Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and methods
Results and discussion
Conclusions
Supplementary Material
Acknowledgements
References
Introduction
Materials and methods
Results and discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(5):1192-1203. doi:10.7150/thno.17881 This issue Cite
Research Paper
Phosphorylcholine-based stealthy nanocapsules enabling tumor microenvironment-responsive doxorubicin release for tumor suppression
Gan Liu1,2*, Hsiang-i Tsai1,2*, Xiaowei Zeng1,2*, Yixiong Zuo1,2, Wei Tao1,2, Jun Han3, Lin Mei1,2 ![]()
1. The Shenzhen Key Lab of Gene and Antibody Therapy and Division of Life and Health Sciences, Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, P. R. China;
2. School of Life Sciences, Tsinghua University, Beijing 100084, PR China;
3. Institute of Biopharmaceutical Research, Liaocheng University, Liaocheng 252059, P.R.China.
* These three authors contributed equally to this work.
Received 2016-10-11; Accepted 2016-12-20; Published 2017-3-5
Citation:
Liu G, Tsai Hi, Zeng X, Zuo Y, Tao W, Han J, Mei L. Phosphorylcholine-based stealthy nanocapsules enabling tumor microenvironment-responsive doxorubicin release for tumor suppression. Theranostics 2017; 7(5):1192-1203. doi:10.7150/thno.17881. https://www.thno.org/v07p1192.htm
Other stylesAbstract

The efficient delivery of anticancer drugs into tumor tissues to improve therapeutic efficacy remains an urgent demand. To satisfy this demand, a drug delivery system based on a stealthy nanocapsule was developed. This nanocapsule was fabricated by encapsulating stealthy cross-linked poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC) and benzaldehyde groups around the protein bovine serum albumin (BSA) followed by conjugation of doxorubicin (Dox) through a pH-responsive benzoic-imine bond. The in vitro results show that the Dox-conjugated nanocapsule (nBSA-Dox) released the drug under an acidic tumor microenvironment (pH ~6.5) and killed HepG2 human liver cancer cells. The half-life of Dox conjugated to nBSA in mice was significantly prolonged, and the area-under-curve of plasma Dox of the mice treated with nBSA-Dox was as much as 242 fold of free Dox. The in vivo results confirmed that this nanocapsule efficiently accumulated in tumor tissue and significantly suppressed the tumor growth.
Keywords: stealthy nanocapsule, tumor suppression, phosphorylcholine, tumor microenvironment, benzoic-imine bond.
Introduction
In the past few years, the efficient delivery of clinically approved drugs using nanoscale vehicles (10-200 nm) for enhanced cancer therapy has attracted much attention, as many types of nanocarriers accumulate within solid tumors because of their enhanced permeability and retention (EPR) effect [1-10]. However, their delivery efficacy is still greatly hampered by several problems. First and foremost, although surface PEGylation is the current gold standard of nanomedicines for evading major clearance mechanisms, such as the mononuclear phagocyte system (MPS) and prolonging half-life [11-15], ~25% patients developed anti-PEG antibody after the first injection of PEGylated nanoparticles [16-19], resulting in the accelerated clearance of nanocarriers from the body. Therefore, stealthier nanomaterials have been investigated to prolong the circulation time for a superior therapeutic efficacy. Second, the delivery efficacy of nanocarriers also relies on their stability. Many widely studied nanocarriers, such as polymeric micelles, are fabricated by the self-assembly of amphiphilic polymers. Although they are very stable above the CMC in vitro, the vast dilution after injecting into the body allows them to disassociate rapidly [20-23], thus necessitating the nanocarriers with robust stability in vivo.
We previously reported a poly (2-methacryloyloxyethyl phosphoryl choline) (PMPC)-based stealthy protein nanocapsule, which was fabricated by the in-situ polymerization of MPC monomer and cross-linker around protein including bovine serum albumin (BSA) [24-27]. This cross-linked PMPC layer-encapsulated nanocapsule was quite stable and proved to significantly prolong half-life and reduce immunogenicity of the proteins because of the excellent antifouling ability of surface PMPC [26, 28]. We assumed that delivering chemotherapeutic agents by this stable stealthy nanocapsule would also effectively evade MPS and prolong half-life of the drugs, resulting in efficient EPR effect for cancer therapy. As far as we know, no such stealthy nanocapsule was reported for efficient delivery of chemotherapeutics into tumor before.
Actually, loading chemotherapeutics efficiently by this PMPC-based stealthy nanocapsule is an unneglectable challenge, because its completely hydrophilic and noble structure could not interact with the hydrophobic drugs. Some previous works reported to attach doxorubicin (Dox) onto BSA through covalent bond [29], whereas the attachable moieties of BSA are quite numbered, severely limiting the drug loading content. Herein, we designed to introduce benzaldehyde group (BzA) into the nanocapsule, which was reported to react with the amino group of Dox to form an acid-responsive benzoic-imine bond [30-33]. We ensure this loading strategy has several advantages, including increase loading content, avoid undesired premature release by covalent conjugation of drugs and achieve tumor microenvironment-responsive drug release.
Therefore, a Dox-conjugated stealthy nanocapsule was designed for enhanced cancer therapy. The PMPC-based BSA nanocapsule with BzA (nBSA-BzA) was fabricated through the in-situ polymerization of the monomers, MPC and methacrylamide benzaldehyde (MA-BzA) and cross-linker Glycerol dimethacrylate (GDA) around BSA, followed by Dox conjugation (Figure 1). The Dox-conjugated nBSA (nBSA-Dox) had a size of ~25 nm with an excellent stability because of its covalent cross-linking structure and significantly prolonged the half-life of Dox in mice. Furthermore, unlike the insensitive bond formed from carboxyl (COOH) and the amino group of Dox, the benzoic-imine bond formed from benzaldehyde and Dox is very stable under physiological conditions but cleaves quickly in an acidic tumor microenvironment. Using HepG2 human liver cancer xenograft-bearing nude mice as the tumor model, nBSA-Dox efficiently accumulated in tumor and exhibited outstanding tumor suppression. Therefore, this nanocapsule, exhibiting a prolonged circulation time, improved tumor accumulation and tumor microenvironment-responsive drug release, has great potential applications in cancer therapy.
Figure 1
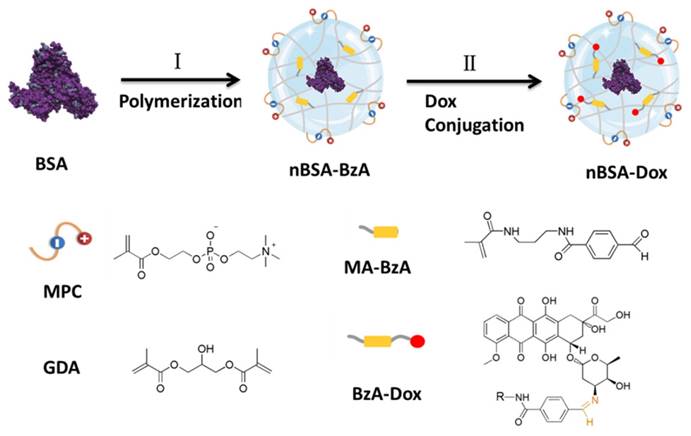
Schematic illustration of the synthesis of Dox-conjugated stealthy nanocapsule. (I) in-situ polymerization of the monomer (MPC and MA-BzA) and degradable cross-linker (GDA) around the BSA to obtain nBSA-BzA, (II) Conjugation of Dox into nBSA-BzA to form nBSA-Dox.
Materials and methods
Materials
BSA, MPC, fluorescein isothiocyanate (FITC), and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Glycerol dimethacrylate (GDA), ammonium persulfate (APS), N,N,N',N'-tetramethylethylenediamine (TEMED) and succinic anhydride (SA) were purchased from Aladdin Industrial (Shanghai, China). N-(3-Aminopropyl) methacrylamide hydrochloride (Apm) was purchased from Polymer Science. N-Succinimidyl 4-forMylbenzoate (SFB) and Dox were purchased from J&K Chemical (Shanghai, China). MA-BzA was synthesized by mixing Apm and SFB under stirring in DMSO for 2 h. Alexa Fluor® 750 NHS Ester was purchased from Thermo Fisher Scientific. All the chemicals were commercially available and used as without further purification. HepG2 cells were purchased from American Type Culture Collection (ATCC). Fetal bovine serum (FBS) was purchased from Lonza Walkerrsville. The Dulbecco's Modified Eagle Medium (DMEM) growth medium and penicillin/streptomycin were purchased from Invitrogen. Female Balb/C nude mice (6 weeks old) were purchased from Guangdong Province Medical Animal Center and maintained in an SPF (specific pathogen free) class experimental animal room.
Preparation of Dox-conjugated nanocapsule nBSA-Dox
The nanocapsule nBSA-BzA was synthesized similar to our previous work [26]. Briefly, in-situ radical polymerization around BSA at 1 mg/mL in phosphate-buffered saline (PBS) was initiated by APS and TEMED. A specific amount of MPC, MA-BzA and GDA (molar ratio of BSA/MPC/MA-BzA/GDA/Aps/TEMED = 1:4700:300:500:500:2000) was dissolved in deoxygenated and deionized water. The reaction was carried out for 60 min in nitrogen atmosphere. Finally, the reaction mixture was dialyzed with PBS to remove the unreacted monomers and initiators. The unencapsulated BSA was removed by ultrafiltration [molecular weight cut-off (MWCO): 100 KDa].
The synthesis and surface charge of nBSA-BzA were verified using agarose gel electrophoresis. Briefly, 10 μL of FITC-labeled BSA or nBSA-BzA with 2 μL loading dye was loaded onto 1 % agarose gel. A voltage of 180V was applied for 15 min before the visualization of the results under a UV lamp.
The BSA concentration in nBSA-BzA was quantified using a BCA microassay. Briefly, a BCA working solution (BWS) was prepared by mixing 50 volume of Reagent A and 1 volume of Reagent B together. A standard curve of BSA was established using native BSA with a series of BSA concentrations (0.125-2 mg/mL). BCA assays were conducted by adding 10 μL of BSA or nBSA solutions into each well of a 96-well plate, followed by the addition of 200 μL of BWS. The plate was then incubated at 60 °C for 30 min. After the solution was cooled to room temperature, the absorbance at 562 nm was determined using a UV-Visible spectrometer.
Then a solution of Dox (10 mg/mL in DI water) was added into nBSA-BzA solution (1 mg/mL, in PBS) (molar ratio of Dox/BSA = 200) and the mixture was stirred overnight at room temperature. The unconjugated Dox was removed by ultrafiltration [MWCO: 10 KDa].
The synthesis of nBSA-COOH-Dox which conjugated Dox through insensitive bond is described in detail in the Supplementary Material. The fabrication of nBSA-Dox and nBSA-COOH-Dox were also verified using agarose gel electrophoresis.
Size, zeta potential and morphology of nanocapsules
The size and zeta potential of nBSA-BzA, nBSA-Dox and nBSA-COOH-Dox in PBS were determined using a Malvern Mastersizer 2000 (Zetasizer Nano ZS90, Malvern Instruments, UK). The data were the average of three times measurements.
A transmission electron microscopy (TEM, Tecnai G2 F30, FEI Company, Hillsboro, Oregon, USA) was used to observe the morphology of the nanocapsules. The TEM sample was prepared by drop-coating 2 μL of nBSA-Dox (1 mg/mL BSA) solution onto a super-thin carbon-coated copper grid. After air drying, the sample was stained by depositing of 1% sodium phosphotungstate (pH 7.0) onto the surface of the sample-loaded grid for 1 min, and then the excess of the samples was removed.
Drug loading content (LC)
To determine the drug LC of nBSA-Dox, 1 mg of lyophilized nBSA-Dox was dissolved in 1 mL 0.1 N HCl solution to cleave the Dox from nBSA. The Concentration of Dox was analyzed using a UV-Visible spectrophotometer at 485 nm using a previously established calibration curve [34]. The drug LC of nBSA-COOH-Dox was estimated by the Dox absorbance at 485 nm (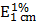 = 196 assumed for Dox) according to the previous work [35].
= 196 assumed for Dox) according to the previous work [35].
X-ray photoelectron spectroscopy
The X-ray photoelectron spectroscopy (XPS) was conducted using a Kratos Axis Ultra DLD spectrometer equipped with monochromatic Al Kα radiation (hν = 1486.58 eV) [36]. The survey and high-resolution spectra were collected at a fixed analyzer pass energy of 160 eV and 20 eV, respectively. The binding energy values were referenced to the Fermi edge, and charge correction was performed by setting the C1s peak at 284.60 eV.
Fluorescence spectroscopy of nBSA-Dox
The Dox fluorescence was obtained by exciting at 485 nm, and the fluorescence spectra were obtained in the wavelength range of 500-700 nm. Both Dox and nBSA-Dox were measured with a Dox concentration of 25 μg/mL.
Stability of Dox-loaded nanocapsule
The stability of nBSA-Dox and nBSA-COOH-Dox in PBS without and with 10% FBS over 14 days and nBSA-BzA at pH 7.4 and 6.5 over 24 h at 37 °C were determined using a Zetasizer Nano ZS90.
In vitro drug release study
The in vitro Dox release profiles of nBSA-Dox and nBSA-COOH-Dox were performed in acetate buffer (pH 5.0 and 6.5) and phosphate buffer (pH 7.4) solutions at 37 °C. Briefly, 3 mL of nBSA-Dox and nBSA-COOH-Dox (with 1 mg/mL Dox) were respectively added to a dialysis bag with a MWCO of 5 kDa and then the bag was immersed in a centrifuge tube containing 27 mL buffer solution. The tubes were maintained in an orbital shaker water bath and vibrated at 120 rpm at 37 °C. At designated time intervals, 1 mL buffer medium was replaced with the same volume of fresh medium and subjected to UV-Visible analysis at 485 nm. The drug release studies for each sample were performed in triplicate.
Cellular uptake study
The cellular uptake behavior of nBSA-Dox was analyzed using confocal laser scanning microscopy (CLSM). For the CLSM studies, HepG2 cells (1x105 cells/well) were seeded in 12-well culture plates and cultured overnight in DMEM with 10% FBS. Fresh DMEM with desired pH was adjusted by adding 0.1 M aqueous HCl or NaOH [37]. On the following day, the cells were washed once with PBS and incubated with free Dox, nBSA-COOH-Dox or nBSA-Dox (all of them with a Dox concentration of 20 μg/mL) in media containing 10% serum for 3 h at pH 7.4 and 6.5, respectively. The cells were observed using a CLSM (Olympus Fluoview FV-1000, Tokyo, Japan) using an imaging software. The fluorescence of Dox was observed at 488 nm excitation using an LP filter of 590 nm.
For the quantitative analysis, HepG2 cells (1x105 cells/well) were seeded in 12-well culture plates and cultured in DMEM with 10% FBS overnight. The cells were equilibrated with Hank's buffered salt solution (HBSS) at 37 °C for 1 h followed by adding free Dox, nBSA-COOH or nBSA-Dox (all of them with a Dox concentration of 20 μg/mL), respectively. After incubation for 3 h, the cells were washed three times with cold PBS and then 50 mL 0.5% Triton X-100 in 0.2 N sodium hydroxide was put into each sample well to lyse the cells [38].
In vitro cytotoxicity
The cells were plated into a 96-well plate (1x104 cells/well) for 24 h confluence. The cell viability of free Dox, nBSA-COOH-Dox, nBSA-Dox and Dox-free nBSA with the same nBSA concentration as nBSA-Dox on the HepG2 cell line were evaluated using the MTT assay. HepG2 cells (1x104 cells/well) were seeded in 96-well culture plates in 100 μL DMEM for 24 h. The cells were then exposed to free Dox nBSA-COOH-Dox and nBSA-Dox with a Dox concentration of 0.25, 2.5, 12.5 and 25 μg/mL and Dox-free nBSA with the same nanocapsule concentration for another 24 h at pH 7.4 and 6.5, respectivley. At each time point, the MTT solution was added, and the cell viability was measured using a Bio-Rad 680 microplate reader by formazan absorbance at 490 nm.
Pharmacokinetics and biodistribution
To measure the pharmacokinetics, nBSA-Dox and Dox (5 mg/kg on a Dox basis) were intravenously injected into female Balb/C mice (n = 3) via the tail vein. A blood sample (10-20 μL) was collected from the tail vein at 0.5,1, 3, 6, 12, 24 and 48 h after the injection and diluted with PBS and heparin (1,000 U/mL). The blood was centrifuged (14,000 rpm, 10 min, 4 °C) and to release the drug 10 μL of diluted plasma was incubated with 490 μL of acidified isopropanol (75 mM HCl, 10% water, 90% isopropanol) overnight at 4 °C in the dark. The isopropanol extract was then centrifuged (14,000 rpm, 10 min, 4 °C) and the supernatant was loaded onto a 96-well plate in triplicate (125 μL per well). The fluorescence was determined using a Victor 3 TM microplate reader (Perkin Elmer; Waltham, MA) at an excitation of 485 nm and an emission of 590 nm. A linear standard curve was created between the logarithm of the background-subtracted fluorescence vs. the logarithm of the Dox concentration for 2000, 500, 125, 31.2, 7.8, 2.0 and 0.5 ng/mL. Assuming a 42% hematocrit for blood, the plasma drug concentration was estimated via this standard.
The plasma Dox concentrations of the free Dox and nBSA-Dox show a biphasic behavior in mice. To obtain the estimates and confidence intervals of the pharmacokinetic parameters describing this behavior, the dataset was fit to a two-compartment pharmacokinetic model using the Origin 8.5 software. The plasma concentration (n = 5) was fitted using Eq. 1, where Cp is the concentration of plasma Dox, and kd and ke are the rate constants for the distribution and elimination processes, respectively.
Cp = A1 e-kdt + A2 e-ket + A3 (1)
From these data and Dose D, other pharmacokinetic parameters were determined as follows:
The initial Dox concentration can be expressed:
Co = A1 +A2 +A3 (2)
The half-lives of the distribution and elimination processes:
t1/2, dist =(3)
t1/2, elim =(4)
The plasma clearance:
Clplasma =(5)
The area-under-curve, AUC:
AUC =(6)
The Dox tissue biodistribution of nBSA-Dox and Dox were also quantified according to the previous work [39]. nBSA-Dox and Dox (5 mg/kg on a Dox basis) were intravenously injected into female Balb/C mice (n = 3) via the tail vein. At 2, 6 and 24 h after injection, tumor, heart, lung, liver, spleen and kidney samples were collected. After weighed, tissues were all suspended in acidified isopropanol (1.0-1.5 mL) and homogenized using 2 mm diameter zirconia beads and a MiniBeadbeater-1TM (Biospec; Bartlesville, OK) at 5000 beats/min for 1 min. After incubated overnight in acidified isopropanol at 4 °C in the dark, all samples were vortexed, and centrifuged (14,000 rpm, 10 min, 4 °C). The supernatant was separated and assayed for fluorescence as described for pharmacokinetic analysis. Untreated control tissues were similarly extracted to correct for tissue autofluorescence. For each sample, following subtracting the background fluorescence, the remaining counts were converted to Dox concentration using the Dox fluorescence standard curve.
In vivo near-IR fluorescence imaging
After the tumor size reached ~100 mm3, the mice were intravenously injected with nBSA-BzA-Dox-Alexa 750 (0.2 mg/kg on an Alexa 750 basis) via the tails (n = 3). The excitation and emission wavelengths for Alexa 750 dye were 750 and 780 nm, respectively. The In vivo images were recorded using a Maestro™ Automated In vivo Imaging system (CRi Maestro™, USA) at 2, 6 and 24 h postinjection. The mice were humanely killed at 24 h postinjection, and then the heart, lung, liver, spleen, kidney and tumor were collected from each mouse without delay. The fluorescence intensity in all the organs was further analyzed by the Maestro™ Automated In vivo Imaging system.
In vivo antitumor efficacy
The mice were randomly divided into five groups (n = 5) and the HepG2 xenograft model was established by the subcutaneous injection of 5x106 HepG2 cells (150 μL) into the right flank of each mouse. The tumor nodules were grown to 100 mm3 in volume before initiating the treatment. Saline, Dox and nBSA-Dox (5 mg/kg on a Dox basis) were injected into the mice intravenously via the tail vein three times on days 0, 4 and 8. The antitumor activity was evaluated in terms of tumor volume (V), which was estimated using the following equation:
V = a × b2/2
where a and b are the major and minor axes of the tumor, respectively, as measured by a caliper. The body weight was measured simultaneously to evaluate the systemic toxicity. The results were considered statistically significant if two-tailed P-values were <0.05.
Statistical methodology
All the experiments were repeated at least three times unless otherwise stated. The results are expressed as mean ± SD, and the statistical significance of all the results was determined by the Student's t-test. p< 0.05 was considered to be statistically significant. *P< 0.05; **P< 0.01; ***P< 0.001.
Results and discussion
Fabrication and characterization of Dox-conjugated nanocapsule nBSA-Dox
The nBSA-BzA was fabricated by the in-situ polymerization of MPC and MA-BzA around BSA similarly as reported [26]. Agarose electrophoresis showed that the nBSA-BzA had slightly negative charge (Figure S1), which was completely different with the BSA, indicating that negative BSA was completely encapsulated. The XPS results showed that both N and P were detected, and the N was slightly more abundant than P (Figure 2C), indicating that nBSA was mainly encapsulated by MPC and relatively slightly MA-BzA. The narrow scan for the O1s peaks also showed that the oxygen atom in the aldehyde group at 531 eV was obvious, directly confirming the successful introduction of BzA [40]. The cellular uptake results showed no uptake of nBSA-BzA by HepG2 cells (Figure S2), thus predicting that the outstanding shielding ability of PMPC would probably maintain the stealthy capability of nBSA-Dox in vivo.
After the Dox loading at pH 7.4, agarose electrophoresis showed that the signal of nBSA-Dox in the light field and fluorescence of FITC-labeled nBSA-Dox were in the same position (Figure S1), indicating the successful conjugation of Dox with nBSA-BzA to form nBSA-Dox. Furthermore, as shown in Figure 2D, the conjugation of Dox with nBSA was also supported by the significant attenuation of the fluorescence observed for nBSA-Dox relative to free Dox at the same concentration. This fluorescence attenuation can be attributed to the fluorescence self-quenching of the Dox after the conjugation with nBSA [41]. Zetasizer identified the size of nBSA-BzA and nBSA-Dox were 15.8 ± 1.6 and 18.6 ± 1.3 nm, respectively (Figure 2A), and their zeta potential values were -2.1 ± 0.2 and -1.8 ± 0.1 mV, respectively. These results indicate that the size of nBSA-Dox increased slightly, whereas its charge maintained weakly negative compared to nBSA-BzA, which is more beneficial to prolonging circulation [26]. The TEM image also directly showed that the nBSA-Dox were nanospheres with a diameter of ~25 nm (Figure 2B), consistent with the Zetasizer results. The UV-Visible analysis showed that the LC of nBSA-Dox was 14.2%. All the above mentioned results confirm that the PMPC-encapsulated and Dox-conjugated nBSA-Dox were obtained. At the other hand, agarose electrophoresis also proved that nBSA-COOH-Dox was successfully fabricated after the negative nBSA-COOH converted to nearly neutral charge by conjugating with Dox (Figure S1). It was proved to have a size as 19.2 ± 1.1 nm and zeta potential as -2.6 ± 0.2 mV with LC as 11.6%.
In vitro stability and drug release profiles
After the fabrication of nBSA-Dox and nBSA-COOH-Dox, their in vitro stability were investigated. As expected, the size of nBSA-Dox and nBSA-COOH-Dox in PBS without and with 10% FBS both maintained almost the same for two weeks (Figure 3A and S4). As a superhydrophilic material, PMPC adsorbs a large amount of water through a strong ionization of water, resulting in the excellent stability and dispersion of PMPC-based nanocapsules.
Figure 2
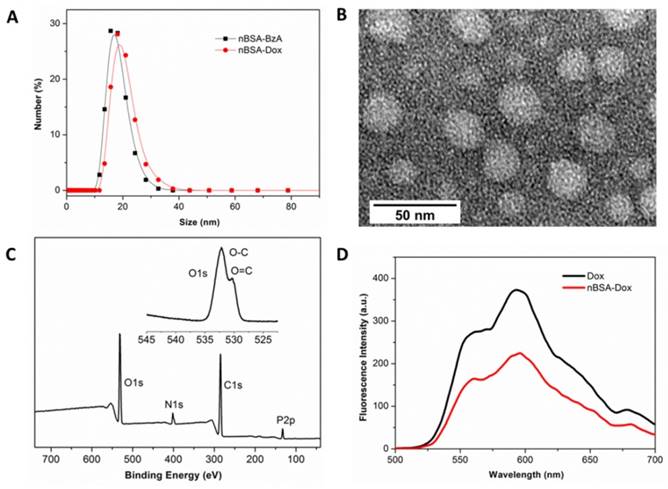
Characterization of nBSA-Dox nanocapsule. (A) Hydrodynamic size distributions of nBSA-BzA and nBSA-Dox in PBS. (B) TEM image of nBSA-Dox. (C) XPS spectrum and narrow scan for the O1s peaks of nBSA-BzA. (D) Fluorescence spectra of Dox and nBSA-Dox with a Dox concentration of 25 μg/mL.
Next, in vitro Dox release from nBSA-Dox and nBSA-COOH-Dox were both monitored. Because many studies reported that the benzoic-imine bond formed from benzaldehyde and amine is very stable at pH 7.4, whereas it would cleave under a weakly acidic environment [37, 42], the drug release of nBSA-Dox was separately measured at pH 7.4, 6.5 and 5.0. As shown in Figure 3B, at pH 7.4, no obvious Dox was detected over 12 h, confirming the robustness of the benzoic-imine bond between nBSA-BzA and Dox. In stark contrast, the amounts of released Dox at pH 6.5 and 5 over 6 h were 60.5% and 83.4 % of the initial content, respectively. This result indicates that nBSA-Dox clearly exhibits an acid-responsive drug release behavior due to the cleavage of benzoic-imine bond under an acidic environment. Because the pH of physiological conditions and tumor microenvironment are approximately 7.4 and 6.5, respectively [43], we expected that nBSA-Dox would be very stable during the blood circulation, whereas the Dox would be rapidly released after accumulating in tumor. This tumor microenvironment-responsive release behavior is absolutely necessary, for not only improving the anticancer efficacy through the tumor-targeting delivery of Dox, but also lowering the cytotoxicity caused by nonspecific Dox distribution. At the other hand, as the control group, nBSA-COOH-Dox did not exhibit any Dox release at three pH conditions (Figure S5), demonstrating the non-responsiveness of the covalent bond conjugating nBSA-COOH and Dox.
Cellular uptake of nBSA-Dox
To investigate whether nBSA-Dox and conjugated Dox could be uptaken by cells, the interaction of Dox, nBSA-COOH-Dox and nBSA-Dox with HepG2 human liver cancer cell line at pH 7.4 and 6.5 after 3 h treatment were observed by CLSM. Dox could be directly observed by CLSM because of its red fluorescence. As shown in Figure 4A, after 3 h treatment with free Dox, a remarkable Dox fluorescence was observed within the cells under both the pH conditions, clearly indicating that Dox entered the cells rapidly. This is consistent with other reports [39, 44]. In contrast, faint Dox fluorescence was observed after treating with nBSA-Dox at pH 7.4. This indicates the difficult release of Dox from nBSA-Dox at pH 7.4. These results are consistent with the Dox release profile in vitro. However, at pH 6.5, significant amounts of Dox were detected within the cells after treating with nBSA-Dox, indicating that the Dox was released from the nBSA-Dox under a weakly acidic environment. As the control, nBSA-COOH-Dox did not show any cellular uptake at both pH, indicating no release of Dox. All the above mentioned results confirm that nBSA-Dox not only has a robust stability and stealthy capability under physiological conditions, but also exhibits tumor microenvironment-responsive release.
In addition, the cellular uptake efficiency of nBSA-Dox was quantified. Similar to the CLSM results, the uptake efficiency of nBSA-Dox at pH 6.5 increased to 6.9 folds of that at pH 7.4 (Figure 4B), directly demonstrating the obvious tumor microenvironment-responsive release.
Figure 3
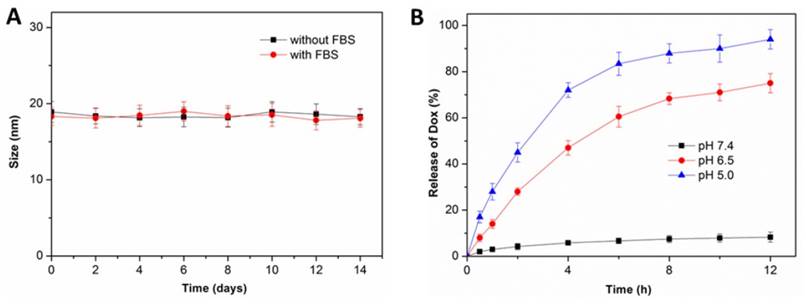
(A) Size change of nBSA-Dox in PBS without and with 10% FBS over two weeks (n = 3). (B) In vitro Dox release profiles of nBSA-Dox in media at different pH.
Figure 4
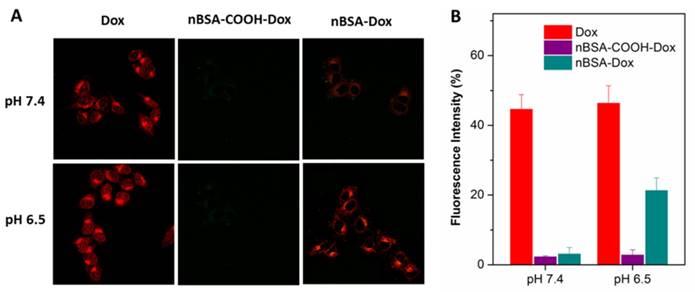
(A) CLSM images of HepG2 cells incubated with Dox, nBSA-COOH-Dox and nBSA-Dox at pH 7.4 and 6.5, respectively, for 3 h. (B) Cellular uptake efficiency of Dox, nBSA-COOH-Dox and nBSA-Dox over 3 h at pH 7.4 and 6.5, respectively. All the samples had a Dox concentration of 20 μg/mL (scale bar: 20 μm).
In vitro cytotoxicity of nBSA-Dox
To evaluate the cytotoxicity of nBSA-Dox, an MTT assay was carried out by treating HepG2 cells with nBSA-Dox at varying Dox concentrations for 24 h at pH 7.4 and 6.5, respectively. Dox and nBSA-COOH-Dox with the same Dox concentration and Dox-free nBSA-BzA with the same nBSA concentration as nBSA-Dox were used as the controls. As shown in Figure 5A, after 24 h incubation at pH 7.4, Dox clearly exhibited cytotoxicity to the cancer cells, inhibiting the cell growth to 54% at 25 μg/mL. However, nBSA-Dox only showed much weaker cytotoxicity than that of Dox. The IC50 of Dox was calculated to be 22.6 μg/mL whereas that of nBSA-Dox was calculated to be 61.4 μg/mL (Table 1). The cytotoxicity of free Dox can be attributed to the fast diffusion of Dox into the cell cytoplasm and then into the nucleus. Dox intercalates the DNA thereby preventing its replication [45]. In contrast, since nBSA-Dox could hardly release Dox at pH 7.4, its much weaker cytotoxicity is reasonable.
On the other hand, after 24 h incubation at pH 6.5, the cytotoxicity of nBSA-Dox (20.9 μg/mL) was almost the same as that of free Dox (19.8 μg/mL) (Figure 5B). Combining the in vitro Dox release and cellular uptake of nBSA-Dox, we assumed that this cytotoxicity results from the triggered Dox release in an acidic medium. Therefore, nBSA-Dox is expected to kill cancer cells efficiently under an acidic tumor microenvironment. In contrast, nBSA-COOH-Dox, conjugated with Dox through insensitive bond, showed almost no cell cytotoxicity both at pH 7.4 and 6.5, indicating no release of Dox as well as the loss of pharmacological activity of Dox [35]. Moreover, the Dox-free nBSA had no effect on the cell viability, indicating its biocompatibility and further potential clinical practice.
Pharmacokinetics study
To study the in vivo pharmacokinetics of nBSA-Dox, the plasma Dox concentrations were monitored over time after the intravenous injection of nBSA-Dox. The Dox concentration was quantified by measuring the fluorescence intensity of Dox and then fitting to a two-compartment pharmacokinetic model [39]. As shown in Figure 6, the plasma level of free Dox was essentially undetectable as early as 2 h postinjection, consistent with the short half-life of doxorubicin (~5 min) in mice [46]. In contrast, nBSA-Dox significantly extended the plasma residence time of Dox as well as increased its concentration within the plasma compartment, with detectable levels remaining even for 48 h after the treatment. The elimination half-life of Dox conjugated to nBSA increased to 38.1 h, much longer than that of conventional PEGylated nanocarriers (~10 h) [14, 39, 46]. Moreover, the plasma AUC for the mice treated with nBSA-Dox was 1428.6 μg h mL-1, as much as 242 fold of free Dox (Table 2). Based on this significant increase in plasma AUC and EPR effect of the nanocarrier, these results anticipated that nBSA-Dox prefers to accumulate in solid tumors compared to free Dox.
Table 1
IC50 values of Dox formulation in the Dox and nBSA-Dox on HepG2 cells following 24 h incubation at pH 7.4 and 6.5, respectively.
| pH | IC50 (μg/mL) | |
|---|---|---|
| Dox | nBSA-Dox | |
| 7.4 6.5 | 22.6 ± 2.6 19.8 ± 1.5 | 61.4 ± 5.0 20.9 ± 2.6 |
Figure 5
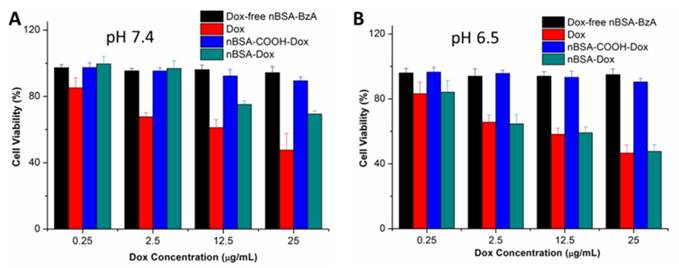
Viability of HepG2 cells cultured with nBSA-Dox compared to that of Dox and nBSA-COOH-Dox at the same Dox dose and that of Dox-free nBSA-BzA with the same nBSA-BzA concentration as nBSA-Dox for 24 h: (A) pH 7.4; (B) pH 6.5.
Figure 6
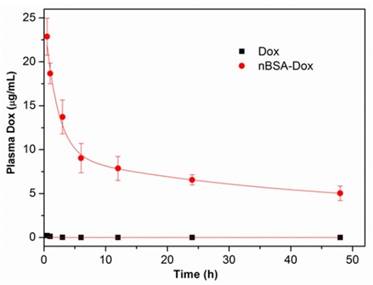
Plasma Dox concentrations of nBSA as a function of time postinjection. A two-compartment model was fitted to the plasma Dox concentration.
Table 2
Pharmacokinetics in SD rats after i.v. administration.
| Parameters | Dox | nBSA-Dox |
|---|---|---|
| Co (μg/mL) | 5.6 | 25.7 |
| kd (h-1) | 11.6 | 0.43 |
| t1/2, dist (h) | 0.06 | 1.6 |
| ke (h-1) | 0.95 | 0.018 |
| t1/2, elim (h) | 0.73 | 38.1 |
| Clplasma (mL h-1 g-1) | 0.85 | 0.0035 |
| AUC 0..∞ (μg h mL-1) | 5.9 | 1428.6 |
Biodistribution analysis of nBSA-Dox
Besides the pharmacokinetics study, the time-varying Dox tissue biodistribution of nBSA-Dox was analyzed by quantifying the fluorescence of Dox in tissues. Free Dox-treated group was used as control. As shown in Figure 7A, opposite with the gradually decreased Dox concentration in tumors of Dox-treated group, that of nBSA-Dox-treated group increased remarkably along with time, as much as 11.8 times of that of free Dox-treated group at 24 h after injection. This result demonstrated that nBSA-Dox shows much superior tumor accumulation capability than free Dox, which would mainly be attributed to the prolonged half-life of nBSA-Dox [47]. Meanwhile, nBSA-Dox significantly decreased the Dox concentration in non-tumor tissues including heart and lung (Figure 7B and 6S). This decreased accumulation is quite important because cardiomyopathy is the dose-limiting side effect of free Dox [48]. Furthermore, unlike conventional nanocarriers, the concentration of nBSA-Dox in liver, spleen and kidney reduced over time (Figure 7B), indicating it would not accumulate in these organs to cause toxicity. Improved tumor accumulation and reduced non-tumor tissue distribution anticipated that nBSA-Dox would enhance the antitumor efficacy.
In vivo near-IR fluorescence imaging of nBSA
A whole animal near-IR (NIR) imaging system was also used to study the in vivo distribution and tumor targeting of nBSA-Dox. Alexa 750 is a very stable NIR fluorescent molecule with a high intensity and widely used for in vivo NIR imaging [49, 50]. When Alexa 750 is conjugated to nBSA-BzA through an insensitive covalent bond, its fluorescence intensity in vivo should be directly proportional to the concentration of nBSA-Alexa 750. The time-varying changes in its fluorescence intensity are shown in Figure 7C. The NIR fluorescence signal was not very strong at the 2 h postinjection of free Alexa 750 and even totally disappeared at 6 h and 24 h. This indicates the rapid clearance of free Alexa 750, quite similar to the small molecule Dox. In stark contrast, at 2 h postinjection of nBSA-Alexa 750, a remarkable fluorescence was observed all over the body of mice including the tumor, as much as 4.2 times of free Alexa 750 at the same dose (Figure S7A). This indicates that nBSA-Alexa 750 was absorbed rapidly by all types of organs and started to accumulate in the tumor. Furthermore, in contrast to the rapid clearance of a small molecule, the fluorescence intensity of nBSA-Alexa 750 at 24 h increased to double of that at 2 h, indicating its continuous accumulation and retention in tumor. These results were consistent with that of nBSA-Dox proved above.
Moreover, the tissue biodistribution at the 24 h postinjection of nBSA-Alexa 750 was also confirmed. As shown in Figure 7D, a strong fluorescence was observed in the tumor, much stronger than that of other nontumor organs including the heart, liver and spleen. The quantitative analysis results also confirm the efficient tumor targeting of nBSA-Alexa 750 (Figure S7B). All these results verify again that nBSA-Dox would improve the anticancer efficacy as well as reduce the side effects.
In vivo antitumor efficacy
In view of the above mentioned positive results, the in vivo antitumor efficacy of nBSA-Dox was evaluated next. HepG2 tumor xenograft-bearing mice were treated with saline, Dox, and nBSA-Dox every four days for three consecutive cycles when their tumor volume became ~100 mm3. Fourteen days after the first treatment, as shown in Figure 8A, the tumor volume of the saline-treated and free Dox-treated groups increased to 879 and 547 mm3, respectively, whereas the tumor volume of nBSA-Dox-treated group instead shrinked slightly. Clearly, the nBSA-Dox formulation outperforms much more than the free drug in tumor suppression, consistent with the in vitro cell cytotoxicity, pharmacokinetics and in vivo imaging. Moreover, in contrast to the significant weight loss of Dox-treated mice, the nBSA-Dox-treated mice did not suffer from weight loss during the study (Figure 8B), indicating a favorable toxicity profile for the nBSA-Dox formulation. After 14 days of treatment, all the mice were sacrificed and the tumors were separated from their bodies. Figure 8C and 8D clearly show the morphology and average weight of the tumors in all the groups, directly confirming an efficient tumor-suppressing efficacy.
Conclusions
A novel drug delivery system for cancer therapy was successfully developed using a stealthy nanocapsule. This stable nanocapsule was easily fabricated and conjugated with an anticancer drug Dox through a pH-responsive weak covalent bond. The in vitro results show that this Dox-conjugated nanocapsule released the drug under the tumor microenvironment and killed cancer cells. Furthermore, the in vivo results confirm that it would significantly prolong the half-life of Dox, efficiently accumulate in tumor and perform outstanding tumor suppression. Herein, this nanocapsule, exhibiting a prolonged circulation time, improved tumor accumulation, tumor microenvironment-responsive drug release and no tissue toxicity, provides a new promising nanomedicine platform for clinical applications.
Figure 7
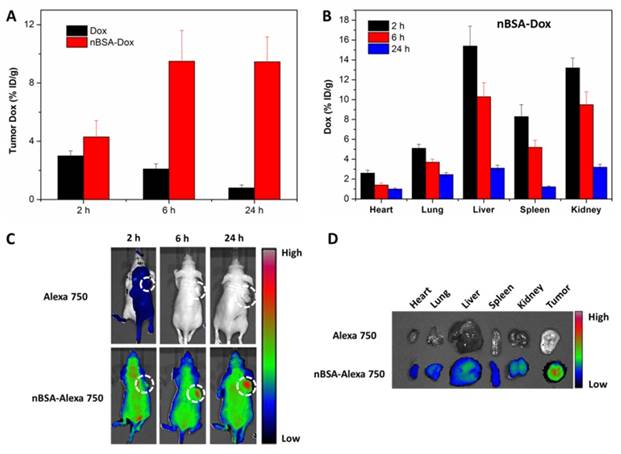
Biodistribution analysis and in vivo imaging of SCID nude mice bearing HepG2 cells xenograft. The Dox concentration in tumor tissue (A) and other organs (B) at 2, 6 and 24 h after the tail vein injection of nBSA-Dox. (C) Time-lapse NIR fluorescence images of nude mice after the tail vein injection of nBSA-Alexa 750. The tumors were circled with a dotted line. (D) NIR fluorescence images of the major organs and tumors after 24 h injection.
Figure 8
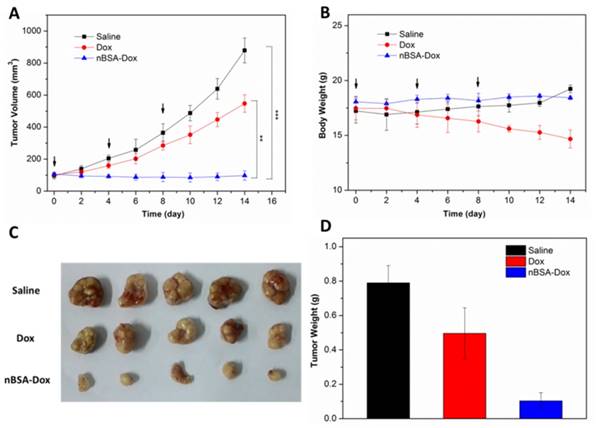
Antitumor efficacy of nBSA-Dox on HepG2 xenograft-bearing nude mice. (A) Plots of tumor volumes after injecting with saline, Dox and nBSA-Dox. (B) Plots of body weights of tumor-bearing mice after injecting with saline, Dox and nBSA-Dox. Images (C) and weights (D) of the tumors of each group taken out from the sacrificed mice at the end point of study. (t-test, **P< 0.01; ***P< 0.001)
Supplementary Material
Supplementary figures.
Acknowledgements
The authors are grateful for the financial support from National Natural Science Foundation of China (31270019), China Postdoctoral Science Foundation (2015M580109), Guangdong Natural Science Funds for Distinguished Young Scholar (2014A030306036), Natural Science Foundation of Guangdong Province (2015A030313848), Guangdong Special Support Program, Science and Technology Planning Project of Guangdong Province (2016A020217001), Science, Technology & Innovation Commission of Shenzhen Municipality (JCYJ20150529164918738 and JCYJ20150430163009479, JCYJ20160301152300347).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Matsumura Y, Maeda H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986;46:6387-92
2. Drummond DC, Meyer O, Hong KL, Kirpotin DB, Papahadjopoulos D. Optimizing Liposomes for Delivery of Chemotherapeutic Agents to Solid Tumors. Pharmacol Rev. 1999;51:691-743
3. Ferrari M. Cancer Nanotechnology: Opportunities and Challenges. Nat Rev Cancer. 2005;5:161-71
4. Lee CC, MacKay JA, Frechet J, Szoka FC. Designing Dendrimers for Biological Applications. Nat Biotechnol. 2005;23:1517-26
5. Duncan R. Polymer Conjugates as Anticancer Nanomedicines. Nat Rev Cancer. 2006;6:688-701
6. Wang S, Huang P, Chen X. Stimuli-Responsive Programmed Specific Targeting in Nanomedicine. ACS Nano. 2016;10:2991-4
7. Chen K, Chen XY. Integrin Targeted Delivery of Chemotherapeutics. Theranostics. 2011;1:189-200
8. Draz MS, Fang BA, Zhang PF, Hu Z, Gu SD, Weng KC. et al. Nanoparticle-Mediated Systemic Delivery of siRNA for Treatment of Cancers and Viral Infections. Theranostics. 2014;4:872-92
9. Kumar R, Kulkarni A, Nagesha DK, Sridhar S. In Vitro Evaluation of Theranostic Polymeric Micelles for Imaging and Drug Delivery in Cancer. Theranostics. 2012;2:714-22
10. Muthu MS, Leong DT, Mei L, Feng S. Nanotheranostics - Application and Further Development of Nanomedicine Strategies for Advanced Theranostics. Theranostics. 2014;4:660-77
11. Knop K, Hoogenboom R, Fischer D, Schubert US. Poly(Ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew Chem Int Edit. 2010;49:6288-308
12. Veronese FM, Mero A. The Impact of PEGylation On Biological Therapies. Biodrugs. 2008;22:315-29
13. Harris JM, Chess RB. Effect of Pegylation On Pharmaceuticals. Nat Rev Drug Discovery. 2003;2:214-21
14. Shi Y, van der Meel R, Theek B, Blenke EO, Pieters EHE, Fens MHAM. et al. Complete Regression of Xenograft Tumors upon Targeted Delivery of Paclitaxel via Pi-Pi Stacking Stabilized Polymeric Micelles. ACS Nano. 2015;9:3740-52
15. Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of Pegylated Liposomal Doxorubicin - Review of Animal and Human Studies. Clin Pharmacokinet. 2003;42:419-36
16. Veronese FM. Peptide and Protein PEGylation: A Review of Problems and Solutions. Biomaterials. 2001;22:405-17
17. Armstrong JK, Hempel G, Koling S, Chan LS, Fisher T, Meiselman HJ. et al. Antibody Against Poly(Ethylene Glycol) Adversely Affects PEG-asparaginase Therapy in Acute Lymphoblastic Leukemia Patients. Cancer-Am Cancer Soc. 2007;110:103-11
18. Ishihara T, Takeda M, Sakamoto H, Kimoto A, Kobayashi C, Takasaki N. et al. Accelerated Blood Clearance Phenomenon upon Repeated Injection of PEG-modified PLA-nanoparticles. Pharm Res-Dordr. 2009;26:2270-9
19. Koide H, Asai T, Hatanaka K, Akai S, Ishii T, Kenjo E. et al. T Cell-Independent B Cell Response is Responsible for ABC Phenomenon Induced by Repeated Injection of PEGylated Liposomes. Int J Pharmaceut. 2010;392:218-23
20. Batrakova EV, Li S, Li YL, Alakhov VY, Elmquist WF, Kabanov AV. Distribution Kinetics of a Micelle-Forming Block Copolymer Pluronic P85. J Controlled Release. 2004;100:389-97
21. Savic R, Azzam T, Eisenberg A, Maysinger D. Assessment of the Integrity of Poly(Caprolactone)-B-Poly(Ethylene Oxide) Micelles Under Biological Conditions: A Fluorogenic-Based Approach. Langmuir. 2006;22:3570-8
22. Liu J, Zeng F, Allen C. In Vivo Fate of Unimers and Micelles of a Poly(Ethylene Glycol)-Block-Poly(Caprolactone) Copolymer in Mice Following Intravenous Administration. Eur J Pharm Biopharm. 2007;65:309-19
23. Cheng TJ, Liu JJ, Ren J, Huang F, Ou HL, Ding YX. et al. Green Tea Catechin-Based Complex Micelles Combined with Doxorubicin to Overcome Cardiotoxicity and Multidrug Resistance. Theranostics. 2016;6:1277-92
24. Yan M, Du JJ, Gu Z, Liang M, Hu YF, Zhang WJ. et al. A Novel Intracellular Protein Delivery Platform Based On Single-Protein Nanocapsules. Nat Nanotechnol. 2010;5:48-53
25. Liang S, Liu Y, Jin X, Liu G, Wen J, Zhang L. et al. Phosphorylcholine Polymer Nanocapsules Prolong the Circulation Time and Reduce the Immunogenicity of Therapeutic Proteins. Nano Res. 2016;9:1022-31
26. Zhang L, Liu Y, Liu G, Xu D, Liang S, Zhu X. et al. Prolonging the Plasma Circulation of Proteins by Nano-Encapsulation with Phosphorylcholine-Based Polymer. Nano Res. 2016;9:2424-32
27. Zhang J, Zhang X, Liu G, Chang D, Liang X, Zhu X. et al. Intracellular Trafficking Network of Protein Nanocapsules: Endocytosis, Exocytosis and Autophagy. Theranostics. 2016;6:2099-113
28. Chen SF, Zheng J, Li LY, Jiang SY. Strong Resistance of Phosphorylcholine Self-Assembled Monolayers to Protein Adsorption: Insights Into Nonfouling Properties of Zwitterionic Materials. J Am Chem Soc. 2005;127:14473-8
29. Ohkawa k, Hatano T, Yamada K, Joh K, Takada K, Tsukada Y. et al. Bovine Serum Albumin-Doxorubicin Conjugate Overcomes Multidrug-Resistance in a Rat Hepatoma. Cancer Res. 1993;53:4238-42
30. Akiyama Y, Nagasaki Y, Kataoka K. Synthesis of Heterotelechelic Poly(Ethylene Glycol) Derivatives Having Alpha-Benzaldehyde and Omega-Pyridyl Disulfide Groups by Ring Opening Polymerization of Ethylene Oxide Using 4-(Diethoxymethyl)benzyl Alkoxide as a Novel Initiator. Bioconjugate Chem. 2004;15:424-7
31. Haag R, Kratz F. Polymer Therapeutics: Concepts and Applications. Angew Chem Int Edit. 2006;45:1198-215
32. Wang Y, Luo Q, Sun R, Zha G, Li X, Shen Z. et al. Acid-Triggered Drug Release From Micelles Based On Amphiphilic Oligo(Ethylene Glycol)-Doxorubicin Alternative Copolymers. J Mater Chem B. 2014;2:7612-9
33. Qiu L, Xu C, Zhong F, Hong C, Pan C. Fabrication of Functional Nano-Objects through RAFT Dispersion Polymerization and Influences of Morphology on Drug Delivery. ACS Appl Mater Interfaces. 2016;8:18347-59
34. Xiao Y, Hong H, Javadi A, Engle JW, Xu W, Yang Y. et al. Multifunctional Unimolecular Micelles for Cancer-Targeted Drug Delivery and Positron Emission Tomography Imaging. Biomaterials. 2012;33:3071-82
35. Hurwitz E, Levy R, Maron R, Wilchek M, Arnon R, Sela M. Covalent Binding of Daunomycin and Adriamycin to Antibodies with Retention of Both Drug and Antibody Activities. Cancer Res. 1975;35:1175-81
36. Tao W, Zeng X, Wu J, Zhu X, Yu X, Zhang X. et al. Polydopamine-Based Surface Modification of Novel Nanoparticle-Aptamer Bioconjugates for in Vivo Breast Cancer Targeting and Enhanced Therapeutic Effects. Theranostics. 2016;6:470-84
37. Ding C, Gu J, Qu X, Yang Z. Preparation of Multifunctional Drug Carrier for Tumor-Specific Uptake and Enhanced Intracellular Delivery through the Conjugation of Weak Acid Labile Linker. Bioconjugate Chem. 2009;20:1163-70
38. Zeng X, Tao W, Mei L, Huang L, Tan C, Feng S. Cholic Acid-Functionalized Nanoparticles of Star-Shaped PLGA-vitamin E TPGS Copolymer for Docetaxel Delivery to Cervical Cancer. Biomaterials. 2013;34:6058-67
39. MacKay JA, Chen M, McDaniel JR, Liu W, Simnick AJ, Chilkoti A. Self-Assembling Chimeric Polypeptide-Doxorubicin Conjugate Nanoparticles that Abolish Tumours After a Single Injection. Nat Mater. 2009;8:993-9
40. Zhou J, Sui Z, Zhu J, Li P, De Chen, Dai Y. et al. Characterization of Surface Oxygen Complexes On Carbon Nanofibers by TPD, XPS and FT-IR. Carbon. 2007;45:785-96
41. Gillies ER, Frechet J. PH-responsive Copolymer Assemblies for Controlled Release of Doxorubicin. Bioconjugate Chem. 2005;16:361-8
42. Gao Y, Yang C, Liu X, Ma R, Kong D, Shi L. A Multifunctional Nanocarrier Based on Nanogated Mesoporous Silica for Enhanced Tumor-Specific Uptake and Intracellular Delivery. Macromol Biosci. 2012;12:251-9
43. Lee ES, Na K, Bae YH. Super pH-sensitive Multifunctional Polymeric Micelle. Nano Lett. 2005;5:325-9
44. Liu J, Bu W, Pan L, Shi J. NIR-Triggered Anticancer Drug Delivery by Upconverting Nanoparticles with Integrated Azobenzene-Modified Mesoporous Silica. Angew Chem Int Edit. 2013;52:4375-9
45. Tewey KM, Reme TC, Yang L, Halligan BD, Liu LF. Adriamycin-Induced Dna Damage Mediated by Mammalian Dna Topoisomerase-Ii. Science. 1984;226:466-8
46. Gabizon A, Tzemach D, Mak L, Bronstein M, Horowitz AT. Dose Dependency of Pharmacokinetics and Therapeutic Efficacy of Pegylated Liposomal Doxorubicin (DOXIL) in Murine Models. J Drug Target. 2002;10:539-48
47. Perrault SD, Walkey C, Jennings T, Fischer HC, Chan W. Mediating Tumor Targeting Efficiency of Nanoparticles through Design. Nano Lett. 2009;9:1909-15
48. Singal PK, Iliskovic N. Doxorubicin-Induced Cardiomyopathy. New Engl J Med. 1998;339:900-5
49. Lowery A, Onishko H, Hallahan DE, Han Z. Tumor-Targeted Delivery of Liposome-Encapsulated Doxorubicin by Use of a Peptide that Selectively Binds to Irradiated Tumors. J Controlled Release. 2011;150:117-24
50. Montet X, Ntziachristos V, Grimm J, Weissleder R. Tomographic Fluorescence Mapping of Tumor Targets. Cancer Res. 2005;65:6330-6
Author contact
![]() Corresponding author: Lin Mei, E-mail: mei.lintsinghua.edu.cn
Corresponding author: Lin Mei, E-mail: mei.lintsinghua.edu.cn
Citation styles
APA
Liu, G., Tsai, H.i., Zeng, X., Zuo, Y., Tao, W., Han, J., Mei, L. (2017). Phosphorylcholine-based stealthy nanocapsules enabling tumor microenvironment-responsive doxorubicin release for tumor suppression. Theranostics, 7(5), 1192-1203. https://doi.org/10.7150/thno.17881.
ACS
Liu, G.; Tsai, H.i.; Zeng, X.; Zuo, Y.; Tao, W.; Han, J.; Mei, L. Phosphorylcholine-based stealthy nanocapsules enabling tumor microenvironment-responsive doxorubicin release for tumor suppression. Theranostics 2017, 7 (5), 1192-1203. DOI: 10.7150/thno.17881.
NLM
Liu G, Tsai Hi, Zeng X, Zuo Y, Tao W, Han J, Mei L. Phosphorylcholine-based stealthy nanocapsules enabling tumor microenvironment-responsive doxorubicin release for tumor suppression. Theranostics 2017; 7(5):1192-1203. doi:10.7150/thno.17881. https://www.thno.org/v07p1192.htm
CSE
Liu G, Tsai Hi, Zeng X, Zuo Y, Tao W, Han J, Mei L. 2017. Phosphorylcholine-based stealthy nanocapsules enabling tumor microenvironment-responsive doxorubicin release for tumor suppression. Theranostics. 7(5):1192-1203.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) license (https://creativecommons.org/licenses/by-nc/4.0/). See http://ivyspring.com/terms for full terms and conditions.