were decreased compared with control cells in HepG2, while anti-CXCL10 mAb treatment in palmitic acid-treated HepG2 cells increased phospho-AMPKα levels (Fig. 5E). Consistent with results from others (17), we found that the phosphorylation of mTOR and its downstream signaling kinases phospho-p70S6K and phospho-4E-BP1, were significantly suppressed in HepG2 cells treated with palmitic acid for 24h compared to HepG2 cells treated with BSA control, indicating the increased autophagosome formation by palmitic acid treatment. However, when palmitic acid-treated HepG2 cells were administered with anti-CXCL10 mAb, the protein expression of phospho-mTOR, phospho-p70S6K and phospho-4E-BP1 was increased (Fig. 5F).
CXCL10 induces mitochondrial dysfunction, ubiquitin-positive inclusions and the accumulation of ER stress in steatohepatitis
As impaired autophagy could result in ROS production due to the accumulation of damaged mitochondria, we evaluated the mitochondria ROS production by CXCL10 in steatohepatitis. MitoTracker ROS staining was performed in HepG2 cells and quantified by flow cytometry in hepatocytes. We found that HepG2 cells challenged with palmitic acid showed significantly increased mitochondrial ROS production as compared to HepG2 cells in control medium (Fig. 6A). Conversely, treatment with anti-CXCL10 mAb significantly abolished mitochondrial ROS level in palmitic acid-exposed HepG2 cells (Fig. 6A). Similarly, we found significantly decreased mitochondrial ROS production as evidenced by reduced MitoTracker fluorescence by flow cytometry in primary hepatocytes isolated from HFHC-fed CXCL10-/- mice versus those from WT mice (P < 0.0001, Fig. 6B). Anti-CXCL10 mAb treatment also reduced the NADP+-to-NADPH ratio in MCD-fed C57BL/6 mice compared to untreated mice (Fig. 6C). As NADPH is essential for redox homeostasis, the accumulation of NADP+ by anti-CXCL10 mAb could result in ROS reduction.
Figure 5
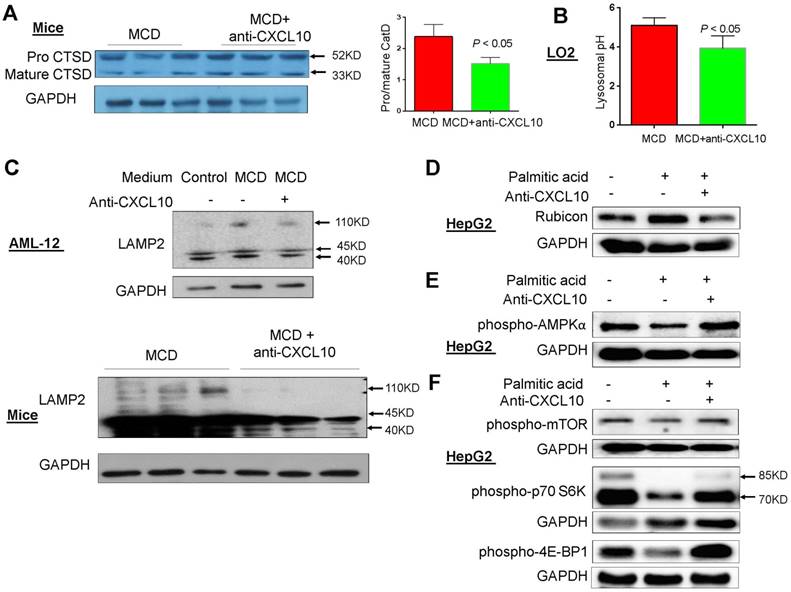
CXCL10 impaired lysosomal acidification. (A) The cleavage of lysosome specific enzyme procathepsin D (Procat D) into mature cathepsin D (mCat D) was restored by anti-CXCL10 mAb treatment in MCD-fed mice; (B) Lysosomal pH value was decreased by anti-CXCL10 mAb treatment in MCD medium cultured LO2 cells; (C) LAMP2 expression was blunted by anti-CXCL10 mAb treatment in both MCD medium cultured AML-12 cells and MCD-fed mice liver tissues; (D) Rubicon expression was blunted by anti-CXCL10 mAb treatment in palmitic acid-challenged HepG2 cells; (E) phospho-AMPKα, (F) phospho-mTOR and phosphor-p70S6 Kinase expression was restored by anti-CXCL10 mAb treatment in palmitic acid-challenged HepG2 cells.
Figure 6
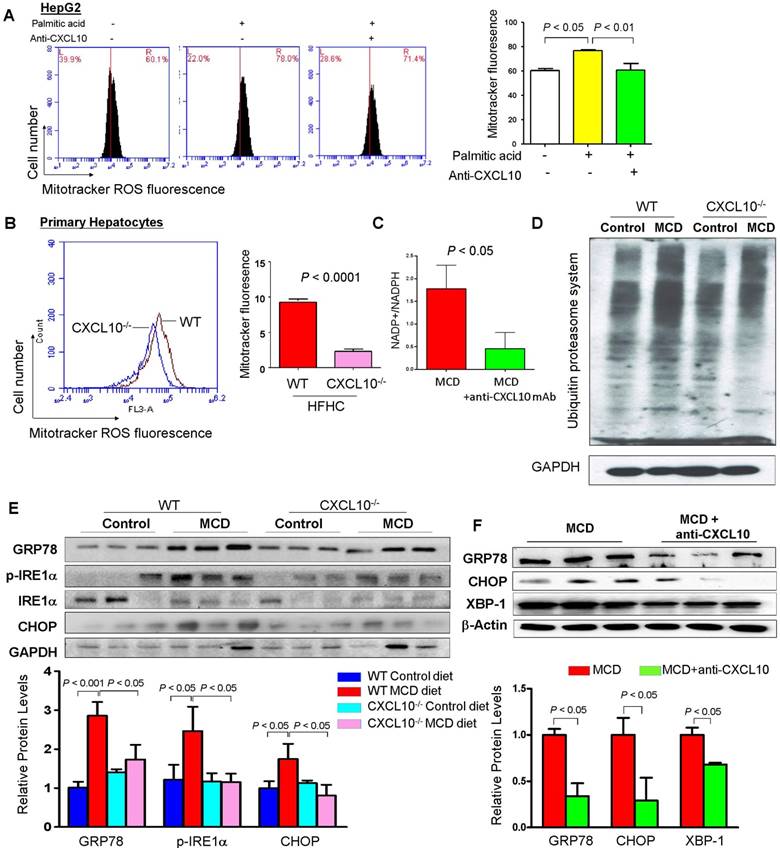
CXCL10 promotes mitochondria dysfunction, polyubiquitinated proteins accumulation and ER stress. (A) Mitochondrial ROS production by Mitotracker staining was abolished by anti-CXCL10 mAb treatment in palmitic acid-treated HepG2 hepatocytes. (B) Mitochondrial ROS production by Mitotracker staining was significantly decreased in primary hepatocytes isolated from HFHC-fed CXCL10-/- mice than those isolated from HFHC-fed WT mice; (C) Anti-CXCL10 mAb decreased the NADP+ to NADPH ratio in mice; (D) The protein levels of ubiquitinated proteins and (E) protein expression of ER stress markers GRP78, p-IRE1α, IRE1α and CHOP in WT and CXCL10-/- mice fed with control or MCD diet for 4 weeks; (F) Protein levels of ER stress markers GRP78, CHOP and XBP-1 were downregulated in MCD-fed WT mice administrated with anti-CXCL10 mAb compared to those administrated with control mAb by Western blot.
The accumulation of ubiquitinated proteins is one of the hallmarks of autophagy deficiency (18). Excessive lipid accumulation in hepatocytes can provoke the formation of ubiquitinated proteins during NASH development. We further assessed hepatic accumulation of ubiquitinated proteins in CXCL10-/- and WT mice using an anti-ubiquitin antibody capable of detecting all ubiquitinated proteins. We found that hepatic accumulation of polyubiquitinated proteins was significantly reduced in CXCL10-/- mice compared to WT mice fed with MCD diet (Fig. 6D), suggesting that CXCL10 induces accumulation of ubiquitin-positive inclusions. Consistent with the decreased levels of polyubiquitinated proteins, MCD-fed CXCL10-/- mice showed significantly reduced protein levels of ER stress markers, including GRP78, p-IRE1α and CHOP compared to MCD-fed WT mice (Fig. 6E). Moreover, CXCL10 neutralization in MCD-fed WT mice suppressed the protein levels of GRP78, XBP-1 and CHOP (Fig. 6F). Collectively, these findings demonstrate that CXCL10 promotes accumulation of polyubiquitinated proteins and ER stress in steatohepatitis.
CXCL10 levels are positively correlated with p62/SQSTM1 levels in human serum
We ascertained the clinical impact of CXCL10 in the regulation of autophagy in NASH patients. Human serum levels of CXCL10 and p62/SQSTM1 were evaluated using an enzyme-linked immunosorbent assay (ELISA)-based assay in a well-established prospective cohort of 66 control subjects and 129 NAFLD patients (Table S1). Our results showed that serum CXCL10 was positively correlated with serum p62/SQSTM1 levels (r=0.30, P < 0.0001) in human subjects (Fig. 7A), giving clinical evidence for the positive relationship of CXCL10 and autophagy impairment in steatohepatitis.
Discussion
Accumulating evidence suggests that autophagy impairment contributes to NASH development in human (9) and mice (19). However, the molecular mechanism underlying autophagic defects in NASH remains obscure and effective therapeutic strategies for correcting autophagic defects in NASH are limited. Our current study suggests that CXCL10 is responsible for autophagic impairment during steatohepatitis development. Upregulated CXCL10 in in vitro and in vivo steatohepatitis inhibits the fusion of autophagosomes and lysosomes, thereby attenuating the autophagic flux. This attenuation leads to the inhibition of autophagic protein degradation and the prominent accumulation of undegraded autophagy substrates, such as p62/SQSTM1 aggregates and ubiquitinated proteins, ultimately resulting in steatohepatitis development.
Figure 7
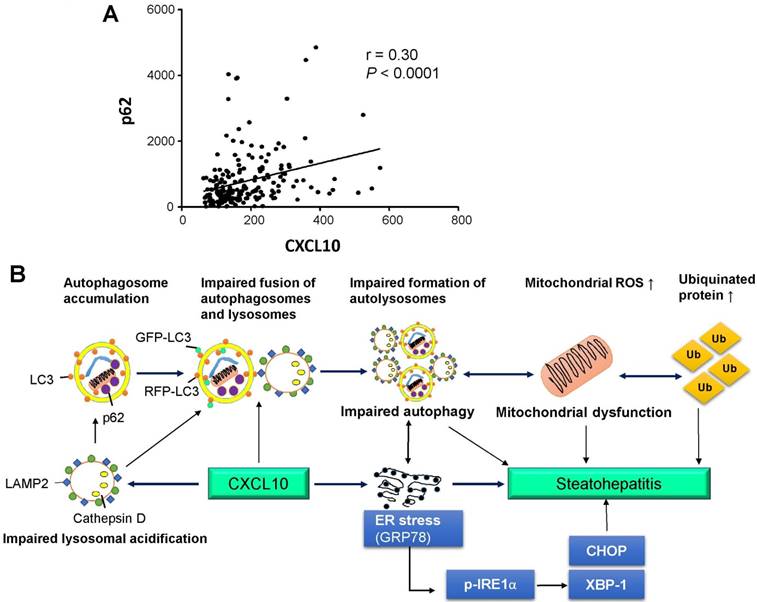
(A) Association of human serum CXCL10 and p62/SQTM1 levels from 66 control subjects and 129 NAFLD patients, including 71 NASH patients; (B) Schematic diagram for the mechanism of CXCL10 in the regulation of the autophagosome-lysosome system in experimental steatohepatitis. CXCL10 impairs the fusion of autophagosomes and lysosomes and the formation of autolysosomes, and induces mitochondria dysfunction, polyubiquitinated proteins accumulation and ER stress, thereby contributing to steatohepatitis development.
Hepatocytes are a major source of CXCL10 in the liver (20). CXCL10 is mainly secreted by hepatocytes in areas of lobular inflammation during liver injury (21). We found that CXCL10 is upregulated by palmitic acid or MCD medium in hepatocytes and MCD diet in mice (12). The increased CXCL10 expression is required for steatohepatitis development (12). In this study, we demonstrated that CXCL10 depletion by CXCL10 knockout or CXCL10 neutralization by anti-CXCL10 mAb blunted the accumulation of p62/SQSTM1 and LC3-II in accordance with the attenuation of steatosis and oxidative injury in hepatocytes and the ameliorated steatohepatitis in mice. The autophagosome-lysosome pathway is the process by which cellular components are delivered to the lysosome for degradation (22). p62/SQSTM1, an indicator of autophagic degradation, facilitates the delivery of ubiquitinated protein aggregates to the autophagic machinery for degradation by a direct interaction with LC3 (23). Increased p62/SQSTM1 level could be caused by either reduced autophagic degradation or transcriptional upregulation by cellular stress (24). In this study, the alteration of p62/SQSTM1 protein levels but not mRNA levels by CXCL10 knockout indicate that the altered p62/SQSTM1 levels were caused by autophagic degradation but not cellular stress. LC3 is a protein marker that can ultimately be degraded by acidic hydrolases (25). Decreased LC3-II levels may result from a decrease in the conversion of LC3-I to LC3-II in the early stage of autophagy, or a rapid degradation via lysosomal turnover in the late stage. The combined accumulation of p62/SQSTM1, LC3-II and autophagosomes reflects a blockade of autophagic flux (13). Our results thus suggest that CXCL10 is involved in the impairment of the autophagic process and subsequent p62/SQSTM1 degradation in steatohepatitis. Ablating CXCL10 by its antibody or genetic depletion restores the autophagic flux by enhancing autophagosome degradation, which contributes to the hepatic improvement of steatohepatitis. Lipid accumulation and autophagy dysfunction are interrelated during NASH development. Autophagy inhibition increased lipid accumulation in cultured hepatocytes and mouse liver. On the other hand, abnormal intracellular lipid accumulation impairs autophagic clearance (4). Therefore, anti-CXCL10 mAb-recovered autophagy function could suppress lipid accumulation, leading to the further recovery of autophagic function that prevents additional lipid retention.
Autophagic flux was further evaluated by LC3-II turnover in the presence and absence of lysosomal degradation inhibitors (bafilomycin A1) and autophagic protein degradation. Bafilomycin A1 is a reversible inhibitor of vacuolar-type H+-ATPase that can prevent lysosomal degradation by neutralizing the lysosomal acidity, thereby blocking fusion of autophagosomes with lysosomes and autolysosome formation (26). We found that in the presence of bafilomycin A1, CXCL10 blockade by its antibody or genetic depletion in AML-12 cells or primary hepatocytes could not change LC3-II levels anymore, suggesting that the restoration of autophagy by CXCL10 inhibition in hepatocytes occurs at the late stage of autophagy, and that CXCL10 inhibits autophagosome-lysosome fusion and/or lysosome-dependent degradation. Another classical assay for the measurement of autophagic flux is the detection of autophagic protein degradation (14). During the autophagic process, long-lived cellular proteins are sequestered by double-membrane autophagosomes, which then fuse with lysosomes for degradation (14). AHA, a surrogate for L-methionine, is incorporated into proteins during de novo protein synthesis. The increased AHA-containing proteins as detected by flow cytometry indicates the impaired autophagic flux. The decreased long-lived protein levels by CXCL10 inhibition and the increased long-lived protein levels by CXCL10 supplement further confirm that CXCL10 inhibits the functional autophagic process.
The inhibitory effect of CXCL10 on autophagosome-lysosome fusion was further evaluated by three assays. The first assay is to visualize autophagosomes and lysosomes using anti-LC3 and anti-LAMP1 antibodies, respectively. The association between autophagosomes and lysosomes could be restored by anti-CXCL10 mAb in HepG2. The second assay involved the autophagic flux indicator RFP-GFP-LC3. The decreased co-localization of GFP-LC3 and RFP-LC3 signals by anti-CXCL10 mAb observed in palmitic acid-challenged HepG2 confirmed the restoration of autolysosome formation (autophagosome-lysosome fusion) by CXCL10 inhibition. We finally utilized TEM to detect autophagosomes and autolysosomes directly. CXCL10 knockout increased autolysosome numbers in mice, suggesting restoration of autophagic flux by CXCL10 depletion in murine steatohepatitis.
Autolysosome acidification is critical to the degradation of autophagosomal cargos for the maintenance of autophagic flux. We discovered that the precursor form of cathepsin D, which is cleaved to its mature form upon acidification in lysosomes, was decreased by anti-CXCL10 mAb treatment in the C57BL/6 mouse steatohepatitis model. In addition, acidic lysosomal pH was restored in anti-CXCL10 mAb-treated steatotic hepatocytes, suggesting that the alterations in lysosome acidification contribute to the impaired autophagic flux in CXCL10-induced steatohepatitis. Our results collectively suggest that CXCL10 impairs lysosomal acidification and suppresses autophagosomes-lysosome fusion, resulting in the accumulation of autophagosomes and increased LC3-II and p62/SQSTM1 in the development of steatohepatitis.
The mechanistic findings of impaired autophagy by CXCL10 in mouse models of steatohepatitis encouraged us to evaluate the correlation between CXCL10 and impaired autophagy in patients with NAFLD. Serum CXCL10 levels were significantly increased in a stepwise fashion from control subjects, patients with simple steatosis to patients with NASH (12). Serum p62/SQTM1 levels were positively associated with CXCL10 levels in 195 human subjects. This result further confirmed our observation that CXCL10 plays an important role in impaired autophagy in the pathogenesis of steatohepatitis.
In conclusion, CXCL10 contributes to impairment of autophagosome-lysosome system by impairing lysosomal acidification in experimental steatohepatitis, which promotes mitochondria dysfunction, the accumulation of polyubiquitinated proteins and ER stress. Blocking this cytokine can ameliorate steatohepatitis through restoring autolysosome formation (Fig. 7B). Our findings provide novel mechanistic insights into CXCL10-mediated autophagic impairment in NASH development in which blockade of CXCL10 may serve as a potential therapeutic approach.
Abbreviations
CXCL10, C-X-C motif chemokine 10; NAFLD, Non-alcoholic fatty liver disease; mAb, monoclonal antibodies; NASH, non-alcoholic steatohepatitis; HCC, hepatocellular carcinoma; ER, endoplasmic reticulum; DMEM, Dulbecco's modified Eagle's medium; BSA, bovine serum albumin; WT, wildtype; CXCL10-/-, CXCL10 knockout; MCD, methionine-and-choline-deficient; HFHC, high-fat-high-cholesterol diet; AHA, L-azidohomoalanine; p70 S6 Kinase, p70 ribosomal protein S6 kinase; LAMP, lysosomal-associated membrane protein; p-IRE1α, phosphorylated inositol-requiring enzyme 1α; CHOP, C/EBP homologous protein; XBP-1, X-box binding protein 1; O.C.T, Optimal Cutting Temperature; PFA, paraformaldehyde; ROS, reactive oxygen species; ELISA, enzyme-linked immunosorbent assay; TBARS, thiobarbituric acid-reactive substances.
Acknowledgements
Financial Support
The project was supported by the RGC-GRF Hong Kong (14106415), (766613); RGC-Early Career Scheme (24115815); National Basic Research Program of China (973 Program, 2013CB531401); CUHK Direct grants for research (4054274); Vice-Chancellor's Discretionary Fund CUHK; RGC-CRF Hong Kong (CUHK3/CRF/12R; HKU3/CRF11R); RGC-Theme-based Research Scheme Hong Kong (T12-403-11); Shenzhen Virtual University Park Support Scheme to CUHK Shenzhen Research Institute.
Authors contributions
XZ was involved in study design, conducted the experiments and drafted the paper; WX, XW, JH, WYL, RW, and KL performed the experiments; KM provided material support; WKKW commented on the study and revised the paper; JY designed, supervised the study and wrote the paper.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wong VW, Chu WC, Wong GL, Chan RS, Chim AM, Ong A, Yeung DK. et al. Prevalence of non-alcoholic fatty liver disease and advanced fibrosis in Hong Kong Chinese: a population study using proton-magnetic resonance spectroscopy and transient elastography. Gut. 2012;61:409-415
2. Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651-662
3. Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861-2873
4. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K. et al. Autophagy regulates lipid metabolism. Nature. 2009;458:1131-1135
5. Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS. et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19:83-92
6. Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11:467-478
7. Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469:323-335
8. Li Y, Zhu H, Wang S, Qian X, Fan J, Wang Z, Song P. et al. Interplay of Oxidative Stress and Autophagy in PAMAM Dendrimers-Induced Neuronal Cell Death. Theranostics. 2015;5:1363-1377
9. Gonzalez-Rodriguez A, Mayoral R, Agra N, Valdecantos MP, Pardo V, Miquilena-Colina ME, Vargas-Castrillon J. et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014;5:e1179
10. Harris J. Autophagy and cytokines. Cytokine. 2011;56:140-144
11. Law AH, Lee DC, Yuen KY, Peiris M, Lau AS. Cellular response to influenza virus infection: a potential role for autophagy in CXCL10 and interferon-alpha induction. Cell Mol Immunol. 2010;7:263-270
12. Zhang X, Shen J, Man K, Chu ES, Yau TO, Sung JC, Go MY. et al. CXCL10 plays a key role as an inflammatory mediator and a non-invasive biomarker of non-alcoholic steatohepatitis. J Hepatol. 2014;61:1365-1375
13. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1-222
14. Wang J, Zhang J, Lee YM, Ng S, Shi Y, Hua ZC, Lin Q. et al. Nonradioactive quantification of autophagic protein degradation with L-azidohomoalanine labeling. Nat Protoc. 2017;12:279-288
15. Zhang J, Wang J, Ng S, Lin Q, Shen HM. Development of a novel method for quantification of autophagic protein degradation by AHA labeling. Autophagy. 2014;10:901-912
16. Ferguson CJ, Lenk GM, Meisler MH. Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum Mol Genet. 2009;18:4868-4878
17. Tanaka S, Hikita H, Tatsumi T, Sakamori R, Nozaki Y, Sakane S, Shiode Y. et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology. 2016;64:1994-2014
18. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y. et al. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011;25:795-800
19. Zhang X, Han J, Man K, Li X, Du J, Chu ES, Go MY. et al. CXC chemokine receptor 3 promotes steatohepatitis in mice through mediating inflammatory cytokines, macrophages and autophagy. J Hepatol. 2016;64:160-170
20. Xu Z, Zhang X, Lau J, Yu J. C-X-C motif chemokine 10 in non-alcoholic steatohepatitis: role as a pro-inflammatory factor and clinical implication. Expert Rev Mol Med. 2016;18:e16
21. Hintermann E, Bayer M, Pfeilschifter JM, Luster AD, Christen U. CXCL10 promotes liver fibrosis by prevention of NK cell mediated hepatic stellate cell inactivation. J Autoimmun. 2010;35:424-435
22. Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823-830
23. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A. et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131-24145
24. Sahani MH, Itakura E, Mizushima N. Expression of the autophagy substrate SQSTM1/p62 is restored during prolonged starvation depending on transcriptional upregulation and autophagy-derived amino acids. Autophagy. 2014;10:431-441
25. Rubinsztein DC, Cuervo AM, Ravikumar B, Sarkar S, Korolchuk V, Kaushik S, Klionsky DJ. In search of an "autophagomometer". Autophagy. 2009;5:585-589
26. Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. 2015;11:1437-1438
Author contact
![]() Corresponding author: Dr. Jun Yu, Institute of Digestive Disease, Department of Medicine and Therapeutics, Prince of Wales Hospital, the Chinese University of Hong Kong, Shatin, NT, Hong Kong. Phone: (852) 37636099, Fax: (852) 21445330, Email: junyuedu.hk
Corresponding author: Dr. Jun Yu, Institute of Digestive Disease, Department of Medicine and Therapeutics, Prince of Wales Hospital, the Chinese University of Hong Kong, Shatin, NT, Hong Kong. Phone: (852) 37636099, Fax: (852) 21445330, Email: junyuedu.hk
Citation styles
APA
Zhang, X., Wu, W.KK., Xu, W., Man, K., Wang, X., Han, J., Leung, W.Y., Wu, R., Liu, K., Yu, J. (2017). C-X-C Motif Chemokine 10 Impairs Autophagy and Autolysosome Formation in Non-alcoholic Steatohepatitis. Theranostics, 7(11), 2822-2836. https://doi.org/10.7150/thno.19068.
ACS
Zhang, X.; Wu, W.KK.; Xu, W.; Man, K.; Wang, X.; Han, J.; Leung, W.Y.; Wu, R.; Liu, K.; Yu, J. C-X-C Motif Chemokine 10 Impairs Autophagy and Autolysosome Formation in Non-alcoholic Steatohepatitis. Theranostics 2017, 7 (11), 2822-2836. DOI: 10.7150/thno.19068.
NLM
Zhang X, Wu WKK, Xu W, Man K, Wang X, Han J, Leung WY, Wu R, Liu K, Yu J. C-X-C Motif Chemokine 10 Impairs Autophagy and Autolysosome Formation in Non-alcoholic Steatohepatitis. Theranostics 2017; 7(11):2822-2836. doi:10.7150/thno.19068. https://www.thno.org/v07p2822.htm
CSE
Zhang X, Wu WKK, Xu W, Man K, Wang X, Han J, Leung WY, Wu R, Liu K, Yu J. 2017. C-X-C Motif Chemokine 10 Impairs Autophagy and Autolysosome Formation in Non-alcoholic Steatohepatitis. Theranostics. 7(11):2822-2836.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.