Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Author Biography
Introduction
Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References
Author Biography
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(5):1225-1244. doi:10.7150/thno.18181 This issue Cite
Research Paper
Recipient Glycemic Micro-environments Govern Therapeutic Effects of Mesenchymal Stem Cell Infusion on Osteopenia
Bing-Dong Sui1,3*, Cheng-Hu Hu1,2*, Chen-Xi Zheng1,3, Yi Shuai1,3, Xiao-Ning He1,2, Ping-Ping Gao1,2, Pan Zhao1,2, Meng Li1, Xin-Yi Zhang1,3, Tao He1,3, Kun Xuan1,3, Yan Jin1,3 ![]()
1. State Key Laboratory of Military Stomatology & National Clinical Research Center for Oral Diseases & Shaanxi International Joint Research Center for Oral Diseases, Center for Tissue Engineering, School of Stomatology, Fourth Military Medical University, Xi'an, Shaanxi, 710032, China;
2. Xi'an Institute of Tissue Engineering and Regenerative Medicine, Xi'an, Shaanxi, 710032, China;
3. Research and Development Center for Tissue Engineering, Fourth Military Medical University, Xi'an, Shaanxi 710032, China.
* Bing-Dong Sui and Cheng-Hu Hu contributed equally to this work.
Received 2016-11-2; Accepted 2016-12-24; Published 2017-3-6
Citation:
Sui BD, Hu CH, Zheng CX, Shuai Y, He XN, Gao PP, Zhao P, Li M, Zhang XY, He T, Xuan K, Jin Y. Recipient Glycemic Micro-environments Govern Therapeutic Effects of Mesenchymal Stem Cell Infusion on Osteopenia. Theranostics 2017; 7(5):1225-1244. doi:10.7150/thno.18181. https://www.thno.org/v07p1225.htm
Other stylesAbstract

Therapeutic effects of mesenchymal stem cell (MSC) infusion have been revealed in various human disorders, but impacts of diseased micro-environments are only beginning to be noticed. Donor diabetic hyperglycemia is reported to impair therapeutic efficacy of stem cells. However, whether recipient diabetic condition also affects MSC-mediated therapy is unknown. We and others have previously shown that MSC infusion could cure osteopenia, particularly in ovariectomized (OVX) mice. Here, we discovered impaired MSC therapeutic effects on osteopenia in recipient type 1 diabetes (T1D). Through intensive glycemic control by daily insulin treatments, therapeutic effects of MSCs on osteopenia were maintained. Interestingly, by only transiently restoration of recipient euglycemia using single insulin injection, MSC infusion could also rescue T1D-induced osteopenia. Conversely, under recipient hyperglycemia induced by glucose injection in OVX mice, MSC-mediated therapeutic effects on osteopenia were diminished. Mechanistically, recipient hyperglycemic micro-environments reduce anti-inflammatory capacity of MSCs in osteoporotic therapy through suppressing MSC interaction with T cells via the Adenosine monophosphate-activated protein kinase (AMPK) pathway. We further revealed in diabetic micro-environments, double infusion of MSCs ameliorated osteopenia by anti-inflammation, attributed to the first transplanted MSCs which normalized the recipient glucose homeostasis. Collectively, our findings uncover a previously unrecognized role of recipient glycemic conditions controlling MSC-mediated therapy, and unravel that fulfillment of potent therapeutic effects of MSCs requires tight control of recipient micro-environments.
Keywords: Mesenchymal stem cells, cell therapy, recipient, glycemic micro-environment, osteopenia, anti-inflammation.
Introduction
Systemic infusion of mesenchymal stem cells (also known as mesenchymal stromal cells, MSCs) has been extensively investigated to treat a variety of human disorders [1-6], through either circulatory immunomodulation/anti-inflammation [7] or local regenerative effects via homing to lesions post infusion [8, 9]. Notably, their therapeutic efficacy was not always achieved [10-12]; in particular cases, MSC transplantation was conditionally beneficial in certain recipient settings [13-17]. It has been reported that recipient inflammatory cytokines play an important role in triggering immunosuppressive capacity of donor MSCs [18, 19]. However, given that degenerative and immunological disorders are often complicated with combined diseased factors such as metabolic alterations [20-22], further elucidating the impacts of recipient micro-environments on therapeutic effects of MSCs and the underlying mechanisms may promote utility of these cells.
Recently, the influences of diseased metabolic micro-environments on MSC function and MSC-mediated therapy have just begun to be noticed. We discovered that diabetic milieu provokes dysfunction of endogenous MSCs in diabetes mellitus-induced osteopenia [23]. Furthermore, donor diabetic hyperglycemic condition has been reported to adversely affect stem cell-mediated therapeutic effects in cardiac repair and angiogenesis [24]. With regard to exogenous MSC application, although pre-clinical and clinical studies have revealed potential benefits of MSC infusion on diabetic hyperglycemia [25, 26], therapeutic effects of MSCs on diabetic complications are limited, which might be attributed to the restricted donor MSC function in in vivo diseased conditions [27, 28]. Nevertheless, detailed investigations of how the recipient diabetic micro-environment affects the therapeutic effects of donor MSCs are still lacking.
We and others have previously established the methodology of using systemic MSC transplantation to cure osteopenias in mice [7, 8, 29, 30]. Current pre-clinical studies have confirmed therapeutic effects of MSCs in ovariectomy (OVX)-induced bone loss [7, 9, 30-33], in which estrogen deficiency exerts pathologic effects through immunologically mediated mechanisms [34], as shown by our findings that administration of Tumor necrosis factor-alpha (TNF-α) neutralizing antibody could rescue OVX-induced skeletal defects [35, 36]. Furthermore, immunomodulatory/anti-inflammatory capacity of MSCs has been recognized as an underlying principle for MSC-mediated therapy in OVX-induced osteopenia [7]. Type 1 diabetes (T1D) is also recognized as a chronic autoimmune disorder characterized by hypoinsulinemia and hyperglycemia [37-39], which develops severe bone loss with endogenous MSC impairments [23, 40, 41]. Whether diseased micro-environments in T1D inhibit the therapeutic effects and anti-inflammatory capacity of exogenous infused MSCs in osteopenia are unknown.
In this study, to investigate the potential effects of recipient diabetic micro-environments on donor MSC therapy, we firstly investigated and compared therapeutic effects of MSC infusion on osteopenia respectively induced by OVX and T1D. We discovered that recipient diabetic milieu impaired therapeutic effects of MSC infusion on osteopenia. We further demonstrated that therapeutic effects of donor MSCs were maintained under glycemic control in recipient T1D but were diminished in glucose injection-induced hyperglycemia in OVX mice. Mechanistically, we showed that hyperglycemic micro-environments reduce anti-inflammatory capacity of MSCs in osteoporotic therapy through suppressing MSC interaction with T cells via the Adenosine monophosphate-activated protein kinase (AMPK) pathway. We further revealed that in diabetic micro-environments, double infusion of MSCs ameliorated osteopenia by anti-inflammation, which was attributed to the first transplanted MSCs which normalized the recipient glucose homeostasis. Collectively, our findings uncover the role of recipient glycemic conditions controlling MSC-mediated therapy, and unravel that fulfillment of potent function of donor MSCs requires tight control of recipient micro-environment.
Methods
Animals
All experiments were approved by Fourth Military Medical University and were performed following the Guidelines of Intramural Animal Use and Care Committee of Fourth Military Medical University. Animal experiments were performed following the ARRIVE guidelines.
12-week-old female wild type (WT) C57BL/6 mice (weight, 20-22g) (Laboratory Animal Center, Fourth Military Medical University, China) and 12-week-old female green fluorescent protein (GFP)+/+ transgenic mice (weight, 20-22g) (C57BL/6 background, Fourth Military Medical University, China) were used, as stated before [8]. WT mice were randomly assigned to experimental groups as donor or recipient samples. GFP+/+ mice were selected as donor samples for MSC tracing. Investigators have been blinded to the sample group allocations. The mice were maintained with good ventilation and a 12-h light/dark cycle, and were kept feeding and drinking ad libitum before being sacrificed.
Osteoporotic modeling
Osteopenias induced by OVX and T1D were modeled according to previous studies [23, 25]. Briefly, for the OVX model and its Sham control, female mice underwent either a bilateral OVX or a Sham operation by the dorsal approach under general anesthesia. For the T1D model and its control (Ctrl), female mice accepted either 50 mg/kg/d multiple low dose of streptozotocin (STZ) (Sigma-Aldrich, USA) for 5 consecutive days (1 injection/d) dissolved in approximately 200-μL 0.1 M citrate buffer (pH 4.5) or equivalent citrate buffer through intraperitoneal injection. Intraperitoneal injections were performed via the right lower quadrant of the abdominal area, 1-cm away from the midabdominal line. Mice were kept at head-down position to ensure that intraperitoneally injected fluid was not accidentally placed in intestine.
Blood glucose quantification
The concentrations of non-fasting blood glucose (GLU) were measured every 3 d throughout the experiments and at indicated time points after insulin (INS) and GLU injections into each recipient mice. Blood GLU concentrations were quantified using a glucometer (Roche Bioproducts, Switzerland) following tail vein-puncture whole blood sampling [23]. Mice with over 250 mg/dL GLU in blood were classified as diabetic or hyperglycemic [25].
MSC isolation, verification and infusion
Isolation, culture and infusion of murine MSCs and MSCsGFP from bone marrow were as previously described [8, 23, 29]. Briefly, murine bone marrow cells were seeded, incubated overnight, and rinsed with phosphate buffer saline (PBS) to remove the non-adherent cells. The adherent cells were cultured with alpha-minimum essential medium supplemented with 20% fetal bovine serum, 2-mM L-glutamine, 100-U/mL penicillin, and 100-g/mL streptomycin (all from Invitrogen, USA) at 37℃ in a humidified atmosphere of 5% CO2. Primary MSC or MSCGFP colonies were applied for infusion after digestion with 0.25% trypsin (MP Biomedicals, USA) to yield appropriate number (2~3×106 MSCs or MSCsGFP could be harvested from one mouse). MSCs were then suspended in PBS at 5×106/mL, and 1×106 MSCs or MSCsGFP in 200-μL PBS or equivalent PBS were administered via caudal vein into each recipient mouse at indicated time points.
MSCs were verified according to previous reports [8, 23, 42]. For colony formation, 1st-passaged MSCs at a density of 1×104 cells/dish in 5-cm culture dishes were cultured for 14 d, fixed with 4% paraformaldehyde and stained with crystal violet (Sigma-Aldrich, USA) for colonies with over 50 cells. For cell morphological evaluation, 1st-passaged MSCs at a density of 5×105 cells/well in 6-well plates were cultured for 1 d, and the cell morphology was evaluated. For osteogenic differentiation, the seeded MSCs were further induced in media containing 100-μg/mL ascorbic acid (MP Biomedicals, USA), 2-mM β-glycerophosphate (Sigma-Aldrich, USA) and 10-nM dexamethasone (Sigma-Aldrich, USA) for 14 d, and alizarin red (Sigma-Aldrich, USA) staining was performed to determine the mineralization. For adipogenic differentiation, the seeded MSCs were induced in media containing 0.5-mM isobutylmethylxanthine (MP Biomedicals, USA), 0.5-mM dexamethasone and 60-mM indomethacin (MP Biomedicals, USA) for 14 d, and oil red O (Sigma-Aldrich, USA) staining was performed to determine the lipid droplet formation. The above photographs were all taken using an inverted optical microscope (CKX41; Olympus, Japan).
For flow cytometric analysis of the surface makers [8, 23, 42], primary MSC colonies were digested and suspended in PBS supplemented with 3% fetal bovine serum at 1×106 cells/mL. 2×105 cells/tube were added with 1-μL FITC-conjugated anti-mouse CD11b antibody, 1-μL PE-conjugated anti-mouse CD29 antibody, 1-μL PE-conjugated anti-mouse CD34 antibody, 1-μL PE-conjugated anti-mouse CD45 antibody, 1-μL PE-conjugated anti-mouse CD106 antibody, and 1-μL FITC-conjugated anti-mouse stem cell antigen-1 (Sca-1) antibody (all from Abcam, UK). Non-immune immunoglobulin of the same isotype was used as the negative control. MSCs were incubated in 4℃ for 30 min in dark, and then washed twice with PBS supplemented with 3% fetal bovine serum. The percentages of positively stained cells were determined with a flow cytometer (FACSAria; BD Biosciences, USA) equipped with the FACSDiva Version 6.1.3 software.
Evaluation of recipient diabetes on MSC therapy
WT mice were divided into 8 groups: the Sham group (n = 6), the OVX group (n = 6), the OVX+PBS group (n = 6), and the OVX+MSC group (n = 6); the Ctrl group (n = 6), the T1D group (n = 6), the T1D+PBS group (n = 6), and the T1D+MSC group (n = 6). Blood GLU levels were determined every 3 d till sacrifice. At D1, Sham/OVX operations and first citrate buffer/STZ injections were carried out. At D25, PBS or MSC infusion was performed according to previous reports on diabetes and osteopenia developments [8, 23, 25]. At D38 and D52 (respectively 16 d and 2 d prior to sacrifice), double calcein labeling were performed, as stated below. At D54, mice were sacrificed, and bone and blood were sampled.
Evaluation of recipient glycemic control on MSC therapy
WT T1D mice were divided into 4 groups: the T1D+Ctrl+PBS group (n = 6), the T1D+Ctrl+MSC group (n = 6), the T1D+INS therapy (INST)+PBS group (n = 6), and the T1D+INST+MSC group (n = 6). Blood GLU levels were determined every 3 d till sacrifice. From D1 to D5, 5 consecutive STZ injections were carried out. INS glargine (Lantus; Sanofi-Aventis, France) was diluted in the recommended buffer: distilled water (pH 4.0) containing 3-mg/L Zn2+ and 1.7-g/L glycerol. From D25 to D54, INS glargine or equivalent buffer (Ctrl) was subcutaneously applied at 20-U/kg INS daily (about 0.4~0.5 U per mouse), corresponding to the maximum tolerable dose used in early animal studies [43-45]. INS doses were also finely adjusted to maintain blood GLU levels in the same range as the non-diabetic mice. At D25, at 30 min after the first buffer or INS treatment, PBS or MSC infusion was performed. At D38 and D52 (respectively 16 d and 2 d prior to sacrifice), double calcein labeling were performed. At D54, mice were sacrificed, and bone and blood were sampled.
Evaluation of recipient transient euglycemia on MSC therapy
WT T1D mice were divided into 4 groups: the T1D+Ctrl+PBS group (n = 6), the T1D+Ctrl+MSC group (n = 6), the T1D+INS+PBS group (n = 6), and the T1D+INS+MSC group (n = 6). Blood GLU levels were determined every 3 d till sacrifice and at indicated time points after a single INS injection. From D1 to D5, 5 consecutive STZ injections were carried out. INS glargine (Lantus; Sanofi-Aventis, France) was diluted in the buffer stated above. At D25, a single subcutaneous injection of INS glargine or equivalent buffer (Ctrl) was applied at 0.2-U/kg INS (about 0.004~0.005 U per mouse), corresponding to the dose used in human trials and based on the dose used in insulin tolerance tests [44, 46, 47]. The INS dose was also adjusted to transiently restore euglycemia in diabetic mice based on our preliminary tests. At D25, at 30 min after buffer or INS injection, PBS or MSC infusion was performed under euglycemia. At D38 and D52 (respectively 16 d and 2 d prior to sacrifice), double calcein labeling were performed. At D54, mice were sacrificed, and bone and blood were sampled.
Evaluation of recipient transient hyperglycemia on MSC therapy
WT OVX mice were divided into 4 groups: the OVX+Ctrl+PBS group (n = 6), the OVX+Ctrl+MSC group (n = 6), the OVX+GLU+PBS group (n = 6), and the OVX+GLU+MSC group (n = 6). Blood GLU levels were determined every 3 d till sacrifice and at indicated time points after a single GLU injection. At D1, OVX operations were carried out. GLU (Sigma-Aldrich, USA) was diluted in distilled water. At D25, a single intraperitoneal injection of GLU or equivalent distilled water (Ctrl) was applied at 2.0-g/kg GLU, corresponding to the dose used in glucose tolerance tests [47]. The GLU dose was also adjusted to induce transiently hyperglycemia in non-diabetic mice based on our preliminary tests. At D25, at 15 min after distilled water or GLU injection, PBS or MSC infusion was performed under hyperglycemia. At D38 and D52 (respectively 16 d and 2 d prior to sacrifice), double calcein labeling were performed. At D54, mice were sacrificed, and bone and blood were sampled.
Evaluation of therapeutic effects of double MSC infusion
WT T1D mice were divided into 3 groups: the T1D+PBS group (n = 6), the T1D+single MSC (sMSC) group (n = 6), and the T1D+double MSC (dMSC) group (n = 6). Blood GLU levels were determined every 3 d till sacrifice. From D1 to D5, 5 consecutive STZ injections were carried out. At D25, PBS or the 1st MSC infusion was performed in all groups. At D39 (14-d later), the 2nd MSC infusion was performed in only the T1D+dMSC group. At D50 and D64 (respectively 16 d and 2 d prior to sacrifice), double calcein labeling were performed. At D66, mice were sacrificed, and bone and blood were sampled.
Micro-computed tomography (Micro-CT) analysis
For trabecular and cortical bone mass and microarchitecture evaluation, a desktop micro-CT system (eXplore Locus SP; GE Healthcare, USA) was employed, as previously documented [8, 23]. At sacrifice, the left femora were removed, fixed overnight in 4% paraformaldehyde, and prepared into 1-mm blocks with the distal femoral metaphysis included. The specimens were scanned at a resolution of 8 μm, a voltage of 80 kV, and a current of 80 μA. Trabecular bone data were obtained at a region of interest in the distal metaphysis, from 0.3 mm to 0.8 mm away from the epiphysis. Cortical region of interest was defined in the midshaft, from 3.3 mm to 3.8 mm away from the epiphysis. Data were analyzed with the Micview V2.1.2 software, and the quantification was performed using parameters of bone volume per tissue volume (BV/TV), bone mineral density (BMD), trabecular thickness (Tb.Th), trabecular number (Tb.N), trabecular separation (Tb.Sp) and cortical thickness (Ct.Th) [8, 48].
Bone histomorphometric analyses
Bone formation rates were examined by double calcein labeling according to previous studies [8, 23]. At 16 d and 2 d prior to sacrifice, mice received intraperitoneal injections of 20 mg/kg calcein (Sigma-Aldrich, USA). Calcein was dissolved at 2 mg/mL in PBS supplemented with 1 mg/mL NaHCO3 (Sigma-Aldrich, USA), and was injected at 10 μL/g each time away from light. Intraperitoneal injections were performed via the right lower quadrant of the abdominal area, 1-cm away from the midabdominal line. Mice were kept at head-down position to ensure that intraperitoneally injected fluid was not accidentally placed in intestine. At sacrifice, right femora were isolated, fixed in 80% ethanol, and embedded in methyl methacrylate. The specimens were sagittally sectioned into 30-μm sections using a hard tissue slicing machine (SP1600; Leica, Germany) away from light. Both double-labeled and single-labeled cortical endosteum surfaces were evaluated by a fluorescence microscope (STP6000; Leica, Germany) with an excitation wavelength of 488 nm. Quantification was performed using the parameters of mineral apposition rate (MAR) and mineralized surface per bone surface (MS/BS) by the ImageJ 1.47 software. Bone formation rate (BFR) was calculated as MAR×MS/BS, according to previous studies [8, 23, 49].
Bone resorption rates were examined by tartrate resistant acid phosphotase (TRAP) staining, as stated before [7, 8]. At sacrifice, tibiae were isolated, fixed with 4% paraformaldehyde, decalcified with 10% ethylene diamine tetraacetic acid (pH, 7.2-7.4), and embedded in paraffin. 5-μm sagittal serial sections of proximal metaphyses were prepared (RM2125; Leica, Germany). TRAP staining was performed on the sections using a commercial kit according to the manufacturers' instructions (387-1A; Sigma-Aldrich, USA). Quantification was determined using the parameters of number of osteoclasts per bone surface (N.Oc/BS) and osteoclast surface per bone surface (Oc.S/BS) by the ImageJ 1.47 software [7, 8].
T-cell culture and co-culture assays with MSCs
As stated [7, 50], murine spleen cells derived from WT C57BL/6 mice were collected and treated with ACK lysis buffer (Lonza, Switzerland) to remove red blood cells. T cells were isolated and stimulated for 2 d with 3-μg/mL plate-bound anti-mouse CD3 antibody (eBioscience, USA) and 2-μg/mL soluble anti-mouse CD28 antibody (eBioscience, USA) in alpha-minimum essential medium supplemented with 20% fetal bovine serum, 2-mM L-glutamine, 100-U/mL penicillin, and 100-g/mL streptomycin (all from Invitrogen, USA) at 37℃ in a humidified atmosphere of 5% CO2. For low glucose (LG) treatment, 5-mM GLU (Invitrogen, USA) was added into culture media. For high glucose (HG) treatment, 25-mM GLU was added [24]. For mannitol (MA) (Invitrogen, USA) treatment as the osmotic control, 20-mM MA was added together with 5-mM GLU [51]. For advanced glycation end products (AGEs) or its solvent treatment, AGE-BSA (BioVision, USA) dissolved in PBS at a 200-μg/ml concentration of AGEs or equivalent PBS was added [52]. For the AMPK activator Metformin (MET) or its solvent treatment, metformin hydrochloride (Sigma-Aldrich, USA) dissolved in PBS at a 2-mM concentration of MET or equivalent PBS was added [53].
T-cell co-culture assays with MSCs were designed as 7 experiments based on previous methods [18]. For T-cell direct co-culture assay with MSCs in different micro-environments, 1st-passaged MSCs were seeded at 5×105 cells/well in 6-well plates for 24 h. Stimulated T cells at 5×106 cells/well were either cultured without MSCs (Blank, BL) or directly added onto MSCs in LG, MA or HG media, or normal media supplemented with PBS or 200-μg/ml AGEs for 6 h. For T-cell direct co-culture assay with MSCs after T-cell pre-treatment, stimulated T cells were pre-treated with HG for 6 h, and were then directly added onto seeded MSCs or not (BL) in HG, MA or LG media for 6 h. For T-cell direct co-culture assay with MSCs after MSC pre-treatment, 1st-passaged MSCs were seeded and pre-treated in LG, MA or HG media for 6 h, or normal media supplemented with PBS or 200-μg/ml AGEs for 6 h. In particular experiments, the MSCs were rescued by PBS or 2-mM MET for another 6 h after HG pre-treatment. Stimulated T cells were then either cultured without MSCs (BL) or directly added onto MSCs in normal media for 6 h. For T-cell direct co-culture assay with serial MSCs, stimulated T cells were firstly directly cultured with MSCs in HG media for 6 h, aspirated, and then cultured without the 2nd batch of MSCs (BL) or directly added onto the 2nd MSCs in HG, MA or LG media for 6 h. For T-cell direct co-culture assay with rescued MSCs, stimulated T cells were firstly directly cultured with MSCs in HG media for 6 h, aspirated, and then cultured without the rescued MSCs (BL) or directly added onto PBS- or 2-mM MET- rescued MSCs in normal media for another 6 h. For T-cell indirect co-culture assay with MSCs, MSCs were seeded in 6-well plates as the bottom of a Transwell system (Corning, USA). Stimulated T cells were either cultured without MSCs (BL) or added into the upper chamber of the Transwell system in LG media for 6 h. For T-cell treatments, stimulated T cells were cultured in LG, MA or HG media, or normal media supplemented with PBS or 200-μg/ml AGEs for 6 h. The media and T cells of the above experiments were collected respectively for TNF-α level and T-cell apoptosis determinations, as stated below.
Enzyme-linked immunosorbent assay (ELISA)
For serum examination, before necropsy, samples of the whole peripheral blood were collected from the retro-orbital venous plexus at 500 μL. For media examination, after T-cell culture and co-culture assays, samples of conditional media were collected at 200 μL. The serum and cell-free media were isolated by centrifuging at 3000 rpm 10 min followed by 12000 rpm 10 min to remove cell debris at 4℃. INS, glycated hemoglobin (HbA1c), hemoglobin, and the bone resorption marker Cross linked C-telopeptide of type 1 collagen (CTX-1) in serum, and inflammatory cytokine TNF-α in both serum and media were detected using murine ELISA kits according to the manufacturers' instructions (R&D Systems, USA) [7, 8].
Apoptosis analysis
After culture and co-culture assays, T cells were harvested and evaluated by FITC-conjugated Annexin V and PI double staining according to the manufacturer's instruction of Annexin V Apoptosis Detection Kit I (BD Biosciences, USA). After incubation, cell apoptosis was detected with a flow cytometer (CytoFLEX; Beckman Coulter, USA) equipped with the CXP 2.1 software. The percentages of early apoptotic (FITC+PI-) plus late apoptotic/necrotic (FITC+PI+) cells were expressed as apoptotic percentages, as stated [23, 54].
Detection of T cells in vivo
Before necropsy of OVX and T1D mice that accepted PBS or MSC infusion, samples of peripheral blood were collected from retro-orbital venous plexus and were treated with ACK lysis buffer (Lonza, Switzerland) to remove red blood cells. 1×106 cells were incubated with 1-μL FITC-labeled anti-mouse CD3 antibody (Abcam, UK) for 40 min on ice. After washing with PBS twice, the percentages of CD3+T cells in peripheral blood mononuclear cells (PBMNCs) were determined with a flow cytometer (CytoFLEX; Beckman Coulter) equipped with the CXP 2.1 software [7, 50].
MSC tracing
Donor MSCs in recipient peripheral blood was determined before and at 6 h, 12 h and 24 h post MSC or MSCGFP infusion (n = 3 for each time point). The existence of infused MSCs based on forward scattering and side scattering was evaluated with a flow cytometer (FACSAria; BD Biosciences, USA) equipped with the FACSDiva Version 6.1.3 software. For further tracing by GFP labeling, the percentages of GFP+ cells in PBMNCs were also determined by flow cytometry at indicated time points [8].
For detection of donor MSC inhabitation in recipient bone marrow, donor MSCsGFP were applied in recipient OVX and T1D models at D25 of the experimental period. At 24 h (n = 3 for each model) and 4 w (n = 3 for each model) post MSCGFP infusion, mice were sacrificed. Femora were isolated, fixed in 4% paraformaldehyde, cryoprotected with 30% sucrose, decalcified with 10% ethylene diamine tetraacetic acid (pH, 7.2-7.4), and embedded in the optimal cutting temperature compound. The specimens were snap-frozen and sectioned into 15-μm sagittal sections (CM1950; Leica, Germany). Sections were blocked with 5% bovine serum albumin (Sigma-Aldrich, USA) dissolved in PBS for 1 h in room temperature. Sections were then stained with a rabbit anti-GFP primary antibody (Cell Signaling Technology, USA) for 2 h in room temperature at a concentration of 1:100, followed by a goat anti-rabbit-FITC secondary antibody (Cell Signaling Technology, USA) for 30 min in room temperature at a concentration of 1:200, and counterstained with Hoechst, as stated [8, 25]. Non-immune immunoglobulin of the same isotype was used as the negative control. The sections were observed under a fluorescence microscopy (DP70; Olympus, Japan). The images were further analyzed using the ImageJ 1.47 software.
Quantitative real-time polymerase chain reaction (qRT-PCR) analysis
Total RNA was collected from MSCs after pre-treatment by LG, MA and HG followed by the co-culture assay with T cells. RNA was extracted by direct adding of Trizol Reagent (Takara, Tokyo, Japan) to the culture plates and purified by phenol-chloroform extraction. cDNA synthesis and PCR procedures were performed as described [23, 55]. The primer sequences of the genes detected in this study were listed in the Supplementary Material (Table S1). Relative expression level of each gene was obtained by normalizing against Gapdh abundance.
Western blot analysis
Western blot was performed as previously described [36, 55]. Whole-cell lysates of MSCs after pre-treatment by LG, MA and HG followed by the co-culture assay with T cells were prepared using the Cell Lysis Buffer (Beyotime, China). Proteins were extracted, loaded on sodium dodecyl sulfate-polyacrylamide gels, transferred to polyvinylidene fluoride membranes (Millipore, USA), and blocked with 5% bovine serum albumin (Sigma-Aldrich, USA) in PBS with 0.1% Tween for 2 h in room temperature. The membranes were incubated overnight at 4℃ with the following primary antibodies: a rabbit anti-mouse primary antibody at a concentration of 1:1000 for Ampk (Cell Signaling Technology, USA); a rabbit anti-mouse primary antibody at a concentration of 1:1000 for p-Ampk (Cell Signaling Technology, USA); a rabbit anti-mouse primary antibody at a concentration of 1:500 for Fas ligand (Fasl) (Santa Cruz Biotechnology, USA); and a rabbit anti-mouse primary antibody at a concentration of 1:4000 for Gapdh (Abcam, UK). The membranes were then incubated with peroxidase-conjugated goat anti-rabbit secondary antibodies (Boster, China) at a concentration of 1:40000 for 1 h in room temperature. The blotted bands were visualized using an enhanced chemiluminescence Kit (Amersham Biosciences, USA) and a gel imaging system (5500; Tanon, China). The grey values and folder changes of the bands were determined using the ImageJ 1.47 software.
Pancreas islet histology
Pancreas islets from Ctrl and T1D mice accepting PBS infusion and T1D mice accepting sMSC or dMSC infusion (n = 6 per group) were examined. At sacrifice, pancreas were rapidly isolated, fixed overnight with 4% paraformaldehyde, and embedded in paraffin. 5-μm serial sections were prepared (RM2125; Leica, Germany) and underwent H&E staining, according to previous reports [56, 57]. Quantification of pancreas islet area over total area was determined using the ImageJ 1.47 software.
Statistical analysis
All the results are represented as the mean ± standard deviation (SD). The data were analyzed using two-tailed Student's t tests (for two group comparisons) or one-way analysis of variance (ANOVA) followed by the Newman-Keuls post-hoc tests (for multiple group comparisons) in the GraphPad Prism 5.01 software. Values of P < 0.05 were considered to be statistically significant.
Results
Recipient diabetes mellitus impairs therapeutic effects of MSC infusion on osteopenia
To investigate the potential effects of recipient micro-environments on MSC therapy, we established osteopenic models with differential pathologies but similar phenotypes in mice [23]: OVX model, in which MSC infusion has been well-documented to prevent or restore bone loss [7, 9, 30-33], and T1D model induced by multiple low dose of STZ injections [23, 25]. We confirmed in the present study that these models differed in the glycemic environments: OVX mice showed euglycemia during the experimental period while T1D mice demonstrated hyperglycemia (Fig. S1A). Nevertheless, OVX and T1D mice exhibited similar activation of systemic inflammation, as shown by common elevation of serum TNF-α levels (Fig. S1B), loss of trabecular and cortical bone mass (Fig. S1C-I), impairments of bone formation rates (Fig. S1J-M) and stimulation of bone resorption rates (Fig. S1N-Q). Therefore, therapeutic effects of MSC infusion on osteopenias and inflammation were compared in these models.
Allogeneic MSCs used in this study were capable of forming colonies and differentiating into multilineage cells (Fig. S2A-D), and were in accordance with surface profiles currently recognized to represent MSCs (Fig. S2E) [8, 23, 42]. MSCs were infused at D25, at which time point hyperglycemia in T1D mice was stable around 400 mg/dL, as shown previously [25] and here by 3 consecutive quantifications (Fig. S1A), and substantial bone loss was reported to occur in both OVX and T1D mice [23]. Post infusion, flow cytometry demonstrated successful transplantation of donor MSCs into recipient circulation (Fig. S3A-E).
We discovered that MSC infusion restored bone mass in OVX mice, but not in T1D mice, as shown by micro-CT analysis (Fig. 1A). Corresponding parameters of BV/TV (Fig. 1B) and BMD (Fig. 1C) confirmed the rescue of trabecular bone loss in OVX but not in T1D model. Quantitative analysis of trabecular bone microarchitecture regarding Tb.Th (Fig. 1D), Tb.N (Fig. 1E) and Tb.Sp (Fig. 1F) further demonstrated therapeutic effects of MSCs on osteopenia in only OVX mice. The impaired MSC therapy in T1D-induced osteopenia was also revealed by cortical bone analysis on Ct.Th (Fig. 1G). To investigate whether the effects of MSC infusion on osteopenias were attributed to interventions on bone remodeling process, we examined bone formation and bone resorption rates in OVX and T1D mice post PBS or MSC transplantation. Double calcein labeling (Fig. 1H) and the corresponding parameters of MAR (Fig. 1I), MS/BS (Fig. 1J) and BFR (Fig. 1K) demonstrated that MSC therapy restored bone formation rates in only OVX mice. TRAP staining (Fig. 1L) and quantification of N.Oc/BS (Fig. 1M) and Oc.S/BS (Fig. 1N) further revealed that suppression of elevated bone resorption rates by MSCs was abolished in T1D mice, which was confirmed by the changes of serum bone resorption marker CTX-1 (Fig. 1O). These data collectively indicated that recipient diabetes mellitus impairs therapeutic effects of MSC infusion on osteopenia.
Figure 1
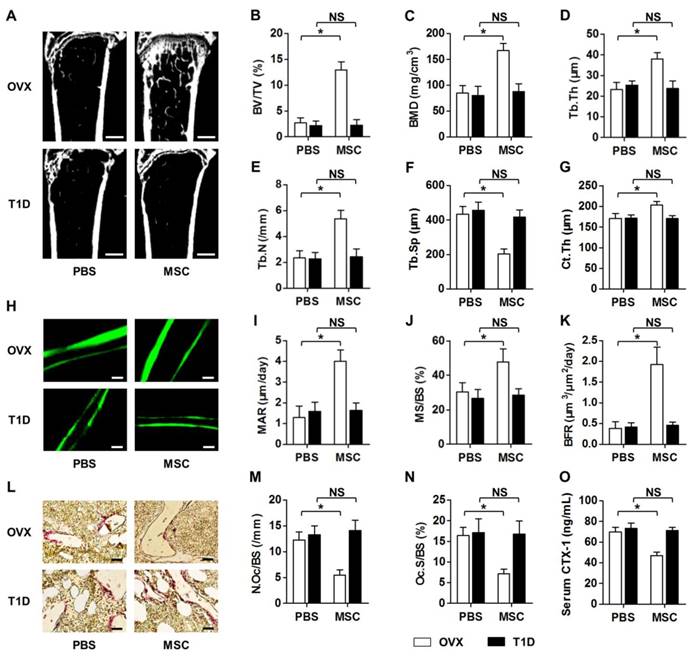
Recipient diabetes mellitus impairs therapeutic effects of MSC infusion on osteopenia. (A-G) Representative micro-CT images (A) and quantitative analysis of trabecular (B-F) and cortical (G) bone microarchitecture in the distal metaphyses of femora, from OVX and T1D mice accepting PBS or MSC infusion. Bars: 500 μm. (H-K) Representative images of calcein double labeling (H) with quantification (I-K) of bone formation rates in distal femora. Bars: 50 μm. (L-N) Representative images of TRAP staining (red) (L) with parameters (M, N) of bone resorption rates in distal femora. Bars: 25 μm. (O) ELISA analysis of bone resorption marker in serum. n = 6 per group. Data represents mean ± SD. *P < 0.05; NS, not significant (P > 0.05). Data were analyzed using ANOVA followed by Newman-Keuls post-hoc tests.
Glycemic control in recipient diabetes maintains therapeutic effects of MSC transplantation on osteopenia
We next examined whether treatment of recipient diabetes by INST could maintain therapeutic effects of donor MSCs on osteopenia. According to previous studies, glycemic control in T1D model was performed by daily subcutaneous application of approximately 20-U/kg INS, which was the maximum tolerable dose used in early animal studies [43-45]. We confirmed that INST rescued hyperglycemia and maintained euglycemia in T1D mice (Fig. 2A). INST also reduced the percentages of HbA1c in T1D model (Fig. 2B). However, INST did not restore bone mass in T1D during the experimental period (Fig. 2C-I), nor did it recover the normal balance between bone formation (Fig. 2J-M) and bone resorption rates (Fig. 2N-Q). Furthermore, despite that MSC infusion could restore normal glycemia and HbA1c percentages both with and without INST (Fig. 2A, B) as reported previously [25], MSC infusion alone could not restore bone mass and bone remodeling process without INST (Fig. 2C-Q), consistent with data in Fig. 1.
Nevertheless, under glycemic control, we discovered that MSC infusion exerted therapeutic effects on osteopenia, as shown by improved bone mass (Fig. 2C) and strengthened trabecular (Fig. 2C-H) and cortical (Fig. 2I) bone microarchitecture. These effects were due to increased bone formation rates (Fig. 2C-I) and decreased bone resorption rates (Fig. 2N-Q) after MSC infusion under glycemic control. These results suggested that glycemic control in recipient diabetes maintains therapeutic effects of MSC transplantation on osteopenia.
Figure 2
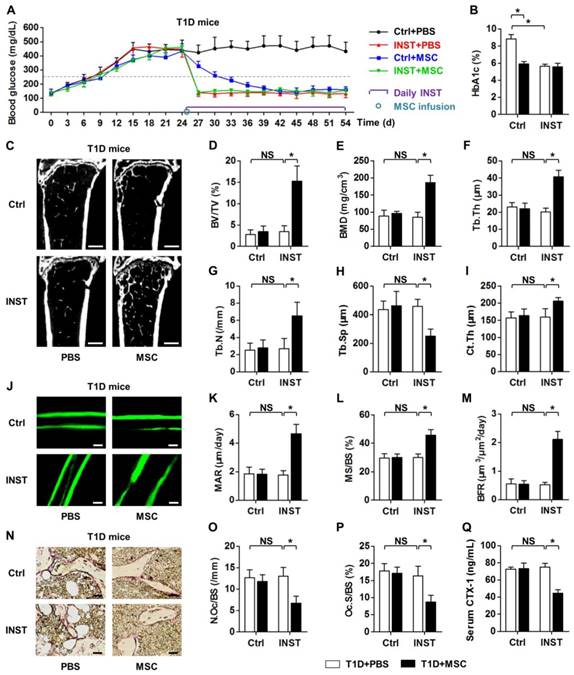
Glycemic control in recipient diabetes maintains therapeutic effects of MSC transplantation on osteopenia. (A) Non-fasting blood glucose levels of T1D mice with buffer (Ctrl) or INST treatments and PBS or MSC infusion. INS was applied daily at 20 U/kg from D25 to D54. MSCs were infused at D25. Grey dashed line indicates diabetic criterion of 250 mg/dL. (B) ELISA analysis of HbA1c percentages after sacrifice at D54. (C-I) Representative micro-CT images (C) and quantification of trabecular (D-H) and cortical (I) bone mass in distal femora, from T1D mice accepting PBS or MSC infusion in Ctrl and during INST. Bars: 500 μm. (J-M) Representative images of calcein double labeling (J) with quantification (K-M) of bone formation rates in distal femora. Bars: 50 μm. (N-P) Representative images of TRAP staining (red) (N) with parameters (O, P) of bone resorption rates in distal femora. Bars: 25 μm. (Q) ELISA analysis of bone resorption marker CTX-1 in serum. n = 6 per group. Data represents mean ± SD. *P < 0.05; NS, not significant (P > 0.05) (ANOVA followed by Newman-Keuls post-hoc tests).
Infusion of MSCs in recipient transient euglycemia rescues diabetes-induced bone loss
Next, we intended to explore whether the maintained therapeutic effects of donor MSCs on osteopenia were attributed to the euglycemic micro-environment restored by INS during infusion. We applied a single injection of INS at 0.2 U/kg, i.e. 1% dose of INS used for intensive glycemic control in mice. This dose was selected in correspondence to the dose used in human trials and based on the dose used in insulin tolerance tests [44, 46, 47]. We found that one-time injection of low-dose INS could transiently reduce hyperglycemia in T1D mice (Fig. 3A) without affecting the long-term glycemic condition (Fig. 3B). Conversely, MSC infusion could not affect glycemia in a short time (Fig. 3A) but could gradually reduce hyperglycemia (Fig. 3B), both with and without a single INS treatment. Importantly, when donor MSCs were infused under euglycemia at 30 min post INS injection, the therapeutic effects on T1D-induced osteopenia were maintained, as demonstrated by micro-CT analysis on both trabecular and cortical bone mass (Fig. 3C-I). The capability of donor MSCs to restore normal balance between bone formation and bone resorption rates was also restored when MSCs were infused under recipient euglycemia (Fig. 3J-Q). These findings revealed that normalizing the recipient glycemic micro-environments prior to MSC transplantation guaranteed the therapeutic effects of donor MSCs on diabetes-induced bone loss.
Figure 3
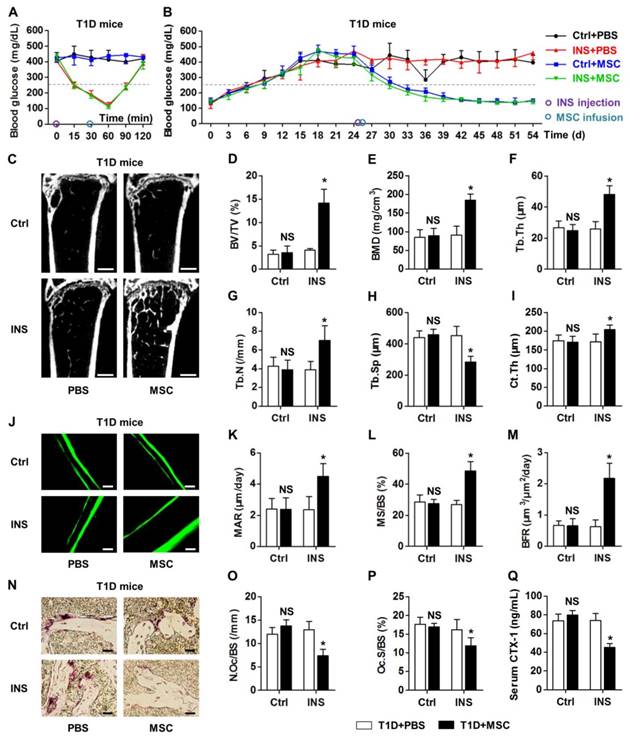
Infusion of MSCs under recipient transient euglycemia rescues diabetes-induced bone loss. (A, B) Non-fasting blood glucose levels of T1D mice which accepted single buffer (Ctrl) or INS injection at 0.2 U/kg at D25 followed by PBS or MSC infusion. MSCs were infused under euglycemia at 30 min after INS injection at D25. Grey line indicates diabetic criterion of 250 mg/dL. (C-I) Representative micro-CT images (C) and quantification of trabecular (D-H) and cortical (I) bone mass in distal femora, from T1D mice accepting PBS or MSC infusion in Ctrl and after INS injection. Bars: 500 μm. (J-M) Representative images of calcein double labeling (J) with quantification (K-M) of bone formation rates in distal femora. Bars: 50 μm. (N-P) Representative images of TRAP staining (red) (N) with parameters (O, P) of bone resorption rates in distal femora. Bars: 25 μm. (Q) ELISA analysis of bone resorption marker CTX-1 in serum. n = 6 per group. Data represents mean ± SD. *P < 0.05; NS, not significant (P > 0.05) (ANOVA followed by Newman-Keuls post-hoc tests).
Recipient hyperglycemia diminishes therapeutic effects of MSC infusion on osteopenia
To clarify whether recipient glycemic micro-environments govern therapeutic effects of MSC infusion on osteopenia, we used one-time injection of 2.0-g/kg GLU to transiently build hyperglycemia in OVX mice. The dose applied was correspondent to the dose used in glucose tolerance tests [47]. We revealed that a single GLU application mimicked hyperglycemic micro-environments from 15 min to 60 min post injection (Fig. 4A), without altering the euglycemia in OVX mice in the long run (Fig. 4B). Besides, MSC infusion did not affect the GLU-induced transient hyperglycemia (Fig. 4A) or the sustained euglycemic condition (Fig. 4B). Notably, GLU injection before MSC infusion impaired the therapeutic effects of donor MSCs on OVX-induced osteopenia, as demonstrated by micro-CT analysis on both trabecular and cortical bone parameters (Fig. 4C-I). Double calcein labeling and TRAP staining also confirmed inhibitory effects of GLU injection on MSC function to restore normal bone remodeling process (Fig. 4J-Q). These findings suggested that recipient hyperglycemia diminishes therapeutic effects of MSC infusion on osteopenia.
Figure 4
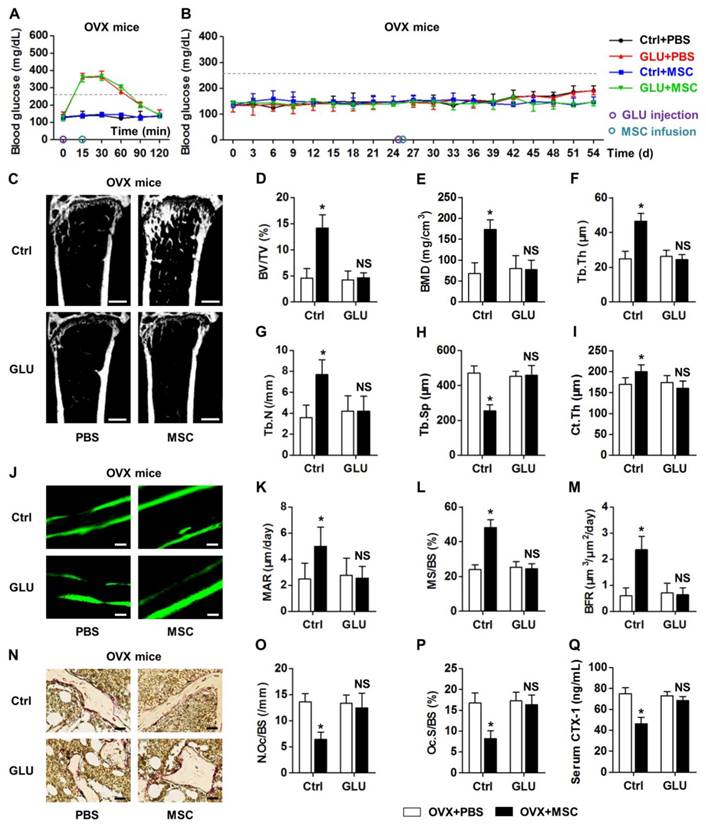
Recipient hyperglycemia diminishes therapeutic effects of MSC infusion on osteopenia. (A, B) Non-fasting blood glucose levels of OVX mice which accepted single distilled water (Ctrl) or 2.0-g/kg GLU injection at D25 followed by PBS or MSC infusion. MSCs were infused under hyperglycemia at 15 min after GLU injection at D25. Grey line indicates diabetic criterion of 250 mg/dL. (C-I) Representative micro-CT images (C) and quantification of trabecular (D-H) and cortical (I) bone mass in distal femora, from OVX mice accepting PBS or MSC infusion in Ctrl and after GLU injection. Bars: 500 μm. (J-M) Representative images of calcein double labeling (J) with quantification (K-M) of bone formation rates in distal femora. Bars: 50 μm. (N-P) Representative images of TRAP staining (red) (N) with parameters (O, P) of bone resorption rates in distal femora. Bars: 25 μm. (Q) ELISA analysis of bone resorption marker CTX-1 in serum. n = 6 per group. Data represents mean ± SD. *P < 0.05; NS, not significant (P > 0.05) (ANOVA followed by Newman-Keuls post-hoc tests).
Hyperglycemic micro-environments reduce anti-inflammatory rather than homing capacity of MSCs in osteoporotic therapy
According to reported data, systemically transplanted MSCs rescue bone loss through either circulatory immunomodulation/anti-inflammation [7] or local effects via homing to recipient bone marrow post infusion [8, 9]. In this study, we confirmed that infused MSCs existed in recipient circulation for less than 24 h by using MSCs from GFP-transgenic donor mice (Fig. S3E). We further confirmed that MSCsGFP homed to recipient bone marrow within 24 h post infusion and inhabited for 4 w (Fig. S3F). However, recipient diabetes mellitus did not impair homing and inhabitation of donor MSCsGFP (Fig. S3G), indicating that the abolished therapeutic effects of MSCs in T1D may not be attributed to the influenced local effects.
Therefore, we investigated whether impairments of systemic immunomodulatory/anti-inflammatory capabilities of donor MSCs occurred in recipient diabetes. T cells have been well-documented as the key targets of MSCs in immunomodulation in treating auto-immune diseases and in osteoporosis [7, 18, 50]. Flow cytometric analysis showed that MSC infusion suppressed the percentage of CD3+T cells in PBMNCs in OVX model, but did not reduce CD3+T-cell percentage in T1D model (Fig. S4A, B). We have previously reported that TNF-α was the key inflammatory cytokine to induce bone loss and endogenous MSC impairments [35, 36]. In this study, we further discovered that TNF-α elevated to similar levels in OVX and T1D mice (Fig. S1B), and that MSC infusion reduced TNF-α level only in OVX mice (Fig. 5A). These data suggested that anti-inflammatory capacity of donor MSCs was impaired in recipient diabetes.
To elucidate the underlying mechanisms, we applied T-cell assays with or without co-culture of MSCs in LG or HG micro-environments. MA was used as the osmotic control of GLU [51]. We mimicked the in vivo experiment in vitro, by directly added T cells onto MSCs in LG, MA or HG media for 6 h (Fig. 5B). The co-culture duration was based on flow cytometric results that most infused MSCs existed in recipient circulation within 6 h (Fig. S3B). ELISA detection on TNF-α level in conditional media showed that compared to the Blank (BL) group, MSC co-culture in LG exerted anti-inflammatory effects, which were abolished by HG but not MA treatments (Fig. 5C). Moreover, these changes were attributed to apoptosis of T cells induced by MSCs in LG but not in HG (Fig. S4C, D). These findings collectively indicated that hyperglycemic micro-environments reduce anti-inflammatory capacity of MSCs in osteoporotic therapy.
High glucose treatments suppress MSC interaction with T cells but not recipient T-cell responses to donor MSCs via the AMPK pathway
To further dissect the mechanisms, we investigated whether the reduced anti-inflammation of MSCs in recipient diabetes was due to altered responses of T cells or impaired function of MSCs. We discovered that INST did not restore the inflammatory conditions, while donor MSC infusion reduced recipient inflammation under both glycemic control and transient euglycemia (Fig. 5D, E). To mimic these in vivo experiments, T cells were pre-treated with HG for 6 h and directly added onto MSCs or not (BL) in HG, MA or LG media for 6 h (Fig. 5F). Data showed that lowering the GLU concentrations in micro-environments could maintain anti-inflammatory capacity of MSCs (Fig. 5G) and the capacity to induce apoptosis of T cells (Fig. S4E, F). Furthermore, these results revealed that pre-treatment in HG did not affect recipient T-cell responses to donor MSCs in LG micro-environment.
Next, we found that hyperglycemia in OVX mice diminished anti-inflammatory capacity of infused MSCs in vivo (Fig. 5H). After in vitro mimicking by MSC pre-treatments in LG, MA or HG media for 6 h (Fig. 5I), we further discovered that HG pre-treatment impaired anti-inflammatory capacity of MSCs when they were co-cultured with T cells thereafter (Fig. 5J). Also, the capacity to induce apoptosis of T cells was inhibited in HG-treated MSCs (Fig. S4G, H). Furthermore, when a 2nd batch of MSCs were applied to T cells in LG for another 6 h (Fig. 5K), TNF-α levels and T-cell numbers were reduced as expected (Fig. 5L; Fig. S4I, J). In addition, we found that anti-inflammation of MSCs was dependent on cell contact with T cells (Fig. S5A-D), and that T-cell survival and TNF-α secretion were not influenced by HG treatment (Fig. S5E-H). Besides, in vitro treatments of MSCs but not T cells with AGEs, which were accumulated with hyperglycemia and were recognized as a detrimental factor of bone and MSCs [41, 52, 58], also impaired anti-inflammatory capacity of MSCs (Fig. S6). These data collectively revealed that diabetic micro-environments inhibited anti-inflammatory capacity of MSCs by suppressing MSC interaction with T cells, but not T-cell responses.
To identify the molecular mechanisms, we screened in MSCs under LG, MA and HG micro-environments by qRT-PCR for the reported immunomodulatory factors [59]: Fasl [7, 50], Hepatocyte growth factor (Hgf) [60], Indoleamine 2,3-dioxigenase (Ido) [61], Interleukin-10 (Il10) [62], Inducible nitric oxide synthase (Inos) [18], Matrix metalloproteinase-2 (Mmp2) [63], Matrix metalloproteinase-9 (Mmp9) [63] and Transforming growth factor-beta 1 (Tgfβ1) [60, 64]. Given the results of T-cell co-culture assays, the expression of putative molecular mediator(s) in MSCs was supposed to be downregulated by HG, but not MA, compared to LG. We discovered that of these candidates, only mRNA expression of Fasl was inhibited by HG but not MA in MSCs, while Hgf and Mmp9 expressions were suppressed by both HG and MA (Fig. 5M). Fasl is documented as the key factor involved in MSC-induction of T-cell apoptosis dependent on cell contact [7, 50], underlying the results obtained in the above T-cell assays. The regulation of Fasl expression by HG in MSCs was further confirmed at the protein level (Fig. 5N).
Figure 5
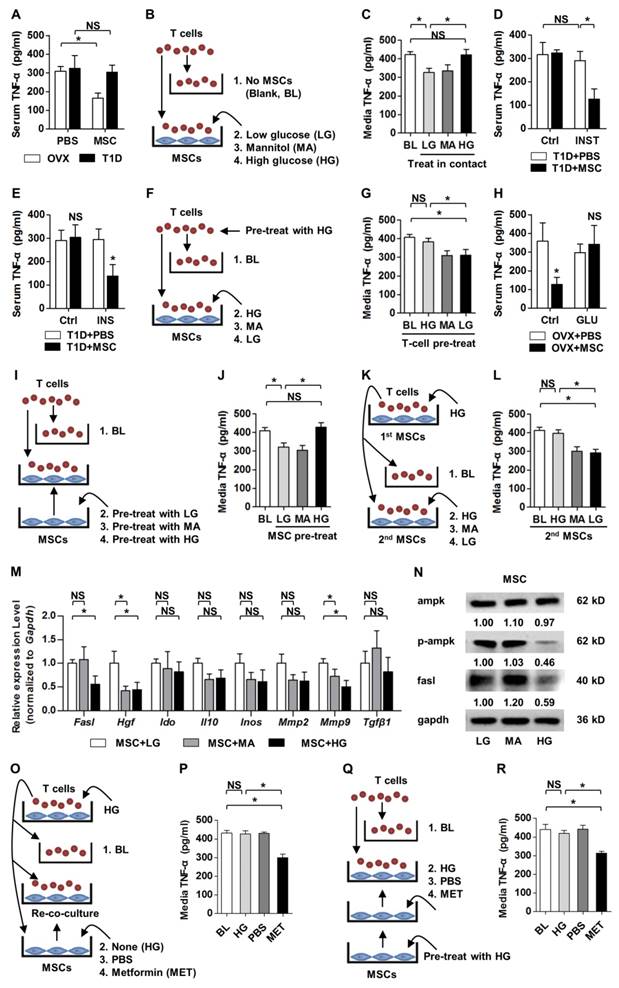
HG micro-environments reduce anti-inflammatory capacity of MSCs via the AMPK pathway. (A, D, E, H) ELISA analysis of inflammatory cytokine TNF-α in serum, respectively from mice analyzed in Figures 1-4. n = 6 per group. (B, F, I, K) Experimental designs to mimick in vivo phenomena in vitro and to clarify HG effects on anti-inflammatory capacity of MSCs or response of T cells. MA was used as the osmotic control. For LG treatment, 5-mM GLU was added. For HG treatment, 25-mM GLU was added. For MA treatment, 20-mM MA was added together with 5-mM GLU. (C, G, J, L) ELISA analysis of TNF-α in media from the respective experiments of B, F, I, K. n = 4 per group. (M) qRT-PCR analysis of mRNA expression profiles of immunomodulatory factors of MSCs after treatments by LG, MA and HG. n = 3 per group. (N) Western blot detection of protein expression levels in MSCs after treatments by LG, MA and HG. Numbers below bands indicate fold changes normalized to gapdh. (O, Q) Experimental designs to rescue HG-induced anti-inflammatory/immunomodulatory impairments of MSCs using the AMPK activator MET after (O) and before (Q) co-culture with T cells. MET was dissolved in PBS and was applied at 2 mM. (P, R) ELISA analysis of TNF-α in media from the respective experiments of O and Q. n = 4 per group. Data represents mean ± SD. *P < 0.05; NS, not significant (P > 0.05) (ANOVA followed by Newman-Keuls post-hoc tests).
For the affected signaling pathways, AMPK, an important intracellular energy sensor, is documented as a downstream effector of HG [21, 65, 66], and AMPK has been reported as a promoter of Fasl expression via stimulating Fasl transcription by phosphorylation [67]. In the present study, we examined and confirmed that HG treatment on MSCs suppressed the p-Ampk protein level without affecting the total Ampk protein level (Fig. 5N), indicating that hyperglycemic suppression of anti-inflammatory capacity of MSCs is possibly via the AMPK pathway. To verify the assumption, we applied a recognized AMPK activator, MET [53, 68], to rescue impaired function of MSCs after HG treatment. We found that, when applied on MSCs after MSC co-culture with T cells in HG micro-environment, MET rescued the immunomodulatory capacity of MSCs, as demonstrated by re-co-culture of MSCs with the HG-treated T cells (Fig. 5O, P; Fig. S4K, L). Furthermore, MET pre-treatment also restored anti-inflammatory capacity of MSCs and the capacity to induce apoptosis of T cells after HG application (Fig. 5Q, R; Fig. S4M, N). Together, the above data suggested that high glucose treatments suppressed anti-inflammation/immunomodulation of MSCs via the AMPK pathway.
A second infusion of MSCs in recipient glucose homeostasis ameliorates diabetic osteopenia by anti-inflammation
The above findings, particularly data from the T-cell co-culture assay with serial MSCs (Fig. 5K, L; Fig. S4I, J), inspired us to determine the therapeutic effects of serial MSC infusion on osteopenia in recipient diabetes mellitus. In consistence with previous reports, both sMSC and dMSC infusion gradually restored normal glycemic micro-environment in T1D mice (Fig. 6A) [25, 56, 69]. These effects were attributed to the regeneration of pancreas islets (Fig. S7A, B), which recovered the INS secretion (Fig. S7C) and reduced the HbA1c percentages in circulation (Fig. S7D). These results prompted us to investigate the therapeutic effects of a second infusion of MSCs in the skeletal complication of diabetes.
The time point regarding the 2nd MSC infusion was set at 14 d post the 1st MSC infusion, based on previous reports [69, 70] and our data in the euglycemia established by the 1st infused MSCs (Fig. 6A). ELISA analysis on serum TNF-α levels demonstrated that dMSC infusion significantly reduced systemic inflammation in T1D mice (Fig. 6B). Micro-CT analysis revealed that dMSC infusion restored both trabecular and cortical bone mass in T1D mice (Fig. 6C), as quantified by corresponding trabecular parameters of BV/TV (Fig. 6D), BMD (Fig. 6E), Tb.Th (Fig. 6F), Tb.N (Fig. 6G) and Tb.Sp (Fig. 6H), and the cortical parameter of Ct.Th (Fig. 6I). Further analysis on bone remodeling rates (Fig. 6J) confirmed the therapeutic effects of dMSC infusion on diabetic osteopenia, in that both bone formation rates (Fig. 6K-M) and bone resorption rates (Fig. 6N, O) were restored. Together, these findings uncovered that a second infusion of MSCs in recipient glucose homeostasis ameliorates diabetic osteopenia by anti-inflammation.
Discussion
Therapeutic effects of MSC infusion have been revealed in various human disorders [1-6], but the role and mechanisms underlying recipient micro-environmental impacts on donor MSC function are not fully understood. In this study, we discovered that recipient diabetes mellitus impaired therapeutic effects of MSC infusion on osteopenia, which was attributed to the hyperglycemic condition reducing anti-inflammatory capacity of MSCs through suppressing MSC interaction with T cells via the AMPK pathway. We have also revealed that restoration of recipient glucose homeostasis prior to MSC transplantation could promote the therapeutic effects of MSC infusion to ameliorate diabetic osteopenia by anti-inflammation. To our knowledge, this is the first study to uncover the role of recipient glycemic conditions controlling MSC-mediated therapy, and to unravel that fulfillment of potent function of donor MSCs requires normalization of recipient micro-environment.
Donor cell-recipient communications are pivotal to MSC-mediated tissue regeneration and therapy [8, 71], while the impacts of recipient comorbidities are only beginning to be noticed [13-17]. Previous findings have pointed out that recipient immunological components, particularly T lymphocytes and inflammatory cytokines TNF-α and Interferon-gamma, actively respond to the transplanted MSCs and initiate the immunomodulatory function of donor MSCs [18, 19, 71]. However, conflicting results still exist regarding therapeutic effects of MSCs on immune disorders [72]; specifically, MSC transplantation could be conditionally therapeutic depending on recipient micro-environmental alterations [14, 17], but detailed investigations of how the diseased recipient micro-milieu affects the donor MSC function are lacking. In this study, we for the first time uncovered that recipient diabetes mellitus impairs MSC-mediated therapy and immunomodulation, despite the onset of inflammation observed here and reported previously [37]. Given that hyperglycemia might combine with degenerative and immunological disorders [20, 22], check of recipient glycemic conditions prior to therapeutic use of MSCs is recommended based on our data. Furthermore, our results suggest that the infusion timing may be vital to MSC therapy, e.g. transplantation upon fasting might be beneficial to therapeutic efficacy of MSCs.
Figure 6
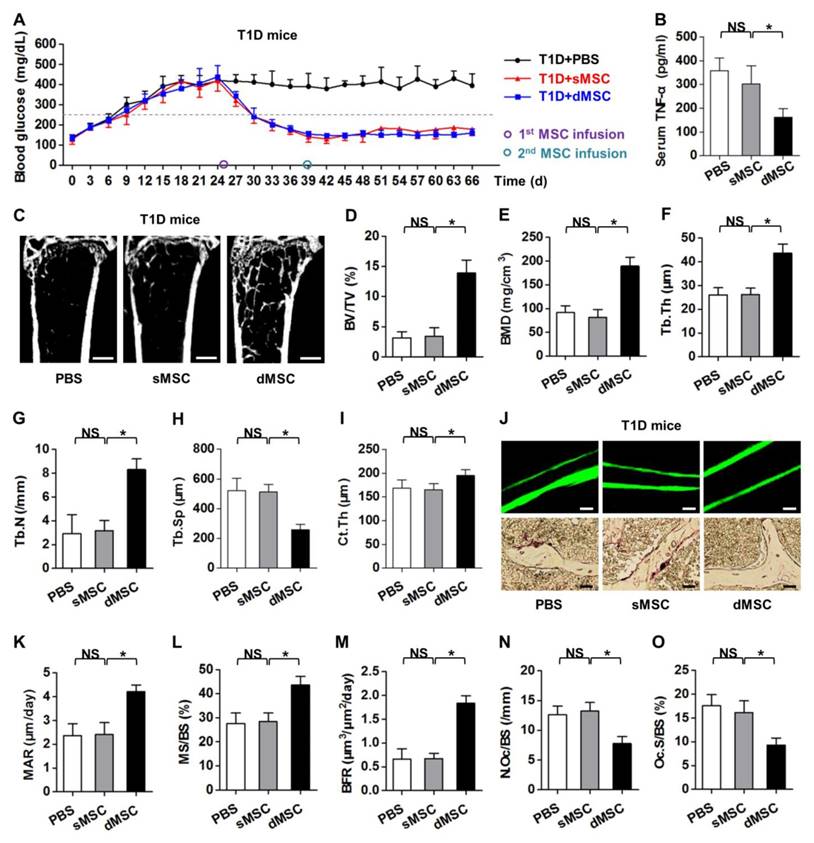
A second infusion of MSCs in recipient glucose homeostasis ameliorates diabetic osteopenia by anti-inflammation. (A) Non-fasting blood glucose levels of T1D mice after PBS, sMSC or dMSC injection. The 1st MSCs were infused at D25. The 2nd MSCs were infused 14 d later at D39. Grey dashed line indicates diabetic criterion of 250 mg/dL. (B) ELISA analysis of inflammatory cytokine TNF-α in serum, from T1D mice accepting PBS, sMSC or dMSC infusion. (C-I) Representative micro-CT images (C) and quantitative analysis of trabecular (D-H) and cortical (I) bone microarchitecture in the distal metaphyses of femora. Bars: 500 μm. (J-O) Representative images of calcein double labeling and TRAP staining (J) with quantification of bone formation rates (K-M) and bone resorption rates (N, O) in distal femora. Bars: 50 μm (up) and 25 μm (bottom). n = 6 per group. Data represents mean ± SD. *P < 0.05; NS, not significant (P > 0.05) (ANOVA followed by Newman-Keuls post-hoc tests).
Diabetes is a metabolic disease of high incidence with various complications affecting cardiovascular, nervous, and skeletal systems. Particularly, severe osteoporosis could develop in T1D, which is also recognized as a chronic autoimmune disorder characterized by hypoinsulinemia and hyperglycemia [37, 38]. MSC therapy based on systemic infusion has been proved effective in reconstructing the damaged islets and reverting hyperglycemia both in animal and in human researches, while little is known about the potential improvements in complicated lesions [25, 26, 73, 74]. Interestingly, despite INS-based intensive glycemic control in T1D, complications may still persist and progress after even transient hyperglycemia [51, 75]. In the present study, we confirmed that INST and sMSC infusion after the onset of T1D could only restore euglycemia without rescue on osteopenia and inflammation, suggesting “tissue/organ-specifically targeted” therapeutic effects. This phenomenon has also been defined as “metabolic memory” and remains poorly understood and a major challenge in treating diabetes [51, 75]. In addition, although elongation of the observing time may provide traces of systemic improvements after hyperglycemic treatments, evidence is lacking. We hereby provide a solution in which pre-normalization of recipient glycemic micro-environment by a single INS or MSC administration could promote the anti-inflammatory and therapeutic effects of the later infused MSCs on complicated lesions. Controversies exist as to whether sMSC infusion alleviates immunological dysfunction in T1D [25, 73, 76-78] and whether multiple administration of MSCs exert more profound therapeutic effects [56, 69, 70, 79, 80]. Based on our findings, donor-recipient interactions may occur in a dynamic pattern, in which recipient glycemic conditions at the infusion time points potently influence the bidirectional balance. Further experiments are now undergoing in OVX mice, in which we apply a 2nd MSC infusion after the transient hyperglycemia fading away. Despite the controversies, we unraveled that fulfillment of potent function of donor MSCs requires tight control of recipient micro-environment.
Our findings also help to clarify the therapeutic mechanisms underlying MSC curing islet damages in T1D. Current understanding of the antidiabetic effects of infused MSCs is unrelated to their transdifferentiation potential but to their paracrine impacts, of which immunomodulation is the research focus [25, 28, 80, 81]. However, current data are controversial in whether MSC infusion could exert systemic immunomodulatory effects [76, 78] or local beneficial effects on pancreas micro-environments [25, 73, 77], which need MSC to migrate to pancreatic lymph nodes [25, 77]. Based on our data, systemic anti-inflammatory capacity of infused MSCs was impaired in hyperglycemia, but the MSC migration capacity post infusion was not affected. Therefore, it would be safe to assume that a portion of infused MSCs could migrate to pancreas and restore the local micro-environments, and that the local trophic effects might account for the major contributions to the improved islets after MSC infusion. Conversely, for complicated lesions of T1D like osteoporosis, the maintenance of systemic immunomodulatory effects may overweigh homing and local effects in preserving MSC therapeutic efficacy. Also in an inflammation-induced osteopenia, the OVX model, previous findings have shown that immunomodulation of MSCs to induce T-cell apoptosis via Fasl is indispensable for their therapeutic effects [7]. Nevertheless, in the anti-inflammatory drug glucocorticoid-induced osteoporosis, homing of infused MSCs is pivotal while no immunomodulatory was observed, as stated in our previous report [8]. The above findings collectively indicate that therapeutic effects of MSCs are micro-environment-dependent; the efficacy differs in organ- and disease- specific patterns upon differential systemic and local niches (for diabetes) and upon different systemic conditions (for osteoporosis). These assumptions should be carefully investigated in future researches and taken into consideration in clinical application of MSCs.
Immunomodulatory/anti-inflammatory capacity of MSCs has been recognized as a significant principle for MSC-mediated therapy in not only autoimmune diseases [50] but also osteoporosis [7]. Despite that immune cells such as dendrite cells, natural killer cells, macrophages and B cells were generally influenced by MSCs [59], T cells are recognized to be the major target of immunosuppressive properties of MSCs [18, 50]. Previous findings have shown that immunosuppression of MSCs is both innate and inducible, respectively depending on the constitutive Fasl-Fas pathway to induce T-cell apoptosis [50], and several cytokines promoted in response to inflammation to suppress T-cell proliferation [18]. Here, we further revealed that anti-inflammation of MSCs is also reducible by recipient hyperglycemic micro-environment. Given that homing capacity of MSCs was not influenced by the diabetic micro-environment, the reduction of anti-inflammatory capacity was key to the impaired therapeutic effects of MSCs in T1D-induced osteoporosis. Furthermore, we screened and discovered that MSC function, but not T-cell responses, is diminished in diabetic micro-environments, which should be attributed to detrimental effects of both high glucose and its metabolic product AGEs [58, 82], considering the complex pathological condition in vivo. Potential contributions of other immune cells might be involved. We have also verified that Fasl, not any of other reported cytokines, may be responsible for the MSC impairment. As far as we know, the high-glucose effects on Fasl expression and the underlying mechanisms are not documented. Fasl expression is primarily regulated by transcription factors [83], and its promotor activity is also reported to be stimulated by phosphorylation of AMPK [67], an important cellular energy sensor [21]. In accordance with previous studies, we revealed that p-Ampk protein expression is down-regulated by high-glucose treatment [65, 66], probably contributing to the inhibition of Fasl. Furthermore, by using the AMPK activator MET, we confirmed that rescuing AMPK signaling could restore anti-inflammatory capacity of MSCs in hyperglycemic micro-environments. As far as we know, this is the first report to reveal that the AMPK pathway plays a necessary role in maintaining immunomodulation of MSCs. Detailed mechanisms are needed to be elucidated in future works. In addition, consistent with previous data [13], we showed that MSCs are not intrinsically immunoprivileged and that in detrimental recipient micro-environments, allogeneic MSCs lose their immunomodulatory capacity but induce a T-cell immunological response. Our findings should be interpreted with specific attention in future utility of MSCs in clinical.
In summary, our findings uncover a previously unrecognized role of recipient glycemic conditions controlling MSC-mediated therapy, and unravel that fulfillment of potent function of donor MSCs requires tight control of recipient micro-environment.
Abbreviations
AGEs: advanced glycation end products;
AMPK: adenosine monophosphate-activated protein kinase;
ANOVA: analysis of variance;
BFR: bone formation rate;
BL: blank;
BMD: bone mineral density;
BV/TV: bone volume per tissue volume;
Ctrl: control;
Ct.Th: cortical thickness;
CTX-1: cross linked C-telopeptide of type 1 collagen;
dMSC: double mesenchymal stem cell;
ELISA: enzyme-linked immunosorbent assay;
Fasl: fas ligand;
GFP: green fluorescent protein;
GLU: glucose;
HbA1c: glycated hemoglobin;
HG: high glucose;
Hgf: hepatocyte growth factor;
Ido: indoleamine 2,3-dioxigenase;
Il10: interleukin-10;
Inos: inducible nitric oxide synthase;
INS: insulin;
INST: insulin therapy;
LG: low glucose;
MA: mannitol;
MAR: mineral apposition rate;
MET: metformin;
Micro-CT: micro-computed tomography;
MMP: matrix metalloproteinase;
MS/BS: mineralized surface per bone surface;
MSCs: mesenchymal stromal cells;
MSCsGFP: green fluorescent protein-labeled mesenchymal stem cells;
N.Oc/BS: number of osteoclasts per bone surface;
Oc.S/BS: osteoclast surface per bone surface;
OVX: ovariectomy;
PBMNCs: peripheral blood mononuclear cells;
PBS: phosphate buffer saline;
qRT-PCR: quantitative real-time polymerase chain reaction;
Sca-1: stem cell antigen-1;
SD: standard deviation;
sMSC: single mesenchymal stem cell;
STZ: streptozotocin;
T1D: type 1 diabetes;
Tb.N: trabecular number;
Tb.Sp: trabecular separation;
Tb.Th: trabecular thickness;
Tgfβ1: transforming growth factor-beta 1;
TNF-α: tumor necrosis factor-alpha;
TRAP: tartrate resistant acid phosphotase;
WT: wild type.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by The National Key Research and Development Program of China (2016YFC1101400 and 2016YFC1102900) and The General Program of National Natural Science Foundation of China (81570937 and 81470710).
Author Contributions
Bing-Dong Sui and Cheng-Hu Hu contributed equally to the study design, experimental work, data analysis, data interpretation and manuscript preparation. Chen-Xi Zheng contributed to the experimental work and revised the manuscript. Yi Shuai, Ping-Ping Gao, Pan Zhao, Xin-Yi Zhang and Tao He contributed to the experimental work. Xiao-Ning He, Meng Li and Kun Xuan contributed to the data interpretation. Yan Jin conceived and supervised the study. All authors have reviewed and approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hare JM, Traverse JH, Henry TD, Dib N, Strumpf RK, Schulman SP. et al. A randomized, double-blind, placebo-controlled, dose-escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. J Am Coll Cardiol. 2009;54:2277-86
2. Kharaziha P, Hellstrom PM, Noorinayer B, Farzaneh F, Aghajani K, Jafari F. et al. Improvement of liver function in liver cirrhosis patients after autologous mesenchymal stem cell injection: a phase I-II clinical trial. Eur J Gastroenterol Hepatol. 2009;21:1199-205
3. Horwitz EM, Gordon PL, Koo WK, Marx JC, Neel MD, McNall RY. et al. Isolated allogeneic bone marrow-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: Implications for cell therapy of bone. Proc Natl Acad Sci U S A. 2002;99:8932-7
4. Duijvestein M, Vos AC, Roelofs H, Wildenberg ME, Wendrich BB, Verspaget HW. et al. Autologous bone marrow-derived mesenchymal stromal cell treatment for refractory luminal Crohn's disease: results of a phase I study. Gut. 2010;59:1662-9
5. Sun L, Akiyama K, Zhang H, Yamaza T, Hou Y, Zhao S. et al. Mesenchymal stem cell transplantation reverses multiorgan dysfunction in systemic lupus erythematosus mice and humans. Stem Cells. 2009;27:1421-32
6. Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I. et al. Mesenchymal stem cells for treatment of steroid-resistant, severe, acute graft-versus-host disease: a phase II study. Lancet. 2008;371:1579-86
7. Liu Y, Wang L, Liu S, Liu D, Chen C, Xu X. et al. Transplantation of SHED prevents bone loss in the early phase of ovariectomy-induced osteoporosis. J Dent Res. 2014;93:1124-32
8. Sui B, Hu C, Zhang X, Zhao P, He T, Zhou C. et al. Allogeneic mesenchymal stem cell therapy promotes osteoblastogenesis and prevents glucocorticoid-induced osteoporosis. Stem Cells Transl Med. 2016;5:1238-46
9. Cho SW, Sun HJ, Yang JY, Jung JY, An JH, Cho HY. et al. Transplantation of mesenchymal stem cells overexpressing RANK-Fc or CXCR4 prevents bone loss in ovariectomized mice. Mol Ther. 2009;17:1979-87
10. Sudres M, Norol F, Trenado A, Gregoire S, Charlotte F, Levacher B. et al. Bone marrow mesenchymal stem cells suppress lymphocyte proliferation in vitro but fail to prevent graft-versus-host disease in mice. J Immunol. 2006;176:7761-7
11. Huang S, Xu L, Sun Y, Lin S, Gu W, Liu Y. et al. Systemic administration of allogeneic mesenchymal stem cells does not halt osteoporotic bone loss in ovariectomized rats. PLoS One. 2016;11:e0163131
12. Fuentes-Julian S, Arnalich-Montiel F, Jaumandreu L, Leal M, Casado A, Garcia-Tunon I. et al. Adipose-derived mesenchymal stem cell administration does not improve corneal graft survival outcome. PLoS One. 2015;10:e0117945
13. Nauta AJ, Westerhuis G, Kruisselbrink AB, Lurvink EG, Willemze R, Fibbe WE. Donor-derived mesenchymal stem cells are immunogenic in an allogeneic host and stimulate donor graft rejection in a nonmyeloablative setting. Blood. 2006;108:2114-20
14. Papadopoulou A, Yiangou M, Athanasiou E, Zogas N, Kaloyannidis P, Batsis I. et al. Mesenchymal stem cells are conditionally therapeutic in preclinical models of rheumatoid arthritis. Ann Rheum Dis. 2012;71:1733-40
15. Salinas Tejedor L, Berner G, Jacobsen K, Gudi V, Jungwirth N, Hansmann F. et al. Mesenchymal stem cells do not exert direct beneficial effects on CNS remyelination in the absence of the peripheral immune system. Brain Behav Immun. 2015;50:155-65
16. Lim R, Milton P, Murphy SV, Dickinson H, Chan ST, Jenkin G. Human mesenchymal stem cells reduce lung injury in immunocompromised mice but not in immunocompetent mice. Respiration. 2013;85:332-41
17. Tisato V, Naresh K, Girdlestone J, Navarrete C, Dazzi F. Mesenchymal stem cells of cord blood origin are effective at preventing but not treating graft-versus-host disease. Leukemia. 2007;21:1992-9
18. Ren G, Zhang L, Zhao X, Xu G, Zhang Y, Roberts AI. et al. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell Stem Cell. 2008;2:141-50
19. Xu G, Zhang Y, Zhang L, Roberts AI, Shi Y. C/EBPbeta mediates synergistic upregulation of gene expression by interferon-gamma and tumor necrosis factor-alpha in bone marrow-derived mesenchymal stem cells. Stem Cells. 2009;27:942-8
20. Sui BD, Hu CH, Zheng CX, Jin Y. Microenvironmental views on mesenchymal stem cell differentiation in aging. J Dent Res. 2016 [Epub ahead of print]
21. Sui B, Hu C, Jin Y. Mitochondrial metabolic failure in telomere attrition-provoked aging of bone marrow mesenchymal stem cells. Biogerontology. 2016;17:267-79
22. Cortes S, Chambers S, Jeronimo A, Isenberg D. Diabetes mellitus complicating systemic lupus erythematosus - analysis of the UCL lupus cohort and review of the literature. Lupus. 2008;17:977-80
23. Sui B, Hu C, Liao L, Chen Y, Zhang X, Fu X. et al. Mesenchymal progenitors in osteopenias of diverse pathologies: differential characteristics in the common shift from osteoblastogenesis to adipogenesis. Sci Rep. 2016;6:30186
24. Molgat AS, Tilokee EL, Rafatian G, Vulesevic B, Ruel M, Milne R. et al. Hyperglycemia inhibits cardiac stem cell-mediated cardiac repair and angiogenic capacity. Circulation. 2014;130(Suppl 1):S70-6
25. Ezquer F, Ezquer M, Contador D, Ricca M, Simon V, Conget P. The antidiabetic effect of mesenchymal stem cells is unrelated to their transdifferentiation potential but to their capability to restore Th1/Th2 balance and to modify the pancreatic microenvironment. Stem Cells. 2012;30:1664-74
26. Skyler JS, Fonseca VA, Segal KR, Rosenstock J, Investigators M-D. Allogeneic mesenchymal precursor cells in type 2 diabetes: A randomized, placebo-controlled, dose-escalation safety and tolerability pilot study. Diabetes Care. 2015;38:1742-9
27. Volarevic V, Arsenijevic N, Lukic ML, Stojkovic M. Concise review: Mesenchymal stem cell treatment of the complications of diabetes mellitus. Stem Cells. 2011;29:5-10
28. Katuchova J, Harvanova D, Spakova T, Kalanin R, Farkas D, Durny P. et al. Mesenchymal stem cells in the treatment of type 1 diabetes mellitus. Endocr Pathol. 2015;26:95-103
29. Liu S, Liu D, Chen C, Hamamura K, Moshaverinia A, Yang R. et al. MSC transplantation improves osteopenia via epigenetic regulation of Notch signaling in lupus. Cell Metab. 2015;22:606-18
30. Shuai Y, Liao L, Su X, Yu Y, Shao B, Jing H. et al. Melatonin treatment improves mesenchymal stem cells therapy by preserving stemness during long-term in vitro expansion. Theranostics. 2016;6:1899-917
31. Lee K, Kim H, Kim JM, Kim JR, Kim KJ, Kim YJ. et al. Systemic transplantation of human adipose-derived stem cells stimulates bone repair by promoting osteoblast and osteoclast function. J Cell Mol Med. 2011;15:2082-94
32. An JH, Park H, Song JA, Ki KH, Yang JY, Choi HJ. et al. Transplantation of human umbilical cord blood-derived mesenchymal stem cells or their conditioned medium prevents bone loss in ovariectomized nude mice. Tissue Eng Part A. 2013;19:685-96
33. Hsiao FS, Cheng CC, Peng SY, Huang HY, Lian WS, Jan ML. et al. Isolation of therapeutically functional mouse bone marrow mesenchymal stem cells within 3 h by an effective single-step plastic-adherent method. Cell Prolif. 2010;43:235-48
34. Clowes JA, Riggs BL, Khosla S. The role of the immune system in the pathophysiology of osteoporosis. Immunol Rev. 2005;208:207-27
35. Yang N, Wang G, Hu C, Shi Y, Liao L, Shi S. et al. Tumor necrosis factor alpha suppresses the mesenchymal stem cell osteogenesis promoter miR-21 in estrogen deficiency-induced osteoporosis. J Bone Miner Res. 2013;28:559-73
36. Wang L, Zhao Y, Liu Y, Akiyama K, Chen C, Qu C. et al. IFN-gamma and TNF-alpha synergistically induce mesenchymal stem cell impairment and tumorigenesis via NFkappaB signaling. Stem Cells. 2013;31:1383-95
37. van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev. 2011;91:79-118
38. Hofbauer LC, Brueck CC, Singh SK, Dobnig H. Osteoporosis in patients with diabetes mellitus. J Bone Miner Res. 2007;22:1317-28
39. McCabe LR. Understanding the pathology and mechanisms of type I diabetic bone loss. J Cell Biochem. 2007;102:1343-57
40. Botolin S, McCabe LR. Bone loss and increased bone adiposity in spontaneous and pharmacologically induced diabetic mice. Endocrinology. 2007;148:198-205
41. Stolzing A, Sellers D, Llewelyn O, Scutt A. Diabetes induced changes in rat mesenchymal stem cells. Cells Tissues Organs. 2010;191:453-65
42. Mei SH, McCarter SD, Deng Y, Parker CH, Liles WC, Stewart DJ. Prevention of LPS-induced acute lung injury in mice by mesenchymal stem cells overexpressing angiopoietin 1. PLoS Med. 2007;4:e269
43. Chen X, Yu QQ, Zhu YH, Bi Y, Sun WP, Liang H. et al. Insulin therapy stimulates lipid synthesis and improves endocrine functions of adipocytes in dietary obese C57BL/6 mice. Acta Pharmacol Sin. 2010;31:341-6
44. Brezar V, Culina S, Gagnerault MC, Mallone R. Short-term subcutaneous insulin treatment delays but does not prevent diabetes in NOD mice. Eur J Immunol. 2012;42:1553-61
45. Atkinson MA, Maclaren NK, Luchetta R. Insulitis and diabetes in NOD mice reduced by prophylactic insulin therapy. Diabetes. 1990;39:933-7
46. Diabetes Prevention Trial-Type 1 Diabetes Study G. Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med. 2002;346:1685-91
47. Wu L, Lu Y, Jiao Y, Liu B, Li S, Li Y. et al. Paternal psychological stress reprograms hepatic gluconeogenesis in offspring. Cell Metab. 2016;23:735-43
48. Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Muller R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res. 2010;25:1468-86
49. Dempster DW, Compston JE, Drezner MK, Glorieux FH, Kanis JA, Malluche H. et al. Standardized nomenclature, symbols, and units for bone histomorphometry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res. 2013;28:2-17
50. Akiyama K, Chen C, Wang D, Xu X, Qu C, Yamaza T. et al. Mesenchymal-stem-cell-induced immunoregulation involves FAS-ligand-/FAS-mediated T cell apoptosis. Cell Stem Cell. 2012;10:544-55
51. Zhao S, Li T, Li J, Lu Q, Han C, Wang N. et al. miR-23b-3p induces the cellular metabolic memory of high glucose in diabetic retinopathy through a SIRT1-dependent signalling pathway. Diabetologia. 2016;59:644-54
52. Yang K, Wang XQ, He YS, Lu L, Chen QJ, Liu J. et al. Advanced glycation end products induce chemokine/cytokine production via activation of p38 pathway and inhibit proliferation and migration of bone marrow mesenchymal stem cells. Cardiovasc Diabetol. 2010;9:66
53. Kasai T, Bandow K, Suzuki H, Chiba N, Kakimoto K, Ohnishi T. et al. Osteoblast differentiation is functionally associated with decreased AMP kinase activity. J Cell Physiol. 2009;221:740-9
54. Mazar J, Thomas M, Bezrukov L, Chanturia A, Pekkurnaz G, Yin S. et al. Cytotoxicity mediated by the Fas ligand (FasL)-activated apoptotic pathway in stem cells. J Biol Chem. 2009;284:22022-8
55. Liao L, Yang X, Su X, Hu C, Zhu X, Yang N. et al. Redundant miR-3077-5p and miR-705 mediate the shift of mesenchymal stem cell lineage commitment to adipocyte in osteoporosis bone marrow. Cell Death Dis. 2013;4:e600
56. Ezquer F, Ezquer M, Simon V, Conget P. The antidiabetic effect of MSCs is not impaired by insulin prophylaxis and is not improved by a second dose of cells. PLoS One. 2011;6:e16566
57. Guo W, Gong Y, Fu Z, Fu J, Sun Y, Ju X. et al. The effect of cholesteryl ester transfer protein on pancreatic beta cell dysfunction in mice. Nutr Metab (Lond). 2016;13:21
58. Kume S, Kato S, Yamagishi S, Inagaki Y, Ueda S, Arima N. et al. Advanced glycation end-products attenuate human mesenchymal stem cells and prevent cognate differentiation into adipose tissue, cartilage, and bone. J Bone Miner Res. 2005;20:1647-58
59. De Miguel MP, Fuentes-Julian S, Blazquez-Martinez A, Pascual CY, Aller MA, Arias J. et al. Immunosuppressive properties of mesenchymal stem cells: advances and applications. Curr Mol Med. 2012;12:574-91
60. Di Nicola M, Carlo-Stella C, Magni M, Milanesi M, Longoni PD, Matteucci P. et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002;99:3838-43
61. Meisel R, Zibert A, Laryea M, Gobel U, Daubener W, Dilloo D. Human bone marrow stromal cells inhibit allogeneic T-cell responses by indoleamine 2,3-dioxygenase-mediated tryptophan degradation. Blood. 2004;103:4619-21
62. Min CK, Kim BG, Park G, Cho B, Oh IH. IL-10-transduced bone marrow mesenchymal stem cells can attenuate the severity of acute graft-versus-host disease after experimental allogeneic stem cell transplantation. Bone Marrow Transplant. 2007;39:637-45
63. Ding Y, Xu D, Feng G, Bushell A, Muschel RJ, Wood KJ. Mesenchymal stem cells prevent the rejection of fully allogenic islet grafts by the immunosuppressive activity of matrix metalloproteinase-2 and -9. Diabetes. 2009;58:1797-806
64. Wu T, Liu Y, Fan Z, Xu J, Jin L, Gao Z. et al. miR-21 modulates the immunoregulatory function of bone marrow mesenchymal stem cells through the PTEN/Akt/TGF-beta1 pathway. Stem Cells. 2015;33:3281-90
65. Szkudelski T, Szkudelska K. Effects of AMPK activation on lipolysis in primary rat adipocytes: studies at different glucose concentrations. Arch Physiol Biochem. 2016:1-7
66. Zheng Z, Chen H, Li J, Li T, Zheng B, Zheng Y. et al. Sirtuin 1-mediated cellular metabolic memory of high glucose via the LKB1/AMPK/ROS pathway and therapeutic effects of metformin. Diabetes. 2012;61:217-28
67. Lee JY, Choi AY, Oh YT, Choe W, Yeo EJ, Ha J. et al. AMP-activated protein kinase mediates T cell activation-induced expression of FasL and COX-2 via protein kinase C theta-dependent pathway in human Jurkat T leukemia cells. Cell Signal. 2012;24:1195-207
68. Chen SC, Brooks R, Houskeeper J, Bremner SK, Dunlop J, Viollet B. et al. Metformin suppresses adipogenesis through both AMP-activated protein kinase (AMPK)-dependent and AMPK-independent mechanisms. Mol Cell Endocrinol. 2016;440:57-68
69. Ho JH, Tseng TC, Ma WH, Ong WK, Chen YF, Chen MH. et al. Multiple intravenous transplantations of mesenchymal stem cells effectively restore long-term blood glucose homeostasis by hepatic engraftment and beta-cell differentiation in streptozocin-induced diabetic mice. Cell Transplant. 2012;21:997-1009
70. Hao H, Liu J, Shen J, Zhao Y, Liu H, Hou Q. et al. Multiple intravenous infusions of bone marrow mesenchymal stem cells reverse hyperglycemia in experimental type 2 diabetes rats. Biochem Biophys Res Commun. 2013;436:418-23
71. Liu Y, Wang L, Kikuiri T, Akiyama K, Chen C, Xu X. et al. Mesenchymal stem cell-based tissue regeneration is governed by recipient T lymphocytes via IFN-gamma and TNF-alpha. Nat Med. 2011;17:1594-601
72. Munir H, McGettrick HM. Mesenchymal stem cell therapy for autoimmune disease: risks and rewards. Stem Cells Dev. 2015;24:2091-100
73. Jurewicz M, Yang S, Augello A, Godwin JG, Moore RF, Azzi J. et al. Congenic mesenchymal stem cell therapy reverses hyperglycemia in experimental type 1 diabetes. Diabetes. 2010;59:3139-47
74. Cai J, Wu Z, Xu X, Liao L, Chen J, Huang L. et al. Umbilical cord mesenchymal stromal cell with autologous bone marrow cell transplantation in established type 1 diabetes: A pilot randomized controlled open-label clinical study to assess safety and impact on insulin secretion. Diabetes Care. 2016;39:149-57
75. Writing Team for the Diabetes C, Complications Trial/Epidemiology of Diabetes I, Complications Research G. Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA. 2002;287:2563-9
76. Hu J, Wang Y, Wang F, Wang L, Yu X, Sun R. et al. Effect and mechanisms of human Wharton's jelly-derived mesenchymal stem cells on type 1 diabetes in NOD model. Endocrine. 2015;48:124-34
77. Fiorina P, Jurewicz M, Augello A, Vergani A, Dada S, La Rosa S. et al. Immunomodulatory function of bone marrow-derived mesenchymal stem cells in experimental autoimmune type 1 diabetes. J Immunol. 2009;183:993-1004
78. Madec AM, Mallone R, Afonso G, Abou Mrad E, Mesnier A, Eljaafari A. et al. Mesenchymal stem cells protect NOD mice from diabetes by inducing regulatory T cells. Diabetologia. 2009;52:1391-9
79. Wang D, Akiyama K, Zhang H, Yamaza T, Li X, Feng X. et al. Double allogenic mesenchymal stem cells transplantations could not enhance therapeutic effect compared with single transplantation in systemic lupus erythematosus. Clin Dev Immunol. 2012;2012:273-91
80. Aali E, Mirzamohammadi S, Ghaznavi H, Madjd Z, Larijani B, Rayegan S. et al. A comparative study of mesenchymal stem cell transplantation with its paracrine effect on control of hyperglycemia in type 1 diabetic rats. J Diabetes Metab Disord. 2014;13:76
81. Abdi R, Fiorina P, Adra CN, Atkinson M, Sayegh MH. Immunomodulation by mesenchymal stem cells: a potential therapeutic strategy for type 1 diabetes. Diabetes. 2008;57:1759-67
82. Yamagishi S. Role of advanced glycation end products (AGEs) in osteoporosis in diabetes. Curr Drug Targets. 2011;12:2096-102
83. Li-Weber M, Krammer PH. Function and regulation of the CD95 (APO-1/Fas) ligand in the immune system. Semin Immunol. 2003;15:145-57
Author Biography
 Prof. Yan Jin is the Chairman of Chinese Society of Tissue Engineering and Regenerative Medicine and Chairman of Chinese Society of Oral Biomedicine. He established the Center for Tissue Engineering of Fourth Military Medical University and he has been serving as the director. Prof. Yan Jin has long been engaged in scientific researches on tissue engineering and regenerative medicine. He leads an accomplished program in China that focuses on the development and industrialization of tissue engineering products, having made great contributions to both basic and applied researches on regeneration of tissues and organs, such as tooth, bone, skin, cornea and nerve. He has also discovered new mechanisms underlying influences of inflammation and aging on stem cells and has uncovered that the abnormalities of mesenchymal stem cells (MSCs) are key to the development of osteoporosis in aging, which provides multiple potential targets for theranostics. He has published over 150 papers in SCI journals, such as Cell Metabolism, Theranostics, Cell Death and Differentiation, Stem Cells, Biomaterials, Journal of Bone and Mineral Research, Molecular Therapy, Journal of Dental Research and Scientific Reports.
Prof. Yan Jin is the Chairman of Chinese Society of Tissue Engineering and Regenerative Medicine and Chairman of Chinese Society of Oral Biomedicine. He established the Center for Tissue Engineering of Fourth Military Medical University and he has been serving as the director. Prof. Yan Jin has long been engaged in scientific researches on tissue engineering and regenerative medicine. He leads an accomplished program in China that focuses on the development and industrialization of tissue engineering products, having made great contributions to both basic and applied researches on regeneration of tissues and organs, such as tooth, bone, skin, cornea and nerve. He has also discovered new mechanisms underlying influences of inflammation and aging on stem cells and has uncovered that the abnormalities of mesenchymal stem cells (MSCs) are key to the development of osteoporosis in aging, which provides multiple potential targets for theranostics. He has published over 150 papers in SCI journals, such as Cell Metabolism, Theranostics, Cell Death and Differentiation, Stem Cells, Biomaterials, Journal of Bone and Mineral Research, Molecular Therapy, Journal of Dental Research and Scientific Reports.
 Dr. Bing-Dong Sui is a doctoral candidate in Fourth Military Medical University, and his supervisor is Prof. Yan Jin. His research focuses on elaborating micro-environmental regulations on mesenchymal stem cells in the development and treatment of osteoporosis. Dr. Bing-Dong Sui has published 9 papers as first or co-first author in SCI journals, including Journal of Dental Research (an Invited Review and an Article), Stem Cells Translational Medicine (selected as Featured Article), Scientific Reports, Mitochondrion and Biogerontology. He is the Laureatee of the First Prize of International Association of Dental Research (IADR) Hatton Divisional Award for Distinguished Young Scholars, the winner of the First Prize of Rising Star Scholarship of Chinese Stomatological Association (CSA), and the winner of the First Prize of New Investigator Award of Society of Oral Biomedicine of CSA (SOBCSA).
Dr. Bing-Dong Sui is a doctoral candidate in Fourth Military Medical University, and his supervisor is Prof. Yan Jin. His research focuses on elaborating micro-environmental regulations on mesenchymal stem cells in the development and treatment of osteoporosis. Dr. Bing-Dong Sui has published 9 papers as first or co-first author in SCI journals, including Journal of Dental Research (an Invited Review and an Article), Stem Cells Translational Medicine (selected as Featured Article), Scientific Reports, Mitochondrion and Biogerontology. He is the Laureatee of the First Prize of International Association of Dental Research (IADR) Hatton Divisional Award for Distinguished Young Scholars, the winner of the First Prize of Rising Star Scholarship of Chinese Stomatological Association (CSA), and the winner of the First Prize of New Investigator Award of Society of Oral Biomedicine of CSA (SOBCSA).
 Dr. Cheng-Hu Hu is a Principal Investigator in the Center for Tissue Engineering of Fourth Military Medical University led by Prof. Yan Jin. He is currently engaged in investigating endogenous stem cell mobilization under pathological micro-environments and the participation of endogenous stem cell mobilization in regeneration of soft and hard tissue damages. Dr. Cheng-Hu Hu is now presiding over a Project of the National Natural Science Foundation of China, and he participates in major researches of Projects of the National Basic Research Program of China, the Innovation Team Development Program of the Ministry of Education of China, and the Key Project on Tissue Engineering Organs granted by the Ministry of Science and Technology of China. He has co-authored 19 well-recognized SCI papers focusing on regulations of mesenchymal stem cell impairments in osteoporosis and the therapeutic solutions, in Journals such as Stem Cells, Journal of Bone and Mineral Research, Biomaterials, Stem Cells Translational Medicine, Scientific Reports, Journal of Dental Research, and Cell Death and Disease. He was also awarded the First Class Prize of the Shaanxi Province Scientific and Technological Progress Award of China.
Dr. Cheng-Hu Hu is a Principal Investigator in the Center for Tissue Engineering of Fourth Military Medical University led by Prof. Yan Jin. He is currently engaged in investigating endogenous stem cell mobilization under pathological micro-environments and the participation of endogenous stem cell mobilization in regeneration of soft and hard tissue damages. Dr. Cheng-Hu Hu is now presiding over a Project of the National Natural Science Foundation of China, and he participates in major researches of Projects of the National Basic Research Program of China, the Innovation Team Development Program of the Ministry of Education of China, and the Key Project on Tissue Engineering Organs granted by the Ministry of Science and Technology of China. He has co-authored 19 well-recognized SCI papers focusing on regulations of mesenchymal stem cell impairments in osteoporosis and the therapeutic solutions, in Journals such as Stem Cells, Journal of Bone and Mineral Research, Biomaterials, Stem Cells Translational Medicine, Scientific Reports, Journal of Dental Research, and Cell Death and Disease. He was also awarded the First Class Prize of the Shaanxi Province Scientific and Technological Progress Award of China.
![]() Corresponding author: Prof. Yan Jin, State Key Laboratory of Military Stomatology, Center for Tissue Engineering, Fourth Military Medical University, No. 145 West Changle Road, Xi'an, Shaanxi 710032, China. E-mail: yanjinedu.cn; Tel: +86-029-84776472; Fax: +86-029-83218039.
Corresponding author: Prof. Yan Jin, State Key Laboratory of Military Stomatology, Center for Tissue Engineering, Fourth Military Medical University, No. 145 West Changle Road, Xi'an, Shaanxi 710032, China. E-mail: yanjinedu.cn; Tel: +86-029-84776472; Fax: +86-029-83218039.
Citation styles
APA
Sui, B.D., Hu, C.H., Zheng, C.X., Shuai, Y., He, X.N., Gao, P.P., Zhao, P., Li, M., Zhang, X.Y., He, T., Xuan, K., Jin, Y. (2017). Recipient Glycemic Micro-environments Govern Therapeutic Effects of Mesenchymal Stem Cell Infusion on Osteopenia. Theranostics, 7(5), 1225-1244. https://doi.org/10.7150/thno.18181.
ACS
Sui, B.D.; Hu, C.H.; Zheng, C.X.; Shuai, Y.; He, X.N.; Gao, P.P.; Zhao, P.; Li, M.; Zhang, X.Y.; He, T.; Xuan, K.; Jin, Y. Recipient Glycemic Micro-environments Govern Therapeutic Effects of Mesenchymal Stem Cell Infusion on Osteopenia. Theranostics 2017, 7 (5), 1225-1244. DOI: 10.7150/thno.18181.
NLM
Sui BD, Hu CH, Zheng CX, Shuai Y, He XN, Gao PP, Zhao P, Li M, Zhang XY, He T, Xuan K, Jin Y. Recipient Glycemic Micro-environments Govern Therapeutic Effects of Mesenchymal Stem Cell Infusion on Osteopenia. Theranostics 2017; 7(5):1225-1244. doi:10.7150/thno.18181. https://www.thno.org/v07p1225.htm
CSE
Sui BD, Hu CH, Zheng CX, Shuai Y, He XN, Gao PP, Zhao P, Li M, Zhang XY, He T, Xuan K, Jin Y. 2017. Recipient Glycemic Micro-environments Govern Therapeutic Effects of Mesenchymal Stem Cell Infusion on Osteopenia. Theranostics. 7(5):1225-1244.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.