Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(4):855-875. doi:10.7150/thno.17558 This issue Cite
Research Paper
A Crucial Role of CXCL14 for Promoting Regulatory T Cells Activation in Stroke
Hsu-Tung Lee1, Shih-Ping Liu2,3, Chen-Huan Lin3, Sophie Wei Lee3, Chung Y. Hsu2, Huey-Kang Sytwu4, Chia-Hung Hsieh2,5 ![]() , Woei-Cherng Shyu2,3
, Woei-Cherng Shyu2,3 ![]()
1. Department of Neurosurgery, Taichung Veterans General Hospital, Taichung, Taiwan 40421
and Graduate Institute of Medical Sciences, National Defense Medical Center, Taipei, Taiwan, Republic of China.
2. Graduate Institute of Biomedical Science, China Medical University, Taichung, Taiwan, Republic of China.
3. Translational Medicine Research Center and Department of Neurology, China Medical University & Hospital, Taichung, Taiwan, Republic of China.
4. Department of Microbiology and Immunology, National Defense Medical Center, Taipei, Taiwan, Republic of China
5. Department of Biomedical Informatics, Asia University, No. 500, Lioufeng Road, Taichung, Taiwan, Republic of China.
Received 2016-9-13; Accepted 2016-11-29; Published 2017-2-8
Abstract

Inflammatory processes have a detrimental role in the pathophysiology of ischemic stroke. However, little is known about the endogenous anti-inflammatory mechanisms in ischemic brain. Here, we identify CXCL14 as a critical mediator of these mechanisms. CXCL14 levels were upregulated in the ischemic brains of humans and rodents. Moreover, hypoxia inducible factor-1α (HIF-1α) drives hypoxia- or cerebral ischemia (CI)-dependent CXCL14 expression via directly binding to the CXCL14 promoter. Depletion of CXCL14 inhibited the accumulation of immature dendritic cells (iDC) or regulatory T cells (Treg) and increased the infarct volume, whereas the supplementation of CXCL14 had the opposite effects. CXCL14 promoted the adhesion, migration, and homing of circulating CD11c+ iDC to the ischemic tissue via the upregulation of the cellular prion protein (PrPC), PECAM-1, and MMPs. The accumulation of Treg in ischemic areas of the brain was mediated through a cooperative effect of CXCL14 and iDC-secreted IL-2-induced Treg differentiation. Interestingly, CXCL14 largely promoted IL-2-induced Treg differentiation. These findings indicate that CXCL14 is a critical immunomodulator involved in the stroke-induced inflammatory reaction. Passive CXCL14 supplementation provides a tractable path for clinical translation in the improvement of stroke-induced neuroinflammation.
Keywords: CXC chemokine 14 (CXCL14), regulatory T cells (Treg), immature dendritic cells (iDC), cerebral ischemia, hypoxia inducible factor 1α (HIF-1α).
Introduction
Cerebral ischemia (CI) leads to a complex interaction between inflammation and immunoregulation[1]. Anti-inflammatory cytokines establish an immunosuppressive environment via the recruitment of regulatory T cells (Treg)[2]. However, the link between ischemia and Treg-mediated immune tolerance in post-stroke recovery remains poorly understood. CXCL14 is a CXC chemokine ligand with unknown physiological function, expressed constitutively in immune cells and throughout the central nervous system [3, 4]. These chemokines also stimulate monocyte, dendritic cell (DC), and natural killer cell migration through an autocrine pathway[5, 6], as well as enhance tumor cell proliferation[7]. Moreover, the homing of CD11c+ immature dendritic cells (iDC) from CD34+ hematopoietic stem cells (HSCs) to epidermal niches is mediated by CXCL14[3]. Thus, CXCL14 might play an anti-inflammatory role in the steady-state functions of DC such as self-antigen presentations for the purpose of eliminating self-reacting T cells and/or induction of Treg[3]. During the process of ischemic brain injury, DC or iDC constitute a substantial portion of infiltrating cells in the temporal dynamics of immune cell accumulation[8]. In addition, iDC coordinate cellular interactions to initiate Treg differentiation[9]. Although the fundamental endogenous counter-regulatory immune mechanisms for the suppression of inflammation after ischemic stroke are poorly understood, these findings reveal a potential mechanism for CXCL14-mediated iDC recruitment, contributing to the immunomodulatory adaptation after stroke.
Tissue hypoxia and ischemic injury can create a specific environmental niche composed of cytokines, chemokines, and growth factors secreted from the adjacent stroma, sensory neurons, and peripheral blood following a stress injury[10]. Neuronal and stromal cells respond to the hypoxic microenvironment through the activity of hypoxia-inducible factors (HIFs)[11]. The HIF-1 signaling pathway has been suggested to have an important role in neurological outcomes following ischemic stroke due to the functions of downstream genes involved in glucose metabolism, angiogenesis, erythropoiesis, and cell survival[12]. The expression of SDF-1α, also known as CXC chemokine ligand 12 (CXCL12), is regulated by the hypoxia-inducible factor-1α (HIF-1α) in ischemic tissue. HIF-1α-induced SDF-1α expression increases the recruitment and homing of bone marrow stem cells and inflammatory cells to ischemic tissue[13, 14]. A large number of chemokines including SDF-1α and CXCL14 are constitutively expressed in the central nervous system[15, 16]. Despite clear evolutionary conservation between SDF-1α and CXCL14[17], it is unclear whether CXCL14 could be upregulated by hypoxia/ischemia through HIF-1 activation.
Previous studies have demonstrated that iDC stimulate the proliferation of Treg[18, 19], which are known to be the major neuroprotective modulator in post-ischemic inflammatory brain injury[20]. Here, we focused on elucidating the molecular mechanism involved in the regulation of CXCL14 expression following ischemia-induced HIF-1α activation. We also tested whether CXCL14 is an ischemia-inducible endogenous factor for mobilization and trafficking in CD34+ HSC-derived iDC from peripheral circulation to ischemic brain tissues. We performed studies aimed at revealing whether CXCL14 exerted anti-inflammatory effects through directly inducing both Treg activation and recruited iDC-mediated Treg differentiation to enhance the cerebroprotective effect in the post ischemic brain.
Materials and Methods
Immunohistochemical Analysis of the Human Brain Following Stroke
Postmortem brain specimens were obtained during autopsies of patients who died from fatal ischemic stroke (massive middle cerebral artery territory infarction with corticle involvement), with disease duration ranging from 1-3 days (courtesy of the Department of Neurology, Buddhist Tzu-Chi General Hospital and China Medical University Hospital). Time to autopsy ranged from 4 -12 hours after death (mean time: < 6 hours). Brain tumor patients who died of non-ischemic causes served as controls (n = 4, two men and two women, age = 60.2 ± 2.5 years) (Supplementary Table S1). The Institutional Review Board of the Buddhist Tzu-Chi General Hospital and China Medical University Hospital approved the study protocol. Informed consent was signed by relatives. Tissue sampling was based on individual infarct topography, which was focused on cerebrovascular anatomy and the most recent MRI scan. Upon autopsy, brain areas with variable degrees of infarction were identified macroscopically and ~1 cm3 cortical samples, including subcortical white matter, were dissected and fixed with formalin prior to embedding in paraffin or freezing at -70oC until analysis, as described previously[21]. CXCL14, CD34, and CD11c immunostaining of brain samples was performed using CXCL14 antibody (1:200, Santa Cruz Biotechnology), CD34 antibody (1:100, BD Pharmingen), and CD11c antibody (1:100, N418, Thermo). Samples from corresponding areas of the contralateral or non-infarcted hemispheres and from the control brains were processed in a similar way. The extent of CXCL14+ cell immunoreactivity was measured as the number of cells per square millimeter (cells/mm2).
In Vivo Ischemia/Reperfusion Brain Model
Adult male Sprague-Dawley rats (weight 250-300 g) were used in this study. All animal experiments were performed using sterile/aseptic techniques in accordance with Institutional Guidelines of Animal Research of China Medical University. Rats were anesthetized with chloral hydrate (0.4 g/kg IP) and subjected to cerebral ischemia. Ligation of the right middle cerebral artery (MCA) and bilateral common carotids arteries (CCAs) were performed by methods described previously[21]. The CCAs were clamped with non-traumatic arterial clips. Blood flow to the right MCA was ligated with a l0-0 nylon suture. After 90 minutes of ischemia, the suture on the MCA and the arterial clips on the CCAs were removed to allow reperfusion. Cortical blood flow (CBF) was measured continuously with a laser Doppler flowmeter (LDF, PF-5010, Periflux system; Perimed AB) in anesthetized animals. The average CBF should be reduced over 95% compared to original level lasting for 90 minutes and returned to normal when it was re-perfused. Core body temperature was monitored with a thermistor probe (Hewlett-Packard Model 21090A probe), and maintained at 37oC with a heating pad during anesthesia. After recovery from anesthesia, rat body temperature was maintained at 37oC with a heat lamp.
In addition, conditional HIF-1α knockout mice (HIF-1α KO mice carrying a loxP-flanked allele of HIF-1α[22]. HIF-1α disruption in the HIF-1α KO mice was induced by feeding doxycycline at a dose of 2 mg/mL in 5% (wt/vol) sucrose solution from embryonic day 15 to postnatal day 1. They were also anesthetized with chloral hydrate (0.3 g/kg, ip) and subjected to right middle cerebral artery (MCA) ligation and right common carotid artery (CCAs) clamping for 120 minutes, which kept average CBF being reduced over 95% compared to original level (both in wild type and HIF-1α KO mice) as described previously[23].
2-Methoxyestradiol Treatment in Vivo and in Vitro
2-Methoxyestradiol (2-ME2, Sigma Aldrich, USA) was dissolved in DMSO to obtain a 10 mM stock solution. For in vivo experiments, the whole procedure was performed as previously described[24]. Rats were treated with an intraperitoneal injection of a liposomal preparation (dioleoylphosphatidylcholine; Avanti Polar Lipids) of 2-ME2 (20 mg/mL) in three concentrations (50, 100 or 150 mg/kg) for 5 days pre- and post-onset of cerebral ischemia. For in vitro experiments with 2-ME2 treatment, PCC were pretreated with different concentrations of 2-ME2 (0.1 μM, 1 μM and 10 μM) for 16 hours as previously described[25].
Total Brain Tissue Protein Extraction for Western Blotting and ELISA
Rats were put under anesthesia and decapitated at 4 h, 12 h, 3 d, or 7 days after reperfusion with 90 minutes MCA ligation. Three rats without MCA ligation were used as controls. Samples of ischemic cerebral cortex were taken from the peripheral region of infarcted brains (penumbral area). Western blot analysis was performed on these samples. Briefly, ischemic brain tissue was homogenized and lysed in the buffer containing 320 mM sucrose, 5 mM HEPES, 1 μg/mL leupeptin, and 1 μg/mL aprotinin. Lysates were centrifuged at 13,000 g for 15 minutes. The resulting pellet was resuspended in sample buffer (62.5 mM Tris-HCl, 10% glycerol, 2% SDS, 0.1% bromophenol blue, and 50 mM DTT) and subjected to SDS-polyacrylamide gel (4-12%) electrophoresis. Then, the gel was transferred to a Hybond-P nylon membrane. This was followed by incubation with appropriately diluted antibodies: CXCL14 (1:200; Santa Cruz Biotechnology), HIF-1α (1:200; Novus Biologicals), HIF-2α (1:200; Novus Biologicals), PrPC (1:200; Santa Cruz Biotechnology), PECAM-1 (1:200; Santa Cruz Biotechnology), selectin (1:200; Santa Cruz Biotechnology), CXCR4 (1:200; R&D Systems), β1-integrin (1:200; Chemicon), β2-integrin (1:200; Chemicon), JAM-A (1:200; Millipore), JAM-C (1:200; Millipore), LFA-1 (1:200; Millipore), ICAM (1:200; Millipore), VCAM-1 (1:200; Millipore), VE-cadherin (1:200; Millipore), CD99 (1:200; Millipore) and β-Actin (1:2000, Santa Cruz Biotechnology). Expression of apoptosis-related proteins (Bcl-2, Bcl-xL, Bax, and Bad; Santa Cruz Biotechnology) in the right cortex and striatum region was also analyzed[26]. Membrane blocking, primary and secondary antibody incubations, and chemiluminescence reactions were conducted for each antibody individually. The intensity of each band was measured using a Kodak Digital Science 1D Image Analysis System (Eastman Kodak). Band intensities were calculated using the internal control. In addition, the total level of CXCL14 (brain lysate) was measured with Quantikine ELISA kit (R&D Systems). Semi-quantitative analysis of CXCL14 levels in brain tissue and plasma were performed. Optical density was measured using a spectrophotometer (Molecular Devices) and standard curves were generated with the program SOFTmax (Molecular Devices).
Measurement of HIF-1α Activity by ELISA
To measure active HIF-1α, 50 μg nuclear extracts were incubated with biotinylated double stranded DNA containing a consensus HIF-1αbinding site from Duo-set ELISA mouse active HIF-1α kit (R&D Systems). HIF-1α activity was expressed by OD (450 nm-540 nm)[27]. The experiments were carried out in triplicate and each experiment was repeated three times unless otherwise mentioned.
Immunohistochemical Analysis of the Rat Brain
Rats were re-anesthetized with chloral hydrate (0.4 g/kg IP), and were decapitated at 4 hours, 12 hours, 3 days, or 7 days following cerebral ischemia. Three non-ischemic rats were used as controls. Rat brains were fixed by transcardial perfusion with saline, followed by perfusion and immersion in 4% paraformaldehyde as previously described[28]. A series of continuous 6-μm sections were cut by cryostat from each tissue block in the coronal plane, stained with H&E, and analyzed by light microscopy (Nikon, E600).
To identify the expression of cell type-specific markers in CXCL14+ cells, double immunofluorescence was performed as previously described[21]. Each coronal section was first stained with primary CXCL14 antibody (1:100, Santa Cruz Biotechnology), followed by treatment additional antibodies: GFAP (1:400, Sigma Aldrich), Tuj-1 (1:200; Chemicon), microtubule-associated protein 2 (MAP-2, 1:200; BM), CD34 (1:100, BD Pharmingen), B220 (1:200, BD Pharmingen), CD11c (1:100, N418, Thermo), CD11b (1:200, BD Pharmingen), MHC-II (1:200, BD Pharmingen), CD80/86 (1:200, BD Pharmingen), CD4 (1:200, BD Pharmingen), CD25 (1:200, BD Pharmingen), Foxp3 (1:300, BD Pharmingen) and HIF-1α (1:200; Novus Biologicals). The tissue sections were analyzed with a Carl Zeiss LSM510 laser-scanning confocal microscope. FITC (green, 1:500; Jackson Immunoresearch), Cy3 (red, 1:500; Jackson Immunoresearch) and Alexa Fluor 680 (blue, 1:1000; Invitrogen) fluorochromes on the immunofluorescence-labeled slides were excited by laser beam at 488 nm, 543 nm, and 680 nm, respectively.
Cerebral Ischemic Animal Model Treated with CXCL14
The cerebral ischemic rat model was established as described above. Beginning 4 hours following MCA ligation, rats were injected intravenously through femoral vein with different dosages of CXCL14 (1, 10 and 100 ng/kg, R&D Systems) for three consecutive days. Two therapeutic groups of CXCL14 (100 ng/kg) and vehicle control were studied further. Core body temperature was monitored with a thermistor probe and maintained at 37oC with a heating pad during anesthesia. For the blocking experiment, 25 mg/kg of a specific matrix metalloproteinase inhibitor (SB-3CT, Sigma) was injected intraperitoneally as previously described[29]. CXCL14 blocking antibody (CXCL14-Ab, 1 μg/kg, R&D Systems) was administered intraperitoneally for 4 consecutive days as previously described[30]. In addition, injection of CD25 blocking antibody (CD25-Ab, 300 μg, BD Pharmingen)[20], CD11c blocking antibody (CD11c-Ab, 500 μg, Millipore)[31] or IL-2 neutralization antibody (IL-2-Ab, 500 μg, R&D Systems)[32] was dosed intraperitoneally.
Triphenyltetrazolium Chloride (TTC) Staining
Three days following ischemic events, animals were perfused with saline intracardially. The brain tissue was removed, immersed in cold saline for 5 minutes, and sliced into 2.0-mm sections (seven slices per rat). The brain slices were incubated in 20-g/L triphenyltetrazolium chloride (TTC; Research Organics Inc), dissolved in saline for 30 minutes at 37oC, and then fixed in a 5% formaldehyde solution. The area of infarction was measured with a digital scanner, as described previously[21].
Neurological Behavioral Measurements
Behavioral assessments were performed 5 days before cerebral ischemia and 1, 7, 14, and 28 days after the treatment. The tests measured (a) body asymmetry and (b) locomotor activity[28]. Further, grip strength was analyzed using Grip Strength Meter (TSE-Systems) as previously described[28]. The baseline-test scores were recorded in order to compare to those taken after cerebral ischemia. The elevated body swing test (EBST) was used to assess body asymmetry after MCA ligation and evaluated quantitatively as previously described[28]. Initially, animals were examined for lateral movement by suspending their bodies by their tails. The frequency of initial head swing contra-lateral to the ischemic side was counted in twenty continuous tests and was normalized, as follows: % recovery = [1 - (lateral swings in twenty tests - 10) / 10 × 100%. Locomotor activity: Rats were subjected to VersaMax Animal Activity monitoring (Accuscan Instruments) for about 2 hours for behavioral recording. The VersaMax Animal Activity monitoring contained 16 horizontal and 8 vertical infrared sensors spaced 87 cm apart. The vertical sensors were situated 10 cm from the floor of the chamber. Motor activity was counted as the number of beams disrupted due to rat movement. Three vertical parameters were calculated over 2 hours at night (from midnight to 2 am): (i) vertical activity, (ii) vertical time, and (iii) number of vertical movements. For grip strength analysis, percentage of improvement in grip strength was measured on each forelimb separately and was calculated as the ratio between the mean strength of 20 pulls on the side contralateral to the ischemia and also the ipsilateral side[21]. The grip strength ratio post-treatment versus baseline were also calculated and presented as percentage of baseline.
Measurement of Infarct Size Using Magnetic Resonance Image (MRI)
MRI was performed on anesthetized rats in the R4 imaging system (GE) at 3.0 T. Brains were scanned in 6-8 coronal image slices, 2 mm thick without gaps. T2-weighted imaging (T2WI) pulse sequences were obtained with the use of a spin-echo technique (repetition time, 4000 ms; echo time, 105 ms). Images were captured sequentially for each animal at 1, 7, and 28 days after cerebral ischemia. To measure the infarction area in the right cortex, we subtracted the noninfarcted area in the right cortex from the total cortical area of the left hemisphere[21]. The area of infarct was drawn manually from slice to slice, and the volume was then calculated by internal volume analysis software (Voxtool, General Electric).
[18F]Fluoro-2-deoxyglucose Positron Emission Tomography (FDG-PET) Examination
To assess the metabolic activity and synaptic density of brain tissue, rats were examined using microPET scanning of FDG to measure relative metabolic activity under the protocol previously described[33]. In brief, 18F was produced by the 18O(p, n)18F nuclear reaction in a cyclotron at China Medical University and Hospital, Taiwan, and 18F-FDG was synthesized as previously described[34] with an automated 18F-FDG synthesis system (Nihon Kokan). Data were collected with a high-resolution small-animal PET scanner (microPET, Rodent R4, Concorde Microsystems). Carmichael et al have described the system parameters previously[35]. After one week of each treatment, animals anesthetized with chloral hydrate (0.4 g/kg, ip), fixed in a customized stereotactic head holder and positioned in the microPET scanner. The animals were given an intravenous bolus injection of 18F-FDG (200-250 μCi/rat) dissolved in 0.5 mL of saline. Data acquisition began upon 18F-FDG administration and continued for 60 minutes using a 3-D acquisition protocol. The image data acquired from microPET were displayed and analyzed by Interactive Data Language (IDL) version 5.5 (Research Systems) and ASIPro ver. 3.2 (Concorde Microsystems) software. FDG-PET images were reconstructed using a posterior-based three-dimensional iterative algorithm[36] and overlaid on MR templates to confirm anatomical location[37]. Coronal sections for striatal and cortical measurements represented brain areas between 0 and +1 mm from bregma, and thalamic measurements represented brain areas between -2 and -3 mm from the bregma, as estimated by visual inspection of the unlesioned side. The relative metabolic activity in regions of interest (ROI) of the striatum was expressed as a percentage deficit as previously described[35].
TUNEL Cytochemistry
Cellular apoptosis was assayed by immuno-histochemistry using a commercial TUNEL staining kit (DeadEnd Fluorimetric TUNEL system; Promega) as previously described[38]. The percentage of TUNEL labeling was expressed as the number of TUNEL-positive nuclei divided by the total number of nuclei stained with DAPI[39].
Blood Cell Isolation and Culture
Peripheral blood mononuclear cells (PBMCs) were isolated from SD rats injected with CXCL14 (100 ng/kg/d, ip for 5 d) or GM-CSF (20 μg/d, ip for 7 d, R&D Systems)[40]. The cells were collected using the Ficoll-Histopaque (Sigma Aldrich) centrifugation method[41], and washed twice with 1 mM EDTA in PBS. Cells were pipetted into a 96-well microtiter plate then blocked for 15 minutes at 4ºC with anti-FC block (BD Pharmingen). Blood cell analysis was conducted by FACS analysis (FACSCalibur, Becton Dickinson) by staining with antibody for CD34, B220, CD11c, CD11b, MHC II, F4/80, CD80/86, c-kit, CD3, CD8, CD19, CD20, CD34, and CD56 from BD Pharmingen as described previously[3]. Total monocytes for transwell assays were isolated from PBMCs by negative depletion of CD3+, CD8+, CD19+, CD20+, CD34+, and CD56+ cells. Levels of nonspecific staining were established by parallel analyses of cells incubated with irrelevant isotype-matched control antibodies.
CD34+ bone marrow derived cells (BMDCs) were separated from 2 × 106 mononuclear cells by a magnetic bead separation method (MACS; Miltenyi Biotec, Gladbach, Germany). Subsequently, CD34+ BMDCs (purity > 95%, 106 cell/mL) were cultured for 72 hours in media (StemSpanTM H3000 and Cytokine Cocktail, StemCell Technologies) at 37oC in a humidified atmosphere of 5% CO2/95% air, and prepared for further experimentation as follows. For the differentiation and proliferation of dendritic cells (DC) and immature DC (iDC), CD34+ BMDCs incubated with or without CXCL14 (1, 10, 100 ng/mL, R&D Systems) were seeded at 35,000 cells/cm2/mL in DMEM medium plus 10 ng/mL IL-6 (R&D Systems), and 10 U/mL SCF (PreproTech)[42]. After 7 d, fresh DMEM medium plus GM-CSF (100 ng/mL, R&D Systems) and IL-4 (500 ng/mL, R&D Systems) was added to iDC cultures[3]. For BrdU labeling and immunocytochemistry, the cells were pulsed with 10 μM BrdU for 4 hours and fixed with 4% paraformaldehyde for 20 minutes[43]. In brief, DNA was denatured by treatment with 2.5 N HCl for 20 minutes at room temperature followed by 0.1 M boric acid treatment to neutralize the cells. Incorporated BrdU was detected with mouse monoclonal anti-BrdU antibody (1:50, BD Biosciences) that was incubated with the cells overnight. The percentage of BrdU-positive cells was determined by counting under a phase contrast microscope (at least 500 cells per sample were scored).
Preparation and Assessment of Treg Activity
CD4+ T cells prepared from spleen cells were isolated by negative selection with immunosorted methods (MACS, Miltenyi Biotec) as described previously (J. Immunol. 2006;176: 3321-3329). CD4+CD25+ and CD4+CD25- cells were obtained by cell sorting labeled with FITC-conjugated anti-CD25 mA (purity 〜98%). To prepare naive and memory CD4+CD25+ and CD4+CD25- cells were labeled with PE-conjugated anti-CD62L for positive and negative selection by anti-PE magnetic beads. Then, CD4+ cells (1 x 106 cells/mL) were treated with anti-CD3/CD28-coated beads (1:5) ± TGF-β (2 ng/mL), ± IL-2 (20 U/mL, R&D Systems) or CXCL14 (100 ng/mL R&D Systems) in AIM-V serum-free medium for various days in 24-well plates (BD). To evaluate the regulatory T cell activity, various doses of CD4+ regulatory cells, immunosorted CD11C+ iDC or control cells were co-cultured to fresh T cells (2 x 105) labeled with CFSE that were activated with anti-CD3 (0.25 μg/ml) in the presence of irradiated APC (2 x 105) in 96-well plates. Cell proliferation was assessed by inhibition of percentages or total CD4+ T cells as described previously (J. Immunol. 2004;172: 5213-5221).
Transwell Migration Assays
Migration analysis was assessed as described previously[44]. In brief, cells were placed in the upper chamber (transwell: 6.5-μm diameter, 5.0-μm pore size) (Corning Costar). CXCL14 (1, 10 and 100 ng/mL) [45], CCL5 (10 nM, R&D Systems), CCL2 (10 nM, R&D Systems), CCL20 (300 nM, R&D Systems) or SDF-1α (100 ng/mL, R&D System, positive control) were placed in the lower chambers. For the blocking studies, anti-CXCL14 blocking antibody (CXCL14-Ab, 10 μg/mL, R&D Systems)[46], or control rat IgG, or specific metalloproteinase inhibitor (SB-3CT, 20 μM, Sigma)[47] were added in the in the lower chambers at 37oC for 1 h. The assays were conducted over a 4-h incubation period at 37oC in a 5% CO2 incubator. Because almost all cells stay at the lower side of the membrane after migration, quantification can be performed by counting cells. Adhered cells at the lower side of the membranes were counted under microscopy[44]. For assessing migrated CD34+ cells or iDC, cells were collected from the bottom of a transwell and assessed by flow cytometry[48].
FACS Analysis for Intracerebral Leukocytes
Isolation of brain-invading leukocytes was performed for FACS analysis as previously described[20]. In brief, ischemic mice were perfused transcardially with 20 mL normal saline and their brains were removed immediately and placed on ice in Hank's balanced salt solution (Gibco). Under a dissecting microscope, the brains were split into two cerebral hemispheres and were then separately homogenized by gentle triturating with frosted glass-slides, and incubated with type II-S Collagenase (600U; Sigma), DNase (450U; Invitrogen), and Dispase II (Roche) for 30 minutes at 37ºC. The cells were then washed by centrifugation and subjected to a 70-37% Percoll gradient centrifugation. The collected interphase mononuclear cells were processed for FACS analysis. The respective single cell suspensions were stained with anti-mouse CD3 (Clone 17A2), CD4 (Clone RM 4-5), CD25 (Clone 7D4), Foxp3 (Clone FJK-16s), IFN-γ (Clone XMG1.2), IL-4 (Clone 11B11), CD11b (Clone M1/70), CD11c, MHC II, or CD80/86, and flow-cytometry was then performed on a Becton Dickinson FACSCalibur and the data analyzed by CellQuest Pro software. A series of three experiments were conducted yielding a total of 6 hemispheres for each condition.
Gene Silencing by RNA Interference
Specific knockdown was achieved by lentiviral delivery of shRNA for CXCL14 (LV-CXCL14-sh; sc-43638-V, Santa Cruz Biotechnology), shRNA for HIF-1α (LV-HIF-1α-sh; sc-35562-V, Santa Cruz Biotechnology), shRNA for HIF-2α (LV-HIF-2α-sh; sc-35316-V, Santa Cruz Biotechnology), Lenti-PrPC shRNA (LV-PrPC-sh, sc-36318-V, Santa Cruz Biotechnology), Lenti-PECAM-1 shRNA (LV-PECAM-1-sh, sc-29445-V, Santa Cruz Biotechnology) and the control shRNA (LV-control-sh; sc-108080, Santa Cruz Biotechnology).
Lentiviral Constructs of CXCL14, CXCL14-Flag, HIF-1α and HIF-2α
Lentiviral constructs were generated by cotransfection of human kidney-derived 293T cells with three plasmids using the calcium phosphate method as previously described[49]. In the transducing vector, an expression cassette with the Rev responsive element and the EF-1α promoter are used to direct the expression of CXCL14 (CXCL14 cDNA, RC202533, OriGene), mouse HIF-1α and HIF-2α (clone ID 4019056 and 5032291, Thermo)[50] and GFP (GFP cDNA; Clontech). Lentiviral vector particles were generated by transient cotransfection of 293T cells with the lentiviral shuttle plasmid from the TRIP GFP plasmid vector[51], an HIV-1-derived packaging plasmid, and a VSV-G envelope-expressing plasmid. Two days after transfection, lentiviral constructs (LV-CXCL14/-Flag, LV-HIF-1α, LV-HIF-2α or LV-GFP) were harvested in the culture medium and concentrated by ultra-centrifugation. Viral titers were quantified by using HIV-1 p24 antigen assay (Beckman Coulter). The p24 concentration was used to determine the vector dose (expressed in nanograms) administered during both in vitro and in vivo experiments. The lentiviral titers were determined by infection of 293T cells seeded in 6-well plates at 1 × 105 cells per well the day before infection by serial dilution of the concentrated viral stock. After overnight incubation, the culture medium was changed and the cells incubated for two more days. GFP fluorescent cells were identified by fluorescent microscopy or by a fluorescent activated cell sorter. Titers ranged from 108 to109 infectious units/mL.
In Vivo Lentiviral Vector Administration
Intracerebral administration was performed in animals under chloral hydrate anesthesia to inject with 1 × 109 viral units of LV-PrPC-shRNA, LV-PECAM-1-shRNA, LV-CXCL14-shRNA, LV-CXCL14/-Flag, LV-GFP, or control shRNA (5 μL) through a 26-gauge Hamilton syringe (Hamilton Company, USA) into three cortical areas, 3.0-5.0 mm below the dura. The approximate coordinates for these sites were l.0-2.0 mm anterior to the bregma and 2.5-3.0 mm lateral to the midline, 0.5-l.5 mm posterior to the bregma and 3.5-4.0 mm lateral to the midline, and 3.0-4.0 mm posterior to the bregma and 4.5-5.0 mm lateral to the midline. The needle was retained in place for 5 minutes after each injection and a piece of bone wax was applied to the skull to prevent leakage of the injected solution. To assess for transgene expression, animals that received an intracerebral injection of lentiviral particles were then killed for histological purposes and western blot quantification of CXCL14, PrPC, and PECAM-1 production in vivo. In in vitro lentiviral vector transduction, a CD11c+ iDC culture was plated in 10-cm dishes at a density of 1 × 105 cells in 5 mL of media per dish. Transductions were carried out in the presence of 8-μg/mL polybrene at an MOI of 5 or 25 for LV-PrPC-shRNA, LV-PECAM-1-shRNA, LV-GFP, LV-CXCL14-shRNA or LV-control-shRNA. After a 24 hours incubation, the transduction media was replaced with fresh original media.
RNA Isolation and Real Time RT-PCR
Total RNA was isolated from ischemic brain with RNA TRIzol (Invitrogen) and reverse transcription was performed with High Capacity cDNA Archive kit (Applied Biosystems) as well as real-time PCR with SYBR-Green assays (Applied Biosystems) on a GeneAmp 5700 SDS from Applied Biosystems as described previously[52]. TaqMan primer/probe sets for various cytokines and housekeeping genes were designed using PRIMER EXPRESS software (Applied Biosystems). Real Time PCR was run using 5 ng template cDNA for each sample. The fast PCR protocol consisted of an initial denaturing step at 95ºC for 20 s. Next, samples were run at 94ºC (denaturation) for 3 s and then 60ºC (annealing) for 30 s (25 cycles). We ran all assays in duplicate. Relative gene expression was calculated using the Ct method with normalization to GAPDH or 18S[52].
Gel Zymography (GZ)
The culture supernatant containing equal amounts of protein was loaded onto a 10% SDS-polyacrylamide gel (gelatin; Bio-Rad, CA). After electrophoresis, gels were washed in 5% (w/v) Triton X-100 and then incubated in MMP assay buffer (Bio-Rad). Bands were visualized with Coomassie Brilliant Blue and destained in 30% methanol with 10% acetic acid.
Preparation of Transgenic GFP-Chimeric Mice
In order to verify the enhancement of bone marrow derived cells (BMDCs) mobilization and homing into brain, a bone marrow niche sample was removed from the long bones of adult male donor mice as previously reported[53]. Both ends of the femur and tibia were penetrated using a syringe with a 25-gauge needle, and the marrow was flushed out with sterile saline. Total marrow from 1 femur was diluted to 1 mL then strained through a 30-μm Spectramesh (Fisher Scientific). Before bone marrow transplantation, recipient wild type (C57BL/6 mice) and CXCL14-/- mice[54] underwent whole-body gamma irradiation with 137Cs using a Gammacell 40 irradiator (MDS Nordion). A total dose of 9 Gy (900 rads) was administered to ablate the whole bone marrow. The mice received rescuing bone marrow transplantations within 24 hours of irradiation. Donor bone marrow was injected into the recipient animal's tail as an 80 μL cell suspension containing 3 × 106 cells. At 3 weeks after transplantation, mice were anesthetized with chloral hydrate (0.3 g/kg, ip) and subjected to right middle cerebral artery (MCA) ligation and bilateral common carotid artery (CCAs) clamping for 120 minutes, as previously described with modification[26]. Mice were injected intraperitoneally with CXCL14 (500 ng/kg), and vehicle control.
In Vitro Primary Cortical Culture (PCCs) Preparation
Primary cortical cultures (PCCs) were prepared from the cerebral cortex of gestation day 17 embryos from C57BL/6 mice as described previously[55]. PCCs were maintained under serum-free conditions in neurobasal medium (Invitrogen), supplemented with B-27 supplement (2%; Invitrogen), glutamine (0.5 mM; Sigma Aldrich), glutamate (25 mM; Sigma Aldrich), penicillin (100 U/mL), and streptomycin (100 mg/mL; Invitrogen). After 4 days in vitro, half of the medium was removed and replaced with fresh medium without glutamate. The cultures were maintained in a humidified incubator at 37oC with 5% CO2. After being cultured in vitro for 7 d, PCCs were used for experimentation.
Hypoxia Procedure
PCCs (1 × 105/mL) cultured at 37°C in 5% CO2-humidified incubators were treated in normoxic (21% O2) or hypoxic conditions (1% O2) for different time points as previously described[56]. Hypoxic cultures were cultivated in a two-gas incubator (Jouan, Winchester, Virginia) equipped with an O2 probe to regulate N2 levels. Cell number and viability were evaluated using a trypan blue exclusion assay.
Immunocytochemical and Western Blot Analysis of PCCs
Following hypoxia (1% O2 for 8 h), PCCs were collected for CXCL14 immunostaining at each time point, cell cultures were washed with PBS and fixed for 30 minutes at room temperature in 4% paraformaldehyde. After being washed with PBS, the fixed cells were treated for 30 minutes with blocking solution (10 g/L BSA, 0.03% Triton X-100, and 4% serum in PBS). PCCs were incubated overnight at 4oC with an antibody against CXCL14 (1:100, Santa Cruz Biotechnology) and then rinsed three times in PBS. The extent of CXCL14+ cell immunoreactivity was measured as the number of cells per square millimeter (cells/mm2). PCC expression of CXCL14 and CCL20 were measured by western blot analyses using appropriately diluted antibodies to CXCL14 (1:100, Santa Cruz Biotechnology) or CCL20 (1:250, R&D Systems).
ChIP Assay
PCCs subjected to 4-h of hypoxia were fixed with 1% formaldehyde for 20 minutes at 37˚C to allow for reversible cross-linkage[57]. The binding of HIF-1α to the promoter of CXCL14 (NCBI Accession number: NC_000005.9) was examined using a commercial kit for ChIP assay (Upstate Biotechnology). DNA-protein complexes were immunoprecipitated with primary antibody against HIF-1α linked to protein A agarose beads, and eluted with 1% SDS, and 0.1 M NaHCO3. The cross-links were reversed by incubation at 650C for 5 h, and the proteins were removed with proteinase K. Isolated DNA was extracted with phenol/chloroform, re-dissolved, and PCR-amplified with CXCL14 promoter primers (E1 primers, PCR product: 109 bp), sense: 5'-TCTTCTGCTTGGGTTTTCTGA-3', and antisense: 5'-TATGGGGAGTGACT TTTGGC-3').
Generation of Promoter Constructs, Transient Transfection, and Reporter Gene Assays
A fragment containing the 5'-flanking region (~2200 bp) of the human CXCL14 gene promoter (NCBI Accession number: NC00005.9) was PCR-generated from human genomic DNA. This product was further cloned into the BamHI and SphI sites of the pGL3-basic vector (Promega), which contained three real HREs. The generated plasmid was designated pCXCL14-luc1. One additional CXCL14 promoter construct (pCXCL14-luc2), which didn't contain the HRE, was generated using the same downstream primer as for pCXCL14-luc1. In the pCXCL14-mutHRE construct, the putative HRE of pCXCL14-luc1 (5'-ACGTG-3') was replaced with 5'-AAAAG-3' using the QuikChange Site-Directed Mutagenesis kit (Stratagene). All constructs were verified by DNA sequencing. 3T3 NIH cells at about 90% confluence in 24-well plates were transiently transfected with reporter plasmid (0.5 μg) using Lipofectamine 2000 (Invitrogen). To correct for variable transfection efficiency, cells were cotransfected with the pRL-SV40 vector (0.05 μg) encoding the Renilla luciferase gene. Cells were lysed and luciferase activity was determined with a multiwell luminescence reader (Molecular Devices), by using the Dual-Luciferase Reporter Assay System (Promega).
Statistical Analysis
The study was designed so that blind measurements were taken. Results were expressed as mean ± SEM. The behavioral scores have been evaluated and adjusted by normal distribution. Two-way ANOVA with appropriate post hoc Newman-Keuls testing was used to evaluate differences between different groups with different treatments. A value of P < 0.05 was deemed significant.
Results
Ischemia/Hypoxia Promotes CXCL14 Expression in Human, and Rodent Brains, As Well As in Primary Cortical Cultures
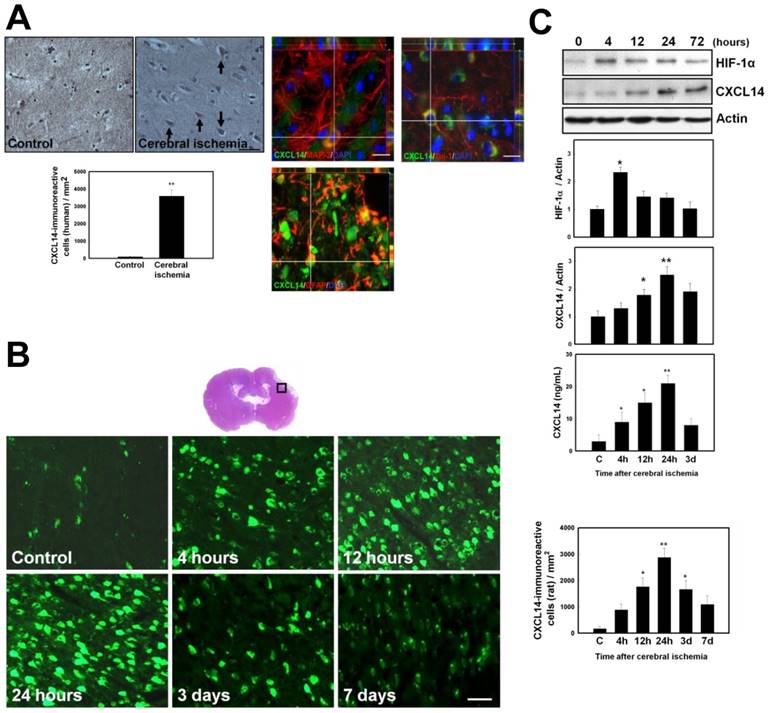
The expression of CXCL14 in human and rat ischemic brains was measured using immunohistochemistry. CXCL14 expression levels were higher in the penumbral region in ischemic brain samples from human stroke patients than in the controls at 1-3 days after cerebral infarction (n = 4) (Fig. 1A and Supplementary Table S1). In post-stroke rat brains, CXCL14 was mainly expressed in the ipsilateral cortex, near the boundary of the infarcted region (Fig. 1B). Western blot analysis showed that CI resulted in an increase in HIF-1α and CXCL14 levels in a time-dependent manner, and reached their peak at 4 hours for HIF-1α and at 24 hours for CXCL14 after CI, which was in agreement with the results obtained by both immunohistochemistry and ELISA (Fig. 1C). Moreover, the immunofluorescence staining of CXCL14 further revealed that CXCL14 expression was colocalized with microtubule-associated protein 2 (MAP-2), neuron-specific class III beta-tubulin (Tuj-1), or glial fibrillary acidic protein (GFAP) in the penumbric region of ischemic brains (both human and rat) (Fig. 1A and S1A), suggesting that both neurons and astrocytes increased CXCL14 expression in response to brain ischemia. Furthermore, primary cortical cells (PCCs) under hypoxic treatment showed a time-dependent increase in CXCL14 levels, peaking at 3 hours after hypoxia, and continuing for 24 hours (Fig. S1, B-C). Taken together, these results indicate that CI is able to promote CXCL14 expression in human and rodent brains.
HIF-1α/Ischemia Induced Upregulation of CXCL14 Expression in human Brain and Rat Brains as well as in Primary Cortical Cultures (PCCs). (A) Figures are representative of stroke patients following ictus. Immunohistochemical studies of the penumbral area showed significantly increased CXCL14 immunoreactivity (upper panel). Quantitative measurement of CXCL14+ cells in the stroke patients' brains showed a significant increase at 1 day and 3 days following cerebral infarction compared to controls (left panel). In addition, some CXCL14+MAP-2+, CXCL14+Tuj-1+ and CXCL14+GFAP+ cells dispersed over the penumbral region of human ischemic brain (right panel). (B) The square-mark (□) shows the immunoreactivity of CXCL14 over the penumbral and perivascular regions of rat brains at different time points (4 hours, 12 hours, 24 hours, 3 days, and 7 days) after ischemia. Quantitative analysis indicated that the increased immunoreactivity of CXCL14 was time-dependent following ischemia compared with non-ischemic rats, which are indicated as “C”. (C) In western blot analysis, HIF-1α and CXCL14 expression was increased in a time-dependent manner after cerebral ischemia. ELISA showed increased CXCL14 levels in ischemic rats compared to non-ischemic controls. The mean ± SEM is shown. *P < 0.05 and **P < 0.01 vs. control. Bar = 50 μm.
HIF-1α Contributes to Ischemia/hypoxia-Induced CXCL14 Expression by Directly Binding to the CXCL14 Promoter
In order to investigate the mechanism of ischemia/hypoxia-induced CXCL14, we first tested whether the upregulation of CXCL14 after ischemia/hypoxia was mediated by HIF-1α. The HIF-1α activity was analyzed in ischemic rats with pharmacological treatment using 2-ME2 (an HIF-1α inhibitor) and in conditional HIF-1α knockout (KO) mice. CI increased nuclear HIF-1α expression and CXCL14 expression, which could be inhibited with a 2-ME2 injection in a dose-dependent manner (Fig. 2, A-B and Fig. S2, A-B). Consistently, brain ischemia-induced CXCL14 upregulation disappeared in conditional HIF-1α KO mice (Fig. 2C). Because HIF-1α upregulated CXCL14 expression, we hypothesized that members of the HIF family might participate directly in this process. To test this hypothesis, we examined CXCL14 levels in PCCs treated with 2-ME2, and HIF-1α or HIF-2α knockdown by lentiviral transduction of HIF-1α or HIF-2α shRNAs. Both HIF-1α translocation and CXCL14 expression were abolished when PCCs were pretreated for 16 hours with HIF-1α inhibitor 2-ME2 (Fig. S2, C-D). Moreover, knockdown of HIF-1α, but not HIF-2α, significantly abrogated HIF-1α-induced CXCL14 expression (Fig. 2D and Fig. S2D). Moreover, overexpression of HIF-1α by lentiviral transduction, but not HIF-2α, significantly enhanced CXCL14 expression (Fig. 2D). These results indicate that HIF-1α is a crucial transcription factor for hypoxia-mediated CXCL14 induction.
Upregulation of CXCL14 After Cerebral Ischemia (CI) and Hypoxia Through Induction of HIF-1α In Vivo and In Vitro. (A) CXCL14 immunoreactivity was decreased in a 2-ME2-dose-dependent manner after CI in comparison with the non-ischemic rats (C). (B) Decreased expression of CXCL14 was present in a 2-ME2-dose-dependent manner. (C) In HIF-1α KO mice, cerebral ischemia could not significantly induce CXCL14 upregulation. (D) Upregulation of CXCL14 by western blot was found in LV-HIF-1α transduction to PCCs, but not in that of LV-HIF-2α (right panel); only LV-HIF-1α-sh transduction to PCCs at 1hoursafter hypoxia downregulated the level of CXCL14 (left panel). (E) Schematic representation of the 5'-flanking CXCL14 gene region: the promoter region (shaded white box), and the first exon (solid block box). HRE binding site: -1895 to -1891 (relative to putative transcription start site +1). Binding of HIF-1α to the CXCL14 promoter was detected in PCCs subjected to hypoxia by the chromatin immunoprecipitation (ChIP) assay. As a negative control, HIF-1α-knockdown by lentiviral delivery of the shRNA (LV-HIF-1α-shRNA) prevented the detection of HIF-1α-to-DNA binding using ChIP. (F) Luciferase activity was significantly enhanced in the hypoxia-treated cells transfected with pCXCL14-luc1 compared to a control construct (pCXCL14-luc2) or an HRE-mutant construct (pCXCL14-mutHRE). n=8 per group. The mean ± SEM is shown. *P < 0.05 and **P < 0.01 vs. control. Bar = 50 μm.
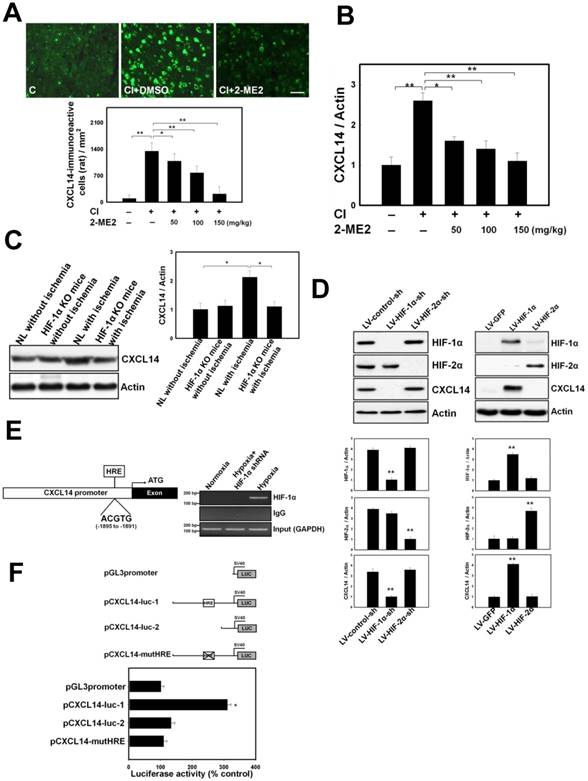
Next, we used bioinformatics to help predict the molecular mechanism by which hypoxia induces CXCL14 expression. This analysis identified one HIF-1α-binding site (hypoxia response element, or HRE) in the CXCL14 promoter sequence from -1895 to -1891 (5'-ACGTG-3'), suggesting that HIF-1α might regulate CXCL14 expression by binding directly to its promoter, which we confirmed using ChIP assays in PCCs subjected to 4 hours of hypoxia. However, this result was not observed in PCCs subjected to 4 hours of normoxia or in PCCs treated with HIF-1α shRNAs with 4 hours of hypoxia (Fig. 2E). In PCC-based reporter assays, the activity of a CXCL14 luciferase reporter construct (pCXCL14-luc1), containing HRE, under hypoxia was much higher than in a control construct (pCXCL14-luc2) (Fig. 2F). To pinpoint the exact binding motifs, point mutations were introduced into the HRE of CXCL14-Luc (pCXCL14-mutHRE). The ablation of HRE on the CXCL14 promoter abrogated hypoxia-mediated CXCL14 induction (Fig. 2F). Collectively, these results suggest that HIF-1α regulates CXCL14 transcription by binding directly to the CXCL14 promoter in a hypoxia-dependent fashion.
Loss of CXCL14 Reduces the Accumulation of iDC and Treg and Increases the Infarct Volume in the Ischemic Brain
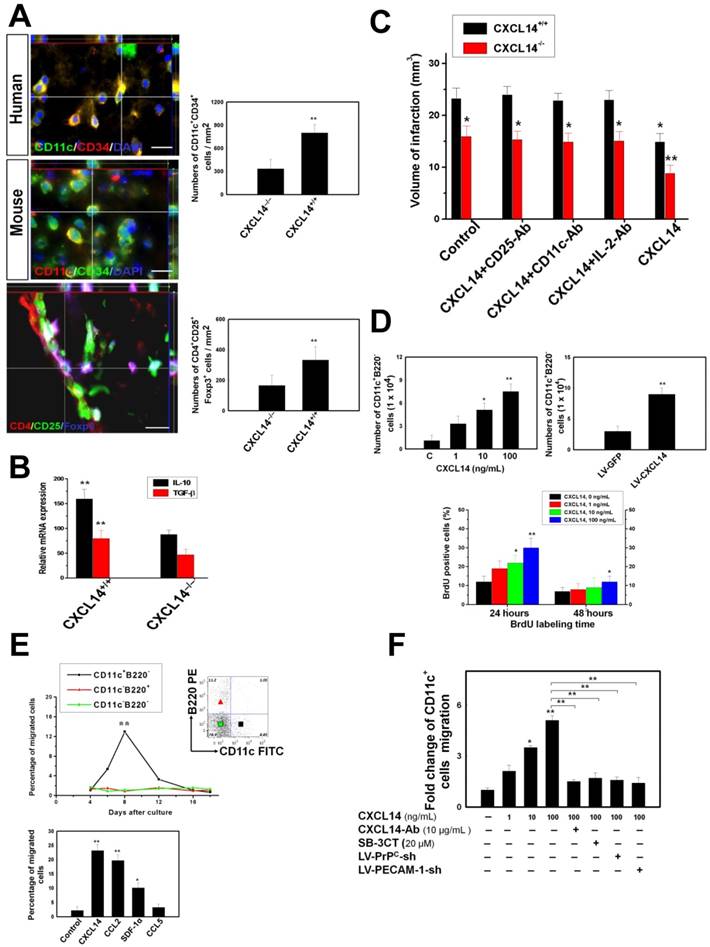
To investigate the impact of CXCL14 in brain ischemia, CI was induced in CXCL14-/- and wild type mice received cerebral ischemia (CI). First, we observed a few CD11c+CD34+ iDC located over the pneumbric area of stroke human brain (Fig. 3A). Next, immunofluorescence imaging in both CXCL14-/- and wild type mice showed that the accumulation of iDC and Treg was significantly increased in the ipsilateral cerebral hemisphere compared with the contralateral cerebral hemisphere. There was a significant decrease in the accumulation of iDC and Treg in CXCL14-/- as compared to wild type mice (Fig. 3A). The enzyme-linked immunosorbent assay (ELISA) of brain homogenates also demonstrated that CXCL14-/- mice exhibited appreciably lower levels of immunosuppressive soluble factors in the ipsilateral hemisphere, such as IL-10 and TGF-beta, compared with wild type mice (Fig. 3B), suggesting that the genetic deficiency of CXCL14-/- decreases CI-mediated immunosuppressive effect. Finally, the 2,3,5-Triphenyltetrazolium chloride (TTC) staining assay also demonstrated that the cortical infarct volume was significantly larger in CXCL14-/- mice as compared to wild type mice (Fig. 3C). Thus, genetic deficiency of CXCL14-/- increased the infarct volume. Taken together, these results suggest CXCL14 plays a role in brain ischemia-induced endogenous immunosuppressive and neuroprotective effects.
CXCL14 Exerts Immunomodulation via Regulating Immature Dendritic Cell (iDC) and Regulatory T Cells (Treg). (A) In the representative 3-D image, some CD11c+CD34+ iDC were observed in the human stroke hemisphere (upper panel). Moreover, increased numbers of CD11c+CD34+ cells were found in the ischemic brain of CXCL14+/+ mice compared to that of CXCL14-/- mice (middle panel). Furthermore, significantly increased numbers of CD4+CD25+Foxp3+ Treg were found in the CXCL14+/+ mice compared to those of CXCL14-/- mice at 5 days after cerebral ischemia (lower panel). (B) ELISA showed a significant decrease in the levels of IL-10 and TGF-beta in brain homogenates of CXCL14-/- mice compared with those of wild type mice. (C) Infarct volume was significantly increased in CXCL14-/- mice compared to that of CXCL14+/+ mice. Furthermore, infarct volume was larger in CD25-Ab-treated, CD11c-Ab-treated and IL-2-Ab-treated CXCL14+/+ mice than in saline-treated CXCL14+/+ mice. (D) Overexpression of CXCL14 significantly increased cell proliferation, as shown by trypan blue, which excluded viable cells and the BrdU labeling index. (E) The transwell migration assay showed that CXCL14 is highly attracted to CD11c+B220- cells, but not to CD11c-B220+ cells or to CD11c-B220- cells. In iDC migration profiles for either chemokine after culturing for 8 days, CD11c+ iDC responded to CXCL14 and to CCL2 chemoattraction. (F) CD11c+ iDC treated with CXCL14 could move across the membrane in a concentration-dependent manner (SDF-1α as a positive control) in the transwell migration assay. In contrast, CXCL14-induced iDC migratory activity could be neutralized by adding the CXCL14-Ab and SB-3CT. LV-PrPC-shRNA or LV-PECAM-1-shRNA transduction also inhibited the CXCL14-induced iDC trafficking. The mean ± SEM is shown. *P < 0.05 and **P < 0.01 vs. control.
CXCL14 Promotes the Proliferation and Migration of iDC
In order to elucidate the mechanism of CXCL14-mediated iDC accumulation in the ischemic brain, we first determined the effect of CXCL14 on the proliferation and migration of HSC-derived iDC in vitro. Trypan blue dye exclusion and BrdU assays confirmed that cell number and BrdU incorporation were significantly increased by the recombinant CXCL14 protein at concentrations from 1 to 100 ng/mL or by the lentiviral transduction of CXCL14 (Fig. 3D), suggesting CXCL14 is able to promote the proliferation of HSC-derived iDC. To address iDC migration, we examined the effect of CXCL14 on their trafficking. First, we generated iDC from circulating CD34+ HSCs after 4 days of in vitro culture, resulting in two nonoverlapping subsets of CD11c+B220- and CD11c-B220+ cells and a third subset of CD11c-B220-cells that have also lost CD34 (Fig. 3E). CXCL14 is highly selective for CD11c+B220- iDC, as is shown by a chemotaxis assay (Fig. 3E). Migration responses peaked 6-10 days into the iDC culture and then returned to the base level by day 12. In contrast, CD11c-B220+ cells and CD11c-B220- cells were poorly attracted by CXCL14 during the entire CD34+ HSC differentiation process (Fig. 3E). After culturing the CD34+ HSC-derived iDC for 8 days, they responded well to the addition of chemokines with known functions in DC migration profiles (Fig. 3E). Importantly, CD11c+ iDC displayed the greatest responses to CXCL14 and to CCL2, the prototypical monocyte chemoattractant (Fig. 3E). The migration of CD11c+ iDC toward CXCL14 was found to be dose-dependent with maximal activity between 10-100 ng/mL (Fig. 3F).
Next, we examined CXCL14-regulated gene candidates that may affect cell migration. We analyzed protein expression of cell adhesion and migration proteins, including: selectin, CXCR4, β1-integrin, β2-integrin, junctional adhesion molecules A and C (JAM-A and JAM-C), lymphocyte function-associated antigen 1 (LFA-1), intercellular adhesion molecule (ICAM), vascular cell adhesion molecule (VCAM), platelet-endothelial cell adhesion molecule type 1 (PECAM-1), vascular endothelial cadherin (VE-cadherin), CD99, and the cellular prion protein (PrPC) in CXCL14-treated iDC. CXCL14 (100 ng/mL) was administered in iDC cultures and control cultures, and the protein was isolated after 10-12 hours of treatment. Among these molecules, we specifically selected two genes, one for PrPC (five folds) and one for platelet-endothelial cell adhesion molecule type 1 (PECAM-1) (seven folds) as CXCL14-regulated genes that were upregulated in CXCL14-treated iDC (Fig. 4A). The induction of PrPC and PECAM-1 by CXCL14 was confirmed in a dose-dependent manner via western blotting analysis (Fig. 4A). However, the upregulation of PrPC and PECAM-1 was inhibited in CXCL14-Ab-treated iDC (data not shown). Furthermore, we determined the effect of CXCL14 on MMP' activity. Overexpression of CXCL14 by lentiviral infection (LV-CXCL14) or by the pharmacological induction of CXCL14 protein stimulates not only the concomitant induction of MMP9 and MMP2 activity by gel zymography, but also the protein biosynthesis of MMP9 and MMP2 (Fig. 4B and S3A) in a dose-dependent manner. In contrast, CXCL14-Ab incubation inhibited the CXCL14 (10 ng/mL)-induced enhancement of MMP9 and MMP2 activity (Fig. 4B). The enhancement of CD11c+ iDC migration in vitro was blocked by the CXCL14 neutralization antibody (CXCL14-Ab, 10 μg/mL), MMP inhibitor (SB-3CT, 10 μM21) or by the knockdown of PrPC or PECAM-1 (Fig. 3F), suggesting that MMP, PrPC, and PECAM-1 play a crucial role as mediators for the CXCL14-induced mobilization of iDC.
CXCL14 Stimulated the Recruitment of iDC and Activation of Treg. (A) Bar graph for the targeted proteins corresponding to the cell adhesion and migration in CXCL14-treated iDC. Higher expression of PECAM-1 and cellular prion protein (PrPC) was induced by administration of CXCL14 in a dose-dependent manner. (B) Gel zymography showed that CXCL14 overexpression by LV-CXCL14 or CXCL14 protein activated MMP9 and MMP2 activity in iDC culture supernatant in a dose-dependent manner (left and middle panel). CXCL14 (10 ng/mL)-induced activation of MMP9 and MMP2 activity was abolished by administration of CXCL14-Ab incubation (C=isotype-control antibody, right panel). (C) There was a significant increase in the percentage of CD11c+ within the GFP+ population, but decrease in the percentage of MHC-II+, CD80+, and CD86+ cells by FACS at 3 days after CXCL14 administration. (D) Systemic injection of CXCL14 induced a higher ratio of brain/plasma CXCL14 levels found in the ipsilateral stroke hemisphere compared to vehicle control in a time-dependent manner (left upper panel). (E) A significant increase was found in the percentage of CD4+CD25+Foxp3+ cells after the treatment with CXCL14- or IL-2 plus CXCL14 compared to the control. (F) The CD4+CD25+ cells showed an increase in the expression of Foxp3 after 4 days of incubation with CXCL14. In addition, the simultaneous administration of CXCL14 and IL-2 induced a synergistic effect on the Foxp3+ cell proliferation. (G) In the co-culture of immunosorted CD11c+ iDC with CD4+CD25+ cells, a significant increase in the expression of Foxp3 was observed. Exogenous CXCL14 administration to the co-culture system further induced Foxp3+ cell proliferation additively. n = 8 per group. The mean ± SEM is shown. *P < 0.05 and **P < 0.01 vs. control. Bar = 50 μm.
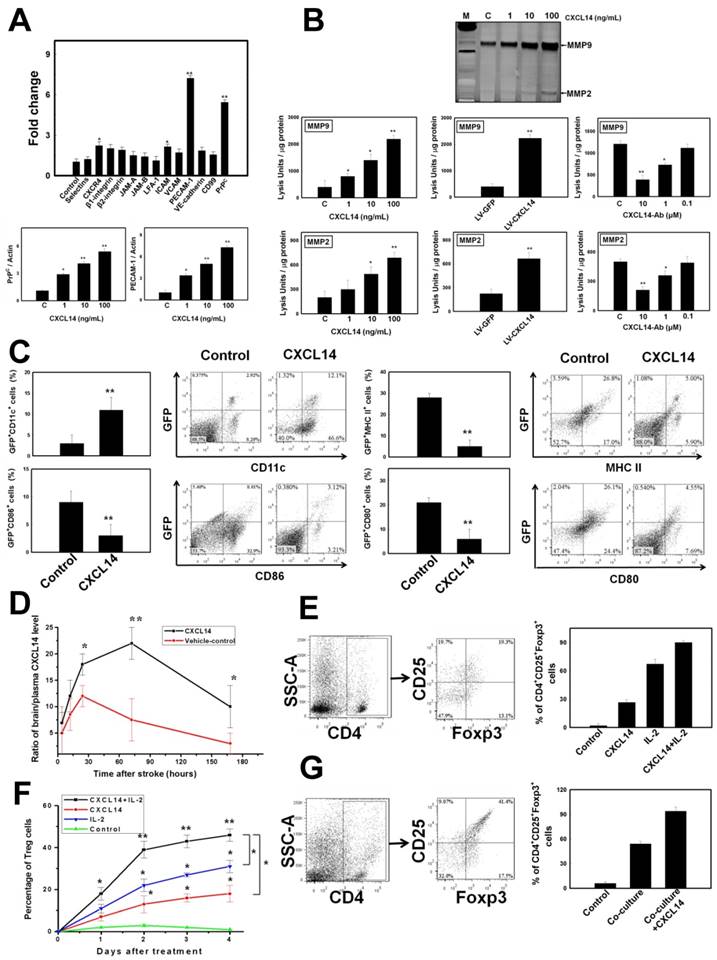
Moreover, the results of in vivo studies revealed that pharmacological CXCL14 protein and LV-CXCL14 injection induced GFP+CD11c+ iDC homing were blocked by the administration of SB-3CT and CXCL14-Ab, or LV-PrPC-shRNA or LV-PECAM-1-shRNA, respectively (Fig. S3C). In contrast, CXCL14 protein and LV-CXCL14 injection decreased the recruitment of GFP+MHC-II+ cells into the ischemic brain (Fig. S3C). By FACS analysis, at 3 days after CXCL14 treatment, the percentage of CD11c+ cells within the GFP+ population was markedly increased (Fig. 4C). However, significant reduced accumulation of MHC-II+, CD80+, and CD86+ cells was found after CXCL14 treatment (Fig. 4C). Systemic injection of CXCL14 upregulated the activity of MMP9 and MMP2, as well as the expression of PrPC and PECAM-1 in the ischemic brain (Fig. S3B). Interestingly, a significantly increased ratio of brain to plasma CXCL14 was detected in the ipsilateral stroke hemisphere compared with the control, and reached its peak level at 3 days following intravenous CXCL14 injection (Fig. 4D). Taken together, it can be concluded that CXCL14-mediated proliferation and migration of iDC contributes to iDC accumulation in the ischemic brain.
CXCL14 Induces CD4+CD25+Foxp3+ Treg Activation and Expansion
To investigate the mechanisms of CXCL14-mediated accumulation of Treg and immunosuppression, we first determined whether CXCL14 could directly induce activation of CD4+ T cells into Treg. We assessed the CD4+CD25+ cells that expressed Foxp3 with or without IL-2 treatment. Higher levels of expression of Foxp3 were noted in the CXCL14- and IL-2 plus CXCL14-treated CD4+CD25+ cells than in the control (medium only) (Fig. 4E). Other than enabling IL-2 to induce Foxp3 expression, CXCL14 was also able to expand these Foxp3+ cells. With CXCL14 treatment, the number of Foxp3+ cells continued to increase among the naive CD4+CD25+ cells (Fig. 4F) after 4 days of incubation. Furthermore, an additive effect on the proliferation of Foxp3+ cells was found in the exogenous administration of CXCL14 plus IL-2 (Fig. 4F), indicating that CXCL14 directly mediated CD4+CD25+Foxp3+ Treg activation and expansion in vitro.
To further determine the role of iDC on Treg proliferation, co-culture of immunosorted CD11c+ iDC with CD4+CD25+ cells was performed to measure the number of Treg. Consistently, iDC co-incubation significantly enhanced the expression of Foxp3, and exogenous CXCL14 additionally promoted the Foxp3+ cells proliferation (Fig. 4G).
CXCL14 Supplementation Provides a Therapeutic Strategy for Cerebral Ischemia. In order to select the most effective treatment dosage of CXCL14, rats were separated into four groups. The infarct volume of the CXCL14-treated rats (100 ng/kg) at 3 days following cerebral ischemia was much smaller than the infarct volumes in other dosage groups (Fig. 5A). As is described in the literature, DCs coordinated the cellular interaction to initiate the proliferation of Treg[18, 19], which then exerted cytokine-modulated neuroprotection in the post-ischemic brain[20]. To elucidate the CXCL14-mediated reduction of infarcted brain volume via Treg activation, we focused on the regulatory relationship between iDC and Treg in the post-ischemic inflammatory brain damage, which targets multiple inflammatory pathways. Importantly, the effect of Treg on infarct volume reduction after focal brain ischemia was abolished in anti-CD25-antibody-treated mice[20]. In our study, at 7 days after rat cerebral ischemia, the infarct volume assessed by MRI was significantly reduced in the CXCL14-treated rats compared to that in CXCL14 plus CD25-Ab-treated, CXCL14 plus CD11c-Ab-treated, CXCL14 plus IL-2-Ab-treated, and saline-treated controls (Fig. 5B). The infarcted area of the largest infarcted slice decreased significantly in CXCL14-treated rats compared to that in CXCL14 plus CD25-Ab-treated, CXCL14 plus CD11c-Ab-treated, CXCL14 plus IL-2-Ab-treated and saline-treated controls (Fig. 5B). In addition, an increase in infarct volume was found in CD25-Ab-treated, CD11c-Ab-treated, and IL-2-Ab-treated CXCL14+/+ mice compared to saline-treated CXCL14+/+ mice. The infarct volume in CXCL14-/- mice was larger than that in CXCL14+/+ mice (Fig. 3C).
CXCL14-Mediated Immunomodulation Induced Neuroprotection on Ischemic Brains. (A) In the TTC staining, the most effective treatment dose of CXCL14 (100 ng/kg) could significantly reduce infarct volume at 3 days after cerebral ischemia compared to the other dosage (G-CSF 50 μg/kg for 5 consecutive days as a positive control). (B) The MRI examination showed that at 7 days after cerebral infarction, the injection of CXCL14 significantly reduced the infarction volume and area of the largest infarcted slice compared to that of control rats. In contrast, CXCL14-induced reduction of infarct volume and area of the largest infarcted slice was blocked by the injection of CD25-Ab, CD11c-Ab and IL-2-Ab. (C) Neurological behavior measurements including body swing tests, locomotor activity tests, and grip strength tests revealed significant improvement in CXCL14-treated rats compared to those in CXCL14 plus CD25-Ab, CXCL14 plus CD11c-Ab, CXCL14 plus IL-2-Ab, and control rats. n = 8 per group. The mean ± SEM is shown. *P < 0.05 and **P < 0.01 vs. control.
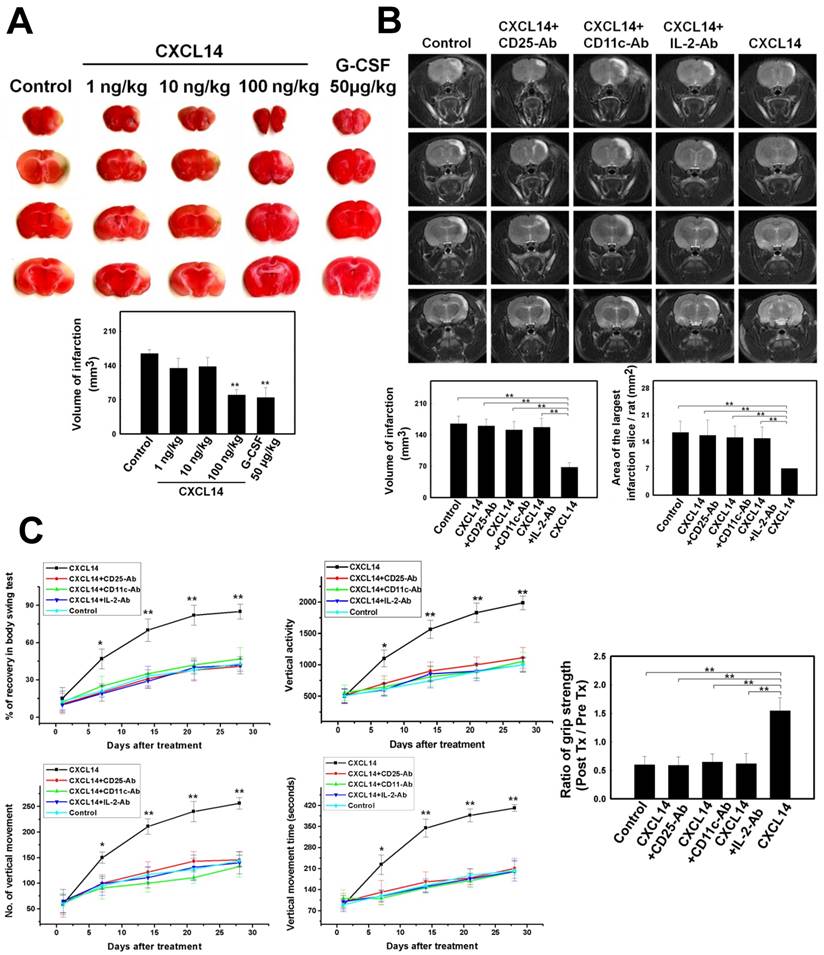
Body asymmetry, locomotor activity, and grip strength measurements were used to analyze the neurological recovery in CXCL14-treated, CXCL14 plus CD25-Ab-treated, CXCL14 plus CD11c-Ab-treated, CXCL14 plus IL-2-Ab-treated, and control rats. In body-swing tests, CXCL14-treated rats revealed more robust recovery than CXCL14+CD25-Ab-treated, CXCL14+CD11c-Ab-treated, CXCL14+IL-2-Ab-treated, or control rats (Fig. 5C). Locomotor activities were significantly better after cerebral ischemia in rats receiving CXCL14 treatment than in other groups (Fig. 5C). In addition, when comparing the state before ischemia and 28 days after ischemia, the CXCL14-treated group had a much better grip strength ratio than any other groups (Fig. 5C).
To demonstrate whether CXCL14 exerted a cerebroprotective effect in response to iDC-induced activation of Treg, a FACS analysis was performed on the post-ischemic rat brain. Results from the FACS study showed that the percentage of CD4+CD25+Foxp3+ Treg and CD11c+B220- iDC was significantly higher in CXCL14-treated mice than in the controls (Fig. 6A). Conversely, CXCL14-induced Treg and iDC activation was blocked by the administration of CXCL14-Ab, CD11c-Ab, CD25-Ab and IL-2-Ab (Fig. 6A). The same in the immunofluorescence colocalization study, a significant increase in CD4+CD25+Foxp3+ Treg proliferation appeared in CXCL14-treated mice compared to saline-treated mice (Fig. 6B). However, CXCL14-induced Treg activation was blocked by the administration of CXCL14-Ab, CD11c-Ab and IL-2-Ab (Fig. 6B). To verify whether CXCL14 administration could enhance metabolic activity, the cortical glucose metabolism was examined by FDG-PET one week after treatment. The microPET images showed a striking increase in FDG uptake over the right cortexes in the CXCL14-treated group, which was higher than in CXCL14 plus CD25-Ab-treated, CXCL14 plus CD11c-Ab-treated, CXCL14 plus IL-2-Ab-treated, or saline-treated rats (Fig. 6C).
CXCL14 Injection Promoted Treg-Modulated Neural Survival in the Post-Stroke Brain. (A) Representative FACS analyses show significant increases in the percentage of CD4+CD25+Foxp3+ Treg (left panel and right upper panel) and CD11c+B220- iDC (right lower panel) that were observed in CXCL14-treated mice compared to the control, but not in CXCL14-Ab-treated, CD11c-Ab-treated, CD25-Ab-treated or IL-2-Ab-treated mice. (B) In immunofluorescence colocalization studies (upper panel), more CD4+CD25+Foxp3+ Treg re-located to the pneumbral region in CXCL14-treated mice than in saline-treated mice, but not in CXCL14-Ab-treated, CD11c-Ab-treated or IL-2-Ab-treated mice. (C) In measurements of relative glucose metabolic activity after stroke, FDG uptake was significantly enhanced in micro-PETs of the right hemisphere (arrows) in CXCL14-treated rats compared to CXCL14 plus CD25-Ab, CXCL14 plus CD11c-Ab, CXCL14 plus IL-2-Ab, and control rats. (D) In real-time PCR analysis for inflammatory cytokine expression, significant increases in the expression of IL-10 and TGF-β were observed at 24 hours post stroke in the CXCL14-treated group compared to control rats. In contrast, injection of CXCL14+CD25-Ab or CXCL14+CD11c-Ab blocked the CXCL14-induced upregulation of IL-10 and TGF-β. (E) Western blot analysis showed significantly increased expression of Bcl-2, but not of Bcl-xL, Bax, or Bad in CXCL14-treated rats at 3 days after cerebral ischemia compared to control rats, but not in CXCL14 plus CD25-Ab-, CXCL14 plus CD11c-Ab-, CXCL14 plus IL-2-Ab-treated rats. (F) Representative images of TUNEL assay (green) and Hoechst 33342 (blue) co-staining. There were fewer TUNEL-positive cells in the brain of CXCL14-treated rats than in those of CXCL14 plus CD25-Ab-, CXCL14 plus CD11c-Ab-, CXCL14 plus IL-2-Ab, or control rats. n = 8 per group. The mean ± SEM is shown. *P < 0.05 and **P < 0.01 vs. control. Bar = 50 μm.
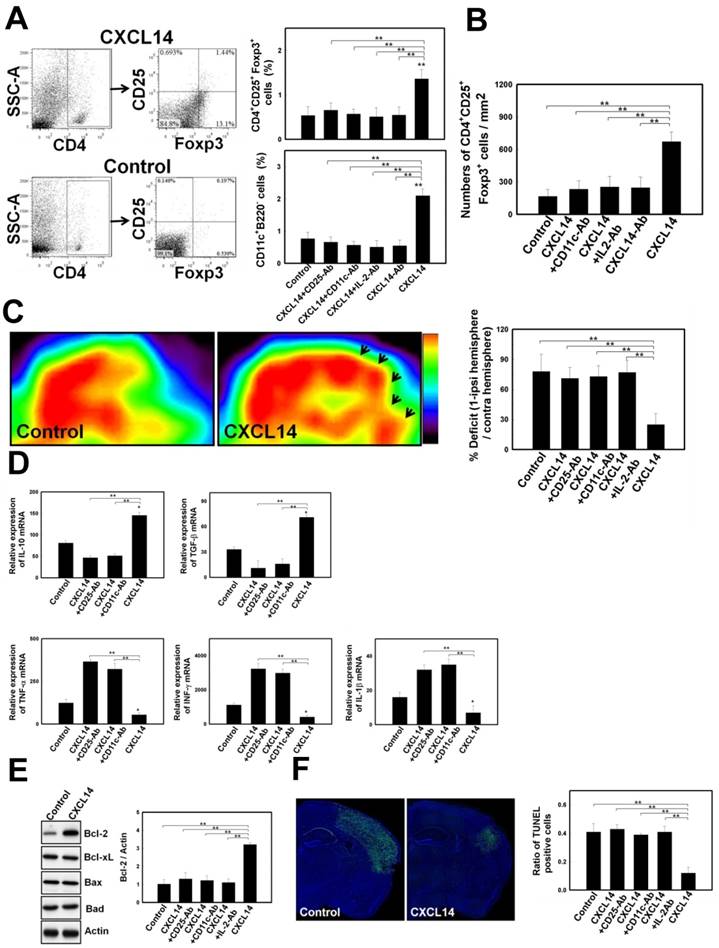
The relative expression of cytokine mRNA was measured in the ischemic hemispheres. Postischemic cerebral expression of the anti-inflammatory cytokines, IL-10 and TGF-β was significantly upregulated at 3 days after ischemic onset in the CXCL14-treated group compared to that in the control, but not in the CXCL14 plus CD25-Ab-treated or CXCL14 plus CD11c-Ab-treated group (Fig. 6D). In addition, expression of pro-inflammatory cytokines TNF-α, INF-γ, and IL-1β was substantially was downregulated at 3 days post ischemia in the CXCL14-treated group compared to the control (Fig. 6D).
The molecular mechanisms underlying the neuroprotective effect of CXCL14 on the ischemic brain were examined by western blot analysis. Bcl-2, an anti-apoptotic factor, was upregulated in CXCL14-treated rats at 24 hours following cerebral ischemia compared to CXCL14 plus CD25-Ab-treated, CXCL14 plus CD11c-Ab-treated, CXCL14 plus IL-2-Ab-treated, or to control rats (Fig. 6E).
Cellular apoptosis in the ischemic rat brain was studied by TUNEL staining. Control animals without stroke had almost no TUNEL staining in any section of their brain. The penumbral region surrounding the ischemic cores of CXCL14-treated rats contained fewer TUNEL+ cells than in CXCL14 plus CD25-Ab-treated, CXCL14 plus CD11c-Ab-treated, CXCL14 plus IL-2-Ab-treated, or control group animals (Fig. 6F).
Discussion
CXCL14 is a member of the CXC chemokine family; however, it lacks the ELR motif typically found at the amino terminus of most CXC members. Although CXCL14 affects the function of monocytes, macrophages, NK, and dendritic cells[6, 58]. it also plays a significant role in insulin signaling and is related to high-fat diet-induced obesity[54, 59]. CXCL also plays a role in tumor biology. For example, increased levels of CXCL14 expression promoted tumor angiogenesis and macrophage migration that promoted prostate cancer growth[7]. To understand the role of CXCL14 in neural injury, we investigated both ischemic in vivo models and hypoxic in vitro models. Increased expression of CXCL14 was induced not only in the human and rat ischemic brain, but also in hypoxic PCCs. CXCL14 expression is localized primarily in neurons and glial cells. We found that this response is mediated by the interaction of the transcription factor HIF-1α via the HRE sequence within the CXCL14 gene promoter. This can be explained with the high expression level of CXCL14 in stress conditions[60]. Such findings prompt speculation that CXCL14 expression is induced in response to stress[61]. In agreement with previous studies, CXCL14 has been shown to possess functions similar to other trophic factors[62], such as the activation of endogenous survival mechanisms to enhance growth and to repair neural tissue[63]. In summary, especially in the cerebral ischemic and hypoxic model, the activation of CXCL14 may be responsible for relaying an important signal to allow neural adaptation to environmental stress.
Although the molecular mechanisms underlying iDC relocation to inflammatory sites are becoming better understood[64], the control of iDC under ischemic conditions, notably in the setting effect of DC development in the post-stroke brain, is uncharacterized. Several factors govern cell trafficking, for example, when cells are in an inflammatory state, CCL20 (the ligand for CCR6) attracts skin-emigrant CD34+ CD14+ HSC-derived iDC [65]. During ischemia, adenosine plays a potent role as a chemotactic stimulus for iDC through an A1 receptor-mediated mechanism[66]. The adenosine-induced migratory effect was comparable to the effect seen by stromal cell-derived factor-1α (SDF-1α), the most potent iDC chemotactic stimulus identified thus far[66]. Here, we found that CXCL14 is upregulated in post-stroke human and rat brains, indicating a homeostatic function for this ill-defined chemokine. CXCL14 is uniquely selective for CD11c+ iDC, generated in vitro from CD34+ HSCs. We provided evidence that enhanced expression of CXCL14 mediates the brain's recruitment of CXCL14-responsive CD11c+ iDC, thereby promoting their in situ differentiation into functional DCs. Cell-cell interactions mediated by adhesion molecules enable and control iDC trafficking. iDC express ICAMs, LFA-1, VCAM, PECAM-1[67], selectin, CXCR4, β1-integrin, β2-integrin, JAM-A, JAM-C, VE-cadherin, CD99, and PrPC[67-73]. Furthermore, MMPs were key enzymes in the mediation of cell migration and the activation of TGF-β to attract iDC [5, 74]. In this study, increased activity and protein expression of MMPs, PrPC, and PECAM-1 appear to be stimulated by CXCL14 in iDC. These factors were knocked down by a specific inhibitor or by an shRNA technique that may in fact block the migratory effect of iDC, indicating that they were responding to the CXCL14-induced CD11c+ iDC relocation to the ischemic brain.
Inflammatory processes characterized by the accumulation of inflammatory cells and inflammatory mediators contributed substantially to ischemic brain damage. Because the mechanism of intrinsic immune system modulation is largely unknown, anti-inflammatory strategies remain of interest in current translational stroke research. One previous study found that cerebral ischemia is accompanied by a systemic increase in Treg numbers[75]. Proliferation of Treg could prevent secondary ischemic damage by counteracting excessive production of proinflammatory cytokines[20]. Further, an increase in Treg numbers diminished the pro-inflammatory response to myelin basic protein in the ischemic brain[76]. Nonetheless, the regulatory mechanism governing the Treg response in the ischemic brain is not well understood. With regard to the relationship between iDC and Treg, iDC played an important role in regulating the proliferation of Treg[18]. Depletion of iDC inhibited the generation of Treg in vivo[18]. In stroke patients, CD14+ iDC that are transiently reduced in systemic circulation might be recruited into the infarcted brain[10]. Relocated iDC activated by a costimulator ligand or by IL-2 were sufficient to induce the proliferation of IL-10-secreting Treg[19, 77]. A Previous report demonstrated that G-CSF stimulated the mobilization of iDC, and further induced the proliferation of Treg[78]. G-CSF treatment augmented the iDC-primed Treg-mediated release of IL-10 and TGF-β[79]. Although the above evidences might be generally accepted, the attenuated inflammatory response to stroke in aged animals including diminished proinflammatory cytokines (IL-6, IL-1β and TNF) may contribute to their smaller infarcts. Therefore, differences between age-based immunological modulation on cerebral microenvironment after stroke need further deep investigation [80]. In the present study, we first found that CXCL14 expressed in the ischemic brain selectively recruited the CD11c+ iDC, and then stimulated the proliferation of IL-10-secreting Treg to induce a cerebroprotectve state.
CXCL14 played an important role in the neuroprotecitive outcome in the ischemic brain. Administration of CXCL14 seemed to augment the survival effect throughout the penumbral area post-stroke. In a previous report, CXCL14 exerted a significant effect on the developmental control of nigral dopaminergic neuronal growth, proliferation, and survival[62]. In addition, CXCL14 modulated this proliferation in cultured Schwann cells[81]. In this study, we discovered that recombinant CXCL14 was able to reduce the infarct volume by increasing the expression of anti-apoptotic protein and diminishing the TUNEL+ cells in the ischemic brain. Based on the data obtained, CXCL14 may be considered a new neuroprotective factor.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank the Dr. Johnson and Dr. Hara provides the conditional HIF-1α knockout mice and CXCL14-/- mice, respectively. This work was supported by research grants from the Chen-Han Foundation for Education; Taiwan Ministry of Health and Welfare Clinical Trial and Research Center of Excellence (MOHW105-TDU-B-212-133019), China Medical University Hospital; Academia Sinica Taiwan Biobank, Stroke Biosignature Project (BM10501010037); NRPB Stroke Clinical Trial Consortium (MOST 105-2325-B-039-005), Tseng-Lien Lin Foundation, Taichung, Taiwan, Taiwan Brain Disease Foundation, Taipei, Taiwan, and Katsuzo and Kiyo Aoshima Memorial Funds, Japan; China Medical University and Hospital (CMU104-S10, CMU104-S-15-03, DMR-104-054), and Taiwan's National Science Council (NSC97-2314-B-039-036-MY3, NSC99-2632-B-039-001-MY3, NSC100-2314-B-039-002-MY3, NSC 101-2321-B-039-004; NSC101-2321-B-039-006; NSC101-2321-B-039-008; NSC102-2325-B-039-006; NSC102-2325-B-039-001; MOST 105-2314-B-039-011 -MY3).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lambertsen KL, Biber K, Finsen B. Inflammatory cytokines in experimental and human stroke. J Cereb Blood Flow Metab. 2012;32:1677-98
2. Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87:779-89
3. Schaerli P, Willimann K, Ebert LM, Walz A, Moser B. Cutaneous CXCL14 targets blood precursors to epidermal niches for Langerhans cell differentiation. Immunity. 2005;23:331-42
4. Huising MO, van der Meulen T, Flik G, Verburg-van Kemenade BM. Three novel carp CXC chemokines are expressed early in ontogeny and at nonimmune sites. Eur J Biochem. 2004;271:4094-106
5. Salogni L, Musso T, Bosisio D, Mirolo M, Jala VR, Haribabu B. et al. Activin A induces dendritic cell migration through the polarized release of CXC chemokine ligands 12 and 14. Blood. 2009;113:5848-56
6. Starnes T, Rasila KK, Robertson MJ, Brahmi Z, Dahl R, Christopherson K. et al. The chemokine CXCL14 (BRAK) stimulates activated NK cell migration: implications for the downregulation of CXCL14 in malignancy. Exp Hematol. 2006;34:1101-5
7. Augsten M, Hagglof C, Olsson E, Stolz C, Tsagozis P, Levchenko T. et al. CXCL14 is an autocrine growth factor for fibroblasts and acts as a multi-modal stimulator of prostate tumor growth. Proc Natl Acad Sci U S A. 2009;106:3414-9
8. Meisel C, Schwab JM, Prass K, Meisel A, Dirnagl U. Central nervous system injury-induced immune deficiency syndrome. Nat Rev Neurosci. 2005;6:775-86
9. Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA. et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40:1849-57
10. Yilmaz A, Fuchs T, Dietel B, Altendorf R, Cicha I, Stumpf C. et al. Transient decrease in circulating dendritic cell precursors after acute stroke: potential recruitment into the brain. Clin Sci (Lond). 2010;118:147-57
11. Zhang Z, Yan J, Chang Y, ShiDu Yan S, Shi H. Hypoxia inducible factor-1 as a target for neurodegenerative diseases. Curr Med Chem. 2011;18:4335-43
12. Shi H. Hypoxia inducible factor 1 as a therapeutic target in ischemic stroke. Curr Med Chem. 2009;16:4593-600
13. Stumm RK, Rummel J, Junker V, Culmsee C, Pfeiffer M, Krieglstein J. et al. A dual role for the SDF-1/CXCR4 chemokine receptor system in adult brain: isoform-selective regulation of SDF-1 expression modulates CXCR4-dependent neuronal plasticity and cerebral leukocyte recruitment after focal ischemia. J Neurosci. 2002;22:5865-78
14. Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME. et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858-64
15. Lazarini F, Tham TN, Casanova P, Arenzana-Seisdedos F, Dubois-Dalcq M. Role of the alpha-chemokine stromal cell-derived factor (SDF-1) in the developing and mature central nervous system. Glia. 2003;42:139-48
16. Klein RS, Rubin JB. Immune and nervous system CXCL12 and CXCR4: parallel roles in patterning and plasticity. Trends Immunol. 2004;25:306-14
17. Huising MO, Stet RJ, Kruiswijk CP, Savelkoul HF, Lidy Verburg-van Kemenade BM. Molecular evolution of CXC chemokines: extant CXC chemokines originate from the CNS. Trends Immunol. 2003;24:307-13
18. Huang Y, Bozulic LD, Miller T, Xu H, Hussain LR, Ildstad ST. CD8α+ plasmacytoid precursor DCs induce antigen-specific regulatory T cells that enhance HSC engraftment in vivo. Blood. 2011;117:2494-505
19. Zou T, Caton AJ, Koretzky GA, Kambayashi T. Dendritic cells induce regulatory T cell proliferation through antigen-dependent and -independent interactions. J Immunol. 2010;185:2790-9
20. Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S. et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15:192-9
21. Shyu WC, Liu DD, Lin SZ, Li WW, Su CY, Chang YC. et al. Implantation of olfactory ensheathing cells promotes neuroplasticity in murine models of stroke. J Clin Invest. 2008;118:2482-95
22. Ryan HE, Poloni M, McNulty W, Elson D, Gassmann M, Arbeit JM. et al. Hypoxia-inducible factor-1alpha is a positive factor in solid tumor growth. Cancer Res. 2000;60:4010-5
23. Lee SD, Lai TW, Lin SZ, Lin CH, Hsu YH, Li CY. et al. Role of stress-inducible protein-1 in recruitment of bone marrow derived cells into the ischemic brains. EMBO Mol Med. 2013;5:1227-46
24. Ricker JL, Chen Z, Yang XP, Pribluda VS, Swartz GM, Van Waes C. 2-methoxyestradiol inhibits hypoxia-inducible factor 1alpha, tumor growth, and angiogenesis and augments paclitaxel efficacy in head and neck squamous cell carcinoma. Clin Cancer Res. 2004;10:8665-73
25. Dai Y, Xu M, Wang Y, Pasha Z, Li T, Ashraf M. HIF-1alpha induced-VEGF overexpression in bone marrow stem cells protects cardiomyocytes against ischemia. J Mol Cell Cardiol. 2007;42:1036-44
26. Shyu WC, Lin SZ, Chiang MF, Chen DC, Su CY, Wang HJ. et al. Secretoneurin promotes neuroprotection and neuronal plasticity via the Jak2/Stat3 pathway in murine models of stroke. J Clin Invest. 2008;118:133-48
27. Ben-Shoshan J, Schwartz S, Luboshits G, Maysel-Auslender S, Barzelay A, Polak-Charcon S. et al. Constitutive expression of HIF-1alpha and HIF-2alpha in bone marrow stromal cells differentially promotes their proangiogenic properties. Stem Cells. 2008;26:2634-43
28. Shyu WC, Lin SZ, Yang HI, Tzeng YS, Pang CY, Yen PS. et al. Functional recovery of stroke rats induced by granulocyte colony-stimulating factor-stimulated stem cells. Circulation. 2004;110:1847-54
29. Gu Z, Cui J, Brown S, Fridman R, Mobashery S, Strongin AY. et al. A highly specific inhibitor of matrix metalloproteinase-9 rescues laminin from proteolysis and neurons from apoptosis in transient focal cerebral ischemia. J Neurosci. 2005;25:6401-8
30. Boni LJ, Ploug KB, Olesen J, Jansen-Olesen I, Gupta S. The in vivo effect of VIP, PACAP-38 and PACAP-27 and mRNA expression of their receptors in rat middle meningeal artery. Cephalalgia. 2009;29:837-47
31. Sadhu C, Ting HJ, Lipsky B, Hensley K, Garcia-Martinez LF, Simon SI. et al. CD11c/CD18: novel ligands and a role in delayed-type hypersensitivity. J Leukoc Biol. 2007;81:1395-403
32. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D. et al. Stability of the regulatory T cell lineage in vivo. Science. 2010;329:1667-71
33. Matsumura A, Mizokawa S, Tanaka M, Wada Y, Nozaki S, Nakamura F. et al. Assessment of microPET performance in analyzing the rat brain under different types of anesthesia: comparison between quantitative data obtained with microPET and ex vivo autoradiography. Neuroimage. 2003;20:2040-50
34. Hamacher K, Coenen HH, Stocklin G. Efficient stereospecific synthesis of no-carrier-added 2-[18F]-fluoro-2-deoxy-D-glucose using aminopolyether supported nucleophilic substitution. J Nucl Med. 1986;27:235-8
35. Carmichael ST, Tatsukawa K, Katsman D, Tsuyuguchi N, Kornblum HI. Evolution of diaschisis in a focal stroke model. Stroke. 2004;35:758-63
36. Kornblum HI, Araujo DM, Annala AJ, Tatsukawa KJ, Phelps ME, Cherry SR. In vivo imaging of neuronal activation and plasticity in the rat brain by high resolution positron emission tomography (microPET). Nat Biotechnol. 2000;18:655-60
37. Brownell AL, Livni E, Galpern W, Isacson O. In vivo PET imaging in rat of dopamine terminals reveals functional neural transplants. Ann Neurol. 1998;43:387-90
38. Li Y, Lu Z, Keogh CL, Yu SP, Wei L. Erythropoietin-induced neurovascular protection, angiogenesis, and cerebral blood flow restoration after focal ischemia in mice. J Cereb Blood Flow Metab. 2007;27:1043-54
39. Shang Y, Wu Y, Yao S, Wang X, Feng D, Yang W. Protective effect of erythropoietin against ketamine-induced apoptosis in cultured rat cortical neurons: involvement of PI3K/Akt and GSK-3 beta pathway. Apoptosis. 2007;12:2187-95
40. Bjorck P. Isolation and characterization of plasmacytoid dendritic cells from Flt3 ligand and granulocyte-macrophage colony-stimulating factor-treated mice. Blood. 2001;98:3520-6
41. Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T. et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964-7
42. Bontkes HJ, De Gruijl TD, Schuurhuis GJ, Scheper RJ, Meijer CJ, Hooijberg E. Expansion of dendritic cell precursors from human CD34(+) progenitor cells isolated from healthy donor blood; growth factor combination determines proliferation rate and functional outcome. J Leukoc Biol. 2002;72:321-9
43. Wilson HL, O'Neill HC. Dynamics of dendritic cell development from precursors maintained in stroma-dependent long-term cultures. Immunol Cell Biol. 2003;81:144-51
44. Shalak V, Kaminska M, Mitnacht-Kraus R, Vandenabeele P, Clauss M, Mirande M. The EMAPII cytokine is released from the mammalian multisynthetase complex after cleavage of its p43/proEMAPII component. J Biol Chem. 2001;276:23769-76
45. Shurin GV, Ferris RL, Tourkova IL, Perez L, Lokshin A, Balkir L. et al. Loss of new chemokine CXCL14 in tumor tissue is associated with low infiltration by dendritic cells (DC), while restoration of human CXCL14 expression in tumor cells causes attraction of DC both in vitro and in vivo. J Immunol. 2005;174:5490-8
46. Kuang H, Chen Q, Zhang Y, Zhang L, Peng H, Ning L. et al. The cytokine gene CXCL14 restricts human trophoblast cell invasion by suppressing gelatinase activity. Endocrinology. 2009;150:5596-605
47. Yao PL, Lin YC, Richburg JH. TNF alpha-mediated disruption of spermatogenesis in response to Sertoli cell injury in rodents is partially regulated by MMP2. Biol Reprod. 2009;80:581-9
48. Plett PA, Frankovitz SM, Wolber FM, Abonour R, Orschell-Traycoff CM. Treatment of circulating CD34(+) cells with SDF-1alpha or anti-CXCR4 antibody enhances migration and NOD/SCID repopulating potential. Exp Hematol. 2002;30:1061-9
49. Salmon P, Oberholzer J, Occhiodoro T, Morel P, Lou J, Trono D. Reversible immortalization of human primary cells by lentivector-mediated transfer of specific genes. Mol Ther. 2000;2:404-14
50. Lin CH, Lee HT, Lee SD, Lee W, Cho CW, Lin SZ. et al. Role of HIF-1alpha-activated Epac1 on HSC-mediated neuroplasticity in stroke model. Neurobiol Dis. 2013;58:76-91
51. Zennou V, Petit C, Guetard D, Nerhbass U, Montagnier L, Charneau P. HIV-1 genome nuclear import is mediated by a central DNA flap. Cell. 2000;101:173-85
52. Qin G, Kishore R, Dolan CM, Silver M, Wecker A, Luedemann CN. et al. Cell cycle regulator E2F1 modulates angiogenesis via p53-dependent transcriptional control of VEGF. Proc Natl Acad Sci U S A. 2006;103:11015-20
53. Hess DC, Hill WD, Martin-Studdard A, Carroll J, Brailer J, Carothers J. Bone marrow as a source of endothelial cells and NeuN-expressing cells After stroke. Stroke. 2002;33:1362-8
54. Nara N, Nakayama Y, Okamoto S, Tamura H, Kiyono M, Muraoka M. et al. Disruption of CXC motif chemokine ligand-14 in mice ameliorates obesity-induced insulin resistance. J Biol Chem. 2007;282:30794-803
55. Wetzel M, Li L, Harms KM, Roitbak T, Ventura PB, Rosenberg GA. et al. Tissue inhibitor of metalloproteinases-3 facilitates Fas-mediated neuronal cell death following mild ischemia. Cell Death Differ. 2008;15:143-51
56. Ivanovic Z, Bartolozzi B, Bernabei PA, Cipolleschi MG, Rovida E, Milenkovic P. et al. Incubation of murine bone marrow cells in hypoxia ensures the maintenance of marrow-repopulating ability together with the expansion of committed progenitors. Br J Haematol. 2000;108:424-9
57. Ponnusamy S, Alderson NL, Hama H, Bielawski J, Jiang JC, Bhandari R. et al. Regulation of telomere length by fatty acid elongase 3 in yeast. Involvement of inositol phosphate metabolism and Ku70/80 function. J Biol Chem. 2008;283:27514-24
58. Kurth I, Willimann K, Schaerli P, Hunziker T, Clark-Lewis I, Moser B. Monocyte selectivity and tissue localization suggests a role for breast and kidney-expressed chemokine (BRAK) in macrophage development. J Exp Med. 2001;194:855-61
59. Takahashi M, Takahashi Y, Takahashi K, Zolotaryov FN, Hong KS, Iida K. et al. CXCL14 enhances insulin-dependent glucose uptake in adipocytes and is related to high-fat diet-induced obesity. Biochem Biophys Res Commun. 2007;364:1037-42
60. Pelicano H, Lu W, Zhou Y, Zhang W, Chen Z, Hu Y. et al. Mitochondrial dysfunction and reactive oxygen species imbalance promote breast cancer cell motility through a CXCL14-mediated mechanism. Cancer Res. 2009;69:2375-83
61. Peterson FC, Thorpe JA, Harder AG, Volkman BF, Schwarze SR. Structural determinants involved in the regulation of CXCL14/BRAK expression by the 26 S proteasome. J Mol Biol. 2006;363:813-22
62. Vadasz C, Smiley JF, Figarsky K, Saito M, Toth R, Gyetvai BM. et al. Mesencephalic dopamine neuron number and tyrosine hydroxylase content: Genetic control and candidate genes. Neuroscience. 2007;149:561-72
63. Lu P, Jones LL, Snyder EY, Tuszynski MH. Neural stem cells constitutively secrete neurotrophic factors and promote extensive host axonal growth after spinal cord injury. Exp Neurol. 2003;181:115-29
64. Sallusto F, Lanzavecchia A. Understanding dendritic cell and T-lymphocyte traffic through the analysis of chemokine receptor expression. Immunol Rev. 2000;177:134-40
65. Larregina AT, Morelli AE, Spencer LA, Logar AJ, Watkins SC, Thomson AW. et al. Dermal-resident CD14+ cells differentiate into Langerhans cells. Nat Immunol. 2001;2:1151-8
66. Schnurr M, Toy T, Shin A, Hartmann G, Rothenfusser S, Soellner J. et al. Role of adenosine receptors in regulating chemotaxis and cytokine production of plasmacytoid dendritic cells. Blood. 2004;103:1391-7
67. Muller WA. Leukocyte-endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol. 2003;24:327-34
68. Muller WA. Mechanisms of transendothelial migration of leukocytes. Circ Res. 2009;105:223-30
69. Ferrero E, Belloni D, Contini P, Foglieni C, Ferrero ME, Fabbri M. et al. Transendothelial migration leads to protection from starvation-induced apoptosis in CD34+CD14+ circulating precursors: evidence for PECAM-1 involvement through Akt/PKB activation. Blood. 2003;101:186-93
70. Torzicky M, Viznerova P, Richter S, Strobl H, Scheinecker C, Foedinger D. et al. Platelet endothelial cell adhesion molecule-1 (PECAM-1/CD31) and CD99 are critical in lymphatic transmigration of human dendritic cells. J Invest Dermatol. 2012;132:1149-57
71. de la Rosa G, Longo N, Rodriguez-Fernandez JL, Puig-Kroger A, Pineda A, Corbi AL. et al. Migration of human blood dendritic cells across endothelial cell monolayers: adhesion molecules and chemokines involved in subset-specific transmigration. J Leukoc Biol. 2003;73:639-49
72. Kaneider NC, Kaser A, Dunzendorfer S, Tilg H, Wiedermann CJ. Sphingosine kinase-dependent migration of immature dendritic cells in response to neurotoxic prion protein fragment. J Virol. 2003;77:5535-9
73. Viegas P, Chaverot N, Enslen H, Perriere N, Couraud PO, Cazaubon S. Junctional expression of the prion protein PrPC by brain endothelial cells: a role in trans-endothelial migration of human monocytes. J Cell Sci. 2006;119:4634-43
74. Opdenakker G, Van den Steen PE, Van Damme J. Gelatinase B: a tuner and amplifier of immune functions. Trends Immunol. 2001;22:571-9
75. Offner H, Subramanian S, Parker SM, Wang C, Afentoulis ME, Lewis A. et al. Splenic atrophy in experimental stroke is accompanied by increased regulatory T cells and circulating macrophages. J Immunol. 2006;176:6523-31
76. Gee JM, Zierath D, Hadwin J, Savos A, Kalil A, Thullbery M. et al. Long term immunologic consequences of experimental stroke and mucosal tolerance. Exp Transl Stroke Med. 2009;1:3
77. Ito T, Yang M, Wang YH, Lande R, Gregorio J, Perng OA. et al. Plasmacytoid dendritic cells prime IL-10-producing T regulatory cells by inducible costimulator ligand. J Exp Med. 2007;204:105-15
78. Rutella S, Bonanno G, Pierelli L, Mariotti A, Capoluongo E, Contemi AM. et al. Granulocyte colony-stimulating factor promotes the generation of regulatory DC through induction of IL-10 and IFN-alpha. Eur J Immunol. 2004;34:1291-302
79. Levings MK, Sangregorio R, Galbiati F, Squadrone S, de Waal Malefyt R, Roncarolo MG. IFN-alpha and IL-10 induce the differentiation of human type 1 T regulatory cells. J Immunol. 2001;166:5530-9
80. Manwani B, Liu F, Scranton V, Hammond MD, Sansing LH, McCullough LD. Differential effects of aging and sex on stroke induced inflammation across the lifespan. Exp Neurol. 2013;249:120-31
81. Barbaria EM, Kohl B, Buhren BA, Hasenpusch-Theil K, Kruse F, Kury P. et al. The alpha-chemokine CXCL14 is up-regulated in the sciatic nerve of a mouse model of Charcot-Marie-Tooth disease type 1A and alters myelin gene expression in cultured Schwann cells. Neurobiol Dis. 2009;33:448-58
Author contact
![]() Corresponding author: Woei-Cherng Shyu, MD. PhD. (shyu9423com) and Chia-Hung Hsieh, PhD. (chhsiehcmucmu.edu.tw).
Corresponding author: Woei-Cherng Shyu, MD. PhD. (shyu9423com) and Chia-Hung Hsieh, PhD. (chhsiehcmucmu.edu.tw).