Theranostics
13.3
Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(5):1277-1289. doi:10.7150/thno.18509 This issue Cite
Research Paper
Hypertension, cerebrovascular impairment, and cognitive decline in aged AβPP/PS1 mice
Maximilian Wiesmann1, Valerio Zerbi2, Diane Jansen1, Dieter Lütjohann3, Andor Veltien4, Arend Heerschap4, Amanda J Kiliaan1 ![]()
1. Radboud university medical center, Donders Institute for Brain, Cognition & Behaviour, Radboud Alzheimer Center, Department of Anatomy, Preclinical Imaging Centre PRIME, Nijmegen, The Netherlands.
2. Neural Control of Movement Lab, Department of Health Sciences and Technology, ETH Zurich, Switzerland.
3. Institute for Clinical Chemistry and Clinical Pharmacology, University of Bonn, Bonn, Germany.
4. Radboud University Medical Center, Department of Radiology & Nuclear Medicine, Preclinical Imaging Centre PRIME, Nijmegen, The Netherlands.
Received 2016-11-26; Accepted 2017-1-18; Published 2017-3-6
Citation:
Wiesmann M, Zerbi V, Jansen D, Lütjohann D, Veltien A, Heerschap A, Kiliaan AJ. Hypertension, cerebrovascular impairment, and cognitive decline in aged AβPP/PS1 mice. Theranostics 2017; 7(5):1277-1289. doi:10.7150/thno.18509. https://www.thno.org/v07p1277.htm
Other stylesAbstract

Cardiovascular risk factors, especially hypertension, are also major risk factors for Alzheimer's disease (AD). To elucidate the underlying vascular origin of neurodegenerative processes in AD, we investigated the relation between systolic blood pressure (SBP) cerebral blood flow (CBF) and vasoreactivity with brain structure and function in a 16-18 months old double transgenic AβPPswe/PS1dE9 (AβPP/PS1) mouse model for AD. These aging AβPP/PS1 mice showed an increased SBP linked to a declined regional CBF. Furthermore, using advanced MRI techniques, decline of functional and structural connectivity was revealed in the AD-like mice coupled to impaired cognition, increased locomotor activity, and anxiety-related behavior. Post mortem analyses demonstrated also increased neuroinflammation, and both decreased synaptogenesis and neurogenesis in the AβPP/PS1 mice. Additionally, deviant levels of fatty acids and sterols were present in the brain tissue of the AβPP/PS1 mice indicating maladapted brain fatty acid metabolism. Our findings suggest a link between increased SBP, decreased cerebral hemodynamics and connectivity in an AD mouse model during aging, leading to behavioral and cognitive impairments. As these results mirror the complex clinical symptomatology in the prodromal phase of AD, we suggest that this AD-like murine model could be used to investigate prevention and treatment strategies for early AD patients. Moreover, this study helps to develop more efficient therapies and diagnostics for this very early stage of AD.
Keywords: Blood pressure, Cerebral blood flow, Alzheimer's disease, Functional connectivity, Neuropathology, Neurodegeneration: experimental models.
Introduction
Dementia has become a public health problem in the aging world population [1, 2]. Alzheimer's disease (AD) is responsible for most dementia cases followed by vascular dementia (VaD) [3, 4]. AD is characterized by brain atrophy and a gradual cognitive decline caused by neuronal death and loss of synapses in brain regions involved in learning and memory processes (e.g. temporal and frontal lobes) [5, 6].
Already the early stages of AD are characterized by the accumulation of Aβ affecting specific brain regions like the forebrain and medial temporal lobe structures like hippocampus, amygdala and entorhinal cortex [7-9]. Almost all insoluble Aβ is accumulated within the neuritic plaques and cerebral vessel walls [5].
Epidemiological and clinical studies revealed that AD and VaD share common vascular related risk factors such as hypertension, diabetes, hyperlipidemia, cerebrovascular disease, and arrhythmia [10-16]. Furthermore, it has been shown that vascular risk factors can influence the development of AD pathology (the vascular hypothesis) [17]. A recent study showed that hypertension affects the expression of tau as well as that of Aβ in an AD mouse model [18]. Further evidence supporting this link between hypertension and AD comes from clinical studies indicating that antihypertensives may reduce the development of AD [19, 20]. Other clinical studies suggested that resting cerebral blood flow (CBF) is decreased among hypertensive patients as opposed to that of normotensive subjects [21-25]. In elderly patients a decreased CBF has been shown to increase hippocampal and amygdalar atrophy [26, 27]. Therefore, an impaired or diminished cerebral perfusion may play a role in the development of AD via decreased delivery of oxygen in ischemia-sensitive brain regions like the hippocampus, inducing neurodegeneration and subsequent cognitive decline [28]. Notably, maladapted cerebral hemodynamics could lead to alteration in structural and functional connectivity in the brain represented by white matter lesions/hyperintensities. Impaired functional connectivity is also found in AD patients [29-31] and additionally, a link between the incidence of white matter lesions and the severity of the underlying AD pathology has been reported [32-34]. Many studies have shown the relationship between increased blood pressure in mid-life and cognitive decline or AD in late-life [35, 36] indicating the relevance of proper blood pressure maintenance.
Therefore, to elucidate the underlying vascular origin of neurodegenerative processes in AD, we investigate the relation between vascular parameters (systolic blood pressure (SBP), cerebrovascular density, cerebral perfusion and vasoreactivity), functional and structural connectivity, and postmortem markers for neuroinflammation, neurogenesis, postsynaptic density, and levels of fatty acids and sterols and, in relation to cognition in the AβPPswe/PS1dE9 (AβPP/PS1) mouse model for AD.
To develop and evaluate potential therapeutic targets for early stages in AD it is necessary to understand underlying (neurovascular) pathological processes in AD by studying well characterized early stage AD animal models.
Materials and methods
Animals
The AβPPswe/PS1dE9 (AβPP/PS1) founder mice were originally obtained from John Hopkins University, Baltimore, MD, USA (D. Borchelt and J. Jankowsky, Dept. of Pathology) [37, 38] and a colony was first established at the University of Kuopio, Finland and thereafter a colony was bred at the Central Animal Facility at Radboud university medical center, The Netherlands. In short, mice were created by co-injection of chimeric mouse/human AβPPswe (mouse AβPP695 harbouring a human Aβ domain and mutations K595N and M596L linked to Swedish familial AD pedigrees) and human PS1-dE9 (deletion of exon 9) vectors controlled by independent mouse prion protein promoter elements. The two transfected genes co-integrate and co-segregate as a single locus [38]. This line was originally maintained on a hybrid background by backcrossing to C3HeJ×C57BL/6J F1 mice (so-called pseudo F2 stage). For the present work, the breeder mice were backcrossed to C57BL/6J for fourteen generations to obtain mice for the current study. Before the actual experiments, animals were housed socially with a maximum of six animals per cage, with room temperature at 21°C, and artificial 12:12h light:dark cycle (lights on at 7 a.m.). Food and water were available ad libitum. The experiments were performed according to Dutch federal regulations for animal protection and were approved by the Veterinary Authority of Radboud university medical center, Nijmegen, The Netherlands, and the Animal Experiment Committee (called the Dierexperimentencommissie or DEC, RU-DEC 2011-058) of the Radboud university, Nijmegen, The Netherlands. The reporting of the animal experiments conforms with the ARRIVE guidelines [39].
For the present experiment, we used a total of 17 16-month-old months old male mice (ten WT littermates, and seven AβPP/PS1 mice); at eighteen months of age, all animals completed the experiments and were euthanized. All behavioural and MRI experiments were performed in the Preclinical Imaging Centre (PRIME) of the Radboud university medical center between 8 a.m. and 6 p.m..
Study design, randomization, blinding, and sample size
This was a single-institution, randomized, and double-blind controlled study (blinded for investigators and outcomes assessor) conducted at the preclinical imaging center (PRIME) of the Radboudumc (Nijmegen, the Netherlands). Per experimental subgroup, the selection of animals was randomized. All animals completed the study, no animals were replaced. The sample size of minimal 6 mice per experimental group was chosen based on formal calculation of power as described in the approved protocols (RU-DEC 2011-058; WP: 110149) and using results from our previous study [40]. At 16, 17, and 18 months of age systolic blood pressure measurements were performed. At 17 months of age (WT: 16,8±0,03; AβPP/PS1: 16,6±0,04) all animals underwent behavioural testing in the open field and Morris water maze (MWM) and subsequently MRI measurements at 18 months of age (WT: 18.0±0,02; AβPP/PS1: 17,9±0,05).
No animals were excluded from analysis. Post mortem immunohistochemical and biochemical procedures were performed on all brains.
Tail-cuff plethysmography
Mice were trained for two consecutive days in the warmed tail-cuff device (IITC Life Scientific Instruments, Woodland Hills, CA) to accustom them to the procedure. Starting from sixteen months of age and being repeated at seventeen and eighteen month of age, SBP was measured for two consecutive days monthly in trained, conscious and preheated mice using computerized tail-cuff plethysmography (IITC Life Scientific Instruments, Woodland Hills, CA), as previously described [41-44].
Behavioral analyses
The results of the behavioral analyses are placed in the supplementary material.
Morris water maze
The Morris water maze (MWM) is used to test spatial learning and memory in rodents. In short, at 17 months of age mice were placed in a circular pool, filled with opaque water, and were trained to find a submerged platform in the northeast (NE) quadrant of the pool by using distant visual cues.
Acquisition (spatial learning)
Mice were trained to find the location of the submerged escape platform in 4 acquisition trials (maximal swimming time 120 s; 30 s on the platform; inter-trial interval 60 min) per day during 4 consecutive days. The latency time (s) to find the hidden platform was scored. Starting positions were south (S), north (N), east (E) and west (W). After the 2 min swim the mice were placed back in their home cage, and a paper towel was available inside the cage for additional drying.
Probe (spatial memory)
At the start of the fifth day, mice performed a single probe trial (starting position: S), in which the platform was removed from the swimming pool. Mice were allowed to swim for 120 s and the time spent swimming and searching in the NE quadrant (where the platform had been located) was recorded.
Open field
Locomotion and explorative behavior were evaluated for 30 minutes in the open field at 17 months of age, as previously described [40, 45, 46]. Using EthoVision XT10.1 (Noldus, Wageningen, The Netherlands), locomotion was automatically recorded. The floor of the arena was divided into center, periphery, and corners. The frequency of entering these zones was measured automatically. In addition, exploration was manually scored (walking, sitting, wall leaning, jumping, rearing, grooming) and analyzed as described previously [47, 48].
MRI protocol
Following the behavioral examinations, at 18 months of age MRI measurements were performed on an 11.7 T BioSpec Avance III small animal MR system (Bruker BioSpin, Ettlingen, Germany) equipped with an actively shielded gradient set of 600 mT/m and operating on the Paravision 5.1 software platform (Bruker, Karlsruhe, Germany). We used a circular polarised volume resonator for radiofrequency transmission and an actively decoupled mouse brain quadrature surface coil with integrated combiner and preamplifier for receive (Bruker BioSpin). For the imaging procedure, the animals were anesthetized with isoflurane (3.5% for fast induction and 1.8% for maintenance) in a 2:1 oxygen and N2O mixture (normal gas condition) or in a 3:0 oxygen and N2O mixture (vasoconstriction), and placed in a stereotactic holder to prevent unwanted movement during the scanning. Body temperature was monitored with a rectal temperature probe and maintained at 37˚C with heated airflow. Respiration of the animal was monitored using a pneumatic cushion respiratory monitoring system (Small Animal Instruments Inc, NY, USA). First gradient echo T2*-weighted images covering the entire mouse brain were acquired in three directions for anatomical reference using previously described acquisition parameters [49].
Cerebral blood flow
MR perfusion data were measured with flow-sensitive alternating inversion recovery (FAIR) MRI techniques; from a series of echo planar imaging (EPI)-images in three different regions of interest (ROI) were evaluated [50, 51] in the cerebral cortex (all cortical regions above corpus callosum), hippocampus, and thalamus according to the Franklin - Paxinos atlas [52]. Twelve images with increasing inversion times (TIs; 40-3000 ms) were obtained for the T1 calculations, accounting to a total scan time of 12 min. Inversion recovery data from the imaging slice were acquired after selective inversion interleaved with nonselective inversion. To evaluate vasoreactivity, a 2:1 oxygen and N2O mixture (normal gas condition) and a 3:0 oxygen and N2O mixture (vasoconstriction) were used. To calculate regional CBF we used the protocol that is described in [49].
Diffusion tensor imaging
Diffusion of water was imaged as described previously [40, 45, 53]. In short, 22 axial slices covering the whole brain were acquired with a four-shot SE-EPI protocol. B0 shift compensation, navigator echoes and an automatic correction algorithm to limit the occurrence of ghosts and artefacts were implemented. Encoding b-factors of 0 s/mm2 (b0 images; 5×) and 1000 s/mm2 were used and diffusion-sensitizing gradients were applied along 30 non-collinear directions in three-dimensional space.
The diffusion tensor was estimated for every voxel using the PATCH algorithm [54]; mean water diffusivity (MD) and fractional anisotropy (FA) were derived from the tensor estimation following a protocol as described elsewhere [45]. MD and FA values were measured in several white matter (WM) and grey matter (GM) areas, manually selected based on an anatomical atlas [52].
Resting state fMRI
Subsequently after the acquisition of the anatomical reference images, resting state fMRI (rsfMRI) datasets were acquired using a single-shot spin-echo sequence with echo-planar readout (SE-EPI) sequence. Six hundred repetitions with a repetition time (TR) of 1.8 s and echo time of 16.9 ms were recorded for a total acquisition time of 18 min. The rsfMRI datasets were first realigned using a least-squares method and rigid-body transformation with Statistical Parametric Mapping (SPM) mouse toolbox (SPM5, University College London; http://www.fil.ion.ucl.ac.uk/spm/; Sawiak et al., 2009). Mean and maximum displacement across the six degrees of freedom (along the x-, y-, and z-axes and on three rotation parameters pitch, roll, and yaw) were measured in each mouse. The mean SE-EPI images for each mouse were then used to generate a study-specific template through linear affine and nonlinear diffeomorphic transformation (ANTs. v1.9; http://picsl.upenn.edu/ANTS/). Visual inspection of the normalised dataset was performed to screen for possible normalization biases. On the template, 12 areas were selected in left and right hemisphere. The selected regions were based on previous work concerning functional connectivity in mice [55], and included: left and right dorsal hippocampus, left and right ventral hippocampus, left and right auditory cortex, left and right motor cortex, left and right somatosensory cortex, and left and right visual cortex. All cortical ROIs were selected 1-2 voxels away from the edge of the cortex, to minimise the impact of susceptibility artefacts, which are more prominent in areas close to tissue interfaces (e.g., near the skull or near the ear canals). In-plane spatial smoothing (0.4 × 0.4 mm), linear detrending, and temporal high-pass filtering (cut-off at 0.01 Hz) were applied to compensate for small across-mouse misregistration and temporal low-frequency noise. FC group comparison between ROIs were calculated from the BOLD time series using total and partial correlation analyses implemented in FSLNets (FSLNets v0.3; www.fmrib.ox.ac.uk/fsl). Pearson's correlation values were Fisher transformed to Z-scores for group comparisons and statistical analysis.
Post mortem brain tissue preparation
Directly following the MR measurements at 18 months of age, anaesthetized mice were sacrificed by transcardial perfusion with 0.1M phosphate buffered saline (PBS) at room temperature. The perfused brains were cut mid-sagittally and the right hemispheres were snap frozen in liquid nitrogen and stored at -80°C, before further biochemical processing. The left hemispheres were immersion fixated for 15h at 4°C in 4% paraformaldehyde fixative and thereafter stored in 0.1M PBS with 0.01% sodium azide at 4°C for immunohistochemical staining.
Immunohistochemistry
Eight series of 30 µm coronal sections were cut through the brain using a sliding microtome (Microm HM 440 E, Walldorf, Germany) equipped with an object table for freeze sectioning at -60°C.
For every staining, one complete series with 240 µm distance between the sections was used. Immunohistochemistry was performed using standard free-floating labeling procedures, as described previously, and was carried out on a shaker table at room temperature. [40].
Postsynaptic density (PSD) was visualized with anti-PSD-95 antibody (1:2000; Abcam, catalog #ab18258, RRID:AB_444362) using one subseries of brain sections per animal. Donkey anti-rabbit biotin 1:1500 (Jackson ImmunoResearch, West Grove, PA, USA) was used as secondary antibody.
To visualize immature neurons we used an anti-doublecortin (DCX) antibody (1:4000; polyclonal goat anti-doublecortin (C18): sc-8066, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) with one subseries of brain sections per animal. Being a microtubule-associated protein, Doublecortin is exclusively found in somata and processes of migrating and differentiating neurons [56, 57]. Donkey-goat biotin 1:1500 (Jackson ImmunoResearch, West Grove, PA, USA) was used as secondary antibody.
To visualize and quantify the amount of macrophages and microglia, an anti-ionized calcium binding adapter molecule 1 (IBA-1) antibody (1:1500; polyclonal goat anti-IBA1 (C18); Abcam Inc., Cambridge, UK) was used with one subseries of brain sections per animal. Donkey-goat biotin 1:1500 (Jackson ImmunoResearch, West Grove, PA, USA) was used as secondary antibody.
The amount of glucose transporter type 1 (GLUT-1) was visualized using an anti-GLUT-1 antibody (1:10.000; rabbit anti GLUT-1 transporter, Chemicon AB 1340, Chemicon International, Inc., Temecula, CA, USA). Donkey-rabbit biotin 1:1500 (Jackson ImmunoResearch, West Grove, PA, USA) was used as secondary antibody.
Results of all immunohistochemical stainings for PSD-95, DCX, IBA-1, and GLUT-1 can be found in the supplementary material.
Quantification - PSD-95
The stained sections were analyzed using a Zeiss Axioskop microscope equipped with hardware and software of Microbrightfield (Williston, VT, USA). Brain regions were based on the mouse brain atlas of Franklin & Paxinos [52] and quantified in five regions of the hippocampus: the inner molecular layer (IML), outer molecular layer (OML), cornu ammonis 1 (CA1), CA2 and CA3. Additionally, two regions in the cortex corresponding to the visual and somatosensory cortex were analyzed. The relevant regions were digitized at 100 times magnification with immersion oil using Stereo Investigator. The quantification of the photographs was performed using Image J (Image J, U. S. National Institutes of Health, and Bethesda, Maryland, USA). The contrast was manually enhanced, following the same procedure for all digitized images, and the amount of tissue stained was measured with a threshold-based approach.
Quantification - DCX, IBA-1 & GLUT-1
Brain sections between bregma -1.46 and -2.30 [52] were preselected for quantification. Quantification was done at a 5x magnification using an Axio Imager (A2, Zeiss Germany). ImageJ (National Institute of Health, Bethesda, MD, USA) was used to analyze the regions of interest: Cortex (IBA-1 & GLUT-1), hippocampus (DCX, IBA-1 & GLUT-1).
Biochemical analyses
Serum and brain sterol levels were measured by gas-chromatography-mass-spectrometry-selected-ion-monitoring (GC-MS-SIM) as described in detail previously [58]. The cerebellum of the right hemisphere was homogenized and sterols were extracted overnight by chloroform/methanol trimethylsilylation prior to GC-MS-SIM analysis [58]. Brain fatty acid analyses were performed with a part of the brain homogenate (olfactory bulb and part of frontal cortex), as described previously [45].
Statistics
For the statistical analysis, IBM SPSS 22 software (IBM Corporation, New York, NY, USA) was used.
Multivariate ANOVA (Repeated measures ANOVA for the MWM and SBP data) with Bonferroni corrections was conducted with between-group-factors genotype to analyze possible differences in all the other parameters. Statistical significance was set at p ≤ 0.05, while a tendency was set at 0.05 < p < 0.08. All data are expressed as mean ± SEM.
Results
Body Weight
18-month-old AβPP/PS1 mice had the same body weight as their WT littermates before both MR scan sessions (Fig. 1A, F(1,15)=2.7, p<0.124).
Systolic blood pressure
Using tail-cuff plethysmography to measure systolic blood pressure (SBP), AβPP/PS1 mice demonstrated an increased SBP compared to their WT littermates from 16 to 18 months of age (Fig. 1B, F(1,15)=5.6, p<0.032). Both AβPP/PS1 and WT mice exhibited a decrease in SBP over time from 16 to 18 months of age (Fig. 1B, F(2,30)=8.1, p<0.002).
Figure 1
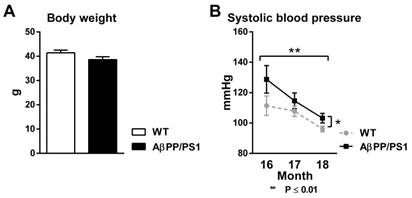
Body weight (A) and systolic blood pressure (B, SBP) of AβPP/PS1 and wild-type (WT) mice. (A) 18 month old AβPP/PS1 mice showed no difference in body weight compared to their WT littermates (p<0.124). (B) SBP was measured each experimental month for two consecutive days in trained, conscious and preheated mice using computerized tail-cuff plethysmography. Here, AβPP/PS1 mice had a higher SBP than their WT littermates from 16 to 18 months of age (p<0.032). SBP of both AβPP/PS1 and WT mice decreased over time from 16 to 18 months of age (p<0.002).
MR measurements
Cerebral blood flow and vasoreactivity
To study the effect of genotype differences on cerebrovascular health, CBF (Fig. 2A) and vasoreactivity (Fig. 2B) were measured with FAIR ASL using normal (200 O2 : 100 N2O mL/min) and vasoconstrictive (300 O2 : 0 N2O mL/min) gas conditions in three regions of interest (ROI): cortex, hippocampus and thalamus. Under normal gas condition a decreased CBF was observed in the cortex (F(1,14)=4.2, p<0.060) and thalamus (F(1,14)=10.5, p<0.007) of AβPP/PS1 mice compared to their WT littermates. In addition, under vasoconstrictive condition our AD model mice had again a lower thalamic CBF (F(1,14)=5.1, p<0.040) than the WT mice. Both AβPP/PS1 and WT mice showed an intact vasoreactivity in the cortex (AβPP/PS1, F(1,6)=6.2, p<0.048; WT, F(1,8)=13.7, p<0.006) and thalamus (AβPP/PS1, F(1,6)=13.2, p<0.011; WT, F(1,8)=35.4, p<0.001). In contrast, in the hippocampus only WT mice (F(1,8)=13.8, p<0.006) revealed an intact vasoreactivity, while AβPP/PS1 mice (F(1,6)=1.6, p<0.253) had an impaired vasoreactivity indicating an incapability to adapt to the vasoconstrictive gas condition.
Diffusion tensor imaging
Quantitative assessment of the diffusion tensor indices fractional anisotropy (FA); and mean diffusivity (MD) was performed for ROIs drawn in several white and gray matter regions to assess effects of AD-like pathology in 18-month-old AβPP/PS1 and WT mice (Fig. 2C+D). AβPP/PS1 mice showed impaired white matter integrity as indicated by a decrease in FA in the visual cortex, not being present in their WT littermates (Fig. 2C, F(1,9)=24.5, p<0.001). No genotype effect was present for the MD (Fig. 2D).
Figure 2
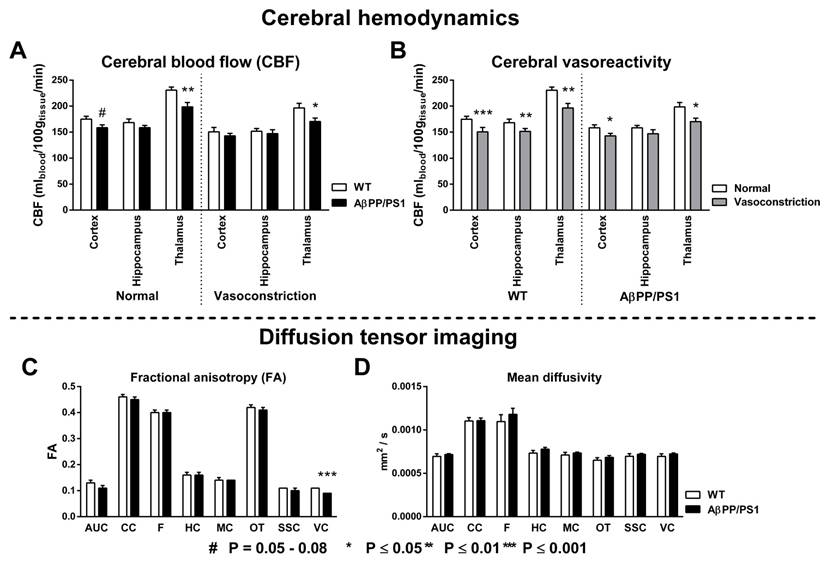
Genotype effects on the cortical, hippocampal and thalamic cerebral blood flow (A, CBF) and vasoreactivity (B) using normal (200 O2 : 100 N2O mL/min) and vasoconstrictive (300 O2 : 0 N2O mL/min) gas conditions in 18-month-old AβPP/PS1 and wild-type (WT) mice. Quantitative assessment of diffusion tensor derived indices was performed for several ROI (AUC= Auditory cortex, CC=Corpus callosum, F=Fornix, HC=Hippocampus, MC=Motor cortex, OT=Optic tract, SSC=Somatosensory cortex, VC=Visual cortex) drawn in white and gray matter to assess effects of AD-like pathology in 18-Month-old AβPP/PS1 and WT mice on fractional anisotropy (C) and mean diffusivity (D). (A) CBF was measured with flow-sensitive alternating inversion recovery MRI technique; from a series of echo planar imaging images. Under normal gas condition, AβPP/PS1 mice had a lower cortical (p<0.060) and thalamic (p<0.007) CBF than their WT littermates. Under vasoconstrictive gas conditions, AβPP/PS1 mice showed again a lower thalamic CBF (p<0.040) than their WT littermates. (B) Both AβPP/PS1 and WT mice showed an intact vasoreactivity in the cortex (AβPP/PS1, p<0.048; WT, p<0.006) and thalamus (AβPP/PS1, p<0.011; WT, p<0.001). In contrast, in the hippocampus only WT mice (p<0.006) revealed an intact vasoreactivity, while AβPP/PS1 mice (p<0.253) had an impaired vasoreactivity indicating an incapability to adapt to the vasoconstrictive gas condition. (C) AβPP/PS1 mice had a lower FA in the VC than WT mice indicating an impaired white matter integrity (p<0.001). (D) No genotype effect was found for the MD in all measured ROI.
Resting state fMRI
To analyze the impact of AD-like pathology on the functional connectivity (FC) patterns at 18 months of age, rsfMRI data were statistically analyzed based on total correlation (Fig. 3A) and partial correlation (Fig. 3B).
Total correlation analyses
AβPP/PS1 mice showed no differences in FC, as analyzed with total correlations compared to their WT littermates (Fig. 3A).
Partial correlation analyses
In comparison with the total correlation analysis, partial correlation analysis accentuates the direct connectivity between two ROI, while regressing the temporal BOLD signal from all other ROI. Resulting connectivity was thresholded at |Z| > 1.0. For the partial correlations we found significant genotype effects at 18 months of age (Fig. 3B). AβPP/PS1 mice showed a disturbed FC between several brain regions. While revealing an increased FC between the left motor cortex to left auditory cortex (F(1,12)=5.5, p<0.037), AβPP/PS1 mice compared to their WT littermates demonstrated a decreased FC between left auditory cortex to left ventral hippocampus (F(1,12)=5.7, p<0.034) and left somatosensory cortex to left visual cortex (F(1,12)=6.3, p<0.028).
Fatty acids in brain tissue
Fatty acid content was determined in frontal cortex (Supplementary table 1).
No genotype effects were found for the brain fatty acids palmitic acid, stearic acid, saturated fatty acid, oleic acid, and mono-unsaturated fatty acid. However, AβPP/PS1 mice showed increased arachidonic acid (F(1,15)=6.8, p<0.020), increased total omega-6 (F(1,15)=4.1, p<0.061), a decreased DHA (F(1,15)=9.6, p<0.008), a decreased overall omega-3 (F(1,15)=10.6, p<0.006), and a resulting decreased omega-3/omega-6 ratio (F(1,15)=8.6, p<0.011).
Sterol levels
Sterol levels were determined in the blood serum (Supplementary table 2) and in the cerebellum of the brain (Supplementary table 3).
Blood serum
No genotype effects were found in levels of cholestanol, lathosterol, campesterol, campestanol, stigmasterol, sitosterol, sitostanol, avenasterol, brassicasterol, lanosterol, desmosterol, dihydro-lanosterol, 24OH-cholesterol, 7αOH-cholesterol, 27OH-cholesterol, and global cholesterol.
Cerebellum
In brain tissue of 18-month-old AβPP/PS1 mice, levels of dihydro-lanosterol (F(1,15)=5.8, p<0.030) were higher than in the brain tissue of their WT littermates. No genotype effects were revealed for the sterol levels of cholestanol, lathosterol, campesterol, stigmasterin, sitosterol, lanosterol, desmosterol, 24OH-cholesterol, 27OH-cholesterol, and global cholesterol.
Figure 3
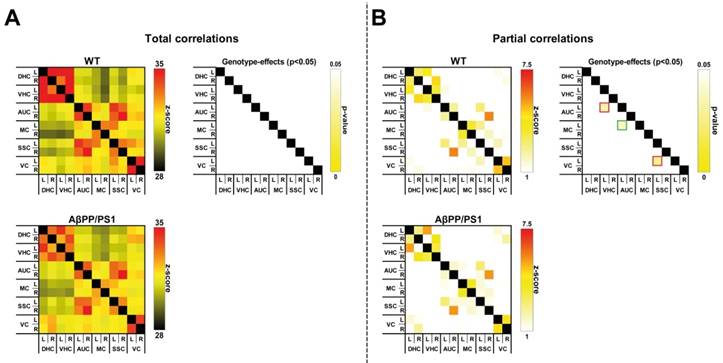
Resting-state functional connectivity (FC) based on total (A) and partial (B) correlation analyses of 12 ROI in the brain of in 18-Month-old AβPP/PS1 and WT mice. (A) For the overall correlations no significant genotype effects were detected in the dorsal (DHC) and ventral hippocampus (VHC), and auditory (AUC), motor (MC), somatosensory (SSC), and visual cortices (VC). (B) AβPP/PS1 mice showed a disturbed FC between several brain regions. Between the left motor cortex to left auditory cortex (p<0.037) AβPP/PS1 mice had a higher FC than their WT littermates, while showing a decreased FC between left auditory cortex to left ventral hippocampus (p<0.034) and left somatosensory cortex to left visual cortex (p<0.028).
Discussion
Hypertension is the most common cardiovascular risk factor [36, 59-65], and associated with all (other) key markers of AD such as presence of amyloid-β (Aβ) plaques, neurofibrillary tangles, and brain atrophy [64]. The increasing prevalence of hypertension due to the aging world population and growing prevalence of obesity could also increase the number of AD patients, since midlife hypertension almost doubles the risk of developing AD in later life [36, 66]. In agreement with this, the AβPP/PS1 AD-like model mice used in the present study, demonstrated an increased systolic blood pressure (SBP) and concomitant decreased regional CBF in the cortical and thalamic areas, decreased vasoreactivity of the circulation of the hippocampus, and impaired cognition. The WT and AβPP/PS1 mice showed a decrease in SBP over time, while the AβPP/PS1 mice always demonstrated a higher SBP than their WT littermates. This is in line with the AGES-Reykjavik study which has shown that midlife hypertension combined with a lower late-life BP is associated with a lowered total brain and gray matter volume [67]. Long-standing hypertension stimulates atherosclerosis and vascular remodeling leading to increases in wall thickness. Arterial stiffness and severe atherosclerosis can lead to an increase in pulse pressure [68]. An increased pulse pressure is correlated with a higher risk of AD in older adults [68]. In the Rotterdam study the presence of atherosclerotic plaques or wall thickening has been associated with dementia and its two major subtypes AD and vascular dementia [66]. In accordance to previous research, this AD-like mouse model suffers from early-impaired cerebrovascular autoregulation and CBF, but also shows abnormalities in the cortical microvasculature [69-72]. These results support the significant reduction in regional and global CBF as identified in studies on MCI and AD patients [73]. A lowered CBF is a common feature in the early phase of AD, which may possibly be caused by an accumulation of Aβ in the vessel walls or in close vicinity of the blood vessels [27, 74, 75]. Furthermore, the impaired vasoreactivity in our transgenic mice is also a clinical hallmark of AD [76]. Aβ has shown to directly enhance the vasoconstriction of the cerebral arteries and to stimulate selected constrictor responses, resulting in a reduced CBF [74]. In addition, deposition of Aβ in the cerebral microvessels promotes vascular pathology and dysfunction [77-79], as embodied by the impaired hippocampal vasoreactivity in the brains of the AβPP/PS1 mice. Hypertension may contribute to this remodeling through increase in smooth muscle tone and damage of endothelial cell function resulting in arterial wall thickening and microvascular rarefaction [80]. In line with our research, Toth et al. demonstrated that in aging mice hypertension could induce an impaired cerebrovascular autoregulation [81], cerebromicrovascular injury and neuroinflammation. In the present study we investigated brain diffusivity with DTI as an imaging biomarker for white and gray matter integrity. FA is a marker of the degree of myelination and fiber density of white matter, while MD characterizes an inverse measure of the membrane density and is sensitive to cellularity, edema, and necrosis in grey matter (GM) [82-84]. In addition to an impaired cerebrovascular function, a reduced FA value of the visual cortex was found in our AD model mice indicating a subtle change in microstructural integrity. This impaired microstructural integrity could imply axonal degeneration or demyelination [84] in the visual cortex. Nevertheless, no genotype effects were found for MD, being considered to be more informative for grey matter regions like the visual cortex. In our previous research using DTI in 12-month-old WT and AβPP/PS1 mice as well, fiber tract volume reduction, loss of axonal neurofilaments, and myelin breakdown in axonal bundles of several white matter regions (i.e. body of the corpus callosum) were detected in AβPP/PS1 mice compared to WT [85]. In this recent study minor genotype effect on FA and no genotype effects on MD were found. One reason for these missing genotype effects could be that already in normal aging significant white matter deterioration occurs due to myelin degeneration [86]. Therefore, myelin degeneration in our 18 months WT mice may mask partly the significant changes regarding structural connectivity in APP/PS1 mice. Nevertheless, in line with the loss of structural connectivity, a disturbed FC pattern in cortical and hippocampal brain regions was revealed in the 18-month-old AβPP/PS1 mice. In support, a mouse model for cerebral amyloidosis demonstrated a compromised FC affecting the sensory motor cortex already in pre-plaque stage [87]. Our latter results are in accordance with clinical studies, in which a decreased FC was also demonstrated in AD patients [29-31].
A limiting factor of the acquired FC results is that the rsfMRI measurements are acquired in isoflurane anesthetized mice. Fortunately, using rsfMRI a general structure of the functional networks transcending levels of consciousness was detected in both preclinical and clinical studies [88-90]. Notably, under isoflurane anaesthesia a bilateral cortical connectivity in several cortical regions was measured [91, 92]. Nevertheless, also contradictory results were detected in isoflurane anesthetized mice showing not the same level of bilateral connectivity [55, 93]. However, in our previous and recent studies using isoflurane as anaesthesia, we confirmed the presence of networks in several well defined cortical and subcortical brain regions in two different murine models for vascular risk factors for AD [94, 95]. In our previous work we confirmed that both FC and CBF are dependent on isoflurane concentrations, and both FC and CBF decline with concentrations of isoflurane >2.2%, but do not further decline below a concentration of 2.2% [95]. Therefore, using the low- dose isoflurane (~1.7%) in this recent experiment will preserve the resting-state networks and will not interfere with the outcome of this study, as all animals were kept under the same low isoflurane concentration.
The combination of impaired cerebral hemodynamics, disturbed structural and functional connectivity, may underlie the impaired spatial learning capability found in the MWM. Here, the AβPP/PS1 mice needed more time to learn the position of the hidden platform. Moreover, these transgenic mice demonstrated a larger swim distance and higher swim velocity indicating an increased usage of non-spatial search strategies instead of hippocampus-dependent search strategies to find the hidden platform in the MWM [96, 97]. The behavior of the APP/PS1 mouse has been well-characterized. Mirroring the cognitive impairment being present in early AD patients, this cognitive deficit is a well-known feature of this AD mouse model. While at 4 and 7 months of age no learning deficit could be determined in AβPP/PS1 mice [98, 99], these AD model mice tend to exhibit an impaired memory capacity at 8 months of age in our earlier studies [48] and a significantly impaired spatial memory at 12 months of age, as demonstrated in studies from Lalonde et al. [100]. In fear-conditioning tests Kilgore et al. showed at already six months of age an impaired contextual memory in the APP/PS1 mouse model [101]. However, Webster et al. performed recently a comprehensive behavioral analysis of a knock-in AβPP/PS1 mouse model [102] using four different age groups (7, 11, 15, and 24 months) in which cognitive deficits in spatial reference memory (radial arm water maze) appeared from 11 months of age and becoming apparent as the disease progressed and recognition memory (novel object recognition) was demonstrated just from 15 months of age [102]. In line with previous results from our lab on younger (8-/12-/15-month-old) AβPP/PS1 mice [40, 48], also our 18-month-old AβPP/PS1 mice exhibited an increased locomotor activity and anxiety-related behavior in the open field. Being linked to elevated anxiety levels, this heightened activity has been observed in many transgenic AD model mice [103-105]. This hyperactivity and anxiety-related behavior resembles restlessness and anxiety-related symptoms found in up to 71% of AD patients [106, 107]. The increased anxiety of our mice could reveal a possible pathological mechanism for the elevation in SBP. This phenotype of anxiety is an expression of a stressful chronic condition. Therefore, it is well known that mental stress is able per se to provoke an increase in blood pressure in mice [108, 109]. The influence of the overexpression of AβPP and PS1 on cognition is well-characterized in several AD mouse models, while the non-cognitive behavior has not been considered as systematically [110]. Pugh et al. demonstrated reduced spontaneous motor activity, disinhibition, heightened frequency and duration of feeding bouts, decreased body weight and, by 10 months, increased activity over a 24h period in both female and male AβPP/PS1 mice [110]. In addition, male mice also expressed a heightened aggression relative to WT controls [110]. In the postmortem part of the study we revealed a decreased postsynaptic density and less DCX+ cells in the hippocampus of AβPP/PS1 mice indicating respectively a lowered synaptogenesis and neurogenesis, which may be the cause of the profound cognitive impairment found in the MWM in this AD-like transgenic mouse model. Notably, in our earlier studies, AβPP/PS1 mice showed decreased neurogenesis combined with a lowered hippocampal CBF already at 12-month of age [40, 49]. Moreover, in these younger 12-month-old AβPP/PS1 mice Zerbi et al. also detected impaired structural connectivity in the corpus callosum, fimbria, and cortex measured also via DTI [49]. This decrease in hippocampal CBF was no longer observed in the 18-month-old AβPP/PS1 mice granting a possible pathological timeframe starting with an impaired hippocampal CBF provoking a decreased synapto- and neurogenesis resulting in an impaired cognition. In another study, Ermini et al. showed as well a decreased neurogenesis in younger (8 months of age) AβPP/PS1 and (5 months of age) AβPP23 mice [111]. In many transgenic mouse models of AD an increased neuroinflammation has been observed reporting modulation of cytokine levels, activation of microglia and in some cases an activation of the complement system [112]. In the early pre-plaque stages of transgenic AD mice and, to a much lesser extent, in old WT mice highly activated microglia have been found [113-117]. In addition, Aβ-plaques in AD are engulfed by activated microglia [117]. To measure neuroinflammation, we immunohistochemically stained the mouse brains with ionized calcium-binding adapter molecule 1 (IBA-1) being a marker for activated microglia [118-120]. Here, in our AD model mice more IBA+ cells were detected in cortical regions compared to their WT littermates. In accordance to the results of Minogue et al., in this mouse model after 14 months of age an age-associated dysregulation of microglial activation is coupled with an enhanced blood-brain barrier permeability [114]. Furthermore, Meadowcroft et al. demonstrated activated microglial cells surrounding Aβ plaques in the brain of AβPP/PS1 mice. In accordance to our previous study, 18-month-old AβPP/PS1 mice did not show any differences in hippocampal capillary density measured via immunohistochemical staining for GLUT-1 [121]. Notably, these mice exhibited the highest level of Aβ in combination with hippocampal atrophy [121]. While 8-month-old AβPP/PS1 mice demonstrated also an increased amount of Aβ deposition in the dentate gyrus, but did not show any differences in hippocampal atrophy [121]. Besides all other AD-like pathological changes in this AD mouse model, we also detected an increase in cerebral omega-6 fatty (n6) acid content (especially arachidonic acid, ARA) combined with a decrease in cerebral omega-3 (n3) fatty acid content (in particular docosahexaenoic acid, DHA) resulting in a pronounced decreased n3/n6-ratio, which is thought to elevate the risk for AD [122, 123]. In accordance to our results, Dutch drug-naïve patients with mild AD (Mini-Mental State Examination (MMSE) = 25.0) showed a decreased relative content of n3 fatty acids (including DHA) and an increased relative content of ARA in erythrocyte membranes compared to a group of Dutch healthy controls (MMSE = 29.0) [124]. In accordance, ARA is a mediator of inflammatory pathways, and its metabolites are involved in the production of Aβ and in the pathogenesis of AD [125]. In our previous research using the same transgenic mouse model, a multi-nutrient diet was able to induce a pronounced shift in n3/n6 ratio in favor of the n3 fatty acids as compared to the control diet [40]. Here, the reduction of the relative n6 content was mainly caused by a decrease in ARA, while the higher n3 content was mainly caused by an increase in DHA. Fabelo et al. revealed that AβPP/PS1 mice show a more rapid lipid raft aging compared to WT, which is related to an increased saturation of phospholipids (higher saturated fatty acids content and decreased content of the long chain unsaturated fatty acids ARA and DHA) and increased sphingomyelin levels rather than to alterations in cholesterol [126]. Moreover, Fabelo et al. demonstrated that both levels of ARA and DHA were gradually lowered with age in AβPP/PS1 mice, with its maximal reduction of approximately 50% at 14 months of age [126]. In our recent study the aged 18-month-old AβPP/PS1 mice demonstrated a pronounced decreased n3/n6-ratio in the frontal cortices due to a decrease in DHA and increased ARA. This contradictory data on ARA levels may be explained by age and the brain area measured, as Fabelo et al. analyzed homogenized cortex of a total hemisphere. Notably, Perez et al. revealed that both male and female 6-month-old AβPP/PS1 mice show neither a change in ARA nor a decrease in DHA brain fatty acid content [127] indicating the pathological role of aging in disturbing cerebral membrane composition. Notably, a novel finding is the increased level of dihydro-lanosterol in our AD-model mice, being an intermediate in the cholesterol synthesis. Nevertheless, no genotype effects on cholesterol levels in blood and brain tissue were found. Decreased brain cholesterol levels were measured in our previous research using 15-month-old AβPP/PS1 mice compared to their WT littermates [48]. In addition, mice fed a high cholesterol-containing typical western diet showed a significant decreased regional cerebral blood volume compared to mice fed a standard control chow diet [48]. Notably, Park et al. showed an increased Aβ synthesis and senile Aβ plaque deposition only in female AD model (Tg2576) mice when using lovastatin (a blood-brain barrier crossing inhibitor of the cholesterol biosynthesis pathway) [128]. Dysregulation of the cerebral cholesterol homeostasis has been increasingly associated with chronic neurodegenerative disorders, including AD, Huntington's disease, and Parkinson's disease [129]. Furthermore, the E4 isoform of apolipoprotein E, being a cholesterol-carrying protein, is linked to an increased risk of developing AD [129].
In conclusion, in this study we detected an increased SBP, impairments in cerebral hemodynamics resulting in an impaired cognition and structural and functional connectivity, increased locomotor activity, and anxiety-related behavior in an AD-like mouse model. In accordance, already in 4.5 months old AβPP/PS1 mice Cifuentes et al. indicated that hypertension is able to accelerate the development of Alzheimer disease-related structural and functional alterations, partially through cerebral vasculature impairment and reduced nitric oxide production [130]. Future research should also consider gender differences in these AβPP/PS1 mice. In detail, male mice develop Aβ plaques more rapidly than female mice, being saturated at nine months of age, while in female mice plaque accumulation does not reach its maximum in female mice until around 12 months of age [131]. Notably, using both male and female 4-, 12-, and 17-month-old AβPP/PS1 mice Wang et al. demonstrated that female AβPP/PS1 mice have a higher hippocampal Aβ40 and Aβ42 levels than male AβPP/PS1 mice at 4 months of age Aβ and female AβPP/PS1 mice had a heavier Aβ burden and higher plaque number compared to male mice of the same age, both at 12 and at 17 months of age [132]. Our results indicate that vascular impairment plays an important role in the very early stage of AD but whether it is a causative factor or just a contributor aggravating the disease progress, remains to be elucidated. Thus, future therapeutic approaches should focus on (cerebro)vascular impairment as a promising strategy for the prevention of AD. The present AD mouse model showing hypertension and cerebral circulation impairment could serve as translational tool for the development of treatments to inhibit neurodegenerative diseases like AD already in the prodromal phase of the disease.
Supplementary Material
Supplementary figures and tables.
Abbreviations
Aβ: Amyloid-β, AβPP/PS1: AβPPswe/PS1dE9, AD: Alzheimer's disease, CA: Cornu ammonis, CBF: Cerebral blood flow, DCX: Doublecortin, FA: Fractional anisotropy, FC: Functional connectivity, GLUT-1: Glucose transporter-1, IBA-1: Ionized calcium-binding adapter molecule 1, IML: Inner molecular layer, MD: Mean diffusivity, MMSE: Mini-Mental State Examination, MWM: Morris water maze, OF: Open field, OML: Outer molecular layer, PSD: Postsynaptic density, rsfMRI: resting state fMRI, SBP: Systolic blood pressure, SL: Stratum lucidum, SR: Stratum radiatum, SSC: Somatosensory cortex, V1: Visual cortex, VaD: Vascular dementia.
Acknowledgements
We would like to thank Bianca Lemmers-van de Weem, Kitty Lemmens-Hermans, Iris Lamers-Elemans, Karin de Haas-Cremers and Henk Arnts for their excellent care for our mice.
Sources of Funding
This study was supported by the EU 7th framework LipidiDiet project (FP7/2007-2013) under grant agreement no211696.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wimo A, Winblad B, Jönsson L. The worldwide societal costs of dementia: Estimates for 2009. Alzheimers Dement. 2010;6:98-103
2. Arevalo-Rodriguez I, Pedraza OL, Rodríguez A, Sánchez E, Gich I, Solà I. et al. Alzheimer's Disease Dementia Guidelines for Diagnostic Testing: A Systematic Review. Am J Alzheimers Dis Other Demen. 2013
3. Morris JC. The Nosology of Dementia. Neurol Clin. 2000;18:773-88
4. Iadecola C, Gorelick PB. Converging Pathogenic Mechanisms in Vascular and Neurodegenerative Dementia. Stroke. 2003;34:335-7
5. Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631-9
6. Albert MS. Cognitive and neurobiologic markers of early Alzheimer disease. Proc Natl Acad Sci U S A. 1996;93:13547-51
7. Braak H, Braak E. Staging of alzheimer's disease-related neurofibrillary changes. Neurobiol Aging. 1995;16:271-8
8. Braak H, Braak E. Diagnostic Criteria for Neuropathologic Assessment of Alzheimer's Disease. Neurobiol Aging. 1997;18:S85-S8
9. Hooijmans CR, Kiliaan AJ. Fatty acids, lipid metabolism and Alzheimer pathology. Eur J Pharmacol. 2008;585:176-96
10. Kilander L, Andrén B, Nyman H, Lind L, Boberg M, Lithell H. Atrial Fibrillation Is an Independent Determinant of Low Cognitive Function. Stroke. 1998;29:1816-20
11. Launer LJ, Ross GW, Petrovitch H, Masaki K, Foley D, White LR. et al. Midlife blood pressure and dementia: the Honolulu-Asia aging study. Neurobiol Aging. 2000;21:49-55
12. Nagata KEN, Sato M, Satoh Y, Watahiki Y, Kondoh Y, Sugawara M. et al. Hemodynamic Aspects of Alzheimer's Disease. Ann N Y Acad Sci. 2002;977:391-402
13. Ott A, Stolk RP, Hofman A, van Harskamp F, Grobbee DE, Breteler MMB. Association of diabetes mellitus and dementia: The Rotterdam Study. Diabetologia. 1996;39:1392-7
14. Posner HB, Tang M-X, Luchsinger J, Lantigua R, Stern Y, Mayeux R. The relationship of hypertension in the elderly to AD, vascular dementia, and cognitive function. Neurology. 2000;58:1175-81
15. Ringman JM, Sachs MC, Zhou Y, Monsell SE, Saver JL, Vinters HV. Clinical predictors of severe cerebral amyloid angiopathy and influence of apoe genotype in persons with pathologically verified alzheimer disease. JAMA Neurol. 2014;71:878-83
16. Wiesmann M, Kiliaan AJ, Claassen JA. Vascular Aspects of Cognitive Impairment and Dementia. J Cereb Blood Flow Metab. 2013;33:1696-706
17. de la Torre JC. Is Alzheimer's disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurol. 2004;3:184-90
18. Diaz-Ruiz C, Wang J, Ksiezak-Reding H, Ho L, Qian X, Humala N. et al. Role of Hypertension in Aggravating Abeta Neuropathology of AD Type and Tau-Mediated Motor Impairment. Cardiovasc Psychiatry Neurol. 2009;2009:107286
19. Haag M, Hofman A, Koudstaal P, Breteler M, Stricker B. Duration of antihypertensive drug use and risk of dementia A prospective cohort study. Neurology. 2009;72:1727-34
20. Keeman J, Mazel J, Zitman F. Het medisch jaar 2008/2009: Bohn Stafleu van Loghum; 2009.
21. Fujishima M, Ibayashi S, Fujii K, Mori S. Cerebral Blood Flow and Brain Function in Hypertension. Hypertens Res. 1995;18:111-7
22. Nobili F, Rodriguez G, Marenco S, De Carli F, Gambaro M, Castello C. et al. Regional cerebral blood flow in chronic hypertension. A correlative study. Stroke. 1993;24:1148-53
23. Rodriguez G, Arvigo F, Marenco S, Nobili F, Romano P, Sandini G. et al. Regional cerebral blood flow in essential hypertension: data evaluation by a mapping system. Stroke. 1987;18:13-20
24. Dai W, Lopez OL, Carmichael OT, Becker JT, Kuller LH, Gach HM. Abnormal Regional Cerebral Blood Flow in Cognitively Normal Elderly Subjects With Hypertension. Stroke. 2008;39:349-54
25. Efimova IY, Efimova NY, Triss SV, Lishmanov YB. Brain Perfusion and Cognitive Function Changes in Hypertensive Patients. Hypertens Res. 2008;31:673-8
26. Hirao K, Ohnishi T, Hirata Y, Yamashita F, Mori T, Moriguchi Y. et al. The prediction of rapid conversion to Alzheimer's disease in mild cognitive impairment using regional cerebral blood flow SPECT. Neuroimage. 2005;28:1014-21
27. Ruitenberg A, den Heijer T, Bakker SLM, van Swieten JC, Koudstaal PJ, Hofman A. et al. Cerebral hypoperfusion and clinical onset of dementia: The Rotterdam study. Ann Neurol. 2005;57:789-94
28. de la Torre JC. Critically attained threshold of cerebral hypoperfusion: the CATCH hypothesis of Alzheimer's pathogenesis. Neurobiol Aging. 2000;21:331-42
29. Allen G, Barnard H, McColl R. et al. Reduced hippocampal functional connectivity in alzheimer disease. Arch Neurol. 2007;64:1482-7
30. Supekar K, Menon V, Rubin D, Musen M, Greicius MD. Network Analysis of Intrinsic Functional Brain Connectivity in Alzheimer's Disease. PLoS Comput Biol. 2008;4:e1000100
31. Wang L, Zang Y, He Y, Liang M, Zhang X, Tian L. et al. Changes in hippocampal connectivity in the early stages of Alzheimer's disease: Evidence from resting state fMRI. NeuroImage. 2006;31:496-504
32. Bronge L, Bogdanovic N, Wahlund LO. Postmortem MRI and Histopathology of White Matter Changes in Alzheimer Brains. Dementia and Geriatric Cognitive Disorders. 2002;13:205-12
33. Bozzali M, Falini A, Franceschi M, Cercignani M, Zuffi M, Scotti G. et al. White matter damage in Alzheimer's disease assessed in vivo using diffusion tensor magnetic resonance imaging. Journal of Neurology, Neurosurgery & Psychiatry. 2002;72:742-6
34. de Groot JC, de Leeuw FE, Oudkerk M, van Gijn J, Hofman A, Jolles J. et al. Cerebral white matter lesions and cognitive function: the Rotterdam Scan Study. Ann Neurol. 2000;47:145-51
35. Kivipelto M, Helkala EL, Laakso MP, Hänninen T, Hallikainen M, Alhainen K. et al. Midlife vascular risk factors and Alzheimer's disease in later life: longitudinal, population based study. BMJ. 2001;322:1447-51
36. Skoog I, Nilsson L, Persson G, Lernfelt B, Landahl S, Palmertz B. et al. 15-year longitudinal study of blood pressure and dementia. Lancet. 1996;347:1141-5
37. Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA. et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159-70
38. Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157-65
39. Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. Improving Bioscience Research Reporting: The ARRIVE Guidelines for Reporting Animal Research. PLoS Biol. 2010;8:e1000412
40. Jansen D, Zerbi V, Arnoldussen IAC, Wiesmann M, Rijpma A, Fang XT. et al. Effects of Specific Multi-Nutrient Enriched Diets on Cerebral Metabolism, Cognition and Neuropathology in AβPPswe-PS1dE9 Mice. PLoS ONE. 2013;8:e75393
41. Capone C, Faraco G, Anrather J, Zhou P, Iadecola C. Cyclooxygenase 1-Derived Prostaglandin E2 and EP1 Receptors Are Required for the Cerebrovascular Dysfunction Induced by Angiotensin II. Hypertension. 2010;55:911-7
42. Capone C, Faraco G, Park L, Cao X, Davisson RL, Iadecola C. The cerebrovascular dysfunction induced by slow pressor doses of angiotensin II precedes the development of hypertension. Am J Physiol Heart Circ Physiol. 2011;300:H397-407
43. Kazama K, Anrather J, Zhou P, Girouard H, Frys K, Milner TA. et al. Angiotensin II Impairs Neurovascular Coupling in Neocortex Through NADPH Oxidase-Derived Radicals. Circulation research. 2004;95:1019-26
44. Kazama K, Wang G, Frys K, Anrather J, Iadecola C. Angiotensin II attenuates functional hyperemia in the mouse somatosensory cortex. American Journal of Physiology - Heart and Circulatory Physiology. 2003;285:H1890-H9
45. Jansen D, Zerbi V, Janssen CF, van Rooij D, Zinnhardt B, Dederen P. et al. Impact of a multi-nutrient diet on cognition, brain metabolism, hemodynamics, and plasticity in apoE4 carrier and apoE knockout mice. Brain Struct Funct. 2014;219:1841-68
46. Janssen CIF, Zerbi V, Mutsaers MPC, de Jong BSW, Wiesmann M, Arnoldussen IAC. et al. Impact of dietary n-3 polyunsaturated fatty acids on cognition, motor skills and hippocampal neurogenesis in developing C57BL/6J mice. The Journal of Nutritional Biochemistry. 2015;26:24-35
47. Streijger F, Oerlemans F, Ellenbroek BA, Jost CR, Wieringa B, Van der Zee CEEM. Structural and behavioural consequences of double deficiency for creatine kinases BCK and UbCKmit. Behavioural Brain Research. 2005;157:219-34
48. Hooijmans CR, Van der Zee CEEM, Dederen PJ, Brouwer KM, Reijmer YD, van Groen T. et al. DHA and cholesterol containing diets influence Alzheimer-like pathology, cognition and cerebral vasculature in APPswe/PS1dE9 mice. Neurobiology of Disease. 2009;33:482-98
49. Zerbi V, Jansen D, Wiesmann M, Fang X, Broersen LM, Veltien A. et al. Multinutrient diets improve cerebral perfusion and neuroprotection in a murine model of Alzheimer's disease. Neurobiology of Aging. 2014;35:600-13
50. Kim SG. Quantification of relative cerebral blood flow change by flow-sensitive alternating inversion recovery (FAIR) technique: Application to functional mapping. Magn Reson Med. 1995;34:293-301
51. Kwong KK, Chesler DA, Weisskoff RM, Donahue KM, Davis TL, Ostergaard L. et al. MR perfusion studies with T1-weighted echo planar imaging. Magn Reson Med. 1995;34:878-87
52. Paxinos G, Franklin KB. The mouse brain in stereotaxic coordinates / George Paxinos, Keith Franklin. London: Academic. 2004
53. Harsan LA, Paul D, Schnell S, Kreher BW, Hennig J, Staiger JF. et al. In vivo diffusion tensor magnetic resonance imaging and fiber tracking of the mouse brain. NMR Biomed. 2010;23:884-96
54. Zwiers MP. Patching cardiac and head motion artefacts in diffusion-weighted images. Neuroimage. 2010;53:565-75
55. Jonckers E, Van Audekerke J, De Visscher G, Van der Linden A, Verhoye M. Functional Connectivity fMRI of the Rodent Brain: Comparison of Functional Connectivity Networks in Rat and Mouse. PLoS ONE. 2011;6:e18876
56. Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet M-C. et al. Doublecortin Is a Developmentally Regulated, Microtubule-Associated Protein Expressed in Migrating and Differentiating Neurons. Neuron. 1999;23:247-56
57. Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin Is a Microtubule-Associated Protein and Is Expressed Widely by Migrating Neurons. Neuron. 1999;23:257-71
58. Lütjohann D, Brzezinka A, Barth E, Abramowski D, Staufenbiel M, von Bergmann K. et al. Profile of cholesterol-related sterols in aged amyloid precursor protein transgenic mouse brain. J Lipid Res. 2002;43:1078-85
59. Elias MF, Wolf PA, D'Agostino RB, Cobb J, White LR. Untreated Blood Pressure Level Is Inversely Related to Cognitive Functioning: The Framingham Study. Am J Epidemiol. 1993;138:353-64
60. Kilander L, Nyman H, Boberg M, Hansson L, Lithell H. Hypertension Is Related to Cognitive Impairment. Hypertension. 1998;31:780-6
61. Launer LJ, Masaki K, Petrovitch H, Foley D, Havlik RJ. The association between midlife blood pressure levels and late-life cognitive function. The Honolulu-Asia Aging Study. JAMA. 1995;274:1846-51
62. Ruitenberg A, Skoog I, Ott A, Aevarsson O, Witteman JCM, Lernfelt B. et al. Blood Pressure and Risk of Dementia: Results from the Rotterdam Study and the Gothenburg H-70 Study. Dement Geriatr Cogn Disord. 2001;12:33-9
63. Sharp SI, Aarsland D, Day S, Sønnesyn H, Alzheimer's Society Vascular Dementia Systematic Review G, Ballard C. Hypertension is a potential risk factor for vascular dementia: systematic review. Int J Geriatr Psychiatry. 2011;26:661-9
64. Skoog I, Gustafson D. Update on hypertension and Alzheimer's disease. Neurol Res. 2006;28:605-11
65. Yoshitake T, Kiyohara Y, Kato I, Ohmura T, Iwamoto H, Nakayama K. et al. Incidence and risk factors of vascular dementia and Alzheimer's disease in a defined elderly Japanese population. Neurology. 1995;45:1161-8
66. Hofman A, Ott A, Breteler M, Bots ML, Slooter AJ, van Harskamp F. et al. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer's disease in the Rotterdam Study. Lancet. 1997;349:151-4
67. Muller M, Sigurdsson S, Kjartansson O, Aspelund T, Lopez OL, Jonnson PV. et al. Joint effect of mid- and late-life blood pressure on the brain: the AGES-Reykjavik study. Neurology. 2014;82:2187-95
68. Qiu C, Winblad B, Viitanen M, Fratiglioni L. Pulse Pressure and Risk of Alzheimer Disease in Persons Aged 75 Years and Older: A Community-Based, Longitudinal Study. Stroke. 2003;34:594-9
69. El Tannir El Tayara N, Delatour B, Volk A, Dhenain M. Detection of vascular alterations by in vivo magnetic resonance angiography and histology in APP/PS1 mouse model of Alzheimer's disease. MAGMA. 2010;23:53-64
70. Niwa K, Kazama K, Younkin SG, Carlson GA, Iadecola C. Alterations in Cerebral Blood Flow and Glucose Utilization in Mice Overexpressing the Amyloid Precursor Protein. Neurobiol Dis. 2002;9:61-8
71. Niwa K, Kazama K, Younkin L, Younkin SG, Carlson GA, Iadecola C. Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J Physiol Heart Circ Physiol. 2002;283:H315-H23
72. Meyer EP, Ulmann-Schuler A, Staufenbiel M, Krucker T. Altered morphology and 3D architecture of brain vasculature in a mouse model for Alzheimer's disease. Proc Natl Acad Sci U S A. 2008;105:3587-92
73. Johnson NA, Jahng G-H, Weiner MW, Miller BL, Chui HC, Jagust WJ. et al. Pattern of Cerebral Hypoperfusion in Alzheimer Disease and Mild Cognitive Impairment Measured with Arterial Spin-labeling MR Imaging: Initial Experience. Radiology. 2005;234:851-9
74. Niwa K, Porter VA, Kazama K, Cornfield D, Carlson GA, Iadecola C. Aβ-peptides enhance vasoconstriction in cerebral circulation. Am J Physiol Heart Circ Physiol. 2001;281:H2417-H24
75. Prohovnik I, Mayeux R, Sackeim HA, Smith G, Stern Y, Alderson PO. Cerebral perfusion as a diagnostic marker of early Alzheimer's disease. Neurology. 1988;38:931
76. Cantin S, Villien M, Moreaud O, Tropres I, Keignart S, Chipon E. et al. Impaired cerebral vasoreactivity to CO2 in Alzheimer's disease using BOLD fMRI. Neuroimage. 2011;58:579-87
77. Iadecola C, Park L, Capone C. Threats to the mind: aging, amyloid, and hypertension. Stroke. 2009;40:S40-4
78. Smith EE, Greenberg SM. Beta-amyloid, blood vessels, and brain function. Stroke. 2009;40:2601-6
79. Pantoni L. Cerebral small vessel disease: from pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010;9:689-701
80. Levy BI, Schiffrin EL, Mourad JJ, Agostini D, Vicaut E, Safar ME. et al. Impaired tissue perfusion: a pathology common to hypertension, obesity, and diabetes mellitus. Circulation. 2008;118:968-76
81. Toth P, Tucsek Z, Sosnowska D, Gautam T, Mitschelen M, Tarantini S. et al. Age-related autoregulatory dysfunction and cerebromicrovascular injury in mice with angiotensin II-induced hypertension. J Cereb Blood Flow Metab. 2013;33:1732-42
82. Le Bihan D, Mangin J-F, Poupon C, Clark CA, Pappata S, Molko N. et al. Diffusion tensor imaging: Concepts and applications. J Magn Reson Imaging. 2001;13:534-46
83. Alexander AL, Hurley SA, Samsonov AA, Adluru N, Hosseinbor AP, Mossahebi P. et al. Characterization of Cerebral White Matter Properties Using Quantitative Magnetic Resonance Imaging Stains. Brain Connect. 2011;1:423-46
84. Feldman HM, Yeatman JD, Lee ES, Barde LHF, Gaman-Bean S. Diffusion Tensor Imaging: A Review for Pediatric Researchers and Clinicians. J Dev Behav Pediatr. 2010;31:346-56
85. Zerbi V, Kleinnijenhuis M, Fang X, Jansen D, Veltien A, Van Asten J. et al. Gray and white matter degeneration revealed by diffusion in an Alzheimer mouse model. Neurobiology of Aging. 2013;34:1440-50
86. Davis SW, Dennis NA, Buchler NG, White LE, Madden DJ, Cabeza R. Assessing the effects of age on long white matter tracts using diffusion tensor tractography. NeuroImage. 2009;46:530-41
87. Grandjean J, Schroeter A, He P, Tanadini M, Keist R, Krstic D. et al. Early Alterations in Functional Connectivity and White Matter Structure in a Transgenic Mouse Model of Cerebral Amyloidosis. J Neurosci. 2014;34:13780-9
88. Vincent JL, Patel GH, Fox MD, Snyder AZ, Baker JT, Van Essen DC. et al. Intrinsic functional architecture in the anaesthetized monkey brain. Nature. 2007;447:83-6
89. Lu H, Zuo Y, Gu H, Waltz JA, Zhan W, Scholl CA. et al. Synchronized delta oscillations correlate with the resting-state functional MRI signal. Proc Natl Acad Sci U S A. 2007;104:18265-9
90. Pan W-J, Billings JCW, Grooms JK, Shakil S, Keilholz SD. Considerations for Resting State Functional MRI and Functional Connectivity Studies in Rodents. Front Neurosci. 2015:9
91. Zhou IY, Liang Y-X, Chan RW, Gao PP, Cheng JS, Hu Y. et al. Brain resting-state functional MRI connectivity: Morphological foundation and plasticity. NeuroImage. 2014;84:1-10
92. Liang Z, King J, Zhang N. Intrinsic Organization of the Anesthetized Brain. J Neurosci. 2012;32:10183-91
93. Guilfoyle DN, Gerum SV, Sanchez JL, Balla A, Sershen H, Javitt DC. et al. Functional connectivity fMRI in mouse brain at 7 T using isoflurane. J Neurosci Methods. 2013;214:144-8
94. Wiesmann M, Zerbi V, Jansen D, Haast R, Lütjohann D, Broersen LM. et al. A Dietary Treatment Improves Cerebral Blood Flow and Brain Connectivity in Aging apoE4 Mice. Neural Plast. 2016;2016:15
95. Zerbi V, Wiesmann M, Emmerzaal TL, Jansen D, Van Beek M, Mutsaers MPC. et al. Resting-State Functional Connectivity Changes in Aging apoE4 and apoE-KO Mice. J Neurosci. 2014;34:13963-75
96. Garthe A, Behr J, Kempermann G. Adult-generated hippocampal neurons allow the flexible use of spatially precise learning strategies. PLoS One. 2009;4:e5464
97. Wiesmann M, Jansen D, Zerbi V, Broersen LM, Garthe A, Kiliaan AJ. Improved spatial learning strategy and memory in aged Alzheimer AbetaPPswe/PS1dE9 mice on a multi-nutrient diet. J Alzheimers Dis. 2013;37:233-45
98. Xiao Q, Shi R, Yang W, Zou Y, Du Y, Zhang M. et al. Time-Dependent Increase of Chitinase1 in APP/PS1 Double Transgenic Mice. Neurochem Res. 2016;41:1604-11
99. Volianskis A, Kostner R, Molgaard M, Hass S, Jensen MS. Episodic memory deficits are not related to altered glutamatergic synaptic transmission and plasticity in the CA1 hippocampus of the APPswe/PS1deltaE9-deleted transgenic mice model of ss-amyloidosis. Neurobiol Aging. 2010;31:1173-87
100. Lalonde R, Kim HD, Maxwell JA, Fukuchi K. Exploratory activity and spatial learning in 12-month-old APP(695)SWE/co+PS1/DeltaE9 mice with amyloid plaques. Neurosci Lett. 2005;390:87-92
101. Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD. et al. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2010;35:870-80
102. Webster SJ, Bachstetter AD, Van Eldik LJ. Comprehensive behavioral characterization of an APP/PS-1 double knock-in mouse model of Alzheimer's disease. Alzheimers Res Ther. 2013;5:28
103. Lalonde R, Fukuchi K, Strazielle C. APP transgenic mice for modelling behavioural and psychological symptoms of dementia (BPSD). Neuroscience & Biobehavioral Reviews. 2012;36:1357-75
104. Lalonde R, Kim HD, Fukuchi K. Exploratory activity, anxiety, and motor coordination in bigenic APPswe + PS1/ΔE9 mice. Neuroscience Letters. 2004;369:156-61
105. Lalonde R, Kim HD, Maxwell JA, Fukuchi K. Exploratory activity and spatial learning in 12-month-old APP695SWE/co + PS1/ΔE9 mice with amyloid plaques. Neuroscience Letters. 2005;390:87-92
106. Ferretti L, McCurry SM, Logsdon R, Gibbons L, Teri L. Anxiety and Alzheimer's Disease. Journal of Geriatric Psychiatry and Neurology. 2001;14:52-8
107. Teri L, Ferretti LE, Gibbons LE, Logsdon RG, McCurry SM, Kukull WA. et al. Anxiety in Alzheimer's Disease: Prevalence and Comorbidity. The Journals of Gerontology Series A: Biological Sciences and Medical Sciences. 1999;54:M348-M52
108. Marvar PJ, Vinh A, Thabet S, Lob HE, Geem D, Ressler KJ. et al. T lymphocytes and vascular inflammation contribute to stress-dependent hypertension. Biol Psychiatry. 2012;71:774-82
109. Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, Zaltsman A. et al. Chronic variable stress activates hematopoietic stem cells. Nat Med. 2014;20:754-8
110. Pugh PL, Richardson JC, Bate ST, Upton N, Sunter D. Non-cognitive behaviours in an APP/PS1 transgenic model of Alzheimer's disease. Behav Brain Res. 2007;178:18-28
111. Ermini FV, Grathwohl S, Radde R, Yamaguchi M, Staufenbiel M, Palmer TD. et al. Neurogenesis and alterations of neural stem cells in mouse models of cerebral amyloidosis. Am J Pathol. 2008;172:1520-8
112. Schwab C, Klegeris A, McGeer PL. Inflammation in transgenic mouse models of neurodegenerative disorders. Biochim Biophys Acta. 2010;1802:889-902
113. Wright AL, Zinn R, Hohensinn B, Konen LM, Beynon SB, Tan RP. et al. Neuroinflammation and neuronal loss precede Abeta plaque deposition in the hAPP-J20 mouse model of Alzheimer's disease. PLoS One. 2013;8:e59586
114. Minogue AM, Jones RS, Kelly RJ, McDonald CL, Connor TJ, Lynch MA. Age-associated dysregulation of microglial activation is coupled with enhanced blood-brain barrier permeability and pathology in APP/PS1 mice. Neurobiol Aging. 2014;35:1442-52
115. Baron R, Babcock AA, Nemirovsky A, Finsen B, Monsonego A. Accelerated microglial pathology is associated with Abeta plaques in mouse models of Alzheimer's disease. Aging cell. 2014;13:584-95
116. Cavanagh C, Colby-Milley J, Bouvier D, Farso M, Chabot JG, Quirion R. et al. betaCTF-correlated burst of hippocampal TNFalpha occurs at a very early, pre-plaque stage in the TgCRND8 mouse model of Alzheimer's disease. J Alzheimers Dis. 2013;36:233-8
117. McGeer PL, McGeer EG. Targeting microglia for the treatment of Alzheimer's disease. Expert Opin Ther Targets. 2015;19:497-506
118. Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun. 1996;224:855-62
119. Ahmed Z, Shaw G, Sharma VP, Yang C, McGowan E, Dickson DW. Actin-binding Proteins Coronin-1a and IBA-1 are Effective Microglial Markers for Immunohistochemistry. J Histochem Cytochem. 2007;55:687-700
120. Streit WJ, Braak H, Xue Q-S, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol. 2009;118:475-85
121. Hooijmans CR, Graven C, Dederen PJ, Tanila H, van Groen T, Kiliaan AJ. Amyloid beta deposition is related to decreased glucose transporter-1 levels and hippocampal atrophy in brains of aged APP/PS1 mice. Brain Res. 2007;1181:93-103
122. Kalmijn S, Feskens EJ, Launer LJ, Kromhout D. Polyunsaturated fatty acids, antioxidants, and cognitive function in very old men. Am J Epidemiol. 1997;145:33-41
123. Cooper JL. Dietary lipids in the aetiology of Alzheimer's disease: implications for therapy. Drugs Aging. 2003;20:399-418
124. Olde Rikkert MG, Verhey FR, Sijben JW, Bouwman FH, Dautzenberg PL, Lansink M. et al. Differences in nutritional status between very mild Alzheimer's disease patients and healthy controls. J Alzheimers Dis. 2014;41:261-71
125. Amtul Z, Uhrig M, Wang L, Rozmahel RF, Beyreuther K. Detrimental effects of arachidonic acid and its metabolites in cellular and mouse models of Alzheimer's disease: structural insight. Neurobiol Aging. 2012;33:831.e21-31
126. Fabelo N, Martin V, Marin R, Santpere G, Aso E, Ferrer I. et al. Evidence for premature lipid raft aging in APP/PS1 double-transgenic mice, a model of familial Alzheimer disease. J Neuropathol Exp Neurol. 2012;71:868-81
127. Perez SE, Berg BM, Moore KA, He B, Counts SE, Fritz JJ. et al. DHA diet reduces AD pathology in young APPswe/PS1 Delta E9 transgenic mice: possible gender effects. Journal of neuroscience research. 2010;88:1026-40
128. Park IH, Hwang EM, Hong HS, Boo JH, Oh SS, Lee J. et al. Lovastatin enhances Abeta production and senile plaque deposition in female Tg2576 mice. Neurobiol Aging. 2003;24:637-43
129. Vance JE. Dysregulation of cholesterol balance in the brain: contribution to neurodegenerative diseases. Dis Model Mech. 2012;5:746-55
130. Cifuentes D, Poittevin M, Dere E, Broqueres-You D, Bonnin P, Benessiano J. et al. Hypertension accelerates the progression of Alzheimer-like pathology in a mouse model of the disease. Hypertension. 2015;65:218-24
131. Ordonez-Gutierrez L, Anton M, Wandosell F. Peripheral amyloid levels present gender differences associated with aging in AbetaPP/PS1 mice. J Alzheimers Dis. 2015;44:1063-8
132. Wang J, Tanila H, Puolivali J, Kadish I, van Groen T. Gender differences in the amount and deposition of amyloidbeta in APPswe and PS1 double transgenic mice. Neurobiol Dis. 2003;14:318-27
Author contact
![]() Corresponding author: Amanda J Kiliaan, PhD., Associate Professor Dpt. of Anatomy, Preclinical Imaging Center PRIME, Donders Institute for Brain Cognition & Behavior, Radboud University Medical Center, Geert Grooteplein 21N, 6525 EZ Nijmegen, The Netherlands Phone +31 24 3614378, Fax +31 24 3613789 E-mail: Amanda.Kiliaannl
Corresponding author: Amanda J Kiliaan, PhD., Associate Professor Dpt. of Anatomy, Preclinical Imaging Center PRIME, Donders Institute for Brain Cognition & Behavior, Radboud University Medical Center, Geert Grooteplein 21N, 6525 EZ Nijmegen, The Netherlands Phone +31 24 3614378, Fax +31 24 3613789 E-mail: Amanda.Kiliaannl
Citation styles
APA
Wiesmann, M., Zerbi, V., Jansen, D., Lütjohann, D., Veltien, A., Heerschap, A., Kiliaan, A.J. (2017). Hypertension, cerebrovascular impairment, and cognitive decline in aged AβPP/PS1 mice. Theranostics, 7(5), 1277-1289. https://doi.org/10.7150/thno.18509.
ACS
Wiesmann, M.; Zerbi, V.; Jansen, D.; Lütjohann, D.; Veltien, A.; Heerschap, A.; Kiliaan, A.J. Hypertension, cerebrovascular impairment, and cognitive decline in aged AβPP/PS1 mice. Theranostics 2017, 7 (5), 1277-1289. DOI: 10.7150/thno.18509.
NLM
Wiesmann M, Zerbi V, Jansen D, Lütjohann D, Veltien A, Heerschap A, Kiliaan AJ. Hypertension, cerebrovascular impairment, and cognitive decline in aged AβPP/PS1 mice. Theranostics 2017; 7(5):1277-1289. doi:10.7150/thno.18509. https://www.thno.org/v07p1277.htm
CSE
Wiesmann M, Zerbi V, Jansen D, Lütjohann D, Veltien A, Heerschap A, Kiliaan AJ. 2017. Hypertension, cerebrovascular impairment, and cognitive decline in aged AβPP/PS1 mice. Theranostics. 7(5):1277-1289.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.