Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(3):647-663. doi:10.7150/thno.16827 This issue Cite
Research Paper
STAT3/NF-κB-Regulated Lentiviral TK/GCV Suicide Gene Therapy for Cisplatin-Resistant Triple-Negative Breast Cancer
Wei-Ying Kuo1, Luen Hwu2, Chun-Yi Wu3, Jhih-Shian Lee1, Chi-Wei Chang4, Ren-Shyan Liu4,5 ![]()
1. Department of Biomedical Imaging and Radiological Sciences, School of Biomedical and Engineering, National Yang-Ming University, Taipei, Taiwan.
2. Molecular and Genetic Imaging Core/Taiwan Mouse Clinic, NRPB, Department of Nuclear Medicine and National PET/Cyclotron Center, Taipei Veterans General Hospital, Taipei, Taiwan.
3. Department of Biomedical Imaging and Radiological Science, China Medical University, Taichung, Taiwan.
4. Department of Nuclear Medicine, Taipei Veterans General Hospital, Taipei, Taiwan.
5. Biomedical Imaging Research Center, Department of Biomedical Imaging and Radiological Sciences, School of Biomedical and Engineering, Institute of Clinical Medicine, School of Medicine, National Yang-Ming University.
Received 2016-7-13; Accepted 2016-10-24; Published 2017-1-15
Abstract

Triple-negative breast cancer (TNBC) represents approximately 20% of all breast cancers and appears resistance to conventional cytotoxic chemotherapy, demonstrating a particularly poor prognosis and a significantly worse clinical outcome than other types of cancer. Suicide gene therapy has been used for the in vivo treatment of various solid tumors in recent clinical trials. In tumor microenvironment, STAT3/NF-κB pathways are constitutively activated in stromal cells as well as in cancer stem cells (CSCs). In this study, we have cloned a novel STAT3/NF-κB-based reporter system to drive the expression of herpes simplex virus thymidine kinase (HSV-TK) against breast cancer. Lentiviral vector expressing HSV-TK under the regulation of STAT3/NF-κB fused response element was developed. In this setting, we exploited the constitutive STAT3/NF-κB activation in tumors to achieve higher transgene expression than that driven by a constitutively active CMV promotor in vivo. An orthotropic MDA-MB-231 triple negative breast cancer mouse model was used for evaluating the feasibility of STAT3-NF-κB-TK/GCV suicide gene therapy system.
The basal promoter activity of Lenti-CMV-TK and Lenti-STAT3-NF-κB-TK in MDA-MB-231 cells was compared by 3H-FEAU uptake assay. The Lenti-CMV-TK showed ~5 fold higher 3H-FEAU uptake then Lenti -STAT3-NF-κB-TK. In clonogenic assay, cells expressing Lenti-CMV-TK were 2-fold more sensitive to GCV than Lenti-STAT3-NF-κB-TK transduced cells. In vitro effect of STAT3-NF-κB-induced transgene expression was determined by 10ng/mL TNF-α induction and confirmed by western blot analysis and DsRedm fluorescent microscopy. In vivo evaluation of therapeutic effect by bioluminescence and [18F]FHBG microPET imaging indicated that Lenti-STAT3-NF-κB-TK showed more tumor growth retardation than Lenti-CMV-TK when GCV (20 mg/kg) was administered. The invasiveness and expression of cancer stem cell markers were both decreased after STAT3/NF-κB-regulated HSV-TK/GCV therapy. Moreover, STAT3/NF-κB signaling targeting could further sensitize tumor cells to cisplatin. This study successfully established a theranositic approach to treat triple-negative breast cancer via STAT3-NF-κB responsive element-driven suicide gene therapy. This platform may also be an alternative strategy to handle with drug-resistant cancer cells.
Keywords: Triple-negative breast cancer, TK/GCV suicide gene therapy, Cisplatin resistance.
Introduction
Triple-negative breast cancer (TNBC) is negative for human epidermal growth factor receptor 2 (HER2), estrogen receptors (ER), and progesterone receptors (PR) [1, 2], presenting a poor prognosis and a high risk of recurrence than other tumor types [3-5]. The main treatments for breast cancer include surgery, radiotherapy, chemotherapy, hormone therapy and targeted therapy. However, there is currently no preferred standard chemotherapy for treated patients with it. There are evidences that initiation, maintenance and metastasis of tumors are through cancer stem cells (CSCs) in tumors [6], and that CSCs are crucial for TNBC maintenance and progression as well [7]. These issues have prompted the development of more effective therapies against CSCs in TNBC, while studying the characteristics of TNBC remains a challenge.
Recent studies revealed that deregulated overexpression of STAT3/NF-κB signaling pathway was associated with self‐renewing and maintenance of CSCs [8, 9], which also appears to confer resistance to tumor therapies as well. The physically and functionally interaction of STAT3 and NF-κB in chemotherapy resistant cells was also observed [10, 11]. Cisplatin has long been used in clinical treatment regiments because of it exerts effectiveness on breast cancer by directly binding to DNA, causing the formation of DNA-adducts with resultant cytotoxicity. However, cisplatin-resistance can eventually develop in patients who relapse after a favorable initial response [12]. Numerous investigations proved that the persistent expression of STAT3 and NF-κB contributes to cisplatin resistance through upregulating anti-apoptotic factors (Bcl-2 and Bcl-xL) and multidrug resistance molecules [13, 14], thus is considered a potential therapeutic target for breast cancer treatment. Although STAT3 inhibition showed antitumor potential, blockade of STAT3 signaling may also activate NF-κB pathway [15]. Regarding to this complex crosstalk signaling exists between them; presently there is no effective strategy to block STAT3/ NF-κB signaling.
One of the most common suicide gene/ prodrug combinations is herpes simplex virus type 1 thymidine kinase (HSV-TK)/ ganciclovir (GCV). The triphosphate GCV produced by the transfected cells is a cytotoxic compound that subsequently induces apoptosis and could have a 'bystander effect' that may increase the proportion of apoptotic cells and minimize the demand to transfect all cancer cells [16]. There are several therapeutic genes could induce apoptosis of cancer cells [17], among them HSV-TK/GCV system is the most constantly used one with a proven therapeutic value in many kinds of cancer [18]. To enhance the tumor selectivity of the suicide gene system, several natural or synthetic promoters which could be induced by radiation, hypoxia or oxidative stress have been used to drive TK expression [19-22]. However, the expressions of suicide gene under these inducible promoters were not sufficient enough to acquire a satisfied therapeutic benefit.
In the current study, we successfully established a theranositic approach to treat TNBC via STAT3-NF-κB responsive element-driven HSV-tk gene/GCV system. [18F]FHBG microPET imaging and bioluminescence-based optical imaging were used to evaluate the transfection efficiency of therapeutic gene and to monitor therapeutic effect in living mice. The results showed a reduction of tumor growth with decreased invasiveness and CSC marker—ALDH expression. Furthermore, we found that this therapeutic strategy could sensitize TNBC to cisplatin. Our study suggests that targeting STAT3/NF-κB pathway has the potential to eradicate not only cancer cells but also CSCs through inhibition of inflammatory cytokines in tumor microenvironment.
Materials and methods
Cell culture
The breast cancer cell line MDA-MB-231 was obtained from the American Type Culture Collection (Manassas, VA, USA) and cells were cultured in ATCC-formulated Leibovitz's L-15 Medium (Catalog No. 30-200) supplemented with 10% fetal bovine serum (Hyclone, Logan, Utah). Human kidney fibroblasts (293T) cells (from Dr Yi-Jang Lee, Yang-Ming University, Taipei, Taiwan) were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin in a 5% CO2 humidified incubator at 37˚C.
Plasmid construction and generation of stable cell lines
Dual Reporter Plasmid, which carries a dual-fused reporter gene controlled by CMV promoter, was constructed following the design of Clontech (pEGFP-Fluc vector, Clontech Laboratories Inc.). To generate MDA-MB-231 cells stably expressing CMV-EGFP-Fluc, cells were transfected with 1μg of plasmid DNA using 1.5 μL of transfection reagent (Bio-Rad Laboratories) in serum-free medium. Two days after the transfection, the cells were replated and 500 cells were seeded in 10-mm culture dishes with continually grown in the presence of 700 μg/mL G418 of (Sigma-Aldrich) for approximately 2 weeks. G418 resistant colonies were isolated and cultured in G418 containing media.
Lentiviral construction and transduction for transgene expression
To specific reflect the gene expression of STAT3, we synthesize palindromic motif with a spacing of 4 bp which selectively binds to STAT3 homodimer as reported by H. M. Seidel et. al [23]. The palindromic sequence elements—[TTCCCGAA]n were synthesis by polymerase chain reaction (PCR) with forward primer STAT3-Up (5'phos-GTGACCTTCCCGAAG-3'), reverse primer STAT3-Dn (5'phos-GTCACCTTCGGGAAG-3') and the proof-reading KOD Hot Start DNA Polymerase (cat. 71086-3, Novagen Inc., USA). We used pE9-FLuc, a reporter construction from our lab as vector, in which we replaced the E9 element with seven tandem repeats of the STAT3 transcription responsive elements by restriction enzyme BstEII and treated the vector molecule with enzyme calf intestinal phosphatase (CIP) (cat. M0290S, New England Biolabs Inc., USA) before ligation to remove the 5'-phosphates to avoid self-ligation. This plasmid identified as pSTAT3-FLuc.
In order to construct the hybrid promotor of STAT3 and NF-κB, first, we used restriction enzyme Acc65I to digest pSTAT3-FLuc and the 3´→5´ exonuclease activity of Klenow DNA Polymerase (cat. M0210S, New England Biolabs Inc., USA) to generate blunt ends from a 3´-overhang. After this, HindIII was used to obtained STAT3 response element from pSTAT3-FLuc as insert. The pSTAT3-NF-κB-luc2P vector was constructed by inserted STAT3 response element into the restriction enzyme Eco47III and HindIII digested NF-κB-luc2P as parent vector from our lab. The dual- fusion reporter gene DsRedm-ttksr39 was then amplified by PCR from pDsRedm-ttksr39 vector and digested with NotI and XbaI along with pSTAT3-NF-κB-luc2P vector to generate pSTAT3-NF-κB-DsRedm-ttksr39. This plasmid was further cloned into a lentivirus vector— CDH-CMV-MCS-EF1-Puro (National RNAi Core Facility of Academia Sinica, Taiwan.) which was referred as Lenti-STAT3-NF-κB-DsRedm-tk. To generate a positive control vector, the STAT3-NF-κB hybrid promotor was replaced by a constitutively active cytomegalovirus (CMV) promotor, referred as Lenti-CMV-DsRedm-tk.
Immunofluorescence staining for HSV-TK expression
For immunofluorescent staining frozen tissues sections were fixed in 4% paraformaldehyde, washed with PBS and permeated with 0.1% Triton X-100 for 5 minutes before blotting with blocking solution (Normal goat serum, #5425, Cell signaling, CA) for 1 h at room temperature, followed by 1 h of incubation with anti-E-cadherin antibody (cat. MABT26, Millipore, MA) and anti-vimentin antibody (cat. MAB3400, Millipore, MA) (diluted 1:100 in 1% BSA in PBS) at 4˚C. Sections were washed three times in PBS, each wash lasting 5 min then stained with Cy5-conjugated goat anti- rat immunoglobulin G (IgG) (1:500, cat. ab6565, Abcam, Cambridge, UK) and Cy5-conjugated goat anti- mouse IgG (1:500, cat. ab6563, Abcam, Cambridge, UK) for 1 h at room temperature, which were used to visualize the location of E-cadherin and vimemtin, respectively. The procedures for cultured cells were the same as that for tissue except that the cells were fixed directly with 4% paraformaldehyde for 20 minutes. Sections were washed three times in PBS and blotting with blocking solution for 45 minutes at room temperature. The slides were then incubated with an anti-HSV-1 Thymidine Kinase antibody (cat. sc-28038, Santa Cruz, CA) in dilution buffer (diluted 1:80 in 1% BSA in PBS) at 37˚C for 1 h followed by three additional PBS washes. The secondary antibody which was a donkey anti-goat IgG that was conjugated with Cy5 (1:500, ab6566, Abcam, Cambridge, UK), and was incubated with slides for 1 h in the dark. Sections were sealed under glass cover slips using VECTASHIELD™ Mounting Medium with DAPI (cat. H-1200, Vector Labs, Burligame, CA) to limit photobleaching and counterstain the cell nuclei. The fluorescence within the samples was analyzed using an Olympus laser scanning confocal microscope (Olympus FV1000, America, Inc.).
Lentiviral particle preparation
Lentivirus was produced from the 293T transfectants with co-transfection of Lenti-STAT3-NF-κB-DsRedm-tk or Lenti-CMV-DsRedm-tk along with pCMV-ΔR8.91 and pMD.G plasmids (Nantioal RNAi Core Facility of Academia Sinica, Taiwan.) (with a ratio of 5:4.5:0.5) using jetPET reagent (DNA:PEI reagent=10μg:20μL) according to the recommended protocol (Polyplus-Transfection, cat. 101-10N, Illkirch, France). Sixteen hours post-transfection, the medium was replaced with regular growth medium. The conditioned medium of the transfected cells containing lentiviral particles were collected at 24 h and 48 h after refreshment.
Clonogenic assay
To examine and compare the cell reproductive death among the two kinds of tk virus, MDA-MB-231 cells were seeded in T25 flasks and infected in parallel with Lenti-STAT3-NF-κB-DsRedm-tk and Lenti-CMV-DsRedm-tk in serum-free medium. Generally, after selecting the appropriate MOIs in pretests, MOIs of 2 were selected for MDA-MB-231 cells. Four hr post transfection, serum-free medium was replaced with full medium with 10% fetal bovine serum and 25 μM ganciclovir (GCV, cat. G2536, Sigma-Aldrich, St. Louis, MO). Cells were incubated in the presence of ganciclovir for 7 days before they were re-seeded at a seeding density of 500 cells/dishes in 60 mm2 dishes. After a two weeks' incubation at 37˚C, the colonies were fixed with 3.7% paraformaldehyde, stained with crystal violet (0.5% w/v) and counted using a stereomicroscope. The data is reported as mean±S.D., n=3.
In vitro 3H-FEAU uptake study
Quantitative evaluation of the uptake of radiotracer in vitro of TK in cells transient transfected with Lenti-CMV-DsRedm-tk and Lenti-STAT3-NF-κB-DsRedm-tk was measured by 3H-FEAU uptake assay as previously described [24]. In brief, MDA-MB-231 cells transfected with equal amount of Lenti-CMV-DsRedm-tk and Lenti-STAT3-NF-κB-DsRedm-tk plasmid with untransfected cells (control) were seeded in 12-well plates (1×105 per well) in triplicate and incubated overnight, followed by 10 ng/mL TNF-α (R&D systems, cat.210-TA-005, Minneapolis, MN, USA) or DMSO treatment for 24 h. Culture medium was then replaced by 0.5 mL of medium containing 29-fluoro-29-deoxyarabinofuranosyl-5-ethyluracil (3H-FEAU) (7.4 kBq [0.2 μCi]), and incubated for 120 minutes at 37˚C with 5% CO2. The medium and 0.5 mL of phosphate-buffered saline used to rinse the wells were then collected in scintillation vials. Cells were lysed by 75 μL of CytoBuster Protein Extraction Reagent (Novagen, US) and washed once with PBS. Then the cells were transferred to 1.5 mL tubes and centrifuged at 12,000 rpm for 1 minute. Supernatant aliquots (100 μL) were transferred to scintillation vials, and the remnant was used to determine total protein concentration. Thereafter, 5 mL of scintillation solution (Perkin-Elmer, Shelton, CT) was added to each vial and radioactivity of 3H-FEAU in samples was measured using a Packard Tri-Carb LS scintillation counter (Perkin-Elmer, Shelton, CT). Radioactivity concentration ratio of the cell pellet versus medium was calculated, and normalized by total protein concentration ([dpm/g of cells]/[dpm/g of medium]/total protein concentration). The ratios of accumulated radiotracer in cells represent the gene expression level of HSV-tk.
Tumor xenograft studies
All the animal experiments in this study were in accordance with the institutional animal welfare guideline of National Yang-Ming University (Taipei, Taiwan). Immunodeficient female mice (BALB/cAnNCrj-nu/nu, six weeks old, 17 gm) were purchased from BioLASCO Taiwan Co. Ltd. Animals were injected of 2×106 MDA-MB-231-EGFP-Fluc cells in 100 μL of PBS:Matrigel (BD Biosciences, cat. 354248, San Jose, CA) mixture (PBS: Matrigel = 1:1) orthotopically into the inguinal mammary fat pads of these mice. When tumor size reached approximately 15 ~ 25 mm3, the mice were divided into the following treatment groups (n=5 tumors/group), and treated with (1) PBS, (2) GCV (20 mg/kg body weight), (3) cisplatin (1 mg/kg body weight), (4) Lenti-CMV-DsRedm-tk with GCV (20 mg/kg body weight), (5) Lenti-STAT3-NF-κB-DsRedm-tk, (6) Lenti-STAT3-NF-κB-DsRedm-tk with GCV (20 mg/kg body weight) and (7) Lenti-STAT3-NF-κB-DsRedm-tk with GCV (20 mg/kg body weight) and cisplatin (1 mg/kg body weight). Tumors were injected twice intratumorally with PBS or vector at a dose of 1×108 PFU in a total volume of 15 μL Opti-MEM® I Reduced Serum Medium (Thermo Fisher Scientific, Waltham, MA, USA, cat. 31985-062). Two days later, mice were given ganciclovir (cat. G2536, Sigma-Aldrich, St. Louis, MO) 20 mg/kg/day intraperitoneally (i.p) once daily for 14 days. Mice injected with Lenti-STAT3- NF-κB-DsRedm-tk, did not receive GCV, were served as an additional control to observe for unspecific Lentivirus effects on tumor growth. Control groups were injected with PBS. After 1 weeks of HSV-tk/GCV gene therapy, mice were treated with cisplatin (1 mg/kg, i.p., 2 times/week) in the combination group. To monitor tumor growth, the longest and shortest diameters of the tumors were measured using a digital caliper twice weekly. Tumor volume (mm3) was computed according to the following formula: tumor volume= (length×width2) × 0.523. Before each treatment, mice were weighed to monitor the systemic toxicity of the treatments.
In vivo Fluc bioluminescence image
Mice were anaesthetized with isoflurane and then received injection of D-luciferin (150 mg/kg body weight diluted in PBS). Fifteen minutes later, mice were placed in the imaging chamber, and photo counts were acquired for 1-5 minutes by the optical imaging system (IVIS 50Imaging System; Xenogen Technology). Signal intensity quantification and analysis were performed using Living Image Software (version 2.50; Xenogen Technology) provided by the manufacturer. Bioluminescent signal was recorded as maximum photons/s/centimeter2/steradian (photon/s/cm2/sr), represented in a pseudo-color photo count manner and superimposed on the photographic image, displaying both bioluminescence intensity and the mice anatomy.
Positron Emission Tomography imaging and image analysis
Positron Emission Tomography (PET) imaging with 9-[4-[18F]fluoro-3-(hydroxymethyl)butyl]guanine ([18F]FHBG) was performed at day 3 and day 10 during the in vivo gene therapy studies, corresponding to before and after GCV treatment. [18F]FHBG is one of the PET report probe for imaging herpes simplex virus type 1 thymidine kinase (HSV1-TK) and mutant HSV1-sr39tk report gene [25]. [18F]FHBG was synthesized by nucleophilic method as described previously [26]. Imaging was performed using a microPET R4 scanner (CTI Concorde Microsystems, Knoxville, TN, USA), equipped with a small-animal PET Manager, (version 2.2.4; Concord Microsystems) for data acquisition and imaging process.One hour prior to imaging, mice were injected i.v. with 150 μCi [18F]-FHBG in 100 μL. Mice were then anesthetized with 2% isoflurane in oxygen at 2L/ min for static imaging in the MicroPET. PET data were acquired for 10 minutes and reconstructed with a filtered background projection probability algorithm. CT images were acquired by using MicroSPECT/CT (Triumph II XOCT®, GE Healthcare, Northridge, CA, USA) preceded by CT scans for anatomic reference. PET and CT images were coregistered by PMOD software. Quantification of PET signal was performed by drawing 3D volume of interest (VOI) using PMOD software (http://www.pmod.com/web/). The maxium intensity of the muscle VOI, based on the percentage of injected dose per gram (%ID/g), was subtracted from each tumor VOI to normalize for background. Images were presented in false-color volumetric renderings generated in PMOD.
Cell invasion assay
Cell invasion assay was performed following the previous literature with a Boyden chamber (pore size: 8 μm, 24-well; BD Biosciences) [27]. Briefly, 2.5×105 cells in serum-free medium were plated on upper transwell chambers percoated with Matrigel (BD Biosciences, cat. 354248, San Jose, CA) (1:3 dilution with medium), and 10% fetal bovine serum-containing medium was added in the lower chamber as a chemoattractant. After 24h, non-invading cells on the upper side of the filter were removed with cotton swabs. The bottom of the chamber insert were fixed in 4% formaldehyde and stained with Coomassie Brilliant Blue. Invading cells were counted under a light microscope.
Histological analysis
Tissue sections were fixed in 4% paraformaldehyde, washed with PBS and permeated with 0.1% Triton X-100. Samples were then incubated with blocking solution (Normal goat serum, cat. 5425, Cell signaling) for 1 h at room temperature. The tumor tissue slides were stained with mouse anti-human E-cadherin (1:100, MABT26, Millipore, MA) and rat anti-human Vimentin (1:100, CBL202, Millipore, MA) at 4 °C overnight. For immunofluorescence, Cy5-conjugated goat anti-mouse immunoglobulin G (IgG) (1:500, cat. A10524, Thermo Fisher Scientific) and Cy5-conjugated goat anti-rat IgG (1:500, cat. A10525, Thermo Fisher Scientific) were used and incubated for 1 h at room temperature. Nucleus is counterstained with DAPI. Images were obtained by Olympus laser scanning confocal microscope (Olympus FV1000, America, Inc.) and analyzed with ImageJ software. For immunohistochemistry, the reaction was visualized by the Dako Liquid DAB+ Substrate-Chromogen Solution (Dako, cat. K3468).
Western blot analysis in tumor tissue extracts and in lysates of tumor cell lines
Cultured cells were placed on ice, washed in ice-cold PBS and lysed using RIPA lysis buffer (cat. 20-188, Millipore, MA) containing Protease Inhibitor Cocktail Set I (cat. 539131, Millipore, MA). Cells were then scraped and incubated for 20 minutes at 4 ˚C. The lysate was spun down at 14,000 g for 20 minutes at 4 °C. The supernatant was removed for protein content measurement. Tissue extracts were prepared from frozen tumor tissues. Briefly, the tissue was cut into smaller pieces, and the total protein was extracted by TOTAL PROTEIN EXTRACTION KIT (cat. 2140, Millipore, MA). The protein content of cell lysates and tissue extracts were measured using Bradford reagent (BIO-RAD, Hercules, CA). A measure of 40 μg of total cell extract was separated on 10% SDS-polyacrylamide gel (30% Acrylamide/Bis Solution, 29:1, cat. 1610157, BIO-RAD, Hercules, CA) and transferred to Poly Screen polyvinylidene difluoride (PVDF) membranes (cat. NEF1002001PK, Perkin-Elmer life sciences, Boston, MA). The blot was blocked in 4% milk in TBST (10 mM Tris-HCl, 150 mM NaCl, and 0.05% Tween 20) for 1 hour, and then incubated with goat anti-HSV-1 Thymidine Kinase antibody (1:200, cat. sc-28038, Santa Cruz, CA), rabbit anti-ALDH1A1 antibody (1:1000, cat. ab9883, Abcam, Cambridge, UK), mouse anti-CD133 antibody (1:1000, Miltenyi Biotec GmbH, Bergisch Gladbach, Gemany), rabbit anti-snail1 (1:500, cat. GTX125918, GeneTex, USA), anti-Akt/PKB antibody (cat. 05-591, Millipore, MA) or anti-phospho-Akt antibody (cat. 05-1003, Millipore, MA) as primary antibody for 4 ˚C overnight. After repeated wash in TBST, the membrane was incubated with secondary anti-goat, anti-rabbit or anti-mouse IgG antibodies conjugated to horseradish peroxidase (1:5000, Abcam, Cambridge, UK) for 1 hour at room temperature. Immunoreactive bands were detected using LumiFast Chemiluminescence Detection Kit (T-Pro Biotechnology, cat. JT96-K001S, Taiwan, ROC). Image capture was performed on an ImageQuant™ LAS 4000 (GE Healthcare Bio-Sciences Corp., USA).
TUNEL assay
Tumor xenografts of MDA-MB-231 cells infected with therapeutic reporter gene driven by STAT3/NF-κB activity or CMV promoter, and treated with GCV alone or combination with cisplatin were used for evaluating the prodrug-induced treatment effects by detecting apoptotic cells. For apoptosis quantification, tissue of each group was frozen in OCT (TissueTek) and sectioned to 7 μm by using Cryomicrotome (CM3050 S, Leica Microsystems, Bensheim, Germany). The xenograft sections were processed for in situ immunocytochemical localization of nuclei exhibiting DNA fragmentation by the technique of terminal deoxynucleotidyl transferase (TdT)-mediated dUTP digoxigenin nick-end labeling (TUNEL) with the use of In Situ Cell Death Detection Kit (POD, Roche Applied Science, cat. 11684817910). The protocols were followed according to the manufacturer's instructions. Briefly, after incubation with the TUNEL reaction mixture containing TdT and fluorescein-dUTP in a chamber at 37 °C for 1 h, a mixture of antidigoxigenin-peroxidase and substrate-chromagen (3′-diaminobenzidine tetrahydrochloride [DAB], Dako, cat. K3468) was used and tissue sections were counterstained with hematoxylin. In this assay, TUNEL controls were performed by incubating tissue slides with 100 μL label solution. The nuclei of apoptotic cells were presented deep brown and counted in ten randomly selected different field areas without necrosis in each slide under a light microscope at ×400 magnification. The apoptosis index (AI) was defined as the percentage of TUNEL-positive cells to all counted cells.
Statistical analyses
Results were presented as mean ± SD. Data was conducted by the one-way analysis of variance (ANOVA) followed by Student's t-test, as appropriate with GraphPad Prism v.5 software. P-value < 0.05 was considered statistically significant.
Results
STAT3-NF-κB-controlled transgene expression
As tumor is exist in an inflammatory micro-environment which constitutively active STAT3 and NF-κB signaling in cancer cells, we designed a reporter system in which we cloned into a Lentivirus vector an hybrid promotor of STAT3 and NF-κB to drive the expression of both the HSV-TK therapeutic protein and the red fluorescence (DsRedm) reporter with genetic engineering methods (Figure 1A). We then performed 3H-FEAU uptake assay to evaluate the basal STAT3/NF-κB activities in MDA-MB-231 cells, by transfecting cells with the lenti-control (Lenti-CMV-DsRedm-tk) or Lenti-STAT3-NF-κB-DsRedm-tk construction. Lenti-CMV-DsRedm-tk transfected MDA-MB-231 cells showed ~10 fold higher 3H-FEAU uptake compared to control MDA-MB-231 cells, and Lenti-STAT3-NF-κB-DsRedm-tk transfected cells showed 4.8 fold lower 3H-FEAU uptake than Lenti-CMV-DsRedm-tk transfected cells (Figure 1B). For further confirmation, we performed immunofluorescence staining with HSV-TK specific antibody in Lenti-CMV-DsRedm-tk or Lenti-STAT3-NF-κB-DsRedm-tk transfected cells (Figure 1C). In MDA-MB-231 cells, the STAT3-NF-κB activity was slightly inducible by NF-κB activator, we then assessed if STAT3-NF-κB-regulated vectors could be used to target suicide gene therapy in breast cancer cells. In this study, we used a successfully established prodrug activating HSV-tk/GCV gene therapy approach [28]. To compare the efficacy of STAT3-NF-κB-regulated HSV-tk/GCV therapy with constitutive HSV-tk/GCV therapy, MDA-MB-231 cells were infected at 2 MOIs and exposed to 25 μM GCV continuously for 5 days. In a clonogenic assay, which was used to determine the proliferation capacity of the surviving cells after treatment, the average cell survival rate of MDA-MB-231 cells treated with GCV (82.1%) was much higher than that of cells transfected with Lenti-STAT3-NF-κB-DsRedm-tk and treated with GCV (9.9%) (Figure 1D, Table 1). There were barely 4.7% of the cells transfected with Lenti-CMV-DsRedm-tk recovered and formed colonies (Figure 1E). The results indicated that constitutive HSV-tk /GCV therapy was more effective than STAT3-NF-κB-regulated HSV-tk /GCV therapy in vitro.
STAT3-NF-κB-controlled transgene expression. (A) Lentivirus vector driving the expression of DsRedm-HSV-TK (B) Forty-eight hr after transducing the MDA-MB-231 cells with Lenti-CMV-tk or Lenti-STAT3-NF-κB-tk vectors, basal TK activity was measured by 3H-FEAU uptake. *p<0.001. Quantified results represent mean ± SD (n=3). Statistics (PBS versus Lenti-CMV-tk or PBS versus Lenti-STAT3-NF-κB-tk) were analyzed by unpaired Student's t-test. (C) The expression of DsRed and TK was confirmed by HSV-TK staining. Scale, 10 μm. (D) The cytotoxic effects of Lenti-CMV-tk/GCV-transfected cells and Lenti-STAT3-NF-κB-tk/GCV-transfected cells were consistent with the TK expression level in MDA-MB-231 cells. (E) Clonogenic assay for the MDA-MB-231 cells infected with two MOIs of each of the two viruses are shown. The pictures of 6 cm2 dishes (3 replicates) with produced MDA-MB-231 colonies (top). Quantification of the percent survival of the MDA-MB-231 cells as presented by a bar chart (bottom). The data is shown as mean±SD (n=3). *p=0.002, Lenti-CMV-TK/GCV versus Lenti-STAT3-NF-κB-TK/GCV by unpaired Student's t-test.
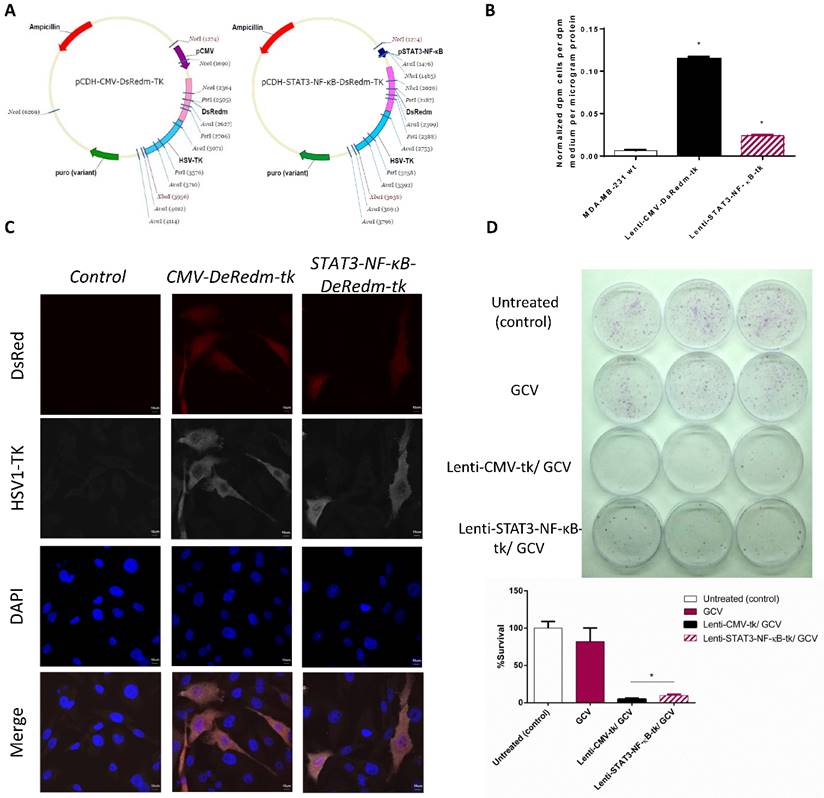
Cytotoxic effects of Lenti-CMV-tk/GCV and Lenti-STAT3-NF-κB-tk/GCV in MDA-MB-231 cells.
| HSV-TK expression level | Survival rate (%) | |
|---|---|---|
| Vehicle Treatment | 0 | 82.1 |
| Lenti-CMV-tk/GCV | 4.8 | 4.7 |
| Lenti-STAT3-NF-κB-tk/GCV | 1 | 9.9 |
STAT3-NF-κB-regulated HSV-TK/GCV therapy is effective in MDA-MB-231 cell line in vitro
We examined whether the STAT3/NF-κB activity could further increase in MDA-MB-231 cells by using their activators TNF-α and IL-6. MDA-MB-231 human breast cancer cells were plated in 24-well plate and infected with lenti-STAT3-NF-κB-DsRedm-tk to measure its inducible STAT3/NF-κB activity. At 48 h, cells were treated with TNF-α (10 ng/ mL) and PBS control, respectively. HSV-TK expression level were measured 6 h later which showed 1.95-fold induction in response to TNF-α (Figure 2A). There are no significant induction of cells transfected with Lentiviral vector expressing HSV-TK under the constitutively active CMV promoter. Since TNF-α is the activator of NF-κB as well as STAT3 [29], we therefore assessed the HSV-TK protein expression in Lenti-STAT3-NF-κB-DsRedm-tk transfected cells after TNF-α administration. In line with the previous reports [30, 31], only few inducible NF-κB and STAT3 activities were observed in MDA-MB-231 cells exhibiting constitutive NF-κB and STAT3 activities (Figure 2B).
In vitro effect of STAT3-NF-κB-induced transgene expression. (A) Comparison of STAT3-NF-κB basal activity and induction in MDA-MB-231 cells. Cells were transduced with Lenti-STAT3-NF-κB-DsRedm-TK and treated with either PBS or TNF-α. At 24h later, TK expressions were measured by 3H-FEAU uptake. Quantified results are represented as mean ± SD (n=3). *p<0.001, Lenti-STAT3-NF-κB-DsRedm-TK versus Lenti-STAT3-NF-κB-DsRedm-TK/TNF-α by unpaired Student's t-test. (B) Immunoblot analysis of HSV-TK in MDA-MB-231 cells transduced with Lenti-CMV-DsRedm-tk or Lenti-STAT3-NF-κB-DsRedm-tk. Total protein (40 μg) was loaded on the gels. β-actin was used as a control of equal loading. (C) The effect of STAT3-NF-κB-induced transgene expression was confirmed by DsRed analysis using confocal microscopy. Scale, 20 μm.
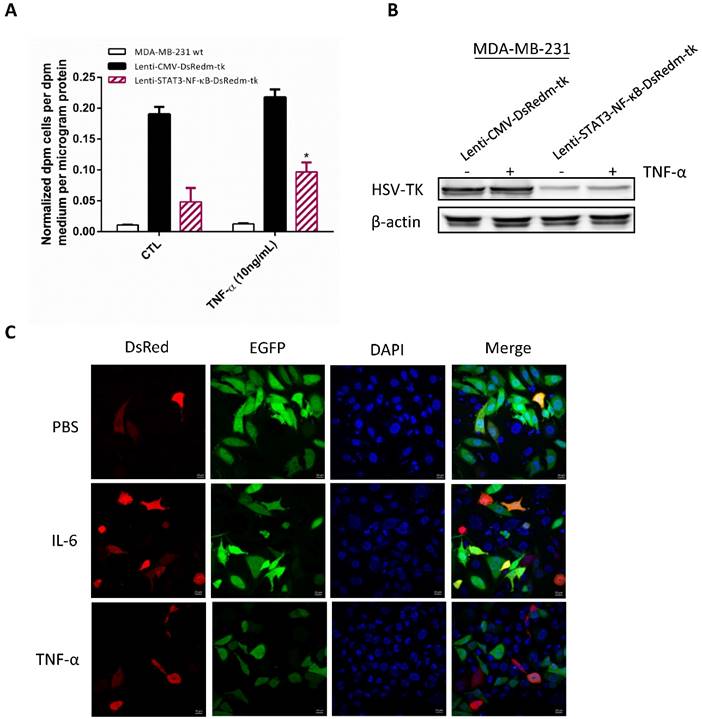
To investigate whether this increase in HSV-TK activity correlates with expression of the red fluorescent protein (DsRedm), MDA-MB-231 cells infected with Lenti-STAT3-NF-κB-DsRedm-tk were treated with TNF-α and IL-6, respectively. After 24 h, the enhanced expression of DsRedm was confirmed by confocal microscopy in response to TNF-α or IL-6 (Figure 2C). The level of HSV-TK in tumor cell correlates with the level of STAT3/NF-κB activity, thereby allowing us to indirectly monitor the therapeutic gene expression. However, the GCV-mediated cell killing and bystander effect is not enhanced in Lenti-STAT3-NF-κB-DsRedm-tk transduced cells with TNF-α induction as measured by clonogenicity assay. Average cell survival rate of Lenti-STAT3-NF-κB-DsRedm-tk transduced MDA-MB-231 cells treated with TNF-α prior to GCV administration (7.5%) was higher than that of cells transduced with Lenti-STAT3-NF-κB-DsRedm-tk with GCV treatment (4.2%) (Figure S1A). It could be the tumorigenic property of TNF-α which results in a further activation of oncogene such as Akt in residual MDA-MB-231 tumor cells [32]. Our result show that TNF-α is able to induce Akt activation in MDA-MB-231 cells (Figure S1B).
Evaluation of the effect of STAT3-NF-κB-regulated suicidal gene therapy in vivo by optical bioluminescence image
Because STAT3-NF-κB-regulated HSV-tk/GCV suicide gene therapy was efficient in MDA-MB-231 cells in vitro, we next evaluated its therapeutic effect in vivo. To study this therapeutic method to a clinically relevant model, MDA-MB-231 tumor cells stably expressing Fluc were implanted orthotopically into nude mice. When the average size of tumors reached approximately 20 mm3, mice were received intratumoral injection of a lentiviral carrying the HSV-tk suicide gene controlled by CMV and STAT3-NF-κB activity, respectively. After 2 days of virus injection, mice were administrated either 20 mg/ kg of GCV or equal volumes of PBS intraperitoneally. Tumor growth was monitored by Fluc-based bioluminescence imaging at indicated date. Control tumors treated with PBS or GCV alone grew faster than other groups in which thymidine kinase was expressed and showed similar exponential growth, revealing that the dosage of GCV we used was not toxic to tumor cells. Primary tumor growth of mice was effectively curbed with an intratumoral injection of Lenti-STAT3-NF-κB-tk followed by GCV treatment (Fig. 3A), the Fluc signal significantly decreased in comparison with Lenti-CMV-tk-infected mice on day 4, and drastically dropped down on day 7 (Fig. 3B), whereas Fluc signal in control group was continuously increasing over time. Histologic analysis of the excised tumor tissue collected at the end of gene therapy was also performed by hematoxylin and eosin (H&E) staining (Supplementary Fig. 2). Body weight of the mice receiving different therapeutic regiments are shown in Figure 3C. There was no significant difference in body weight among treated groups compared with the PBS-treated group during the treatment course of HSV-tk/GCV gene therapy.
In vivo suicide gene therapy in an STAT3-NF-κB-controlled tumor environment. Orthotropic breast tumor-bearing mice were giving GCV (20 mg/kg) or PBS over the course of tumor progression. (A) Tumor growth curve of the orthotropic MDA-MB-231 breast tumor-bearing mice treated with STAT3-NF-κB-guided suicide gene therapy, mice were giving GCV (20 mg/kg) or PBS over the course of tumor progression. Data are presented as the mean tumor volume ± SD of the surviving animals in each group. All groups contained five mice. *p=0.015, Comparison of tumor volume in control (PBS) versus Lenti-STAT3-NF-κB-tk/GCV treated group by unpaired Student's t test. (B) Tumor burden was monitored with IVIS at day 0, day 4 and day 7. Representative bioluminescent images and quantification shows enhanced tumor killing effect in Lenti-STAT3-NF-κB-tk/ GCV treated mice. Representative images of tumor tissues treated with PBS, GCV, Lenti-CMV-tk/ GCV and Lenti-STAT3-NF-κB-tk/ GCV, respectively. The strongest inhibition rate was observed in the mice treated with Lenti-STAT3-NF-κB/GCV. (C) The body weight of mice during the period of monitoring was not significantly different between groups.
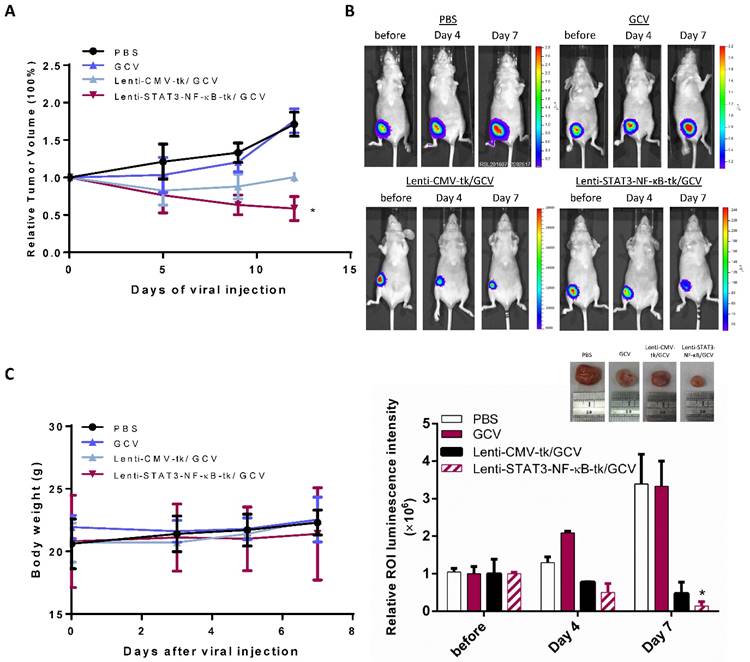
An efficient therapeutic potential by STAT3-NF-κB- regulated HSV- TK/GCV therapy was observed in MDA-MB-231 orthotopic xenograft model
In our design, HSV-TK expression is regulated by STAT3 and NF-κB activity which are constitutively activated by tumor environment. We performed [18F]FHBG PET imaging to non-invasively detect HSV-TK expression in cellular/gene therapy and evaluate its therapeutic effect. Animals were subjected to microPET imaging prior to GCV administration and 8 days post-drug administration. The expression of HSV-TK with [18F]FHBG was clearly imaged in Lenti-CMV-tk- and Lenti-STAT3-NF-κB-tk-infected tumors of living animals before starting GCV treatment. In contrast, GCV treated mice showed decreased [18F]FHBG accumulation within the tumors at 8 days post treatment (Fig. 4). Uptake in Lenti-CMV-tk-infected tumors of treated mice decreased from 1.73 ± 0.41 (n=3) to 0.75 ± 0.14 (n=3) %ID/ g, while uptake in Lenti-STAT3-NF-κB-tk-infected tumors of treated mice decreased from 1.36 ± 0.30 (n=3) to 0.33 ± 0.12 (n=3) %ID/ g (Table 2). Tumors in treated mice not only exhibited reduce in [18F]FHBG accumulation, but also underwent shrinkage after HSV-tk/GCV therapy, while in untreated mice the tumor kept growing constantly.
Evaluation of the effect of Lenti-STAT3-NF-κB-tk/ GCV gene therapy by µPET/CT imaging. Mice bearing TNBC tumors were imaged with µPET/CT at 60 min after i.v. injection with [18F]FHBG on day 2 post intratumoral injection of tk-expression virus. Representative PET/CT images of animals before treatment with the GCV showed uptake of [18F]FHBG in Lenti-CMV-tk and Lenti-STAT3-NF-κB-tk-infected tumors, respectively (upper panel). At eight days post GCV treatment, the radiotracer uptake in Lenti-CMV-tk or Lenti-STAT3-NF-κB-tk-infected tumors decreased in contrast to increased uptake in Lenti-CMV-tk-infected tumor without GCV treatment (lower panel).
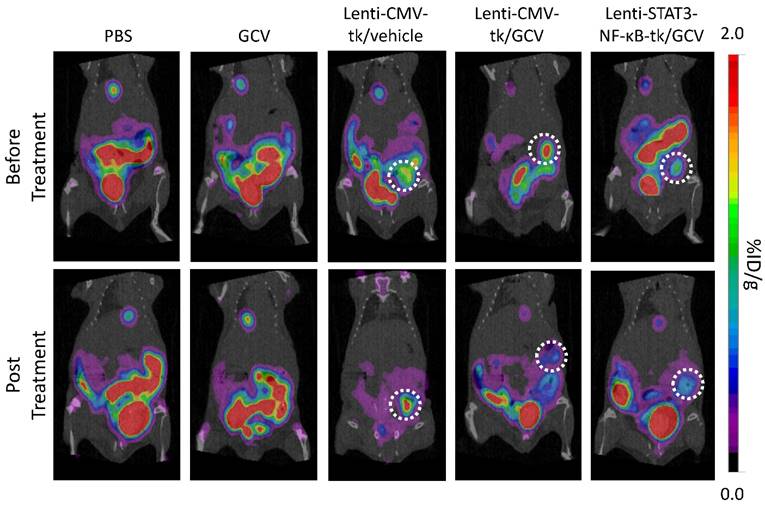
[18F]FHBG uptake in Lenti-CMV-tk or Lenti-STAT3-NF-κB-tk infected MDA-MB-231 tumors pre- and post-treatment with GCV.
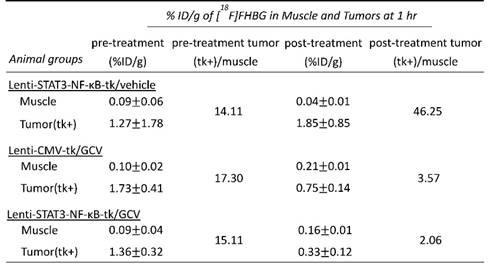
Diminished invasive ability and CSC markers in triple negative breast cancer cells sorted from STAT3-NF-κB-regulated HSV-tk/GCV-treated xenografts
To determine the metastatic capacity of these cells after HSV-tk/GCV treatment, bulk tumor cell suspensions from mouse xenografts were prepared based on the previously described [27] .Cells from xenografts of each group were further examined by Matrigel invasion assay. Cells from Lenti-STAT3-NF-κB-tk/GCV-treated group displayed lower invasive potential than that of the Lenti-CMV-tk/GCV-treated group. As shown in Fig. 5A, the percentage of cells from Lenti-STAT3-NF-κB-tk/GCV-treated group which were capable of invasion through Matrigel was at least 3-fold lower than that of the cells from Lenti-CMV-tk/GCV-treated controls (P< 0.05). Consistent with this observation, immunostaining with antibodies against EMT markers including the decreased E-cadherin and an increased vimentin expression, showed a remarkable decrease in EMT after Lenti-STAT3-NF-κB-tk/GCV therapy in tumor tissues (Fig. 5B and C). Moreover, the expression of CSC markers was also decreased by Lenti-STAT3-NF-κB-tk/GCV therapy, as judged by immunofluorescent staining and immunoblotting for ALDH and CD133, respectively (Fig. 5D and E). The results suggest that targeting STAT3/NF-κB-controlled tumor environment not only attenuates tumor growth but also suppress invasiveness of residual tumor cells.
Reduced invasive ability and expression of CSC markers in MDA-MB-231 cells after Lenti-STAT3-NF-κB-tk/ GCV therapy. (A) Coomassie blue staining results of lower surface filters showed the cells invading the Matrigel (×100) (top). The number of cells that invaded through the chamber was evaluated in 3 fields for each experimental group and averaged. The invading cells of each experimental group was counted as the average of the sum of 10 fields of vision under a microscope. *p<0.001 compared with untreated group (bottom). (B) Immunofluorescence staining for E-cadherin, showing increased membrane integrity of epithelial cancer cells after Lenti-STAT3-NF-κB-tk/GCV therapy in MDA-MB-231 TNBC-bearing mice. (C) Vimentin staining, showing decreased EMT in Lenti-STAT3-NF-κB-tk/GCV treated cells. (D) Immunofluorescence staining of tumor sections showing that ALDH and CD133 expression decreases after Lenti-STAT3-NF-κB-tk/ GCV therapy. Scale bar, 20 μm. (E) Western blots showing downregulation of CD133, ALDH and Snail1 after therapy. Loading control, GAPDH.
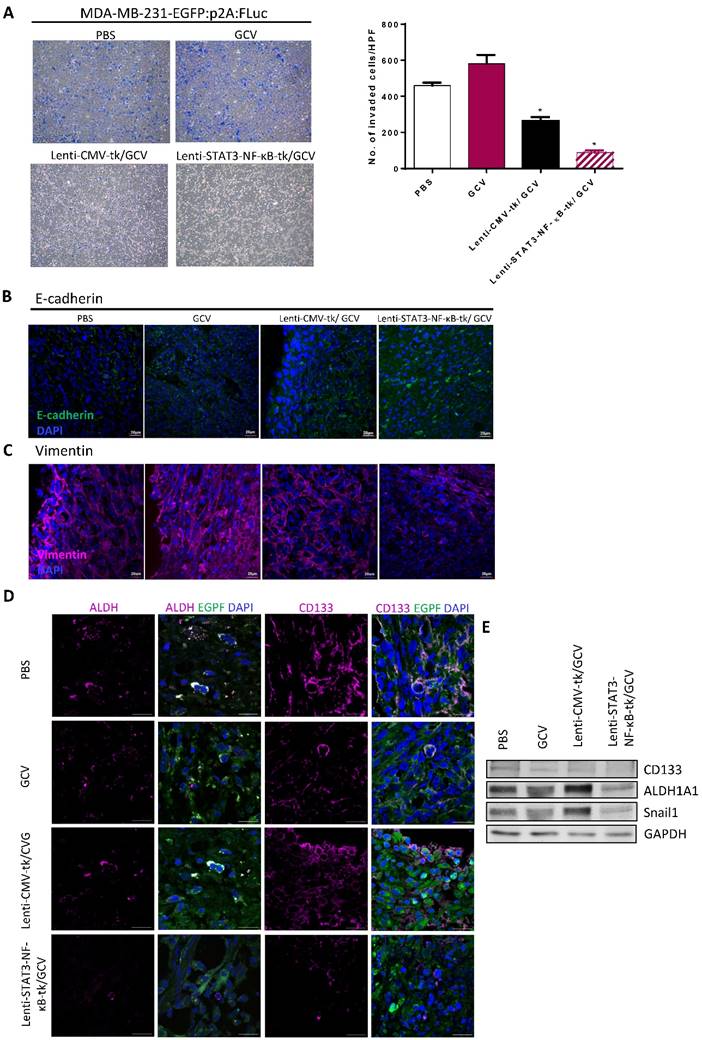
Combination of Lenti-STAT3-NF-κB-TK/GCV therapy with cisplatin enhanced the tumor-killing effect in vivo
To evaluate whether the efficacy of STAT3-NF-κB-tk /GCV therapy in triple negative breast cancer could be further enhanced by combining with platinum-based chemotherapeutic agent, cisplatin (1mg/kg) was injected intraperitoneally into mice twice per week; starting at 5-weeks post-implantation when STAT3-NF-κB-regulated HSV-tk/GCV gene therapy was finished (Fig. 6A). Tumor regression was monitored for 2 months post implantation. Treatment with cisplatin alone for 31 days was also carried out and the results showed slight inhibition of primary tumor growth of MDA-MB-231, while Lenti-CMV-tk/GCV gene therapy only induced a moderate, but significant inhibition in comparison with PBS treated controls. The growth inhibition effect was more pronounce in Lenti-STAT3-NF-κB-HSV-tk/GCV treated mice than that of Lenti-CMV-tk/GCV treated mice. Notably, longitudinal measurement of tumor burden revealed that growth inhibition effect persisted for at least 2 weeks (Fig. 6B). However, most prominent anti-tumor effect was observed on the animals received the treatment regimen of Lenti-STAT3-NF-κB-DsRedm-tk/GCV plus cisplatin. The cisplatin-induced tumor regression was drastically enhanced in Lenti-STAT3-NF-κB- DsRedm-tk/GCV treated mice as assessed by in vivo imaging of tumor burden (Fig. 6C). Tumor burden in mice received Lenti-STAT3-NF-κB-DsRedm-tk/GCV and cisplatin combination treatment was about 2.6-fold less than that in mice only received STAT3-NF-κB-regulated HSV-tk/GCV therapy. The tumor size in both the mice received STAT3-NF-κB-regulated HSV-tk/GCV treatment and combination treatment were significantly smaller than that in controls. There were almost no visible tumors left in mice receiving combination treatment; whereas tumors can be detected in all other groups at the end of the study. In light of these results, the inhibitory effect of combination treatment presumably because of the high expression of transgene which is constantly induced by tumor microenvirnment, which can specifically target the STAT3/NF-κB activated tumor cells, subsequently suppress EMT process and finally result in cisplatin sensitization [33, 34]. Body weight changes of the mice receiving different treatment are shown in Figure 6D. There were no significant differences in body weight among treated groups compared with the PBS-treated group.
In vivo suicidal gene therapy enhances cisplatin sensitivity in a STAT3/NF-κB-controlled tumor environment. (A) Schematic diagram for MDA-MB-231 orthotopic animal model. 2×106 cancer cells were implanted into nude mice. 26 days post-tumor implantation, mice were given intratumoral injection of Lenti-STAT3-NF-κB-tk or Lenti-CMV-tk. 2 days later, mice were received GCV (20mg/kg) for 7 days. Cisplatin (1 mg/kg, ip., twice weekly) was administered at 3-weeks post-gene therapy. (B) Quantification of tumor volume, showing combination of STAT3-NF-κB-tk/GCV gene therapy and cisplatin suppressed tumor growth in MDA-MB-231 TNBC-bearing mice. Data represent mean ± SD (n=4-5/group). *p=0.049, CMV-tk/GCV versus STAT3-NF-κB-tk/GCV; **p=0.002, STAT3-NF-κB-tk/GCV versus STAT3-NF-κB-tk/GCV+CIS by Student's t-test. (C) Tumor burden of each group at a whole body level was monitored with IVIS at indicated time points. Representative images showing increased cisplatin sensitivity and therapeutic effect in Lenti-STAT3-NF-κB-tk/ GCV-treated mice. Tumor burden at the end point was determined and represented as mean ±SD (n=5/group). *p=0.0055, Lenti-STAT3-NF-κB-tk/GCV versus Lenti-STAT3-NF-κB-tk/GCV+CIS by unpaired Student's t-test.
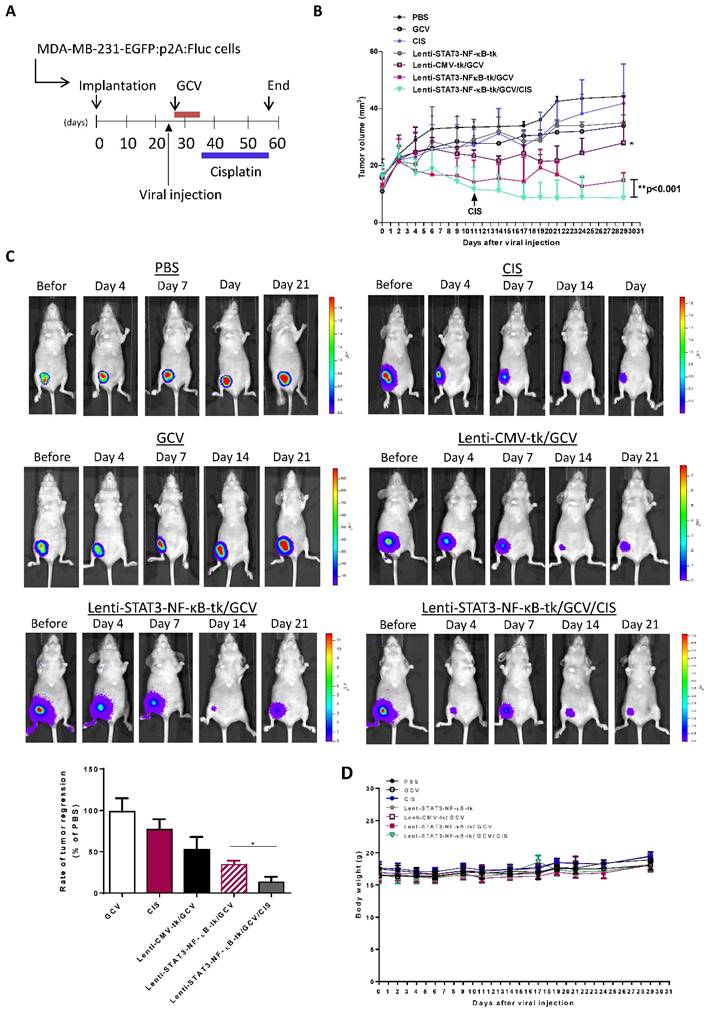
Lenti-STAT3-NF-κB-TK/GCV gene therapy reduces tumor cell proliferation, EMT phenotype and increases cisplatin-induced apoptosis in vivo
Tumor samples obtained after therapy from each group were examined by H&E staining to detect the presence of tumor cells. The IHC staining of Ki 67 and TUNEL assay were performed to investigate the proliferative activity and to detect early DNA fragmentation associated with apoptosis within the tumor. The results showed that the mice receiving a low dosage of cisplatin (1 mg/kg, i.p. twice a week) displayed significantly enhanced staining of the cell proliferation marker Ki67 as compared to other treatment groups. However, this enhanced nuclear staining of Ki67 in response to cisplatin administration was significantly reduced when STAT3-NF-κB-regulated gene therapy was added prior to cisplatin treatment (Fig. 7A and B).
Combined STAT3-NF-κB-tk/ GCV and cisplatin treatments reduced proliferative and increased apoptotic rates (A) Tumor samples obtained from experiment groups of PBS, cisplatin (CIS), Lenti-CMV-tk/ GCV, Lenti-STAT3-NF-κB-tk/ GCV and Lenti- STAT3-NF-κB-tk/ GCV/CIS, respectively were subjected to H&E stain or analyzed for proliferation (Ki67) and apoptosis (TUNEL). All representative samples are tumors collected after the final cisplatin or vehicle treatment. Black arrow denotes positive staining nuclei in Ki67 samples. White arrow denotes positive staining on TUNEL assay samples (× magnification 200). Graphs of the Ki67 expression (B) and TUNEL expression (C) as percentage of positive cells (5 random fields, *p < 0.05).
We also evaluated the cell death of tumor tissues. TUNEL-positive cells with different staining levels in each group were observed. All the treatment groups exhibited enhanced tumor cell apoptosis, compared with the control group. There was an apparent increase in the number of apoptotic cells within the tumor tissues treated with STAT3-NF-κB-HSV-tk/GCV therapy plus cisplatin combination therapy, compared with the other three treatment groups (Fig. 7A and C).
Discussion
High constitutive expression of STAT3 and NF-κB genes play a pivotal role in chemoresistance of breast cancer [35] as well as lung, pancreatic and colorectal cancers [36-39]. In addition to the established role in tumor cell proliferation, survival, invasion and immunosuppression, STAT3 and NF-κB signaling also promote cancer progression through microRNAs modulation, cancer stem cells maintenance and the pre-metastatic niche [40-42]. Treatment approaches which could specifically target these dangerous liaisons and could be translated into clinical applications are highly desirable.
Ideally, enzyme-based gene directed therapy with a prodrug to target one cellular mechanism requires a strong delivered transgene expression for an efficient therapeutic response, but this is a difficult challenge to be overcome in most gene delivery systems. In addition, gene therapy should affect merely the malignant cells, having limited or no effect on normal cells. Although the constitutively active cytomegalovirus (CMV) promoter have been widely used to drive the expression of toxic gene in gene therapy, a concern about this kind of promotor might be harmful toward healthy tissues surrounding the tumor exists. Translational targeting is an effective way in which a better selectivity is applied by using promoter that is primarily induced in the environment within tumors. For HSV-tk/GCV suicide gene therapy, tumor microenvironment-specific promoters, such as redox-responsive promoter [21] and the NF-κB-based promoter system [43] have been designed for this goal. The signaling pathways are rather complicated. McFarland et al. disclosed the phenomenon of signaling crosstalk between STAT3 and NF-κB and elucidated that the NF-κB signaling activation triggers the IL-6 expression, which ultimately results in STAT3 activation [44]. Inflammatory situation can also induce transcriptional synergy between STAT3 and NF-κB to boost FAT10 production, which suppresses the transcriptional activity of p53 and accelerates tumorigenesis [45]. McFarland et al has also reported that the inhibition of NF-κB in tumor did not further suppress the STAT3 activity in glioblastoma, but the mechanisms are still unknown. [46]. Grivennikov et al. explored that crosstalk signaling between NF-κB and STAT3 was the key player in immunosuppression which ultimately causes obstacle in cancer treatment [47], for instance, inhibition of STAT3 could increase the levels of TNF-α which is major source of NF-κB activation within tumors. This stimulation further attracts more immune/inflammatory cells including macrophages, neutrophils and myeloid-derived suppressor cells (MDSCs) into tumor location [48]. However, some of these cells, although designed to help promote tumor scavenging, can conversely assist in the promotion and expansion of tumor growth through secreted cytokines including TNF-α and IL-6, which can positively feedback to induce STAT3 activation in both tumor cells and tumor-infiltrating immune cells. STAT3 has dual roles in tumor inflammation and immunity; activation of STAT3 negatively regulates the immuno-potency of natural killer cells and cytotoxic T-lymphocytes, inhibits the maturation of dendritic cells, but promotes the expansion of MDSCs and regulatory T-cells functions in the tumor microenvironment, resulting in a state of immunosuppression [49, 50]. Therefore, targeting a wider STAT3/NF-κB population such as tumor-associated macrophages may serve as a target for subverting the tumor immunologic microenvironment to benefit cancer therapies. Inspiringly, several recent studies showed that simply disruption of the crosstalk between STAT3 and NF-κB in tumors presents some significant therapeutic effects [11, 36, 51]. Thus, strategies aimed at the co-inhibiting of STAT3/NF-κB activation seems more promising to combat cancers.
In our model, mice received an intratumoral injection of lentiviral vectors for delivering therapeutic genes to target STAT3/NF-κB activation and crosstalk, exhibited marked reduction of tumor growth and diminished expression of EMT and CSCs phenotype (Fig. 5). In order to evaluate the transduction efficiency of transgene at tumor site and monitor therapeutic value, we applied in vivo optical imaging and PET imaging using a radioactive thymidine kinase specific tracer, [18F]FHBG [26]. Significant reduction of Fluc signal was observed 4 days after two doses of prodrug treatment in the mice infected with Lenti-STAT3-NF-κB-tk as compared with the mice infected with Lenti-CMV- tk or treated with the same amount of GCV and PBS alone. Interestingly, we found a diminished protein expression level of ALDH and the breast CSC marker in tumor tissue from Lenti-STAT3-NF-κB-tk/GCV treated mice [Fig. 12C]. Our results support and expand the previous reports, showing that both STAT3 and NF-κB are crucial factors in EMT process [52, 53]. It might be the reason why the STAT3/NF-κB-direct targeted therapy could markedly reduce tumor progression due to inhibition of proliferation and stemness simultaneously.
CSCs possess the ability to resist conventional radio-chemotherapy, and to generate tumor relapse and progression of breast cancer [54]. STAT3 and NF-κB signaling is known to be selectively activated in breast CSC populations [55, 56], emerging them an important therapeutic avenue to be considered. Inhibition of STAT3 and NF-κB influences the growth and survival of tumor cells with mutation and drug resistance. TNBC is the most aggressive type of breast cancer which has heterogenic nature, multi-drug resistance, and uncontrolled metastasis. [57]. Although cisplatin has been well established in clinical treatment regiments due to its effectiveness on breast cancer management. Cisplatin triggers the formation of DNA-adducts, which leads to cell cycle arrest and apoptosis [58]. An acquired cisplatin resistance is a major clinical drawback for patients who relapse after initial favorable response [12]. In the current study, we have demonstratedthe beneficial effects of STAT3/NF-κB inhibition within tumors, which are linked to EMT and CSC markers downregulation. We also disclosed the specific inhibition of STAT3/NF-κB activity by tk/GCV suicide gene therapy can sensitize TNBC to cisplatin treatment. Lenti-STAT3-NF-κB-tk/GCV-treated tumors were more sensitive to cisplatin (Fig. 6B, Lenti-STAT3-NF-κB-tk/GCV vs. Lenti-STAT3-NF-κB-tk/GCV/CIS), and mice that received an intratumoral injection of Lenti-STAT3-NF-κB-tk plus cisplatin showed a clear delay in tumor growth when compared with those treated with Lenti-CMV-tk/GCV, GCV or PBS alone (Fig. 6C). The results suggest that destroying the STAT3 and NF-κB crosstalk firstly can attenuate the expression of EMT phenotype and CSC markers, and make the subsequently cisplatin treatment more efficiently. Our results also support the report from Park et al [34], that cisplatin resistance in TNBC were associated with EMT process.
Although our results showed an enormous therapeutic potential of Lenti-STAT3-NF-κB-tk/ GCV gene therapy in combination with cisplatin, there are also several limitations. First, the lentivirus system used as therapeutic gene delivery vectors into tumors; targets both dividing and non-dividing cells, and integrates randomly throughout the entire gene. A clinical trial using lentiviral vector conducted in 2009 showed a promise therapeutic effect but also revealed its potential risk of proto-oncogene activation [59]. Second, our therapeutic strategy is demonstrated in local tumor treatment rather than metastatic breast cancer model. Therefore, development of effective vectors that can systemically deliver therapeutic gene into tumor bulk and sites of cancer metastasis is warranted.
In summary, we have provided in vivo evidences supporting that specific targeting STAT3/NF-κB signaling within tumor led to TNBC shrinkage with decreased expression of EMT phenotype and CSC markers. Furthermore, we found that this therapeutic strategy could further sensitizes TNBC to cisplatin, suggesting that inhibition of STAT3/NF-κB may exhaust the chemoresistant cell pool within the tumor thus enhance the cisplatin sensitivity. These results also highlighted the beneficial effects of molecular targeted suicide gene therapy in cancer treatment, and provided a potential gene therapy system against cancers.
Supplementary Material
Supplementary figures.
Acknowledgements
This research was supported by the grants: NSC 102-2314-B-010-038-MY3, NSC 102-2627-M-010-003, MOST 105-2314-B-010-034-MY2 (Ministry of Science and Technology), MOHW105-TDU-B- 211-134-003 (Department of Health), V105C-038 (Taipei Veterans General Hospital), 105AC-BI1 (BMIRC, National Yang-Ming University). The authors thank the technical support from Molecular and Genetic Imaging Core, Taiwan Mouse Clinic (MOST 104-2325-B-001-011) which is funded by the National Research Program for Biopharmaceuticals (NRPB) at the Ministry of Science and Technology (MOST) of Taiwan, and Ms. Tsuey-Ling Jan for the assistance of preparing the manuscript.
References
1. Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H. et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10869-74
2. Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A. et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8418-23
3. O'Shaughnessy J, Osborne C, Pippen JE, Yoffe M, Patt D, Rocha C. et al. Iniparib plus chemotherapy in metastatic triple-negative breast cancer. The New England journal of medicine. 2011;364:205-14
4. Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA. et al. Triple-negative breast cancer: clinical features and patterns of recurrence. Clinical cancer research: an official journal of the American Association for Cancer Research. 2007;13:4429-34
5. Rakha EA, El-Sayed ME, Reis-Filho J, Ellis IO. Patho-biological aspects of basal-like breast cancer. Breast cancer research and treatment. 2009;113:411-22
6. Malanchi I, Santamaria-Martinez A, Susanto E, Peng H, Lehr HA, Delaloye JF. et al. Interactions between cancer stem cells and their niche govern metastatic colonization. Nature. 2012;481:85-9
7. Atkinson RL, Yang WT, Rosen DG, Landis MD, Wong H, Lewis MT. et al. Cancer stem cell markers are enriched in normal tissue adjacent to triple negative breast cancer and inversely correlated with DNA repair deficiency. Breast cancer research: BCR. 2013;15:R77
8. Wolfson B, Eades G, Zhou Q. Adipocyte activation of cancer stem cell signaling in breast cancer. World journal of biological chemistry. 2015;6:39-47
9. Burnett JP, Korkaya H, Ouzounova MD, Jiang H, Conley SJ, Newman BW. et al. Trastuzumab resistance induces EMT to transform HER2(+) PTEN(-) to a triple negative breast cancer that requires unique treatment options. Scientific reports. 2015;5:15821
10. Cioce M, Canino C, Pulito C, Muti P, Strano S, Blandino G. Butein impairs the protumorigenic activity of malignant pleural mesothelioma cells. Cell cycle. 2012;11:132-40
11. Canino C, Luo Y, Marcato P, Blandino G, Pass HI, Cioce M. A STAT3-NFkB/DDIT3/CEBPbeta axis modulates ALDH1A3 expression in chemoresistant cell subpopulations. Oncotarget. 2015;6:12637-53
12. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O. et al. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869-83
13. Gu F, Ma Y, Zhang Z, Zhao J, Kobayashi H, Zhang L. et al. Expression of Stat3 and Notch1 is associated with cisplatin resistance in head and neck squamous cell carcinoma. Oncology reports. 2010;23:671-6
14. Morais C, Gobe G, Johnson DW, Healy H. Inhibition of nuclear factor kappa B transcription activity drives a synergistic effect of pyrrolidine dithiocarbamate and cisplatin for treatment of renal cell carcinoma. Apoptosis: an international journal on programmed cell death. 2010;15:412-25
15. McFarland BC, Gray GK, Nozell SE, Hong SW, Benveniste EN. Activation of the NF-kappaB pathway by the STAT3 inhibitor JSI-124 in human glioblastoma cells. Molecular cancer research: MCR. 2013;11:494-505
16. Kagawa S, He C, Gu J, Koch P, Rha SJ, Roth JA. et al. Antitumor activity and bystander effects of the tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) gene. Cancer research. 2001;61:3330-8
17. Vyas VK, Chintha C, Pandya MR. Biology and medicinal chemistry approaches towards various apoptosis inducers. Anti-cancer agents in medicinal chemistry. 2013;13:433-55
18. Fillat C, Carrio M, Cascante A, Sangro B. Suicide gene therapy mediated by the Herpes Simplex virus thymidine kinase gene/Ganciclovir system: fifteen years of application. Current gene therapy. 2003;3:13-26
19. Shibata T, Giaccia AJ, Brown JM. Hypoxia-inducible regulation of a prodrug-activating enzyme for tumor-specific gene therapy. Neoplasia. 2002;4:40-8
20. Greco O, Joiner MC, Doleh A, Powell AD, Hillman GG, Scott SD. Hypoxia- and radiation-activated Cre/loxP 'molecular switch' vectors for gene therapy of cancer. Gene therapy. 2006;13:206-15
21. Policastro LL, Ibanez IL, Duran HA, Soria G, Gottifredi V, Podhajcer OL. Suppression of cancer growth by nonviral gene therapy based on a novel reactive oxygen species-responsive promoter. Molecular therapy: the journal of the American Society of Gene Therapy. 2009;17:1355-64
22. Leinonen HM, Ruotsalainen AK, Maatta AM, Laitinen HM, Kuosmanen SM, Kansanen E. et al. Oxidative stress-regulated lentiviral TK/GCV gene therapy for lung cancer treatment. Cancer research. 2012;72:6227-35
23. Seidel HM, Milocco LH, Lamb P, Darnell JE Jr, Stein RB, Rosen J. Spacing of palindromic half sites as a determinant of selective STAT (signal transducers and activators of transcription) DNA binding and transcriptional activity. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:3041-5
24. Hsieh YJ, Hwu L, Chen YC, Ke CC, Chen FD, Wang HE. et al. P21-driven multifusion gene system for evaluating the efficacy of histone deacetylase inhibitors by in vivo molecular imaging and for transcription targeting therapy of cancer mediated by histone deacetylase inhibitor. Journal of nuclear medicine: official publication, Society of Nuclear Medicine. 2014;55:678-85
25. Yaghoubi SS, Jensen MC, Satyamurthy N, Budhiraja S, Paik D, Czernin J. et al. Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nature clinical practice Oncology. 2009;6:53-8
26. Chang CW, Lin M, Wu SY, Hsieh CH, Liu RS, Wang SJ. et al. A high yield robotic synthesis of 9-(4-[18F]-fluoro-3-hydroxymethylbutyl) guanine ([18F]FHBG) and 9-[3-[18F]fluoro-1-hydroxy-2-propoxy)methyl]guanine([18F] FHPG) for gene expression imaging. Applied radiation and isotopes: including data, instrumentation and methods for use in agriculture, industry and medicine. 2007;65:57-63
27. Kuo WY, Wu CY, Hwu L, Lee JS, Tsai CH, Lin KP. et al. Enhancement of tumor initiation and expression of KCNMA1, MORF4L2 and ASPM genes in the adenocarcinoma of lung xenograft after vorinostat treatment. Oncotarget. 2015;6:8663-75
28. Maatta AM, Tenhunen A, Pasanen T, Merilainen O, Pellinen R, Makinen K. et al. Non-small cell lung cancer as a target disease for herpes simplex type 1 thymidine kinase-ganciclovir gene therapy. International journal of oncology. 2004;24:943-9
29. Chakilam S, Gandesiri M, Rau TT, Agaimy A, Vijayalakshmi M, Ivanovska J. et al. Death-associated protein kinase controls STAT3 activity in intestinal epithelial cells. The American journal of pathology. 2013;182:1005-20
30. Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ Jr, Sledge GW Jr. Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Molecular and cellular biology. 1997;17:3629-39
31. Shin DS, Kim HN, Shin KD, Yoon YJ, Kim SJ, Han DC. et al. Cryptotanshinone inhibits constitutive signal transducer and activator of transcription 3 function through blocking the dimerization in DU145 prostate cancer cells. Cancer research. 2009;69:193-202
32. Jin H, Lee WS, Eun SY, Jung JH, Park HS, Kim G. et al. Morin, a flavonoid from Moraceae, suppresses growth and invasion of the highly metastatic breast cancer cell line MDA-MB231 partly through suppression of the Akt pathway. International journal of oncology. 2014;45:1629-37
33. Chung SS, Aroh C, Vadgama JV. Constitutive activation of STAT3 signaling regulates hTERT and promotes stem cell-like traits in human breast cancer cells. PloS one. 2013;8:e83971
34. Park J, Morley TS, Scherer PE. Inhibition of endotrophin, a cleavage product of collagen VI, confers cisplatin sensitivity to tumours. EMBO molecular medicine. 2013;5:935-48
35. Yi EH, Lee CS, Lee JK, Lee YJ, Shin MK, Cho CH. et al. STAT3-RANTES autocrine signaling is essential for tamoxifen resistance in human breast cancer cells. Molecular cancer research: MCR. 2013;11:31-42
36. Nan J, Du Y, Chen X, Bai Q, Wang Y, Zhang X. et al. TPCA-1 is a direct dual inhibitor of STAT3 and NF-kappaB and regresses mutant EGFR-associated human non-small cell lung cancers. Molecular cancer therapeutics. 2014;13:617-29
37. Snyder M, Huang J, Huang XY, Zhang JJ. A signal transducer and activator of transcription 3.Nuclear Factor kappaB (Stat3.NFkappaB) complex is necessary for the expression of fascin in metastatic breast cancer cells in response to interleukin (IL)-6 and tumor necrosis factor (TNF)-alpha. J Biol Chem. 2014;289:30082-9
38. Surh YJ, Bode AM, Dong Z. Breaking the NF-kappaB and STAT3 alliance inhibits inflammation and pancreatic tumorigenesis. Cancer Prev Res (Phila). 2010;3:1379-81
39. De Simone V, Franze E, Ronchetti G, Colantoni A, Fantini MC, Di Fusco D. et al. Th17-type cytokines, IL-6 and TNF-alpha synergistically activate STAT3 and NF-kB to promote colorectal cancer cell growth. Oncogene. 2015;34:3493-503
40. Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell. 2009;139:693-706
41. Sherry MM, Reeves A, Wu JK, Cochran BH. STAT3 is required for proliferation and maintenance of multipotency in glioblastoma stem cells. Stem cells. 2009;27:2383-92
42. Guo L, Chen C, Shi M, Wang F, Chen X, Diao D. et al. Stat3-coordinated Lin-28-let-7-HMGA2 and miR-200-ZEB1 circuits initiate and maintain oncostatin M-driven epithelial-mesenchymal transition. Oncogene. 2013;32:5272-82
43. Badr CE, Niers JM, Morse D, Koelen JA, Vandertop P, Noske D. et al. Suicidal gene therapy in an NF-kappaB-controlled tumor environment as monitored by a secreted blood reporter. Gene therapy. 2011;18:445-51
44. McFarland BC, Hong SW, Rajbhandari R, Twitty GB Jr, Gray GK, Yu H. et al. NF-kappaB-induced IL-6 ensures STAT3 activation and tumor aggressiveness in glioblastoma. PloS one. 2013;8:e78728
45. Choi Y, Kim JK, Yoo JY. NFkappaB and STAT3 synergistically activate the expression of FAT10, a gene counteracting the tumor suppressor p53. Molecular oncology. 2014;8:642-55
46. McFarland BC, Ma JY, Langford CP, Gillespie GY, Yu H, Zheng Y. et al. Therapeutic potential of AZD1480 for the treatment of human glioblastoma. Molecular cancer therapeutics. 2011;10:2384-93
47. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S. et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer cell. 2009;15:103-13
48. Zhao X, Rong L, Zhao X, Li X, Liu X, Deng J. et al. TNF signaling drives myeloid-derived suppressor cell accumulation. The Journal of clinical investigation. 2012;122:4094-104
49. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883-99
50. Gotthardt D, Putz EM, Straka E, Kudweis P, Biaggio M, Poli V. et al. Loss of STAT3 in murine NK cells enhances NK cell-dependent tumor surveillance. Blood. 2014;124:2370-9
51. Lin C, Wang L, Wang H, Yang L, Guo H, Wang X. Tanshinone IIA inhibits breast cancer stem cells growth in vitro and in vivo through attenuation of IL-6/STAT3/NF-kB signaling pathways. Journal of cellular biochemistry. 2013;114:2061-70
52. Avtanski DB, Nagalingam A, Bonner MY, Arbiser JL, Saxena NK, Sharma D. Honokiol inhibits epithelial-mesenchymal transition in breast cancer cells by targeting signal transducer and activator of transcription 3/Zeb1/E-cadherin axis. Molecular oncology. 2014;8:565-80
53. Huber MA, Azoitei N, Baumann B, Grunert S, Sommer A, Pehamberger H. et al. NF-kappaB is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. The Journal of clinical investigation. 2004;114:569-81
54. Hu Y, Fu L. Targeting cancer stem cells: a new therapy to cure cancer patients. American journal of cancer research. 2012;2:340-56
55. Marotta LL, Almendro V, Marusyk A, Shipitsin M, Schemme J, Walker SR. et al. The JAK2/STAT3 signaling pathway is required for growth of CD44(+)CD24(-) stem cell-like breast cancer cells in human tumors. The Journal of clinical investigation. 2011;121:2723-35
56. Shostak K, Chariot A. NF-kappaB, stem cells and breast cancer: the links get stronger. Breast cancer research: BCR. 2011;13:214
57. Andre F, Zielinski CC. Optimal strategies for the treatment of metastatic triple-negative breast cancer with currently approved agents. Annals of oncology: official journal of the European Society for Medical Oncology / ESMO. 2012;23(Suppl 6):vi46-51
58. Kelland L. The resurgence of platinum-based cancer chemotherapy. Nature reviews Cancer. 2007;7:573-84
59. Kaiser J. Gene therapy. Beta-thalassemia treatment succeeds, with a caveat. Science. 2009;326:1468-9
Author contact
![]() Corresponding author: Prof. Ren-Shyan Liu, M.D., Department of Biomedical Imaging and Radiological Sciences, School of Biomedicine and Engineering, National Yang-Ming University. Phone: +886-2-28757301 ext. 299. Fax: +886-2-28749431 E-mail: rsliugov.tw.
Corresponding author: Prof. Ren-Shyan Liu, M.D., Department of Biomedical Imaging and Radiological Sciences, School of Biomedicine and Engineering, National Yang-Ming University. Phone: +886-2-28757301 ext. 299. Fax: +886-2-28749431 E-mail: rsliugov.tw.