Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
Supplementary Material
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(11):2806-2821. doi:10.7150/thno.19081 This issue Cite
Research Paper
Non-invasive in vivo imaging of tumour-associated cathepsin B by a highly selective inhibitory DARPin
Lovro Kramer1, 2, Miha Renko1, Janja Završnik1, 2, Dušan Turk1, 3, Markus A. Seeger4, Olga Vasiljeva1, Markus G. Grütter5, Vito Turk1, Boris Turk1, 3 ![]()
1. Jozef Stefan Institute, Department of Biochemistry and Molecular and Structural Biology, Ljubljana, Slovenia;
2. International Postgraduate School Jozef Stefan, Ljubljana, Slovenia;
3. Center of Excellence CIPKEBIP, Ljubljana, Slovenia;
4. Institute of Medical Microbiology, University of Zürich, Zürich, Switzerland;
5. Department of Biochemistry, University of Zürich, Zürich, Switzerland.
Received 2017-1-6; Accepted 2017-4-15; Published 2017-7-8
Abstract

Cysteine cathepsins often contribute to cancer progression due to their overexpression in the tumour microenvironment and therefore present attractive targets for non-invasive diagnostic imaging. However, the development of highly selective and versatile small molecule probes for cathepsins has been challenging. Here, we targeted tumour-associated cathepsin B using designed ankyrin repeat proteins (DARPins). The selective DARPin 8h6 inhibited cathepsin B with picomolar affinity (Ki = 35 pM) by binding to a site with low structural conservation in cathepsins, as revealed by the X-ray structure of the complex. DARPin 8h6 blocked cathepsin B activity in tumours ex vivo and was successfully applied in in vivo optical imaging in two mouse breast cancer models, in which cathepsin B was bound to the cell membrane or secreted to the extracellular milieu by tumour and stromal cells. Our approach validates cathepsin B as a promising diagnostic and theranostic target in cancer and other inflammation-associated diseases.
Keywords: protease, cathepsin B, designed ankyrin repeat protein (DARPin), non-invasive diagnostic imaging, tumour microenvironment.
Introduction
Non-invasive whole-body monitoring of tumour-associated proteins is essential for developing a proper understanding of the involvement of proteins in disease and can lead to the development of new diagnostic and therapeutic approaches [1, 2]. Among these proteins, tumour-associated proteases have gained attention in non-invasive imaging and as drug targets due to their high up-regulation and accumulation within the tumour microenvironment. Their excessive proteolysis critically contributes to tumorigenesis by affecting tumour cell invasiveness, angiogenesis, immune cell infiltration and metastasis [3-5]. In the last decade, cysteine cathepsins in particular have been recognized as major players in cancer biology [5]. Although there are 11 human cathepsins known at the gene level, they do not all contribute equally to cancer. Based on animal and clinical studies, the major contributors are cathepsins (Cat) B, S, K, L and X. CatB and CatS have been found to be heavily up-regulated in cancer [6, 7], where they are primarily secreted into the tumour microenvironment by immune cells [8-11], including tumour-associated macrophages. Moreover, CatB was found to be one of the most abundantly secreted proteins in the macrophage secretome [12]. This tumour-specific translocation to the cell membrane and tumour interstitium [13] makes CatB particularly interesting as a tumour-associated antigen in contrast to classical targets such as cell membrane receptors or cell-adhesion molecules [14].
Therefore, it is not surprising that members of the cathepsin family of proteases were among the first enzymes targeted in vivo by activatable fluorescent reagents [15]. Activity-based approaches have been the method-of-choice and have led to the development of several NIR fluorescent substrates [16, 17] and activity-based probes (ABPs) [18-20]. These tools are particularly useful since they provide low-background signals until they are processed, although substrate-based probes have the downside of diffusing from the target enzyme after cleavage, resulting in rapid clearance of the signal from the target location. This has, in part, been overcome by adding a homing component such as a lipid moiety to the substrate [21]. On the other hand, ABPs have the unique characteristic of irreversible covalent association with the target enzyme after their activation (ABPs are suicide inhibitors). This is of particular use in pre-clinical research, as it allows proteomic-based identification and quantification of targets [22, 23], but does not enable any signal amplification, which is problematic with low-abundance enzymes. A major limitation of optical diagnostic imaging in humans based on fluorescence is the relatively poor tissue penetration of NIR light, which led to modifications of ABPs with PET tracers. In this manner, the issue of tissue penetration for imaging has been solved, but the quenching principle has been lost [24, 25].
Central to all activity-based tools is targeting the enzyme active site, which is highly conserved in cathepsins and makes the development of truly selective tools challenging. Discrimination among different family members is difficult to achieve due to the exclusive focus on the active site while completely disregarding the structural variability of other regions. Furthermore, even minor modifications to small-molecule probes can severely affect their selectivity and bioavailability [26-28]. We therefore explored the possibility of targeting CatB by designed ankyrin repeat proteins (DARPins). In this approach, there is no inherent bias to target the structurally conserved active site region of a protease, as the targeting ligand from the DARPin library is obtained solely on the basis of strong and selective protein-protein interactions [29, 30]. These small and rigid antibody mimetics are obtained completely in vitro by ribosome or phage display and can be expressed as recombinant proteins in E. coli with high yields. Each of the repeat motives in DARPins is composed of 33 amino acid residues folded into a β-turn, followed by two antiparallel α-helices and a loop connecting to the next repeat, which yields a highly stable structure. The selectivity for their respective targets comes from the 7-10 randomized amino acids in each of the internal repeats. Moreover, DARPins offer the possibility of multiple site-specific modifications for pharmacokinetic fine-tuning and imaging label conjugation (i.e., NIR fluorescent dye, PET/SPECT tracer) without the loss of selectivity and affinity [31, 32]. They also have certain advantages in imaging over conventional antibodies, including higher tissue penetration and fast blood clearance following systemic administration [33].
Here, we present the development of DARPin 8h6, which binds to CatB with picomolar affinity at a site of high structural variability in the family of cysteine cathepsins. This resulted in complete selectivity over other important cancer-related cathepsins (S, K, L and X), confirming the rationale for selective targeting via binding to a variable surface separate from the protease active site Cys residue. The extremely high affinity between DARPin 8h6 and CatB proved to be critical for successful localization of CatB in vivo in tumours of two mouse breast cancer models, in which CatB was confirmed to be an excellent target with the potential to be used as a diagnostic marker.
Materials and methods
DARPin selection and production
DARPins 8h6 and 81 were selected by ribosome display from the recently developed DARPin 2.0 library [34] as described in the supplementary material, expressed from the pQE30 vector in E. coli XL-1 Blue cells and purified by Ni2+ affinity and size-exclusion chromatography. When C-terminally labelled DARPins were used, a C-terminal Cys, which is the only Cys in the DARPin sequence, was introduced by mutagenesis. Biotin or cyanine5.5 (Cy5.5) tags were covalently attached via the maleimide linker, and the labelling efficiency was confirmed as described in the supplementary material.
Solid-phase affinity assay
Cathepsins (200 nM) were immobilized to a 96-well MaxiSorp plate (Nunc) in PBS (20 mM sodium phosphate, 150 mM NaCl, pH 7.0) by incubation overnight at 4 °C. After blocking with 3 % BSA in TBS (50 mM Tris/HCl, 500 mM NaCl, pH 7.0) with 0.05 % Tween-20 (TBST), C-terminally biotinylated DARPins were incubated for 1 h at 4 °C in TBST, followed by three washes with TBST and subsequent incubation with streptavidin-HRP (1:10000, Thermo Fisher) for 30 min. Following four washes with TBST, 1 mg mL-1 2,2'-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid (ABTS) and 0.06 % H2O2 were added in 0.1 M citric acid-Na2HPO4, pH 4.5, for 30 min, after which the absorbance at 405 nm was measured using a Tecan Infinite M1000 plate reader (Tecan, Austria).
Inhibition of cathepsin activity
Recombinant cathepsins (human CatB 1 nM, mouse CatB 1 nM, human CatL 1 nM, human CatK 1 nM, human CatS 10 nM, mouse CatS 10 nM, human CatX 250 nM) were incubated for 15 min at 37 °C with 10 μM DARPins in 0.1 M phosphate buffer, pH 6.0, containing 5 mM DTT, 1 mM EDTA, 0.1 % PEG6000 and 0.1 % BSA. Enzymatic activity was determined by the addition of 20 μM substrate (z-Arg-Arg-AMC for CatB, z-Phe-Arg-AMC for other cathepsins) and measuring the linear slopes (initial rates) of the fluorescence of the liberated product (em/ex = 370/460 nm) over time using the Tecan Infinite M1000 plate reader. Residual activity was defined as the fraction (%) of activity in the absence of DARPin.
The kinetics of the inhibition of CatB by DARPins was performed essentially as described previously [35]. Briefly, the inhibition type was determined according to the general modifier scheme (Fig. S4a) [36] with 1 nM CatB by varying the concentrations of DARPin 8h6 (25-400 nM) and the substrate z-Arg-Arg-AMC (75-600 μM, KM (human CatB) = 173 μM, KM (mouse CatB) = 262 μM) in 0.1 M phosphate buffer, pH = 6.0, containing 5 mM DTT, 1 mM EDTA and 0.1 % PEG6000. Graphical analysis with Prism5 (GraphPad Software) was used to determine the parameter α, which corresponds to the ratio between competitive and uncompetitive Ki, and β, which represents incomplete inhibition at saturating inhibitor concentrations (i.e., hyperbolic inhibition). To determine the Ki values, the initial velocities at fixed CatB (50 pM) and z-Arg-Arg-AMC (30 μM) concentrations were measured in the presence of increasing DARPin concentrations (50 pM - 5 nM). The velocities were fitted considering the tight-binding, hyperbolic, mixed-mode inhibition as described previously [37].
Structure determination
Complexes between human CatB and DARPin 8h6 or human CatB and DARPin 81 were formed by mixing the proteins CatB:DARPin in a ratio of 1:2 (M/M) in 20 mM sodium phosphate, 300 mM NaCl, pH 7.0, and isolated using a Superdex S200 size-exclusion column, followed by concentrating via Amicon Ultra-15 Centrifugal Filter Units with a 10 kDa cut-off (EMD Millipore) to approximately 10 mg mL-1. Crystals were grown using the sitting drop method in 0.1 M sodium citrate, pH = 4.0 with 15 % PEG6000 (DARPin 8h6 complex) or in 0.1 M MES, 1.8 M AmSO4 (DARPin 81 complex). The crystals were soaked in mother liquor supplemented with 25 % glycerol and frozen in liquid N2 prior to data collection. Diffraction data were collected on MX14.1 at Synchrotron Desy (Berlin, Germany) and processed using XDSAPP/XDS. Molecular replacement was performed using the crystal structures of CatB (PDB ID 3AI8) and DARPin E3_5 (PDB ID 4J7W). Refinement was performed using REFMAC [38] and MAIN [39]. Model building was performed using MAIN. Structural figures were prepared using PyMOL (www.pymol.org).
Cell cultures
Primary PyMT cells were prepared from tumours of 14-week-old FVB/N-TgN(MMTVPyMT) mice as described previously [40], whereas 4T1 cells were obtained from ATCC (ATCC CRL-2539). Both types of cells were grown in DMEM medium containing 10 % FBS, 1 % PS and 1 % Glutamax; differentiated bone-marrow derived macrophages (dBMMs) were prepared from FVB/N mice as described previously [41]. Briefly, the isolated bone marrow progenitors were differentiated into macrophages in the presence of M-CSF by 7 day incubation with DMEM containing 20 % heat-inactivated FBS, 1 % PS, 1 % Glutamax and 15 % L929 conditioned medium. U87-MG (ECACC 89081402) and HeLa cells (ECACC 93021013) were obtained from ECACC and grown in MEM medium containing 10 % FBS, 1 % PS, 1 % non-essential amino acids and 1 % Glutamax.
Detection of cell membrane-associated CatB
Cells (40,000 per well) were seeded onto black 96-well plates with transparent bottoms and grown overnight. Active endocytosis was initially blocked by a 1 h incubation at 4 °C; to prevent any additional endocytosis, all subsequent steps were also performed at 4 °C. Goat anti-mouse CatB polyclonal antibodies (1:1000, AF965, R&D), mouse anti-human CatB monoclonal antibodies 3E1 (100 nM, [6]) or biotinylated DARPins (100 nM) were next added in PBS supplemented with 3 % BSA and incubated for 1 hour. After washing with PBS, the Alexa488-conjugated secondary anti-goat antibodies (1:1000, Thermo Fisher Scientific), anti-mouse antibodies (1:1000, Thermo Fisher Scientific) or Alexa488-conjugated streptavidin (1:1000, Thermo Fisher Scientific) was added for 1 hour. After washing, the cell membrane-associated fluorescence (ex/em = 488/520 nm) was monitored in a Tecan Infinite M-1000 plate reader by measuring nine locations in each well. The mean background fluorescence (where only secondary antibody or only streptavidin was incubated) was subtracted from the measurements. In the control experiments in which unlabelled competitor DARPins were used to block cell membrane-associated CatB binding to biotinylated DARPin 8h6, the competitor DARPins (1 μM) were incubated with cells during the last 30 min of active endocytosis blocking.
Measurement of cell membrane-associated CatB activity
The measurement was performed as described previously [42]. Briefly, 10,000 cells were seeded onto black 96-well plates with transparent bottoms and grown overnight. Cells were then incubated with Hank's balanced salt solution lacking sodium bicarbonate and containing 0.6 mM CaCl2, 0.6 mM MgCl2, 2 mM L-cysteine, 25 mM PIPES, pH 7.0 for 30 min at 37 °C. The solution was then replaced with fresh buffer containing 50 μM z-Arg-Arg-AMC in the absence or presence of DARPin (100 nM) and fluorescence (em/ex = 370/460 nm) was monitored over time using the Tecan Infinite M-1000 plate reader. Data analysis was performed using Prism5 (GraphPad Software).
Three-dimensional cell invasion assay
Multicellular tumour spheroids (MCTS) were prepared as described previously [43]. MCTSs were then transferred to a U-bottom 96-well plate coated with 25 μL Matrigel (5 mg/mL) in serum-free DMEM and additionally covered with 25 μL Matrigel (5 mg/mL) in serum-free DMEM. After 1 h, 100 μL of complete medium was added and the spheroid size was imaged 24 h later using Olympus IX 81 inverted microscope (Olympus). The volume of the spheroids was calculated as V = π/6 × (spheroid length) × (spheroid width)2. Where noted, inhibitors were added at the designated concentrations to both the Matrigel and the culture medium.
In situ binding to native tumour tissues
Cryopreserved PyMT tumour tissues were cut into 5 μm thick slices and thawed at 4 °C. To prevent tissue damage, all subsequent steps were also performed at 4 °C. In the next step, slices were incubated with PBS containing 3 % BSA for 15 min, followed by a 30 min incubation with C-terminally biotinylated DARPins (1 μM). After three washing steps with PBS, Alexa488-conjugated streptavidin (1:1000) was added for 30 min. After additional washing with PBS, slices were mounted with Prolong Gold antifade reagent containing DAPI and imaged on IX81 microscope (Olympus) with Imaging Software for Life Science Microscopy Cell.
In situ zymography
Cryopreserved PyMT tumour tissues were cut into 5-μm thick slices and thawed at room temperature. The slices were incubated for 1 hour in 20 mM sodium phosphate, 137 mM NaCl, pH 7.0 with 2.7 mM L-cysteine, 0.25 mM EDTA, 1 mM DTT, 1 mM 5-nitrosalicylaldehyde and 1 mg mL-1 z-Arg-Arg-4MβNA. DARPins E3_5 (1 μM), 8h6 (1 μM) or CA074 (1 μM) were added where indicated. After 1 h, the slices were mounted with Prolong Gold antifade reagent containing DAPI and microscopy was performed as described above. In the procedure 5-nitrosalicylaldehyde forms a Schiff base with proteolytically released 4-methoxy-β-naphthylamine to generate a yellow fluorescent final reaction product [44].
In vivo and ex vivo imaging
Mice were used in accordance with the protocols approved by the Veterinary Administration of the Republic of Slovenia (VARS) and the government Ethical Committee. Procedures for animal care and use were in accordance with the “PHS Policy on Human Care and Use of Laboratory Animals” and the “Guide for the Care and Use of Laboratory Animals” (NIH publication 86-23, 1996).
In the tumour imaging study we used a congenic PyMT model [45] and a syngeneic 4T1 model [21]. Briefly, PyMT and 4T1 cells were grown to subconfluency as described above, detached by trypsinization and resuspended in DMEM at a concentration of 5×106 cells mL-1 and 1×106 cells mL-1, respectively. One hundred microliters of the cell suspension was then injected into the left inguinal mammary gland of the recipient mouse (congenic FVB/N mice for PyMT cells; Balb/c strain for 4T1 cells) and tumours were allowed to grow for 2-3 weeks until they reached 100-150 mm3.
Mice (n=3) received 1 nmol (approx. 20 μg) of C-terminally Cy5.5 labelled DARPins in 100 μL of PBS by intravenous (i.v.) administration. At designated time-points (30 min, 3 h, 6 h, 24 h, 48 h, 72 h), mice were anesthetized with 2 % isofluorane and transferred to the IVIS Spectrum small animal imaging system (Perkin Elmer), where isofluorane anaesthesia was maintained for the duration of imaging. Imaging was performed using the ex/em = 675/720 nm bandpass filters. Image analysis was performed in Living Image Software 4.3.1 (Perkin Elmer), where false blue-hot images of radiant efficiency (radiance (photons per second per square centimetre per steradian) per incident excitation power (microwatt per square cm)) were overlaid on bright-light images. For quantification of radiant efficiency, equally sized round regions of interest (ROI) were selected at the site of the tumour and contralateral mammary gland. In case of pretreatment with unlabelled DARPin 8h6, 10 nmol of the unlabelled DARPin in 100 μL of PBS were administered i.v. 1 h prior to Cy5.5-labelled DARPin administration.
For ex vivo analysis of organ biodistribution, mice (n = 4) were sacrificed at 3 h or 72 h time-points post DARPin administration. Organs (tumour, liver, lungs, heart, kidneys, spleen and intra-abdominal lymph nodes) were collected and immediately imaged by IVIS Spectrum as described above.
Immunohistochemistry
Cryopreserved PyMT tumours from mice with administered Cy5.5 labelled DARPin 8h6 were cut into 5-μm sections and air-dried for 20 min, followed by fixation in ice-cold methanol for 5 min and acetone (-20 °C) for 2 min. Slices were rehydrated in PBS for 15 min and labelled with anti-F4/80 antibody (1:200, ab6640, Abcam) and Alexa488-labelled anti-rat secondary antibody (Thermo Fisher Scientific). Slices were mounted with Prolong Gold antifade reagent containing DAPI and microscopy was performed as described above.
Streptavidin pull-down
Fourteen-week-old FVB/N-TgN (MMTVPyMT) mice with developed tumours were used in the experiment. Mice were anesthetised with 2 % isofluorane and C-terminally biotinylated DARPins (5 nmol) were intratumourally injected into one tumour per mouse. After thirty minutes, mice were sacrificed, tumours were collected and tumour extracts were obtained by tissue (50 μg) disruption using Ultrathurrax (IKA, Staufen, Germany) in 200 μL of tissue lysis buffer (0.25 M Tris-HCl, pH 7.0, 5 mM EDTA, 1 % Triton X-100). The extracts were incubated with 10 μL of Pierce™ Streptavidin Agarose (Thermo Fisher Scientific) for 30 min on 4 °C. After three washes with PBS, the beads were resuspended in SDS-PAGE loading buffer and boiled for 5 min, followed by SDS-PAGE and immunoblotting using anti-CatB antibodies or streptavidin-HRP.
Statistical analysis
Statistical analysis was performed by a two-tailed t-test for two groups and one-way analysis of variance with Turkey's post-hoc test for multiple groups using Prism 5 (GraphPad Software). A p value < 0.05 was considered statistically significant.
Results
DARPin 8h6 and CatB interact with high selectivity and affinity
DARPin 8h6 was selected using ELISA screening after eight successive rounds of ribosome display against both human and mouse recombinant CatB from a recently described DARPin library [34]. In order to achieve site-specific chemical labelling with biotin or Cy5.5, a unique cysteine residue was introduced at the C-terminus that allowed conjugation with maleimide linkers (Figs. S1-S3). The selectivity of DARPin 8h6 binding to CatB was determined in a solid-phase affinity assay in which only human and mouse CatB bound to biotinylated DARPin 8h6 in a concentration-dependent manner. There was no signal for cathepsins L, K, S or X with up to 1 μM of DARPin (Fig. 1a).
DARPin 8h6 selectivity and affinity. (a) Colorimetric affinity assay with immobilized recombinant cathepsins (50 nM). Increasing concentrations of C-terminally biotinylated DARPin 8h6 were incubated in 96-well plates with immobilized recombinant cathepsins. After washing, the biotin tag was detected by streptavidin-HRP. (b) Residual proteolytic activity (% of uninhibited) of recombinant cathepsins (human CatB 1 nM, mouse CatB 1 nM, human CatL 1 nM, human CatK 1 nM, human CatS 10 nM, mouse CatS 10 nM, human CatX 250 nM) in the presence of 10 µM DARPin 8h6; z-Arg-Arg-AMC was used for measuring CatB activity and z-Phe-Arg-AMC for other cathepsins. The experiment was performed in triplicate and is presented as the mean ± standard error. (c) Inhibition of human CatB (50 pM) and mouse CatB (50 pM) by DARPin 8h6 (50 pM - 5 nM). The solid line represents the least-squared fit for the hyperbolic tight-binding inhibition, as described in Materials and methods. The proteolytic activity was measured by z-Arg-Arg-AMC (30 µM).
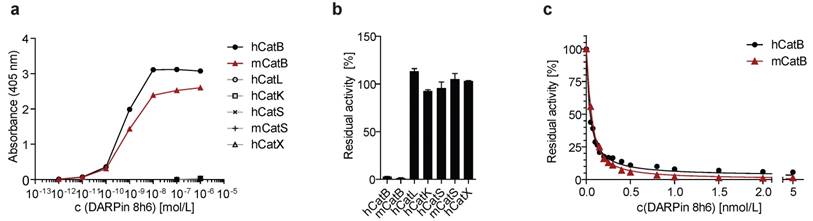
Next, we found that DARPin 8h6 almost completely inhibited both human and mouse CatB activities, whereas other cathepsins were not inhibited (Fig. 1b). A detailed kinetic analysis of the DARPin 8h6-CatB interaction revealed fast, tight-binding characteristics, with inhibition constants of 34 pM and 35 pM for human and mouse CatB, respectively (Fig. 1c, Fig. S4b-e). In both cases, the mechanism was mixed-mode inhibition with a very strong competitive component, as represented by the parameter α>1 (Fig. S4a). This indicated that DARPin 8h6 competes for binding with the z-Arg-Arg-AMC substrate, although the ternary enzyme-inhibitor-substrate complex can still form (i.e., uncompetitve inhibition, where α•Ki is the uncompetitive inhibition constant), but with roughly six-fold lower affinity (Ki = 34 pM, α•Ki = 224 pM for human CatB; Ki = 35 pM, α•Ki = 214 pM for mouse CatB). Furthermore, the ternary complex was partially productive for the substrate (β > 0), although with substantially lower efficiency, suggesting that DARPin 8h6 does not directly bind to the active site of CatB. As subsequent surface plasmon resonance (SPR) experiments revealed, both the fast association (ka = 2.1 × 106 M-1 s-1 for human CatB, ka = 7.5 × 105 M-1 s-1 for mouse CatB) and very slow dissociation (kd = 6.7 × 10-5 s-1 for human CatB, kd = 1.5×10-4 s-1 for mouse CatB) of DARPin 8h6 contributed to the high affinity (Fig. S5, Table 1). The interaction with the zymogen form of CatB, proCatB, was substantially weaker (KD = 7.1 nM) due to the much slower association of the complex (ka = 1.4 × 104 M-1 s-1), whereas the dissociation was not affected significantly (kd = 1.0 × 10-4 s-1) (Fig. S6a, Table 1).
Binding characterization of DARPin 8h6 to human and mouse CatB as determined by enzyme inhibition (Fig. 1c and Fig. S4) and surface plasmon resonance (Figs. S5, 6).
| Inhibition | SPR | ||||||
|---|---|---|---|---|---|---|---|
| Ki [pM] | α | β | ka [M-1 s-1] | kd [s-1] | KD [nM] | χ2 [%Rmax] | |
| Human CatB | 34 ± 1 | 6.6 | 0.15 | 2.1 × 106 | 6.6 × 10-5 | 0.031 | 2.2 % |
| Mouse CatB | 35 ± 3 | 6.1 | 0.064 | 7.5 × 105 | 1.5 × 10-4 | 0.20 | 12.5 % |
| Human proCatB | 1.4 × 104 | 1.0 × 10-4 | 7.1 | 3.6 % | |||
In the selection process, another CatB-inhibitory DARPin, DARPin 81, was also obtained and characterized (Fig. S7b, c). DARPin 81 shared 15 of the 27 variable site residues with DARPin 8h6 and had 90.1 % overall sequence identity (Fig. S1a). Although DARPin 81 had an analogous inhibitory profile with DARPin 8h6, it was found to have a substantially lower affinity for CatB in the low nanomolar range (Ki= 4.1 nM; Fig. S4f-i). However, this was still a sufficiently high affinity to enable the use of DARPin 81 in some of the experiments as a competitive binder of DARPin 8h6 for CatB. Non-selective DARPin E3_5 [46] did not bind to CatB, nor did it have any effect on CatB activity, and was used as a control DARPin throughout the experiments (Fig. S7a, c).
Extensive interactions with the CatB left domain account for the tight binding of DARPin 8h6
To understand the interactions between CatB and DARPins 8h6 and 81, crystal structures of the complexes were determined at 2.76 Å and 1.81 Å resolutions, respectively. The DARPin 8h6 complex crystallized with two pairs of molecules in the asymmetric unit, whereas DARPin 81 crystallized with one. The high-quality diffraction data enabled us to unambiguously build models of DARPin 81 and CatB apart from their termini. By contrast, the lower resolution crystals yielded a weaker diffraction of the DARPin 8h6 complex, which enabled us to build the structures along the whole chain length. However, the refinement and density maps in part pulled the local geometry out of the minima that would correspond to a model that scored well according to the validation criteria (Table S1).
The CatB structures of the two DARPin 8h6 complexes from the asymmetric unit superimpose onto the CatB structure from the DARPin 81 complex with a RMSD over all of the CA atoms of 0.55 Å and 0.62 Å, respectively, whereas among them the RMSD is 0.61 Å. The relative positions of both DARPin 8h6 structures are shifted > 3 Å away from the DARPin 81 structure using the CatB superimposition parameters, whereas the superimposition of the DARPins over 161 residues yielded a RMSD of 1.7 Å and 1.8 Å, respectively. While the structures indicate the same binding mode, the rather large shift of 3 Å indicates considerable differences among the binding contacts.
The concave binding surface of both DARPins includes the variable site residues on the β-turns of the internal repeats and C-cap; this surface forms the majority of interactions with CatB on the enzyme's interdomain interface as well as the surface of the L-domain, leaving the active site Cys-His dyad exposed to solvent (Fig. 2a). The central interactions of both DARPins are formed with the CatB S65 - A77 loop that builds the left side of the surface of the S2 and S3 substrate-binding sites (Fig. 2b). Intriguingly, both DARPins share the variable site residues Y46, R48, W78, Y80, E89 and R111, which contribute to interactions with CatB in this area of low structural conservation in the cathepsin family (Fig. S8), providing an explanation for the specificity. However, DARPin 8h6 forms additional contacts in this area through W122 and interacts with R85-K86 at the end of the third CatB alpha helix on the L-domain through W56 (Fig. 2c), whereas DARPin 81 has S122 and N56 at these positions, which do not substantially contribute to binding. The consequence is a slight shift of DARPin 8h6 towards the CatB L-domain compared to DARPin 81, as well as a substantial increase in the interaction surface. The average interface area of the DARPin 8h6-CatB complexes is 973 Å2 compared to 860 Å2 for the DARPin 81-CatB complex (as calculated by the PDBePISA server, http://www.ebi.ac.uk/pdbe/pisa/), which correlates well with the significantly higher affinity of DARPin 8h6. Both interaction surfaces and affinities are consistent with the previously reported DARPin-target protein complex [35].
Structure of human CatB in complex with DARPin 8h6. (a) Superimposition of complexes between CatB (grey) and DARPins 8h6 (red) and 81 (blue). (b) Close-up of the binding interface (top view) and (c) (side-view). The CatB active site, S65-A77 loop and R85-K86 residues are in yellow, light-green and dark-green, respectively. DARPin 8h6 (left) is shown in red and DARPin 81 (right) in blue. Variable site DARPin residues are shown as sticks. (d) Superimposition of DARPin 8h6-CatB complex with the crystal structure of proCatB (PDB ID 3PBH). In all figures, superimposition was performed using CatB residues 1-254 (mature enzyme numbering). All figures were prepared in PyMOL.
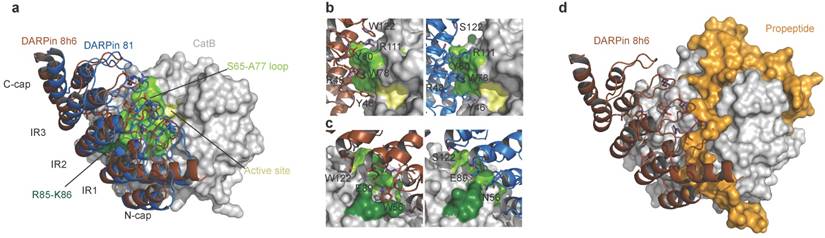
Next, we compared the crystal structures of the DARPin 8h6-CatB complex with the crystal structure of proCatB [47]. Superimposition of the structures showed no considerable differences in the CatB structure apart from the occluding loop region, which is displaced in proCatB. The partial steric overlap between DARPin 8h6 and the N-terminal propeptide in proCatB, which occupies the entire active site cleft, suggests that the much higher affinity of CatB for DARPin 8h6 than for the propeptide (35 pM vs. ~1 nM; ref [48]) could lead to the displacement of the propeptide by the DARPin (Fig. 2d). This is consistent with the substantially lower affinity of DARPin 8h6 for proCatB than for the mature CatB (Table 1). The formation of the complex between DARPin 8h6 and proCatB was confirmed by size-exclusion chromatography and by streptavidin pull-down with a biotinylated DARPin 8h6, using a cell extract from the human monocytic cell line THP-1 (Fig. S6b, c), although the difference in the rates of association and affinities results in a clear preference for binding of DARPin 8h6 to the mature form of the enzyme.
DARPin 8h6 selectively interacts with membrane-associated and secreted cathepsin B, and inhibits cancer cell invasion
Having shown selective binding of DARPin 8h6 to CatB in vitro, we next tested whether it can also bind to CatB translocated to the membrane or secreted into the extracellular space by tumour and other cells including macrophages. As models we selected human glioblastoma cell line U-87 MG and human cervical adenocarcinoma cell line HeLa, primary mouse PyMT cells and bone marrow-derived macrophages (dBMM), and mouse 4T1 cells. The mouse cells were selected because of the PyMT and 4T1 mouse breast cancer models used in further studies, and in which CatB is known to have a role in tumour development by contributing to pericellular proteolysis in both tumour and stromal cells, particularly macrophages [49, 50]. The main reason to select the two human cell lines was their dramatically different expression of CatB (http://www.proteinatlas.org/ENSG00000164733-CTSB/cell), which makes them suitable for evaluating the selectivity of DARPin 8h6.
In both PyMT primary cells and 4T1 cultures, CatB was found to be abundantly secreted to the extracellular space in the zymogen form. By contrast, the enzyme was secreted as both the zymogen and mature enzyme from the primary dBMM, which were found to express considerably more CatB than tumour cells (Fig. 3a). In U-87 MG human cells the secreted CatB was found to be a mixture of the mature (single chain (scCatB) and heavy chain (hcCatB)) and the zymogen form (proCatB), whereas in cell extracts the majority of CatB was in the mature single chain form. By contrast, no secreted CatB was observed in HeLa cells, and only a very low amount of primarily mature CatB was observed in cell extracts (Fig. 3a). Next, we evaluated the levels and activity of pericellular CatB in different cells. Higher levels of membrane-associated CatB in dBMMs compared to tumour cells were confirmed by detecting CatB in live cells with CatB-specific antibodies (Fig. 3b). Consistent with this, a significantly higher pericellular CatB activity was detected in dBMMs compared to the tumour cells (Fig. 3c). Significant amounts of membrane-associated CatB and pericellular CatB activity were also observed in U-87 MG cells. However, no CatB was detected in HeLa cells, consistent with very low levels of the enzyme in these cells (Fig.3b, c).
DARPin 8h6 interacts with membrane-associated and secreted CatB in mouse cells. (a) Western-blot detection of the active CatB and zymogen (proCatB) protein levels in whole cell extracts (CE) and conditioned media (CM) of tumour cells (PyMT, 4T1, U-87 MG, HeLa) as well as differentiated bone-marrow derived macrophages (dBMM). Actin was used as a loading control. (b) Immunodetection of cell membrane-associated CatB. An equal numbers of cells were grown on a 96-well plate. Active endocytosis was blocked by cooling cells to 4 °C for 1 hour, followed by immunodetection with antibodies against mouse CatB for PyMT, 4T1 cells and dBMMs or against human CatB for U-87 MG and HeLa (1 h incubation) and Alexa488-conjugated secondary antibodies (1 h incubation). Cell-associated fluorescence after washing was measured by a plate reader (Tecan Infinite) with ex/em = 488/520 nm at nine separate locations in each well. Background fluorescence from secondary antibodies was subtracted from the measurements. The experiment was performed in triplicate and is presented as the mean ± standard error of the relative fluorescence intensity. (c) Cell membrane-associated CatB activity. Equals number of cells on a 96-well plate were assayed with 50 µM z-Arg-Arg-AMC for CatB activity as described in the Materials and methods. CatB activities are presented as the mean ± standard error of relative fluorescence units per second. (d) Detection of CatB by DARPin 8h6 on the cell membrane of cells. The experiment was performed as in b with biotinylated DARPin 8h6 (100 nM) or nonselective DARPin E3_5 (100 nM) and Streptavidin-Alexa488 used instead of antibodies. Unlabelled competitor DARPins (1 µM) were preincubated with cells for 30 min where indicated. (e) DARPin 8h6 inhibition of cell membrane-associated CatB activity. The experiment was performed as in c, with the addition of a preincubation step for cells with 100 nM DARPins. (f) Cell invasion of U-87 MG spheroids in the presence of 5 µM CA074 (or DMSO control) or 1 µM DARPins. Quantification of spheroid volumes after 24 h of invasion and representative images of spheroids are shown. Scale bar = 200 µm. * p < 0.05 ** p < 0.01 *** p < 0.0001
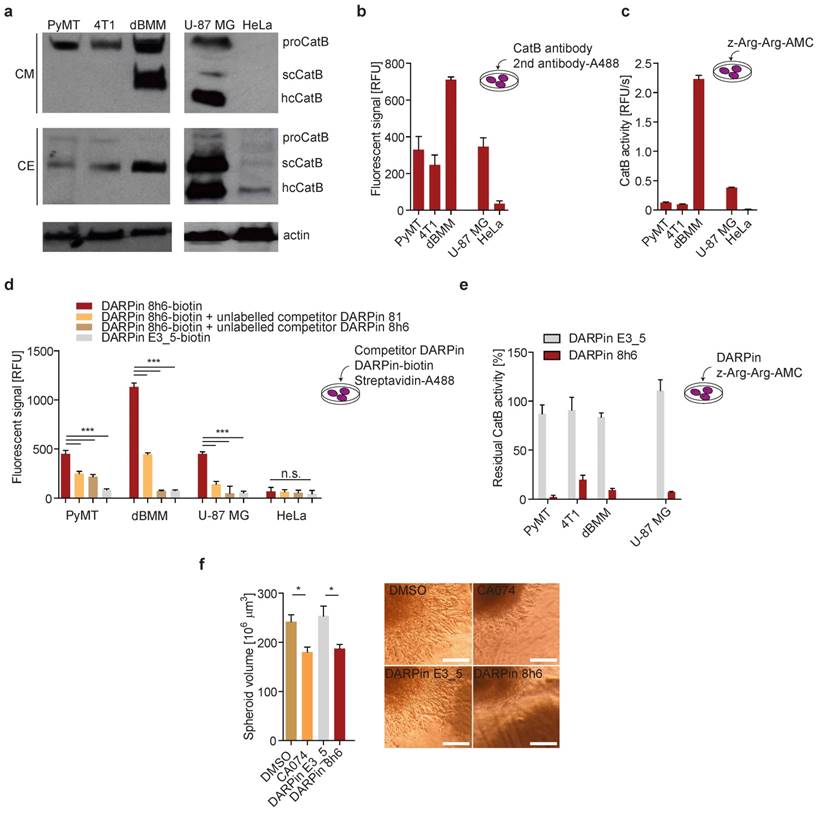
We next evaluated whether DARPin 8h6 can interact with membrane-associated CatB. Since DARPins are known to be endocytosed [51], endocytosis was blocked prior to the experiment. We used a biotinylated form of DARPin 8h6 and indeed detected the membrane-associated CatB in the U-87 MG cells, while there was no binding to the CatB-negative HeLa cells (Fig. 3d), confirming selectivity of this DARPin. In the control experiment, only minimal unspecific binding by the nonselective control DARPin E3_5 was observed. The selective nature of binding to CatB was confirmed in a competition experiment in which cells were preincubated with unlabelled DARPin 8h6 or the competitor DARPin 81. In both cases, preincubation significantly reduced the binding of the labelled DARPin 8h6, pointing to a selective interaction of DARPin 8h6 with membrane-associated CatB (Fig. 3d). As shown in Fig. 3d, a significantly higher association of the biotinylated DARPin 8h6 to dBMMs was detected compared to PyMT tumour cells, reflecting the differences in the CatB expression patterns and membrane association in these two cell types. As with the U-87 MG cells, preincubation with unlabelled DARPin 8h6 or competitor DARPin 81 reduced the signal in dBMMs and PyMT cells as well.
Due to the inhibitory nature of DARPin 8h6, we also evaluated the potential to block pericellular CatB activity. This activity was completely inhibited by DARPin 8h6 already at 100 nM in U-87 MG, dBMM, PyMT and 4T1 cells, while the nonselective control DARPin E3_5 had no significant effect on CatB activity under the same conditions (Fig. 3e). Since proteolysis by CatB contributes to cancer cell invasion, we tested how DARPin 8h6 affects invasion of U-87 MG cell spheroids through Matrigel. DARPin 8h6 substantially inhibited invasion, comparably to the irreversible covalent CatB inhibitor CA074, while the nonselective DARPin E3_5 had no significant effect (Fig. 3f).
DARPin 8h6 binds to CatB in the tumour microenvironment
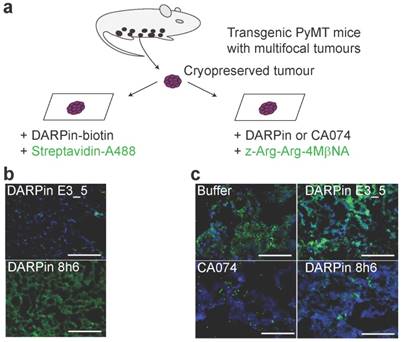
Next, the interaction of DARPin 8h6 with CatB was tested in the PyMT tumour microenvironment. We performed a two-step histochemical test on native tumour tissues that consisted of (i) an evaluation of DARPin binding to CatB and (ii) in situ zymography using a CatB-selective substrate z-Arg-Arg-4MβNA in tissue slices (Fig. 4a). Biotinylated DARPin 8h6 bound to CatB in the tissue slices, while the nonselective control DARPin E3_5 did not (Fig. 4b). However, no binding was observed in fixed histological samples, most likely due to the destruction of the extended three-dimensional epitope on CatB during the fixation procedure (data not shown). In addition, inhibition of CatB enzymatic activity by DARPin 8h6 was observed, whereas DARPin E3_5 did not have an effect. DARPin 8h6 exhibited a similar inhibitory efficacy as CA074 (Fig. 4c).
DARPin 8h6 selectively interacts with CatB in PyMT tumour tissues. (a) Schematic representation of the in situ imaging of CatB localization and activity in native cryopreserved tumour tissue. (b) C-terminally biotinylated nonselective DARPin E3_5 (1 μM) or CatB-selective DARPin 8h6 (1 μM) were incubated for 30 min with native PyMT tumour tissue slices, followed by detection by streptavidin-A488 (green). Cell nuclei were stained by DAPI (blue). (c) Native PyMT tumour tissue slices were incubated for 1 h with the fluorogenic CatB-selective substrate z-Arg-Arg-4MβNA (green) and DARPins E3_5 (1 μM) or 8h6 (1 μM), or the irreversible covalent inhibitor CA074 (1 μM). Cell nuclei were stained with DAPI (blue; scale bar, 50 μm).
In vivo tumour imaging with fluorescently labelled DARPin 8h6
In the final set of experiments, we evaluated the in vivo imaging potential of DARPin 8h6 in mice. Since in vivo imaging requires appropriately labelled DARPins, we next evaluated whether Cy5.5-fluorescently labelled DARPin 8h6, conjugated at the same site on the C terminus as the biotin tag, could also bind CatB in cells. Indeed, Cy5.5-labelled DARPin 8h6 interacted with CatB at the cell membrane after blocking endocytosis, while the nonselective DARPin E3_5 did not (Fig. S9).
Since DARPin 8h6 binding to serum proteins could significanlty influence the biodistribution profile of the DARPin in vivo, we next incubated biotinylated DARPin 8h6 for 24 h at 37 °C in human serum, separated the mixture on a size-exclusion column and analysed the collected fractions. Western-blotting analysis revealed that the DARPin remained a monomeric protein and was eluted in the same fractions as purified DARPin in buffer (Fig. S10), supporting its in vivo use. In the next step we examined the pharmacokinetic profile of Cy5.5-labelled DARPins 8h6 and E3_5 in healthy mice. Both DARPins had fast biphasic blood clearance with the fast half-life below 2 min and the slow half-life of 78 min (DARPin E3_5) and 48 min (DARPin 8h6), respectively. More than 90 % of DARPin 8h6 was cleared from blood in the first two hours and less than 0.5 % was retained after 24 h (Fig. S11).
Finally, we evaluated whether the DARPin 8h6 interaction with CatB could be monitored in vivo in tumour-bearing mice. We administered Cy5.5-labelled DARPin 8h6 i.v. and followed its biodistribution using non-invasive whole-body imaging and ex vivo imaging of isolated organs. Initially we evaluated DARPin 8h6 in the orthotopic PyMT tumour model. In this model, DARPin 8h6 had already rapidly localized to tumours 30 min post administration and was selectively retained at the tumour site for up to 72 h post administration (Fig. 5b, c, Fig. S12). By contrast, the nonselective control DARPin E3_5 only displayed a very minor elevated fluorescence signal in tumours at early time-points and was not retained after the 3 h time point, likely due to the enhanced permeability and retention (EPR) effect in the leaky tumour vasculature. Moreover, when the ratios of the fluorescence signals between the tumour and contralateral mammary gland in individual mice were examined, DARPin 8h6 showed significantly elevated ratios at all time-points compared to DARPin E3_5, with a peak at 3 h post administration (Fig. 5d). The selective nature of DARPin 8h6 was further evaluated by an in vivo competition assay, where an excess of unlabelled DARPin 8h6 was administered to tumour-bearing mice 1 h before the administration of Cy5.5-labelled DARPin 8h6. This resulted in a significant reduction of the tumour signal as well as the ratios of the fluorescent signals between the tumours and contralateral mammary glands 3 h after administration (Fig. S13b, c, d). The lower-affinity DARPin 81 was also selectively retained in tumours after i.v. administration, although the tumour signals were significantly lower than with DARPin 8h6 (Fig. S14), likely due to the lower affinity of this DARPin for CatB.
Non-invasive optical tumour imaging by fluorescently labelled DARPin 8h6 in PyMT tumour model. (a) Schematic representation of the imaging experiment outline with the orthotopic PyMT tumour model. (b) PyMT cells were injected into the left inguinal fat pads of FVB/N mice and allowed to grow until 100-150 mm3. C-terminally Cy5.5-labelled DARPin 8h6 or nonselective DARPin E3_5 was injected i.v. (1 nmol in 100 μL) and epi-fluorescent images were taken at designated time-points after administration. False blue-hot colouring of the radiant efficiency (radiance (photons per second per square centimetre per steradian) per incident excitation power (microwatt per square cm)) is overlaid on bright-field images. (c) Quantification of the average radiant efficiency from b in tumours and contralateral mammary glands as the means ± standard error. Regions of interest (ROI): tumour - red circle, contralateral mammary gland - black circle. (d) Tumour-to-contralateral mammary gland ratios of the average radiant efficiency in individual mice from b represented as the means ± standard error. *** p<0.001, ** p<0.01 (e, f) DARPin distribution in isolated organs 3 h and (g, h) 72 h after administration. False blue-hot images of the radiance efficiency are overlaid on bright-field images. Representative images of four mice per group are shown. The average radiant efficiency from isolated organs is quantified as the mean ± standard error. Li = liver, S = spleen, Ln = lymph nodes, T = tumour, H = heart, Lu = lungs, K = kidneys. *** p<0.001, ** p<0.01, * p<0.05 (i) Immunohistochemical analysis of cryopreserved tumours 3 h after i.v. administration of Cy5.5-labelled DARPin 8h6 (red). The tissue sections were stained with macrophage marker F4/80 (green) and for cell nuclei (DAPI, blue). Scale bar, 20 μm; inlet scale bar, 10 μm. (j) DARPin pull-down of CatB from tumour tissue 30 minutes after intratumoural administration of 5 nmol of C-terminally biotinylated DARPins. The streptavidin-bound fraction was separated using SDS-PAGE and immunoblotted against CatB. The presence of DARPins was confirmed by detection with streptavidin-HRP. Total extract from tumour (50 μg) was added to visualize different forms of CatB in the tumours. proCatB = procathepsin B, scCatB = single chain CatB, hcCatB = heavy chain CatB.
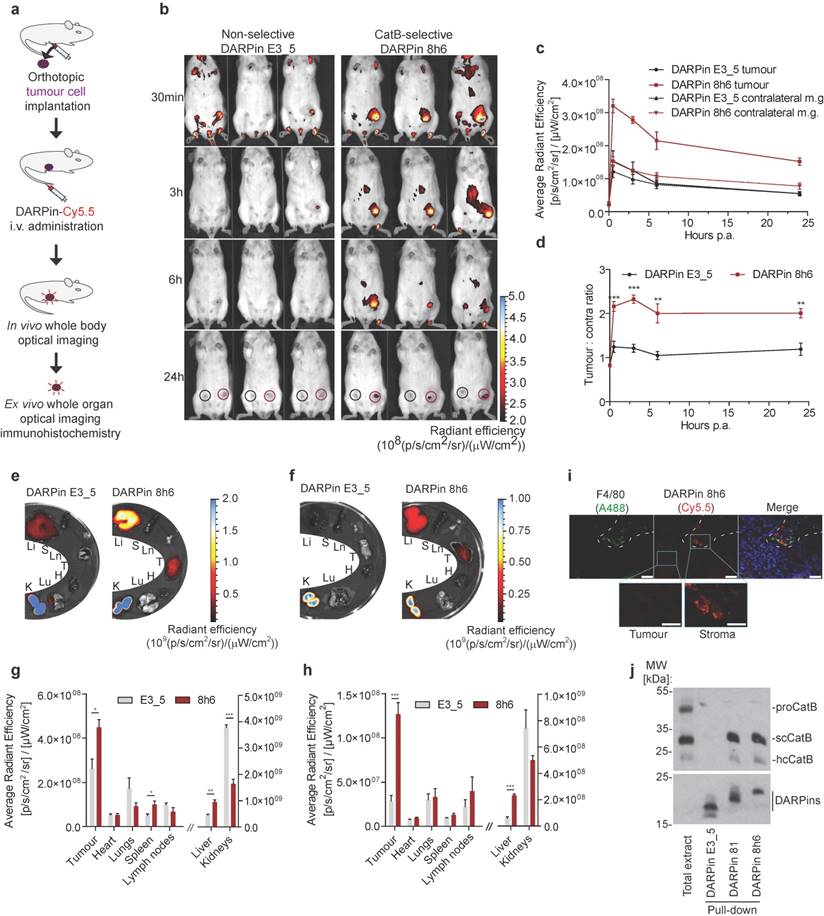
Next, we examined the DARPin distribution in isolated organs 3 h and 72 h post i.v. administration. The fluorescence signal from DARPin 8h6 was significantly higher in tumours than in other organs at both time-points, except for the liver and kidneys, which are involved in DARPin secretion from the body (Fig. 5e-h). Notably, there was specific up-regulation of DARPin 8h6 in comparison with the nonselective DARPin E3_5 in the tumour, as well as in the liver and spleen, in agreement with the high CatB content in these tissues [52]. Mice that were pre-treated with unlabelled DARPin 8h6 had significantly reduced tumour and liver signals after Cy5.5-labelled DARPin 8h6 administration, pointing to selective binding of DARPin 8h6 to CatB in these tissues (Fig. S13e, f). Considerable amounts of DARPin 8h6 were detected in tumours even after 72 h (1.5 % ± 0.3 % of the injected dose, compared to 5.4 % ± 0.7 % at 3 h, as determined from the fluorescence ratio of the tumour tissue and the injected DARPin 8h6-Cy5.5 solution (average radiant fluorescence = 8.3 × 109 (p/s/cm²/sr) / (µW/cm²))), indicative of substantial DARPin uptake by cells in the tumour microenvironment. This was additionally confirmed by immunohistochemical analysis of tumours following DARPin 8h6 administration, which showed that DARPin co-localized with CatB (Fig. S15) and was taken up by both tumour cells and F4/80-positive tumour-associated macrophages (Fig. 5i). To further validate the interaction between DARPin 8h6 and CatB in the tumour microenvironment, we performed streptavidin pull-down assay of tumour tissue after intratumoural application of biotinylated DARPin 8h6 (Fig. 5j). The DARPin interacted only with the mature form of CatB and not with the zymogen, which is in agreement with the significantly higher affinity of the DARPin for this form of CatB. DARPin 81 also bound only mature CatB, while there was no interaction with the nonselective control DARPin E3_5.
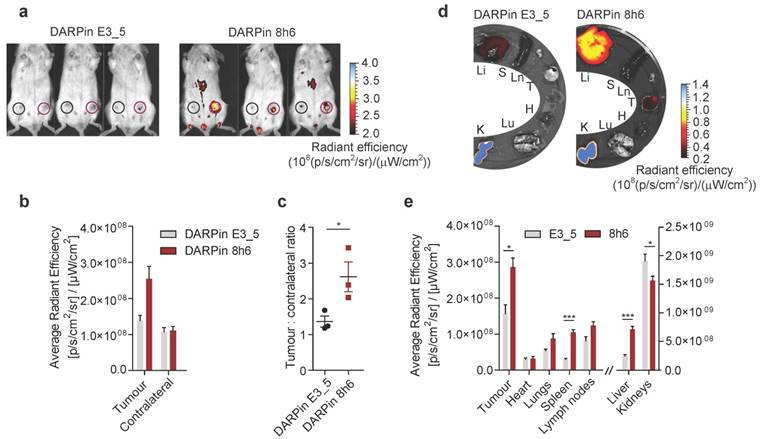
To investigate whether DARPin 8h6 can generally be used for imaging secreted CatB in the tumour microenvironment, we applied the 4T1 model, which is a highly aggressive model of breast cancer. Again, DARPin 8h6 selectively localized to the mammary gland tumours, as confirmed by both in vivo whole-body imaging and ex vivo imaging of isolated organs 3 h after administration. In comparison with the nonselective control DARPin E3_5, DARPin 8h6 displayed higher accumulation at the tumour site (Fig. 6a, b) as well as a significantly higher ratio of tumour-to-contralateral mammary gland (Fig. 6c). Ex vivo analysis confirmed that there were elevated levels of fluorescence in tumours, as well as liver and spleen, from mice administered DARPin 8h6 compared to DARPin E3_5 (Fig. 6d, e, Fig. S15), consistent with high CatB levels in these tissues. As in the PyMT model, the DARPin 8h6 signals in tumours were significantly higher compared to other organs, except for the liver and kidneys, which are involved in DARPin secretion. This evidence supports the broad usefulness of DARPin 8h6 in in vivo diagnostic imaging of CatB.
Non-invasive optical tumour imaging by fluorescently labelled DARPin 8h6 in 4T1 mouse tumour model. (a) 4T1 cells were injected into the left inguinal fat pads of Balb/c mice and allowed to grow until 100-150mm3. C-terminally Cy5.5-labelled DARPin 8h6 or nonselective DARPin E3_5 was injected i.v. (1 nmol in 100 µL), and epi-fluorescent images were taken 3 h after administration. False blue-hot colouring of the radiant efficiency (radiance (photons per second per square centimetre per steradian) per incident excitation power (microwatt per square cm)) is overlaid on bright-field images. (b) Quantification of the average radiant efficiency from a in tumours and contralateral mammary glands is shown as the means ± standard error. Regions of interest (ROI): tumour - red circle, contralateral mammary gland - black circle (c) Tumour-to-contralateral mammary gland ratios of the average radiant efficiency in individual mice from a represented as the means ± standard error. * p<0.05 (d, e) DARPin distribution in isolated organs 3 h after administration. False blue-hot images of the radiance efficiency are overlaid on bright-field images. Representative images of four mice per group are shown. The average radiant efficiency from each organ is quantified below as the mean ± standard error. Li = liver, S = spleen, Ln = lymph nodes, T = tumour, H = heart, Lu = lungs, K = kidneys *** p<0.001, * p<0.05.
Discussion
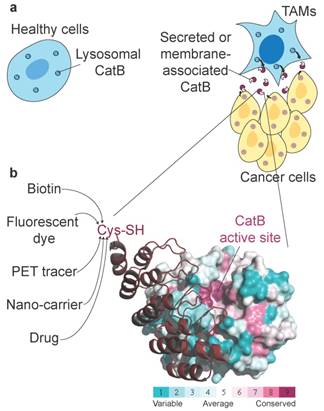
Non-invasive imaging of tumour-associated proteases has been widely investigated in the past, primarily because several proteases exhibit up-regulated expression and activity in tumour tissues and can therefore potentially be used in cancer diagnostics. Among these tumour-associated proteases, cysteine cathepsins have been particularly well investigated, with numerous fluorescent ABPs and substrates reported that showed good selectivity for target proteases and enabled in vivo detection of tumours in several mouse models as well as ex vivo detection in human biopsy samples [21, 53]. One important enzyme is CatB (Fig. 7a), which has already been proposed as a diagnostic target in preclinical tumour imaging, particularly by employing quenched NIR fluorescent substrates [54, 55] and a nanoparticulate CatB-targeting system [12]. To overcome the low penetration of fluorescence light and therefore its very limited use in human non-invasive diagnostics, we applied a new concept for highly selective targeting of CatB that could be applied to other proteases using the small and rigid non-immunoglobulin DARPin scaffold to obtain highly selective binders. The main advantage of this approach is that DARPins can be modified by various imaging tags without affecting their binding properties, including fluorescent and PET/SPECT tracers [33, 56] as well as other functional tags such as polymers or serum albumin to modify pharmacokinetics [31, 32]. A further advantage is that the interaction site is not limited to the active site cleft, as in ABPs and substrates (Fig. 7b). Although this approach has found substantial use in developing extremely selective binders to various receptors, including for potential therapeutic applications [57, 58], it has not been used in cellular or non-invasive diagnostic imaging of proteases. Moreover, the only protease-targeting DARPins were developed as selective inhibitors of a tobacco virus protease [59] or caspases [35, 60] to help elucidate the molecular basis of their specificity.
| Unconventional targeting of an emerging tumour target. (a) Schematic illustration of dysregulated CatB secretion from cancer and stromal cells (i.e., tumour-associated macrophages, TAMs) in the tumour microenvironment. In healthy tissues, CatB is contained intracellularly in endo-lysosomal vesicles. (b) Structural conservation (determined by ConSurf (http://consurf.tau.ac.il/)) within the cathepsin family in human CatB. Cathepsins have highly conserved active site clefts, making truly selective targeting using activity-based approaches difficult, whereas other regions with high structural variability provide uncharted territory for probe design with affinity probes such as DARPin 8h6. Engineering a unique cysteine residue at the C terminus of DARPins enables site-specific labelling with various chemical tags for the purpose of imaging, pull-down or drug delivery. The image was produced in PyMOL (www.pymol.org).
Using this approach, we developed a subset of DARPins that bound to both human and mouse CatB. This is of particular importance, as it allows immediate transfer of DARPins from mouse to human systems without affecting their binding properties and selectivity. We further characterized DARPins 8h6 and 81, with a major focus on DARPin 8h6. This DARPin selectively binds and inhibits only human and mouse CatB with very high affinity in the picomolar range (Ki ~ 35 pM), but not the structurally related cathepsins L, K, S or X. This is a substantially higher affinity than the affinity of cystatin C (Ki ~ 150 pM; ref [61]), which is the best endogenous inhibitor of CatB. Both the specificity and high affinity can be explained on a structural basis, since the DARPin binds to an extended region on the left CatB domain, which is poorly conserved in the family of cathepsins, as well as to a part of the active site cleft S2 and S3 subsites, but does not clash with the occluding loop residues, thereby differing from cystatins [62]. By contrast, DARPin 81, which binds to essentially the same region on CatB and was also specific for CatB, exhibited affinity that was lower by two orders of magnitude (Ki ~ 4.1 nM) due to the absence of two important tryptophan residues in the sequence. Extremely high affinity is vital for successful in vivo targeting, as demonstrated previously using differently modified HER2-targeting monovalent DARPins as a model. In that study, the high-affinity DARPin G3 (Kd ~90 pM) led to significantly higher tumour targeting and accumulation than low-affinity mutant DARPins (Kd >1 nM), which lack critical interface residues that are involved in binding [33]. In agreement with this, the high affinity-binding DARPin 8h6 exhibited substantially higher tumour accumulation than the low-affinity DARPin 81 in the PyMT mammary gland mouse model (Fig. 5, S14). Substantial accumulation of DARPin 8h6 in the tumour was observed after only 30 min, with a peak value of the signal ratio between the tumour and contralateral mammary gland after 3 h, which is comparable to quenched ABPs [19]. The signal ratio between the tumour and contralateral mammary gland for DARPin 8h6 remained very high even at 24 h, 48 h and 72 h time points, indicating that reliable in vivo imaging of tumours can be achieved for a very long time after initial low unspecific background signal is washed away. Moreover, the pharmacokinetic behaviour and unspecific background signals generated by the DARPin might be further improved by using alternative imaging reporters (i.e., different fluorophores, PET or SPECT reporters) or replacing the N-terminal His-tag with His-Glu-tag [56].
Moreover, our study also confirmed the presence of high levels of CatB in the tumour microenvironment. Although this tumour CatB, which predominantly originated in macrophages, was secreted as both the zymogen and mature form from dBMM in the culture (Fig. 3a), DARPin 8h6 only labelled the mature form in the tumour and not the zymogen (Fig. 5j), consistent with the preferential binding of the DARPin to this CatB form due to a steric clash with the propeptide that results in a diminished rate of association and affinity for the zymogen. This is in agreement with the recent results of a proteomic analysis of PyMT tumour interstitial fluids, which showed that at least part of the secreted CatB exists in the zymogen proCatB form [63]. Our results therefore support previous findings, where a significant proportion of the secreted zymogen has been found to be activated in the acidic extracellular milieu in tumours [64, 65]. Importantly, the DARPin signal in the tumour was clearly detectable at least until 72 hours post-administration (Fig. S12), which provides an excellent opportunity for theranostic applications based on CatB targeting. The DARPin can also be used as the targeting moiety in an antibody-drug conjugate (ADC)-like approach, especially since conjugation is achieved via the C-terminus and thereby does not affect the interaction with the target. However, DARPin 8h6 is not really suitable for imaging liver tumours. DARPin 8h6 namely partially accumulated in the liver as a consequence of binding to CatB (Fig. S13), which is highly abundant in this organ [52]. Nevertheless, masking the binding region with polymers such as PEG might reduce the CatB-specific liver signal. Moreover, conjugating the polymer to the DARPin via a linker that is cleavable by another tumour-specific protease, might result in selective tumour targeting even in the liver and improve the overall imaging quality as has been recently suggested [66].
DARPin 8h6 is not just an excellent diagnostic agent for non-invasive cancer imaging based on CatB but is also an excellent tool for studying CatB function in cells. A significant amount of pericellular and membrane-attached CatB was detected in dBMM, consistent with previous findings [9, 10, 12]. Moreover, using the DARPin 8h6 inhibitory potential we successfully reduced tumour cell invasion, where pericellular proteolysis of extracellular matrix by CatB importantly contributes to cancer spreading [43]. Furthermore, DARPin 8h6 is the best macromolecular inhibitor of CatB found to date, thereby offering numerous additional possibilities, ranging from selective inhibition of CatB in cellular studies to identification of the physiological ligands of CatB using affinity pull-down as well as subsequent identification of the ligands via mass spectrometry. In addition, selective inhibition of individual intracellular cathepsins, including CatB, is very difficult to achieve using small molecule inhibitors due to their limited selectivity, including the most widely used CatB cell-permeable inhibitor CA074-OMe [67]. Therefore, transgenic expression of DARPin 8h6 in mammalian cells could serve as a powerful tool in biomedical research.
Conclusions
In summary, the use of engineered macromolecules, such as DARPins, in non-invasive diagnostic imaging is a new approach in targeting tumour-associated proteases that is completely different from the standard small molecule-based approaches. The strategy is applicable to secreted or membrane-associated proteases, particularly where complete selectivity in the active site is difficult to achieve. In addition, such highly selective DARPins provide a major opportunity for in vivo validation of the therapeutic compounds targeting these proteases. Moreover, the DARPins can be used both as inhibitors and imaging agents, thereby combining the properties of small molecule inhibitors and ABPs for various theranostic applications.
Abbreviations
DARPin: designed ankyrin repeat protein; CatB: cathepsin B; scCatB: single chain cathepsin B; hcCatB: heavy chain cathepsin B; proCatB: procathepsin B; dBMM: differentiated bone-marrow macrophages; MCTSs: multicellular tumour spheroids; ABP: activity-based probe; NIR: near infra-red; ADC: antibody-drug conjugate; PET: positron emission tomography; SPECT: single-photon emission computed tomography; MRI: magnetic resonance imaging; HRP: horse-radish peroxidase; RMSD: Root-mean-square deviation.
Acknowledgements
We thank Robert Vidmar, Reto Zbinden, Stefan Schauer and the Functional Genomic Center Zürich for technical and methodological assistance. This work was supported by Slovene Research Agency (grant no. P1-0140 and J1-6739 to B.T., P1-0048 to D.T.), by the EC's Seventh Framework Program LIVIMODE (to B.T. and M.G.G.) and P-CUBE (under grant agreement 227764 to M.G.G.), the Swiss National Science Foundation Grant (310030B_138673 to M.G.G.) and the Swiss National Foundation (SNF Professorship PP00P3_144823, to M.A.S.).
Author contributions
L.K. and B.T. conceived and designed the experiments. L.K., J.Z. and M.R. performed the experiments. L.K., M.R., D.T., O.V. and B.T. analysed the data. M.A.S. contributed the DARPin library and reagents for ribosome display. M.G.G., V.T. and B.T. supervised the project. L.K. and B.T. wrote the manuscript. All authors discussed the results and commented on the manuscript.
Supplementary Material
Supplementary figures and tables.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Deu E, Verdoes M, Bogyo M. New approaches for dissecting protease functions to improve probe development and drug discovery. Nature structural & molecular biology. 2012;19:9-16
2. Weissleder R, Pittet MJ. Imaging in the era of molecular oncology. Nature. 2008;452:580-9
3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
4. Sevenich L, Joyce JA. Pericellular proteolysis in cancer. Genes & development. 2014;28:2331-47
5. Olson OC, Joyce JA. Cysteine cathepsin proteases: regulators of cancer progression and therapeutic response. Nature reviews Cancer. 2015;15:712-29
6. Strojnik T, Kos J, Zidanik B, Golouh R, Lah T. Cathepsin B immunohistochemical staining in tumor and endothelial cells is a new prognostic factor for survival in patients with brain tumors. Clinical cancer research. 1999;5:559-67
7. Thomssen C, Schmitt M, Goretzki L, Oppelt P, Pache L, Dettmar P. et al. Prognostic value of the cysteine proteases cathepsins B and cathepsin L in human breast cancer. Clinical cancer research. 1995;1:741-6
8. Shree T, Olson OC, Elie BT, Kester JC, Garfall AL, Simpson K. et al. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes & development. 2011;25:2465-79
9. Mohamed MM, Cavallo-Medved D, Rudy D, Anbalagan A, Moin K, Sloane BF. Interleukin-6 increases expression and secretion of cathepsin B by breast tumor-associated monocytes. Cellular physiology and biochemistry. 2010;25:315-24
10. Gocheva V, Wang H-W, Gadea BB, Shree T, Hunter KE, Garfall AL. et al. IL-4 induces cathepsin protease activity in tumor-associated macrophages to promote cancer growth and invasion. Genes & development. 2010;24:241-55
11. Yan D, Wang H-W, Bowman Robert L, Joyce JA. STAT3 and STAT6 Signaling Pathways Synergize to Promote Cathepsin Secretion from Macrophages via IRE1α Activation. Cell Reports. 2016;16:2914-27
12. Mikhaylov G, Klimpel D, Schaschke N, Mikac U, Vizovišek M, Fonović M. et al. Selective targeting of tumor and stromal cells by a nanocarrier system displaying lipidated cathepsin B inhibitor. Angewandte Chemie (International ed in English). 2014;53:10077-81
13. Roshy S, Sloane BF, Moin K. Pericellular cathepsin B and malignant progression. Cancer and Metastasis reviews. 2003;22:271-86
14. Sörensen J, Sandberg D, Sandström M, Wennborg A, Feldwisch J, Tolmachev V. et al. First-in-human molecular imaging of HER2 expression in breast cancer metastases using the 111In-ABY-025 affibody molecule. Journal of nuclear medicine. 2014;55:730-5
15. Weissleder R, Tung C-H, Mahmood U, Bogdanov A. In vivo imaging of tumors with protease-activated near-infrared fluorescent probes. Nature Biotechnology. 1999;17:375-8
16. Watzke A, Kosec G, Kindermann M, Jeske V, Nestler HP, Turk V. et al. Selective activity-based probes for cysteine cathepsins. Angewandte Chemie (International ed in English). 2008;47:406-9
17. Hilderbrand SA, Weissleder R. Near-infrared fluorescence: application to in vivo molecular imaging. Current Opinion in Chemical Biology. 2010;14:71-9
18. Sanman LE, Bogyo M. Activity-Based Profiling of Proteases. Annual Review of Biochemistry. 2014;83:249-73
19. Blum G, Von Degenfeld G, Merchant MJ, Blau HM, Bogyo M. Noninvasive optical imaging of cysteine protease activity using fluorescently quenched activity-based probes. Nature Chemical Biology. 2007;3:668-77
20. Verdoes M, Oresic Bender K, Segal E, van der Linden WA, Syed S, Withana NP. et al. Improved quenched fluorescent probe for imaging of cysteine cathepsin activity. Journal of the American Chemical Society. 2013;135:14726-30
21. Hu HY, Vats D, Vizovišek M, Kramer L, Germanier C, Wendt KU. et al. In vivo imaging of mouse tumors by a lipidated cathepsin S substrate. Angewandte Chemie (International ed in English). 2014;53:7669-73
22. Nomura DK, Dix MM, Cravatt BF. Activity-based protein profiling for biochemical pathway discovery in cancer. Nature reviews Cancer. 2010;10:630-8
23. Li N, Kuo C-L, Paniagua G, van den Elst H, Verdoes M, Willems LI. et al. Relative quantification of proteasome activity by activity-based protein profiling and LC-MS/MS. Nature Protocols. 2013;8:1155-68
24. Ren G, Blum G, Verdoes M, Liu H, Syed S, Edgington LE. et al. Non-Invasive Imaging of Cysteine Cathepsin Activity in Solid Tumors Using a 64Cu-Labeled Activity-Based Probe. PloS one. 2011;6:e28029
25. Withana NP, Ma X, McGuire HM, Verdoes M, van der Linden WA, Ofori LO. et al. Non-invasive Imaging of Idiopathic Pulmonary Fibrosis Using Cathepsin Protease Probes. Scientific Reports. 2016;6:19755
26. Caglič D, Globisch A, Kindermann M, Lim N-H, Jeske V, Juretschke H-P. et al. Functional in vivo imaging of cysteine cathepsin activity in murine model of inflammation. Bioorganic & medicinal chemistry. 2011;19:1055-61
27. Verdoes M, Edgington LE, Scheeren FA, Leyva M, Blum G, Weiskopf K. et al. A nonpeptidic cathepsin S activity-based probe for noninvasive optical imaging of tumor-associated macrophages. Chemistry & biology. 2012;19:619-28
28. Jaffer FA, Kim D-E, Quinti L, Tung C-H, Aikawa E, Pande AN. et al. Optical visualization of cathepsin K activity in atherosclerosis with a novel, protease-activatable fluorescence sensor. Circulation. 2007;115:2292-8
29. Binz HK, Stumpp MT, Forrer P, Amstutz P, Pluckthun A. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. Journal of Molecular Biology. 2003;332:489-503
30. Binz HK, Amstutz P, Kohl A, Stumpp MT, Briand C, Forrer P. et al. High-affinity binders selected from designed ankyrin repeat protein libraries. Nature Biotechnology. 2004;22:575-82
31. Simon M, Zangemeister-Wittke U, Pluckthun A. Facile double-functionalization of designed ankyrin repeat proteins using click and thiol chemistries. Bioconjugate Chemistry. 2012;23:279-86
32. Simon M, Frey R, Zangemeister-Wittke U, Pluckthun A. Orthogonal assembly of a designed ankyrin repeat protein-cytotoxin conjugate with a clickable serum albumin module for half-life extension. Bioconjugate Chemistry. 2013;24:1955-66
33. Zahnd C, Kawe M, Stumpp MT, de Pasquale C, Tamaskovič R, Nagy-Davidescu G. et al. Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: effects of affinity and molecular size. Cancer Research. 2010;70:1595-605
34. Seeger MA, Zbinden R, Flutsch A, Gutte PG, Engeler S, Roschitzki-Voser H. et al. Design, construction, and characterization of a second-generation DARP in library with reduced hydrophobicity. Protein science. 2013;22:1239-57
35. Schweizer A, Roschitzki-Voser H, Amstutz P, Briand C, Gulotti-Georgieva M, Prenosil E. et al. Inhibition of caspase-2 by a designed ankyrin repeat protein: specificity, structure, and inhibition mechanism. Structure. 2007;15:625-36
36. Baici A. The specific velocity plot. A graphical method for determining inhibition parameters for both linear and hyperbolic enzyme inhibitors. European journal of biochemistry. 1981;119:9-14
37. Szedlacsek SE, Ostafe V, Serban M, Vlad MO. A re-evaluation of the kinetic equations for hyperbolic tight-binding inhibition. The Biochemical Journal. 1988;254:311-2
38. Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallographica Section D: Biological Crystallography. 2011;67:355-67
39. Turk D. MAIN software for density averaging, model building, structure refinement and validation. Acta Crystallographica Section D: Biological Crystallography. 2013;69:1342-57
40. Vasiljeva O, Papazoglou A, Kruger A, Brodoefel H, Korovin M, Deussing J. et al. Tumor cell-derived and macrophage-derived cathepsin B promotes progression and lung metastasis of mammary cancer. Cancer Research. 2006;66:5242-50
41. Weischenfeldt J, Porse B. Bone Marrow-Derived Macrophages (BMM): Isolation and Applications. CSH Protocols. 2008;2008:pdb.prot5080
42. Hulkower KI, Butler CC, Linebaugh BE, Klaus JL, Keppler D, Giranda VL. et al. Fluorescent microplate assay for cancer cell-associated cathepsin B. European Journal of Biochemistry. 2000;267:4165-70
43. Mirković B, Markelc B, Butinar M, Mitrovič A, Sosič I, Gobec S. et al. Nitroxoline impairs tumor progression in vitro and in vivo by regulating cathepsin B activity. Oncotarget. 2015;6:19027-42
44. Ruettger A, Wiederanders B. Microplate assay for cathepsin detection in viable cells using derivatives of 4-methoxy-beta-naphthylamide. Current Protocols in Protein Science. 2007 Chapter 21: Unit 21
45. Mikhaylov G, Mikac U, Magaeva AA, Itin VI, Naiden EP. et al. Ferri-liposomes as an MRI-visible drug-delivery system for targeting tumours and their microenvironment. Nature Nanotechnology. 2011;6:594-602
46. Kohl A, Binz HK, Forrer P, Stumpp MT, Plückthun A, Grütter MG. Designed to be stable: crystal structure of a consensus ankyrin repeat protein. Proceedings of the National Academy of Sciences. 2003;100:1700-5
47. Podobnik M, Kuhelj R, Turk V, Turk D. Crystal structure of the wild-type human procathepsin B at 2.5 Å resolution reveals the native active site of a papain-like cysteine protease zymogen. Journal of Molecular Biology. 1997;271:774-88
48. Quraishi O, Nagler DK, Fox T, Sivaraman J, Cygler M, Mort JS. et al. The occluding loop in cathepsin B defines the pH dependence of inhibition by its propeptide. Biochemistry. 1999;38:5017-23
49. Bengsch F, Buck A, Gunther SC, Seiz JR, Tacke M, Pfeifer D. et al. Cell type-dependent pathogenic functions of overexpressed human cathepsin B in murine breast cancer progression. Oncogene. 2014;33:4474-84
50. Withana NP, Blum G, Sameni M, Slaney C, Anbalagan A, Olive MB. et al. Cathepsin B inhibition limits bone metastasis in breast cancer. Cancer Research. 2012;72:1199-209
51. Stefan N, Martin-Killias P, Wyss-Stoeckle S, Honegger A, Zangemeister-Wittke U, Plückthun A. DARPins recognizing the tumor-associated antigen EpCAM selected by phage and ribosome display and engineered for multivalency. Journal of Molecular Biology. 2011;413:826-43
52. Shuja S, Sheahan K, Murnane MJ. Cysteine endopeptidase activity levels in normal human tissues, colorectal adenomas and carcinomas. International Journal of Cancer. 1991;49:341-6
53. Withana NP, Garland M, Verdoes M, Ofori LO, Segal E, Bogyo M. Labeling of active proteases in fresh-frozen tissues by topical application of quenched activity-based probes. Nature Protocols. 2016;11:184-91
54. Yhee JY, Kim SA, Koo H, Son S, Ryu JH, Youn IC. et al. Optical imaging of cancer-related proteases using near-infrared fluorescence matrix metalloproteinase-sensitive and cathepsin B-sensitive probes. Theranostics. 2012;2:179-89
55. Ryu JH, Kim SA, Koo H, Yhee JY, Lee A, Na JH. et al. Cathepsin B-sensitive nanoprobe for in vivo tumor diagnosis. Journal of Materials Chemistry. 2011;21:17631-4
56. Goldstein R, Sosabowski J, Livanos M, Leyton J, Vigor K, Bhavsar G. et al. Development of the designed ankyrin repeat protein (DARPin) G3 for HER2 molecular imaging. European Journal of Nuclear Medicine and Molecular Imaging. 2015;42:288-301
57. Münch RC, Muth A, Muik A, Friedel T, Schmatz J, Dreier B. et al. Off-target-free gene delivery by affinity-purified receptor-targeted viral vectors. Nature Communications. 2015;6:6246
58. Tamaskovič R, Schwill M, Nagy-Davidescu G, Jost C, Schaefer DC, Verdurmen WP. et al. Intermolecular biparatopic trapping of ErbB2 prevents compensatory activation of PI3K/AKT via RAS-p110 crosstalk. Nature Communications. 2016;7:11672
59. Kawe M, Forrer P, Amstutz P, Pluckthun A. Isolation of intracellular proteinase inhibitors derived from designed ankyrin repeat proteins by genetic screening. The Journal of Biological Chemistry. 2006;281:40252-63
60. Flütsch A, Schroeder T, Barandun J, Ackermann R, Bühlmann M, Grütter MG. Specific targeting of human caspases using designed ankyrin repeat proteins. Biological Chemistry. 2014;395:1243-52
61. Abrahamson M, Mason RW, Hansson H, Buttle DJ, Grubb A, Ohlsson K. Human cystatin C. Role of the N-terminal segment in the inhibition of human cysteine proteinases and in its inactivation by leucocyte elastase. Biochemical Journal. 1991;273:621-6
62. Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B. et al. Cysteine cathepsins: from structure, function and regulation to new frontiers. Biochimica et biophysica acta. 2012;1824:68-88
63. Gomez-Auli A, Hillebrand LE, Biniossek ML, Peters C, Reinheckel T, Schilling O. Impact of cathepsin B on the interstitial fluid proteome of murine breast cancers. Biochimie. 2016;122:88-98
64. Rozhin J, Sameni M, Ziegler G, Sloane BF. Pericellular pH affects distribution and secretion of cathepsin B in malignant cells. Cancer Research. 1994;54:6517-25
65. Rothberg JM, Bailey KM, Wojtkowiak JW, Ben-Nun Y, Bogyo M, Weber E. et al. Acid-Mediated Tumor Proteolysis: Contribution of Cysteine Cathepsins. Neoplasia. 2013;15:1125-37
66. Stefan N, Zimmermann M, Simon M, Zangemeister-Wittke U, Pluckthun A. Novel prodrug-like fusion toxin with protease-sensitive bioorthogonal PEGylation for tumor targeting. Bioconjugate Chemistry. 2014;25:2144-56
67. Greenbaum D, Medzihradszky KF, Burlingame A, Bogyo M. Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chemistry & Biology. 2000;7:569-81
Author contact
![]() Corresponding author: boris.turksi
Corresponding author: boris.turksi