Impact Factor
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(5):1204-1213. doi:10.7150/thno.17069 This issue Cite
Research Paper
Aptamer Internalization via Endocytosis Inducing S-Phase Arrest and Priming Maver-1 Lymphoma Cells for Cytarabine Chemotherapy
Huan Li1,2, Shuanghui Yang2,3, Ge Yu2,4, Liangfang Shen1, Jia Fan5, Ling Xu2, Hedong Zhang5, Nianxi Zhao2, Zihua Zeng2, Tony Hu5, Jianguo Wen2 ![]() , Youli Zu2
, Youli Zu2 ![]()
1. Department of Oncology, Xiangya Hospital, Central South University, Changsha, Hunan Province, China.
2. Department of Pathology and Genomic Medicine, Houston Methodist Hospital, Houston, TX, USA.
3. Department of Hematology, Xiangya Hospital, Central South University, Changsha, Hunan Province, China.
4. Department of Hepatology, The First Hospital of Jilin University, Changchun, Jilin Province, China.
5. Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX, USA.
Received 2016-8-1; Accepted 2016-11-4; Published 2017-3-6
Abstract

The goal of precision therapy is to efficiently treat cancer without side effects. Aptamers are a class of small ligands composed of single-stranded oligonucleotides that bind to their targets with high affinity and specificity. In this study, we identified an ssDNA aptamer specifically targeting Maver-1 lymphoma cells with high binding affinity (Kd = 70±8 pmol/L). Interestingly, cellular cycle studies revealed that exposure of Maver-1 cells to synthetic aptamers triggered S-phase arrest of 40% of the cells (vs. 18% baseline). Confocal microscopy confirmed specific cell binding of aptamers and the resultant endocytosis into Maver-1 cells. Subsequent functional assays validated the fact that aptamer internalization into targeted cells is a prerequisite for Maver-1 cell growth inhibition. Importantly, aptamer-induced S-phase arrest induced enhanced chemotherapeutic results involving cytarabine, which primarily kills lymphoma cells at S-phase. Combination treatments revealed that aptamer re-exposure considerably primed Maver-1 cells for cytarabine chemotherapy, thus achieving a synergistic killing effect by reaching cell death rates as high as 61% (vs. 13% or 14% induced by aptamer or cytarabine treatment alone). These findings demonstrated that aptamers do not only act as molecular ligands but can also function as biotherapeutic agents by inducing S-phase arrest of lymphoma cells. In addition, logical combination of aptamer and cytarabine treatments ushers the way to a unique approach in precision lymphoma chemotherapy.
Keywords: aptamer, biotherapeutics, lymphoma, synergistic chemotherapy, S-phase arrest
Introduction
Aptamers are a class of low molecular weight ligands composed of short, single-stranded oligonucleotides (RNA or ssDNA) that are able to specifically recognize and bind to their targets with high affinity (1). Target-specific aptamer sequences are developed by the process of Systematic Evolution of Ligands by Exponential Enrichment (SELEX) (2, 3). As targeting ligands, aptamers offer several advantages for clinical use as compared to protein antibodies because they: (i) can be chemically synthesized (4), (ii) possess the capacity for faster cancer-cell binding (5), (iii) exhibit potential for deeper tumor-tissue penetration (6), and (iv) are easily conjugated to various functional molecules (7, 8). Biomedical studies revealed that aptamers are useful for in vitro detection of cancer cells, staining tumor tissues (9, 10), in vivo tumor imaging (11), and targeted delivery and therapy (12).
Non-Hodgkin's lymphoma (NHL) is the most common hematological malignancy in adults, with B-cell lymphomas accounting for 85% of all NHLs (13). Most patients with B-cell lymphomas are presently treated with cytotoxic chemotherapeutic agents combined with the anti-CD20 monoclonal antibody (mAb) rituximab (14). However, as a chimeric mAb, rituximab commonly induces allergic reactions (15). In addition, rituximab also reacts with and depletes normal B cells, which express CD20, and are required for humoral immune responses in patients (16). It is well known that B cell lymphoma is a clonal disease and thus, lymphoma cells express only one type of surface immunoglobulin (Ig) light chain (kappa or lambda). Currently, clonal Ig light chains are the gold standard biomarkers for B-cell lymphoma diagnosis. They also provide a potential target for precision lymphoma therapy with fewer adverse effects on normal B cells (17, 18).
Aiming for precision lymphoma therapy, we developed a ssDNA aptamer sequence that specifically targets Maver-1 lymphoma cells and immunoglobulin lambda-like polypeptide 5. Selective internalization of synthetic aptamers into Maver-1 lymphoma cells triggered cell cycle S-phase arrest and thus inhibited cell growth. Importantly, the aptamer-induced S-phase arrest primed Maver-1 lymphoma cells for cytarabine treatment, achieving a synergistic killing effect. These findings clearly showed the enhanced value of aptamers in biotherapy by cell cycle regulation and their synergistic killing of lymphoma cells in combination with chemotherapy.
Materials and methods
Reagents and cell lines
The cell lines, including B lymphoma Maver-1, mantle cell lymphoma Jeko-1, Burkitt's lymphoma CA46, acute erythroid leukemia HEL, diffuse histiocytic lymphoma SU-DHL-1 and breast cancer MDA-MB-468 were purchased from ATCC (Manassas, VA). All cell suspensions were cultured in RPMI 1640 medium (Thermo Fisher Scientific, Rockford, IL) containing 10% Fetal Bovine Serum (FBS, Atlanta Biologicals, Lawrenceville, GA), and 100 IU/mL penicillin-streptomycin. All adhesion cell lines were cultured with DMEM (Atlanta Biologicals, Lawrenceville, GA) containing 10% FBS and 100 IU/mL penicillin-streptomycin. The washing buffer used during aptamer enrichment contained 4.5 g/L glucose and 5 mmol/L MgCl2 in Dulbecco's Phosphate-Buffered Saline (DPBS, Sigma Aldrich, St. Louis, MO). Binding buffer was prepared by adding Bovine Serum Albumin (1 mg/mL; BSA, Thermo Fisher Scientific) with 0.1 mg/mL t-RNA (Sigma Aldrich) to the washing buffer.
Cell binding assay
The Cy3-labeled ssDNA pool or individual aptamers were incubated with 5 × 105 cells at the indicated concentrations for 30 min at room temperature (RT). Cells were washed once with washing buffer and re-suspended in binding buffer. The resultant cell binding was quantified with a FACScan cytometer (LSRII, BD Biosciences, San Jose, CA) by counting 10,000 events. The Cy3-labeled ssDNA library was used as a negative control. The dissociation constants (Kd) were calculated from the cell binding results detected by flow cytometry.
For competition studies, cells were pre-incubated with 1 μmol/L unlabeled aptamer sequences for 30 min and treated with fluorophore-labeled anti-Ig lambda light chain antibody. Changes in cell binding of the antibody were then quantified by flow cytometry. The ssDNA library was used as a non-specific control.
Cell staining
To observe cell binding, cells were treated with Cy3-labeled aptamers (50 nmol/L) at RT for 1 h, and examined under a fluorescent microscope (Olympus America, Melville, NY). Cell nuclei were also stained with 2 µg/mL Hoechst 33342 dye for tracking purposes. To confirm binding specificity, a cell mixture was prepared by mixing equal numbers of target Maver-1 and control Jeko-1 lymphoma cells, which were used for the negative selection of aptamers. For identification, control cells were pre-stained with 25 nmol/L of carboxyfluorescein diacetate succinimidyl ester (CFSE, Life Technologies, Carlsbad, CA) at RT for 10 min. The Cy3-labeled aptamer probe was added to the cell mixture (50 nmol/L) in RPMI 1640 medium containing 10% FBS, and incubated at RT for 30 min. After washing twice, cell smears were prepared and examined under a fluorescent microscope.
Immunoprecipitation and mass spectrometry
Freshly cultured cells (1.0 x 107) were lysed with cell lysis buffer (Thermo Fisher Scientific) for 30 min on ice, and centrifuged at 14,000×g. The supernatants were collected and incubated with biotin-labeled aptamers (100 nmol/L) overnight at 4°C and then with 100 μL of streptavidin beads (Thermo Fisher scientific) for another hour. After washing twice with Phosphate-Buffered Saline (PBS, Sigma Aldrich) buffer, the streptavidin beads were suspended in 60 μL Laemmli Sample buffer (Bio-rad, Hercules, CA) and then boiled for 5 min; the precipitated proteins were separated on 4-20% Mini Protein Gels (Life Technologies). The gels were stained using the silver stain kit (Thermo Fisher Scientific). The stained protein bands of interest were recovered from the gel, and analyzed by liquid chromatography-mass spectrometry (LC-MS) for identification (HMRI Proteomics Programmatic Core, Houston, TX).
Biostability assays of aptamers
Synthetic ssDNA aptamers were incubated with RPMI-1640 medium containing 10% human serum for 24 h at 37°C, and the residual reaction products were collected at different time points using the phenol-chloroform extraction method as previously described (19). The aptamer products were analyzed on a 3% agarose gel and quantified by the ImageJ software.
Cell function assays
To evaluate cell proliferation, the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay was performed. Briefly, cells were treated daily with aptamers or the ssDNA library as a control in RPMI 1640 medium containing 10% serum for up to 72 h. At each set time point, 10 μL of MTT solution was added to each well and incubated for 4 h, followed by addition of isopropanol/HCl (100 μL) and incubation for 30 min. The absorbance at 570 nm was measured with a microplate reader as described previously (20, 21).
Cell growth was also detected by the CFSE staining assay. Cells were pre-stained with CFSE (6 μmol/L) and then treated with 1.2 µmol/L aptamer HL-1 or the control ssDNA library. After culturing the cells for 3 days, they were collected and the changes in cellular fluorescence were measured by flow cytometry.
For the cell cycle study, cells (1x106) were treated with 1.2 µmol/L aptamer HL-1 or the control ssDNA library for 3 days in RPMI 1640 medium containing 10% serum. Cells were then collected and washed with cold PBS buffer without Mg2+ ions, and fixed in 70% ethanol at -20°C for 1 h. Subsequently, the cells were incubated with propidium iodide solution: 10 µg/mL PI, 0.2 mg/mL RNase A (Sigma Aldrich) for 30 min at RT. After washing with cold PBS buffer, the cells were suspended in 500 µL cold PBS buffer, and cell cycle analysis was performed by flow cytometry (FACS caliber, BD Biosciences, San Jose, CA, USA) with Cellquest software.
To rule out non-specific toxicity, normal T cells were isolated from healthy donor blood samples with CD3 antibody-conjugated magnetic beads (Miltenyi Biotec, Auburn, CA) using AutoMACS (Miltenyi Biotec) as previously described (21). T cells were then treated with aptamer (0.3 µmol/L) or the same amount of ssDNA library for 3 days. Cell proliferation rates were measured by the MTT assay.
Cell endocytosis study
To investigate aptamer internalization, cells were treated with 0.6 µmol/L Cy3-labeled aptamer HL-1 for 2 h, and subsequently membrane-stained by CellMask™ Green Plasma Membrane Stain (Thermo Fisher Scientific) for 30 min in DPBS buffer. After washing twice, slides of cell smears were prepared, and intracellular aptamer signals were examined under a FluoViewTM FV1000 confocal microscope (Olympus). For confirmation, cells were pre-treated with 80 µmol/L dynasore (Sigma Aldrich), an endocytosis inhibitor, in DPBS buffer for 30 min. Cells were then subjected to aptamer treatment and membrane staining, and examined under the confocal microscope as described above.
In addition, to evaluate the effect of endocytosis on cellular function, cells were treated with or without dynasore in DPBS for 30 min and then exposed to 0.6 µmol/L aptamer treatment for 2 h as described above. After washing 3 times, cells were suspended in RPMI 1640 medium containing 10% serum, and then cells were seeded in a 96-well plate (5000 cells/well, 5 wells/condition). Daily aptamer treatments were conducted for the next two days as described above. After culturing the cells for 3 days, the MTT assay for cell proliferation was performed.
Combination treatment of aptamer and cytarabine
Maver-1 cells were pre-treated with 0.15 µmol/L of aptamers daily for 3 days and then exposed to cytarabine chemotherapy at different final concentrations for 2 additional days. Control groups were exposed to aptamer and cytarabine treatment only for 3 and 2 days, respectively. Untreated cells were used for comparison. Changes in cell proliferation rates were determined by the MTT assay as described above.
To investigate cell viability, the treated cells were stained in wells with Acridine Orange/Ethidium Bromide (AO/EB) staining solution at RT for 30 min (22). Cells were then examined under the microscope, and a total of five hundred stained cells were counted. Viable cells showed uniform green fluorescence. Apoptotic cells displayed dim orange fluorescence signal on condensed chromatin, and dead cells displayed red fluorescence (23). Finally, the percentages of viable, apoptotic and dead cells were calculated in individual groups subjected to different treatments.
Statistics
In the binding affinity graphs, standard deviation values have been depicted with error bars; the average readings from three different experiments were used. The graphs were plotted using MS-Excel 2010, and the data were analyzed with the integral Solver tool pack.
Results
High affinity of ssDNA aptamer for Maver-1 lymphoma cells
To develop ssDNA aptamers specific for Maver-1 cells, a hybrid SELEX approach including cell- and biomarker-based selections was conducted (Fig. S1). It included 7 rounds of enrichment with Maver-1 cells and counter elimination with Jeko-1 cells, and 2 subsequent rounds of section with purified Ig lambda light chain proteins. Evolution of aptamer selection was monitored by quantifying specific cell binding of aptamer pools from the 4th, 7th, and 9th rounds of SELEX. Flow cytometry showed that the aptamer pools gradually gained binding capacity to Maver-1 cells, but did not react with Jeko-1 lymphoma cells that do not express Ig lambda proteins (Fig. S2A). Sequencing of the final aptamer pool revealed a top dominant sequence accounting for 36.62% out of a total of 5 million reads (Fig. S2B). In addition, cell-binding evolution of the top 10 dominant aptamer sequences (%) was compared for the 4th, 7th, and 9th rounds of SELEX (Fig. S2C).
Notably, although the top two dominant aptamers (named HL-1 and HL-2) differed in the center of the core region by a single base only (Fig. 1A), their secondary structures, predicated by Unpack software, differed significantly (Fig. 1B). For the cell binding studies, aptamers HL-1 and HL-2 were synthesized and labeled with the Cy3 fluorochrome reporter. Cultured cells were treated with aptamers at different concentrations, and the resultant cell binding was quantified by flow cytometry analysis. The data plotted in Fig. 1C revealed that aptamers specifically bound to Maver-1 lymphoma cells with high affinities (HL-1 and HL-2 had Kd = 70±8 pmol/L and 472±104 pmol/L, respectively) but did not react with Jeko-1 control cells under the same conditions. To examine the cell-binding specificity of the aptamers, multiple cell lines were tested, and anti-Ig lambda light chain antibody was used as a control. Flow cytometry showed that aptamers HL-1 and HL-2 specifically targeted Maver-1 lymphoma cells with a nearly identical binding pattern to that detected for the antibody (Fig. S2D, Fig. 1D) but did not react with the other cells.
To determine the minimal functional sequence, truncated forms of aptamer HL-1 were synthesized, including sequences with full-length, 3'del, 5'del, Core region, h'del, and Loop region (Fig. 1E). Maver-1 cells were treated with individual aptamer forms at a final concentration of 50 nmol/L for 30 min. Quantitative cell binding assays, conducted by flow cytometry, indicated that the full-length sequence had the highest binding ability for Maver-1 cells, and that all sequence truncations resulted in partial or complete abrogation of the cell binding capacity (Fig. 1E). These findings indicated that the full-length sequence was required for high-affinity cell binding of aptamer HL-1 and therefore, it was used for the ensuing studies.
Specific binding of aptamer to Maver-1 cells with high affinity
For further confirmation, cells were treated with Cy3-labeled aptamer and nuclear stained for tracking purposes. Fluorescence microscopy revealed that cytoplasmic red fluorescence signal arised from aptamer HL-1 within Maver-1 cells but not from off-target Jeko-1 cells (Fig. 2A). Similarly, a cell mixture containing both Maver-1 and Jeko-1 cells was also treated with aptamer and nuclear-stained. The images in Fig. 2B showed that the aptamer HL-1 selectively stained Maver-1 cells, and did not react with off-target Jeko-1 cells (pre-stained with green fluorescence) in the same cell mixture. Detection of cytoplasmic signal indicated that the aptamer HL-1 specifically bound to and was selectively internalized into Maver-1 cells.
To test binding specificity, a competitive cell-binding assay was performed using anti-Ig lambda light chain antibody. Maver-1 cells were simultaneously treated with FITC-labeled antibody and aptamer HL-1, or an equimolar ssDNA library as a control. The data in Fig. 2C showed that antibody binding to Maver-1 cells was significantly inhibited by the presence of aptamer HL-1, but was not affected by the ssDNA library under the same conditions. For further validation, immunoprecipitation of cell lysates was performed with the aptamers, and the obtained cellular proteins were analyzed by PAGE electrophoresis. Silver staining of the gel revealed a cellular protein band with MW ~26 kDa due to Maver-1 cell products, which were immunoprecipitated by aptamers HL-1 and HL-2 (Fig. 2D, upper panel). The detected ~26 kDa protein bands were then recovered from the gel for amino acid sequencing. Analysis by mass spectrometry identified an 8-amino acid peptide (VTVLGQPK), which was matched to Accession ID: B9A064.2 in UniProtKB/Swiss-Plot database, and was consistent with the immunoglobulin lambda-like polypeptide 5. Notably, although a weak band was found with a control aptamer specific for CD38 (12), no lambda-like polypeptide sequence was identified by mass spectrometry. In addition, no cellular proteins with MW ~26 kDa were immunoprecipitated from off-target Jeko-1 cells (Fig. 2D, lower panel).
Aptamer endocytosis resulted in growth inhibition of target cells
Sequence analysis by QGRS Mapper program predicted that aptamer HL-1 likely forms G-quadruplex structures with two flat planes (Fig. 3A), which have been reported to be associated with aptamer stability and cellular functions (24-26). The G-quadruplex structure in aptamer HL-1 was further confirmed with Thioflavin T staining (Fig. S3). To test biostability, aptamer HL-1 was incubated in culture medium containing 10% human serum at 37°C, and residual products were recovered at different time points and analyzed by gel electrophoresis. As shown in Fig. 3B, 87% and 65% intact aptamer remained 8 and 24 h post incubation, respectively.
High affinity binding of aptamer HL-1 to Maver-1 lymphoma cells. (A) Sequences of the developed aptamers HL-1 and HL-2. (B) Predicted secondary structures of aptamer sequences. (C) High affinity binding of aptamers to Maver-1 lymphoma cells with no reaction to off-target control Jeko-1 cells. Cultured cells were treated with Cy-3 labeled aptamers at different final concentrations, and the resultant cell binding was quantified by flow cytometry. (D) Specificities of aptamers were validated at 20 nmol/L in multiple cultured cell lines with anti-lambda light chain antibody as a control. Cell binding was examined by flow cytometry. (E) Function characterization of aptamer sequences. Aptamer HL-1 was truncated and tested with or without primer region(s) and/or an intact core. Binding of each truncated sequence to Maver-1 lymphoma cells at 50 nmol/L final concentration was quantified by flow cytometry. Relative cell binding capacities (%) of truncated aptamers were normalized to the full-length aptamer HL-1 (used as baseline).
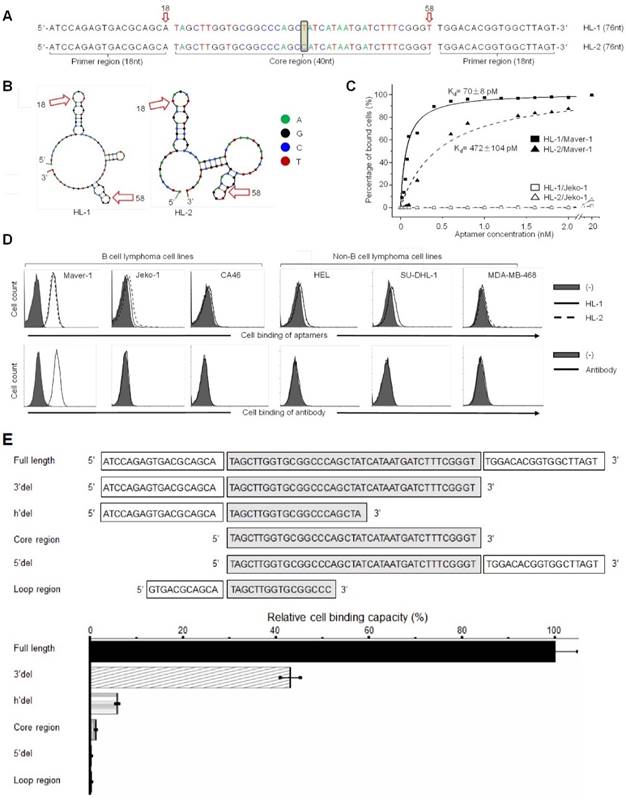
Specific targeting of Maver-1 lymphoma cells and immuno-precipitation of immunoglobulin lambda-like polypeptide 5. (A) Specific cell staining of aptamer. Culture cells were stained with Cy3-labeled aptamer HL-1 followed by nuclear staining with dye Hoechst 33342 (2 µg/mL). Stained cells were observed under the fluorescence microscope. (B) Specific cell targeting of aptamer. Cell mixture of Maver-1 and pre-stained Jeko-1 cells were treated with aptamer HL-1. Cell binding was monitored under a fluorescence microscope by detecting Maver-1 cells targeted by aptamer (red fluorescence) and control Jeko-1 cells (green fluorescence). (C) Competitive cell-binding. Cells were treated with FITC-labeled anti-Ig lambda light chain antibody with or without the presence of synthetic aptamer HL-1 (1 μmol/L) or ssDNA library as a control. Changes in cell binding of the antibody were quantified by flow cytometry. (D) Immunoprecipitation of cellular proteins by aptamers. Maver-1 and Jeko-1 cell lysates were immunoprecipitated by 100 nmol/L aptamer HL-1 or control aptamer CD38. Cellular proteins were then separated on SDS-PAGE and visualized by silver staining. Protein bands with molecular weight ~26 kDa were recovered for mass spectrometry analysis and matched to immunoglobulin lambda-like polypeptide 5 (UniProtKB/Swiss-Plot database).
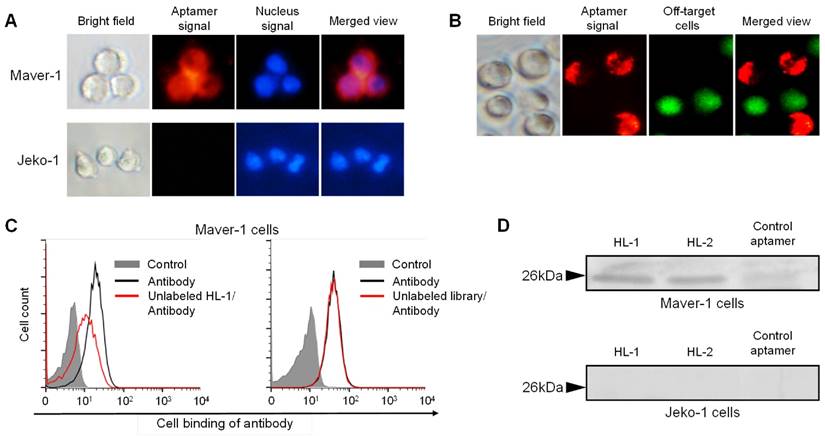
Subsequently, to test potential cellular functions, lymphoma cells were exposed to aptamer HL-1 at different final concentrations for 3 days, and the resultant changes in cell proliferation rates were monitored by MTT assay. The data in Fig. 3C showed that aptamer treatment resulted in significant growth inhibition of Maver-1 lymphoma cells 3 days post treatment, but had no effect on off-target Jeko-1 lymphoma cells under the same conditions. In control experiments, the synthetic ssDNA library showed no effects on cell growth.
For further confirmation, Maver-1 lymphoma cells were pre-stained with CFSE, and then treated with aptamer HL-1 or ssDNA library for 3 days as shown in Fig. 3C. Changes in cellular fluorescence, correlated to cell proliferation, were quantified by flow cytometry. Similarly, aptamer treatment had an inhibitory effect on cell proliferation evident at day 3, but no effect was detected during the first two days (Fig. 4A). To rule out side effects on off-target cells, T cells were isolated from blood of healthy donors and treated with the aptamer under the same conditions. The plot in Fig. 4B showed that aptamer treatment had no detectable effect on normal T cell proliferation. Taken together, these findings demonstrate that, in addition to specific cell binding to Maver-1 lymphoma cells, aptamer HL-1 also plays a role in regulating cell proliferation.
To dissect the potential mechanism involved in aptamer cellular functions, we initially studied whether the aptamer acts on the cell surface or within targeted cells. To this end, Maver-1 lymphoma cells were membrane-stained and treated with Cy3-labeled aptamer simultaneously. Confocal microscopy revealed green fluorescent cell membranes and abundant intracellular red fluorescence, indicating rapid endocytosis of the aptamer into the target cells (Fig. 4C upper panel). For the validation studies, cells were pretreated with the endocytosis inhibitor dynasore (27) to block aptamer internalization (28), and then treated with Cy3-labeled aptamer, as well as fluorescent dye for cell membrane staining. Confocal microscopy showed green fluorescence in the cell membranes, but no intracellular aptamer signal (red fluorescence) in the merged image (Fig. 4C lower panel), confirming that the resultant internalization of aptamer was mediated by cell endocytosis.
Aptamer induced cell growth inhibition. (A) Predicted G-quadruplex structures with two flat planes in aptamer HL-1. (B) Biostability assay. Aptamer HL-1 was incubated in culture medium containing 10% human serum at 37°C, and residual products were measured by gel electrophoresis at different time points. (C) Cell proliferation analysis. Cells were treated with aptamer HL-1 at different final concentrations or synthetic ssDNA library as control, and changes in cell growth were monitored by MTT proliferation assay. The aptamer induced significant growth inhibition of Maver-1 cells 3 days post treatment, but had no effect on off-target Jeko-1 cells.
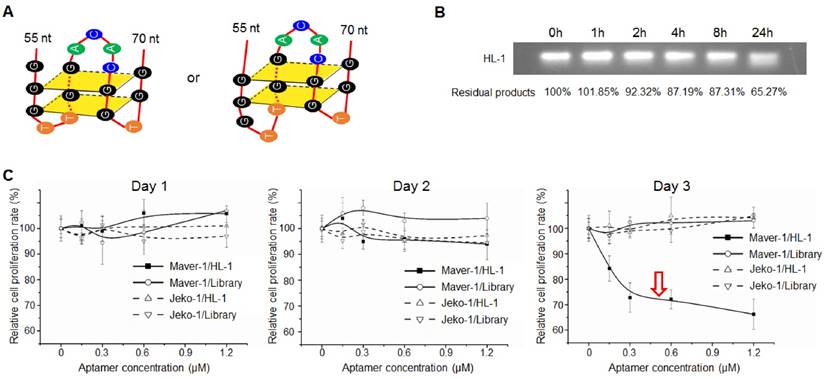
For the cell function studies, Maver-1 lymphoma cells were treated with the aptamer with or without pre-exposure to the endocytosis inhibitor under the same conditions as described above. Changes in cell growth were examined by MTT cell proliferation assays 3 days post treatment. In comparison to the non-treatment control group, aptamer treatment induced significant inhibition of cell proliferation, and importantly, blockage of cell endocytosis by pre-exposure to the inhibitor completely eliminated the inhibitory effect of the aptamer on cell proliferation (Fig. 4D). Notably, the observed variation in the combination treatment group was due to the adverse effect of the inhibitor dynasore (13% inhibition induced by dynasore alone vs. 11% inhibition observed in combination treatment with dynasore and aptamer). These findings demonstrated that aptamer internalization specifically into target cells via cell endocytosis is indispensable for inhibition of Maver-1 cell growth.
Aptamer induced S-phase arrest and primed Maver-1 lymphoma cells for synergistic killing via cytarabine chemotherapy
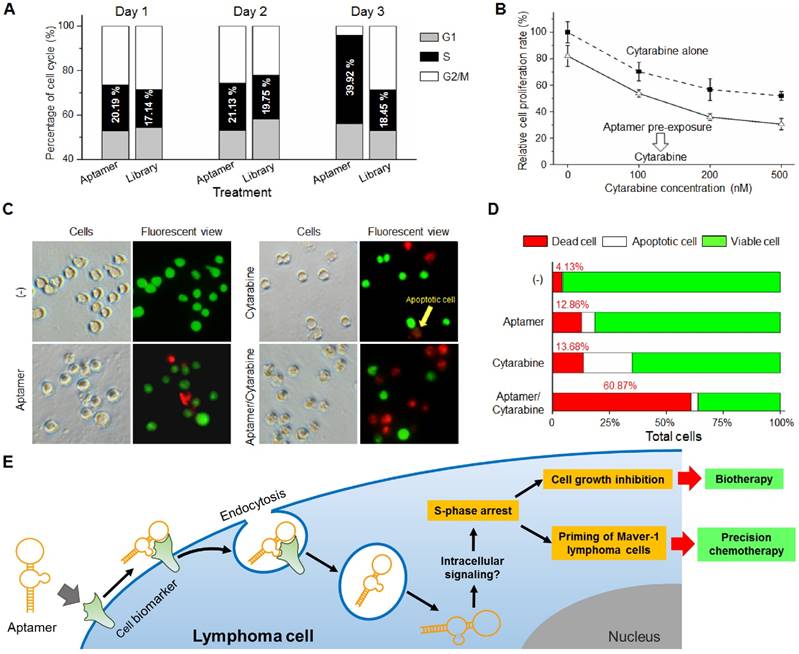
To understand the potential cellular events involving growth inhibition, Maver-1 cells were exposed to aptamer HL-1 and cell cycle analysis was performed. Quantification analysis by flow cytometry indicated that aptamer treatment induced S-phase arrest of Maver-1 cells, reaching about 39.92% on day three (vs. 18.45% in the control experiment with ssDNA library treatment) and resulted in a significantly reduced number of cells that entered G2/M phase (Fig. 5A). Notably, similarly to the observed growth inhibition (Fig. 3C), aptamer treatment had no cellular effects on the first two days and selectively induced cell S-phase arrest on day 3.
Because aptamer treatment can cause S-phase arrest of Maver-1 lymphoma cells, it was reasonable to hypothesize that the treated cells could become more sensitive to treatment in the presence of an S-phase-specific chemotherapeutic agent. Cytarabine is a common chemotherapeutic drug for lymphoma/leukemia, and selectively acts on cells at S-phase (29). To test the potential synergistic effect, Maver-1 lymphoma cells were pre-exposed to aptamer HL-1 and then treated with cytarabine. Cell proliferation assays showed that cytarabine treatment alone induced growth inhibition in a dose dependent pattern (Fig. 5B). Interestingly, aptamer pre-exposure primed Maver-1 lymphoma cells to cytarabine treatment, resulting in additional inhibition in cell proliferation.
Although both aptamer and cytarabine collectively showed an inhibitory effect on Maver-1 cell growth, they may have different functions in regulating cell viability, apoptosis, and death. To address this question, Maver-1 lymphoma cells were treated with aptamer or cytarabine alone. In addition, cells were also primed by aptamer pre-exposure for 3 days and were then treated with cytarabine for 2 additional days. After treatment, cells were collected and stained with AO/EB solution for fluorescent microscopic examination of viable cells, apoptotic cells, and dead cells, which were detected under the fluorescent microscope as shown in Fig. 5C. For quantification, in each experiment, five hundred cells were counted, and the percentage of viable, apoptotic, and dead cells were calculated. Fig. 5D revealed that aptamer pre-exposure significantly primed cells for cytarabine treatment, achieving a synergetic killing effect of 60.87% cell death (vs. 12.86% by aptamer treatment alone or 13.7% by cytarabine treatment alone).
Discussion
In this study, we demonstrated that in addition to specific cell-binding, aptamers may also act as biotherapeutic agents by regulating cellular functions, thus opening a new avenue for precision therapy. As depicted in Fig. 5E, aptamer HL-1 specifically binds Maver-1 lymphoma cells and triggers rapid internalization into target cells via endocytosis. Although the exact molecular mechanism is unknown, the internalized aptamer may regulate intracellular signaling pathway(s) and subsequently induce S-phase arrest of target cells, resulting in growth inhibition. Importantly, the induced S-phase arrest by aptamer exposure will prime target cells for chemotherapeutic agents, such as cytarabine, which selectively kills tumor cells at S-phase. Therefore, logical combination of aptamer priming and chemotherapy will achieve a synergistic killing effect on targeted tumor cells, providing an additional platform for precision cancer therapy.
Notably, aptamer treatment induced a delayed response in targeted Maver-1 lymphoma cells, leading to S-phase arrest and growth inhibition 3 days post treatment. These findings suggest that aptamer-induced cellular effects may not be due to rapid changes in activation status of cellular signaling pathways, such as those regulated by protein phosphorylation or de-phosphorylation. The delayed cellular effects most likely result from slow changes in gene expression and/or protein synthesis, which control cell cycles. Therefore, to dissect the potential molecular mechanism involved in aptamer cellular functions, Maver-1 lymphoma cells were treated with aptamer for 3 days as described above. Cellular signaling pathways mediated via cyclin D1, cyclin D3, CDK2, CDK4, CDK6, p27 Kip1 and p21 Waf1/Cip1 were examined by Western blot assays, and showed no changes in the expression levels of these proteins with or without aptamer treatment (data not shown).
Internalization of aptamer via endocytosis into target cells is indispensable for cell growth inhibition. (A) Cell growth study. Maver-1 lymphoma cells were pre-stained with CFSE dye and then treated with 1.2 µmol/L aptamer HL-1 or ssDNA library. Flow cytometry revealed a delayed cell growth/division by aptamer treatment on day 3. (B) MTT proliferation assay showed no effect on normal T cell growth 3 days post aptamer treatment (1.2 µmol/L). (C) Aptamer internalization via cell endocytosis. Maver-1 lymphoma cells were treated with 0.6 µmol/L aptamer HL-1 and also had their membranes stained green. Confocal microscopy revealed that the intracellular signal, derived from internalized aptamer (red fluorescence), was completely blocked in the presence of endocytosis inhibitor dynasore. (D) Cell proliferation assay indicated that the growth inhibition induced by aptamer treatment (0.6 µmol/L) was prevented by endocytosis inhibitor dynasore, although dynasore alone showed mild toxicity background. Data are depicted as the mean ± s.d. of three independent experiments (*p<0.05).
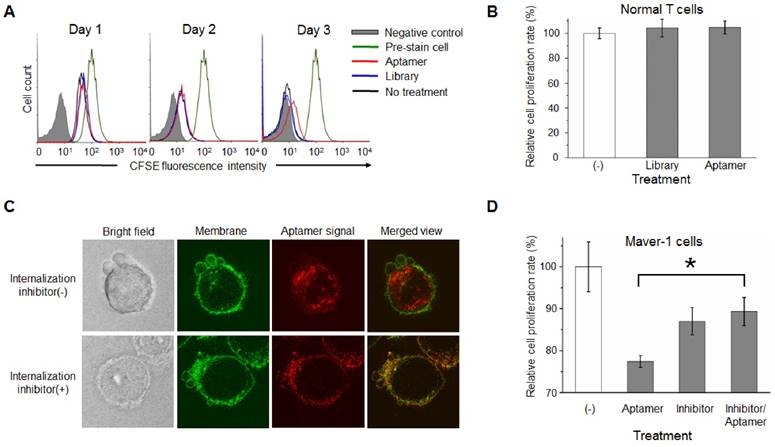
Aptamer induced S-phase arrest and primed Maver-1 lymphoma cells for cytarabine chemotherapy. (A) Cell S-phase arrest. Maver-1 lymphoma cells were treated with 1.2 µmol/L aptamer HL-1 or synthetic ssDNA library as control. Cell cycle analysis revealed a significantly increased cell S-phase arrest 3 days post aptamer treatment. (B) Enhanced effect by aptamer/cytarabine combination treatment. Maver-1 lymphoma cells were pre-exposed to 0.15 µmol/L aptamer or not exposed to it, and then treated with cytarabine, a chemotherapeutic agent that primarily kills tumor cells at S-phase. Aptamer pre-exposure enhanced cytarabine-induced growth inhibition at all tested doses. Data are shown as the mean ± s.d. of three independent experiments. (C) Different cellular effects of aptamer and cytarabine. To dissect cellular effects, Maver-1 lymphoma cells were treated with aptamer alone or cytarabine with or without aptamer pre-exposure. Cells were collected and stained with AO/EB solution for fluorescent microscopic examination 3 days after each treatment. Resultant viable cells (green fluorescence), apoptotic cells (dim orange), and dead cells (red fluorescence) were examined under a fluorescent microscope. (D) Synergistic cell killing effect by aptamer pre-exposure and cytarabine combination treatment. For quantitative analysis, five hundred cells in each experiment in C were counted under the fluorescent microscope and the percentage of viable, apoptotic, and dead cells were calculated. Aptamer pre-exposure primed Maver-1 lymphoma cells for cytarabine treatment, achieving a synergistic killing effect with 60.87% cell death (vs. 12.86% by aptamer alone and 13.7% by cytarabine alone). (E) Depiction of aptamer-mediated biotherapy and precision chemotherapy. Specific cell-binding resulting in rapid aptamer internalization into cells via endocytosis. Through unknown intracellular signaling pathway(s), aptamer induces S-phase arrest and growth inhibition of targeted cells, leading to a new biotherapeutic method by regulating cellular functions without use of toxic agent. In addition, the aptamer inducing S-phase arrest will prime targeted cells for chemotherapeutic agents that principally kill S-phase tumor cells, thus providing a unique precision cancer chemotherapeutic agent.
On the other hand, the presence of G-quadruplex structures or G-rich sequences has been associated with aptamer cellular functions (24-26). As shown in Fig. 3A, aptamer HL-1 contains complete G-quadruplex structures with two flat planes (nucleotides 55'-70'). For validation, Thioflavin T staining assay was conducted by incubating synthetic aptamers HL-1 with Thioflavin T dye (30). High fluorescence signals were detected in reaction, confirming the presence of G-quadruplex structure in aptamer HL-1 sequence (Fig. S3). Partial deletion of the G-quadruplex showed more than 55% reduction of aptamer cell-binding capacity (aptamer 3'del shown in Fig. 1E), although this deletion may not have an effect on its functional structure predicted in Fig. 1A. Surprisingly, cell assays revealed that the aptamer 3'del had very similar capacity to inhibit Maver-1 cell growth as compared to the full length aptamer HL-1 (data not shown), suggesting that the full length aptamer is critical for cell binding and that the aptamer 3'del sequence contains essential structural features for cell inhibition. It's reported that the aptamer AS1411, which contains G-quadruplex (31, 32), could induce cell cycle arrest of cancer cells by targeting nucleolin (31, 33). To determine whether aptamer HL1 binds to nucleolin proteins, immunoprecipitation of Maver-1 cell lysates were performed with aptamer HL-1. Western blot analysis of precipitated cellular proteins revealed that aptamer HL-1 did not react to nucleolin (Fig. S4).
Further studies to elucidate the molecular mechanism and sequence structure involved in the function of the aptamers reported herein are currently underway, and the findings will be reported in the near future.
Conclusions
Our study demonstrated that in addition to specific binding to Maver-1 lymphoma cells, aptamer HL-1 also functions intracellularly via endocytosis and induces S-phase arrest of cells, thereby inhibiting growth of the targeted lymphoma cells by regulating tumor cell function. Moreover, aptamer-induced S-phase arrest can also prime tumor cells for S phase-specific chemotherapy, resulting in synergistic toxicity in targeted cells. Our findings clearly showed the enhanced value of aptamer HL-1 as a unique “precision cancer biotherapeutic” and its synergistic killing effect of lymphoma cells in combination with chemotherapy.
Supplementary Material
Supplementary figures.
Acknowledgements
This project was supported in part by NIH grants R01CA151955 and R33CA173382 to Y.Z.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Banerjee J, Nilsen-Hamilton M. Aptamers: multifunctional molecules for biomedical research. Journal of molecular medicine. 2013Dec;91(12):1333-42
2. Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990Aug30;346(6287):818-22
3. Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990Aug3;249(4968):505-10
4. Jayasena SD. Aptamers: an emerging class of molecules that rival antibodies in diagnostics. Clinical chemistry. 1999Sep;45(9):1628-50
5. Shangguan D, Li Y, Tang Z, Cao ZC, Chen HW, Mallikaratchy P. et al. Aptamers evolved from live cells as effective molecular probes for cancer study. Proceedings of the National Academy of Sciences of the United States of America. 2006Aug8;103(32):11838-43
6. Sun H, Zhu X, Lu PY, Rosato RR, Tan W, Zu Y. Oligonucleotide aptamers: new tools for targeted cancer therapy. Molecular therapy Nucleic acids. 2014;3:e182
7. Que-Gewirth NS, Sullenger BA. Gene therapy progress and prospects: RNA aptamers. Gene therapy. 2007Feb;14(4):283-91
8. Sundaram P, Kurniawan H, Byrne ME, Wower J. Therapeutic RNA aptamers in clinical trials. European journal of pharmaceutical sciences: official journal of the European Federation for Pharmaceutical Sciences. 2013Jan23;48(1-2):259-71
9. Zeng Z, Tung CH, Zu Y. A cancer cell-activatable aptamer-reporter system for one-step assay of circulating tumor cells. Molecular therapy Nucleic acids. 2014;3:e184
10. Zeng Z, Zhang P, Zhao N, Sheehan AM, Tung CH, Chang CC. et al. Using oligonucleotide aptamer probes for immunostaining of formalin-fixed and paraffin-embedded tissues. Modern pathology: an official journal of the United States and Canadian Academy of Pathology, Inc. 2010Dec;23(12):1553-8
11. Zeng Z, Parekh P, Li Z, Shi ZZ, Tung CH, Zu Y. Specific and sensitive tumor imaging using biostable oligonucleotide aptamer probes. Theranostics. 2014;4(9):945-52
12. Wen J, Tao W, Hao S, Iyer SP, Zu Y. A unique aptamer-drug conjugate for targeted therapy of multiple myeloma. Leukemia. 2016Apr;30(4):987-91
13. Dotan E, Aggarwal C, Smith MR. Impact of Rituximab (Rituxan) on the Treatment of B-Cell Non-Hodgkin's Lymphoma. P & T: a peer-reviewed journal for formulary management. 2010Mar;35(3):148-57
14. Cheah CY, Fowler NH, Wang ML. Breakthrough therapies in B-cell non-Hodgkin lymphoma. Ann Oncol. 2016 Jan 22
15. Reff ME, Carner K, Chambers KS, Chinn PC, Leonard JE, Raab R. et al. Depletion of B cells in vivo by a chimeric mouse human monoclonal antibody to CD20. Blood. 1994Jan15;83(2):435-45
16. Manzur S, Cohen S, Haimovich J, Hollander N. Enhanced therapeutic effect of B cell-depleting anti-CD20 antibodies upon combination with in-situ dendritic cell vaccination in advanced lymphoma. Clinical and experimental immunology. 2012Dec;170(3):291-9
17. de Martini RM, Turner RR, Boone DC, Lukes RJ, Parker JW. Lymphocyte immunophenotyping of B-cell lymphomas: a flow cytometric analysis of neoplastic and nonneoplastic cells in 271 cases. Clinical immunology and immunopathology. 1988Dec;49(3):365-79
18. Fukushima PI, Nguyen PK, O'Grady P, Stetler-Stevenson M. Flow cytometric analysis of kappa and lambda light chain expression in evaluation of specimens for B-cell neoplasia. Cytometry. 1996Dec15;26(4):243-52
19. Parekh P, Kamble S, Zhao N, Zeng Z, Portier BP, Zu Y. Immunotherapy of CD30-expressing lymphoma using a highly stable ssDNA aptamer. Biomaterials. 2013Nov;34(35):8909-17
20. Wen J, Cheng HY, Feng Y, Rice L, Liu S, Mo A. et al. P38 MAPK inhibition enhancing ATO-induced cytotoxicity against multiple myeloma cells. Br J Haematol. 2008Jan;140(2):169-80
21. Wen J, Feng Y, Bjorklund CC, Wang M, Orlowski RZ, Shi ZZ. et al. Luteinizing Hormone-Releasing Hormone (LHRH)-I antagonist cetrorelix inhibits myeloma cell growth in vitro and in vivo. Mol Cancer Ther. 2011Jan;10(1):148-58
22. Curcic MG, Stankovic MS, Mrkalic EM, Matovic ZD, Bankovic DD, Cvetkovic DM. et al. Antiproliferative and proapoptotic activities of methanolic extracts from Ligustrum vulgare L. as an individual treatment and in combination with palladium complex. Int J Mol Sci. 2012;13(2):2521-34
23. Baskic D, Popovic S, Ristic P, Arsenijevic NN. Analysis of cycloheximide-induced apoptosis in human leukocytes: fluorescence microscopy using annexin V/propidium iodide versus acridin orange/ethidium bromide. Cell Biol Int. 2006Nov;30(11):924-32
24. Garcia-Recio EM, Pinto-Diez C, Perez-Morgado MI, Garcia-Hernandez M, Fernandez G, Martin ME. et al. Characterization of MNK1b DNA Aptamers That Inhibit Proliferation in MDA-MB231 Breast Cancer Cells. Molecular therapy Nucleic acids. 2016;5:e275
25. Xu XH, Hamhouyia F, Thomas SD, Burke TJ, Girvan AC, McGregor WG. et al. Inhibition of DNA replication and induction of S phase cell cycle arrest by G-rich oligonucleotides. Journal of Biological Chemistry. 2001Nov16;276(46):43221-30
26. Ireson CR, Kelland LR. Discovery and development of anticancer aptamers. Molecular cancer therapeutics. 2006Dec;5(12):2957-62
27. Kirchhausen T, Macia E, Pelish HE. Use of dynasore, the small molecule inhibitor of dynamin, in the regulation of endocytosis. Methods in enzymology. 2008;438:77-93
28. Opazo F, Eiden L, Hansen L, Rohrbach F, Wengel J, Kjems J. et al. Modular Assembly of Cell-targeting Devices Based on an Uncommon G-quadruplex Aptamer. Molecular therapy Nucleic acids. 2015;4:e251
29. Hamada A, Kawaguchi T, Nakano M. Clinical pharmacokinetics of cytarabine formulations. Clinical pharmacokinetics. 2002;41(10):705-18
30. Renaud de la Faverie A, Guedin A, Bedrat A, Yatsunyk LA, Mergny JL. Thioflavin T as a fluorescence light-up probe for G4 formation. Nucleic Acids Res. 2014Apr;42(8):e65
31. Bates PJ, Kahlon JB, Thomas SD, Trent JO, Miller DM. Antiproliferative activity of G-rich oligonucleotides correlates with protein binding. J Biol Chem. 1999Sep10;274(37):26369-77
32. Hanakahi LA, Sun H, Maizels N. High affinity interactions of nucleolin with G-G-paired rDNA. J Biol Chem. 1999May28;274(22):15908-12
33. Soundararajan S, Chen W, Spicer EK, Courtenay-Luck N, Fernandes DJ. The nucleolin targeting aptamer AS1411 destabilizes Bcl-2 messenger RNA in human breast cancer cells. Cancer Res. 2008Apr1;68(7):2358-65
Author contact
![]() Corresponding authors: jwenorg (J. Wen). yzuorg (Y. Zu).
Corresponding authors: jwenorg (J. Wen). yzuorg (Y. Zu).