Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(4):805-813. doi:10.7150/thno.17117 This issue Cite
Research Paper
Preclinical Melanoma Imaging with 68Ga-Labeled α-Melanocyte-Stimulating Hormone Derivatives Using PET
Chengcheng Zhang1, Zhengxing Zhang1, Kuo-Shyan Lin1,2, Jinhe Pan1, Iulia Dude1, Navjit Hundal-Jabal1, Nadine Colpo1, François Bénard1,2, ![]()
1. Department of Molecular Oncology, BC Cancer Agency, Vancouver, BC, Canada
2. Department of Radiology, University of British Columbia, Vancouver, BC, Canada
Received 2016-8-5; Accepted 2016-11-10; Published 2017-2-8
Citation:
Zhang C, Zhang Z, Lin KS, Pan J, Dude I, Hundal-Jabal N, Colpo N, Bénard F. Preclinical Melanoma Imaging with 68Ga-Labeled α-Melanocyte-Stimulating Hormone Derivatives Using PET. Theranostics 2017; 7(4):805-813. doi:10.7150/thno.17117. https://www.thno.org/v07p0805.htm
Other stylesAbstract

It is estimated that melanoma accounted for 76,380 new cases and 10,130 deaths in the United States in 2016. The melanocortin 1 receptor (MC1R) is highly expressed in the vast majority of melanomas, which makes it an attractive target for molecular imaging and radionuclide therapy. Lactam bridge-cyclized α-melanocyte-stimulating hormone (Ac-Nle4-cyclo[Asp5-His-D-Phe7-Arg-Trp-Lys10]-NH2, or Nle-CycMSHhex) analogues have been successfully developed and studied for MC1R-targeted imaging, predominantly with single-photon emission computed tomography (SPECT). The goal of this study was to design and evaluate novel peptides for melanoma imaging with positron emission tomography (PET). We designed and synthesized three peptides, DOTA-PEG2-Nle-CycMSHhex (CCZ01047), DOTA-4-amino-(1-carboxymethyl) piperidine (Pip)-Nle-CycMSHhex (CCZ01048), and DOTA-Pip-Pip-Nle-CycMSHhex (CCZ01056). All three peptides exhibited high binding affinity to MC1R with sub-nanomolar Ki values, rapid internalization into B16F10 melanoma cells and high in vivo stability with more than 93% remaining intact at 15 min post-injection (p.i.) in blood plasma. All three 68Ga-labeled tracers produced high contrast PET images in C57BL/6J mice bearing B16F10 tumors, and their respective tumor uptakes were 8.0 ± 3.0, 12.3 ± 3.3, and 6.5 ± 1.4 %ID/g at 1 h p.i. Minimal normal organ activity was observed at 1 h p.i., except for kidneys (5.1 ± 1.4, 4.7 ± 0.5, and 6.2 ± 2.0 %ID/g, respectively), and thyroid (4.1 ± 0.6 %ID/g for CCZ01047 and 2.4 ± 0.6 %ID/g for CCZ01048). Due to high accumulation at tumor sites and rapid background clearance of 68Ga-CCZ01048, we further evaluated it at 2 h p.i., and a tumor uptake of 21.9 ± 4.6 %ID/g was observed, with background activity further decreased. Exceptional image contrast was also achieved, i.e. tumor-to-blood, tumor-to-muscle, tumor-to-bone and tumor-to-kidney ratios were 96.4 ± 13.9, 210.9 ± 20.9, 39.6 ± 11.9 and 4.0 ± 0.9, respectively. A blocking study was also performed by co-injection of excess amount of non-radioactive Ga-coupled of CCZ01048, which confirmed that the tumor uptake was MC1R mediated. In conclusion, the introduction of a cationic Pip linker to Nle-CycMSHhex, CCZ01048, not only improved tumor uptake, but also generated high tumor-to-normal tissue contrast with PET imaging in a preclinical melanoma model. Therefore, CCZ01048 is a promising candidate for PET imaging of melanoma, and potentially as a theranostic agent for radionuclide therapy of melanoma when labeled with α or β emitters.
Keywords: Melanocortin 1 receptor, Melanoma, α-melanocyte-stimulating hormone, PET imaging, Ga-68.
Introduction
It is estimated that melanoma accounted for 76,380 new cases and 10,130 deaths in the United States in 2016 [1]. Despite the fact that novel therapies such as immune checkpoint inhibitors [2], BRAF inhibitors [3], adoptive T-cell therapies [4] and oncolytic viruses [5] have shown promising results, the majority of patients with advanced metastatic melanoma will eventually die from disease progression. Therefore, accurate staging remains a key aspect in managing melanoma. With high sensitivity and spatial resolution, positron emission tomography (PET) has emerged as a powerful imaging technique for such purpose. While 2-[18F]Fluorodeoxyglucose PET coupled with computed tomography (CT) is highly accurate in detecting metastatic disease in cutaneous melanomas [6], this modality lacks sensitivity for early nodal metastases, and liver metastases in uveal melanomas [7, 8].
The melanocortin 1 receptor (MC1R) is highly expressed in malignant melanomas. It has been shown to be expressed in 83% of melanoma cell lines, all tissue samples from primary and metastatic melanomas (n = 26) [9], and 95% of uveal melanomas [10]. In addition, low levels of MC1R expression in normal tissues make this protein an attractive target for molecular imaging. Indeed, radiolabeled peptide ligands based on the sequence of α-melanocyte-stimulating hormone (α-MSH) have been studied extensively for MC1R-targeted imaging, summarized in a review article [11]. α-MSH, a tridecapeptide, is prone to proteolysis by peptidase in vivo. Unnatural amino acid substitutions at position 4 with Nle and position 7 with D-Phe not only increased in vivo stability, but also improved binding affinity to MC1R. Furthermore, His-Phe-Arg-Trp has been identified as a key sequence for biological activity and binding with MC1R. With this core sequence, cyclized α-MSH analogues have been designed, which further improved both stability and affinity. The most successful analogues to date are based on lactam bridge-cyclized α-MSH (Ac-Nle4-cyclo[Asp5-His-D-Phe7-Arg-Trp-Lys10]-NH2, or Nle-CycMSHhex), which have shown promising results in animal models predominately with single-photon emission computed tomography (SPECT) over the past few years [12-20].
The goal of this study was to develop and optimize Nle-CycMSHhex-based agents and evaluate their potential for PET imaging of melanoma. We designed a series of imaging probes by attaching a PEG2 linker, a cationic linker (4-amino-(1-carboxymethyl) piperidine, or Pip), or dual Pip linkers to Nle4. A chelator, 1, 4, 7, 10-tetraazacyclododecane-1, 4, 7, 10-tetraacetic acid (DOTA) was then coupled to enable radiometal coupling. In this paper, we report the synthesis of these peptides, namely CCZ01047, CCZ01048 and CCZ01056 respectively, radiolabeling with 68Ga, in vitro receptor binding and internalization assay, and in vivo biodistribution and PET imaging results in a preclinical melanoma model.
Materials and Methods
Peptide synthesis
Solid phase peptide synthesis was performed via standard Fmoc chemistry on an Endeavor 90 peptide synthesizer (Aapptec). Fmoc-Rink-Amide-MBHA resin was swelled in dichloromethane (DCM), and the Fmoc protecting group was removed by treating the resin with 20% piperidine in dimethylformamide (DMF). Fmoc-protected amino acids (3 equivalents), Fmoc-Lys(Mtt)-OH, Fmoc-Trp(Boc)-OH, Fmoc-Arg(Pbf)-OH, Fmoc-D-Phe-OH, Fmoc-His(Trt)-OH, and Fmoc-Asp(O-2-PhiPr)-OH, were coupled to the resin in presence of HBTU (3 equivalents), HOBt (3 equivalents) and DIEA (6 equivalents). Before deprotection of the Fmoc on the Asp, the Mtt group on the Lys and the O-2-PhiPr group on the Asp were removed by 2.5% trifluoroacetic acid (TFA). Subsequently, the Lys and the Asp were cyclized in presence of benzotriazole-1-yl-oxy-trispyrrolidinophosphonium hexafluorophosphate (PyBOP, 4 equivalents) and DIEA (4 equivalents). Then, the Fmoc protecting group was removed, and Fmoc-Nle-OH was coupled, and the Fmoc was removed. For CCZ01047, CCZ01048 and CCZ01056, Fmoc-PEG2-OH, Fmoc-Pip-OH, and two sequential Fmoc-Pip-OH linkers were coupled, respectively. Finally, the last Fmoc was removed, and the DOTA chelator (3 equivalents) was coupled, which was pre-activated with N-hydroxysuccinimide (3.6 equivalents) and N,N'-Dicyclohexylcarbodiimide (DCC, 3 equivalents). The peptide was simultaneously deprotected and cleaved from the resin by incubating with 90/2.5/2.5/2.5/2.5 TFA/phenol/H2O/triisopropylsilane/1, 2-ethanedithiol for 3 h at room temperature. The solution was filtered and the peptide was precipitated in diethyl ether, and purified on a semi-preparative column using 23% acetonitrile containing 0.1% TFA at a flow rate of 4.5 mL/min using high-performance liquid chromatography (HPLC, Agilent). The HPLC eluate was collected and lyophilized, and the purity of the peptides was > 97%. Mass analysis was performed on a 5600 mass spectrometer (AB/Sciex). For gallium complexation, the purified peptide and GaCl3 (5 equivalents) in sodium acetate buffer (0.1 M, pH 4.0) was incubated at 80°C for 15 min. The mixture was purified by HPLC using the same condition as described above.
Radiochemistry
68Ga was obtained from a 68Ge/68Ga generator (iThemba Labs), by eluting with 0.6 M HCl. The eluate was mixed with concentrated metal-free 34-37% HCl. The mixture was passed through a DGA resin column (Eichrom), and washed by 5 M HCl. After the column was air-dried, 68Ga was eluted off with deionized water. The purified 68Ga solution was mixed with HEPES buffer (2 M, pH 5.0) and the DOTA conjugated peptides. The radiolabeling reaction was carried out under microwave heating for 1 min as described previously [21]. HPLC purification was then performed to remove free 68Ga, and to separate radiolabeled product from precursor to ensure high specific activity of the final product for preclinical imaging studies. The reaction mixture was separated by HPLC using a semi-preparative column eluted with 24% acetonitrile containing 0.1% TFA for 68Ga-CCZ01047, 21% acetonitrile containing 0.1% TFA for 68Ga-CCZ01048 and 25% acetonitrile in phosphate-buffered saline (PBS) solution for 68Ga-CCZ01056, at a flow rate of 4.5 mL/min. The retention times were 17.6, 24.4 and 18.7 min, respectively. The collected radiolabeled peptides were diluted with deionized water and retained on C18 SepPak cartridges (Waters) to remove acetonitrile and TFA/PBS. The purified peptides were eluted with ethanol and diluted with 0.9% NaCl saline. Quality control was performed on analytical column eluted with 24% acetonitrile containing 0.1% TFA for 68Ga-CCZ01047, 23% acetonitrile containing 0.1% TFA for 68Ga-CCZ01048 and 25% acetonitrile in PBS solution for 68Ga-CCZ01056, at a flow rate of 2 mL/min. The retention times were 11.1, 7.0 and 6.7 min, respectively. Peptides with radiochemical purity of > 95% were used for subsequent experiments.
In vivo stability in mouse plasma
In vivo stability was characterized according to previously published procedures [22]. Briefly, mice were sedated with 2% isoflurane, and 8-12 MBq of 68Ga-labeled peptide was injected via tail vein. At 5 or 15 min p.i., mice were euthanized and the blood was promptly withdrawn, which was mixed with equal volume of acetonitrile. After centrifugation for 15 min, the supernatant was collected and analyzed using radio-HPLC.
LogD7.4 measurements
LogD7.4 values of 68Ga-labeled peptides were assessed using published procedures [23].
Cell culture
The B16F10 melanoma cell line (Mus musculus) used in the tumor model was obtained commercially from ATTC (CRL-6475). The cell line was confirmed pathogen-free by the IMPACT 1 mouse profile test (IDEXX BioResearch). Cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM, StemCell Technologies) supplemented by 10% FBS, 100 U/mL penicillin and 100 μg/mL streptomycin at 37°C in a humidified incubator containing 5% CO2. Cells grown to approximately 90% confluence were washed with sterile 1× PBS (pH 7.4), followed by trypsinization.
Receptor binding assays
In vitro competitive binding assays were performed using B16F10 cells. 500,000 cells/well were seeded onto a 24 well poly-D-lysine coated plate (Corning) overnight. Growth media was removed, and reaction buffer containing 4.8 mg/mL HEPES, 100 U/mL penicillin, 1,000 µg/mL streptomycin and 2 mg/mL BSA was added, and allowed to incubate with cells at 37°C for at least an hour. Non-radioactive Ga-coupled peptides and 125I-[Nle4, D-Phe7]-alpha-MSH (PerkinElmer) were added to each well. The binding of 125I-[Nle4, D-Phe7]-alpha-MSH was competed by the ligand of interest at increasing concentrations from 0.5 pM to 5 µM. The reaction mixture was incubated at 25°C with gentle agitation for 1 h. After the incubation, the reaction mixture was removed, and cells were washed with ice-cold PBS twice. 0.25% Trypsin solution was used to harvest the cells and radioactivity was measured on a WIZARD 2480 gamma counter (PerkinElmer).
Internalization assay
In vitro internalization assays were performed using B16F10 cells on 24 well poly-D-lysine coated plate (Corning) according to previously published procedures [24]. Briefly, approximately 500,000 cpm of 68Ga-labeled peptide was added to the cells and allowed to incubate for 30, 60, 90 and 120 min at 37°C. After the incubation, cells were washed with ice-cold PBS. Two acid washes (0.2M Acetic acid, 0.5M NaCl, pH 2.6) were performed and collected to measure the membrane-bound fraction. The cells were subsequently trypsinized and collected to measure the internalized fraction. These fractions were counted for radioactivity using a WIZARD 2480 gamma counter (PerkinElmer).
Tumor implantation
All animal experiments were conducted according to the guidelines established by Canadian Council on Animal Care and approved by Animal Ethics Committee of the University of British Columbia. Mice were housed under pathogen-free conditions and kept on twelve-hour light and dark cycles in the Animal Resource Centre, BC Cancer Research Centre, Vancouver, Canada. PET imaging and biodistribution studies were performed using male C57BL/6J mice. For tumor implantation, mice were anesthetized by inhalation with 2% isoflurane in 2.0 L/min of oxygen, and 1×106 B16F10 cells were implanted subcutaneously on the right back at the level of the forelimbs. Mice were imaged or used in biodistribution studies once the tumor grew to reach 8-10 mm in diameter in 8-10 days.
Preclinical PET/CT imaging
PET/CT imaging studies were carried out on a microPET/CT scanner (Inveon, Siemens) following the same procedures as previously published [21]. Briefly, for static PET scans, each tumor bearing mouse was injected with 4-6 MBq of 68Ga-labeled peptides for the one hour time point, or 8-10 MBq for the two hour time point, via the caudal lateral tail vein under isoflurane sedation. After injection, the mice were allowed to recover and roam freely in their cages. After 1 or 2 hours, the mice were sedated again and positioned in the scanner. A baseline CT scan was obtained for localization and attenuation correction. This was followed by a 10 min static PET scan. For dynamic PET scans, a 60 min list-mode acquisition was started at the time of intravenous injection with 4-6 MBq of 68Ga-labeled peptides following a baseline CT scan. The mice were kept warm by a heating pad during acquisition. The mice were euthanized using CO2 inhalation after static PET imaging followed by biodistribution. The PET images were reconstructed using the ordered subset expectation maximization and maximum a posteriori algorithm (OSEM3D/MAP), using 2 OSEM3D iterations followed by 18 MAP iterations, with a requested resolution of 1.5 mm.
Biodistribution studies
Mice not used for imaging studies were anesthetized by 2% isoflurane inhalation, and injected with 1-2 MBq of 68Ga-labeled peptides. For blocking studies, 100 μg of non-radioactive Ga-coupled peptides was co-injected with the radioactive compound. After injection, the mice were allowed to recover and roam freely in their cages, and euthanized by CO2 inhalation 1 h or 2 h later. Blood was promptly withdrawn, and the organs of interest were harvested, rinsed with 1× PBS (pH 7.4), and blotted dry. Each organ was weighed and the radioactivity of the collected tissue was measured using a WIZARD 2480 (PerkinElmer) or a Cobra II gamma counter (Packard), normalized to the injected dose using a standard curve and expressed as the percentage of the injected dose per gram of tissue (%ID/g).
Autoradiography
Mice were sedated by 2% isoflurane inhalation, and injected with 6-8 MBq of 68Ga-CCZ01048 with or without co-injection of 100 μg of non-radioactive Ga-coupled peptides. At 1 h p.i., mice were euthanized, and tumor, muscle and thyroid were harvested and frozen in isopentane/dry-ice bath. Autoradiography was performed according to previously published procedures [24]. Ten μm-thick sections were acquired for the organs of interest using a cryostat (Leica), thaw-mounted onto Superfrost Plus microscope slides (Fisherbrand). These sections along with a set of 68Ga-peptide standards, were exposed to a phosphor screen overnight and imaged on a Typhoon FLA 9500 scanner (GE Healthcare).
Statistical analysis
Data analysis was performed using GraphPad Prism 6.0h. Multiple t tests were performed for all organs in the biodistribution studies; multiple comparisons were corrected using the Holm-Sidak method. Outliers were removed using the ROUT method with Q = 1%. The difference was considered statistically significant when the adjusted p value was < 0.05. Blood half-life of radiotracers was calculated using non-linear regression assuming one phase decay.
Results
Chemical properties and in vitro binding affinity
All three α-MSH analogues, CCZ01047, CCZ01048 and CCZ01056, were prepared with high purity (> 97%, Figure S1), and the molecular weight was verified by mass spectrometry (Table 1, Figure S2). The chemical structures of the tracers are shown in Figure 1. The inhibition constant (Ki) of these α -MSH analogues to MC1R was measured using competitive binding assays. All three peptides showed sub-nanomolar Ki (Figure 2, Table 1).
Table 1
Analytical data for α-MSH analogues and affinities for MC1R.
| Peptide | Mass calculated | Mass found | Purity (%) | Ki (nmol/L) |
|---|---|---|---|---|
| natGa-CCZ01047 | 1593.71 | 1595.12 (M+2H) | > 99% | 0.73 ± 0.08 |
| natGa-CCZ01048 | 1574.71 | 1576.12 (M+2H) | > 99% | 0.31 ± 0.06 |
| natGa-CCZ01056 | 1714.81 | 1715.69 (M+1H) | > 97% | 0.16 ± 0.10 |
Radiochemical characteristics, in vivo plasma stability and in vitro internalization
The radiochemical data for the three tracers are summarized in Table 2. The 68Ga-labeled CCZ01047, CCZ01048 and CCZ01056 were prepared with > 96% radiochemical purity in average decay-corrected radiochemical yields ranging from 50-61%. High specific activity was achieved with the average > 236.8 MBq / nmol. All three 68Ga-labeled tracers were fairly hydrophilic with average LogD7.4 values ranging from -3.14 to -3.07. Furthermore, all three tracers were shown to be considerably stable in vivo with > 93% remaining intact at 15 min p.i. in blood plasma in mice (Table 2, Figure S3). In addition, all three tracers were shown to be rapidly internalized into B16F10 cells with 36%-52% of total bound activity being internalized after 30 min incubation, and increased to 55%-62% after 120 min (Figure S4).
Figure 1
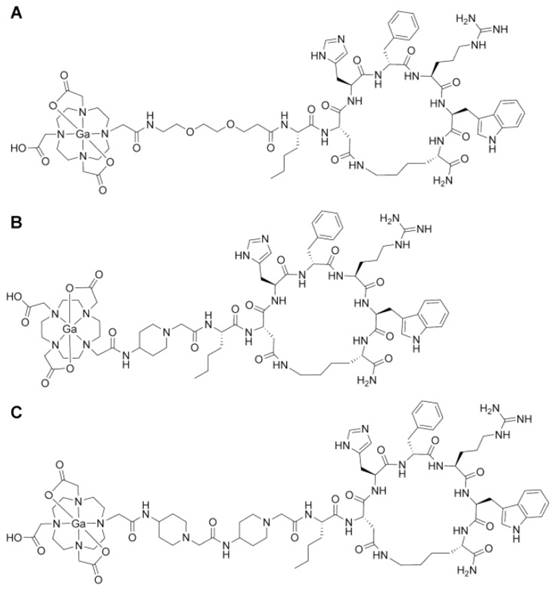
Chemical structures of gallium-labeled A. CCZ01047, B. CCZ01048, and C. CCZ01056.
Figure 2
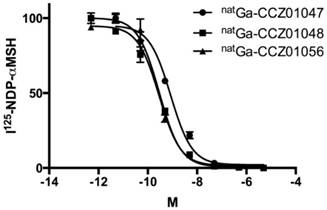
Representative competitive binding curves for the natural gallium-labeled A. CCZ01047, B. CCZ01048, and C. CCZ01056.
Table 2
Radiochemistry data for 68Ga-labeled CCZ01047, CCZ01048 and CCZ01056.
| Peptide | Radiochemical yield (%, decay-corrected) | Radiochemical purity (%) | Specific activity (MBq / nmol) | LogD7.4 | In vivo plasma stability (%) | |
|---|---|---|---|---|---|---|
| 5 min | 15min | |||||
| 68Ga-CCZ01047 | 61 ± 0.7 | 96 ± 1.4 | 288.6 ± 170.2 | -3.10 ± 0.04 | > 99 | > 98 |
| 68Ga-CCZ01048 | 58 ± 3.1 | 98 ± 0.3 | 236.8 ± 66.6 | -3.14 ± 0.04 | > 96 | > 93 |
| 68Ga-CCZ01056 | 50 ± 12.8 | 99 ± 0.1 | 270.1 ± 29.6 | -3.07 ± 0.06 | > 96 | > 94 |
Figure 3
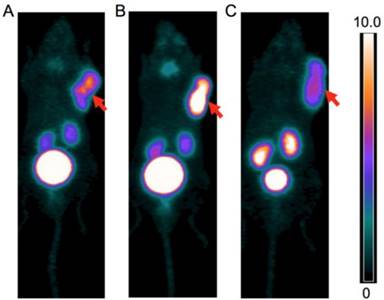
PET images of A. 68Ga-CCZ01047 B. 68Ga-CCZ01048 and C. 68Ga-CCZ01056 at 1 h p.i., in mice bearing B16F10 tumor. The scale bar unit is %ID/g. Tumors are indicated by arrows.
Figure 4
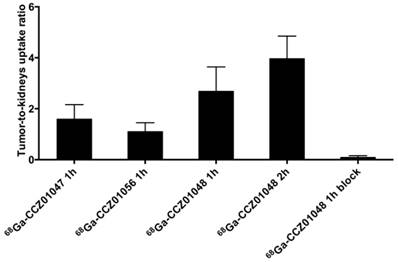
Tumor-to-kidneys uptake ratios for 68Ga-CCZ01047 and 68Ga-CCZ01056 at 1 h p.i., and 68Ga-CCZ01048 at 1 h p.i. with and without co-injection of 100 μg of non-radioactive Ga-labeled CCZ01048 and at 2 h p.i.
PET/CT imaging, biodistribution and autoradiography
For all three tracers, tumors were clearly visualized with high contrast on the PET images at 1 h p.i. (Figure 3). Biodistribution studies revealed tumor uptake at 1 h p.i. for 68Ga-labeled CCZ01047, CCZ01048 and CCZ01056 was 8.0 ± 3.0, 12.3 ± 3.3, and 6.5 ± 1.4 %ID/g, respectively (Table 3). Minimal normal organ activity was observed at 1 h p.i., except for kidney (5.1 ± 1.4, 4.7 ± 0.5, and 6.2 ± 2.0 %ID/g, respectively), and thyroid (4.1 ± 0.6 %ID/g for CCZ01047 and 2.4 ± 0.6 %ID/g for CCZ01048). Tumor-to-kidney uptake ratios were 1.6 ± 0.6, 2.7 ± 0.9 and 1.1 ± 0.3, respectively (Figure 4). Dynamic PET scans were performed to characterize the time activity curves of the three tracers. For CCZ01047 and CCZ01048, tumor uptake was sustained and increased over time; but for CCZ01056, tumor uptake slightly decreased (Figure 5). Moreover, activity clearance was much faster in normal tissues with CCZ01048 compared to CCZ01047. Therefore, CCZ01048 was further evaluated at 2 h p.i., and a higher tumor uptake of 21.9 ± 4.6 %ID/g was observed, with background activity further decreased compared to the one hour time point (Figure 6, Table 3). Excellent image contrast was achieved for CCZ01048 at 2 h p.i. Tumor-to-blood, tumor-to-muscle, tumor-to-bone and tumor-to-kidney ratios were all increased compared to 1 h p.i., reaching 96.4 ± 13.9, 210.9 ± 20.9, 39.6 ± 11.9, and 4.0 ± 0.9, respectively. A blocking study was performed for CCZ01048 at 1 h p.i. with co-injection of 100 µg of non-radioactive Ga-labeled CCZ01048 (Figure 6C, Table 3). Tumor uptake significantly decreased (p < 0.001) by more than 80%, to 2.3 ± 1.1 %ID/g, which was comparable to the blood activity at 2.8 ± 1.3 %ID/g. Kidney accumulation was higher in the blocking study, at 24.4 ± 13.5 %ID/g. Autoradiography was also performed to further evaluate the in vivo radioactivity distribution of 68Ga-CCZ01048 at 1 h p.i. (Figure S5). Strong intensity was observed for tumor slices, and minimal intensity for background muscle and thyroid slices. Moreover, with co-injection of 100 µg of non-radioactive Ga-labeled CCZ01048, tumor slices showed largely reduced signal intensity and comparable to that of muscle slices.
Table 3
Biodistribution data of 68Ga-labeled CCZ01047, CCZ01048 and CCZ01056 in tumor-bearing C57BL/6J mice (Multiple t tests, multiple comparisons corrected using Holm-Sidak method, *** p < 0.001, n ≥ 6, &n ≥ 4).
| Tissue | 68Ga-CCZ01047 | 68Ga-CCZ01056 | 68Ga-CCZ01048 | ||
|---|---|---|---|---|---|
| 1 h p.i. unblock | 1 h p.i. unblock | 1 h p.i. unblock | 2 h p.i. unblock | 1 h p.i. block | |
| Tumor | 8.0±3.0 | 6.5±1.4 | 12.3±3.3 | 21.9±4.6 | 2.3±1.1*** |
| Blood | 0.5±0.2 | 0.5±0.2 | 0.9±0.5 | 0.2±0.0 | 2.8±1.3 |
| Fat | 0.1±0.1 | 0.1±0.0 | 0.2±0.1 | 0.2±0.1 | 0.5±0.4 |
| Seminal | 0.2±0.1 | 0.1±0.1 | 0.3±0.1 | 0.4±0.1 | 0.4±0.2 |
| Testes | 0.2±0.1 | 0.2±0.0 | 0.3±0.1 | 0.4±0.1 | 0.6±0.3 |
| Intestine | 0.3±0.1 | 0.7±0.5 | 0.7±0.5 | 0.4±0.1 | 1.2±0.6 |
| Spleen | 0.3±0.1 | 0.2±0.0 | 0.3±0.1 | 0.3±0.0 | 1.1±0.5 |
| Pancreas | 0.1±0.0 | 0.1±0.0 | 0.1±0.1 | 0.1±0.0 | 0.7±0.5 |
| Stomach | 0.4±0.2 | 2.8±3.8 | 0.5±0.2 | 0.8±0.2 | 1.1±0.6 |
| Liver | 0.6±0.1 | 0.5±0.1 | 0.6±0.2 | 0.7±0.0 | 1.4±0.6 |
| Adrenal Glands | 0.3±0.2 | 0.2±0.1 | 0.5±0.2 | 0.4±0.2 | 1.5±1.4 |
| Kidney | 5.1±1.4 | 6.2±2.0 | 4.7±0.5 | 5.5±0.4 | 24.4±13.5*** |
| Heart | 0.2±0.1 | 0.2±0.0 | 0.3±0.1 | 0.1±0.0 | 1.0±0.5 |
| Lungs | 0.6±0.3 | 0.5±0.1 | 0.6±0.2 | 0.4±0.1 | 2.8±1.6 |
| Thyroid& | 4.1±0.6 | 0.7±0.3 | 2.4±0.6 | 4.7±1.1 | 1.3±1.5 |
| Bone | 0.2±0.1 | 0.2±0.0 | 0.4±0.1 | 0.6±0.2 | 1.1±0.5 |
| Muscle | 0.1±0.0 | 0.1±0.0 | 0.2±0.0 | 0.1±0.0 | 0.9±0.5 |
| Brain | 0.0±0.0 | 0.0±0.0 | 0.1±0.0 | 0.0±0.0 | 0.1±0.0 |
| Tumor/Blood | 15.8±7.2 | 13.6±3.1 | 21.5±14.6 | 96.4±13.9 | 0.8±0.2 |
| Tumor/Muscle | 70.2±30.6 | 53.3±13.0 | 73.6±30.5 | 210.9±20.9 | 2.8±0.9 |
| Tumor/Bone | 40.3±10.6 | 40.8±5.2 | 35.4±15.3 | 39.6±11.9 | 2.4±1.0 |
| Tumor/Kidney | 1.6±0.6 | 1.1±0.3 | 2.7±0.9 | 4.0±0.9 | 0.1±0.1 |
Figure 5
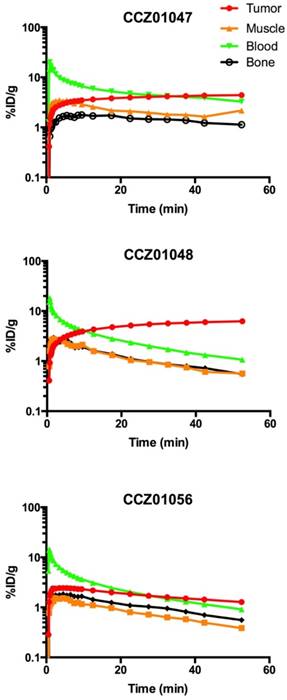
Time activity curves of A. 68Ga-CCZ01047, B. 68Ga-CCZ01048 and C. 68Ga-CCZ01056 using region-of-interest located around the tumor, muscle, blood (from left ventricle), and bone. %ID/g is in log scale. The blood half-life of the radiotracers was 9.0, 4.0 and 4.1 min, respectively.
Figure 6
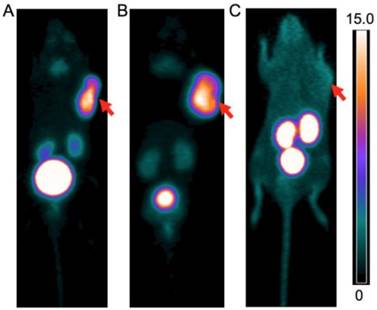
PET images of 68Ga-CCZ01048 at A. 1 h p.i., B. 2 h p.i. and C. 1 h p.i. with co-injection of 100 μg of non-radioactive Ga-labeled CCZ01048, in mice bearing B16F10 tumors. The scale bar unit is %ID/g. Tumors are indicated by arrows.
Discussion
MC1R has been studied extensively for melanoma imaging in the preclinical setting, based on the sequence of α-MSH peptide, predominantly for radiolabeling using single photon emitters. A few generations of tracers have been designed and evolved from linear to cyclized peptides, which were further improved by shortening the ring to six amino acids, i.e. CycMSHhex. Guo and colleagues introduced 111In-DOTA-Nle-CycMSHhex, which showed tumor uptake of 19.4 ± 1.7 %ID/g, with tumor-to-kidneys uptake ratio of 2.0 at 2 h p.i. [19]. Introduction of a -GlyGly- or a -GlyGlu- linker between the DOTA and the Nle-CycMSHhex resulted in tumor uptake of 19.1 ± 5.0 and 9.0 ± 1.9 %ID/g at 2 h p.i., respectively [16]. Moreover, 111In-DOTA-GlyGly-Nle-CycMSHhex had a slightly increased tumor-to-kidney uptake ratio of 2.8 at 2 h p.i. Interestingly, the addition of the negatively charged amino acid glutamate in the linker significantly decreased tumor uptake. In previous studies, we observed that introduction of a cationic Pip linker not only improved receptor binding affinity, but also generated approximately two-fold increase in tumor uptake for radiotracers targeting the bradykinin 1 receptor [22], similar to observations made by others with gastrin-releasing peptide receptor radioligands [25]. Therefore, in this study, based on the previously reported core structure, DOTA-Nle-CycMSHhex, we synthesized three new tracers with a neutral PEG2 linker (CCZ01047), a cationic Pip linker (CCZ01048), and dual Pip linkers (CCZ01056).
All three peptides exhibited high binding affinity, with sub-nanomolar inhibition constants to MC1R on B16F10 cells, which is an established model for melanoma imaging [26-28]. For CCZ01048, which has a single cationic Pip linker, the inhibition constant improved more than 2-fold compared to CCZ01047 with the neutral PEG2 linker. The peptide with the dual Pip linkers (CCZ01056) further improved the binding affinity approximately 2-fold. Also, all 68Ga-labeled peptides were fairly stable in vivo with more than 93% remaining intact at 15 min p.i. in mice. Due to the fast blood clearance of the radiotracers in vivo, stability tests at longer time points resulted in low radioactivity of the remaining peptides, which were comparable to background, and thus cannot be quantified. Moreover, all three tracers were rapidly internalized into cells with 55%-62% after 120 min incubation. The high in vivo stability and high level of internalization is consistent with a 99mTc(CO)3-labeled peptide, which has the same core structure of Nle-CycMSHhex [18].
Tumors were clearly visualized with high tumor-to-background contrast for all three tracers in static PET scans. Also, both 68Ga-CCZ01047 and 68Ga-CCZ01048 showed increased and sustained tumor uptake in dynamic PET scans, with 68Ga-CCZ01048 showing faster background clearance as well as faster accumulation at tumor sites. Moreover, 68Ga-CCZ01048 produced the highest tumor uptake at 12.3 ± 3.3 %ID/g at 1 h p.i. in biodistribution studies, compared to 8.0 ± 3.0 %ID/g for 68Ga-CCZ01047 and 6.5 ± 1.4 %ID/g for 68Ga-CCZ01056. In addition, 68Ga-CCZ01048 showed the lowest activity accumulation in kidneys at 4.7 ± 0.5 %ID/g at 1 h p.i., compared to 5.1 ± 1.4 %ID/g for 68Ga-CCZ01047 and 6.2 ± 2.0 %ID/g for 68Ga-CCZ01056. Therefore, 68Ga-CCZ01048 was selected as the best candidate and evaluated further. Interestingly, moderate thyroid accumulation was also observed for 68Ga-CCZ01047 and 68Ga-CCZ01048. Similar uptake at the region of thyroid was shown for another PET tracer, 64Cu-DOTA-GlyGly-Nle-CycMSHhex [20]. However, no thyroid uptake was reported for CycMSHhex-based radiopeptides labeled with SPECT isotopes. Furthermore, the decrease in thyroid uptake was not statistically significant following co-injection of excess amount of non-radioactive Ga-coupled CCZ01048. Nonetheless, activity accumulation in thyroid was moderate and lower than kidneys in all conditions tested; therefore, kidneys remain to be the dose-limiting organ for radiotherapy using this peptide. Potential adverse effects due to thyroid damage would still need to be closely monitored. There is extensive experience with radioiodine in diagnosis and treatment of thyroid diseases. Radiation exposure to the thyroid gland can readily be managed with thyroid hormone supplementation. This would be minor in the context of advanced metastatic melanoma treatment.
Blocking studies via PET imaging, biodistribution and autoradiography all confirmed that 68Ga-CCZ01048 tumor uptake was MC1R mediated. The excess amount of the non-radioactive counterpart slowed down the clearance of the 68Ga-CCZ01048, and thus higher background radioactivity level, including the kidneys, was observed in the static PET image, biodistribution and autoradiography data, which was expected. We further investigated 68Ga-CCZ01048 at 2 h p.i., which led to higher tumor uptake at 21.9 ± 4.6 %ID/g, and moderate kidneys uptake of 5.5 ± 0.4 %ID/g. The average tumor-to-kidney uptake ratio of 4.0 at 2 h p.i. is the highest reported to date for all radiopeptides based on the CycMSHhex. The low kidney uptake is particularly advantageous, since kidney is commonly a major dose-limiting organ when peptide receptor radionuclide therapy is employed. Furthermore, the 21.9 ± 4.6 %ID/g tumor uptake value is the highest among all PET tracers targeting MC1R. This is a notable improvement from 13.4 ± 1.6 %ID/g of 64Cu-labeled NOTA-GlyGly-Nle-CycMSHhex at 2 h p.i. [20]. Therefore, the combination of the rapid internalization, high tumor uptake and excellent tumor-to-normal organ uptake ratios make CCZ01048 a promising candidate for radionuclide therapy, by coupling the same peptide with 177Lu or 213Bi/225Ac.
In conclusion, we designed and evaluated three 68Ga-labeled peptides based on CycMSHhex, and observed that the introduction of a cationic Pip linker, CCZ01048, not only improved tumor uptake, but also generated high tumor-to-normal tissue contrast with PET imaging in a preclinical melanoma model. Therefore, CCZ01048 is a promising candidate for PET imaging of malignant melanoma, and potentially as a theranostic compound for radionuclide therapy of melanoma using α or β emitters.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported in part by the Canadian Institutes of Health Research (MOP-119361), the BC Cancer Foundation, and the BC Leading Edge Endowment Fund.
Competing Interests
The authors have declared that no competing interest exists.
References
1. American Cancer Society. http://www.cancer.org/cancer/skincancer-melanoma/detailedguide/melanoma-skin-cancer-key-statistics
2. Postow MA, Callahan MK, Wolchok JD. Immune checkpoint blockade in cancer therapy. J Clin Oncol. 2015;33:1974-82
3. Johansson CH, Brage SE. BRAF inhibitors in cancer therapy. Pharmacol Ther. 2014;142:176-82
4. Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257:56-71
5. Andtbacka RH, Kaufman HL, Collichio F. et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol. 2015;33:2780-8
6. Reinhardt MJ, Joe AY, Jaeger U. et al. Diagnostic performance of whole body dual modality 18F-FDG PET/CT imaging for N-and M-staging of malignant melanoma: experience with 250 consecutive patients. J Clin Oncol. 2006;24:1178-87
7. Strobel K, Bode B, Dummer R. et al. Limited value of 18F-FDG PET/CT and S-100B tumour marker in the detection of liver metastases from uveal melanoma compared to liver metastases from cutaneous melanoma. Eur J Nucl Med Mol Imaging. 2009;36:1774-82
8. Servois V, Mariani P, Malhaire C. et al. Preoperative staging of liver metastases from uveal melanoma by magnetic resonance imaging (MRI) and fluorodeoxyglucose-positron emission tomography (FDG-PET). Eur J Surg Oncol. 2010;36:189-94
9. Salazar-Onfray F, López M, Lundqvist A. et al. Tissue distribution and differential expression of melanocortin 1 receptor, a malignant melanoma marker. Br J Cancer. 2002;87:414-22
10. López MN, Pereda C, Ramirez M. et al. Melanocortin 1 receptor is expressed by uveal malignant melanoma and can be considered a new target for diagnosis and immunotherapy. Invest Ophthalmol Vis Sci. 2007;48:1219-27
11. Raposinho PD, Correia JD, Oliveira MC. et al. Melanocortin-1 receptor-targeting with radiolabeled cyclic α-melanocyte-stimulating hormone analogs for melanoma imaging. Biopolymers. 2010;94:820-9
12. Guo H, Gallazzi F, Miao Y. Gallium-67-labeled lactam bridge-cyclized alpha-MSH peptides with enhanced melanoma uptake and reduced renal uptake. Bioconjug Chem. 2012;23:1341-8
13. Guo H, Gallazzi F, Miao Y. Design and evaluation of new Tc-99m-labeled lactam bridge-cyclized alpha-MSH peptides for melanoma imaging. Mol Pharm. 2013;10:1400-8
14. Guo H, Miao Y. Melanoma targeting property of a Lu-177-labeled lactam bridge-cyclized alpha-MSH peptide. Bioorg Med Chem Lett. 2013;23:2319-23
15. Guo H, Miao Y. Introduction of an 8-Aminooctanoic Acid Linker Enhances Uptake of 99mTc-Labeled Lactam Bridge-Cyclized α-MSH Peptide in Melanoma. J Nucl Med. 2014;55:2057-63
16. Guo H, Yang J, Gallazzi F. et al. Effects of the Amino Acid Linkers on the Melanoma-Targeting and Pharmacokinetic Properties of 111In-Labeled Lactam Bridge-Cyclized α-MSH Peptides. J Nucl Med. 2011;52:608-16
17. Morais M, Oliveira BL, Correia JoD. et al. Influence of the bifunctional chelator on the pharmacokinetic properties of 99mTc (CO) 3-labeled cyclic α-melanocyte stimulating hormone analog. J Med Chem. 2013;56:1961-73
18. Raposinho PD, Xavier C, Correia JD. et al. Melanoma targeting with α-melanocyte stimulating hormone analogs labeled with fac-[99mTc (CO) 3]+: effect of cyclization on tumor-seeking properties. J Biol Inorg Chem. 2008;13:449-59
19. Guo H, Yang J, Gallazzi F. et al. Reduction of the ring size of radiolabeled lactam bridge-cyclized α-MSH peptide, resulting in enhanced melanoma uptake. J Nucl Med. 2010;51:418-26
20. Guo H, Miao Y. Cu-64-labeled lactam bridge-cyclized α-MSH peptides for PET imaging of melanoma. Mol Pharm. 2012;9:2322-30
21. Lin K-S, Pan J, Amouroux G. et al. In vivo radioimaging of bradykinin receptor B1, a widely overexpressed molecule in human cancer. Cancer Res. 2015;75:387-93
22. Amouroux G, Pan J, Jenni S. et al. Imaging Bradykinin B1 Receptor with 68Ga-Labeled [des-Arg10] Kallidin Derivatives: Effect of the Linker on Biodistribution and Tumor Uptake. Mol Pharm. 2015;12:2879-88
23. Lin K-S, Amouroux G, Pan J. et al. Comparative Studies of Three 68Ga-Labeled [Des-Arg10] Kallidin Derivatives for Imaging Bradykinin B1 Receptor Expression with PET. J Nucl Med. 2015;56:622-7
24. Zhang C, Pan J, Lin K-S. et al. Targeting the neuropeptide Y1 receptor for cancer imaging by positron emission tomography using novel truncated peptides. Mol Pharm. 2016 [Epub ahead of print]
25. Mansi R, Wang X, Forrer F. et al. Development of a potent DOTA-conjugated bombesin antagonist for targeting GRPr-positive tumours. Eur J Nucl Med Mol Imaging. 2011;38:97-107
26. Cheng Z, Xiong Z, Subbarayan M. et al. 64Cu-labeled alpha-melanocyte-stimulating hormone analog for microPET imaging of melanocortin 1 receptor expression. Bioconjug Chem. 2007;18:765-72
27. Ren G, Liu S, Liu H. et al. Radiofluorinated rhenium cyclized α-MSH analogues for PET imaging of melanocortin receptor 1. Bioconjug Chem. 2010;21:2355-60
28. Ren G, Liu Z, Miao Z. et al. PET of malignant melanoma using 18F-labeled metallopeptides. J Nucl Med. 2009;50:1865-72
Author contact
![]() Corresponding author: Dr. François Bénard. Address: Department of Molecular Oncology, BC Cancer Agency, 675 West 10th Avenue, Rm 14-111, Vancouver, BC V5Z 1L3, Canada. Phone: 604-675-8206. Fax: 604-675-8218. E-mail: fbenardca.
Corresponding author: Dr. François Bénard. Address: Department of Molecular Oncology, BC Cancer Agency, 675 West 10th Avenue, Rm 14-111, Vancouver, BC V5Z 1L3, Canada. Phone: 604-675-8206. Fax: 604-675-8218. E-mail: fbenardca.
Citation styles
APA
Zhang, C., Zhang, Z., Lin, K.S., Pan, J., Dude, I., Hundal-Jabal, N., Colpo, N., Bénard, F. (2017). Preclinical Melanoma Imaging with 68Ga-Labeled α-Melanocyte-Stimulating Hormone Derivatives Using PET. Theranostics, 7(4), 805-813. https://doi.org/10.7150/thno.17117.
ACS
Zhang, C.; Zhang, Z.; Lin, K.S.; Pan, J.; Dude, I.; Hundal-Jabal, N.; Colpo, N.; Bénard, F. Preclinical Melanoma Imaging with 68Ga-Labeled α-Melanocyte-Stimulating Hormone Derivatives Using PET. Theranostics 2017, 7 (4), 805-813. DOI: 10.7150/thno.17117.
NLM
Zhang C, Zhang Z, Lin KS, Pan J, Dude I, Hundal-Jabal N, Colpo N, Bénard F. Preclinical Melanoma Imaging with 68Ga-Labeled α-Melanocyte-Stimulating Hormone Derivatives Using PET. Theranostics 2017; 7(4):805-813. doi:10.7150/thno.17117. https://www.thno.org/v07p0805.htm
CSE
Zhang C, Zhang Z, Lin KS, Pan J, Dude I, Hundal-Jabal N, Colpo N, Bénard F. 2017. Preclinical Melanoma Imaging with 68Ga-Labeled α-Melanocyte-Stimulating Hormone Derivatives Using PET. Theranostics. 7(4):805-813.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.