Theranostics
13.3
Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2017; 7(4):987-1001. doi:10.7150/thno.17322 This issue Cite
Research Paper
Obatoclax and LY3009120 Efficiently Overcome Vemurafenib Resistance in Differentiated Thyroid Cancer
Wei-Jun Wei*, Zhen-Kui Sun*, Chen-Tian Shen, Hong-Jun Song, Xin-Yun Zhang, Zhong-Ling Qiu ![]() , Quan-Yong Luo
, Quan-Yong Luo ![]()
Department of Nuclear Medicine, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, Shanghai Jiao Tong University School of Medicine, 600# Yishan Road, 200233, Shanghai, China.
* Contributed equally to this work.
Received 2016-8-23; Accepted 2016-12-31; Published 2017-2-23
Citation:
Wei WJ, Sun ZK, Shen CT, Song HJ, Zhang XY, Qiu ZL, Luo QY. Obatoclax and LY3009120 Efficiently Overcome Vemurafenib Resistance in Differentiated Thyroid Cancer. Theranostics 2017; 7(4):987-1001. doi:10.7150/thno.17322. https://www.thno.org/v07p0987.htm
Other stylesAbstract

Although the prognosis of differentiated thyroid cancer (DTC) is relatively good, 30-40% of patients with distant metastases develop resistance to radioactive iodine therapy due to tumor dedifferentiation. For DTC patients harboring BRAFV600E mutation, Vemurafenib, a BRAF kinase inhibitor, has dramatically changed the therapeutic landscape, but side effects and drug resistance often lead to termination of the single agent treatment. In the present study, we showed that either LY3009120 or Obatoclax (GX15-070) efficiently inhibited cell cycle progression and induced massive death of DTC cells. We established that BRAF/CRAF dimerization was an underlying mechanism for Vemurafenib resistance. LY3009120, the newly discovered pan-RAF inhibitor, successfully overcame Vemurafenib resistance and suppressed the growth of DTC cells in vitro and in vivo. We also observed that expression of anti-apoptotic Bcl-2 increased substantially following BRAF inhibitor treatment in Vemurafenib-resistant K1 cells, and both Obatoclax and LY3009120 efficiently induced apoptosis of these resistant cells. Specifically, Obatoclax exerted its anti-cancer activity by inducing loss of mitochondrial membrane potential (ΔΨm), dysfunction of mitochondrial respiration, reduction of cellular glycolysis, autophagy, neutralization of lysosomes, and caspase-related apoptosis. Furthermore, the cancer killing effects of LY3009120 and Obatoclax extended to two more Vemurafenib-resistant DTC cell lines, KTC-1 and BCPAP. Taken together, our results highlighted the potential value of LY3009120 for both Vemurafenib-sensitive and -resistant DTC and provided evidence for the combination therapy using Vemurafenib and Obatoclax for radioiodine-refractory DTC.
Keywords: Thyroid cancer, Drug resistance, Vemurafenib, Obatoclax, LY3009120.
Introduction
The first-line therapeutic methods for differentiated thyroid cancer (DTC) consist of thyroidectomy, radioactive iodine treatment, and levothyroxine replacement therapy. However, we and others have shown that in about 30-40% of patients with metastatic DTC these traditional treatment methods are not curative. The tumor dedifferentiation or concomitant loss of thyroid-specific genes regulating iodide concentration, especially the gene encoding human sodium iodide symporter (SLC5A5), expose DTC patients to high risk of disease persistence or recurrence [1-3].
With increasing understanding of the cellular and molecular mechanisms of thyroid cancer, the BRAFV600E mutation was identified as one of the most important mechanisms involved in DTC pathogenesis [4]. Although targeted therapy inhibiting BRAFV600E mutation has achieved preliminary success, side effects and resistance to single agent BRAFV600E inhibitor often lead to termination of the targeted therapy [5-7]. For BRAFV600E-positive papillary thyroid cancer (PTC) patients, who were refractory to radioactive iodine and had never been treated with a multikinase inhibitor, a BRAF kinase inhibitor, Vemurafenib, was recently tested in a phase 2 clinical trial. Results showed the antitumor activity of Vemurafenib with a partial response rate of 38.5%, but the grade 3 or 4 adverse events rate was as high as 65% [8]. While intermittent therapy may delay resistance to Vemurafenib, using next-generation BRAF inhibitor or drug combinations to prevent mitogen-activated protein kinase (MAPK) pathway reactivation may further improve clinical efficacy over BRAF inhibition alone [9, 10]. Furthermore, mechanisms causing Vemurafenib resistance in DTC still need to be elucidated and strategies overcoming drug resistance are urgently needed in clinical practice. The potential combination with inhibitors antagonizing anti-apoptotic proteins, such as ABT-737, proved to be effective in vitro in several cancer types including melanoma, lymphoma, small-cell lung carcinoma, and hepatoma [11-14]. However, our previous study reported that the combined usage of Vemurafenib and ABT-737 only caused cell cycle arrest in BRAFV600E-positive K1 and BCPAP cells without inducing apoptosis, indicating that this concomitant therapy was ineffective in thyroid cancer [15]. This prompted us to further investigate the potential therapeutic and synergistic effects of LY3009120, a next-generation pan-RAF inhibitor, and Obatoclax, which has shown an excellent anti-cancer effect in esophageal carcinoma and leukemia [16-18].
In this study, we found that LY3009120 was more potent than Vemurafenib and induced massive apoptosis of DTC cells. We also discovered that Obatoclax efficiently killed DTC cells as a single agent and enhanced the cytotoxicity of Vemurafenib through several mechanisms. We cultured Vemurafenib-resistant K1 cells to give an insight into the mechanism of resistance occurrence, and verified the inhibitory effects of LY3009120 and Obatoclax in Vemurafenib resistant K1 cells. The efficacy of these two inhibitors were further validated in two other Vemurafenib resistant BCPAP and KTC-1 DTC cell lines, both of which are BRAFV600E mutation-positive. Finally, we used an in vivo study to confirm the tumor-suppressing property of LY3009120 as a single agent and the combination therapy consisting of Vemurafenib and Obatoclax.
Method
Cell lines, compounds, and transfection
PTC cell lines K1, BCPAP, and KTC-1 were purchased from Institute of Biochemistry and Cell Biology (SIBS, CAS, Shanghai, China). All cell lines have been confirmed for BRAFV600E mutation, Short Tandem Repeat (STR) and amelogenin authentication as described in ANSI Standard by the ATCC Standards Development Organization (SDO). BHP27 cell line was a gift from Prof. Li-Bo Chen [19]. All cell lines were maintained in RPMI 1640 medium (Gibco, Cat. # 11875-093) supplemented with 10% FBS (Gibco, Cat. # 16000-044) in 5% CO2 - 95% air atmosphere at 37 °C. Additional 1% NEAA (ThermoFisher, Cat. #11140050) was supplemented when culturing BCPAP cells. Vemurafenib-resistant cells were created as previously reported and maintained in complete medium containing 5 µM or 10 µM Vemurafenib [20]. All compounds used in the study were obtained from Selleck and were dissolved in DMSO and used at the indicated concentrations. Pre-made lentiviral particles expressing a fusion target of GFP-RFP-LC3 were purchased from Genomeditech Co. Ltd and transfected into K1 cells according to the manufacturer's protocol. Short hairpin RNA sequence targeting human Mcl-1 gene was inserted into hU6-MCS-CMV-EGFP vector to generate Mcl-1-si plasmid. The sequence silencing Mcl-1 expression was 5'-CAGGAATGTGCTGCTGGCTTT-3'. Plasmid transfected with scramble siRNA (5'-TTCTCCGAACGTGTCAC-3') was used as a negative control (synthesized by Genechem, Shanghai, China). The knock-down efficiency was confirmed by fluorescence microscope and Western blotting.
Cell counting kit-8 and colony formation assay
Survival curves of K1, resistant K1, BCPAP, and KTC-1 cells were assessed using cell counting kit-8 (CCK8) assay (Yeasen, Shanghai, China). Briefly, cells were seeded in 96-well plates at a density of 1x104 cells per well, allowed to adhere for 24 h, and then incubated for 48 h with increasing concentrations of the compounds under study. Control groups were allowed to grow in the absence of any inhibitors. The samples were assayed in sextuplet at least in three independent experiments and the mean value for each experiment was calculated. For clonogenic survival studies, 300 cells were plated per well in a six-well plate and treated with Vemurafenib (5 µM and 10 µM), LY3009120 (5 µM and 10 µM) and Obatoclax (1 µM and 5 µM) for 48 h. Then the media were changed and cells were cultured for another 15 days before crystal violet staining and imaging.
Western blotting and immunoprecipitation
Western blotting was carried out as described previously [15, 21, 22]. Anti-Bcl-2, anti-Mcl-1, anti-PARP1, anti-cytochrome C, anti-ERK, anti-p-ERK, anti-Bim, anti-TSHR, anti-HK2, anti-Bax, anti-cyclin D1, and anti-GAPDH antibodies were purchased from ProteinTech (Chicago, IL, USA); anti-GLUT1 (NB110-39113SS) antibody was acquired from Novusbio. Anti-Beclin1 (#3738), anti-p62 (#8025), anti-p27 Kip1 (#3688), anti-Bcl-xl (#2764), anti-LC3A/B (#12741), anti-Caspase-3 (#9662), anti- Caspase-9 (#9502), anti-Caspase-8 (#9746) antibodies were purchased from Cell Signaling Technology. For immunoprecipitation, cells were treated for 24 hours with Vemurafenib before harvesting. NP40 lysates and BRAF immunoprecipitates were prepared according to the previous publication using rabbit anti-BRAF antibody (Santa Cruz Biotechnology, sc-5284) and analyzed by immunoblot using mouse anti-CRAF antibody (Santa Cruz Biotechnology, sc-227) or inversely [23].
Flow cytometric analysis and Seahorse XF24 assay
For apoptosis analysis, DTC cells were treated for 24 h with increasing concentrations of Vemurafenib, LY3009120, or Obatoclax before flow cytometric apoptosis analysis. At each concentration, half a million cells were collected, washed two times with cold PBS. Then cells were stained with FITC-Annexin V/PI or APC-Annexin V/7-AAD (Affymetrix eBioscience). For cell cycle analysis, 106 cells were treated with different concentrations of Vemurafenib, LY3009120, or Obatoclax for 24 h. Cells were trypsinized and centrifuged at 800 rpm for 5 min. Cell pellets were washed and resuspended in 300 µL ice-cold PBS and fixed overnight after adding 700 µL ethanol to each sample. After fixation, cell pellets were resuspended and incubated in 300 µL PBS (containing 20 µL RNase A) at 37˚C for 30 min. Subsequently, 400 µL PI solution (Yeasen, Shanghai, China) was added and samples were analyzed. Mitochondrial membrane potential was determined using the fluorogenic, lipophilic cation JC-1 (Yeasen, Shanghai, China) by confocal microscopy, fluorescent microscopy, and flow cytometric analysis (Beckman Coulter, Brea, CA, USA). For Seahorse experiments, K1 cells and Mcl-1-silenced K1 cells were seeded overnight in 24-well culture plate at a density 10,000 cells per well in the RPMI 1640 medium. The following day, the cell culture medium was removed and replaced with fresh medium containing 2 μM Vemurafenib, LY3009120, or Obatoclax for 36 h and incubated at 37 °C. Per the manufacturer's protocol, Seahorse injection ports were loaded with glucose, rotenone, FCCP, oligomycin, and 2-deoxy glucose. After incubation, oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured by SeaHorse XF24 Extracellular Analyzer from SeaHorse Bioscience.
Confocal microscopy and Transmission electron microscopy
GFP-RFP-LC3-transfected K1 cells were grown on coverslips and treated with LY3009120 or Obatoclax for indicated times. Fluorescent microscopy and transmission electron microscopy (TEM) were used for the analysis of changes in various autophagic structures. For TEM assays, samples were fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer. Subsequently, the samples were post-fixed in 1% osmium tetroxide and 2% uranyl acetate, dehydrated through a graded series of ethanol, and embedded in LX112 resin (LADD Research Industries). Ultrathin sections were cut on a Reichert Ultracut E, stained with uranyl acetate followed by lead citrate and viewed at 80 kV.
Animal Studies
All protocols involving mice were evaluated and approved by our Institutional Animal Care and Use Committee and performed under veterinary supervision. 4-week-old nude mice were injected subcutaneously with 5x106 normal K1 or Mcl-1 knockdown K1 cells in RPMI 1640 in the right flank. When tumors reached 50 mm3, mice were injected subcutaneously with PBS, Vemurafenib, LY3009120, Obatoclax, or combination of Vemurafenib and Obatoclax (n=6 per group) up to 21 days (at a dose of 10 mg/kg/day for the single agent or 20 mg/kg/day for combination therapy). Tumor growth was monitored every two days and tumor volume was calculated (volume=length×width2/2). Statistical analyses of tumor volumes between the normal control group and drug-treated group or between different drug-treated groups at day 21 were determined by a one-way ANOVA test. The resected tumor specimens were fixed in 10% neutral buffered formalin and embedded in paraffin. Sections were cut on a microtome and mounted on glass slides. Sections were de-waxed and hydrated in graded alcoholic solutions and then distilled water. For immunohistochemical staining, two slices were stained with terminal TUNEL (Yeasen, Shanghai, China) according to the manufacturer's protocol. The cell nuclei were counterstained with 40, 6- diamidino-2-phenylindole (DAPI, Sigma). TUNEL-stained slides were visualized under a fluorescence microscope (Zeiss Axio Observer.Z1).
Cancer Genome Atlas (TCGA) analysis of Bcl-2 family expressions
The cBioPortal for Cancer Genomics web was used to explore the gene expression levels of Bcl-2 members in thyroid cancer according to the protocol [24].
Statistical analysis
Statistical analyses were performed using the Statistical Package for the Social Sciences, version 20.0 (SPSS, Inc., Chicago, IL, USA) and GraphPad Prism version 5.0 (GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was considered to indicate a statistically significant result.
Results
LY3009120 as a next-generation BRAF inhibitor was more potent than Vemurafenib
First, we confirmed our previous observation that Vemurafenib suppressed the expression of p-ERK in BRAFV600E mutant K1 cells but had little impact on the p-ERK expression in BRAF wild-type (WT) BHP27 cells (Fig.1A) [15]. However, Vemurafenib was unable to induce apoptosis of K1 cells as supported by the unchanged levels of caspase-3, PARP1, and its cleaved fragments. We investigated the redifferentiation effect of Vemurafenib and found that it suppressed GLUT1 in a dose-dependent manner without reducing HK2 or increasing TSHR (Fig.1B). Using Memorial Sloan Kettering Cancer Center cBioPortal (http://www.cbioportal.org/public-portal/study.do?cancer_study_id=thca_tcga), we performed a comprehensive evaluation of The Cancer Genome Atlas (TCGA) thyroid cancer data where tumor and normal tissues from 496 PTCs were profiled for gene expression using multi-platforms [25]. Our analysis revealed that Bcl-2 mRNA was overexpressed more than two-fold in 7% of DTC tissues when compared with adjacent normal thyroid tissue (Fig.1C). A previous study had shown that anti-apoptotic Bcl-2 governed outcome of the targeted therapy and addition of ABT-737 to Vemurafenib sensitized melanoma cells to the mitochondrial pathway of apoptosis [12].
Figure 1
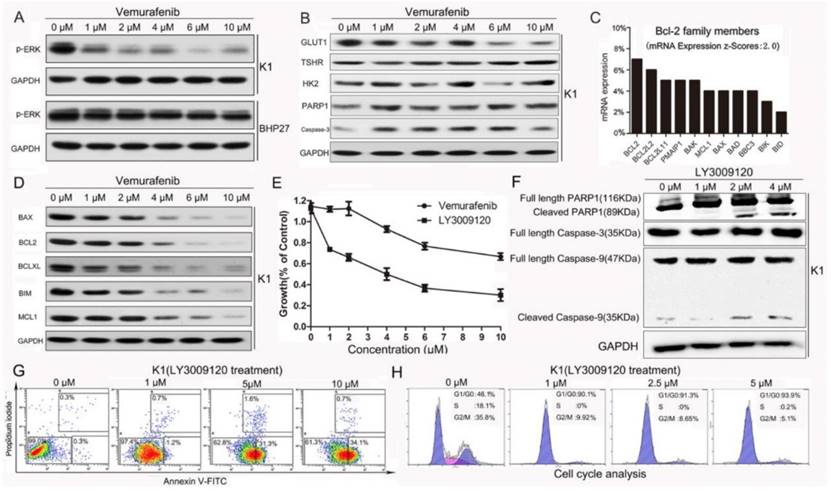
The effect of Vemurafenib and LY3009120 in BRAFV600E or BRAFwide type DTC cells. (A) Vemurafenib suppressed expression of p-ERK in BRAFV600E mutant K1 cells but not in the wild-type BHP27 cells. (B) Vemurafenib failed to increase the cleaved fractions of Caspase-3 and PARP1, which are markers of typical Caspase-dependent apoptosis. (C)The mRNA levels of Bcl-2 family members were analyzed using TCGA thyroid cancer database (fold change>2 when compared with adjacent normal thyroid tissue). (D) Change of Bcl-2 family proteins following Vemurafenib treatment for 24 h. (E) Survival curves of K1 cells following Vemurafenib and LY3009120 treatment, the latter is a novel BRAF inhibitor undergoing clinical trial. (F) LY3009120 is a more potent BRAF inhibitor against K1 cells as demonstrated by appearance of cleaved Caspase-9 and PARP1 fragments. (G) Flow cytometry revealed increased apoptosis of K1 cells following LY3009120 treatment. (H) LY3009120 efficiently induced cell cycle arrest of K1 cells.
However, in our study neither ABT-737 nor the combination of ABT-737 and Vemurafenib resulted in profound synergism and extensive tumor cell death (Supplemental Fig. 1A&B). The confounding levels of Bcl-2 family members following Vemurafenib intervention further strengthened its inability to induce apoptosis (Fig. 1D). We then tested the efficacy of a newly discovered next-generation BRAF inhibitor LY3009120 [26]. The proliferation rates of K1 cells treated with increasing doses of Vemurafenib and LY3009120 for 24 h revealed that LY3009120 was a much more potent inhibitor with IC50 of 4 μM (Fig.1E). Following LY3009120 treatment for 24 h, the appearance of PARP1 and Caspase-9 fragments (Fig.1F) and dose-dependent increase in the number of apoptotic K1 cells (apoptosis rates from 0 to 10 μM: 0.6%, 1.9%, 32.9%, 34.8%) (Fig.1G) were further evidence for the cytotoxicity of LY3009120. Also, the LY3009120-induced apoptosis was accompanied by cell cycle arrest (Fig.1H).
Obatoclax enhanced cytotoxicity of Vemurafenib through inducing mixed cell death forms, loss of MOMP, suppression of mitochondrial respiration, and cellular glycolysis
In vitro proliferation assays revealed that IC50 values of Obatoclax in three DTC cell lines varied. K1 cells depicted a relatively higher IC50 of 3.18 μM, whereas BCPAP and KTC-1 cells showed comparable IC50 values (0.85 μM for BCPAP and 0.76 μM for KTC-1, data not shown). Contrary to Vemurafenib, Obatoclax induced strong cytotoxic effects in two PTC cell lines (K1 and BCPAP) in vitro. Flow cytometry revealed a dose-dependent increase in Annexin Ⅴ-FITC/PI double positive apoptotic cells (apoptosis rates from 0 to 10 μM: 5.0%, 30.4%, 89.6%, 97.7%, 99.9%, 100% for K1 and 4.6%, 21.3%, 62.4%, 94.0%, 99.1%, 99.7% for BCPAP, respectively) following Obatoclax treatment for 24 h (Fig. 2A&B). Although Vemurafenib as a single agent was insufficient to induce apoptosis, Obatoclax pretreatment (1 μM for 2 h) sensitized both cell lines to the cytotoxicity of Vemurafenib as evidenced by the increased apoptosis rates (Fig. 2C&D). Considering the fact that Obatoclax produces autofluorescence (absorbance peak, 490 nm; emission peak, 550 nm), which interferes with both FITC and PI excitation/emission channels [27], we used 7-AAD (excitation peak, 546 nm; emission peak, 647 nm) as a nuclear exclusion dye and Annexin V conjugated to APC (absorbance peak, 650 nm; emission peak, 660 nm) to avoid the interference.
Figure 2
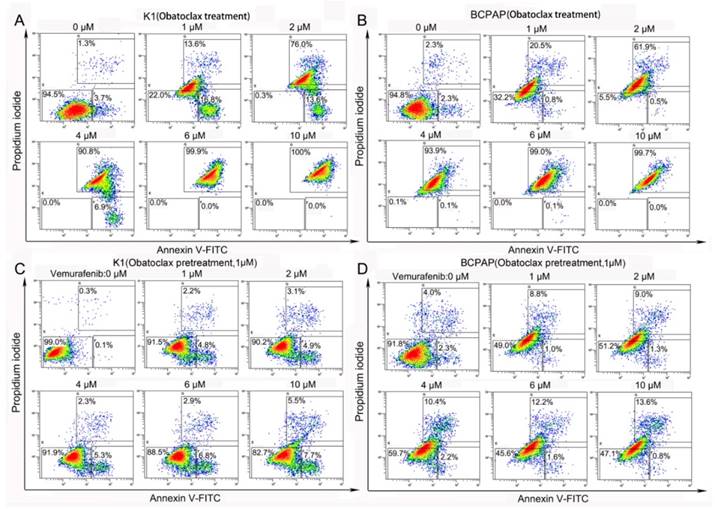
Obatoclax alone or Obatoclax plus Vemurafenib powerfully induced cell death in K1 and BCPAP cells (assessed by Annexin V-FITC/PI flow cytometry). (A, B) Obatoclax induced massive apoptosis in K1 and BCPAP cells in a concentration-dependent manner. (C, D) Although Vemurafenib was unable to induce apoptosis in K1 cells as a single agent, pretreatment using 1 µM Obatoclax for 2 h enhanced the cytotoxic effect of Vemurafenib in both K1 and BCPAP cells.
Figure 3
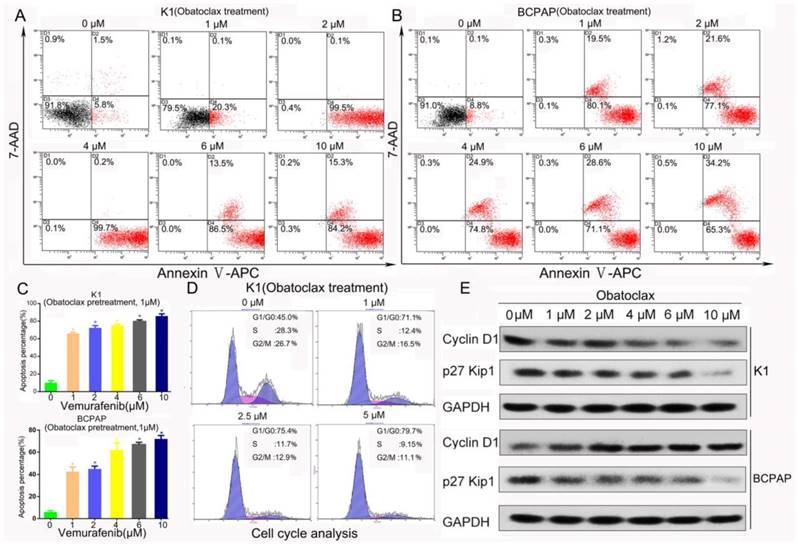
Obatoclax alone or Obatoclax plus Vemurafenib induced cell death (assessed by Annexin V-APC/PI flow cytometry) and cell cycle arrest in K1 and BCPAP cells. (A, B) The cancer cell killing effects of Obatoclax in K1 and BCPAP cells were further evaluated using Annexin V-APC/7-AAD. Flow cytometry results comfirmed the cytotoxicity of Obatoclax in K1 and BCPAP cells. (C) Annexin Ⅴ-APC/7-AAD apoptosis assays confirmed the efficacy of combination therapy where K1 cells were pretreated using Obatoclax of 1 µM for 2 h and then subjected to Vemurafenib treatment for another 24 h. (D) Cell cycle analysis revealed that Obatoclax inhibited cell cycle progression of K1 cells dose-dependently. (E) Western blotting showed that Obatoclax impacted cell cycle distribution through lowering cyclin D1 in K1 cells but in BCPAP cells, it impacted expression of p27 Kip1 rather than cyclin D1.
As displayed in Fig. 3A&B, independent of autofluorescence interference, Obatoclax was capable of killing K1 and BCPAP cells in a dose-dependent manner (apoptosis rates from 0 to 10 μM: 7.3%, 20.4%, 99.6%, 99.9%, 100%, 99.5% for K1 and 8.9%, 99.6%, 98.7%, 99.7%, 99.7%, 99.5% for BCPAP, respectively). We also confirmed the efficacy of synergistic therapy containing Obatoclax and Vemurafenib using Annexin V-APC/7-AAD reagents (Fig.3C, supplemental Fig.1C&D). Because of the limited number of Annexin V-FITC positive cells, it was easy to discern from Fig. 2B&D death of BCPAP cells as necroptosis or necrosis rather than typical apoptosis, which was possibly caused by autofluorescence of Obatoclax. Hence, proper usage of fluorescent dyes like Annexin V-APC/7-AAD is important when assessing the therapeutic effect of Obatoclax. Also, exposure of K1 cells to Obatoclax resulted in enrichment of cells in the G0/G1 phase accompanied by a decrease in the S and G2/M phases in a concentration-dependent manner (Fig. 3D), and cell cycle arrest was correlated with reduced expression of cyclin D1 in K1 cells. Although Obatoclax inhibited cell cycle progression of BCPAP cells as well (data not shown), a decrease in p27 Kip1 rather than cyclin D1 was observed (Fig. 3E).
As shown in Fig. 4A, pretreatment of K1 cells with a general caspase inhibitor Z-VAD-FMK (50 μM) failed to reverse Obatoclax-induced cell death, indicating that Obatoclax induced mixed forms of cell death in DTC cells other than Caspase-related apoptosis. In other words, concomitant cell death types occurred after treatment with Obatoclax. To better understand the relationship between mitochondrial response and inhibition of Bcl-2 anti-apoptotic members, we fractionated K1 cells into mitochondrial proteins and cytosolic proteins and subjected these fractions to Western blotting analysis for cytochrome C. Obatoclax treatment resulted in marked redistribution of cytochrome C from mitochondria to the cytoplasm (Fig.4B). Consistently, we detected the appearance of cleaved Caspase-3 and Caspase-9 in Obatoclax-treated samples (Fig.4C). The phenomenon that Z-VAD-FMK failed to reverse cancer cell killing effect of Obatoclax prompted us to further elucidate other potential Caspase-independent mechanisms. Considering Obatoclax occupies a hydrophobic pocket within the BH3-binding groove of Bcl-2 in the intact mitochondrial outer membrane [28], we examined the mitochondrial membrane potential of both K1 cells and resistant K1 cells. We used the dye JC-1, which can facilitate discrimination of energized and de-energized mitochondria by forming red fluorescent aggregates when concentrated in energized mitochondria in response to the higher membrane potential. From the cytometry-based analyses, we observed a gradual loss of membrane potential in the energized mitochondria as there was a transition from red fluorescence to green fluorescence following Obatoclax intervention (Fig. 5A). Confocal microscopy confirmed that in healthy K1 cells both green and red signals were strong, indicating the presence of both cytoplasmic JC-1 monomer and mitochondrial J-aggregates. However, regions of high mitochondrial polarization indicated by red fluorescence were replaced by the green fluorescence of the JC-1 monomers after Obatoclax treatment (Fig. 5B, Supplemental Fig. 2). Although Obatoclax is believed to impair lysosomal function [16], we failed to observe the lysosome neutralization effect of Obatoclax due to its autofluorescence. It is of note that Vemurafenib and LY3009120 had no obvious effect on K1 lysosomes as indicated by Lysotracker Red DND-99 staining (Supplemental Fig.3).
As most of the ATP in cancer cells comes from mitochondria [29], we measured two major energy pathways in K1 cells, mitochondrial respiration, and cellular glycolysis, using Seahorse XF24 Analyzer [30]. Basal respiration responds to the ATP demand and can alter dramatically depending on the presence or absence of different factors. Furthermore, respiration is controlled very strongly by proton leak kinetics and partially by substrate oxidation. In contrast to the observation that Obatoclax did not induce any appreciable changes in ATP production in anaplastic thyroid cancer cells [31], Fig. 5C showed that although 2 µM Obatoclax did not affect the basal respiratory rate, it augmented proton leak and reduced ATP production significantly after treatment for 36 h. While 2 µM of Vemurafenib unexpectedly up-regulated mitochondrial respiration, the same concentration of LY3009120 suppressed basal respiration rate, ATP production, as well as cellular glycolysis.
Figure 4
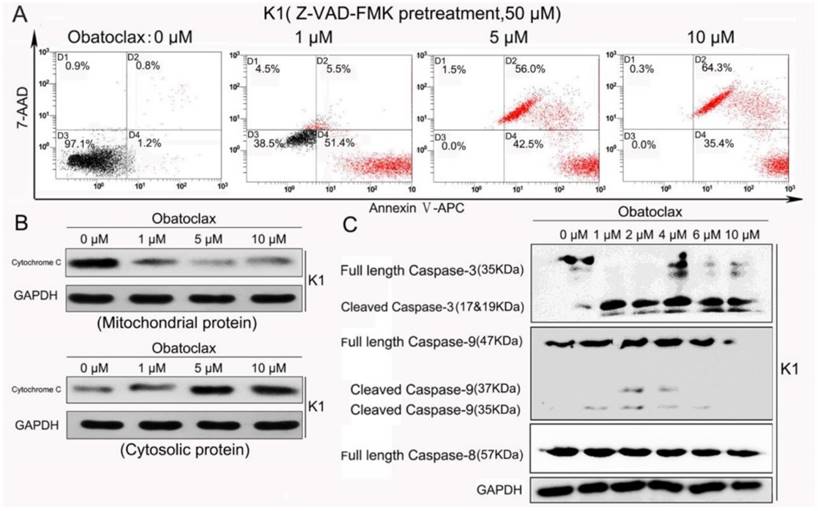
Redistribution of cytochrome C and significant Caspase-3/-9 enzymatic activity were observed in K1 cells treated with Obatoclax. (A) Pretreatment with Z-VAD-FMK failed to reverse Obatoclax-induced apoptosis in K1 cells, indicating that Obatoclax may induce mixed cell death forms. (B) After Obatoclax treatment for 24 h, Western blotting of cytochrome C in the fractionated cytosol and heavy membranes revealed redistribution of cytochrome C from mitochondria to cytoplasm. (C) Redistribution of cytochrome C subsequently triggered cleavage of Caspase-9 and PARP1 in K1 cells as demonstrated by Western blotting.
Figure 5
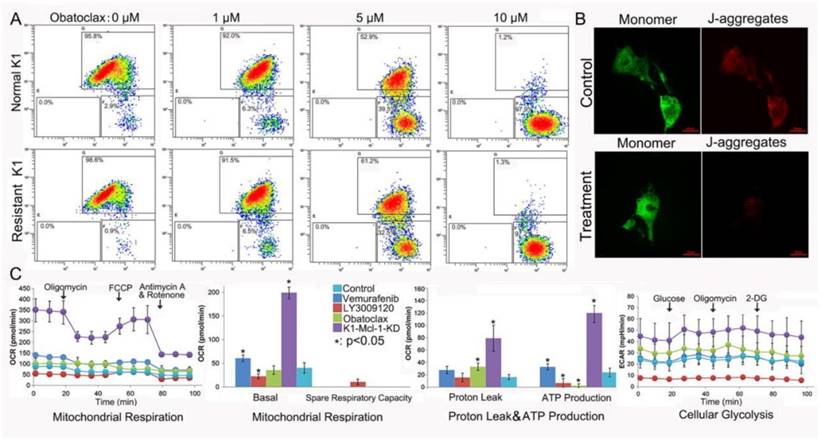
Obatoclax reduced mitochondrial outer membrane permeabilization (MOMP) and ATP production in K1 cells. (A) Bivariate JC-1 analysis of mitochondrial membrane potential in K1 cells (top panel) and Vemurafenib-resistant K1 cells (bottom panel) by flow cytometry. Distinct populations of cells with different extents of mitochondrial depolarization were detectable following Obatoclax treatment with 0 µM, 1 µM, 5 µM and 10 µM for 24 h. The number in each right bottom quadrant represent the percentage of cells with reduction in ΔΨm. (B) Mitochondrial depolarization was indicated by a transition in the red/ green fluorescence intensity ratio under confocal microscopy. While both red and green signals were detectable in normal K1 cells (top panel) the red signal nearly disappeared in Obatoclax-treated K1 cells (bottom panel). (C) Detection of mitochondrial respiration and cellular glycolysis in Mcl-1-silenced K1 cells or in K1 cells following treatments using 2 μM of Vemurafenib, LY3009120, or Obatoclax for 36h.
Obatoclax and LY3009120 induced autophagy through different mechanisms
Anti-apoptotic Bcl-2 family members participate in the inhibition of autophagy and there is an overlap between proteins that are essential for both apoptosis and autophagy [32, 33]. It is well established that cellular oncogenic signaling including BRAF and MAP2K pathways are connected with autophagy, which indicates a well differentiated state in thyroid cancer [34-37]. Therefore, it is plausible that Obatoclax and LY3009120 may modulate autophagy activity. We transfected K1 cells using GFP-RFP-LC3 lentiviral particles and monitored autophagy under various conditions. Depending on the extent of autophagy, LC3 proteins have different subcellular localizations. LC3-I is a constitutively expressed protein involved in autophagosome assembly. LC3-II is localized to the cytosolic and luminal surfaces of mature, double-membraned autophagic vesicles that microscopically appear as intracellular puncta [38]. After incubating with Obatoclax for 48 hours, images were obtained by oil immersion confocal microscopy. A remarkable increase in GFP-RFP-LC3-positive autophagic puncta was observed in K1 cells at a concentration as low as 1 μM (Fig.6A). Most K1 cells lost viability when incubated at a higher concentration of Obatoclax (5 μM). TEM images showed that mitochondria of normal K1 cells were surrounded by an outer, smooth membrane and an inner, folded membrane forming the cristae filling the interior of the organelle (Fig.6B1). Autophagosomes formed following Obatoclax (5 µM) treatment for 48 h as indicated by red arrows (Fig. 6B2-6), and colocalized with lysosomes (Fig. 6B4, red arrow). Consistent with decreased mitochondrial permeability and mitochondrial dysfunction, we observed swollen mitochondria characterized by a decrease or disappearance of cristae (Fig. 6B3, 5, blue arrows). Although we failed to track changes of lysosomes using Lysotracker Red DND-99, damaged lysosomes or large vacuoles (black arrows) increased significantly in TEM scanning indirectly indicating the neutralization of lysosomes by Obatoclax.
We then detected LC3 conversion and p62 (SQSTM1) by immunoblot analysis and observed that both LC3-II and p62 increased following Obatoclax intervention (Fig.6C). This was inconsistent with the basic tenet that upregulation in LC3-II is accompanied by a decrease in p62, as the former serves as a reliable method for monitoring autophagy while the latter acts as a readout of autophagic degradation [38, 39]. Western blotting of apoptosis- and autophagy-related proteins showed that there was a reverse correlation between the expressions of Mcl-1 and Beclin-1 after incubation with Obatoclax (Fig. 6D&E). Previous studies validated that suppression of Mcl-1 could mediate Beclin-1-associated autophagy [33]. We verified whether this kind of interaction contributed to the activation of autophagy in thyroid cancer cells by suppressing the Mcl-1 level in K1 cells using lentivirus. Fluorescence microscopy and Western blotting confirmed decreased Mcl-1 in the cells transfected with the lentivirus VS control. Interestingly, suppression of Mcl-1 in K1 cells led to increased expression of Beclin-1 (Fig.7A), suggesting that the Mcl-1/Beclin-1 signaling axis might play a role in the Obatoclax-induced autophagy. Previous studies showed that Obatoclax potentiated accumulation of LC3-II by interrupting the autophagosome-lysosome fusion or inhibiting lysosome-mediated proteolysis [16, 31, 39], Obatoclax also induced caspase-independent cell death through triggering Beclin-1-mediated autophagy [40]. Our results suggest that Obatoclax impact not only the induction but also the degradation phase of autophagy in DTC cells.
As shown in Fig 7B, Mcl-1 knock-down alone resulted in early apoptosis in K1 cells (17.4%) without blocking Obatoclax-induced apoptosis. Importantly, Mcl-1-deletion rendered K1 cells highly susceptible to cell death stimuli exerted by Vemurafenib as revealed by flow cytometry results (apoptosis rates from 0 to 10 μM: 12.9%, 13.5%, 16.8%, 17.4%, 18.3%, 28.4%) (Fig.7 C). LY3009120-induced autophagy was concentration-dependent without a reverse correlation between Mcl-1 and Beclin-1 (Fig. 7E). We also found that LY3009120 inhibited phosphorylation of ERK more strongly than Vemurafenib and even suppressed expressions of BRAF and HRAS (Fig. 7F), another unique property of LY3009120 as reported in melanoma [9].
Dimerization of RAF protein accounted for Vemurafenib resistance which could be overcome by LY3009120 and Obatoclax
We exposed resistant K1 cells to increasing concentrations of Vemurafenib (5 μM and 10 μM). These cells were resistant to Vemurafenib treatment when compared to normal K1 cells. Notably, Vemurafenib-resistant K1 cells were quite sensitive to LY3009120 (Fig. 8A). Cell colonies decreased significantly in the LY3009120- and Obatoclax-treated groups as depicted in Fig. 8B.
Figure 6
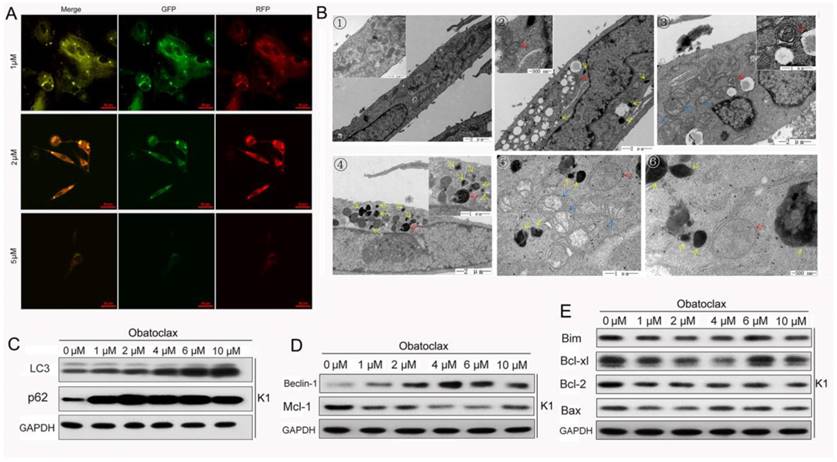
Obatoclax regulated both the induction and degradation phases of authophagy in K1 cells. (A) In K1 cells Obatoclax, at a concentration of 1 µM and 2 µM, induced formation of cytoplasmic LC3-II puncta while a higher concentration of Obatoclax (5 µM) inversely decreased LC3-II-positive puncta. (B) Transmission electron microscopy is regarded as the gold standard for visualizing autophagy. In the control K1 cells relatively well-preserved mitochondria were observed (B①). Autophagosomes (red arrows) and large vacuoles (black arrows) formed following Obatoclax treatment for 48 h (B②-⑥). Colocalization of autophagosomes with lysosomes (red arrow, B④) and swollen mitochondria (blue arrows, B③ and B⑤) were also observed. Scale bars are included within the micrographs. (C, D) Change of autophagy and apoptosis related proteins in K1 cells following Obatoclax treatment.
Figure 7
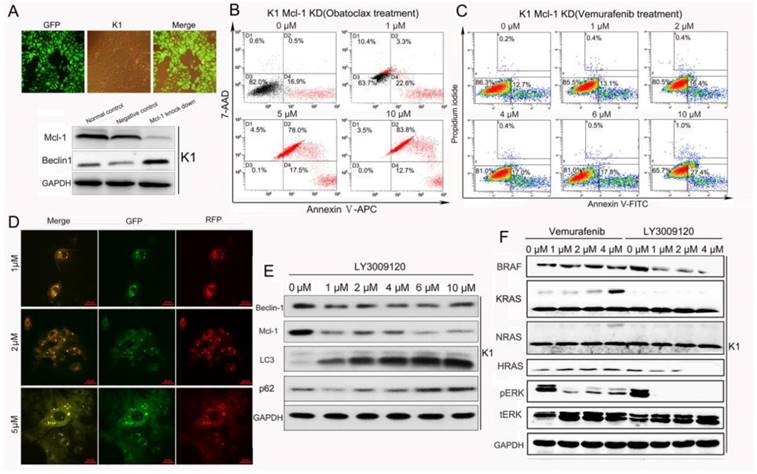
Obatoclax but not LY3009120 promoted Mcl-1/Beclin-1 dependent autophagy in K1 cells. (A) Knockdown of Mcl-1 in K1 cells was confirmed by fluorescence microscopy and Western blotting. Knockdown of Mcl-1 led to increased expression of Beclin-1. (B) Reduction of Mcl-1 alone failed to reverse Obatoclax-induced death of K1 cells. (C) Apoptosis analysis revealed that suppression of Mcl-1 by lenti-virus enhanced the cancer cell killing effect of Vemurafenib. (D) Live-cell confocal microscopic images of mRFP-GFP/LC3-transfected K1 cells showed LY3009120-induced autophagy in a concentration-dependent manner without causing obvious cell death. (E) LY3009120 treatment of K1 cells increased the conversion of LCⅠ to LC3Ⅱ without influencing the expression of Beclin-1. (F) Western blotting further indicated the advantageous propery of LY3009120 over Vemurafenib in suppressing p-ERK, BRAF and HRAS.
Figure 8
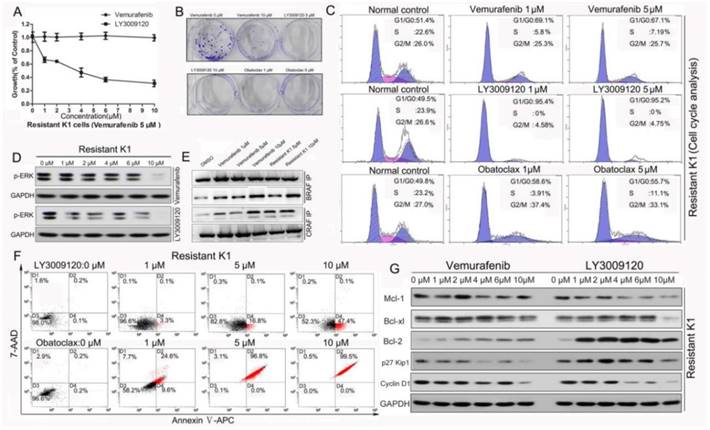
LY3009120 and Obatoclax retained activity in Vemurafenib-resistant K1 cells. (A) Survival curves of resistant K1 cells treated with either Vemurafenib or LY3009120. (B) Colony formation assay of resistant K1 cells under different conditions. (C) Flow cytometry analysis of cell cycle distribution of resistant K1 cells following LY3009120 and Obatoclax treatment for 24 h. (D) Western blotting of p-ERK in resistant K1 cells following treatment with either Vemurafenib or LY3009120. (E) Immunoprecipitation of BRAF-CRAF or CRAF-BRAF proteins in resistant K1 cells and normal K1 cells receiving Vemurafenib intervention. (F) Vemurafenib resistant K1 cells were still sensitive to the cancer cell killing effect of Obatoclax and LY3009120 as supported by apoptosis analysis. (G) Expression changes of cell cycle and apoptosis related proteins in resistant K1 cells following Vemurafenib and LY3009120 treatment.
Cell cycle analyses showed that there was no significant G2/M phase change in response to Vemurafenib treatment in the resistant K1 cells, but LY3009120 acutely suppressed cell cycle progression of resistant K1 cells. Obatoclax suppressed cell cycle distribution of resistant K1 cells mainly by reducing S-phase (Fig. 8C). This was different from a previous study which reported suppression of esophageal cancer cell viability by Obatoclax with concomitant G1/G0-phase cell cycle arrest [41]. Furthermore, while the p-ERK level in the Vemurafenib-resistant K1 cells did not change as the concentration of Vemurafenib increased, it decreased significantly in the LY3009120-treated group (Fig. 8D). It has been reported that dimerization of RAF proteins accounted for one of the potential reasons for Vemurafenib resistance and that RAF dimers are insensitive to clinical RAF inhibitors due to negative cooperativity [42]. Given that A-Raf showed weak dimerization upon RAS activation, we investigated RAF dimerization focusing on BRAF and CRAF. Our results showed that binding between endogenous BRAF-CRAF and CRAF-BRAF was strongly induced not only in normal K1 cells following Vemurafenib intervention but also in Vemurafenib-resistant K1 cells (Fig. 8E). These findings indicated that LY3009120 may be of potential clinical value due to its ability to overcome resistance by inhibiting mutant BRAF monomers and dimers [9, 26]. In line with the cell cycle inhibitory effect, LY3009120 induced early phase apoptosis while Obatoclax induced massive cell death in resistant K1 cells in a dose-dependent manner (Fig.8F). The cell cycle arresting effect of LY3009120 was associated with decreased cyclin D1 (Fig. 8G). In contrast to changes of Bcl-2 family members following Vemurafenib treatment in normal K1 cells (Fig.1D), we observed that Bcl-2 increased in resistant K1 cells (Fig. 8G). This phenomenon not only indicated increased anti-apoptotic proteins as another potential factor for Vemurafenib resistance but also further clarified the reason why resistant K1 cells were still sensitive to Obatoclax.
To investigate the tumor-suppressing functions of LY3009120 and Obatoclax, we cultured two more Vemurafenib-resistant DTC cell lines, BCPAP and KTC-1, both of which are BRAFV600E mutation positive. As expected, results of the cell cycle analysis showed that Vemurafenib-resistant BCPAP and KTC-1 responded minimally to Vemurafenib intervention. However, LY3009120 substantially prevented cell cycle progression of resistant BCPAP and KTC-1 by inducing G1/G0 arrest and considerable reduction in the S-phases. Similar results were observed in the Obatoclax treated samples (Fig. 9A&B).
Figure 9
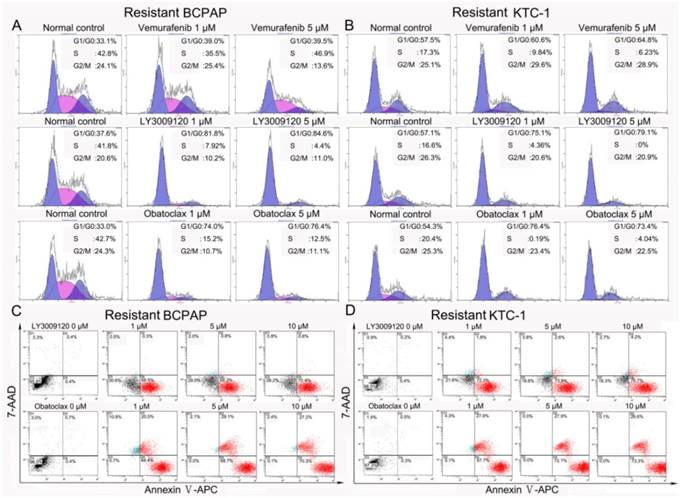
The effect of LY3009120 and Obatoclax extended to BRAFV600E mutation positive Vemurafenib resistant DTC cell lines. (A, B) Flow cytometry cell cycle analyses of Vemurafenib-resistant BCPAP and KTC-1 cells; both cell lines harbor BRAFV600E mutation. BCPAP cells (A) and KTC-1 cells (B) were treated by increasing concentrations of Vemurafenib, LY3009120 and Obatoclax for 24 h. (C, D) Flow cytometry apoptosis analyses of Vemurafenib-resistant BCPAP and KTC-1 cells after being exposed to different concentrations of LY3009120 and Obatoclax for 24 h.
Furthermore, LY3009120 and Obatoclax induced abundant apoptosis of the resistant BCPAP and KTC-1 cells. After treatment for 24 h, LY3009120 induced a dose-dependent apoptosis of the resistant cells with Annexin Ⅴ-APC positive rates as high as 70.4% for resistant BCPAP and 76.7% for resistant KTC-1 at the concentration of 10 μM. Obatoclax induced more powerful death of Vemurafenib resistant BCPAP and KTC-1 as manifested by both early phase and late phase apoptosis (Fig. 9C&D).
In vivo animal studies
We investigated the efficacy of these compounds on the growth of K1 xenografts in vivo. At the last follow up, tumor volumes of the control group (96.95±1.09 mm3) were statistically larger than that of the compounds treated group or Mcl-1 knock down group. Tumor volumes of Vemurafenib, LY3009120, Obatoclax, Mcl-1 knock down, Vemurafenib plus Obatoclax treated group were 38.4±0.8 mm3, 12.3±0. 38 mm3, 37.5±0. 92 mm3, 38.0±0.67 mm3, 15.2±0. 76 mm3, respectively. Although tumor volumes of the Vemurafenib-treated group at the last follow up were smaller than that of the control group, they were statistically larger than that of the LY3009120 intervention group and Vemurafenib-Obatoclax combination therapy group (38.4 mm3 VS 12.3 mm3, p<0.05 and 38.4 mm3 VS 15.2 mm3, p<0.05, respectively) (Fig.10A-B).
Figure 10
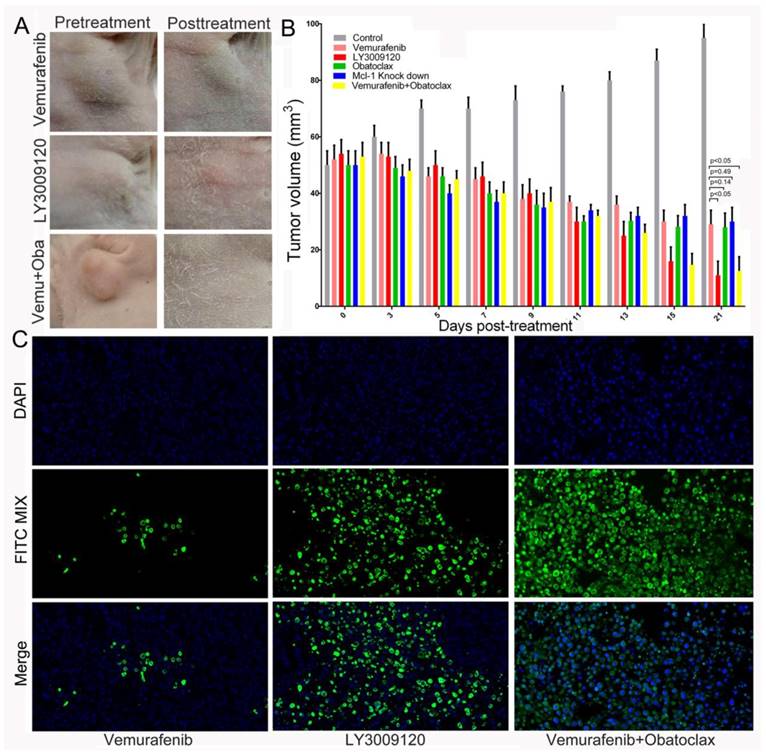
The LY3009120 monotherapy or Obatoclax+Vemurafenib combination therapy retarded tumor growth more thoroughly in subcutaneous xenograft model of thyroid cancer. (A) Images of thyroid tumors before and after treatment with Vemurafenib, LY3009120, and Vemurafenib plus Obatoclax for up to 21 days. (B) Change in tumor volumes in each group as treatment extended up to 21 days. Statistical results comparing tumor volumes of Vemurafenib treated group with that of other groups except normal control group were shown. (C) Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL, D) of specific tumor tissues at 21 days post-treatment. Green fluorescence indicated TUNEL-positive apoptotic cells; blue fluorescence indicated DAPI-stained nuclei.
In addition, tumor volumes of the LY3009120 or combinational therapy group were still statistically smaller than tumor volumes of the Obatoclax treated group or Mcl-1 knock down group. Although tumor volumes in the Vemurafenib-treated group, Obatoclax-treated group and Mcl-1 knock down group shrunk, there was not any statistical differences within these three groups. It was noteworthy that mean body weights of the drug-treated mice were not statistically significant with that of the control group, suggesting limited toxicity of single agent or drug combination in the short-term. Consistently, terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) staining were in agreement with the above results and showed much more FITC-positive apoptotic cells in the LY3009120-treated group and Vemurafenib plus Obatoclax treated group, validating the therapeutic efficacies of LY3009120 as a single agent and synergistic therapy using Vemurafenib and Obatoclax in DTCs (Fig.10C).
Discussion
Vemurafenib is an ATP-competitive, promising BRAFV600E inhibitor for the treatment of thyroid cancers expressing BRAFV600E mutation. Under normal signaling conditions, RAF dimerization is RAS dependent and occurs at the plasma membrane where the RAFs are activated. The RAF dimers are subsequently disrupted by ERK-mediated feedback phosphorylation, which also disrupts the RAS-RAF interaction and attenuates RAF signaling. However, dimerization is not needed for the function of BRAFV600E which alone has a high catalytic activity [42, 43]. In the current study, we observed that Vemurafenib could promote BRAF/CRAF dimerization causing paradoxical ERK activation in Vemurafenib-resistant DTC cells. Also, binding of Vemurafenib to one protomer in the dimer significantly reduced the affinity for binding to the second protomer. Although Vemurafenib was able to induce rapid tumor regression [5-7, 44], the activity could not be sustained as prolonged application of Vemurafenib led to drug resistance and toxicity. In this regard, a next generation pan-RAF inhibitor, LY3009120 [9, 26], currently undergoing Phase I clinical trial (NCT02014116), exerted powerful inhibitory effect against both normal DTC cells and Vemurafenib-resistant DTC cells. As shown in Fig. 7F and Fig. 8D, LY3009120 efficiently blocked the activity of BRAFV600E monomer and induced dimers. The superior efficacy of LY3009120 was further validated by its ability to suppress the growth of BRAFV600E-positive thyroid tumors in vivo much more efficiently than Vemurafenib. LY3009120 prevented cell cycle progression via suppressing cyclin D1; this was consistent with the recent observation that LY3009120 suppressed G0/G1 transition through downregulating cyclin D1 [45]. It has been reported that simultaneous inhibition of RAF and CDK4/6 signaling using LY3009120 and Abemaciclib may exert synergistic effects for patients harboring KRAS, NRAS or BRAF mutations [45]. It is of note that LY3009120 restrained ATP production and basal respiration rate in DTC cells, reinforcing its advantageous anti-cancer property over Vemurafenib, as sustained treatment of DTC cells with Vemurafenib induced addiction to oxidative phosphorylation by altering mitochondrial pool sizes (Fig. 5C). The more detailed interactions between mitochondrial homeostasis and BRAF inhibitor resistance are still under investigation in our laboratory. Vemurafenib resistance also limits the benefits of long term targeted therapy for patients with malignant melanomas harboring BRAFV600E mutation. Similarly, Heidorn et al. reported that BRAF binding to CRAF in the presence of oncogenic RAS was a mechanism for melanoma tumorigenesis [23]. Besides, Sinnberg et al. reported that β-catenin was involved in the mediation of Vemurafenib resistance in melanoma by interacting with Stat3 [46]. Therefore, sustained inhibition of BRAFV600E/ RAF dimers using LY3009120 and/or suppression of Stat3 using Stattic may facilitate better management of BRAFV600E mutation positive melanoma patients as well.
We also studied a proapoptotic small-molecule pan-Bcl-2 inhibitor, Obatoclax, and demonstrated that mixed cell death forms including typical apoptosis, Caspase-independent cell death as well as autophagy occur simultaneously in response to Obatoclax treatment. We observed that Obatoclax affected the generation of ATP, production of reactive oxygen species, the function of lysosomes, mitochondrial membrane potential, and release of cytochrome C from mitochondria to cytoplasm. Redistribution of cytochrome C triggered Caspase-3- and Caspase-9-mediated apoptosis, as detected by flow cytometry and Western blotting (Fig. 2, Fig. 3, and Fig. 4). Furthermore, electron microscopy revealed swelling of mitochondria and loss of cristae as well as the formation of vacuoles in the majority of DTC cells. These endoplasmic stress response exerted by Obatoclax may also facilitate Caspase-independent cell death induction by alternative pathways. Obatoclax induced atypical death forms have already been described by McCoy et al. in NSCLC cells and by Vogler et al. in CLL cells [47, 48]. While Obatoclax significantly reduced ATP production in DTC cells, we discovered, for the first time, that basal respiration rate and ATP production in the Vemurafenib-treated DTC cells were increased. Previous studies suggested one factor limiting the efficacy of BRAF inhibitor was increased mitochondrial biogenesis or heightened rates of oxidative phosphorylation [49, 50]. Thus, strategies targeting mitochondrial function are likely to be lethal when delivered in combination with BRAF inhibitors. Indeed, our results revealed that Obatoclax efficiently sensitized DTC cells to the cytotoxicity of Vemurafenib in vitro and in vivo. Unexpectedly, knockdown of Mcl-1 elevated the levels of mitochondrial bioenergetics in DTC cells, which was contrary to a previous study demonstrating that Mcl-1-deletion decreased both basal and maximal OCR compared to normal control [51].
Autophagy and apoptosis share molecular regulatory mechanisms and signaling pathways primarily governed by multidomain anti-apoptotic Bcl-2 family proteins [32]. We found that Obatoclax could modulate apoptosis and autophagy concomitantly in DTC cells. Similar findings were also reported by other groups in leukemia cells in which Obatoclax was found to induce apoptosis, autophagy and even necroptosis in an apoptosis-deficient, drug-resistant condition [52-54]. In the current study, we also found that silencing of Mcl-1 significantly induced apoptosis and upregulated Beclin-1 expression in K1 cells. These results were consistent with a previous publication which reported that Mcl-1 could coordinately regulate apoptosis and autophagy in neurons [33]. Recently Plantinga et al. reported a significant association between diffuse LC3-I intensity and LC3-II-positive puncta in non-medullary thyroid cancer patients, and that the LC3-II was closely correlated with clinical response to RAI therapy [35]. Although the role of autophagy in thyroid cancer initiation and development is still in debate, proper modulation of autophagy may optimize treatment strategies for DTC patients [55].
In summary, dimerization of BRAF/CRAF kinases acts as one of the main mechanisms underlying Vemurafenib resistance and can be successfully overcome by next-generation RAF inhibitor LY3009120. Also, in the future, synergistic inhibition of mutated BRAF and Bcl-2 anti-apoptotic proteins using Vemurafenib and Obatoclax may further provide an optimal therapeutic strategy for DTC patients.
Supplementary Material
Supplementary figures.
Acknowledgements
We thank Qiu-Fang Liu and Ping Miao from Renji Hospital Affiliated to Shanghai Jiao Tong University School of Medicine for their professional guidance when carrying out Seahorse XF24 assay.
This study was sponsored by the National Natural Science Foundation of China (No: 81271611).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Song HJ, Qiu ZL, Shen CT, Wei WJ, Luo QY. Pulmonary metastases in differentiated thyroid cancer: efficacy of radioiodine therapy and prognostic factors. Eur J Endocrinol. 2015;173:399-408
2. Qiu ZL, Song HJ, Xu YH, Luo QY. Efficacy and survival analysis of 131I therapy for bone metastases from differentiated thyroid cancer. J Clin Endocrinol Metab. 2011;96:3078-86
3. Ahn BC. Sodium iodide symporter for nuclear molecular imaging and gene therapy: from bedside to bench and back. Theranostics. 2012;2:392-402
4. Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013;13:184-99
5. Kim KB, Cabanillas ME, Lazar AJ, Williams MD, Sanders DL, Ilagan JL. et al. Clinical responses to vemurafenib in patients with metastatic papillary thyroid cancer harboring BRAFV600E mutation. Thyroid. 2013;23:1277-83
6. Dadu R, Shah K, Busaidy NL, Waguespack SG, Habra MA, Ying AK. et al. Efficacy and tolerability of vemurafenib in patients with BRAFV600E -positive papillary thyroid cancer: M.D. Anderson Cancer Center off label experience. J Clin Endocrinol Metab. 2015;100:E77-81
7. Carneiro RM, Carneiro BA, Agulnik M, Kopp PA, Giles FJ. Targeted therapies in advanced differentiated thyroid cancer. Cancer Treat Rev. 2015;41:690-8
8. Brose MS, Cabanillas ME, Cohen EE, Wirth LJ, Riehl T, Yue H. et al. Vemurafenib in patients with BRAFV600E-positive metastatic or unresectable papillary thyroid cancer refractory to radioactive iodine: a non-randomised, multicentre, open-label, phase 2 trial. Lancet Oncol. 2016;17:1272-82
9. Peng SB, Henry JR, Kaufman MD, Lu WP, Smith BD, Vogeti S. et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell. 2015;28:384-98
10. Cheng W, Liu R, Zhu G, Wang H, Xing M. Robust Thyroid Gene Expression and Radioiodine Uptake Induced by Simultaneous Suppression of BRAFV600E and Histone Deacetylase in Thyroid Cancer Cells. J Clin Endocrinol Metab. 2016;101:962-71
11. Cragg MS, Jansen ES, Cook M, Harris C, Strasser A, Scott CL. Treatment of B-RAF mutant human tumor cells with a MEK inhibitor requires Bim and is enhanced by a BH3 mimetic. J Clin Invest. 2008;118:3651-9
12. Serasinghe MN, Missert DJ, Asciolla JJ, Podgrabinska S, Wieder SY, Izadmehr S. et al. Anti-apoptotic BCL-2 proteins govern cellular outcome following B-RAFV600E inhibition and can be targeted to reduce resistance. Oncogene. 2015;34:857-67
13. Hikita H, Takehara T, Shimizu S, Kodama T, Shigekawa M, Iwase K. et al. The Bcl-xL inhibitor, ABT-737, efficiently induces apoptosis and suppresses growth of hepatoma cells in combination with sorafenib. Hepatology. 2010;52:1310-21
14. Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA. et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677-81
15. Wei WJ, Shen CT, Song HJ, Qiu ZL, Luo QY. Propranolol sensitizes thyroid cancer cells to cytotoxic effect of vemurafenib. Oncol Rep. 2016;36:1576-84
16. Yu L, Wu WK, Gu C, Zhong D, Zhao X, Kong Y. et al. Obatoclax impairs lysosomal function to block autophagy in cisplatin-sensitive and -resistant esophageal cancer cells. Oncotarget. 2016;7:14693-707
17. Champa D, Russo MA, Liao XH, Refetoff S, Ghossein RA, Di Cristofano A. Obatoclax overcomes resistance to cell death in aggressive thyroid carcinomas by countering Bcl2a1 and Mcl1 overexpression. Endocr Relat Cancer. 2014;21:755-67
18. Xie C, Edwards H, Caldwell JT, Wang G, Taub JW, Ge Y. Obatoclax potentiates the cytotoxic effect of cytarabine on acute myeloid leukemia cells by enhancing DNA damage. Mol Oncol. 2015;9:409-21
19. Ruan M, Liu M, Dong Q, Chen L. Iodide- and glucose-handling gene expression regulated by sorafenib or cabozantinib in papillary thyroid cancer. J Clin Endocrinol Metab. 2015;100:1771-9
20. Hanly EK, Bednarczyk RB, Tuli NY, Moscatello AL, Halicka HD, Li J. et al. mTOR inhibitors sensitize thyroid cancer cells to cytotoxic effect of vemurafenib. Oncotarget. 2015;6:39702-13
21. Shen CT, Wei WJ, Qiu ZL, Song HJ, Luo QY. Afamin promotes glucose metabolism in papillary thyroid carcinoma. Mol Cell Endocrinol. 2016;434:108-15
22. Shen CT, Qiu ZL, Song HJ, Wei WJ, Luo QY. miRNA-106a directly targeting RARB associates with the expression of Na+/I- symporter in thyroid cancer by regulating MAPK signaling pathway. J Exp Clin Cancer Res. 2016;35:101
23. Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N. et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209-21
24. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1
25. Cancer Genome Atlas Research N. Integrated genomic characterization of papillary thyroid carcinoma. Cell. 2014;159:676-90
26. Henry JR, Kaufman MD, Peng SB, Ahn YM, Caldwell TM, Vogeti L. et al. Discovery of 1-(3,3-dimethylbutyl)-3-(2-fluoro-4-methyl-5-(7-methyl-2-(methylamino)pyrido[2,3- d]pyrimidin-6-yl)phenyl)urea (LY3009120) as a pan-RAF inhibitor with minimal paradoxical activation and activity against BRAF or RAS mutant tumor cells. J Med Chem. 2015;58:4165-79
27. Denisova OV, Kakkola L, Feng L, Stenman J, Nagaraj A, Lampe J. et al. Obatoclax, saliphenylhalamide, and gemcitabine inhibit influenza a virus infection. J Biol Chem. 2012;287:35324-32
28. Nguyen M, Marcellus RC, Roulston A, Watson M, Serfass L, Murthy Madiraju SR. et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc Natl Acad Sci U S A. 2007;104:19512-7
29. Sharpley MS, Marciniak C, Eckel-Mahan K, McManus M, Crimi M, Waymire K. et al. Heteroplasmy of mouse mtDNA is genetically unstable and results in altered behavior and cognition. Cell. 2012;151:333-43
30. Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. 2011;435:297-312
31. Champa D, Orlacchio A, Patel B, Ranieri M, Shemetov AA, Verkhusha VV. et al. Obatoclax kills anaplastic thyroid cancer cells by inducing lysosome neutralization and necrosis. Oncotarget. 2016;7:34453-71
32. Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4:600-6
33. Germain M, Nguyen AP, Le Grand JN, Arbour N, Vanderluit JL, Park DS. et al. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J. 2011;30:395-407
34. Martinez-Lopez N, Athonvarangkul D, Mishall P, Sahu S, Singh R. Autophagy proteins regulate ERK phosphorylation. Nat Commun. 2013;4:2799
35. Plantinga TS, Tesselaar MH, Morreau H, Corssmit EP, Willemsen BK, Kusters B. et al. Autophagy activity is associated with membranous sodium iodide symporter expression and clinical response to radioiodine therapy in non-medullary thyroid cancer. Autophagy. 2016:1-11
36. Wang J, Whiteman MW, Lian H, Wang G, Singh A, Huang D. et al. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J Biol Chem. 2009;284:21412-24
37. Roy B, Pattanaik AK, Das J, Bhutia SK, Behera B, Singh P. et al. Role of PI3K/Akt/mTOR and MEK/ERK pathway in Concanavalin A induced autophagy in HeLa cells. Chem Biol Interact. 2014;210:96-102
38. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1-222
39. Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol. 2008;445:77-88
40. Brem EA, Thudium K, Khubchandani S, Tsai PC, Olejniczak SH, Bhat S. et al. Distinct cellular and therapeutic effects of obatoclax in rituximab-sensitive and -resistant lymphomas. Br J Haematol. 2011;153:599-611
41. Zhong D, Gu C, Shi L, Xun T, Li X, Liu S. et al. Obatoclax induces G1/G0-phase arrest via p38/p21(waf1/Cip1) signaling pathway in human esophageal cancer cells. J Cell Biochem. 2014;115:1624-35
42. Yao Z, Torres NM, Tao A, Gao Y, Luo L, Li Q. et al. BRAF Mutants Evade ERK-Dependent Feedback by Different Mechanisms that Determine Their Sensitivity to Pharmacologic Inhibition. Cancer Cell. 2015;28:370-83
43. Wee EJ, Wang Y, Tsao SC, Trau M. Simple, Sensitive and Accurate Multiplex Detection of Clinically Important Melanoma DNA Mutations in Circulating Tumour DNA with SERS Nanotags. Theranostics. 2016;6:1506-13
44. Prager GW, Koperek O, Mayerhoefer ME, Muellauer L, Wrba F, Niederle B. et al. Sustained Response to Vemurafenib in a BRAFV600E-Mutated Anaplastic Thyroid Carcinoma Patient. Thyroid. 2016;26:1515-6
45. Gong X, Wu W, Chio L-C, Pratt S, King C, Webster Y. et al. An unbiased tumor cell panel profiling method to identify drug-drug interactions reveals synergy between the CDK4 and CDK 6 inhibitor abemaciclib and the Raf dimer and pan-Raf inhibitor LY3009120 in Ras mutant cancers. Cancer Research. 2016;76:2818 -
46. Sinnberg T, Makino E, Krueger MA, Velic A, Macek B, Rothbauer U. et al. A Nexus Consisting of Beta-Catenin and Stat3 Attenuates BRAF Inhibitor Efficacy and Mediates Acquired Resistance to Vemurafenib. EBioMedicine. 2016
47. McCoy F, Hurwitz J, McTavish N, Paul I, Barnes C, O'Hagan B. et al. Obatoclax induces Atg7-dependent autophagy independent of beclin-1 and BAX/BAK. Cell Death Dis. 2010;1:e108
48. Vogler M, Weber K, Dinsdale D, Schmitz I, Schulze-Osthoff K, Dyer MJ. et al. Different forms of cell death induced by putative BCL2 inhibitors. Cell Death Differ. 2009;16:1030-9
49. Lito P, Rosen N, Solit DB. Tumor adaptation and resistance to RAF inhibitors. Nat Med. 2013;19:1401-9
50. Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S. et al. Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest. 2016;126:1834-56
51. Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M. et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012;14:575-83
52. Wei Y, Kadia T, Tong W, Zhang M, Jia Y, Yang H. et al. The combination of a histone deacetylase inhibitor with the Bcl-2 homology domain-3 mimetic GX15-070 has synergistic antileukemia activity by activating both apoptosis and autophagy. Clin Cancer Res. 2010;16:3923-32
53. Heidari N, Hicks MA, Harada H. GX15-070 (obatoclax) overcomes glucocorticoid resistance in acute lymphoblastic leukemia through induction of apoptosis and autophagy. Cell Death Dis. 2010;1:e76
54. Bonapace L, Bornhauser BC, Schmitz M, Cario G, Ziegler U, Niggli FK. et al. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J Clin Invest. 2010;120:1310-23
55. Pietrocola F, Pol J, Vacchelli E, Baracco EE, Levesque S, Castoldi F. et al. Autophagy induction for the treatment of cancer. Autophagy. 2016;12:1962-4
Author contact
![]() Corresponding authors: Quan-Yong Luo, Email: lqynnet, Zhong-Ling Qiu, Email: qiuzhongling123com. Address: Department of Nuclear Medicine, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600# Yishan Road, 200233, Shanghai, China.
Corresponding authors: Quan-Yong Luo, Email: lqynnet, Zhong-Ling Qiu, Email: qiuzhongling123com. Address: Department of Nuclear Medicine, Shanghai Jiao Tong University Affiliated Sixth People's Hospital, 600# Yishan Road, 200233, Shanghai, China.
Citation styles
APA
Wei, W.J., Sun, Z.K., Shen, C.T., Song, H.J., Zhang, X.Y., Qiu, Z.L., Luo, Q.Y. (2017). Obatoclax and LY3009120 Efficiently Overcome Vemurafenib Resistance in Differentiated Thyroid Cancer. Theranostics, 7(4), 987-1001. https://doi.org/10.7150/thno.17322.
ACS
Wei, W.J.; Sun, Z.K.; Shen, C.T.; Song, H.J.; Zhang, X.Y.; Qiu, Z.L.; Luo, Q.Y. Obatoclax and LY3009120 Efficiently Overcome Vemurafenib Resistance in Differentiated Thyroid Cancer. Theranostics 2017, 7 (4), 987-1001. DOI: 10.7150/thno.17322.
NLM
Wei WJ, Sun ZK, Shen CT, Song HJ, Zhang XY, Qiu ZL, Luo QY. Obatoclax and LY3009120 Efficiently Overcome Vemurafenib Resistance in Differentiated Thyroid Cancer. Theranostics 2017; 7(4):987-1001. doi:10.7150/thno.17322. https://www.thno.org/v07p0987.htm
CSE
Wei WJ, Sun ZK, Shen CT, Song HJ, Zhang XY, Qiu ZL, Luo QY. 2017. Obatoclax and LY3009120 Efficiently Overcome Vemurafenib Resistance in Differentiated Thyroid Cancer. Theranostics. 7(4):987-1001.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.