Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(6):1689-1704. doi:10.7150/thno.18125 This issue Cite
Research Paper
Tumor Penetrating Theranostic Nanoparticles for Enhancement of Targeted and Image-guided Drug Delivery into Peritoneal Tumors following Intraperitoneal Delivery
Ning Gao1*, Erica N. Bozeman1*, Weiping Qian1, Liya Wang2, Hongyu Chen3, Malgorzata Lipowska2, Charles A Staley1, Y. Andrew Wang3, Hui Mao2, Lily Yang1, 2 ![]()
1. Department of Surgery, Emory University School of Medicine, Atlanta, GA 30322;
2. Department of Radiology and Imaging Sciences, Emory University School of Medicine, Atlanta, GA 30322;
3. Ocean Nanotech, LLC, San Diego, CA 92126.
* Authors contributed equally to this study
Received 2016-10-28; Accepted 2017-2-15; Published 2017-4-10
Abstract

The major obstacles in intraperitoneal (i.p.) chemotherapy of peritoneal tumors are fast absorption of drugs into the blood circulation, local and systemic toxicities, inadequate drug penetration into large tumors, and drug resistance. Targeted theranostic nanoparticles offer an opportunity to enhance the efficacy of i.p. therapy by increasing intratumoral drug delivery to overcome resistance, mediating image-guided drug delivery, and reducing systemic toxicity. Herein we report that i.p. delivery of urokinase plasminogen activator receptor (uPAR) targeted magnetic iron oxide nanoparticles (IONPs) led to intratumoral accumulation of 17% of total injected nanoparticles in an orthotopic mouse pancreatic cancer model, which was three-fold higher compared with intravenous delivery. Targeted delivery of near infrared dye labeled IONPs into orthotopic tumors could be detected by non-invasive optical and magnetic resonance imaging. Histological analysis revealed that a high level of uPAR targeted, PEGylated IONPs efficiently penetrated into both the peripheral and central tumor areas in the primary tumor as well as peritoneal metastatic tumor. Improved theranostic IONP delivery into the tumor center was not mediated by nonspecific macrophage uptake and was independent from tumor blood vessel locations. Importantly, i.p. delivery of uPAR targeted theranostic IONPs carrying chemotherapeutics, cisplatin or doxorubicin, significantly inhibited the growth of pancreatic tumors without apparent systemic toxicity. The levels of proliferating tumor cells and tumor vessels in tumors treated with the above theranostic IONPs were also markedly decreased. The detection of strong optical signals in residual tumors following i.p. therapy suggested the feasibility of image-guided surgery to remove drug-resistant tumors. Therefore, our results support the translational development of i.p. delivery of uPAR-targeted theranostic IONPs for image-guided treatment of peritoneal tumors.
Keywords: Targeted theranostic nanoparticles, tumor penetration, intraperitoneal drug delivery, image-guided therapy, pancreatic cancer.
Introduction
Conventional chemotherapy for the treatment of tumors in the peritoneal cavity, such as pancreatic, gastric, colon, liver, and ovarian cancers, presents clinical challenges due to diagnosis at the advanced stage, high incidence of tumor recurrence, and chemoresistance (1, 2). Peritoneal tumors account for approximately 250,000 new cancer cases annually in the USA (3). A large percentage of cancer patients have local tumor invasion or intraperitoneal (i.p.) metastatic tumors at presentation, or develop i.p. recurrent tumors shortly after surgery resulting in high mortality rates among patients. Therefore, the development of novel, localized treatment for i.p. metastatic tumors should improve the therapeutic efficacy and survival of cancer patients.
Based on the presence of the peritoneal-plasma barrier, regional therapy by i.p. infusion of chemotherapeutics offers a means of facilitating drug delivery to the tumor-containing peritoneal cavity (1). Hyperthermic intraperitoneal chemotherapy administered following cytoreductive surgery has demonstrated clinical/survival benefits in cancer patients (4, 5). However, chemotherapy drugs used for i.p. therapy are quickly absorbed through the peritoneal capillaries and enter into systemic circulation, which significantly reduce their residence time in the peritoneal cavity and thus limit their therapeutic efficacy (2). Drug delivery into tumors by passive diffusion can only enter a few cell layers or up to 3 mm in depth in bulky tumors (6). To improve drug delivery, different drug formulations are under investigation (3). Microparticle or nanoparticle formulations have been used to improve drug's pharmacokinetic performances including longer retention time in the peritoneal fluid (3, 7). It has been shown that pH sensitive, polymeric nanoparticles carrying paclitaxel were retained in the peritoneal cavity for 7 days after i.p. delivery in a mouse peritoneal tumor model (8). PEGylated liposomal nanoparticles encapsulated with doxorubicin (Dox) have been used for i.p. therapy of patients with gastrointestinal or gynecologic malignancies and demonstrated survival benefit among patients (9).
To enhance the specificity of cancer therapy, targeted nanoparticles have been developed by conjugating targeting ligands that bind to cell receptors highly expressed in tumor cells (10-13). Increasing evidence showed that targeted delivery of nanoparticle-drug increases drug accumulation in tumors and therapeutic effects while minimizing systemic toxicity. Urokinase plasminogen activator receptor (uPAR) is overexpressed by tumor cells and active stromal cells (14-16). 70% of pancreatic (15), 80% of ovarian (17), 85% of colon (18), and 37% of gastric cancers (19) express high levels of uPAR. uPAR expression is further increased along the invasive tumor edge and in metastatic tumors (20, 21). We have previously demonstrated that uPAR targeting enhanced internalization of nanoparticles by tumor and stromal cells, making uPAR a viable target for effective drug delivery into peritoneal metastatic tumors with a dense surface stromal barrier (22, 23). Furthermore, we have shown the therapeutic efficacy as well as magnetic resonance imaging (MRI) capability of uPAR targeted theranostic iron oxide nanoparticles (IONPs) carrying gemcitabine in a human pancreatic cancer xenograft model following intravenous (i.v.) administration (12). In this study, we investigated and compared targeting efficiency and biodistribution of uPAR targeted delivery of IONPs with or without chemotherapy drugs (cisplatin (Cis) or Dox) into peritoneal tumors following i.v. or i.p. delivery in an orthotopic mouse pancreatic tumor (PANC02) model. The therapeutic efficacy of i.p. delivery of uPAR targeted theranostic nanoparticles was also demonstrated in this highly aggressive tumor model.
Materials and Methods
Mouse peritoneal tumor model
The PANC02 mouse pancreatic cancer cell line (24) was kindly provided by Dr. Keping Xie (M.D. Anderson Cancer Center, Houston). We verified that the PANC02 cells had the same morphology and biomarker expression as previously published (25, 26). For establishment of an orthotopic pancreatic tumor model, 6 to 8 week-old female, C57BL/6 mice (Harlan Laboratories, IN) were anesthetized by intramuscular injection of a mixture of 95 mg/kg ketamine hydrochloride and 5 mg/kg xylazine of body weight in sterile saline. Following local shaving and disinfection, an abdominal incision was made to expose the spleen and the pancreas. PANC02 cells (5x105) mixed with BD Matrigel were injected directly into the pancreas. Incision was closed using suture and surgical clamps. Mice were allowed to recover and treated with pain medication for 2 days after the surgical procedure.
For comparison of the effect of targeted delivery of IONPs into subcutaneous (s.c.) and orthotopic tumors after i.v. or i.p. injection, the same mice received tumor cell injections into the pancreas as well as on the hind flank. Such an approach enabled accurate evaluation of nanoparticle delivery without the effect of variations in blood concentrations of nanoparticles among different mice.
Production of targeting ligand conjugated nanoparticles
Recombinant amino terminal fragment (ATF) peptides containing 135 amino acids of the receptor-binding domain of mouse uPA was produced in a bacterial expressing system and used as uPAR targeting ligands (12, 27). Bovine serum albumin (BSA) was used as a non-targeted control for nanoparticle drug delivery mediated by the enhanced permeability and retention effect (EPR) (28).
Magnetic IONPs with 10 nm core size functionalized with an amphiphilic copolymer containing carboxyl groups were prepared by Ocean Nanotech, LLC (San Diego, CA) using an established protocol (29, 30). Amine PEG carboxyl (MW2000, Biomatrik, Zhejiang, China) was conjugated to the surface carboxyl groups of the amphiphilic polymer coating to generate PEG-IONPs with surface carboxyl groups in order to avoid non-specific uptake by macrophages present within the peritoneal cavity. Mouse ATF or control BSA proteins were conjugated to IONPs by cross-linking carboxyl to amino groups of ATF mediated by 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDAC) at a ratio of ATF : IONP of 15:1. For near infrared (NIR) optical imaging, a NIR 830-maleimide dye was labeled to the free thiol group on the cysteine of the peptides or proteins using a standard protocol prior conjugating ligands onto the surface of PEGylated IONPs (31, 32) (Fig. 1A). Excitation wavelength of NIR-830 dye is 800 nm and emission wavelength is 830 nm. The final nanoparticle conjugates were purified using a Nanosep 100k filter column (Pall Corp, MI). Nanoparticles were characterized by transmission electron microscopy (TEM). Hydrodynamic sizes of nanoparticles were determined using Zetasizer Nano (Malvern Instruments Inc., MA).
Development and characterization of uPAR targeted theranostic IONPs. A, Schematic illustration of the production of uPAR targeted ATF-PEG-IONP and theranostic IONPs carrying chemotherapeutic drugs, Cisplatin (Cis) or Doxorubicin (Dox). B, Transmission electron microscopy (TEM) images of non-targeted PEG-IONP-Cis and ATF-PEG-IONP-Cis. Hydrodynamic sizes of both IONPs were determined using the dynamic light scattering (DLS) method. C, Targeted delivery of IONPs into the PANC02 mouse pancreatic cancer cells using mouse ATF conjugated PEG-IONP. The amount of IONP uptake was quantified by chemical analysis of iron concentration in cells. Bar figure shows the mean O.D. values of three repeat samples.
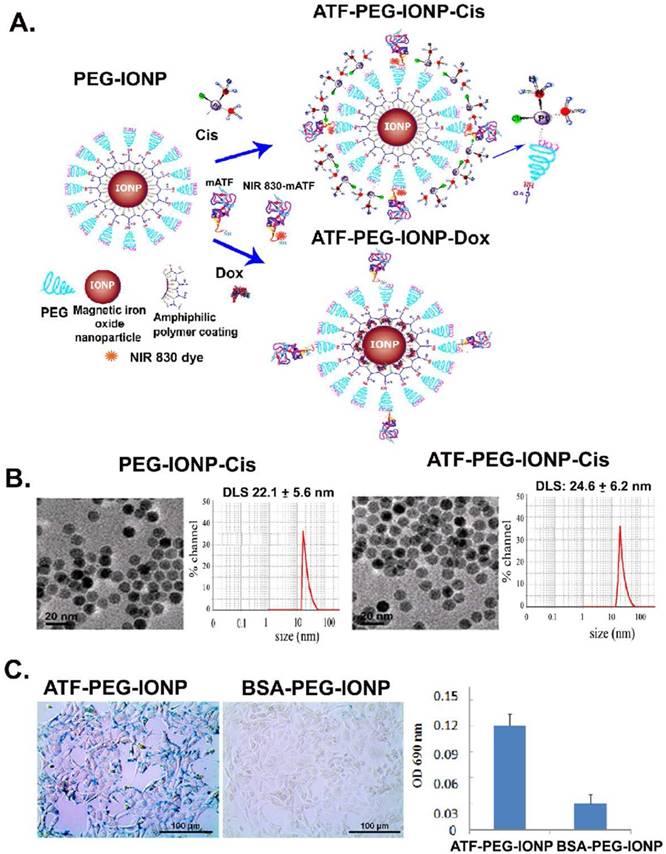
Target specificity of ATF-PEG-IONP was determined on uPAR expressing mouse pancreatic cancer PANC02 cells following incubation with 50 picomolar (pmol) of IONP-equivalent BSA-PEG-IONPs or ATF-PEG-IONPs in 6 well plates for 4 hours at 37°C. Prussian blue staining was then performed (12). Uptake of the IONPs in cells was further quantified by measuring the absorbance at O.D. 690 nm in cell lysates.
Loading chemotherapy drugs on IONPs
Cisplatin (Cis) (Polymed Therapeutics, Inc., TX) was conjugated to the surface of uPAR targeted ATF-PEG-IONP by mixing targeting ligand conjugated IONPs with Cis at a ratio of 1 mg Cis to 1 mg of iron equivalent PEG-IONPs in H2O, pH 8.5 at room temperature for 4 hours. The positive charge of platinum (Pt+) interacts with the negative charge of carboxylate group (O=C-O-, Lewis base) on the surface of IONP via a coordinate bond which is very stable at pH > 7.4. Unconjugated cisplatin was removed by washing through purified Nanosep 100 K column, resulting in ATF-PEG-IONP-Cis theranostic nanoparticles (Fig. 1A). The amount of Cis on the IONPs was determined by phenylenediamine (OPDA) colorimetric assay at O.D. 702 nm (33). The conjugation ratio of Cis versus IONPs was 0.2 -0.3 mg of Cis/ Fe mg of IONPs. The ability of Cis release from IONPs under pH 5.0 was determined in vitro in solution.
Dox (Polymed Therapeutics, Inc) was encapsulated into the hydrophobic space in the amphiphilic polymer coating by mixing ATF-PEG-IONP with Dox in 10 mM borate buffer (pH 8.5) at a ratio of 1 mg of Dox and 2 mg of iron equivalent of PEG-IONP at room temperature for 4 hours (22) (Fig. 1A). The amount of encapsulated Dox was determined based on optical density measurements at Dox absorbance wavelength of 490 nm and IONP absorbance of 400 nm and four standard curves (using free Dox and IONPs). The Dox encapsulation is approximately 0.3 mg to 0.5 mg of Dox per mg of iron equivalent PEG-IONPs.
In vivo nanoparticle delivery protocols
Mice bearing orthotopic PANC02 tumors were administered with 300 pmol of ATF-PEG-IONPs or control BSA-PEG-IONPs without or with Cis via the tail vein (i.v.) or i.p. delivery. Biodistribution analysis of IONP accumulation after a single injection was determined 24 hours following IONP delivery. For i.p. treatment, tumor bearing mice received four injections of ATF-PEG-IONP-Cis, BSA-PEG-IONP-Cis or free Cis (5 mg/kg equivalent dose) twice per week. An additional i.p. treatment study used ATF-PEG-IONP-Dox and free Dox (10 mg/kg of Dox equivalent dose) twice per week for four injections.
NIR optical imaging and MRI
Optical imaging was performed 48 hours after the injection of NIR-830 dye labeled targeted IONPs using the Kodak FX In Vivo imaging system. Regions of Interests (ROIs) were selected for measuring the mean intensities of tumors and ex vivo images of tumors and organs.
MRI was performed on mice before, 24, and 48 hours following the i.p. injection of different IONPs using a 4.7T animal scanner (Varian Unity, Agilent, CA). T2-weighted fast spin echo imaging sequence was used to acquire images. MRI contrast in the tumor was analyzed using the ROI method and Image J software (National Institutes of Health, Bethesda, MD). Averaged signal intensities of the ROI were obtained from all tumor areas traced from MR images and the signal intensity of the muscle was used as a baseline to normalize the signal intensity in the tumor. The percentage of the mean signal change as the result of the targeted accumulation of IONPs was calculated from comparing the mean signal intensity of the tumors in the mice before and 24 and 48 hours after received i.p. delivery.
Prussian blue staining
Fixed cells or 5 μm thick frozen tissue sections were incubated with Prussian blue staining solution containing equal parts of 10% hydrochloric acid and 10% potassium ferrocyanide for 2 to 4 hours. For tissue sections, nuclear fast red was used for counterstaining.
Measurement of iron concentration in tumor and normal tissues
Tissues were digested in 2M HNO3 (0.2 g tissue/ml HNO3) at 50°C overnight. Supernatants were collected and adjusted to pH 7.4 using NaOH. 40 µL K4Fe(CN)6 and 10 µL HNO3 (1N) were added to the supernatants for a final volume of 200 µL. The mixtures were incubated at 37°C for 1-2 hours and the absorbance at 690 nm was measured. A standard curve was created at the same condition using Fe standard (Ocean Nanotech, LLC) to calculate the iron concentration in tissues.
Immunofluorescence staining
Frozen sections of tumor and normal tissues were subjected to immunofluorescence staining. Anti-CD68 antibody (Biorad, CA) and anti-CD163 antibody (Santa Cruz Biotechnology, Inc., CA) were used to identify macrophages. Anti-CD31 antibody (BD Bioscience, NJ) was used to identify endothelial cells in tumor blood vessels, and anti-Ki67 antibody (eBioscience, CA) was used to label proliferating cells. Alexa-Fluor 488 or Alexa-Fluor 555-labeled secondary antibodies (Invitrogen, CA) were used to visualize biomarker positive cells.
Statistical analysis
Student's t-test was used for the determination of statistically significant differences between experimental groups. A p value <0.05 is considered statistically significant.
Results
Characterization of uPAR-targeted ATF-PEG-IONP and theranostic ATF-PEG-IONPs
Three types of targeted nanoparticles, including uPAR-targeted ATF-PEG-IONP and theranostic PEG-IONPs, ATF-PEG-IONP-Cis and ATF-PEG-IONP-Dox, were prepared (Fig. 1A). In addition, BSA-PEG-IONPs were used as non-targeted controls. TEM images indicated that all IONPs maintained uniform core size after surface functionalization with targeting ligands and drugs (Fig. 1B). Unconjugated PEG-IONP-Cis has a hydrodynamic particle size around 22 nm whereas ATF conjugated PEG-IONP-Cis slightly increased the nanoparticle size to ~25 nm (Fig. 1B).
The binding and uptake of ATF-PEG-IONPs into uPAR-expressing cancer cells were determined in the PANC02 mouse pancreatic cancer cell line. Prussian blue staining of cells following incubation with the nanoparticles showed that ATF-PEG-IONPs bound to and were internalized by tumor cells, whereas cells treated with non-targeted BSA-PEG-IONPs had a minimal level of IONP staining (Fig. 1C). Quantification of IONP concentration in those cell lysates showed that cells treated with ATF-PEG-IONP had five times higher level of iron than that of the BSA-PEG-IONP incubated cells, indicating more efficient binding and uptake of uPAR targeted IONPs (Fig. 1C).
Significantly higher level of IONP accumulation in the orthotopic tumor following i.p. delivery of ATF-PEG-IONPs relative to i.v. delivery
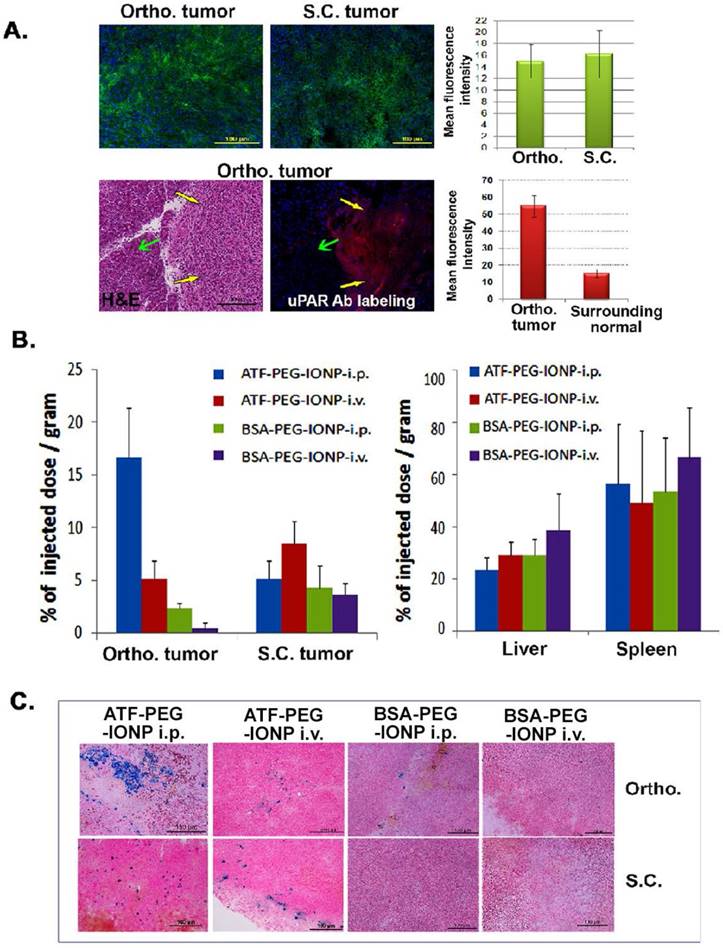
Efficiency of targeted delivery of nanoparticles following i.p. or i.v. delivery was compared in mice bearing a subcutaneous (s.c.) and an orthotopic tumor in the pancreas on the same mice. Immunofluorescence labeling of PANC02 tumor tissue sections showed a similar level of uPAR expression in s.c. and orthotopic tumors. A high level of uPAR was detected in the pancreatic tumor but not in surrounding normal pancreatic tissues (Fig 2A). 24 hours after a single injection of IONPs via i.p. or i.v. delivery, normal and tumor tissues were collected for chemical analysis and Prussian blue staining to quantify the amount of IONP delivery. We found that i.p. delivery of ATF-PEG-IONPs led to an accumulation of 17% of the total injected IONP dose (TID)/gram of tissue in the orthotopic tumors while i.v. delivery of the same amount of ATF-PEG-IONPs resulted in only 5% of the TID/gram of tissue in the orthotopic tumors (Fig. 2B). There was a three-fold higher level of IONPs in orthotopic tumors compared to that in s.c. tumors following i.p. delivery in the same mice (Fig. 2B). In contrast, s.c. tumors had a slightly higher level of IONP accumulation (8% of TID) after i.v. delivery of ATF-PEG-IONP compared with that of i.p. delivery of the same IONPs (5% of TID). Overall, the highest level of IONP accumulation was detected in orthotopic tumors treated with i.p. delivery. Our results also indicated that increased delivery efficiency using uPAR-targeted nanoparticles was due to active targeting since there was a seven fold higher level of the IONPs in orthotopic tumors following i.p. delivery of ATF-PEG-IONPs compared to the tumor that received i.p. delivery of non-targeted BSA-PEG-IONPs (Fig. 2B). Consistent with our previous observations, i.v. delivery of ATF-PEG-IONPs also resulted in a two-fold increase in the amount of IONPs in s.c. tumors compared to that the s.c. tumors treated with BSA-PEG-IONP (Fig. 2B). There was no significant difference in IONP accumulation in the liver and spleen among the mice that received i.p. or i.v. delivery of ATF-PEG-IONPs, or the mice treated with uPAR-targeted or non-targeted IONPs (Fig. 2B). Histological analysis of frozen tumor tissues further confirmed the presence of a higher level of IONP positive cells in the orthotopic tumors obtained from mice that received i.p. delivery of ATF-PEG-IONP compared to i.v. delivery of the nanoparticles (Fig. 2C). However, s.c. tumors had a similar level of IONP positive cells after i.v. or i.p. delivery of ATF-PEG-IONPs. Very low levels of IONP positive cells were found in the tumor tissue section in BSA-PEG-IONP treated mice (Fig. 2C). As expected, we found IONPs in the liver and spleen tissue sections following i.v. or i.p. delivery (Fig. S1) but no apparent acute toxicity.
Comparison of targeted delivery and biodistribution of uPAR targeted and non-targeted nanoparticles following i.p. or i.v. delivery in mice bearing both s.c. and orthotopic tumors. A, Detection of uPAR expression in PANC02 tumors obtained from s.c. and orthotopic (Ortho.) sites in tumor bearing mice using immunofluorescence labeling. Upper panel: uPAR was labeled as green fluorescence. Lower panel: uPAR was labeled as red fluorescence in an orthotopic tumor. Yellow arrows: orthotopic tumor; Green arrows: adjacent normal tissue based on H&E staining. The mean fluorescence intensity was determined from three separate tissue sections. There was no significant difference between the level of uPAR expression in s.c. and orthotopic tumors (p=0.09). However, a significant difference between the levels of uPAR expression was seen in the tumor tissues and adjacent normal tissues (p=2.0 x10-10). B, Efficiency of targeted delivery and biodistribution of i.p. or i.v. injection of the nanoparticles in s.c. and Ortho. tumors by chemical iron analysis. Tumors and normal tissue lysates from the same mice collected 24 hours after an i.v. or i.p. delivery of 300 pmol of ATF-PEG-IONP or BSA-PEG-IONP were examined for the amount of iron concentration by chemical analysis, which correlated with the IONP delivery. The amount of the total iron in tumors or normal organs from the mice without receiving the IONP injection was used as the baseline iron level. The percentage of the total injected IONP dose (TID) was converted based on each gram of tissue weight for tumors and normal organs. The bar figure shows the mean value of three mice. There was a significant difference between the amount of iron accumulation in the orthotopic pancreatic cancer between the mouse group that received i.p. delivery of ATF-PEG-IONP and the mouse group treated with i.v. delivery of the same IONPs (p=0.0166). C, Prussian blue staining of s.c. tumor and ortho. tumors for determination of the levels and locations of the IONP-positive cells (blue) in s.c. and Ortho. tumors. Nuclear fast red was used as a background staining.
Detection of targeted delivery of ATF-PEG-IONP or theranostic ATF-PEG-IONP-Cis into orthotopic tumors following i.p. or i.v. delivery by noninvasive imaging
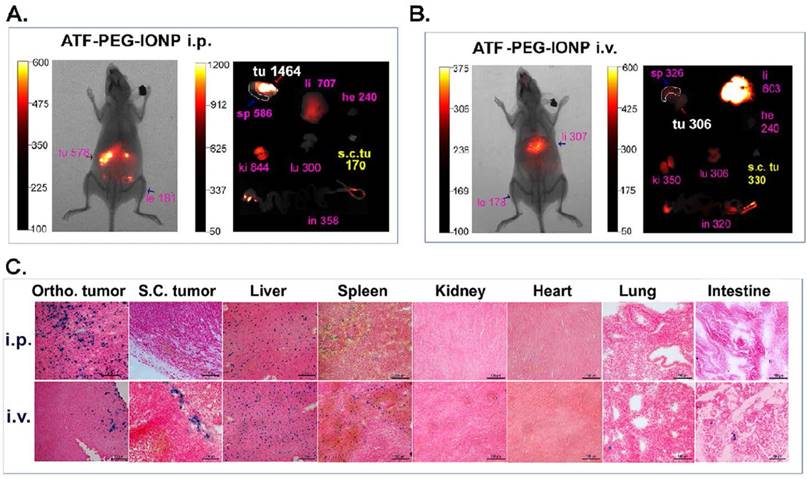
To determine whether noninvasive optical imaging could detect targeted delivery of ATF-PEG-IONP into peritoneal tumors, tumor bearing mice received i.v. or i.p. administration of NIR-830 dye-ATF-PEG-IONPs. Consistent with the above biodistribution results, i.p. delivery of ATF-PEG-IONPs led to strong optical signals in peritoneal tumors with a tumor signal /background ratio of 3.19, which was detectable by in vivo NIR imaging (Fig. 3A). Ex vivo NIR imaging of collected tumors and normal tissues further supported the notion that i.p. delivery of the targeted IONPs enhanced delivery into the orthotopic tumor. A low level of signal was found in the s.c. tumor of the same mouse that received i.p. nanoparticle delivery (Fig. 3A). Prussian blue staining revealed the presence of a high level of IONP positive cells throughout the entire tumor in the orthotopic tumor but a few IONP positive cells in the s.c. tumor in mice that received i.p. delivery of ATF-PEG-IONP (Fig. 3C). Signal in the s.c. tumor was slightly increased in the mice that received i.v. nanoparticle delivery compared with the s.c. tumor after i.p. delivery (Fig. 3A and B). Ex vivo NIR imaging also showed a low signal in the orthotopic tumor after i.v. delivery (Fig. 3B). Prussian blue staining of tumors from the mice received i.v. delivery showed a low level of IONP positive cells in the peripheral areas of the orthotopic and s.c. tumors (Fig. 3C). The accumulation of targeted nanoparticles in normal organs showed similar patterns between i.p. and i.v. delivery, with the most being found in the liver and spleen but a very little or none in the heart, kidney, intestine, and lung (Fig. 3C).
Comparison of targeted delivery of uPAR targeted IONPs into orthotopic and s.c. tumors following i.p. or i.v. delivery by optical imaging and histological analysis. Non-invasive mouse whole body optical imaging and ex vivo organ imaging 24 hours after a single i.p. (A) or i.v. (B) dose of NIR-830-ATF-PEG-IONP. Mean signal intensity of orthotopic pancreatic tumor (Ortho. tu, white) and the mean signal intensity of subcutaneous tumor (s.c. tu, yellow) as well as liver (li), spleen (sp), heart (he), kidney (ki), lung (lu) and intestine (in) (pink). C, Prussian blue staining of tissues from tumor and normal tissues following i.p or i.v. delivery.
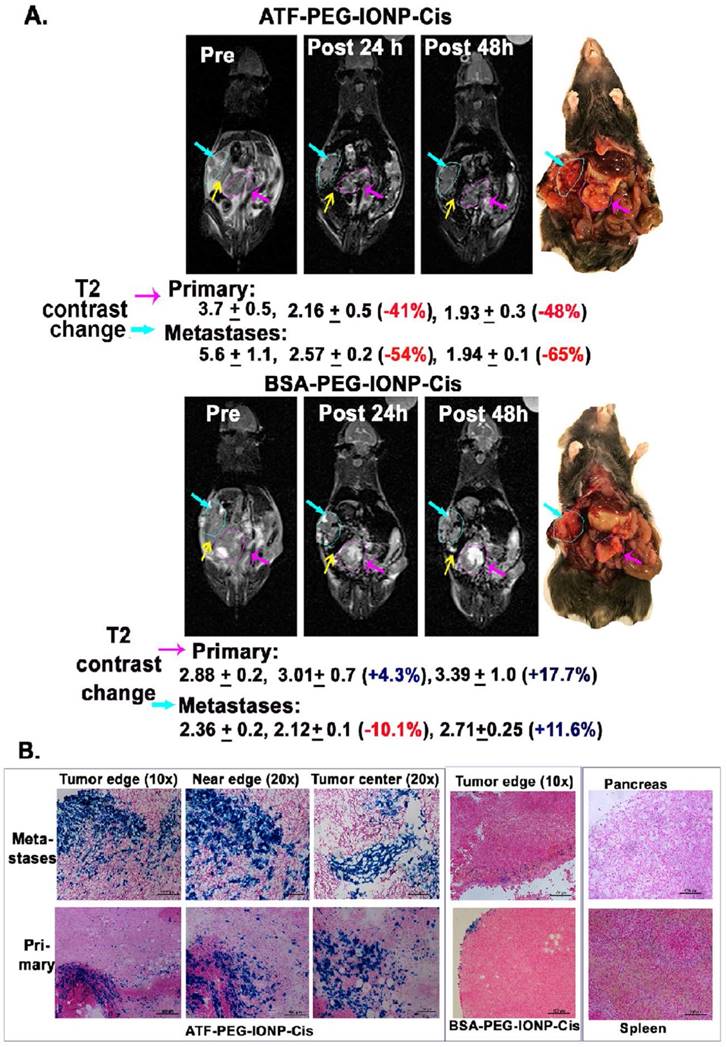
Next, we examined the feasibility of non-invasive MRI of theranostic IONP carrying Cis (ATF-PEG-IONP-Cis) in the orthotopic pancreatic tumor model (Fig. 4). T2-weighted MRI showed 41% and 48% signal decreases in the primary tumor at 24 and 48 hours, respectively, after i.p. delivery of ATF-PEG-IONP-Cis (Fig. 4A). 54% and 65% T2 signal decreases were also detected in peritoneal metastatic tumors 24 and 48 hours following the i.p. delivery (Fig. 4A). However, there was no obvious T2 contrast change in the tumor after i.p. delivery of non-targeted BSA-PEG-IONP-Cis (Fig. 4A). Thus efficient and targeted delivery of IONPs into the peritoneal tumors through i.p. delivery was able to produce sufficient MRI contrasts for the evaluation of the IONP-drug accumulation in both primary and metastatic tumors. Importantly, results of optical imaging, MRI and histological analysis all consistently showed that the nonspecific accumulation of the i.p. delivered nanoparticles in the peritoneal cavity was significantly reduced 48 hours after the IONP administration since we did not observe strong, nonspecific signal in the peritoneum and on the intestines (Fig. 4A and 3C). Prussian blue staining further confirmed the selective accumulation of the IONPs in the primary pancreatic tumors and metastatic tumor lesions, but not in adjacent normal pancreatic tissues, following i.p. delivery of uPAR targeted IONP-Cis (Fig. 4B). We observed a higher level of clusters of IONP positive cells in the tumor center of the metastatic tumors compared to the primary tumor collected in the pancreas (Fig. 4B). However, a low level of IONP positive cells was only detected in the tumor edge after i.p. delivery of non-targeted BSA-IONP-Cis (Fig. 4B). Therefore, results of imaging and histological analysis supported the ability of ATF-PEG-IONP-Cis theranostic nanoparticles in targeted delivery into peritoneal tumors following i.p. administration.
Non-invasive MRI detection of targeted delivery of theranostic IONPs into peritoneal primary and metastatic tumors following i.p. delivery. A, T2-weighted MR images taken pre, 24 hour and 48 hour post an i.p. delivery of the targeted ATF-PEG-IONP-Cis or non-targeted BSA-PEG-IONP-Cis. Pink line and arrows: Orthotopic primary pancreatic tumor. Blue line and arrows: metastatic lesions. Yellow arrow: spleen. Black numbers shown are the mean value and standard deviation of MRI T2 contrast from the entire tumor obtained from ROI analysis using Image J software. The percentage of MRI T2-signal decrease (red numbers) following i.p. nanoparticle delivery was calculated using pre-scan T2 signal in the tumor as 100%. Mouse photos showed the location of tumors in the peritoneal cavity. B, Prussian blue staining of tissue sections of the primary and metastatic tumors showed the presence of high levels of IONP positive cells in the tumor edge and center of primary and metastatic tumors in the mice that received i.p. delivery of ATF-PEG-IONP but not BSA-PEG-IONP. Normal pancreas did not have nonspecific accumulation of IONPs. Low to intermediate levels of IONP positive cells were seen in the spleen.
Infiltration of uPAR-targeted theranostic IONPs from tumor peripheral areas into tumor center regions
To gain insight into the mechanism by which a high delivery efficiency of uPAR targeted IONPs into the orthotopic tumors after i.p. delivery was achieved, histological and immunofluorescence labeling were performed on tumor tissue sections. First, we found a high level of uPAR expression in tumor cells and CD68 positive tumor associated macrophages of the PANC02 tumor tissue (Fig 5A). Similar level and distribution of CD68 positive macrophages were found in s.c. and orthotopic tumors (Fig 5A). Next, we examined location and cell types taking up IONPs in tumor tissues using dual Prussian blue staining and immunofluorescence labeling. Our results showed that i.p. delivery of ATF-PEG-IONPs led to the delivery of high levels of the nanoparticles not only in tumor peripheral areas but also extensive infiltration into the center of orthotopic tumors with sizes of 8 to 10 mm in diameter. IONP containing cells were found both in non-macrophage cells and CD68 positive macrophages, despite the use of the PEG-coating to reduce nonspecific macrophage uptake (Fig. 5B and Fig. S3). However, a large portion of IONP positive cells seen in the tumor center were not co-localized with CD68 positive macrophages, suggesting that ATF-PEG-IONPs were capable of infiltrating from the tumor edge where they initially interacted with the tumors to the tumor center through a macrophage independent mechanism (Fig. 5B and C). In the tumor peripheral areas, more IONPs were found in CD68 positive macrophages (Fig 5B and C). It seemed that the accumulation of the ATF-PEG-IONPs in the tumor center was not due to the nanoparticles entering the systemic circulation and subsequently being delivered into the tumor center through blood vessels since we found a very low level of IONP positive cells in the tumor center following i.v. delivery of ATF-PEG-IONPs (Fig. 5B). We also found IONP positive cells in mostly CD68 negative cells in the s.c. tumor of mice that received i.p. delivery of ATF-PEG-IONPs, indicating that some of the nanoparticles could enter the blood circulation, likely via the lymphatic system, and retained targeting ability (Fig. 5B).
Histological analysis of intratumoral delivery mechanism and distribution of uPAR targeted nanoparticles following i.p. delivery. A, Immunofluorescence labeling identifies uPAR expressing cells in the PANC02 tumors. uPAR: red fluorescence, macrophage marker (CD68): green fluorescence. B, Dual immunofluorescence labeling and Prussian blue staining for identification of IONP positive cells. Frozen tissue sections were labeled with an anti-CD68 antibody (green) and then subjected to Prussian blue staining. Fluorescent images were overlaid with bright-field images from the same field. A higher level of IONP-positive cells was detected in the orthotopic tumor, especially in the tumor center, obtained from mice that received i.p. delivery of ATF-PEG-IONP. C, Localization of IONP positive cells after four i.p. deliveries of either ATF-PEG-IONP-Cis or ATF-PEG-IONP-Dox theranostic nanoparticles. For B & C, most of IONP positive cells in the tumor center were not co-localized with CD68 positive macrophages (yellow arrow). White arrows show IONP and CD68 positive cells in the tumor edge. D, Detection of intratumoral localization of tumor vessels, macrophages, and ATF-PEG-IONP-Cis following four i.p. deliveries. Tumor tissue sections were dual-labeled with endothelial cell marker CD31 (red) and another macrophage marker CD163 (green), and then stained with Prussian blue solution. Pink arrow: tumor stromal vessel and macrophage area; Yellow arrow: IONP positive cells. Paired tumor center and edge images of merged dual-CD31 and CD163 with Prussian blue (first image) and only CD31 and CD163 (second image) were shown. Blue: Hoechst 33342 nuclear staining.
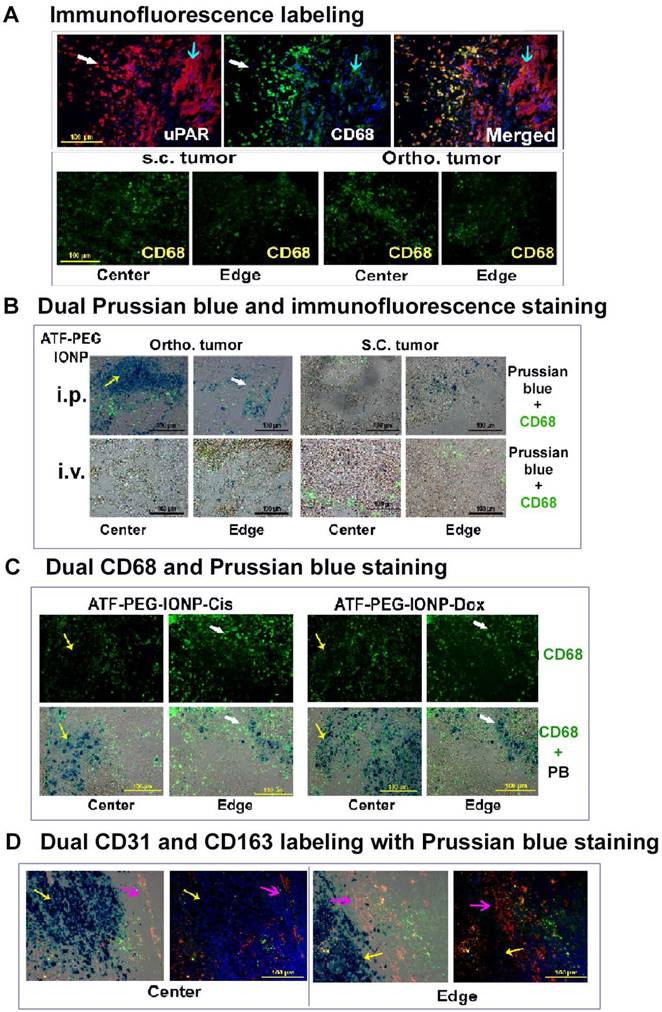
We further examined intratumoral distribution of uPAR targeted theranostic IONPs following repeated i.p. delivery. Consistent with the observation using the targeted IONPs without drugs, we found a higher level of IONP positive cells in the tumor center compared to the tumor peripheral areas in the orthotopic tumors obtained from mice that received four i.p. deliveries of either ATF-PEG-IONP-Cis or ATF-PEG-IONP-Dox (Fig. 5C). Again, the majority of IONP containing cells in the tumor center were not macrophages. Peripheral tumor areas had IONPs in both CD68 positive and negative cell populations (Fig. 5C). To further demonstrate that the accumulation of uPAR targeted theranostic IONPs was largely mediated by tissue penetration, tumor tissue sections were dual labeled with a tumor endothelial cell marker, CD31 (red), and another macrophage marker, CD163 (green) and subsequently stained with Prussian blue solution. We found that repeated i.p. deliveries of ATF-PEG-IONP-Cis resulted in extensive IONP positive cells in the tumor cells that were away from CD31 positive tumor vessels in both tumor center and edge areas, suggesting a tumor vessel independent delivery (Fig. 5D).
Our results further supported the notion that targeting to peritoneal tumors and nanoparticle penetrating into the tumor center is the result of active targeting of uPAR in tumor tissues since a low level of IONP positive cells were detected in the tumor edge but not in the tumor center after i.p. delivery of non-targeted BSA-PEG-IONPs (Fig. S2). Most IONP positive cells were also CD68 positive macrophages in tumor tissues obtained from BSA-PEG-IONP treated mice. Although it was likely that non-targeted BSA-PEG-IONPs could be delivered into tumors through leaky tumor vasculatures after i.v. delivery, they were found in the outer layers of the tumor which typically are enriched in active stromal macrophages (Fig. S2).
Significant growth inhibition of orthotopic tumors following i.p. delivery of uPAR targeted theranostic IONPs and optical imaging detection of drug resistant tumors
The potential for the development of targeted cancer therapy using i.p. delivery of ATF-PEG-IONP-Cis was evaluated in the orthotopic mouse pancreatic tumor model. Cell proliferation assay showed that over 60% of tumor cells were viable after free Cis treatment at a concentration as high as 3.3 µM, indicating a low sensitivity to Cis. Targeted treatment using ATF-PEG-IONP-Cis improved the inhibitory effect with 40% of viable cells remained at 3.3 µM Cis equivalent concentration (Fig. S4).
We then examined the in vivo effect of i.p. delivery of ATF-PEG-IONP-Cis or ATF-PEG-IONP-Dox. Tumor bearing mice received i.p. treatment as shown in Fig. 6A. We found that i.p. delivery of ATF-PEG-IONP-Cis significantly reduced the total peritoneal tumor volume of the orthotopic primary and metastatic tumors in the mice relative to untreated control mice (Fig. 6B, student's t-test, p=0.0088). There was also a significant difference in the tumor weight between the mice treated with ATF-PEG-IONP-Cis and BSA-PEG-IONP-Cis (student's t-test, p=0.016). Relative to the no treatment control group, ATF-PEG-IONP-Cis treatment led to a 40% inhibition of tumor growth. Free Cis alone and BSA-PEG-IONP-Cis treated mice exhibited only 15% tumor growth inhibition (Fig. 6B). Based on body weights of the mice, there was no apparent systemic toxicity following the treatments of 5 mg/kg of Cis equivalent dose in C57/BL6 mice (Fig. S5).
I.p. delivery of uPAR targeted theranostic nanoparticles carrying cisplatin inhibited tumor growth in the orthotopic mouse pancreatic tumor model. A, Schematic illustration of the treatment protocol for ATF-PEG-IONP-Cis. C57BL/6 mice bearing orthotopic pancreatic (PANC02) tumors received 5mg/kg of Cis equivalent concentration of different IONP-Cis theranostic nanoparticles via i.p. injections twice per week for 2 weeks. B, Anti-tumor growth effect of ATF-PEG-IONP-Cis. Total tumors including primary and peritoneal metastatic tumors from each of the mice were collected and weighed. The mean tumor weight and the percentage of tumor growth inhibition after the treatment compared those of the no treatment control are shown. Student's t test: No treatment control or Cis vs. ATF-PEG-IONP-Cis, p<0.01 (n=7 mice). C, NIR optical imaging of drug-resistant residual tumors in the peritoneal cavity. Three days following the 4th i.p. treatment with NIR-830-ATF-PEG-IONP-Cis, mice were sacrificed and optical imaging was performed on the exposed abdominal cavity. Strong optical signals were found in the residual tumors (Ortho. tu, red arrow). Intermediate levels of optical signals were found in the liver (pink arrow). The numbers are mean fluorescence intensity of tumor or normal tissues. Ex vivo organ imaging further confirmed the presence of strong optical signals in the tumor. Liver (li), Spleen (sp), Heart (he), Kidney (ki), Lung (lu) and Intestine (in). D, Prussian blue staining showed a substantial accumulation of IONPs (blue) following ATF-PEG-IONP-Cis treatment along the tumor edge and tumor center relative to BSA-PEG-IONP-Cis treatment. E-F, Immunofluorescence labeling of frozen tumor tissue sections for (E) cell proliferation marker (Ki67, red) and (F) endothelial cells marker (CD31, red). Counterstaining: Hoechst 33342 (blue). Results of quantitative analysis of the percentage of proliferating cells (Ki67+) or tumor vessel density were obtained from six randomly selected microscopic fields of tumor sections and analyzed using Image J software. ATF-PEG-IONP-Cis treatment significantly inhibited ki67 positive proliferating cell population compared with no treatment control (p<0.01). There were statistically significant differences in the number of CD31 positive tumor vessels between no treatment control and ATF-PEG-IONP-Cis treated group, Cis and ATF-PEG-IONP-Cis treated group, or ATF-PEG-IONP-Cis and BSA-PEG-IONP-Cis (p< 0.005 for all groups).
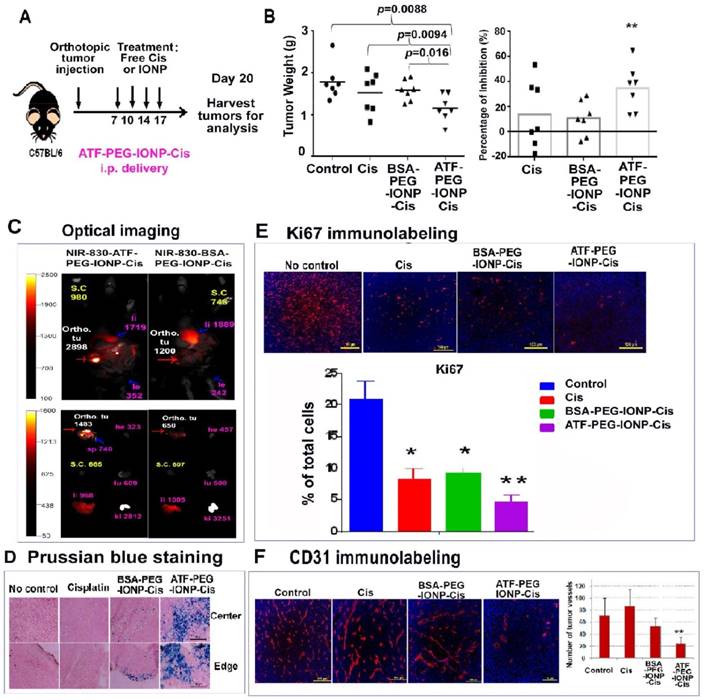
A potential benefit of targeted therapy using theranostic IONPs is the ability to detect drug-resistant tumors using imaging approaches. Following four i.p. injections of NIR-830-ATF-PEG-IONP-Cis, mice were sacrificed and the abdominal cavity was opened for NIR imaging, which mimicked the situation of intraoperative imaging. We detected strong NIR signals in the residual tumors with a mean signal intensity of 2898 at 48 hours following the last injection (Fig. 6C). On the other hand, mice that received the same dose and schedule i.p. treatment of non-targeted NIR-830-BSA-PEG-IONP-Cis had the mean optical signal of 1200 in tumors (Fig. 6C). Background signals in the peritoneal cavity were low at 48 hours for both targeted and non-targeted IONP-Cis even after multiple i.p. injections. Ex vivo imaging of tumor and normal tissues showed strong NIR signals in the residual tumors obtained from the mice treated with ATF-PEG-IONP-Cis but not BSA-PEG-IONP-Cis (Fig. 6C). Histological analysis further supported targeted delivery of a high level of ATF-PEG-IONP-Cis theranostic nanoparticles into both the tumor edge and central areas following four i.p. treatments (Fig. 6D).
To determine the mechanisms mediating tumor growth inhibition, we examined the effects of ATF-PEG-IONP-Cis treatment on tumor cells and tumor vessels since uPAR is expressed by both tumor cells and angiogenic tumor endothelial cells (16). Results of immunofluorescence labeling using an antibody for cell proliferation marker (Ki67) indicated a marked decrease in the number of Ki-67+ proliferating cells in tumors treated with ATF-PEG-IONP-Cis, especially in the tumor center, compared with no treatment (student's t-test, p=0.0086) and BSA-PEG-IONP-Cis treated tumors (p=0.0327) (Fig. 6E). Although there was a significant decrease in the level of Ki-67+ cells in tumors treated with Cis compared with that of no treatment control (p=0.04), there was only a marginal anti-tumor effect observed in the mice.
Additionally, we observed a significant reduction in CD31+ tumor vessels in the tumor tissues following four treatments of ATF-PEG-IONP-Cis (Fig. 6F, student's t-test, p=0.004 vs control, p=0.0016 vs free Cis, and p=0.003 vs BSA-PEG-IONP-Cis). However, there was no significant difference in the blood vessel density in the tumors obtained from the mice treated with free Cis (p=0.32) or BSA-PEG-IONP-Cis (p=0.175) compared with no treatment control (Fig 6F).
PANC02 cells also had a relatively low sensitivity to Dox treatment in vitro with 0.85 µM of Dox resulting in approximately 50% tumor cell growth inhibition (Fig. S4). Tumor cell growth inhibition (90%) was observed at a very high Dox concentration (8.5 µM). The effect of targeted i.p. therapy using theranostic nanoparticles was also demonstrated using ATF-PEG-IONP Dox (Fig. 7). We found that the total tumor weight in the mice that received ATF-PEG-IONP-Dox was significantly lower than that of the no treatment control or ATF-PEG-IONP without carrying Dox (Fig. 7 A&B) (student's t-test, vs no treatment control: p=0.0019, vs ATF-PEG-IONP: p=0.006). There was 71.5% of tumor growth inhibition in the mice that received i.p. delivery of ATF-PEG-IONP-Dox compared with 47.8% of tumor growth inhibition seen in the free Dox treated group (Fig. 7B). Although the tumor bearing mice treated with ATF-PEG-IONP without carrying Dox showed 25% of tumor growth inhibition compared with no treatment control, there was no statistical significance (p=0.074)
Evaluation of the effects of ATF-PEG-IONP-Dox treatment on cell proliferation and tumor angiogenesis in orthotopic mouse pancreatic tumors A, Treatment protocol for ATF-PEG-IONP-Dox. PANC02 tumor bearing mice received 10 mg/kg of Dox equivalent concentration of different IONP-Dox theranostic nanoparticles i.p. twice per week for 2 weeks. B, Anti-tumor effect of ATF-PEG-IONP-Dox. ATF-PEG-IONP-Dox treated group had significantly lower tumor weight compared with either no treatment control (p<0.005) or conventional Dox treatment (p<0.05). n=6 (control group), n=4 (Dox and ATF-PEG-IONP-Dox). The mean tumor weight of the mouse group is shown as a line and individual tumor weight distributions are shown as symbols (Left). The percentages of tumor growth inhibition after the treatment are also shown as the mean and individual tumors in each mouse group (right). C, Prussian blue staining of tumor tissue sections showed accumulation of IONPs (blue). Immunofluorescence labeling of tumor sections for cell proliferation marker (Ki67, red). Counterstaining: Hoechst 33342 (blue). D and E, Quantitative analysis of Ki67 positive cells from six randomly selected microscopic fields of tumor sections by Image J analysis. Significant difference was found between no treatment control and ATF-PEG-IONP-Dox groups (p<0.001), Dox and ATF-PEG-IONP-Dox groups (p< 0.005), or ATF-PEG-IONP and ATF-PEG-IONP-Dox (p<0.005). F and G, Immunofluorescence labeling for endothelial cell marker (CD31, red) and quantitative analysis of CD31 positive tumor vessels. Significance was determined using student's t-test between no treatment control and ATF-PEG-IONP-Dox groups (p<0.05), or Dox and ATF-PEG-IONP-Dox groups (p<0.01).
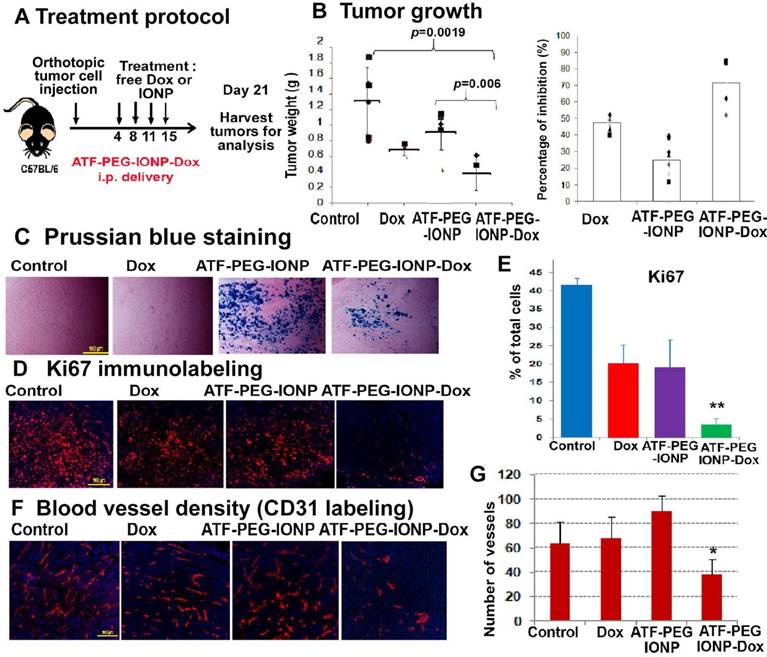
Histological analysis using Prussian blue staining showed high levels of IONP positive cells in the tumor tissues treated with ATF-PEG-IONP or ATF-PEG-IONP-Dox (Fig 7C). Immunofluorescence labeling using an anti-Ki67 antibody showed that ATF-PEG-IONP-Dox treatment significantly inhibited cell proliferation compared with the control groups (Student's t-test, vs no treatment control: p=6.17 x10-11, vs Dox: p=0.001, vs ATF-PEG-IONP: p=0.003) (Fig. 7D). Furthermore, ATF-PEG-IONP-Dox treatment significantly reduced tumor vessel intensity compared with no treatment and Dox control groups, suggesting a strong anti-angiogenic effect (Student's t-test, vs no treatment control: p=0.013, vs Dox p=0.001) (Fig. 7E).
Moreover, we found that i.p. therapy using uPAR targeted theranostic nanoparticles markedly reduced the amount of ascites production with 74.5% to 81% inhibition in the mouse group treated with ATF-PEG-IONP-Dox or ATF-PEG-IONP-Cis (Fig. S6). Taken together, our results indicate that i.p. administration of uPAR targeted nanoparticle drug delivery platform is a promising approach for enhanced efficacy of targeted therapy of peritoneal tumors.
Discussion
For the effective treatment of peritoneal cancers, a local-regional approach offers clinical advantages. I.p. delivery allows for high drug concentrations to be delivered to the primary and metastatic tumor sites within the peritoneum which can mediate enhanced therapeutic responses (34). However, the infusion of a high dose of drugs into the peritoneal cavity of patients leads to local toxicity and fast absorption of drug molecules into the blood circulation causing systemic toxicity (35). Conventional i.p. therapy is ineffective in the treatment of bulky tumors (>1 cm) due to inability of drug penetration into the tumor center. As a result, a number of delivery platforms, including microparticles and nanoparticles, have been investigated to enhance drug delivery and retention after i.p. administrations (3, 7, 36). Results of preclinical and clinical studies have shown that size and other physical properties of drug carriers determine drug retention time, intratumoral delivery, pharmacokinetic, and elimination rate in the peritoneal cavity. Although microparticles with sizes of 50 to 200 µm were retained in the peritoneal cavity to release drug for 3 weeks (37, 38), those drug carriers had a poor distribution inside the peritoneal cavity with a high concentration of the particles in the lower abdomen cavity, resulting in a low efficiency in drug delivery into peritoneal tumors and inflammatory responses in the normal tissues (39). Several polymeric nanoparticle drug carriers have also been shown to increase peritoneal retention time and therapeutic effect (3). Nanoparticles (<50 nm) are able to pass through the lymphatic system to enter systemic circulation, and thus can be cleared out of the peritoneum within 24 to 48 hours (7, 40). Therefore, the development of tumor targeted nanoparticle drug carriers that selectively deliver large amount of therapeutic agents into peritoneal tumors while unbound nanoparticle-drugs can be eliminated from the peritoneal cavity at a desired time period is critical for the improvement of therapeutic efficacy and decrease in local and systemic toxicities.
One of the major advantages of i.p. administration of nanoparticle-drugs is avoiding nonspecific uptake of the nanoparticle by the reticuloendothelial system in the liver and spleen following the initial infusion, which is a major problem for i.v. delivery. The theranostic IONPs developed in this study have a hydrodynamic size of 25 nm, which has relatively longer retention in the peritoneum before entering the systemic circulation through the lymphatic system. The use of PEG-coated IONPs further prevented nonspecific uptake by macrophages in the peritoneal cavity.
Conventional i.p. chemotherapy is not effective in pre-operative treatment of peritoneal tumors with local invasion and peritoneal metastasis due to the limitation in the depth of drug delivery in bulking tumors and a dense stroma surrounding tumor nodules that creates a physical barrier. Since uPAR is highly expressed in active tumor stromal cells, nanoparticles targeting uPAR could directly bind to the tumor surface enriched in stromal cells to increase retention in the tumor site. Targeting both stromal and tumor cells also facilitated the penetration of uPAR targeted nanoparticles into tumor center areas. As the result of a high level of nanoparticle delivery into tumor peripheral and center areas, significant tumor growth inhibition was observed in the mouse pancreatic tumor model. Importantly, uPAR targeted i.p. therapy using theranostic nanoparticles markedly inhibited both tumor cell proliferation and angiogenesis. Therefore, i.p. delivery of uPAR targeted theranostic nanoparticles has the potential for translational development of novel preoperative therapy for cancer patients with local invasion or peritoneal metastatic tumors that are currently considered as un-resectable. Effective i.p. therapy may reduce tumor burdens in the peritoneal cavity thus making it possible for curative surgery.
The development of image-guided and targeted therapeutic approaches provides a means to address an unmet clinical challenge in the treatment of cancer patients with highly heterogeneous tumors by assessing drug delivery and tumor response in a timely manner. MRI provides good anatomic information and imaging depth and resolution for evaluation of nanoparticle-drug delivery in peritoneal tumors. Highly sensitive NIR optical imaging allows intraoperative detection and removal of small drug-resistant tumor lesions. We demonstrated that uPAR targeted theranostic IONPs offered high capacity and biodegradable drug carriers with MRI capability for non-invasive tumor imaging of drug delivery. We found that the appropriate time for MRI is around 48 hours following i.p. delivery when unbound IONPs were cleared out from the peritoneal cavity. Moreover, the ability of optical imaging detection of drug-resistant tumors following i.p. therapy will be useful for the development of image-guided surgery.
Our study using a mouse pancreatic tumor model with both orthotopic and s. c. tumors offers a unique opportunity to compare the efficiency of uPAR-targeted delivery of theranostic IONPs following i.v. or i.p. delivery. Results of this study provided new information for understanding the mechanism of uptake of uPAR targeted and PEGylated IONPs in the i.p. tumors and distant s.c. tumors after i.p. delivery. It is likely that increased bioavailability in the peritoneal cavity and active targeting to uPAR expressing pancreatic cancer and tumor stromal cells contributed to the high delivery efficiency (17% of TID/gram) observed in the orthotopic tumors following i.p. delivery. At present, the mechanism of efficient tumor tissue penetration from the peripheral to central tumor area following i.p. delivery of ATF-PEG-IONPs is under investigation. Results of our study suggested that the improved intratumoral delivery and distribution as well as the ability of ATF-PEG-IONPs to penetrate into the tumor center is not dependent on macrophage uptake and infiltration since a high level of IONP positive cells in the tumor center were not macrophages. The majority of IONP containing macrophages were found in the peripheral tumor areas. One potential mechanism of intratumoral distribution and tumor penetration of ATF-PEG-IONPs is through cell to cell transport of the nanoparticle. It has been shown that nanoparticles conjugated with tumor penetrating peptides, such as iRGD, were able to penetrate tumor tissue through cell to cell transport mediated by a unique endocytic pathway (41-43). Our finding that IONP positive cells presented as clusters and away from tumor blood vessels supports this mechanism.
Although a considerable level of i.p. delivered IONPs entered into the systemic circulation as a normal mechanism of drug clearance, those IONPs were taken up by the RES in the liver and spleen where they could be degraded by macrophages. There were no apparent systemic and local toxicities found in the mice after the targeted i.p. therapy. Therefore, uPAR targeted theranostic IONPs carrying Cis or Dox have the potential for the treatment of peritoneal metastatic tumors derived from several tumor types, such as pancreatic, ovarian, and colon. Platinum-based drugs have been used to treat ovarian, colon and pancreatic cancer. Although Dox is not commonly used for the treatment of the above cancers due to its cardiotoxicity, encapsulation of Dox into nanoparticle drug carriers reduces systemic toxicity. Targeted i.p. delivery of Cis or Dox using theranostic nanoparticles may offer an opportunity to treat cancer patients with drug resistant peritoneal metastatic tumors who have likely been treated by the first- and second-line chemotherapy drugs. Taken together, results of our studies provide strong evidence to support further development of i.p. delivery of uPAR-targeted theranostic IONPs for the image-guided treatment of peritoneal tumors.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank Dr. Keping Xie for providing the PANC02 mouse pancreatic cancer cell line and Dr. Yuan Kai for detection of uPAR level in tumors by immunofluorescence labeling. We also thank research funding supports from NIH/NCI, including U01CA151810 (Yang and Mao) and R01CA202846-01(Yang, Mao and Wang), SBIR Phase II Contract NO. HHSN261201200029C (Wang, Yang and Mao), and F32CA189633 (Bozeman), and the Nancy Panoz Endowed Chair Funds (Yang).
Competing Interests
Dr. Y. Andrew Wang is the President and Principal Scientist, and Dr. Hongyu Chen is a Senior Scientist at Ocean Nanotech, LLC., San Diego, CA. All other authors do not have any conflicts of interest.
References
1. Sadeghi B, Arvieux C, Glehen O, Beaujard AC, Rivoire M, Baulieux J. et al. Peritoneal carcinomatosis from non-gynecologic malignancies: results of the EVOCAPE 1 multicentric prospective study. Cancer. 2000;88:358-63
2. Cannistra SA. Cancer of the ovary. N Engl J Med. 2004;351:2519-29
3. Lu Z, Wang J, Wientjes MG, Au JL. Intraperitoneal therapy for peritoneal cancer. Future Oncol. 2010;6:1625-41
4. Bakrin N, Bereder JM, Decullier E, Classe JM, Msika S, Lorimier G. et al. Peritoneal carcinomatosis treated with cytoreductive surgery and Hyperthermic Intraperitoneal Chemotherapy (HIPEC) for advanced ovarian carcinoma: a French multicentre retrospective cohort study of 566 patients. Eur J Surg Oncol. 2013;39:1435-43
5. Spiliotis J, Halkia E, Lianos E, Kalantzi N, Grivas A, Efstathiou E. et al. Cytoreductive surgery and HIPEC in recurrent epithelial ovarian cancer: a prospective randomized phase III study. Ann Surg Oncol. 2015;22:1570-5
6. Los G, Mutsaers PH, Lenglet WJ, Baldew GS, McVie JG. Platinum distribution in intraperitoneal tumors after intraperitoneal cisplatin treatment. Cancer Chemother Pharmacol. 1990;25:389-94
7. Van der Speeten K, Stuart OA, Sugarbaker PH. Pharmacology of perioperative intraperitoneal and intravenous chemotherapy in patients with peritoneal surface malignancy. Surg Oncol Clin N Am. 2012;21:577-97
8. Colson YL, Liu R, Southard EB, Schulz MD, Wade JE, Griset AP. et al. The performance of expansile nanoparticles in a murine model of peritoneal carcinomatosis. Biomaterials. 2011;32:832-40
9. Harrison LE, Bryan M, Pliner L, Saunders T. Phase I trial of pegylated liposomal doxorubicin with hyperthermic intraperitoneal chemotherapy in patients undergoing cytoreduction for advanced intra-abdominal malignancy. Ann Surg Oncol. 2008;15:1407-13
10. McCarthy JR, Weissleder R. Multifunctional magnetic nanoparticles for targeted imaging and therapy. Adv Drug Deliv Rev. 2008;60:1241-51
11. Li C. A targeted approach to cancer imaging and therapy. Nat Mater. 2014;13:110-5
12. Lee GY, Qian WP, Wang L, Wang YA, Staley CA, Satpathy M. et al. Theranostic nanoparticles with controlled release of gemcitabine for targeted therapy and MRI of pancreatic cancer. ACS Nano. 2013;7:2078-89
13. Zhou H, Qian W, Uckun FM, Wang L, Wang YA, Chen H. et al. IGF1 Receptor Targeted Theranostic Nanoparticles for Targeted and Image-Guided Therapy of Pancreatic Cancer. ACS Nano. 2015;9:7976-91
14. Chen Y, Zheng B, Robbins DH, Lewin DN, Mikhitarian K, Graham A. et al. Accurate discrimination of pancreatic ductal adenocarcinoma and chronic pancreatitis using multimarker expression data and samples obtained by minimally invasive fine needle aspiration. Int J Cancer. 2007;120:1511-7
15. Cantero D, Friess H, Deflorin J, Zimmermann A, Brundler MA, Riesle E. et al. Enhanced expression of urokinase plasminogen activator and its receptor in pancreatic carcinoma. Br J Cancer. 1997;75:388-95
16. Mazar AP. Urokinase plasminogen activator receptor choreographs multiple ligand interactions: implications for tumor progression and therapy. Clin Cancer Res. 2008;14:5649-55
17. Kenny HA, Leonhardt P, Ladanyi A, Yamada SD, Montag A, Im HK. et al. Targeting the urokinase plasminogen activator receptor inhibits ovarian cancer metastasis. Clin Cancer Res. 2011;17:459-71
18. Boonstra MC, Verbeek FP, Mazar AP, Prevoo HA, Kuppen PJ, van de Velde CJ. et al. Expression of uPAR in tumor-associated stromal cells is associated with colorectal cancer patient prognosis: a TMA study. BMC Cancer. 2014;14:269
19. Taniguchi K, Yonemura Y, Nojima N, Hirono Y, Fushida S, Fujimura T. et al. The relation between the growth patterns of gastric carcinoma and the expression of hepatocyte growth factor receptor (c-met), autocrine motility factor receptor, and urokinase-type plasminogen activator receptor. Cancer. 1998;82:2112-22
20. Hemsen A, Riethdorf L, Brunner N, Berger J, Ebel S, Thomssen C. et al. Comparative evaluation of urokinase-type plasminogen activator receptor expression in primary breast carcinomas and on metastatic tumor cells. Int J Cancer. 2003;107:903-9
21. Li Y, Shen Y, Miao Y, Luan Y, Sun B, Qiu X. Co-expression of uPAR and CXCR4 promotes tumor growth and metastasis in small cell lung cancer. Int J Clin Exp Pathol. 2014;7:3771-80
22. Yang L, Cao Z, Sajja HK, Mao H, Wang L, Geng H. et al. Development of Receptor Targeted Magnetic Iron Oxide Nanoparticles for Efficient Drug Delivery and Tumor Imaging. J Biomed Nanotechnol. 2008;4:439-49
23. Yang L, Mao H, Cao Z, Wang YA, Peng X, Wang X. et al. Molecular imaging of pancreatic cancer in an animal model using targeted multifunctional nanoparticles. Gastroenterology. 2009;136:1514-25 e2
24. Corbett TH, Roberts BJ, Leopold WR, Peckham JC, Wilkoff LJ, Griswold DP Jr. et al. Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res. 1984;44:717-26
25. Torres MP, Rachagani S, Souchek JJ, Mallya K, Johansson SL, Batra SK. Novel pancreatic cancer cell lines derived from genetically engineered mouse models of spontaneous pancreatic adenocarcinoma: applications in diagnosis and therapy. PLoS One. 2013;8:e80580
26. Huang C, Qiu Z, Wang L, Peng Z, Jia Z, Logsdon CD. et al. A novel FoxM1-caveolin signaling pathway promotes pancreatic cancer invasion and metastasis. Cancer Res. 2012;72:655-65
27. Yang E, Qian W, Cao Z, Wang L, Bozeman EN, Ward C. et al. Theranostic nanoparticles carrying doxorubicin attenuate targeting ligand specific antibody responses following systemic delivery. Theranostics. 2015;5:43-61
28. Jain RK, Stylianopoulos T. Delivering nanomedicine to solid tumors. Nat Rev Clin Oncol. 2010;7:653-64
29. Yu WW, Falkner JC, Yavuz CT, Colvin VL. Synthesis of monodisperse iron oxide nanocrystals by thermal decomposition of iron carboxylate salts. Chem Commun (Camb). 2004:2306-7
30. Chen K, Xie J, Xu H, Behera D, Michalski MH, Biswal S. et al. Triblock copolymer coated iron oxide nanoparticle conjugate for tumor integrin targeting. Biomaterials. 2009;30:6912-9
31. Yang L, Mao H, Cao ZH, Wang YA, Peng XH, Wang XX. et al. Molecular Imaging of Pancreatic Cancer in an Animal Model Using Targeted Multifunctional Nanoparticles. Gastroenterology. 2009;136:1514-25
32. Zhou Z, Chen H, Lipowska M, Wang L, Yu Q, Yang X. et al. A dual-modal magnetic nanoparticle probe for preoperative and intraoperative mapping of sentinel lymph nodes by magnetic resonance and near infrared fluorescence imaging. J Biomater Appl. 2013;28:100-11
33. Moura MJG. et al. Delivery of cisplatin from thermosensitive co-cross-linked chitosan hydrogels. European Polymer Journal. 2013;49:2504-10
34. Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S. et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354:34-43
35. Walker JL, Armstrong DK, Huang HQ, Fowler J, Webster K, Burger RA. et al. Intraperitoneal catheter outcomes in a phase III trial of intravenous versus intraperitoneal chemotherapy in optimal stage III ovarian and primary peritoneal cancer: a Gynecologic Oncology Group Study. Gynecol Oncol. 2006;100:27-32
36. Montagner IM, Merlo A, Zuccolotto G, Renier D, Campisi M, Pasut G. et al. Peritoneal tumor carcinomatosis: pharmacological targeting with hyaluronan-based bioconjugates overcomes therapeutic indications of current drugs. PLoS One. 2014;9:e112240
37. Sugiyama T, Kumagai S, Nishida T, Ushijima K, Matsuo T, Yakushiji M. et al. Experimental and clinical evaluation of cisplatin-containing microspheres as intraperitoneal chemotherapy for ovarian cancer. Anticancer Res. 1998;18:2837-42
38. Hagiwara A, Takahashi T, Sawai K, Sakakura C, Tsujimoto H, Imanishi T. et al. Pharmacological effects of 5-fluorouracil microspheres on peritoneal carcinomatosis in animals. Br J Cancer. 1996;74:1392-6
39. Armstrong DK, Fleming GF, Markman M, Bailey HH. A phase I trial of intraperitoneal sustained-release paclitaxel microspheres (Paclimer) in recurrent ovarian cancer: a Gynecologic Oncology Group study. Gynecol Oncol. 2006;103:391-6
40. Hirano K, Hunt CA. Lymphatic transport of liposome-encapsulated agents: effects of liposome size following intraperitoneal administration. J Pharm Sci. 1985;74:915-21
41. Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR. et al. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science. 2010;328:1031-5
42. Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci U S A. 2009;106:16157-62
43. Pang HB, Braun GB, Friman T, Aza-Blanc P, Ruidiaz ME, Sugahara KN. et al. An endocytosis pathway initiated through neuropilin-1 and regulated by nutrient availability. Nat Commun. 2014;5:4904
Author contact
![]() Corresponding author: Dr. Lily Yang, Department of Surgery, Emory University School of Medicine, Clinic C, Room C-4088, 1365 C Clifton Road, NE, Atlanta, GA 30322. Telephone: 404-778-4269; Fax: 404-778-5530. E-mail address: Lyang02edu
Corresponding author: Dr. Lily Yang, Department of Surgery, Emory University School of Medicine, Clinic C, Room C-4088, 1365 C Clifton Road, NE, Atlanta, GA 30322. Telephone: 404-778-4269; Fax: 404-778-5530. E-mail address: Lyang02edu