Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(3):664-676. doi:10.7150/thno.15162 This issue Cite
Research Paper
miR-17-3p Contributes to Exercise-Induced Cardiac Growth and Protects against Myocardial Ischemia-Reperfusion Injury
Jing Shi1*, Yihua Bei2*, Xiangqing Kong1 ![]() , Xiaojun Liu3, Zhiyong Lei4, Tianzhao Xu2, Hui Wang1, Qinkao Xuan1, Ping Chen2, Jiahong Xu5, Lin Che5, Hui Liu1, Jiuchang Zhong6, Joost PG Sluijter4, Xinli Li1, Anthony Rosenzweig3, Junjie Xiao2
, Xiaojun Liu3, Zhiyong Lei4, Tianzhao Xu2, Hui Wang1, Qinkao Xuan1, Ping Chen2, Jiahong Xu5, Lin Che5, Hui Liu1, Jiuchang Zhong6, Joost PG Sluijter4, Xinli Li1, Anthony Rosenzweig3, Junjie Xiao2 ![]()
1. Department of Cardiology, The First Affiliated Hospital of Nanjing Medical University, Nanjing 210029, China;
2. Cardiac Regeneration and Ageing Lab, School of Life Science, Shanghai University, Shanghai 200444, China.
3. Massachusetts General Hospital Cardiovascular Division and Harvard Medical School, Boston, MA 02115, USA.
4. Laboratory of Experimental Cardiology, University Medical Centre Utrecht, Utrecht 3508GA, The Netherlands.
5. Department of Cardiology, Tongji Hospital, Tongji University School of Medicine, Shanghai 200065, China.
6. State Key Laboratory of Medical Genomics & Shanghai Institute of Hypertension, Ruijin Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China.
* These authors contributed equally to this work.
Received 2016-2-1; Accepted 2016-11-4; Published 2017-1-15
Citation:
Shi J, Bei Y, Kong X, Liu X, Lei Z, Xu T, Wang H, Xuan Q, Chen P, Xu J, Che L, Liu H, Zhong J, Sluijter JPG, Li X, Rosenzweig A, Xiao J. miR-17-3p Contributes to Exercise-Induced Cardiac Growth and Protects against Myocardial Ischemia-Reperfusion Injury. Theranostics 2017; 7(3):664-676. doi:10.7150/thno.15162. https://www.thno.org/v07p0664.htm
Other stylesAbstract

Limited microRNAs (miRNAs, miRs) have been reported to be necessary for exercise-induced cardiac growth and essential for protection against pathological cardiac remodeling. Here we determined members of the miR-17-92 cluster and their passenger miRNAs expressions in two distinct murine exercise models and found that miR-17-3p was increased in both. miR-17-3p promoted cardiomyocyte hypertrophy, proliferation, and survival. TIMP-3 was identified as a direct target gene of miR-17-3p whereas PTEN was indirectly inhibited by miR-17-3p. Inhibition of miR-17-3p in vivo attenuated exercise-induced cardiac growth including cardiomyocyte hypertrophy and expression of markers of myocyte proliferation. Importantly, mice injected with miR-17-3p agomir were protected from adverse remodeling after cardiac ischemia/reperfusion injury. Collectively, these data suggest that miR-17-3p contributes to exercise-induced cardiac growth and protects against adverse ventricular remodeling. miR-17-3p may represent a novel therapeutic target to promote functional recovery after cardiac ischemia/reperfusion.
Keywords: Exercise, Cardiac growth, microRNA.
Introduction
Cardiovascular diseases (CVD) are increasing causes of morbidity and mortality worldwide, accounting for the largest proportion of deaths due to non-communicable diseases [1]. As populations age, the burden of CVD is likely to increase [2]. Current pharmacological treatments for heart failure are far from satisfactory as they are largely inhibitors of adverse cardiac remodeling caused by pathological stress [3]. Developing novel therapeutic strategies for heart failure is urgently needed.
Exercise training has been recommended as an adjunctive intervention for the prevention and treatment of CVD [4]. Exercise training can lead to physiological cardiac growth including an increase in cardiomyocyte size and markers of proliferation. However, given the limited regenerative capacity of the heart, it remains unclear whether this results in cardiomyogenesis. Understanding how exercise induces cardiac growth may help identify novel therapeutic targets to mitigate the adverse cardiac remodeling in response to pathological stress. Whether these pathways may enhance the limited cardiac regenerative capacity, and could thereby help compensate for the loss of cardiomyocytes, remains an exciting but unproven possibility [5].
Exercise-induced physiological cardiac growth is different from pathological hypertrophy even at an early stage when the two are indistinguishable in function and structure [6]. The IGF-1-PI3K(p110α)-Akt pathway is necessary for physiological hypertrophy while the Gαq pathway regulates pathological hypertrophy [7]. Previous work has identified that the reduction of C/EBPβ contributed to cardiac growth induced by exercise, which was also protective against pathological cardiac remodeling [5, 8]. MicroRNAs (miRNAs, miRs) have been reported to be central regulators of gene expression, and their dysregulation can lead to CVD [9, 10]. Nevertheless, the roles of miRNAs in exercise-induced cardiac growth are largely unclear.
The miR-17-92 cluster is among the most studied miRNA clusters, which has six members including miR-17, -18a, -19a, -20a, -19b-1 and -92a [11]. The miR-17-92 cluster is necessary and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts [11]. Interestingly, the passenger miRNAs of the miR-17-92 cluster have recently been found to play roles in the regulatory networks of miRNA target molecules [12, 13]. However, the role of any of these miRNAs in cardiac response to exercise is unknown.
Here we found that miR-17* (miR-17-3p, a passenger miRNA of miR-17) was consistently increased in two distinct murine exercise models. miR-17-3p increased cardiomyocyte proliferation and cell size in vitro by targeting metallopeptidase inhibitor 3 (TIMP3) and acting upstream of the PTEN-AKT pathway, respectively. miR-17-3p contributed to exercise-induced cardiac growth in vivo, and therapeutic delivery of miR-17-3p agomir was sufficient to protect against adverse remodeling after myocardial ischemia-reperfusion injury (IRI).
Materials and Methods
Mice Exercise Models
All mice were purchased and raised at the Experimental Animal Center of Nanjing Medical University (Nanjing, China) or Shanghai University (Shanghai, China). All procedures with animals were in accordance with guidelines on the use and care of laboratory animals for biomedical research published by National Institutes of Health (No. 85-23, revised 1996), and the experimental protocol was reviewed and approved by the ethical committees of Nanjing Medical University or Shanghai University or Massachusetts General Hospital. For ramp swimming model, 8-week-old male C57BL/6 mice swam in water tanks as our previously described, starting from 10 minutes twice daily with 10 minutes increase each day until 90 minutes reached [14]. Mice were sacrificed after finishing a total of 21 days of swimming. For voluntary wheel exercise, 8-week-old male C57BL/6 mice ran with wheels in a cage for 21 days [14]. At the endpoint, mice ventricle samples were harvested and snap frozen in liquid nitrogen until further analysis.
Ventricular Cardiomyocyte Isolation, Culture, and Treatment
Cardiomyocytes and non-cardiomyocytes were isolated and identified from adult swum or control mice as our previously described [14]. Primary neonatal rat ventricular cardiomyocytes (NRCMs) were prepared as previously described [14] and purified by Percoll gradient centrifugation. micrON® miRNA mimic (50 nM), micrOFF® miRNA inhibitor (100 nM) or their negative controls (RiboBio, Guangzhou, China) were transfected using Lipofectamine 2000 (Invitrogen, MA, USA) according to the manufacturer's instruction. Transfection of siRNAs for TIMP3 or PTEN (75 nM) (Invitrogen, MA, USA) were carried out using Lipofectamine 2000 as recommended by the manufacturer. To explore the downstream mechanism of TIMP3 in regulating cardiomyocyte proliferation, NRCMs were respectively treated with 10 μM JNK inhibitor (SP-600125, Beyotime Biotechnology, China), 10 μM EGFR inhibitor (PD-168393, Selleck Chemicals, USA), and 10 nM SP-1 inhibitor (mithramycin A, R&D systems, Minneapolis, MN, USA), in the presence of TIMP3 siRNA or negative control.
Cell Proliferation Assay
For EdU incorporation assay, 24 h post-transfection of miR-17-3p mimic (1% serum), inhibitor (10% serum) or their negative controls, NRCMs were labeled with EdU 24 h before harvesting and Click-iT® EdU Alexa Fluor® 555 Imaging Kit (Thermo Scientific, MA, USA) was used to reveal EdU incorporation. For Ki-67, NRCMs were incubated with primary antibodies overnight: Ki-67 (1:100, ab16667, Abcam, Cambridge, UK), and α-actinin (1:100, A7811, Sigma-Aldrich, St. Louis, MO, USA). Nuclei were counterstained with DAPI. EdU-, Ki-67-positive α-actinin-labeled cardiomyocytes were calculated to determine cell proliferation.
In Vitro Oxygen-Glucose Deprivation and Reperfusion Model and TUNEL Assay
The oxygen-glucose deprivation and reperfusion (OGD/R) model was established as previously described [15]. Briefly, NRCMs were subjected to 8 h OGD followed by 16 h recovery. To achieve OGD, glucose-free DMEM was used and NRCMs were cultured in an air-tight chamber with a humidified hypoxic atmosphere containing 5% CO2 and 95% N2 at 37 °C. After exposure to hypoxia, the medium was replaced with the glucose-containing DMEM to terminate OGD and NRCMs were transferred to a normal incubator for recovery. Terminal deoxynucleotidyl transferase-mediated dUTP in situ nick-end labeling (TUNEL) was used to detect apoptotic nuclei by fluorescence microscopy in NRCM as previously described [16] using In Situ Cell Death Detection Kit, Fluorescein (Roche, Basel, Switzerland).
Quantitative Reverse Transcription Polymerase Chain Reactions (RT-PCRs)
Total RNAs were isolated with the RNeasy Mini Kit (Qiagen, Hilden, Germany) and then reverse transcribed into cDNA with iScript cDNA synthesis kit (Bio-Rad Laboratories, CA, USA). For quantitative miRNA analysis, the Bulge-LoopTM miRNA qPCR Primer Set (RiboBio) was used to determine the expression levels of miRNAs with Takara SYBR Premix Ex TaqTM (TliRNaseH Plus) in ABI-7900 Real-Time PCR Detection System (7900HT, Applied Biosystems, CA, USA). U6 was used as an internal control. For mRNA analysis, cDNA was synthesized using Bio-Rad iScriptTM cDNA Synthesis Kit (Bio-Rad) and was subjected to 40 cycles of quantitative PCR with Takara SYBR Premix Ex TaqTM (Tli RNaseH Plus, Japan) in ABI-7900 Real-Time PCR Detection System. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. Primer sequences (forward and reverse) used in the present study are as follows: ANP, AGCCGTTCGAGAACTTGTCTT and CAGGTTATTGCCACTTAGGTTCA; BNP, GAGGTCACTCCTATCCTCTGG and GCCATTTCCTCCGACTTTTCTC; α-SMA, GTCCCAGACATCAGGGAGTAA and TCGGATACTTCAGCGTCAGGA; Collagen I, GCTCCTCTTAGGGGCCACT and CCACGTCTCACCATTGGGG; Collagen III, CTGTAACATGGAAACTGGGGAAA and CCATAGCTGAACTGAAAACCACC; GAPDH, TGGATTTGGACGCATTGGTC and TTTGCACTGGTACGTGTTGAT. The relative expression level was calculated using the 2-ΔΔCt method.
Western Blot
Equal amounts of protein were subjected to standard SDS-PAGE and transferred onto PVDF membranes by an electroblot apparatus. Antibodies against TIMP3 (1:1000; 10858-1-AP, Proteintech, Chicago, IL, USA), BCl-2 (1:1000; 2876, Cell Signaling Technology, Boston, MA, USA), Bax (1:1000; 2772, Cell Signaling Technology), Caspase 3 (1:1000; 9661, Cell Signaling Technology), PTEN (1:1000; 9552, Cell Signaling Technology), Akt (1:1000; 4685, Cell Signaling Technology), Phospho-Akt (Thr308) (1:1000; 2965, Cell Signaling Technology), Phospho-Akt (Ser473) (1:1000; 4060, Cell Signaling Technology), α-actinin (1:1000; 11313-2-AP, Proteintech), cTnI (1:1000; 21652-1-AP, Proteintech), Desmin (1:1000; 16520-1-AP, Proteintech), EGFR (1:1000; 18986-1-AP, Proteintech), Phospho-EGFR (1:1000; 3777, Cell Signaling Technology), JNK (1:1000; 9252, Cell Signaling Technology), Phospho-JNK (Thr183/Tyr185) (1:1000; 4668, Cell Signaling Technology) and Phospho-SP1 (Thr739) (1:1000; 341484, zen-bioscience, Chengdu, China) were used as primary antibodies. Mouse or rabbit IgG antibodies coupled to horseradish peroxidase were used as secondary antibodies. β-Actin was used as loading control.
Luciferase Reporter Assay
A pmiR-RB-REPORT™ vector was constructed by inserting a fragment of the 3'-UTR of TIMP3 mRNA containing the putative miR-17-3p binding site as followed: h-TIMP3-3UTR-F: GCGCTCGAGACCTCACCATCTCCCAGACC and h-TIMP3- 3UTR-R: AATGCGGCCGCTCAAACAAGCAACGACAACA, using the XhoI and NotI sites. As a mutated control vector, a 3'-UTR fragment, with mutations in seed binding sites was generated using the following primers: h-TIMP3-mut-F: GCTGCCACACGTTGTCCCAACCAGACTGTG and h-TIMP3-mut-R: TTGGGACAACGTGTGGCAGCTCCTGGTGTA. For reporter assays, 293T cells were transfected with wild-type or mutant reporter plasmid and miR-17-3p mimic or negative control. Firefly and Renilla luciferase activities were measured 48 h post-transfection using the Dual-Glo™ Luciferase Assay System (Promega).
Murine models of Transverse Aortic Constriction (TAC) and Cardiac Ischemia-Reperfusion Injury (IRI)
Ventricles in chronic failing heart were collected from mouse transverse aortic constriction (TAC) model (4 weeks). The mouse myocardial ischemia-reperfusion injury (IRI) model was established by ligation of the left anterior descending artery (LAD) with 7-0 silk suture. Following 30 min occlusion, the LAD ligature was released followed by a 4-week reperfusion [14].
miRNA Agomir and Antagomir Injections in mice
miR-17-3p agomir (2'OME+5'chol modified) and antagomir (2'OME+5'chol modified) (RiboBio) were used. To determine if miR-17-3p is sufficient to induce physiological cardiac growth, adult mice were injected via tail vein with 30 mg/kg agomir or the scramble control for 3 consecutive days and harvested after 2 weeks. To check if miR-17-3p contributes to exercise-induced cardiac growth, mice subjected to swimming training received a tail vein injection with 200 mg/kg antagomir or the scramble control for 3 consecutive days. For myocardial IRI, mice were injected via tail vein with 10 mg/kg agomir or the scramble control every 3 days for 4 weeks starting 24 h after reperfusion.
Echocardiography
Echocardiography was performed by Vevo2100 (VisualSonics, Ontario, Canada) as previously described [17]. Parameters were measured from M-mode images taken from the parasternal short-axis view at papillary muscle level: fractional shortening (FS) and ejection fraction (EF).
Wheat Germ Agglutinin (WGA), Masson's Trichrome, and Immunofluorescent Staining
EdU (50 mg/kg) was intraperitoneally injected daily for 2 days before harvesting. Ventricles were snap frozen in liquid nitrogen with OCT on short axis at 8 μm. For staining, sections were fixed in 4% paraformaldehyde followed by washing with PBS. Sections were blocked with 3% BSA and then incubated with primary antibodies (all 1:100) overnight: phospho-HistoneH3 (pHH3) (1:100, ab74297, Abcam, Cambridge, UK), α-actinin (Sigma) and Ki-67 (Abcam). To measure cell size, frozen sections were stained with WGA (1:50, Sigma). Click-iT® Plus EdU Alexa Fluor® 555 Imaging Kit and Click-iT® TUNEL Alexa Fluor® Imaging Assay (Invitrogen) were performed to label EdU positive cells and apoptotic cells. For fibrosis measurement, Masson's Trichrome staining was performed.
Dilated Cardiomyopathy and Heart Failure Patients
All human investigation conformed to the principles outlined in the Declaration of Helsinki and was approved by the institutional review committees. All participants gave written informed consent before enrollment. Left ventricular tissue samples were collected from patients with dilated cardiomyopathy (DCM) undergoing cardiac transplantation and healthy donors. Serum of chronic heart failure patients underwent a symptom-limited cardiopulmonary exercise test were from the cohort as reported [14].
Statistical Analysis
Results were presented as mean±SEM. An unpaired, two-tailed Student's t test was used for comparisons between two groups. Two-way ANOVA test was performed to compare multiple groups followed by Bonferroni's post hoc test. All analyses were performed using GraphPad Prism 5.0. Differences were considered significant with p<0.05.
Results
miR-17-3p Is Increased in Exercise-Induced Physiological Hypertrophy
Expression levels of miR-17-92 cluster members and their passenger miRNAs were determined by qRT-PCR using ventricular samples from swum mice in comparison to sedentary controls. As shown in Figure 1A and B, healthy cardiac growth was induced by swimming. miR-17-3p and miR-18a-3p were each increased ~1.5-fold in swum hearts (Figure 1C). miR-17-3p was also upregulated ~1.4-fold in hearts from mice after voluntary wheel running while miR-18a-3p remained unchanged (Figure 1D). Interestingly, miR-17-3p was specifically increased in adult cardiomyocytes from swum mice, but a non-significant trend toward decreased level of miR-17-3p was found in non-cardiomyocytes from swum mice (Figure 1E). Thus, among all members and passenger miRNAs of the miR-17-92 cluster, only miR-17-3p was dynamically regulated in both exercise models and thus was selected for in-depth study.
To exclude the possibility that the induction of miR-17-3p in exercise is a general systemic response, we checked its expression in skeletal muscle of exercised mice and found that miR-17-3p was unchanged (Figure 1F). Of note, an examination of the tissue expression distribution of miR-17-3p demonstrated that it was most highly expressed in the myocardium (Figure 1G), suggesting a possible functional role in the heart.
Next, we examined miR-17-3p expression in mice with TAC-induced pathological hypertrophy as well as in human DCM cardiac samples. Interestingly, miR-17-3p was decreased in both models (Figure 1H and I), indicating that the induction of miR-17-3p is specific to exercise-induced hypertrophy and could potentially contribute to the protective effect of exercise.
Serum miR-17-3p Increases after Exercise in Heart Failure Patients
As miR-222 is increased in serum from chronic heart failure patients after exercise [14], we were interested in determining if miR-17-3p also had a similar change. Using the same cohort, we found that after a cardiopulmonary exercise test, serum miR-17-3p was increased by 5.36-fold (p=0.002, Figure 1J).
miR-17-3p Promotes Cardiomyocyte Hypertrophy, Proliferation, and Survival in Vitro
In NRCMs, miR-17-3p gain- and loss-of-function experiments were performed to determine its roles in regulating cardiomyocyte hypertrophy, proliferation, and survival. miR-17-3p overexpression increased (Figure 2D), while its inhibition decreased cardiomyocyte size (Figure 2G), indicating that miR-17-3p is a positive regulator for cardiomyocyte hypertrophy. In addition, miR-17-3p overexpression induced an increase in markers of cardiomyocyte proliferation and cell number (Figure 2A-C), while miR-17-3p inhibition had opposite effects (Figure 2E and F), suggesting that miR-17-3p also positively regulates cardiomyocyte proliferation. Moreover, miR-17-3p overexpression suppressed cardiomyocyte apoptosis induced by oxygen-glucose deprivation and reperfusion (OGD/R) (Figure 2H).
Figure 1
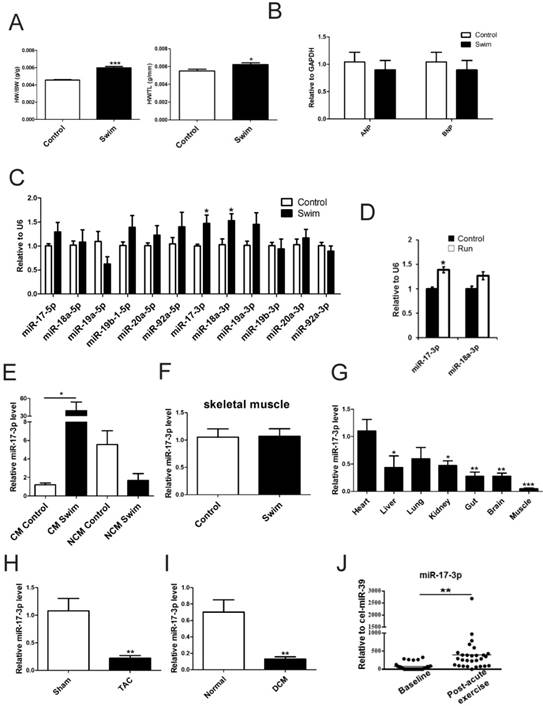
miR-17-3p Is Increased in Exercise-Induced Physiological Hypertrophy. (A) Heart weight/body weight (HW/BW) and heart weight/tibia length (HW/TL) ratios of sedentary (control) and swum (swim) mice. n=6 per group. (B and C) qRT-PCRs for ANP and BNP (B, n=6 per group), members of the miR-17-92 cluster, and their passenger microRNAs (C, n=5 per group) in sedentary and swum mice. (D) qRT-PCRs for miR-17-3p and miR-18a-3p in sedentary (control) and voluntary wheel-running (run) mice. n=4 per group. (E) qRT-PCRs for miR-17-3p in isolated adult cardiomyocytes (CM) and non-cardiomyocytes (NCM) from sedentary and swum mice. n=4 per group. (F) qRT-PCRs for miR-17-3p in skeletal muscle from sedentary and swum mice. n=5 per group. (G) qRT-PCRs of tissue distribution of miR-17-3p. n=5 per group. (H and I) qRT-PCRs for miR-17-3p in hearts from transverse aortic constriction (TAC)-induced pathological hypertrophy and human dilated cardiomyopathy (DCM) heart samples. n=5 for mice per group and n=4 for human per group. (J) qRT-PCRs for miR-17-3p in serum from chronic heart failure patients (n=28) before and after acute exercise training. *, P<0.05, **, P<0.01, ***, P<0.001 versus respective control.
Figure 2
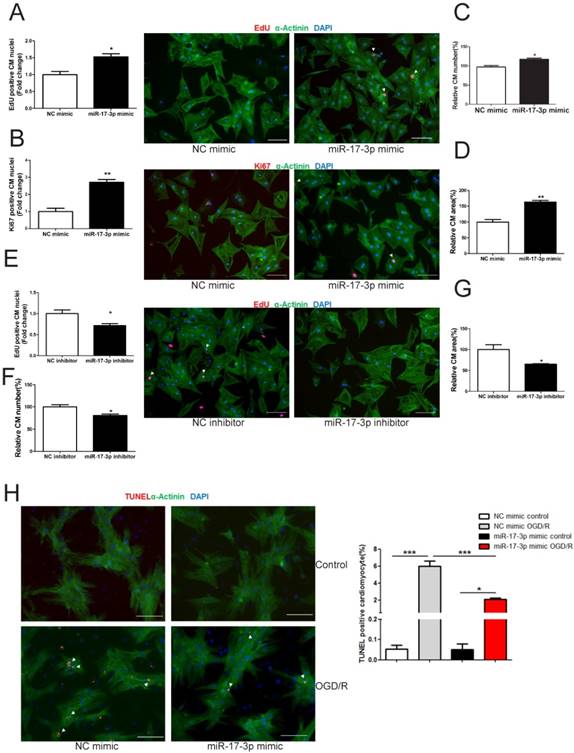
miR-17-3p Promotes Cardiomyocyte Hypertrophy, Proliferation, and Survival in Vitro. (A-D) Immunohistochemical staining for sarcomeric α-actinin and EdU (A) or Ki-67 (B) followed by quantification of cell number (C) and cardiomyocyte area (D) in neonatal rat ventricular myocytes (NRVMs) transfected with control (NC mimic) or miR-17-3p mimic. (E-G) Immunohistochemical staining for α-actinin and EdU (E) followed by quantification of cell number (F) and cardiomyocyte area (G) in NRVMs transfected with control (NC inhibitor) or miR-17-3p inhibitor. (H) TUNEL staining for NRVMs treated with control (NC mimic) or miR-17-3p mimic. Apoptosis was induced by oxygen-glucose deprivation and reperfusion (OGD/R). At least 2000 cells were quantified for each group. Data are shown as mean±SEM and reflect at least three independent experiments. Scale bar: 100 μm. *, P<0.05, **, P<0.01, ***, P<0.001 versus respective control.
miR-17-3p Controls Cardiomyocyte Proliferation and Hypertrophy by Targeting TIMP3 and the PTEN-Akt pathway
Targetscan showed that TIMP3 was a potential target of miR-17-3p (Figure 3A). Luciferase reporter assay confirmed that miR-17-3p led to a reduction in luciferase activity for the wild-type 3'UTR construct, but had no effect when the miR-17-3p binding site was mutated (Figure 3A), indicating that TIMP3 is a direct target gene of miR-17-3p. miR-17-3p mimic downregulated, while its inhibitor increased TIMP3 expression level in cardiomyocytes (Figure 3B and C), indicating that miR-17-3p could negatively regulate endogenous TIMP3 expression. In addition, we examined TIMP3's contribution to the functional role of miR-17-3p in cardiomyocytes by using siRNA-mediated knockdown of TIMP3. TIMP3 siRNA induced an increase in EdU incorporation, without affecting cell size of cardiomyocytes (Figure 3D). However, co-transfection of TIMP3 siRNA and miR-17-3p mimic did not exert an additive effect in increasing EdU incorporation (Figure 3D), confirming that inhibition of TIMP3 likely mediates the pro-proliferative effect of miR-17-3p in cardiomyocytes.
Figure 3
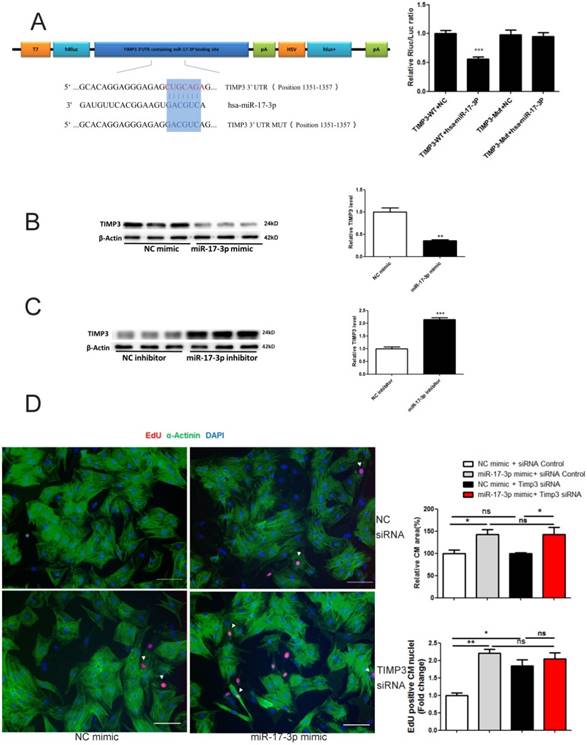
miR-17-3p Controls Proliferation of Cardiomyocytes in Vitro by Targeting TIMP3. (A) RNAhybrid and Luciferase assays identified TIMP3 as a direct target of miR-17-3p. n=6 per group. (B and C) Western blot showed that miR-17-3p negatively regulated TIMP3 in cardiomyocytes. n=3 per group. (D) Immunohistochemical staining for α-actinin and EdU followed by quantification of cardiomyocyte area showed that TIMP3 knockdown induced an increase in EdU incorporation without affecting cell size, while co-transfection of TIMP3 siRNA and miR-17-3p mimic did not exert an additive effect. At least 2000 cells were quantified in each group. Data are shown as mean±SEM and reflect at least three independent experiments. Scale bar: 100 μm. *, P<0.05, **, P<0.01, ***, P<0.001 versus respective control.
TIMP3 has previously been reported to inhibit neonatal mouse cardiomyocyte proliferation via EGFR/JNK/SP-1 signaling [18]. Interestingly, here we also found that EGFR expression/phosphorylation levels, as well as JNK and SP-1 phosphorylation levels, were enhanced by overexpression of miR-17-3p (Figure 4A). Furthermore, functional rescue experiments showed that inhibition of EGFR/JNK/SP-1 signaling via specific inhibitors could abolish the effect of TIMP-3 siRNA in promoting cardiomyocyte proliferation (Figure 4B). These data provide strong evidence that overexpression of miR-17-3p and subsequent inhibition of TIMP3 promotes cardiomyocyte proliferation via EGFR/JNK/SP-1 signaling.
The PI3K(p110α)-Akt activation is necessary for physiological hypertrophy [7], while PTEN is a natural inhibitor of this pathway [19]. We thus determined the effect of miR-17-3p in regulating the PTEN-Akt pathway. Our results showed that PTEN was down-regulated by miR-17-3p mimic while phosphorylation of Akt at Ser473 and Thr308 was enhanced (Figure 5A). No binding site for miR-17-3p was identified in the 3'UTR of PTEN, suggesting that PTEN is not a direct target of miR-17-3p. To determine whether PTEN contributes to the functional role of miR-17-3p in cardiomyocytes, we used siRNA to knockdown PTEN and found that PTEN siRNA induced an increase in the size of cardiomyocytes, without affecting their EdU incorporation (Figure 5B). However, co-transfection of PTEN siRNA and miR-17-3p mimic did not further increase cardiomyocyte size (Figure 5B), suggesting that inhibition of PTEN contributes to the hypertrophic effect of miR-17-3p in cardiomyocytes.
Collectively, miR-17-3p directly targets TIMP3 to enhance cardiomyocyte proliferation, and indirectly regulates PTEN to promote cardiomyocyte hypertrophy.
miR-17-3p Contributes to Exercise-induced Cardiac Growth
To investigate if miR-17-3p contributes to exercise-induced cardiac growth, mice were injected with miR-17-3p antagomir or scrambled control for 3 days prior to exercise. miR-17-3p antagomir was efficient to decrease miR-17-3p expression level in hearts from both sedentary and swum mice (Figure 6A). miR-17-3p inhibition at least partly blocked exercise-induced cardiac growth, as evidenced by less increase in cardiac mass, including the ratios of heart weight to body weight (HW/BW) or to tibia length (HW/TL), respectively (Figure 6B). In addition, WGA-stained heart sections revealed that miR-17-3p inhibition partly blocked the increase in cardiomyocyte size after exercise (Figure 6C). Previous study suggested that exercise could increase proliferation markers in the cardiomyocyte lineage [14], which is confirmed here by increased EdU incorporation and phospho-HistoneH3 (pHH3) staining in cardiomyocytes from exercised, untreated mice (Figure 6D). Interestingly, these increases were partly decreased by miR-17-3p antagomir (Figure 6D). Meanwhile, we analyzed contractile proteins including α-actinin and cardiac Troponin I (cTnI), as well as cytoskeleton protein Desmin using Western blot. Our results showed that exercise-induced increase in α-actinin and cTnI, but not Desmin, was totally abolished by treatment with miR-17-3p antagomiR, indicating that exercise-associated elevation of contractile protein levels was also attenuated by miR-17-3p inhibition (Supplemental Figure 1). Collectively, these data indicate that miR-17-3p is necessary for exercise-induced cardiac growth in vivo.
Figure 4
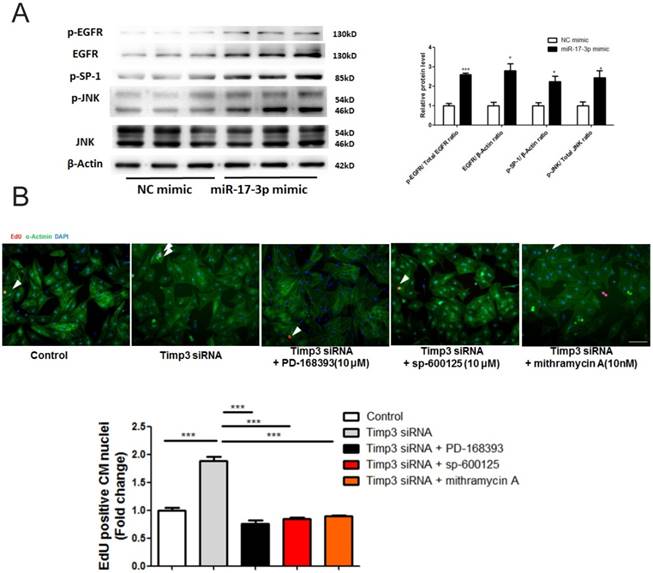
TIMP3 knockdown promotes cardiomyocyte proliferation via activating EGFR/JNK/SP-1 signaling. (A) Western blot showed that miR-17-3p overexpression activated EGFR/JNK/SP-1 signaling. (B) Immunohistochemical staining for α-actinin and EdU showed that EGFR inhibitor (PD-168393), JNK inhibitor (sp-600125), and SP-1 inhibitor (mithramycin A) could abolish the effect of TIMP-3 siRNA in promoting cardiomyocyte proliferation. n=3 per group for Western blot. At least 2000 cells were quantified in each group. Data are shown as mean±SEM and reflect at least three independent experiments. Scale bar: 100 μm. *, P<0.05, ***, P<0.001 versus respective control.
Figure 5
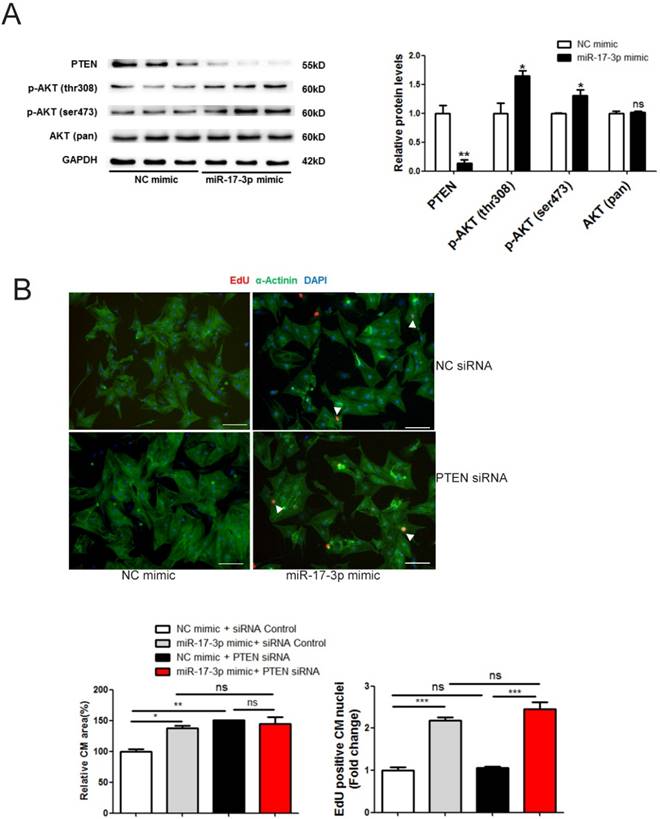
miR-17-3p Controls Growth of Cardiomyocytes in Vitro through the PTEN-Akt pathway. (A) Western blot showed that PTEN was downregulated and phosphorylation of Akt at Ser473 and Thr308 were enhanced by miR-17-3p mimic. (B) Immunohistochemical staining for α-actinin and EdU followed by quantification of cardiomyocyte area showed that PTEN knockdown induced an increase in cell size of cardiomyocytes without affecting EdU incorporation, while co-transfection of PTEN siRNA and miR-17-3p mimic did not exert an additive effect. n=3 per group for Western blot. At least 2000 cells were quantified in each group. Data are shown as mean±SEM and reflect at least three independent experiments. Scale bar: 100 μm. *, P<0.05, **, P<0.01, ***, P<0.001 versus respective control.
Moreover, our results showed that PTEN and TIMP3 were downregulated while Akt phosphorylation was enhanced in exercised heart (Figure 6E), which could be at least partly attenuated by miR-17-3p antagomiR treatment (Figure 6E). These data provide evidence that miR-17-3p might also be able to regulate PTEN and TIMP3 in vivo.
miR-17-3p Protects against Myocardial Ischemia-Reperfusion Injury
miR-17-3p induced by agomir at a comparable level to exercise led to an elevation of miR-17-3p in heart (~1.7 fold), but failed to induce an increase in heart weight, cell size, and proliferation of cardiomyocytes (Supplemental Figure 2). Therefore, although miR-17-3p contributes to exercise-induced cardiac growth, increasing miR-17-3p expression in the heart to a similar level as seen in exercised mice, is not sufficient to recapitulate the phenotype observed in exercised heart, which is similar to what was seen with miR-222 [14].
Since exercise protects against adverse cardiac remodeling [14, 20], we were interested in assessing the potential role of miR-17-3p in reducing adverse cardiac remodeling. We subjected mice to IRI induced by 30 min of coronary artery ligation and reperfusion for four weeks, and treated mice with miR-17-3p agomir starting 24 h after reperfusion. miR-17-3p agomir was efficient to increase miR-17-3p expression level in hearts from both sham and IRI mice (Figure 7A). miR-17-3p agomir preserved cardiac function as indicated by fractional shortening (FS) and ejection fraction (EF) (Figure 7B). Strikingly, cardiac apoptosis was also attenuated by miR-17-3p agomir in IRI group as determined by TUNEL staining (Figure 7C) and the ratio of Bcl-2/Bax and cleaved caspase 3 level (Figure 7D). Moreover, cardiac fibrosis was mitigated by miR-17-3p agomir in IRI group, as evaluated by Masson's Trichrome staining and expression levels of α-SMA, Collagen I, and Collagen III (Figure 7E). Interestingly, treatment with miR-17-3p agomir in IRI mice was associated with further increased Ki-67 positive cardiomyocyte nuclei, suggesting enhanced cardiomyocyte proliferation with chronic forced expression of miR-17-3p (Figure 7F). Collectively, increasing miR-17-3p is able to protect against myocardial IRI and might serve as a novel therapeutic strategy for pathological cardiac remodeling.
Figure 6
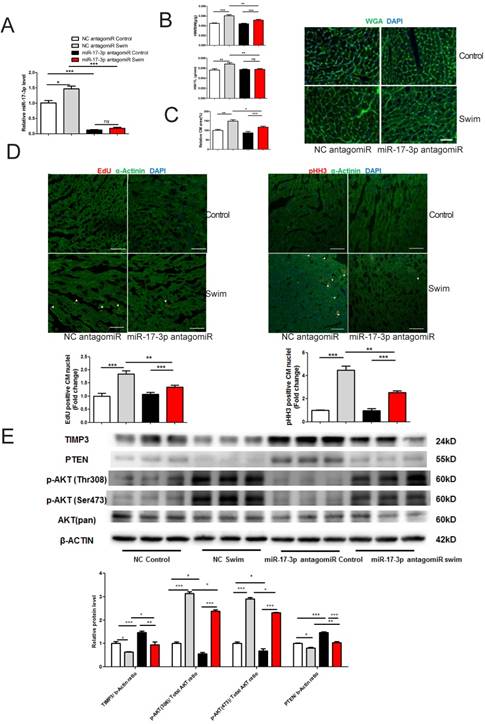
miR-17-3p Is Required for Exercise-induced Cardiac Growth. (A) miR-17-3p antagomiR significantly decreased miR-17-3p in hearts from both sedentary control and swum mice. n=7 per group. (B) Heart weight/body weight (HW/BW) and heart weight/tibia length (HW/TL) ratios were shown from sedentary control and swum mice. n=6 per group. (C) Wheat germ agglutinin (WGA) staining for determination of cardiomyocyte area showed that miR-17-3p antagomiR partly blocked exercise-induced cardiomyocyte hypertrophy. n=4 per group. (D) Immunohistochemical staining for EdU and phospho-histoneH3 (pHH3) indicated that miR-17-3p antagomiR partly reduced markers of cardiomyocyte proliferation. n=4 for control and n=8 for swim group. (E) Western blot showed that exercise-induced downregulation of TIMP3 and PTEN, and activation of AKT were partly altered by miR-17-3p antagomiR in vivo. n=3 per group. At least 4000 cells were quantified in each group. Data are shown as mean±SEM. Scale bar: 100 μm. *, P<0.05, **, P<0.01, ***, P<0.001 versus respective control.
Figure 7
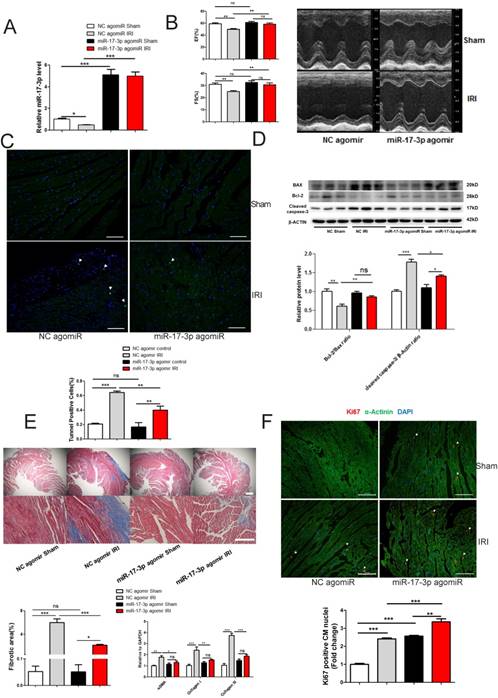
miR-17-3p Protects Against Myocardial Ischemia-Reperfusion Injury. (A) miR-17-3p agomiR significantly increased miR-17-3p in hearts from both sham control and IRI mice. n=6 per group. (B) miR-17-3p agomiR preserved fractional shortening (FS) and ejection fraction (EF) in IRI mice (n=6-7 for control and n=10-11 for IRI mice per group). (C and D) miR-17-3p agomiR attenuated cardiac apoptosis as determined by TUNEL staining (C) and the ratio of Bcl-2/Bax and cleaved caspase 3 (D). At least 4000 cells were quantified in each group. n=3 per group for Western blot. (E) miR-17-3p agomiR attenuated cardiac fibrosis as evidenced by Masson's Trichrome staining (n=3 per group) and expression levels of α-SMA, Collagen I, and Collagen III (n=6 per group). (F) miR-17-3p agomiR further increased the marker of cardiomyocyte proliferation (Ki-67) in IRI mice as determined by immunohistochemical staining. n=3 per group. Data are shown as mean±SEM. Scale bar: 100 μm. *, P<0.05, **, P<0.01, ***, P<0.001 versus respective control.
Discussion
Exercise training has been recommended as an important component of therapy for chronic heart failure patients [21]. Nevertheless, the underlying molecular mechanisms responsible for the benefit of exercise are still far from clear. miRNAs have been reported to be regulated by exercise in heart [22]. However, their functional relevance in exercise-induced cardiac growth is unclear. Recently, we (X.L., J.X., L.C., A.R.) have found that cardiac miR-222 was robustly increased in both ramp swimming and voluntary wheel running exercise models using TaqMan-based miRNA arrays. miR-222 is able to induce cardiomyocyte hypertrophy and proliferation by targeting P27, HIPK1, and HMBOX1. Interestingly, forced expression of miR-222 is sufficient to protect the heart from IRI [14]. Here we show that miR-17-3p contributes to exercise-induced cardiac growth and protects against myocardial IRI. This study has several unique points. Firstly, the use of unformulated synthetic oligonucleotide-based approaches to increase miRNA levels has not been well explored and this study provides promising data that miR-17-3p agomir protects against adverse remodeling after myocardial IRI. Secondly, the idea that promoting endogenous cardiomyocyte proliferation to regenerate and repair the heart after injury is worth pursuing and this study provides direct evidence that miR-17-3p promotes cardiomyocyte proliferation. Lastly, this study indicates the functional role of a passenger strand miRNA (miR-17-3p) that was once thought to be degraded and thereby inactivated.
Precursor miRNAs can be processed by Dicer and produce a mature miRNA and a passenger strand [12]. The passenger strand miRNA (previously named miRNA*) was thought to be degraded and inactivated, however, it is now clear that at least some passenger miRNAs can regulate gene expressions and play functional roles [23]. Actually, mature sequences of miRNAs are currently denoted by the miRNA-5p (5′-arm) and miRNA-3p (3′-arm) [23]. The miR-17-92 cluster has been shown to be required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts [11]. However, their roles in cardiac response to exercise are unexplored. Of note, passenger strand miRNAs of miR-17-92 members were not included in the miRNA array we have used in previous study [14]. Here, we first provide direct evidence that only miR-17-3p of the miR-17-92 cluster is dynamically regulated in two models of exercise. Second, miR-17-3p contributes to exercise-induced cardiac growth and can help mitigate myocardial IRI. Interestingly, though increasing miR-17-3p was not sufficient to induce physiological cardiac growth, it did protect against adverse cardiac remodeling including attenuating cardiac apoptosis, decreasing fibrosis, and preserving cardiac function.
The loss of cardiomyocytes (apoptosis and necrosis) is a hallmark in the early phase after myocardial IRI. In addition to the increased antioxidant capacity and angiogenesis [24, 25], exercise has also been demonstrated to reduce myocardial apoptosis and increase cardiomyocyte renewal which contributes to the benefit of exercise in IRI [14, 26]. Considering the fact that miR-17-3p has been reported to promote mouse cardiac fibroblast self-renewal [27], we speculated that the decrease of cardiac fibrosis in this study was an associated phenomenon of the protective effect of miR-17-3p against cardiomyocyte death as miR-17-3p was specifically increased in adult cardiomyocytes from swum mice, but a non-significant trend toward decreased level of miR-17-3p was found in non-cardiomyocytes from swum mice. Impressively, we did observe a decrease in cardiomyocyte apoptosis by miR-17-3p overexpression both in vivo and in vitro. Meanwhile, forced expression of miR-17-3p could enhance cardiomyocyte proliferation in vitro and further increase Ki-67-positive cardiomyocyte nuclei in IRI mice in vivo. Collectively, these data indicate that improving cardiomyocyte survival is a major contributor for the benefit that we observed for miR-17-3p.
Similar to the role of miR-222 in exercise-induced cardiac growth [14], overexpressing miR-17-3p is not sufficient to induce an increase in the heart size though miR-17-3p contributes to exercise-induced cardiac growth. Therefore, other signals such as miR-222 may be needed together to recapitulate the cardiac growth response induced by exercise, thus it would be interesting to carry out co-forced expression of miR-17-3p and miR-222 in vivo in the future though beyond the scope of the present study.
Based on bioinformatic analysis and experimental validation, TIMP3 was identified as a target gene of miR-17-3p in cardiomyocytes. Functional studies in cardiomyocytes further indicated that reduction of TIMP3 was responsible for the pro-proliferation effect of miR-17-3p in cardiomyocytes. In fact, the regulatory effect of TIMP3 in cardiomyocyte proliferation was previously reported [18]. The present study further demonstrates that the increased miR-17-3p and subsequent inhibition of TIMP3 leads to enhanced proliferation of cardiomyocytes via activating EGFR/JNK/SP-1 signaling. Besides that, PTEN and Akt were identified to be indirectly regulated by miR-17-3p in cardiomyocytes, and the inhibition of PTEN and activation of Akt were responsible for the pro-hypertrophy effect of miR-17-3p in cardiomyocytes. Meanwhile, miR-17-3p could also regulate TIMP3 and PTEN/Akt signalings in exercised heart in vivo. However, it would be of interest to further determine their in vivo functional roles for miR-17-3p each or combined based on gain-of-function and loss-of-function studies.
Last but not least, the clinical relevance of miR-17-3p is confirmed by the fact that circulating miR-17-3p was increased by exercise in heart failure patients. Of note, miR-17-3p was also found to be decreased in pressure overload-induced pathological hypertrophy and in human DCM heart samples. Previous work by others has suggested that factors responsible for physiological hypertrophy may be antagonistic to pathological cardiac growth. Such facts have been demonstrated with Akt and C/EBPβ [8, 28]. Indeed, the functional roles of miR-17-3p in pathological hypertrophy deserves further exploration.
In conclusion, miR-17-3p is required for exercise-induced cardiac growth and protects against myocardial IRI. miR-17-3p might serve as a novel therapeutic target for enhancing cardiac survival and regeneration.
Supplementary Material
Supplementary figures.
Acknowledgements
This work was supported by the grants from National Natural Science Foundation of China (81570362, 91639101, and 81200169 to JJ Xiao, 81370332 and 81170201 to XL Li, 81400647 to Y Bei, 81370362 to JC Zhong), NIH (R21HL114352, 1UH2TR000901, R01HL110733 to A Rosenzweig), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD20102013 to XL Li), the Netherlands Cardiovascular Research Initiative (CVON): the Dutch Heart Foundation, Dutch Federation of University Medical Centers, the Netherlands Organization for Health Research and Development, and the Royal Netherlands Academy of Science (to JPG Sluijter), the National Basic Research Program of China (2014CB542300), the National Major Research Plan Training Program (91339108), Shanghai Municipal Education Commission-Gaofeng Clinical Medicine Grant (20152509). Dr XQ Kong is a Fellow and Dr XL Li is an Associate Fellow at the Collaborative Innovation Center For Cardiovascular Disease Translational Medicine.
Author Contributions
J. S and Y. B contributed equally to this work. J.S. performed functional experiments in vitro and gain-of-function experiments in vivo. Y.B performed the experiments of miR-17-3p target and loss-of-function experiments in vivo. J.X. designed the study, instructed all experiments and drafted the manuscript. X.K participated in the design of the study and coordination of the whole work.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hunter DJ, Reddy KS. Noncommunicable diseases. N Engl J Med. 2013;369:1336-43
2. Pagidipati NJ, Gaziano TA. Estimating deaths from cardiovascular disease: a review of global methodologies of mortality measurement. Circulation. 2013;127:749-56
3. Braunwald E. The war against heart failure: the Lancet lecture. Lancet. 2015;385:812-24
4. Fleg JL, Cooper LS, Borlaug BA, Haykowsky MJ, Kraus WE, Levine BD. et al. Exercise training as therapy for heart failure: current status and future directions. Circ Heart Fail. 2015;8:209-20
5. Bostrom P, Mann N, Wu J, Quintero PA, Plovie ER, Panakova D. et al. C/EBPbeta controls exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell. 2010;143:1072-83
6. Weeks KL, McMullen JR. The athlete's heart vs. the failing heart: can signaling explain the two distinct outcomes? Physiology (Bethesda). 2011;26:97-105
7. McMullen JR, Shioi T, Zhang L, Tarnavski O, Sherwood MC, Kang PM. et al. Phosphoinositide 3-kinase(p110alpha) plays a critical role for the induction of physiological, but not pathological, cardiac hypertrophy. Proc Natl Acad Sci U S A. 2003;100:12355-60
8. Zou J, Li H, Chen X, Zeng S, Ye J, Zhou C. et al. C/EBPbeta knockdown protects cardiomyocytes from hypertrophy via inhibition of p65-NFkappaB. Mol Cell Endocrinol. 2014;390:18-25
9. Tao L, Bei Y, Chen P, Lei Z, Fu S, Zhang H. et al. Crucial role of miR-433 in regulating cardiac fibrosis. Theranostics. 2016;6:2068-83
10. Boon RA, Dimmeler S. MicroRNAs in myocardial infarction. Nat Rev Cardiol. 2015;12:135-42
11. Chen J, Huang ZP, Seok HY, Ding J, Kataoka M, Zhang Z. et al. mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res. 2013;112:1557-66
12. Mah SM, Buske C, Humphries RK, Kuchenbauer F. miRNA*: a passenger stranded in RNA-induced silencing complex? Critical reviews in eukaryotic gene expression. 2010;20:141-8
13. Yang X, Du WW, Li H, Liu F, Khorshidi A, Rutnam ZJ. et al. Both mature miR-17-5p and passenger strand miR-17-3p target TIMP3 and induce prostate tumor growth and invasion. Nucleic Acids Res. 2013;41:9688-704
14. Liu X, Xiao J, Zhu H, Wei X, Platt C, Damilano F. et al. miR-222 is necessary for exercise-induced cardiac growth and protects against pathological cardiac remodeling. Cell Metab. 2015;21:584-95
15. Wu WY, Wang WY, Ma YL, Yan H, Wang XB, Qin YL. et al. Sodium tanshinone IIA silate inhibits oxygen-glucose deprivation/recovery-induced cardiomyocyte apoptosis via suppression of the NF-kappaB/TNF-alpha pathway. Br J Pharmacol. 2013;169:1058-71
16. Gonzalez-Juanatey JR, Iglesias MJ, Alcaide C, Pineiro R, Lago F. Doxazosin induces apoptosis in cardiomyocytes cultured in vitro by a mechanism that is independent of alpha1-adrenergic blockade. Circulation. 2003;107:127-31
17. Tao L, Shen S, Fu S, Fang H, Wang X, Das S. et al. Traditional Chinese Medication Qiliqiangxin attenuates cardiac remodeling after acute myocardial infarction in mice. Sci Rep. 2015;5:8374
18. Hammoud L, Burger DE, Lu X, Feng Q. Tissue inhibitor of metalloproteinase-3 inhibits neonatal mouse cardiomyocyte proliferation via EGFR/JNK/SP-1 signaling. Am J Physiol Cell Physiol. 2009;296:C735-45
19. Worby CA, Dixon JE. Pten. Annu Rev Biochem. 2014;83:641-69
20. McMullen JR, Amirahmadi F, Woodcock EA, Schinke-Braun M, Bouwman RD, Hewitt KA. et al. Protective effects of exercise and phosphoinositide 3-kinase(p110alpha) signaling in dilated and hypertrophic cardiomyopathy. Proc Natl Acad Sci U S A. 2007;104:612-7
21. Achttien RJ, Staal JB, van der Voort S, Kemps HM, Koers H, Jongert MW. et al. Exercise-based cardiac rehabilitation in patients with chronic heart failure: a Dutch practice guideline. Neth Heart J. 2015;23:6-17
22. Ooi JY, Bernardo BC, McMullen JR. The therapeutic potential of miRNAs regulated in settings of physiological cardiac hypertrophy. Future Med Chem. 2014;6:205-22
23. Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A. et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124:2136-46
24. Bei Y, Zhou Q, Sun Q, Xiao J. Telocytes in cardiac regeneration and repair. Semin Cell Dev Biol. 2016;55:14-21
25. Gomes EC, Silva AN, de Oliveira MR. Oxidants, antioxidants, and the beneficial roles of exercise-induced production of reactive species. Oxid Med Cell Longev. 2012;2012:756132
26. Zhang KR, Liu HT, Zhang HF, Zhang QJ, Li QX, Yu QJ. et al. Long-term aerobic exercise protects the heart against ischemia/reperfusion injury via PI3 kinase-dependent and Akt-mediated mechanism. Apoptosis. 2007;12:1579-88
27. Du WW, Li X, Li T, Li H, Khorshidi A, Liu F. et al. The microRNA miR-17-3p inhibits mouse cardiac fibroblast senescence by targeting Par4. J Cell Sci. 2015;128:293-304
28. Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard M. et al. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;104:330-5
Author contact
![]() Corresponding authors: Dr. Xiangqing Kong, Department of Cardiology, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing 210029, China. Tel:0086-25-84352775; Fax:0086-25-84352775; E-mail:xiangqingkong_njcom Dr. Junjie Xiao Cardiac Regeneration and Ageing Lab, School of Life Science, Shanghai University, 333 Nan Chen Road, Shanghai 200444, China. Tel: 0086-21-66138131; Fax: 0086-21-66138131; E-mail: junjiexiaoedu.cn.
Corresponding authors: Dr. Xiangqing Kong, Department of Cardiology, The First Affiliated Hospital of Nanjing Medical University, 300 Guangzhou Road, Nanjing 210029, China. Tel:0086-25-84352775; Fax:0086-25-84352775; E-mail:xiangqingkong_njcom Dr. Junjie Xiao Cardiac Regeneration and Ageing Lab, School of Life Science, Shanghai University, 333 Nan Chen Road, Shanghai 200444, China. Tel: 0086-21-66138131; Fax: 0086-21-66138131; E-mail: junjiexiaoedu.cn.
Citation styles
APA
Shi, J., Bei, Y., Kong, X., Liu, X., Lei, Z., Xu, T., Wang, H., Xuan, Q., Chen, P., Xu, J., Che, L., Liu, H., Zhong, J., Sluijter, J.PG., Li, X., Rosenzweig, A., Xiao, J. (2017). miR-17-3p Contributes to Exercise-Induced Cardiac Growth and Protects against Myocardial Ischemia-Reperfusion Injury. Theranostics, 7(3), 664-676. https://doi.org/10.7150/thno.15162.
ACS
Shi, J.; Bei, Y.; Kong, X.; Liu, X.; Lei, Z.; Xu, T.; Wang, H.; Xuan, Q.; Chen, P.; Xu, J.; Che, L.; Liu, H.; Zhong, J.; Sluijter, J.PG.; Li, X.; Rosenzweig, A.; Xiao, J. miR-17-3p Contributes to Exercise-Induced Cardiac Growth and Protects against Myocardial Ischemia-Reperfusion Injury. Theranostics 2017, 7 (3), 664-676. DOI: 10.7150/thno.15162.
NLM
Shi J, Bei Y, Kong X, Liu X, Lei Z, Xu T, Wang H, Xuan Q, Chen P, Xu J, Che L, Liu H, Zhong J, Sluijter JPG, Li X, Rosenzweig A, Xiao J. miR-17-3p Contributes to Exercise-Induced Cardiac Growth and Protects against Myocardial Ischemia-Reperfusion Injury. Theranostics 2017; 7(3):664-676. doi:10.7150/thno.15162. https://www.thno.org/v07p0664.htm
CSE
Shi J, Bei Y, Kong X, Liu X, Lei Z, Xu T, Wang H, Xuan Q, Chen P, Xu J, Che L, Liu H, Zhong J, Sluijter JPG, Li X, Rosenzweig A, Xiao J. 2017. miR-17-3p Contributes to Exercise-Induced Cardiac Growth and Protects against Myocardial Ischemia-Reperfusion Injury. Theranostics. 7(3):664-676.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.