Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(5):1177-1191. doi:10.7150/thno.17895 This issue Cite
Research Paper
Tumor Hypoxia Regulates Forkhead Box C1 to Promote Lung Cancer Progression
Yu-Jung Lin1, 2, Woei-Cherng Shyu3, Chi-Wei Chang4, Chi-Chung Wang5, Chung-Pu Wu6, Hsu-Tung Lee7, Liang-Jwu Chen1 ![]() , Chia-Hung Hsieh2, 8, 9
, Chia-Hung Hsieh2, 8, 9 ![]()
1. Institute of Molecular Biology College of Life Science, National Chung Hsing University, Taiwan;
2. Graduate Institute of Biomedical sciences, China Medical University, Taiwan;
3. Department of Neurology, Center for Neuropsychiatry, and Graduate Institute of Immunology, China Medical University and Hospital, Taichung, Taiwan;
4. National PET/Cyclotron Center and Department of Nuclear Medicine, Taipei Veterans General Hospital, Taipei, Taiwan;
5. Graduate Institute of Biomedical and Pharmaceutical science, Fu Jen Catholic University, New Taipei, Taiwan;
6. Department of Physiology and Pharmacology, Chang Gung University, Tao-Yuan, Taiwan;
7. Department of Neurosurgery, Taichung Veterans General Hospital, Taichung, Taiwan;
8. Department of Medical Research, China Medical University Hospital, Taichung, Taiwan;
9. Department of Biomedical Informatics, Asia University, Taichung, Taiwan.
Received 2016-10-12; Accepted 2017-1-3; Published 2017-3-5
Abstract

Forkhead box C1 (FOXC1) is a member of the forkhead family of transcription factors that are characterized by a DNA-binding forkhead domain. Increasing evidence indicates that FOXC1 is involved in tumor progression. However, the role of tumor hypoxia in FOXC1 regulation and its impact on lung cancer progression are unclear. Here, we report that FOXC1 was upregulated in hypoxic areas of lung cancer tissues from rodents or humans. Hypoxic stresses significantly induced FOXC1 expression. Moreover, hypoxia activated FOXC1 transcription via direct binding of hypoxia-inducible factor-1α (HIF-1α) to the hypoxia-responsive element (HRE) in the FOXC1 promoter. FOXC1 gain-of-function in lung cancer cells promoted cell proliferation, migration, invasion, angiogenesis, and epithelial-mesenchymal transition in vitro. However, a knockdown of FOXC1 in lung cancer cells inhibited these effects. Notably, knockdown of tumor hypoxia-induced FOXC1 expression via HIF-1-mediated FOXC1 shRNAs in lung cancer xenograft models suppressed tumor growth and angiogenesis. Finally, systemic delivery of FOXC1 siRNA encapsulated in lipid nanoparticles inhibited tumor growth and increased survival time in lung cancer-bearing mice. Taken together, these data indicate that FOXC1 is a novel hypoxia-induced transcription factor and plays a critical role in tumor microenvironment-promoted lung cancer progression. Systemic FOXC1 blockade therapy may be an effective therapeutic strategy for lung cancer.
Keywords: lung cancer, tumor hypoxia, fork head box C1, hypoxia-inducible factor-1α.
Introduction
Each year, lung cancer is the leading cause of cancer-related deaths worldwide. There is a high risk of mortality in patients with recurrences and metastases [1]. Compared to the other cancers, such as liver, breast, and prostate cancers, the clinical outcomes of conventional therapies, including radiotherapy and chemotherapy, remain poor despite major efforts to improve treatment methods over the past years. Almost 85% of lung cancers are classified as non-small-cell lung cancer (NSCLC), which is the most common lung cancer with only a 15.9% five-year survival rate [2]. The strategy for treating NSCLC is to slow down its progression using surgery in early stages (I-II), or chemotherapy, as well as radiotherapy, in late stage (III) [3]. More recent therapeutic strategies employ anti-angiogenesis agents (Bevacizumab) and anti-epidermal growth factor receptor agents (Gefitinib). However, the overall survival rate remains low because NSCLC progression is rapid and there is no effective therapeutic target for NSCLC.
Tumor hypoxia is a fundamental characteristic of various tumors [4]. High levels of hypoxia in lung tumor patients are associated with higher diagnostic grades [5]. HIF-1α is a potent transcription factor which can regulate tumor cell proliferation, growth, metastasis, and apoptosis [6]. HIF-1α has been reported to have different expression profiles in NSCLC and in other types of lung cancer. HIF-1α expression was 75.8% positive in NSCLC and 45.5% positive in small-cell lung cancer [7]. HIF-1α was also reported to have a positive association with various genes in NSCLC, such as epidermal growth factor receptor (EGFR), matrix metalloproteinase 9 (MMP)-9, and p53, which are considered to be cancer-related genes [8]. Tumor hypoxia also promoted NSCLC metastasis through M2 macrophages by ERK signaling [9]. Moreover, the hypoxic environment triggering HIF-1α expression can also lead to the growth of tumor vasculature in lung tumors, which contributes to their metastases by interacting with vascular endothelial growth factor (VEGF) [10]. Nevertheless, the contribution of tumor hypoxia in regulating the putative, main target gene that promotes lung cancer progression remains unclear. Therefore, it is primordial to find the major indicator that can be used as a diagnostic or therapeutic target in NSCLC.
Forkhead Box C1 (FOXC1) is a transcription factor that plays an important role in regulating ocular development in the embryonic stage [11]. It has been pointed out that mice homozygous for either a spontaneous mutation in FOXC1, or an engineered null mutation, die prenatally or perinatally with identical phenotypes. These defects include hemorrhagic hydrocephalus, genitourinary and cardiovascular defects, as well as the intervention, or narrowing, of the aortic arch, ventricular septal defects (VSD), and pulmonary valve dysplasia [12]. These results show that FOXC1 is important in embryonic development. Besides, FOXC1 is also required for arterial specification and lymphatic sprouting during vascular development [13]. FOXC1 and FOXC2 induced the expression of arterial markers in vitro and worked in conjunction, at an earlier stage, to precisely control the proper process of arterial specification, by regulating the expression of Notch signaling genes, such as Dll4. FOXC1 and FOXC2, and also directly interacted with the VEGF signaling pathway to promote arterial gene expression [14]. Over the past decade, studies have shown that FOXC1 is highly expressed, and promotes metastases, in several cancers [15, 16]. FOXC1 participates in breast cancer, hepatocellular carcinoma, pancreatic ductal adenocarcinoma, and lung cancer progression with poor clinical prognoses [17, 18]. FOXC1 has been implicated in promoting both tumor cell proliferation and metastasis. To date, the precise mechanism by which FOXC1 expression is upregulated in cancers is still unknown.
In this study, we hypothesize that FOXC1 is one of the hypoxia-responsive genes and plays a role in tumor hypoxia-enhanced progression effects in the tumor microenvironment. We show that FOXC1 expression is regulated by HIF-1α in tumor hypoxia conditions and it contributes to lung cancer progression. Lung cancer cells stably transduced with HRE-driven FOXC1 shRNAs exhibit significant growth inhibition in vivo. Moreover, systemic delivery of FOXC1 siRNA encapsulated in lipid nanoparticles (LNPs) as an oligonucleotide drug was sufficient to blunt lung cancer progression and constituted a translational path for the clinical treatment of lung cancer patients.
Materials and Methods
Cell culture
A549, CL1-0 and CL1-5 human lung cells were cultured in RPMI (Life Technologies) supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES, and 1% penicillin-streptomycin (PS). Cells were maintained at 37°C in a humidified incubator containing 5% CO2 and 20% O2 in air.
In vitro hypoxic treatments
Cells were treated in Biospherix C-Chamber (Biospherix) inside a standard culture chamber by means of exhausting and gassing with 95% N2 and 5% CO2 to produce oxygen concentrations of 0.5 to 1% at 37 °C to achieve hypoxic condition as previous described [19, 20].
Vector constructions and viral transduction
The lentiviral vector pLKO AS2 (National RNAi Core Facility, Taiwan) was used as the backbone to generate a lentiviral reporter vector. The multiple cloning sites (MCS) of pTA-Luc vector (Clontech) was inserted with the cDNA fragment bearing -2100 to + 1 bp FOXC1 promoter to drive the expression of firefly luciferase gene. The FOXC1 promoter driven reporter gene cassette was amplified from promoter to SV40 ploy A on the constructed pTA-Luc vector using PCR and inserted into pLKO AS2 as pLKO AS2-FOXC1-p by XhoI and MluI restriction enzymes. The mutant of hypoxia response element (HRE) on FOXC1 promoter was generated in the pLKO AS2-FOXC1-p as template by Quick Change Site-directed Mutagenesis Kit (Stratagene). Full-length human FOXC1 cDNA was amplified in a reaction with Platinum Taq DNA polymerase (Invitrogen) and was subcloned into pAS2.EYFP.puro (National RNAi core facility, Academia Sinica, Taiwan) at the NheI and EcoRI sites. The pGreenFire1-SFFV [21] was used to generate lung cancer reporter cells bearing SFFV promoter-driven a dual optical reporter gene encoding both green fluorescence protein (GFP) and luciferase (Luc). Lentiviral vectors carrying short hairpin RNAs (shRNA)-targeting HIF-1α (5'- TGCTCTTTGTGGTTGGATCTA-3'), HIF-2α (5'-CCATGAGGAGATTCGTGAGAA-3') and FOXC1 (5'-GCCGCACCATAGCCAGGGCTT-3' for FOXC1 shRNA-1; 5'-CCACTGCAACCTGCAAGCCAT-3' for FOXC1 shRNA-2) and scrambled shRNA (http://rnai.genmed.sinica.edu.tw/file/vector/C6-7/17.1.pLAS.Void.pdf) were provided by National RNAi core facility, Academia Sinica in Taiwan. To generate a hypoxia-driven FOXC1 knockdown system, the cytomegalovirus (CMV) promoter in the lentiviral vectors carrying FOXC1 shRNAs (FOXC1 shRNA-2) were replaced with 8 repeats of the HREs derived from p8xHRE-TKGFP plasmids described elsewhere [22]. Lentivirus production and cell transduction were carried out according to protocols described elsewhere [23, 24]. All constructs were confirmed by DNA sequencing.
Promoter analysis and Luciferase assays
rVISTA software was used to identify one putative hypoxia response elements (HRE) within the human and mouse FOXC1 proximal promoter region (-2100 to + 1). To determine the role of HIF-1α in hypoxia-induced transcriptional activation of FOXC1, the stably FOXC1 promoter-driven luciferase reporter-transfected A549, CL1-0, CL1-5 and HEK293 cells were pretreated with or without scramble or HIF-1α shRNAs for 48 hours and then exposed to in vitro hypoxic stress for 24 hours or cells were incubated with YC-1 (10 μM; Sigma-Aldrich) together with in vitro hypoxic stress for 24 hours. Besides, cells were transfected with control plasmids (pcDNA3) or pcDNA3-HIF-1α with oxygen-dependent degradation domain (ODD) deletion mutant (HIF-1α/ΔODD) for 48 hours under normoxic conditions. To assay the reporter activities of hypoxia response element (HRE) mutations in FOXC1 promoter, HRE mutant and wild-type constructs of FOXC1 promoter were co-transfected with phRL-SV40 containing the Renilla luciferase gene (Promega) into A549 cells, and the cells were treated with or without hypoxia for 24 hours. Firefly luciferase activities were assayed and normalized to those of Renilla luciferase. Luciferase activity was determined by mixing 10 μL of extracts from 1 ×105 cells and 100 μL of luciferase assay reagent (Promega) according to the manufacturer's instructions.
Chromatin immunoprecipitation
Chromatin immunoprecipitation assays were performed using Imprint Chromatin Immunoprecipitation Kit (Sigma-Aldrich) according to the manufacturer's protocol using an anti-HIF-1α antibody (Novus). For tissue chromatin immunoprecipitation assays, frozen human tumor tissues were collected and followed by the Tissue Chromatin Immunoprecipitation Kit (Abcam). PCR for the HRE in the FOXC1 promoter was performed with specific primers: HRE (F) 5'-ACCCAGGAAGTCTGCGCGAA-3' and (R) 5'-AGCCCCGCGGTCCTCCGGCC-3' were used for the input DNA PCR product.
Western blot assay
Cells were lysed and extracts were prepared as described previously [22]. HIF-1α, HIF-2α, FOXC1, VEGF, E-cadherin and Fibronectin proteins in human cells were detected in 150 µg of cell extract using monoclonal anti-HIF-1α antibody (diluted 1:650; Novus), anti-HIF-2α antibody (diluted 1:700; Cell Signaling Technology), anti-FOXC1 antibody (1:1000; Novus), anti-VEGF antibody (1:2000; Genetex), E-cadherin antibody (diluted 1:1000; Novus) and Fibronectin antibody (diluted 1:1000; Novus). Western blots were normalized using a monoclonal anti-β-actin antibody (diluted 1:10,000; Sigma-Aldrich).
Real-time quantitative PCR
Q-PCR analysis was performed as described previously [22]. The gene primers were: VEGF (F) 5'-TGCCCGCTGCTGTCTAAT-3' and (R) 5'-TCTCCGCTCTGAGCAAGG-3' ; FOXC1 (F) 5'-TTACCGGTAAGCCTAGATTAGGCC-3' and (R) 5'-TTGAATTCGGTAACATTATTGGTT-3' ;β-catenin (F) 5'-GCTTTCAGTTGAGCTGACCA-3' and (R) 5'- CAAGTCCAAGATCAGCAGTCTC -3' ; E-cadherin (F) 5'-GGTCTGTCATGGAAGGTGCT -3' and (R) 5'-GATGGCGGCATTGTAGGT-3'; Vimentin (F) 5'-TGGTCTAACGGTTTCCCCTA-3'and (R) 5'-GACCTCGGAGCGAGAGTG-3'; Fibronectin (F) 5'-GACGCATCACTTGCACTTCT-3' and (R) 5'-GATGCACTGGAGCAGGTTTC-3'; GAPDH (F) 5'-GCACAAGAGGAAGAGAGAGACC-3' and (R) 5'-AGGGGAGATTCAGTGTGGTG-3'.
Enzyme-linked immunosorbent assay (ELISA)
Antibody sandwich ELISAs were used to evaluate VEGF levels in the CM (Sigma-Aldrich) according to the manufacturer's instructions.
Cell growth analysis
Cell proliferation and viability were determined by the trypan blue dye exclusion test. Briefly, cells were seeded in 24-well plates (5×104cells/well). Cells were harvested in indicated days by trypsin/EDTA treatment and counted by microscope after trypan blue staining, respectively.
Cell migration and invasion assays
Cell migration was measured using the 24-well cell transwell from (Corning Costar Corporation) and following manufacturer's instructions. The lung cancer cells were trypsinized, washed by PBS, and suspended in medium without FBS. The lower wells of the chambers were added medium with 10% FBS. Upper wells were filled with serum-free medium with lung tumor cells (20,000 cells per well). Then, the chamber was placed into incubator. Assays were stopped by removal of the medium from the upper wells and careful removal of the filters after 12 hours. Filters were then fixed with methanol by submersion and were briefly wiped on the cells on the upper side using the Q-tip. Evaluation of migrating cells was performed under the microscope were examined (four fields per filter) for the presence of cells at the lower membrane side. The lung cancer cell invasive ability was also performed by transwell pre-coated with Matrigel (2.5 mg/ml; BD Biosciences Discovery Labware). Cells (3×104) were seeded onto the upper wells of transwells and incubated for 16 hours. Evaluation of invading cells was as same as the protocol of migration assay. The experiments were performed thrice in triplicate.
Tube formation assay
The standard Matrigel assay was utilized to evaluate in vitro angiogenesis activity by quantifying the tube formation of HUVECs. To examine the role of FOXC1 on lung cancer cell-induced tube formation of HUVECs, 48-well culture plates were coated with 200 µl of Matrigel (BD) per well then allowed to polymerize for 30 minutes at 37°C. The CM derived from CL1-5 cells with or without FOXC1 knockdown or CL1-0 cells with or without FOXC1 overexpression was added to HUVEC suspension 30 minutes before seeding. Cell suspensions of 150 µl (1.5 × 104 cells) were seeded on polymerized Matrigel. After incubation at 37°C for 8 hours, each culture was photographed using a Zeiss observer A1 axio microscope (Zeiss). For quantitative measurements of capillary tube formation, Matrigel wells were digitized under a ×4 objective for measurement of total tubes and tube length of capillary tube formation. Tracks of HUVEC organized into networks of cellular cords (tubes) were counted and averaged in randomly selected three microscopic fields.
Animal models
Eight-week-old male nude mice (Balb/c nu/nu) were purchased from the Animal Facility of the National Science Counsel (NSC) and were used to establish lung cancer animal models. For in vivo tumor growth assay, 3×106 lung cancer cells with luciferase reporter were injected subcutaneously into the dorsal aspects of anterior or posterior limbs of mice. For in vivo metastatic assay, ~1×106 lung cancer cells with luciferase reporter were injected intravenously via tail vein. Tumor growth and metastasis were assayed by bioluminescent imaging (BLI). All animal studies were conducted according to Institutional Guidelines of China Medical University and approved by the Institutional Animal Care and Use Committees of China Medical University.
Bioluminescent imaging (BLI)
Mice were imaged with the IVIS Imaging System 200 Series (Caliper) to record bioluminescent signal emitted from the engrafted tumors. Mice were anesthetized with isoflurane from the imaging system and received intraperitoneal injection of D-Luciferin (Caliper) at a dose of 250 µg/g body weight. Imaging acquisition was performed at 15 minutes after luciferin injected. For BLI signal quantitative analysis, regions of interest encompassing the intracranial area of signal were defined using the software (Living Image 2.60.1) and the total number of photons per second per steradian per square centimeter were recorded.
FOXC1 siRNA encapsulated in lipid nanoparticles
FOXC1 siRNA encapsulated in lipid nanoparticles were generated according to previously published methods [25]. Briefly, FOXC1 siRNA was synthesized in a large scale by Integrated DNA Technologies and contained 2'-O-methyl (2'-OMe) modifications. Modified target siRNAs were encapsulated into LNP. Scramble siRNA molecules were used for control experiments.
MicroPET imaging
The [18F] fluoromisonidazole (FMISO) tracer was produced as previously reported [26]. Each subject was injected with 3.9 MBq of 18F-FMISO. At 120 minutes after injection, mice were scanned on a small-animal positron emission tomography (PET) scanner (microPET; Concorde Microsystems) under isoflurane anesthesia. Static images (10 minutes) were obtained with a zoom factor of 2 in a 256 × 256 matrix. Calculations were corrected for radiation decay of 18F and the amount of injected dose, and the consistent color scale was applied to all PET images.
Tissue immunofluorescence imaging
For hypoxic area studies, the hypoxia marker, pimonidazole (70 mg/kg, intraperitoneal; HPI) and perfusion marker, Hoechst 33342 (1 mg/mouse; intravenous; Sigma) were administered 3 hours and 5 minutes prior to tumor excision, respectively. Tumor tissues were frozen in the OCT embedding matrix (Shandon Lipshaw). Frozen tissue sections (10 µm) were obtained with an OTF cryomicrotome (Bright-Hacker), fixed in ice-cold methanol for 10 minutes, and washed with PBS. Tumor sections were first incubated with FITC-conjugated anti-pimonidazole monoclonal antibody (1: 25; Chemicon International) for 1 hour at room temperature. Tumor sections were then co-stained for FOXC1 by including FOXC1 monoclonal antibody (Novus Biologicals) at a final concentration of 10 µg/mL. Sections were washed 3 times in PBS, each wash lasting 5 minutes. For FOXC1 staining, sections were incubated with DyLght 649-conjugated goat anti-rabbit antibody (1∶100 dilution; Molecular Probes) and washed again. For human specimen staining studies, frozen human lung cancer and normal lung tissue sections were incubated with primary antibodies, FOXC1 (1:200 dilution; Novus Biologicals) or/and HIF-1α (1:150 dilution; Novus Biologicals) overnight at 4 °C and secondary antibodies, DyLght 649-conjugated goat anti-rabbit antibody (1:100 dilution; Molecular Probes) or Dylight 488-conjugated goat-anti-mouse antibody (1:100 dilution; Abcam). For angiogenesis studies, HIF-1α (1:100 dilution; Novus Biologicals) and vWF (1:100 dilution; Cell Signaling Technology) double staining were also followed the same procedures. At the end of the staining periods, the sections were sealed with 90% glycerin in PBS containing antifade medium DABCO (25 mg/mL) and DAPI (0.5 mg/mL). Tissue fluorescence was visualized with the Axio Observer A1 digital fluorescence microscope system (ZEISS).
Cell immunofluorescence imaging
Cells were fixed for 15 minutes with 4% of paraformaldehyde at room temperature and then washed three times with PBS. The samples were then incubated overnight at 4°C with the respective anti-E-cadherin antibody (1:100 dilution; Novus Biologicals), anti-Vimentin antibody (1:100 dilution; Novus Biologicals) in PBS. The samples were subsequently rinsed with PBS three times and incubated for 1hour at room temperature with the appropriate dye-conjugated Alexa Fluor® 594 and Alexa Fluor® 488 secondary antibodies (1:100 dilution; Jackson ImmunoResearch). Finally, the samples were washed again with PBS and sealed with 90% glycerin in PBS containing antifade medium DABCO (25 mg/mL) and DAPI (0.5 mg/mL). Control sample were performed without primary antibody. All of images were observed with the Axio Observer A1 digital fluorescence microscope system (ZEISS).
Immunohistochemical staining
Immunohistochemical staining was performed on human lung cancer tissue arrays (Biomax) using the avidin-biotin-peroxidase complex method. The paraffin sections were deparaffinized, and rehydrated in a graded series of ethanol (50-100%). Endogenous peroxidase activity was blocked using 0.3% hydrogen peroxidefor for 15 minutes. The sections were incubated overnight at 4°C with primary antibody (1:100 dilution) followed by conjugation to the secondary antibody (1:100 dilution) then counterstained with Delafield's haematoxylin, dehydrated and mounted. Negative controls were stained without primary antibody.
Fluorescence-activated cell-sorting (FACS) analyses
Tumor tissues were disaggregated with an enzyme cocktail containing collagenase type III (Sigma), hyaluronidase (Sigma), and collagenase type IV (Sigma), washed several times, and resuspended in phosphate-buffered saline (PBS) to produce a single cell suspension. Prior to flow cytometry, cells were incubated with monoclonal FOXC1 antibody in cold fluorescence-activated cell sorting (FACS) buffer (PBS, 0.5% BSA) on ice for 30 minutes. After washing in FACS buffer, cells were incubated with DyLght 649-conjugated goat anti-rabbit antibody. After the final washing step, fluorescence was measured using a FACScalibur instrument and FACSDiva 6.0 software (BD Bioscience). The hypoxic subpopulations were further gated or isolated based on the analysis of Hoechst 3342 and pimonidazole in dot plots. The control cells are derived from disaggregated the A549 xenografts with Hoechst 33342 and pimonidazole treatments, which are both Hoechsr 3342 and pimonidazole-negative, and were set in the lower left quadrant of the plot. Same setting conditions were used thereafter, cell populations located outside of this quadrant of the plot were defined as either Hoechst 3342- and pimonidazole + cells (chronic hypoxic cells), Hoechst 3342+ and pimonidazole + cells (cycling hypoxic cells) or Hoechst 3342+ and pimonidazole- cells (Normoxic cells). FOXC1 expression was further evaluated in these subpopulations.
Statistical Analysis
All experiments were done at least in triplicates. All data are given as mean ± SD. Statistical analyses were performed with SPSS 18.0 software using unpaired Student's t test and ANOVA with Bonferroni's or Tukey's multiple comparison post hoc tests, where appropriate. P < 0.05 was considered significant.
Results
FOXC1 is highly expressed in areas of tumor hypoxia
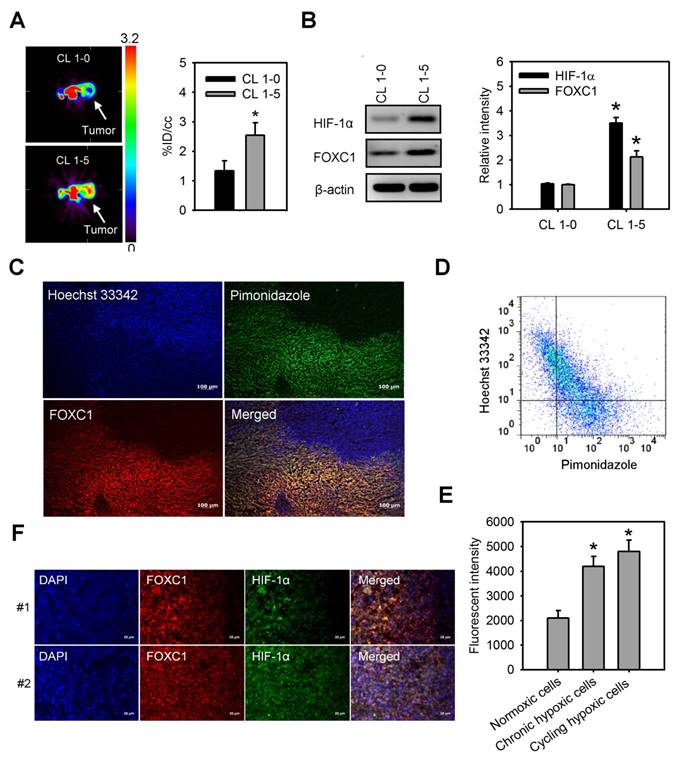
We first observed tumor hypoxia in lung cancer xenografts by PET imaging with 18F-FMISO tracer (Fig. 1A). There was a significant increase in the accumulation of 18F-FMISO in high aggressive CL1-5 xenografts as compared to low aggressive CL1-0 xenografts, indicating that hypoxia levels in CL1-5 xenografts were higher than in CL1-0 xenografts. Moreover, western blotting of xenograft lysates confirmed that both HIF-1α and FOXC1 expression were significantly higher in CL1-5 xenografts as compared to CL1-0 xenografts (Fig. 1B). To better verify endogenous tumor hypoxia-mediated FOXC1 expression in the solid tumor, staining with Hoechst 33342, a perfusion marker, and pimonidazole, a hypoxia marker, together, with immunofluorescence imaging and flow cytometry, were utilized to identity hypoxic tumor subpopulations from A549 xenografts. Immunofluorescence imaging and flow cytometry analyses showed highly heterogeneous Hoechst 33342 and pimonidazole staining in A549 xenografts (Fig. 1C and 1D). The FOXC1 staining indicated that FOXC1 expression occurs in the cycling hypoxic (Hoechst 3342+ and pimonidazole +) and chronic hypoxic (Hoechst 3342- and pimonidazole+) areas of tumors (Fig. 1C). FOXC1 expression in cycling hypoxic cells (Hoechst 3342+ and pimonidazole + cells) and chronic hypoxic cells (Hoechst 3342- and pimonidazole+) was higher than in normoxic cells (Hoechst 3342+ and pimonidazole-) (Fig. 1E). In addition, the results from immunofluorescence staining of HIF-1α and FOXC1 human lung cancer specimens also demonstrate that FOXC1 expression co-localizes with high expression of HIF-1α (Fig. 1F). Taken together, these results suggest that hypoxia may regulate FOXC1 expression in tumor microenvironments.
FOXC1 highly expresses in tumor hypoxic areas. (A) [18F] fluoromisonidazole (FMISO) PET imaging of CL1-0 and CL1-5 xenografts. Data are means ± SD (n=6), *P < 0.01 compared to CL1-0 xenografts, unpaired Student t-test. (B) HIF-1α and FOXC1 protein levels in homogenised CL1-0 and CL1-5 xenografts after PET imaging. Data are means ±SD (n=6). *P < 0.001 compared to CL1-0 xenografts, unpaired Student t-test. (C) Representative immunofluorescence images of microscopic A549 xenografts. Top left, fluorescence image of perfusion marker, Hoechst 33342 (blue). Top right, fluorescence image of hypoxia marker, pimonidazole (green). Bottom left, fluorescence image of FOXC1 expression (red). Bottom right, fluorescence overlay image of Hoechst 33342 (blue), pimonidazole (green), and FOXC1 (red). Yellow color represents the co-localization of pimonidazole and FOXC1. Bars = 100 μm. (D) Scatterplots by 2-color staining with Hoechst 3342 and pimonidazole. (E) Mean channel fluorescence of FOXC1 staining was determined in cycling hypoxic cells (Hoechst 3342+ and pimonidazole+), chronic hypoxic cells (Hoechst 3342- and pimonidazole+), and normoxic cells (Hoechst 3342+ and pimonidazole-) as gated in scatterplots by Hoechst 3342 and pimonidazole staining. Data are means ±SD (n=6). *P < 0.0001 compared to normoxic cells, one-way ANOVA with Tukey multiple comparison test. (F) Immunofluorescence imaging of FOXC1 and HIF-1α expression in two human lung cancer tissue samples. Bars = 20 μm.
FOXC1 is upregulated and correlates with HIF-1α expression in human lung cancer
In order to understand the clinical significance of FOXC1 in lung cancer patients, FOXC1 expression was first examined in the lung specimens of five NSCLC patients and their paired normal background tissues by immunofluorescence imaging and western blotting analysis. Higher protein levels of FOXC1 were observed in tumor tissues than in the paired normal background tissues (Fig. 2A and 2B).
FOXC1 is upregulated and correlated with HIF-1α expression in human lung cancer. (A) Immunofluorescence imaging of FOXC1 and HIF-1α expression in the human lung specimen and its paired normal background tissue. Bars = 20 μm. (B) FOXC1 protein levels in homogenised human lung cancer tissues and its paired normal background tissues. Data are means ±SD (n=5). *P < 0.0001 compared to normal background tissues, unpaired Student t-test. (C) Immunohistochemical staining of FOXC1 expression in human lung cancer specimens (n=64) and unpaired normal tissue specimens (n=15). Data are means ±SD. *P < 0.0001 compared to normal background tissues, unpaired Student t-test. (D) The association between HIF-1α expression and tumor grade in 64 lung cancer specimens. (E) The association between FOXC1expression and tumor grade in 64 lung cancer specimens. (F) The association between HIF-1α and FOXC1expression in 64 lung cancer specimens. (G) Survival analysis of the lung cancer patients with FOXC1 high and low expression using the Kaplan-Meier Plotter website for lung cancer. (H and I) Immunohistochemical staining of FOXC1 expression in human lung cancer specimens with or without lymph node and distant metastasis. Data are means ±SD. *P < 0.001 compared to human lung cancer specimens without lymph node and distant metastasis, unpaired Student t-test.
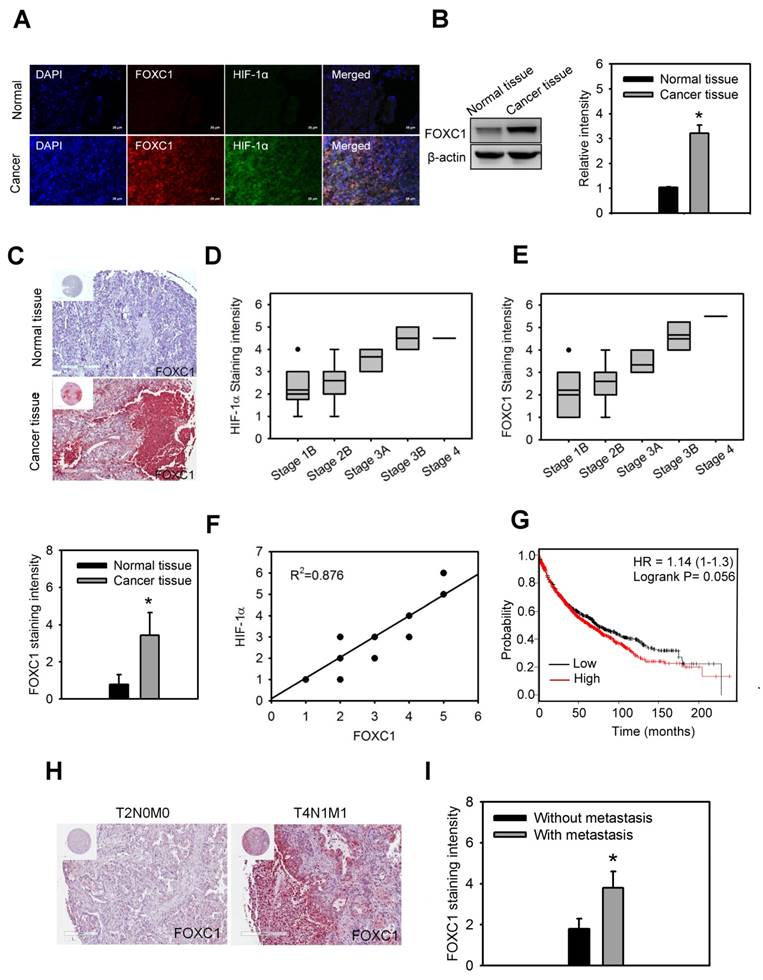
Moreover, immunohistochemical staining of FOXC1 expression in the lung specimens of 64 NSCLC patients and 15 unpaired normal tissues also showed that
FOXC1 is highly expressed in lung cancer tissues, but not in unpaired normal tissues (Fig. 2C). The expression of FOXC1 and HIF-1α in late stage lung cancer samples was significantly higher than in early stages (Fig 2D and 2E). In addition, there was a linear correlation between FOXC1 and HIF-1α expression in lung cancer samples (R2=0.876) (Fig 2F). We also used the web-based Kaplan-Meier Plotter analysis tool to investigate the association between FOXC1 expression and survival outcomes in lung cancer patients. Although the P value did not reach statistical significance, lung cancer patients with high FOXC1 levels had poor overall survival rates compared to those with low FOXC1 levels (P=0.056, HR=1.14) in a total 1,926 patients (Fig. 2G). Furthermore, the expression of FOXC1 in lung cancers with lymph node and distant metastasis was higher than that in lung cancers without lymph node and distant metastasis (Fig. 2H and 2I). These findings suggest that the expression of FOXC1 was significantly higher in human lung cancer tissues. The expression of FOXC1 is closely associated with the tumor grade, HIF-1α expression, and poor prognosis of lung cancer.
Hypoxia-induced FOXC1 expression is dependent on HIF-1α
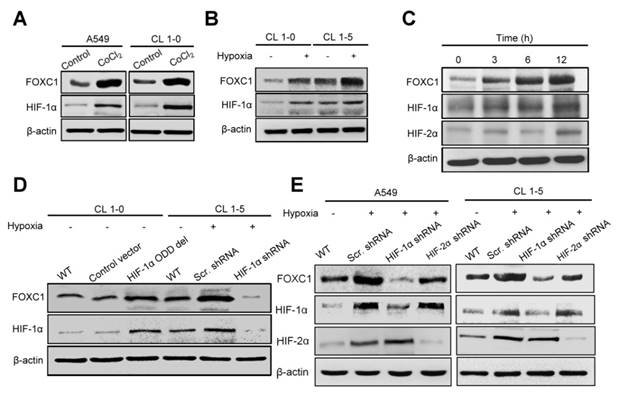
To determine the role of hypoxia in FOXC1 regulation, we examined FOXC1 expression in A549 and CL1-5 cells after treatment with cobalt chloride (CoCl2), a chemical inducer of HIF-1. FOXC1 expression was clearly induced by CoCl2 compared to controls in mRNA and protein levels (Fig. 3A, S1A, S2A and S2B). Moreover, FOXC1 mRNA and protein levels were increased in CL1-0 and CL1-5 cells at 24 hours after hypoxic treatment (<1% O2) (Fig. 3B, S1B, S2C and S2D). We next analyzed the time course of FOXC1, HIF-1α, and HIF-2α expression in A549 cells with hypoxic stress (<1% O2). FOXC1 expression was induced 3 hours after hypoxic treatment and was accompanied by HIF-1α expression (Fig. 3C and S2E). The expression of VEGF was examined as positive control in CoCl2 and hypoxia chamber treatment (Fig. S1C and S1D). However, HIF-2α expression was induced at 12 hours after hypoxic treatment. FOXC1 and HIF-1α had similar dynamic expression patterns, suggesting that HIF-1α may regulate FOXC1 expression. To identify the role of HIF-1α in the regulation of FOXC1 expression, CL1-0 cells were transfected with HIF-1α-oxygen-dependent degradation domain (ODD) deletion mutant plasmids (Fig. 3D, S2F and S2G). HIF-1α overexpression in CL1-0 cells significantly increased FOXC1 expression under normoxic conditions. In contrast, knockdowns of HIF-1α in CL1-5 and A549 cells, via lentiviral transduction with shRNA against HIF-1α, fully inhibited hypoxia-induced FOXC1 expression (Fig. 3D, 3E, S2H and S2I). However, knockdowns of HIF-2α in CL1-5 and A549 cells did not have a significant effect on hypoxia-induced FOXC1 expression. These results indicate that HIF-1α is the major regulator of hypoxia-induced FOXC1 expression.
Hypoxia-induced FOXC1 expression is dependent on HIF-1α. (A) The protein levels of FOXC1 and HIF-1α in A549 and CL1-5 cells with incubation of cobalt chloride (50 μM), which mimics hypoxia, for 24 hours. (B) The protein levels of FOXC1 and HIF-1α in CL1-0 and CL1-5 cells at 24 hours after normoxic and hypoxic treatment (<1% O2). (C) Time course of FOXC1 and HIF-1α expression after exposure of A549 cells to <1% O2. (D) The protein levels of FOXC1 and HIF-1α in CL1-0 cells transfected with control or HIF-1α-ODD deletion mutant plasmids for 48 hours and in CL1-5 cells with or without HIF-1α knockdown at 24 hours after hypoxic treatment (<1% O2). (E) The protein levels of FOXC1, HIF-1α and HIF-2α in A549 and CL1-5 cells with or without HIF-1α or HIF-2α knockdown at 24 hours after hypoxic treatment (<1% O2).
HIF-1α binds directly to the FOXC1 promoter and regulates its expression
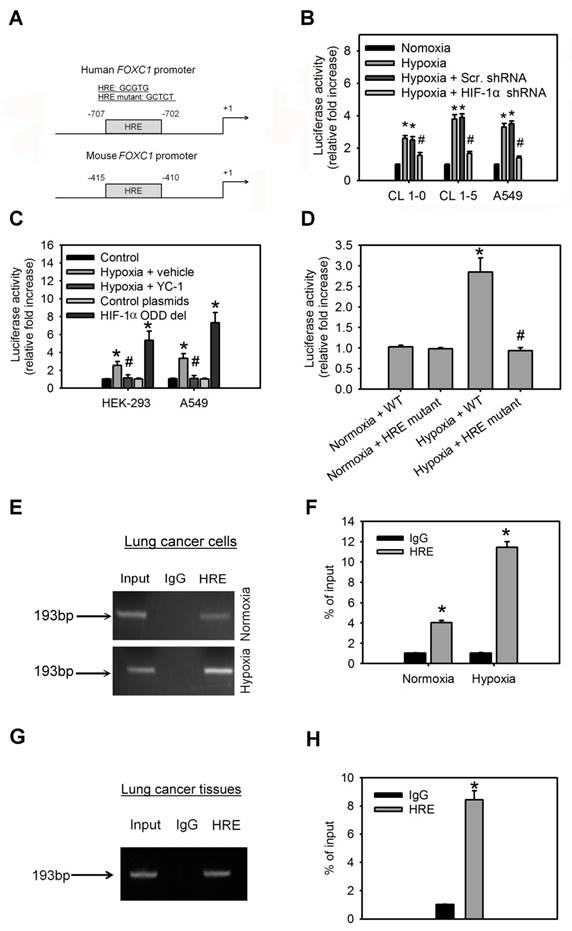
To investigate the molecular mechanism by which hypoxia induces FOXC1 expression, bioinformatics analysis identified one HIF-1α-binding site in the human and mouse FOXC1 promoter sequence from bases -2000 to +1 (Fig. 4A), suggesting that HIF-1α might regulate FOXC1 expression by directly binding to its promoter. In the reporter assays, FOXC1 promoter reporter plasmids (FOXC1-Luc) responded to hypoxia stimulation in CL1-0, CL1-5 and A549 cells (Fig. 4B). However, knockdown of HIF-1α in these cells inhibited hypoxia-induced FOXC1 promoter activation. Moreover, treatment with YC-1 (an HIF-1α inhibitor) also abrogated hypoxia-mediated FOXC1 promoter activities (Fig. 4C). Coexpression of HIF-1α-oxygen-dependent degradation domain (ODD) deletion mutant and FOXC1-Luc significantly enhanced the reporter activity, but control plasmids did not. To pinpoint the exact binding motifs, we introduced the mutations into the HRE of FOXC1-Luc (Fig. 4A). The ablation of HRE, on the FOXC1 promoter, abrogated hypoxia-mediated FOXC1 induction (Fig. 4D). ChIP assays also confirmed the binding of HIF-1α to the FOXC1 promoter in cells (Fig. 4E, 4F) as well as in human lung cancer tissues (Fig. 4G, 4H). Collectively, these results suggest that HIF-1α regulates FOXC1 transcription by directly binding to the FOXC1 promoter in a hypoxia-dependent fashion.
HIF-1α binds directly to the FOXC1 promoter to regulate its expression. (A) Graphic representation of the putative and mutant FOXC1 promoter. One putative hypoxia response element (HRE) was identified in human and mouse FOXC1 promoter. (B) The reporter activities of FOXC1 promoter in CL1-0, CL1-5 and A549 cells with or without HIF-1α knockdown at 24 hours after hypoxic treatment (<1% O2). Data are means ±SD (n=9). *P < 0.0001 compared to normoxic cells; # P <0.001 compared to scramble (scr.) shRNA, one-way ANOVA with Tukey multiple comparison test. (C) The reporter activities of FOXC1 promoter in HEK-293 and A549 cells cultured in hypoxia for 24 hours in the absence or presence of YC-1 or in cells co-transfected with control or HIF-1α-ODD deletion mutant plasmids for 48 hours. Data are means ±SD (n=9). *P < 0.0001 compared to control cells; # P <0.001 compared to vehicle, one-way ANOVA with Tukey multiple comparison test. (D) Luciferase reporter plasmids carrying the wild type (WT) or mutant FOXC1 promoter regions were co-transfected with the Renilla luciferase reporter plasmid into A549 cells; and the cells were treated with or without hypoxia (<1% O2) for 24 hours. (E and F) Chromatin immunoprecipitation followed by real-time PCR (ChIP-qPCR) assay of HIF-1α binding in FOXC1 promoter in response to hypoxia (<1% O2) for 24 hours. Results are expressed as percentage of input. Data are means ±SD (n=9). *P < 0.001 compared to non-specific IgG, unpaired Student t-test. (G and H) Chromatin immunoprecipitation followed by real-time PCR (ChIP-qPCR) assay of HIF-1α binding in FOXC1 promoter in frozen human lung cancer tissues. Results are expressed as percentage of input. Data are means ±SD (n=9). *P < 0.001 compared to non-specific IgG, unpaired Student t-test.
FOXC1 promotes cell growth and malignancy in lung cancer cells
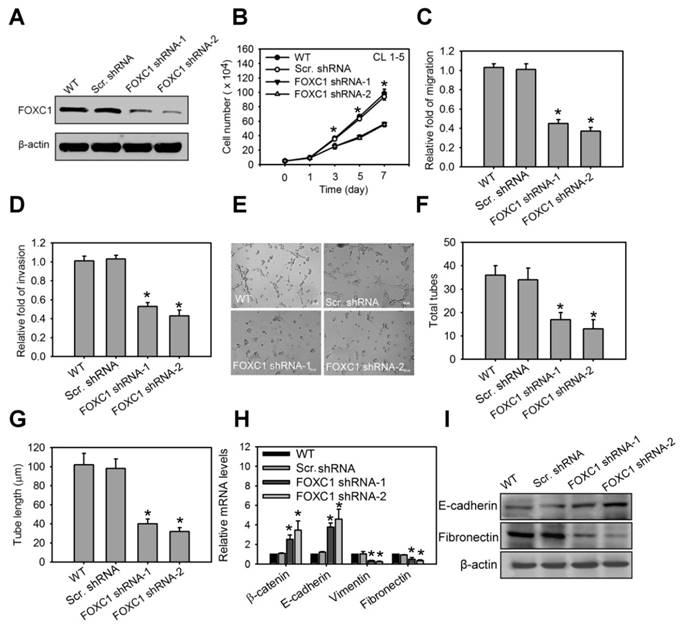
We next investigated the functional role of FOXC1 in lung cancer cell progression. In the high aggressive CL1-5 cells expressing high levels of FOXC1 endogenously, FOXC1 was knocked down using two independent FOXC1 target shRNAs via a lentiviral-based system. Western blot analysis confirmed successful knockdown as manifested by significantly decreased FOXC1 expression (Fig. 5A and S2J). Knockdown of FOXC1 in CL1-5 cells significantly inhibited cell growth, migration and invasion (Fig. 5B, 5C and 5D), suggesting that FOXC1 is essential in contributing to cell growth and malignancy in lung cancer cells. Moreover, the conditional medium (CM) from CL1-5 cells, with or without a FOXC1 knockdown, was collected. Human umbilical vein endothelial cells (HUVECs), cultured in CM from CL1-5 cells with a FOXC1 knockdown, exhibited a decrease in tube formation, compared to HUVECs cultured with CM from wild-type CL1-5 cells (Fig. 5E, 5F and 5G). The expression and secretion of VEGF were also decreased in A549 and CL 1-5 cells with FOXC1 knockdown (Fig. S3A and S3B). Therefore, tumor FOXC1 plays a role in angiogenesis via the secretion of angiogenic factors. We also examined the epithelial-mesenchymal transition (EMT) by observing the expression of EMT markers such as β-catenin, E-cadherin, vimentin, and fibronectin. Q-PCR and western blotting analysis revealed that FOXC1 knockdown in CL1-5 cells increased the expression of β-catenin and E-cadherin, but decreased the expression of vimentin and fibronectin (Fig. 5H, 5I, S2K and S2L), suggesting that cells with a FOXC1 knockdown undergo a mesenchymal to epithelial transition-like process. In order to further test whether FOXC1 gain-of-function is able to promote lung cancer cell progression, the low aggressive CL1-0 cells that express low levels of FOXC1 endogenously were stably transduced using recombinant lentiviruses expressing FOXC1 (Fig. S4A). Compared with wild-type CL1-0 cells, or with control lentiviral vector-infected CL1-0 cells, FOXC1 overexpressing CL1-0 cells exhibited increased cell growth, migration, and invasion (Fig. S4B, S4C and S4D). Moreover, the CM collected from CL1-0 cells with FOXC1 gain-of-function significantly promoted the capillary tube-like formation of HUVECs, compared to CM from wild-type CL1-0 cells, or to the control lentiviral vector-infected CL1-0 cells (Fig. S4E, S4F and S4G). FOXC1 overexpression in CL1-0 cells also increased the expression of vimentin and fibronectin, but decreased the expression of β-catenin and E-cadherin (Fig. S4H). Moreover, FOXC1 overexpression induced a mesenchymal spindle-like morphology in A549 cells under a phase-contrast microscope (Fig. S5A and S5B). Immunofluorescence imaging analysis also demonstrated that A549 cells with FOXC1 gain-of-function exhibited an upregulation of vimentin, but a downregulation of E-cadherin (Fig. S5C). However, a knockdown of FOXC1 in A549 cells generated the opposite results. Collectively, these findings indicate that FOXC1 promotes cell growth, migration, invasion, angiogenesis, and EMT in lung cancer cells.
FOXC1 promotes cell growth and malignancy in lung cancer cells. (A) Western blot analysis of FOXC1 knockdown in CL1-5 cells via the lentiviral-based FOXC1 shRNA. (B) Cell growth of CL1-5 cells with or without FOXC1 knockdown for 7 days. Cell growth was examined by trypan blue staining. The migration (C) and invasion (D) of CL1-5 cells with or without FOXC1 knockdown were measured by in vitro migration and invasion assay, respectively. The morphology characteristics (E) and quantification of total tubes (F) and tube length (G) for in vitro tube formation of HUVECs incubated with conditional medium from CL1-5 cells with or without FOXC1 knockdown. (H) The transcript levels of epithelial-mesenchymal transition (EMT) markers in CL1-5 cells with or without FOXC1 knockdown. (I) The protein levels of E-cadherin and fibronectin in CL1-5 cells with or without FOXC1 knockdown. Data are means ±SD (n=9). *P < 0.001 compared to wild type (WT) cells, one-way ANOVA with Tukey multiple comparison test.
Blockage of hypoxia-induced FOXC1 inhibits tumor growth in lung cancer xenografts
To investigate the impact of tumor hypoxia-induced FOXC1 in lung cancer progression, we first created a novel conditional FOXC1 knockdown platform using lentivirus vectors encoding 8× HRE-driven FOXC1 shRNAs (Fig. S6A). A549 cells stably expressing luciferase were transduced with lentivirus vectors carrying HRE-driven FOXC1 shRNAs or scramble shRNAs. Western blotting confirmed FOXC1 knockdown under hypoxic condition in the A549 cells (Fig. S6B). These cells were implanted subcutaneously into nude mice. Bioluminescent imaging (BLI) was utilized to assess tumor growth. A549 cells with a hypoxia-induced FOXC1 knockdown displayed significant growth inhibition in vivo, compared to wild-type cells or to control cells (Fig. 6A and 6B). These xenografts were further examined for the expression of HIF-1α, FOXC1 and Von Willebrand factor (vWF), an endothelial cell marker for angiogenesis, by immunofluorescence staining. HIF-1α and FOXC1 double-staining indicated that FOXC1 expression co-localized with HIF-1α expression in wild-type or in control lung cancer xenografts (Fig. 6C). In the lung cancer xenografts with the HIF-1α-induced FOXC1 knockdown, however, FOXC1 expression disappeared in HIF-1α-expressed areas, indicating that tumor hypoxia-mediated FOXC1 knockdown really works in tumor microenvironment. Moreover, the HIF-1-induced FOXC1 knockdown also largely decreased the vWF expression (Fig. 6C), indicating genetic depletion of FOXC1 inhibits tumor angiogenesis in vivo. These results show that tumor hypoxia-induced FOXC1 plays an important role in tumor growth and angiogenesis. Furthermore, we determined the role of FOXC1 in lung cancer metastasis. CL1-5 cells, with or without a FOXC1 knockdown, were lentivirally transduced with the luciferase reporter gene and intravenously injected into nude mice. The FOXC1 knockdown in CL1-5 cells caused a significant reduction in the number of mice exhibiting lung metastases (Fig. 6D), suggesting that FOXC1 contributes to lung cancer metastasis.
Blockage of hypoxia-induced FOXC1 inhibits tumor growth in lung cancer xenografts. (A) Bioluminescent images of tumor growth in A549 xenografts with or without conditional FOXC1 knockdown using lentivirus vectors encoding 8X HRE-driven FOXC1 shRNAs. (B) Tumor growth derived from luminescent signal (photon flux) in above experiments. Data are means ±SD (n=6). *P < 0.01 compared to wild type (WT) xenografts, two-way ANOVA with Bonferroni's post hoctest. (C) representative immunofluorescence images from frozen sections of tumor tissues in above experiments immunostained to detect the HIF-1α (green), FOXC1 (red), vWF (red) and nuclear DNA (blue). (D) Bioluminescent images and metastatic probability of mice intravenously injected with CL1-5 cells with or without FOXC1 knockdown via tail vein. (E) FOXC1 protein levels in homogenised CL1-5 xenografts from mice with systemic delivery of empty LNPs (NP-1), scramble siRNA LNPs (NP-2) or FOXC1 siRNA LNPs (NP-3) at a dose of 2 mg/kg for 3 days. Data are means ±SD (n=6). *P < 0.001 compared to NP-1, one-way ANOVA with Tukey multiple comparison test. (F) Bioluminescent images from mice received NP-1, NP-2 or NP-3 treatment on day 21 after tumor implantation. Mice were injected at a dose of 2 mg/kg at 7, 10, 13 and 16 days via tail vein. (G) The mean normalized BLI values associated with longitudinal monitoring of tumor growth for each treatment group. Data are means ±SD (n=6~8). *P < 0.001 compared to NP-1, two-way ANOVA with Bonferroni's post hoctest. (H) The corresponding survival curves of CL1-5 xenograft-bearing mice for each treatment group. (I) Tumor weight of CL1-5 xenografts from mice received NP-3 treatment at different dosages on day 21. Data are means ±SD (n=6~8). *P < 0.01 compared to control mice without treatment. one-way ANOVA with Tukey multiple comparison test.
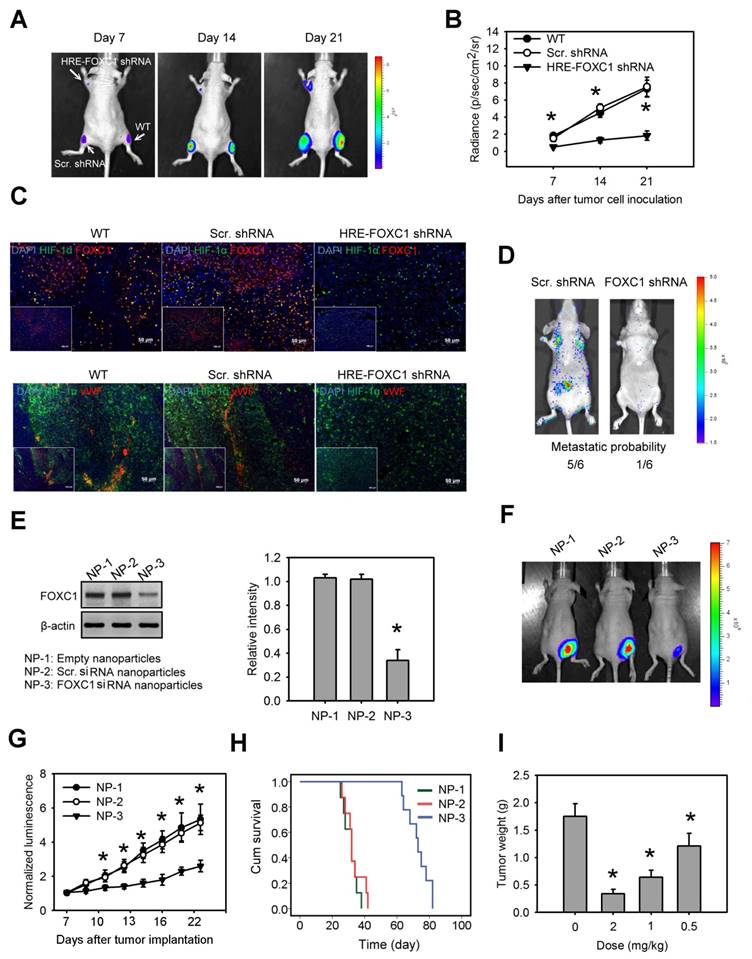
Finally, we investigated whether the targeted inhibition of FOXC1 by the systemic delivery of FOXC1 siRNA represents a viable means to inhibit tumor growth in lung cancer xenografts. To enhance siRNA stability in vivo, and prevent unwanted immune activation, native FOXC1 siRNA was chemically modified by selective incorporation of 2'-O-methyl (2'OMe)-uridine or -guanosine nucleosides into one strand of the siRNA duplex, and encapsulated into LNPs. Mice receiving systemic delivery of FOXC1 siRNA LNPs displayed a significant inhibition of FOXC1 expression and tumor growth in CL1-5 xenografts, compared to mice receiving systemic delivery of empty LNPs or scramble siRNA LNPs (Fig. 6E, 6F and 6G). We also performed the Kaplan-Meier analysis for animals treated with FOXC1 siRNA LNPs, scramble siRNA LNPs, and empty LNPs. FOXC1 siRNA LNP-treated mice showed significantly longer morbidity-free survival times (Fig. 6H). Moreover, FOXC1 siRNA LNPs-mediated tumor growth inhibition was dose-dependent (Fig. 6I). Taken together, these results show that FOXC1 is an important regulator of lung cancer growth and that specific targeting of FOXC1 by RNAi could be a novel therapeutic modality for lung cancer.
Discussion
Hypoxia is a common feature of the tumor microenvironment in solid tumors and it contributes to malignant tumor progression [27-29]. In lung cancer, tumor hypoxia is generally associated with disease progression and poor prognosis [30]. Moreover, growing evidence suggests that tumor hypoxia is able to promote lung cancer growth and metastasis [9, 10]. In mechanisms of hypoxia-promoted tumor progression, HIF-1 has been recognized as a master regulator that controls proliferation, apoptosis, differentiation, energy metabolism, angiogenesis and metastasis [31]. Many effector genes involved in these physiological processes have one or more HRE motifs, and therefore, HIF-1 is able to directly regulate their expression and function by binding to their promoters [32, 33]. However, HIF-1 has been found to interact with a variety of transcription factors and to interfere with their transcriptional activities. The mechanisms by which HIF-1-regulated transcription factors and their transcriptional activities act are through the modulation of expression level in activator, repressor or transcription factor itself or the interference of transcription factor and its associated protein interaction [34, 35]. The current study identified a novel HIF-1α interacting transcription factor, FOXC1, which plays an important role in lung cancer progression. HIF-1α directly binds to the FOXC1 promoter and acts to increase its transcriptional activity. Therefore, FOXC1 is a hypoxia-responsive gene, and the protein it encodes acts as another regulator to modulate hypoxia-response genes even if they lack the HRE motif.
Although it is widely known that FOXC1 plays important roles in both, normal biological processes and tumor progression [15, 36], the conditions and mechanisms involved in the regulation of FOXC1 expression are less well understood. In tumor progression, the microRNAs, miR-639 and miR-548I, modulate FOXC1 expression via post-transcriptional gene regulation [37, 38]. Epidermal growth factor receptor (EGFR) activates FOXC1 transcription through the ERK and Akt pathways in basal-like breast cancer [39]. Moreover, interleukin-8 (IL-8) induces FOXC1 expression through activation of phosphoinositide 3-kinase signaling [40], suggesting that tumor inflammation is an induction condition for FOXC1 expression. Besides, FOXC1 expression is regulated by BMP4 in early osteogenic differentiation [41]. In the present study, we show that hypoxia induces FOXC1 expression in the tumor microenvironment. FOXC1 upregulation is found in chronic or cycling tumor hypoxic areas, and is associated with hypoxia and HIF-1α levels in lung cancer xenograft models. Our data also provide a molecular mechanism to explain how hypoxia can regulate FOXC1 expression. HIF-1α directly binds to HRE motif in the FOXC1 promoter and further increases its transactivation. These findings not only highlight the fact that hypoxia is a critical stressor for FOXC1 induction in the tumor microenvironment, but also suggest that other pathological conditions, such as stroke or myocardial infarction, may also promote FOXC1 upregulation and function. Further studies are needed to confirm this hypothesis and elucidate the potential role of hypoxia-induced FOXC1 in these ischemic diseases.
Recent studies have shown that the high expression of FOXC1 is associated with poor clinical outcome in non-small cell lung cancer patients [18]. However, the impact of FOXC1 upregulation in lung cancer progression is still unclear. In this study, our results indicate that FOXC1 plays an important role in lung cancer progression. The knockdown of FOXC1 in CL1-5 cells with high FOXC1 expression and invasiveness inhibited cell proliferation, migration, invasion, angiogenesis, and EMT. By contrast, overexpression of FOXC1 in CL1-0 cells with low FOXC1 expression and invasiveness has opposite effects. These observations are in line with reports linking FOXC1 to other cancers, such as nasopharyngeal cancer, liver cancer, stomach cancer, pancreatic cancer, and melanoma [16, 42-47], suggesting that FOXC1 promotes tumor growth and malignancy. Most importantly, we show that tumor hypoxia-induced FOXC1 contributes to lung cancer progression. Hypoxia-driven FOXC1 knockdown in CL1-5 cells suppressed their growth and angiogenesis in vivo, indicating that FOXC1 is a critical mediator involved in tumor microenvironment-mediated tumor growth and malignancy. These findings suggest that FOXC1 is a potential therapeutic target in lung cancer treatment.
In order to translate our findings derived from basic research into clinical applications, we used RNAi-based LNP as a treatment strategy to allow for effective silencing of a target RNA in tumor cells in vivo, while reducing unwanted immune responses, because a FOXC1 inhibitor has not yet been developed. Here, we used FOXC1 siRNA chemically modified by selective incorporation of 2'OMe uridine or guanosine nucleosides and formulated with LNP to protect the systemically administered siRNA from serum nucleases, thus extending the siRNA in vivo half-life, according to previous studies [48-50]. Our results clearly demonstrate that the treatment of lung cancer-bearing mice with 2'OMe-modified FOXC1 siRNA encapsulated in LNP was sufficient to mediate potent silencing of the target mRNA and to effectively suppress lung tumor growth and angiogenesis in vivo. Moreover, this compound inhibited tumor growth in a dose-dependent manner and extended the survival time in these mice. Although more work is needed to optimize the therapeutic dose and to address the problem of potential side-effects, these findings indicate that there is clinical utility of FOXC1 siRNA targeting for the treatment of lung cancer disease with high therapeutic efficiency.
In conclusion, this is the first study to report that tumor hypoxia regulates FOXC1 expression and that this mechanism contributes to lung cancer progression. In addition, our findings provide preclinical proof-of-concept for FOXC1 blockade as a systemic therapy for lung cancer. Targeting of FOXC1 with RNAi-based nanoparticles allowed a therapeutic benefit in a preclinical model of lung cancer, providing an attractive therapeutic modality for lung cancer.
Supplementary Material
Supplementary figures.
Abbreviations
FOXC1: Forkhead box C1; HIF-1α: hypoxia-inducible factor-1α; HRE: hypoxia responsive element; NSCLC: non-small-cell lung cancer; EGFR: epidermal growth factor receptor; MMP: matrix metalloproteinase; VEGF: vascular endothelial growth factor; PET: positron emission tomography; CM: conditional medium; EMT: epithelial-mesenchymal transition; IL-8: interleukin-8.
Acknowledgements
We thank the National RNAi Core Facility and Molecular and Genetic Imaging Core/Taiwan Mouse Clinic, Taiwan, for technical support. Grant support was provided by the Ministry of Science and Technology of the Republic of China (Grant No. 103-2325-B-039-010; 105-2628-B-039 -009 -MY3), grants CMU103-BC-6 and DMR-105-089 from China Medical University and Hospital.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. Cancer statistics, 2003. CA Cancer J Clin. 2003;53:5-26
2. Ettinger DS, Akerley W, Borghaei H, Chang AC, Cheney RT, Chirieac LR. et al. Non-small cell lung cancer, version 2.2013. J Natl Compr Canc Netw. 2013;11:645-53 quiz 53
3. Bareschino MA, Schettino C, Rossi A, Maione P, Sacco PC, Zeppa R. et al. Treatment of advanced non small cell lung cancer. J Thorac Dis. 2011;3:122-33
4. Harris AL. Hypoxia-a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38-47
5. Meng X, Kong FM, Yu J. Implementation of hypoxia measurement into lung cancer therapy. Lung Cancer. 2012;75:146-50
6. Weidemann A, Johnson RS. Biology of HIF-1alpha. Cell Death Differ. 2008;15:621-7
7. Karetsi E, Ioannou MG, Kerenidi T, Minas M, Molyvdas PA, Gourgoulianis KI. et al. Differential expression of hypoxia-inducible factor 1alpha in non-small cell lung cancer and small cell lung cancer. Clinics (Sao Paulo). 2012;67:1373-8
8. Swinson DE, Jones JL, Cox G, Richardson D, Harris AL, O'Byrne KJ. Hypoxia-inducible factor-1 alpha in non small cell lung cancer: relation to growth factor, protease and apoptosis pathways. Int J Cancer. 2004;111:43-50
9. Zhang J, Cao J, Ma S, Dong R, Meng W, Ying M. et al. Tumor hypoxia enhances Non-Small Cell Lung Cancer metastasis by selectively promoting macrophage M2 polarization through the activation of ERK signaling. Oncotarget. 2014;5:9664-77
10. Jackson AL, Zhou B, Kim WY. HIF, hypoxia and the role of angiogenesis in non-small cell lung cancer. Expert Opin Ther Targets. 2010;14:1047-57
11. Aldinger KA, Lehmann OJ, Hudgins L, Chizhikov VV, Bassuk AG, Ades LC. et al. FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat Genet. 2009;41:1037-42
12. Kume T. The cooperative roles of Foxc1 and Foxc2 in cardiovascular development. Adv Exp Med Biol. 2009;665:63-77
13. Akbari OS, Bousum A, Bae E, Drewell RA. Unraveling cis-regulatory mechanisms at the abdominal-A and Abdominal-B genes in the Drosophila bithorax complex. Dev Biol. 2006;293:294-304
14. Hayashi H, Kume T. Foxc transcription factors directly regulate Dll4 and Hey2 expression by interacting with the VEGF-Notch signaling pathways in endothelial cells. PLoS One. 2008;3:e2401
15. Ray PS, Wang J, Qu Y, Sim MS, Shamonki J, Bagaria SP. et al. FOXC1 is a potential prognostic biomarker with functional significance in basal-like breast cancer. Cancer Res. 2010;70:3870-6
16. Xu ZY, Ding SM, Zhou L, Xie HY, Chen KJ, Zhang W. et al. FOXC1 contributes to microvascular invasion in primary hepatocellular carcinoma via regulating epithelial-mesenchymal transition. Int J Biol Sci. 2012;8:1130-41
17. Nagel S, Meyer C, Kaufmann M, Drexler HG, MacLeod RA. Deregulated FOX genes in Hodgkin lymphoma. Genes Chromosomes Cancer. 2014;53:917-33
18. Wei LX, Zhou RS, Xu HF, Wang JY, Yuan MH. High expression of FOXC1 is associated with poor clinical outcome in non-small cell lung cancer patients. Tumour Biol. 2013;34:941-6
19. Hsieh CH, Chang HT, Shen WC, Shyu WC, Liu RS. Imaging the impact of Nox4 in cycling hypoxia-mediated U87 glioblastoma invasion and infiltration. Mol Imaging Biol. 2012;14:489-99
20. Hsieh CH, Lin YJ, Wu CP, Lee HT, Shyu WC, Wang CC. Livin contributes to tumor hypoxia-induced resistance to cytotoxic therapies in glioblastoma multiforme. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:460-70
21. Hsieh CH, Chang HT, Shen WC, Shyu WC, Liu RS. Imaging the Impact of Nox4 in Cycling Hypoxia-mediated U87 Glioblastoma Invasion and Infiltration. Mol Imaging Biol. 2011
22. Hsieh CH, Kuo JW, Lee YJ, Chang CW, Gelovani JG, Liu RS. Construction of mutant TKGFP for real-time imaging of temporal dynamics of HIF-1 signal transduction activity mediated by hypoxia and reoxygenation in tumors in living mice. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2009;50:2049-57
23. Serganova I, Doubrovin M, Vider J, Ponomarev V, Soghomonyan S, Beresten T. et al. Molecular imaging of temporal dynamics and spatial heterogeneity of hypoxia-inducible factor-1 signal transduction activity in tumors in living mice. Cancer Res. 2004;64:6101-8
24. Szulc J, Aebischer P. Conditional gene expression and knockdown using lentivirus vectors encoding shRNA. Methods Mol Biol. 2008;434:291-309
25. Judge AD, Bola G, Lee AC, MacLachlan I. Design of noninflammatory synthetic siRNA mediating potent gene silencing in vivo. Molecular therapy: the journal of the American Society of Gene Therapy. 2006;13:494-505
26. Liu RS, Chou TK, Chang CH, Wu CY, Chang CW, Chang TJ. et al. Biodistribution, pharmacokinetics and PET imaging of [(18)F]FMISO, [(18)F]FDG and [(18)F]FAc in a sarcoma- and inflammation-bearing mouse model. Nuclear medicine and biology. 2009;36:305-12
27. Hockel M, Vaupel P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 2001;93:266-76
28. Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437-47
29. Vaupel P. The role of hypoxia-induced factors in tumor progression. Oncologist. 2004;9(Suppl 5):10-7
30. Li C, Lu HJ, Na FF, Deng L, Xue JX, Wang JW. et al. Prognostic role of hypoxic inducible factor expression in non-small cell lung cancer: a meta-analysis. Asian Pac J Cancer Prev. 2013;14:3607-12
31. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70
32. Greijer AE, van der Groep P, Kemming D, Shvarts A, Semenza GL, Meijer GA. et al. Up-regulation of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor 1 (HIF-1). J Pathol. 2005;206:291-304
33. Liu W, Shen SM, Zhao XY, Chen GQ. Targeted genes and interacting proteins of hypoxia inducible factor-1. Int J Biochem Mol Biol. 2012;3:165-78
34. Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ. et al. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat Cell Biol. 2008;10:295-305
35. Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD. et al. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604-13
36. Lambers E, Arnone B, Fatima A, Qin G, Wasserstrom JA, Kume T. Foxc1 Regulates Early Cardiomyogenesis and Functional Properties of Embryonic Stem Cell Derived Cardiomyocytes. Stem Cells. 2016;34:1487-500
37. Lin Z, Sun L, Chen W, Liu B, Wang Y, Fan S. et al. miR-639 regulates transforming growth factor beta-induced epithelial-mesenchymal transition in human tongue cancer cells by targeting FOXC1. Cancer Sci. 2014;105:1288-98
38. Medina-Trillo C, Aroca-Aguilar JD, Ferre-Fernandez JJ, Mendez-Hernandez CD, Morales L, Garcia-Feijoo J. et al. The Role of hsa-miR-548l Dysregulation as a Putative Modifier Factor for Glaucoma-Associated FOXC1 Mutations. Microrna. 2015;4:50-6
39. Jin Y, Han B, Chen J, Wiedemeyer R, Orsulic S, Bose S. et al. FOXC1 is a critical mediator of EGFR function in human basal-like breast cancer. Ann Surg Oncol. 2014;21(Suppl 4):S758-66
40. Huang W, Chen Z, Zhang L, Tian D, Wang D, Fan D. et al. Interleukin-8 Induces Expression of FOXC1 to Promote Transactivation of CXCR1 and CCL2 in Hepatocellular Carcinoma Cell Lines and Formation of Metastases in Mice. Gastroenterology. 2015;149:1053-67 e14
41. Hopkins A, Mirzayans F, Berry F. Foxc1 Expression in Early Osteogenic Differentiation Is Regulated by BMP4-SMAD Activity. J Cell Biochem. 2016;117:1707-17
42. Han B, Qu Y, Jin Y, Yu Y, Deng N, Wawrowsky K. et al. FOXC1 Activates Smoothened-Independent Hedgehog Signaling in Basal-like Breast Cancer. Cell Rep. 2015;13:1046-58
43. Xu Y, Shao QS, Yao HB, Jin Y, Ma YY, Jia LH. Overexpression of FOXC1 correlates with poor prognosis in gastric cancer patients. Histopathology. 2014;64:963-70
44. Wang L, Gu F, Liu CY, Wang RJ, Li J, Xu JY. High level of FOXC1 expression is associated with poor prognosis in pancreatic ductal adenocarcinoma. Tumour Biol. 2013;34:853-8
45. Wang J, Li L, Liu S, Zhao Y, Wang L, Du G. FOXC1 promotes melanoma by activating MST1R/PI3K/AKT. Oncotarget. 2016
46. Xia L, Huang W, Tian D, Zhu H, Qi X, Chen Z. et al. Overexpression of forkhead box C1 promotes tumor metastasis and indicates poor prognosis in hepatocellular carcinoma. Hepatology. 2013;57:610-24
47. Ou-Yang L, Xiao SJ, Liu P, Yi SJ, Zhang XL, Ou-Yang S. et al. Forkhead box C1 induces epithelialmesenchymal transition and is a potential therapeutic target in nasopharyngeal carcinoma. Mol Med Rep. 2015;12:8003-9
48. Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M. et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173-8
49. Xu CF, Wang J. Delivery systems for siRNA drug development in cancer therapy. Asian J Pharm Sci. 2015;10:1-12
50. Pai SI, Lin YY, Macaes B, Meneshian A, Hung CF, Wu TC. Prospects of RNA interference therapy for cancer. Gene Ther. 2006;13:464-77
Author contact
![]() Corresponding authors: Chia-Hung Hsieh, China Medical University and Hospital, No. 91, Hsueh-Shih Road, Taichung 404, Taiwan. Phone: 886-4-22052121; Fax: 886-4-22333641; E-mail: chhsiehcmucmu.edu.tw; Liang-Jwu Chen, National Chung Hsing University, No.145 Xingda Rd, South Dist., Taichung 402, Taiwan. Phone: 886-4-22840485; Fax: 886-4-22874879; E-mail: ljchennchu.edu.tw
Corresponding authors: Chia-Hung Hsieh, China Medical University and Hospital, No. 91, Hsueh-Shih Road, Taichung 404, Taiwan. Phone: 886-4-22052121; Fax: 886-4-22333641; E-mail: chhsiehcmucmu.edu.tw; Liang-Jwu Chen, National Chung Hsing University, No.145 Xingda Rd, South Dist., Taichung 402, Taiwan. Phone: 886-4-22840485; Fax: 886-4-22874879; E-mail: ljchennchu.edu.tw