Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results and Discussion
Conclusion
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results and Discussion
Conclusion
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(3):614-623. doi:10.7150/thno.17381 This issue Cite
Research Paper
Radionuclide I-131 Labeled Albumin-Paclitaxel Nanoparticles for Synergistic Combined Chemo-radioisotope Therapy of Cancer
Longlong Tian1,2‡, Qian Chen2‡, Xuan Yi1, Guanglin Wang1, Jie Chen1, Ping Ning1, Kai Yang1 ![]() , Zhuang Liu2
, Zhuang Liu2 ![]()
1. School of Radiation Medicine and Protection & School for Radiological and Interdisciplinary Sciences (RAD-X), Collaborative Innovation Center of Radiation Medicine of Jiangsu Higher Education Institutions, Soochow University, Suzhou, Jiangsu, 215123, China
2. Institute of Functional Nano & Soft Materials (FUNSOM), Collaborative Innovation Center of Suzhou Nano Science and Technology, Soochow University, Suzhou, Jiangsu, 215123, China
‡These authors contributed equally to this work
Received 2016-8-29; Accepted 2016-11-1; Published 2017-1-12
Citation:
Tian L, Chen Q, Yi X, Wang G, Chen J, Ning P, Yang K, Liu Z. Radionuclide I-131 Labeled Albumin-Paclitaxel Nanoparticles for Synergistic Combined Chemo-radioisotope Therapy of Cancer. Theranostics 2017; 7(3):614-623. doi:10.7150/thno.17381. https://www.thno.org/v07p0614.htm
Other stylesAbstract

Development of biocompatible/biodegradable materials with multiple functionalities via simple methods for cancer combination therapy has attracted great attention in recent years. Herein, paclitaxel (PTX), a popular anti-tumor chemotherapeutic drug, is used to induce the self-assembly of human serum albumin (HSA) pre-labeled with radionuclide I-131, obtaining 131I-HSA-PTX nanoparticles for combined chemotherapy and radioisotope therapy (RIT) of cancer. Such 131I-HSA-PTX nanoparticles show prolonged blood circulation time, high tumor specific uptake and excellent intra-tumor penetration ability. Interestingly, as revealed by in vivo photoacoustic imaging and ex vivo immunofluorescence staining, PTX delivered into the tumor by HSA-nanoparticle transportation can remarkably enhance the tumor local oxygen level and suppress the expression of HIF-1α, leading to greatly relieved tumor hypoxia. As the results, the combined in vivo chemotherapy & RIT with 131I-HSA-PTX nanoparticles in the animal tumor model offers excellent synergistic therapeutic efficacy, likely owing to the greatly modulated tumor microenvironment associated with PTX-based chemotherapy. Therefore, in this work, a simple yet effective therapeutic agent is developed for synergistic chemo-RIT of cancer, promising for future clinic translations in cancer treatment.
Keywords: albumin, paclitaxel, radioisotope therapy, chemotherapy, hypoxia
Introduction
Despite the severe toxic side effects and non-ideal efficacy of radiotherapy and chemotherapy, both two types of cancer therapies are still the most wildly used methods in current cancer therapy [1-3]. To achieve better therapeutic outcomes, radiotherapy and chemotherapy are often combined in clinic for cancer treatment [4, 5]. As far as the combination of chemotherapy with radioisotope therapy (RIT) is concerned, however, the separated delivery of chemotherapeutic drug and radioactive isotopes often leads to inconsistent pharmacokinetics and tumor homing profiles, possibility unfavorable for achieving the optimized therapeutic results. In the past few years, nanomedicine based on multi-functional nanomaterials has been proposed to realize combined radiotherapy and chemotherapy simultaneously [6-10]. Therefore, the development of biocompatible / biodegradable nano-platforms with multiple functionalities to combine chemo-radioisotope therapy would have significant potential for clinical cancer treatment.
Human serum albumin (HSA), an abundant serum protein with inherent biocompatibility, has been demonstrated to be an ideal drug carrier [11, 12]. As a compelling case, nanoparticles of HSA bound with paclitaxel (PTX), a potent anti-cancer drug, has been approved by US Food and Drug Administration (FDA) for cancer treatment under the trade name of Abraxane® [13, 14]. Recently, a number of groups including ours have developed many new types of theranostic nanoparticles by utilizing HSA as the versatile biocompatible carrier to load hydrophobic imaging and/or therapeutic molecules, for biomedical imaging and imaging-guided cancer therapies [15-17]. In addition to the utilization of the hydrophobic pocket in HSA for non-covalent drug loading, the functional groups (e.g. carboxyl, amino, phenolic hydroxyl, and thiol groups) of HSA are also available for covalent functionalization and radiolabeling [18]. Radionuclides (such as 188Re, 99mTc, 125I and 177Lu) labeled HSA has thus been explored for cancer diagnoses and therapy [19, 20]. However, using the versatile role of HSA for combined chemo-radioisotope therapy has not yet been reported to our best knowledge.
In this work, we develop a chemo-RIT nanomedicine by simply mixing 131I-labeled HSA with PTX, the latter of which could induce the self-assembly of 131I-HSA to form 131I-HSA-PTX nanoparticles with an average size of ~100 nm. In this system, while 131I emitting strong beta and gamma rays could serve as RIT and gamma imaging agent, PTX would act an effective anti-tumor drug for cancer chemotherapy. Compared with 131I-HSA alone and free 131I, 131I-HSA-PTX nanoparticles exhibit prolonged blood circulation time, enhanced tumor specific uptake and excellent intra-tumor penetration ability. Interestingly, although the synergistic effect in the combined chemo-RIT with 131I-HSA-PTX at the in vitro level appears to be not that significant, greatly improved tumor growth inhibition effect is observed in our in vivo tumor model experiment with 131I-HSA-PTX, which offers much better therapeutic efficacy compared to the predicted additive effect. Such an obvious synergistic in vivo therapeutic outcome is found to be attributed to the PTX-induced tumor microenvironment modulation by greatly relieving tumor hypoxia [21, 22], which is known to have a crucial role in radiotherapy resistance [23-26]. Therefore, a biocompatible therapeutic nanoagent based on HSA is developed via a simple and reliable approach in this work, to achieve chemo-RIT with high in vivo synergistic efficacy, promising for cancer combination therapy and showing substantial potential for clinical translation.
Materials and Methods
131I labeling HSA
HSA was labeled with radionuclide 131I (purchased from Shanghai GMS Pharmaceutical Co., Ltd) through a standard chloramine-T oxidation method. In brief, 200 μCi of 131I and 10 μL of chloramine T (10 mg mL-1) were added into the solution of HSA (2 mg mL-1, 1 mL). The mixture was then reacted in a pH 7.5 phosphate buffer (50 mM) for 20 min at room temperature. Excess 131I and chloramine T were removed by centrifugation filtration through Amicon filters (MWCO =10 kDa) and washed several times with PBS until no detachable radioactivity in the filtration solution.
Synthesis of HSA-PTX and 131I-HSA-PTX nanoparticles
10 ml of HSA (2 mg mL-1) without or with 131I labeling were added with 130 μl PTX pre-dissolved in ethanol (20 mg mL-1) at a mole ratio 1:10 (HSA: PTX) and stirred overnight at room temperature to prepare HSA-PTX or 131I-HSA-PTX nanoparticles. The samples were centrifuged at 14800 rpm for 10 min to remove excess PTX. The morphology of HSA-PTX after being stained by phosphotungstic acid (1 wt. %) was characterized by transmission electron microscopy (TEM) using a FEI Tecnai F20 transmission electron microscope. The loading efficiency of PTX into the nanoparticles was measured by high performance liquid chromatography (HPLC) with a UV-vis detector at 227 nm. Acetonitrile was used as the mobile phase. The PTX content in this HSA-PTX formulation was measured to 6.5% (w/w), and the mole ratio of HSA to PTX was calculated to be ~1:5.
Radiolabeling stability of 131I-HSA and 131I-HSA-PTX
5 μl of 131I-HSA or 131I-HSA-PTX was incubated with mouse serum (~50 μl) at 37 oC. After incubation for different periods of time (1, 2, 4, 12, and 24 h), 5 μl of the mixture was taken out and centrifuged through Amicon filters (MWCO=10 kDa) to remove the detached 131I. The radioactivity was detected by Gamma counter. The radiolabeling stability was then calculated by the supernatant counts dividing by the total counts (n=3).
Cy5.5 labeling HSA
5 μl of NHS-Cy5.5 (10 mg/mL) was added into 2 ml of HSA solution (5 mg/ml) and stirred overnight at room temperature in dark. Free Cy5.5 was removed by centrifugation filtration through Amicon filters (MWCO=10 kDa) and washed several times with deionized water until no detachable color in the filtration solution.
Cellular experiments
4T1 murine breast cell line was originally obtained from American Type Culture Collection (ATCC) and cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin in a humidified atmosphere containing 5% CO2 at 37 °C. For the in vitro cytotoxicity assay, 4T1 cells were firstly seeded into 96-well plates at a density of 2000 cells per well overnight, and then incubated with different concentrations of 131I-HSA, HSA-PTX or 131I-HSA-PTX for 72 h. Cell counting kit-8 (CCK-8) assay was then conducted to determine the relative cell viabilities.
Gamma imaging
Nude mice bearing 4T1 tumors (~200 mm3) were intravenously (i.v.) injected with free 131I (200 μCi of 131I), 131I-HSA(200 μCi of 131I) or 131I-HSA-PTX (200 μCi of 131I corresponding to 200 μg of PTX), and imaged by in vivo animal imaging system (Kodak, FX Pro) at various time points post injection.
Blood circulation and biodistribution
Healthy nude mice were i.v. injected with 131I-HSA-PTX (200 μCi of 131I corresponding to 200 μg of PTX). For blood circulation measurement, ~20 μL of blood was drawn from one side of orbital venous plexus at different time points post injection. The radioactivity in blood samples were measured by a gamma counter (LB211, Berthold Technologies Gmbh & Co.KG). In order to investigate the tumor uptake of 131I-HSA-PTX, nude mice bearing 4T1 tumors were i.v. injected with 131I-HSA-PTX (200 μCi of 131I and 200 μg of PTX per mouse) and sacrificed at 24 h post injection. Major organs including liver, spleen, kidney, heart, lung, stomach, intestine, skin, muscle, bone, brain, and tumor were collected, weighed and measured by the gamma counter.
Location of nanoparticles in tumor tissue
Nude mice bearing 4T1 tumors (~200 mm3) were i.v. injected with Cy5.5-HSA or Cy5.5-HSA-PTX. 24 h after injection, tumors were then surgically excised for frozen sections. The blood vessels were stained by incubation with rat anti-CD31 mouse monoclonal antibody (dilution 1:200, Biolegend) and Rhodamine-conjugated donkey anti-rat secondary antibody (dilution 1:200, Jackson).
Photoacoustic imaging
Nude mice bearing 4T1 tumors (~200 mm3) were i.v. injected with HSA-PTX (200 μg of PTX), and imaged by in vivo Photoacoustic Imaging System (Vevo LAZR) at 0, 12, 24 and 48 h post injection. The tumor deoxygenated hemoglobin (DO2) and oxygenated hemoglobin (HO2) were detected at 750 nm and 850 nm, respectively. Tumor saturated O2 (sO2) directly revealed variations in tissue hypoxia, and was calculated by sO2 = HO2/TO2 (TO2 = DO2+ HO2).
Immunofluorescence images
Nude mice bearing 4T1 tumors were i.v. injected with HSA or HSA-PTX (200 μg of PTX, 3 mg of HSA). At 24 h post injection, mice were then i.v. injected with pimonidazole hydrochloride (60 mg/kg) (Hypoxyprobe-1 plus kit, Hypoxyprobe Inc). 90 min later, tumors on those mice were then surgically excised for frozen sections. The tumor slices were incubated with mouse anti-pimonidazole antibody (dilution 1:200, Hypoxyprobe Inc.) and Alex 488-conjugated goat anti-mouse secondary antibody (dilution 1:200, Jackson Inc.) following the kits' instructions. The blood vessels were stained by incubation with rat anti-CD31 mouse monoclonal antibody (dilution 1:200, Biolegend) and Rhodamine-conjugated donkey anti-rat secondary antibody (dilution 1:200, Jackson). Cell nuclei were stained with 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride (DAPI) (dilution 1:5000, Invitrogen). The images were captured with a confocal fluorescence microscopy (Leica SP5). For HIF-1α staining, mouse anti-HIF-1α monoclonal antibody (Abcam113642, 200 times dilution) was used as the primary antibody, while goat-anti-mouse IgG antibody conjugated with FITC (Biolegend) was used as the secondary antibody.
Combination therapy
Nude mice bearing 4T1 tumors (~120 mm3) were randomly divided into four groups: 1) PBS, 2) 131I-HSA (200 μCi of 131 I), 3) HSA-PTX (200 μg of PTX), 4) 131I-HSA-PTX (200 μCi of 131I corresponding to 200 μg of PTX). The above agents were i.v. injected every 4 days at day 0, 4, 8, and 12. The tumor volumes were monitored by a caliper every the other day, and calculated according to the following formula: Volume (mm3) = length (mm) × width2 (mm2)/2. The combination index (CI) was calculated by CI = AB/(A×B). AB was the ratio of the combination group to the control group. A or B was the ratio of the single treatment group (chemotherapy or RIT) to the control group.
Results and Discussion
In this work, we prepared 131I-HSA-PTX nanoparticles in the aqueous solution (Figure 1a) by simply mixing 131I-labeled HSA with PTX, the latter of which would induce assembly of multiple proteins to form nanoparticles via hydrophobic interactions [27, 28]. In brief, HSA was firstly labeled with 131I, which has been widely used in clinic for cancer radioisotope therapy [29, 30], by a standard chloramine-T oxidation method, yielding radiolabeled 131I-HSA. Afterwards, PTX as a typical anticancer drug was added into the solution of 131I-HSA at the mole ratio of 10:1 (PTX: HSA) to induce the self-assembly of HSA [31], forming 131I-HSA-PTX nanoparticles. As revealed by transmission electron microscope (TEM) image, HSA-PTX nanoparticles without radiolabeling showed uniform morphology (Figure 1b). Dynamic light scattering (DLS, Figure 1c) showed the average sizes of HSA with or without 131I labeling to be ~8 nm, while the average sizes of HSA-PTX and 131I-HSA-PTX (PDI = 0.18) were both measured to be ~100 nm, owing to the self-assembly of multiple HSA after PTX binding to form nanoparticles. The PTX content in this HSA-PTX formulation was measured to 6.5% (w/w) by high-performance liquid chromatography (HPLC). The radioactive stability of 131I-HSA and 131I-HSA-PTX was further measured in mouse serum at 37 oC. It was found that 131I-labeled HSA/HSA-PTX nanoparticles exhibited great radiolabeling stability in mouse serum after incubation for 24 h (Figure 1d).
Figure 1
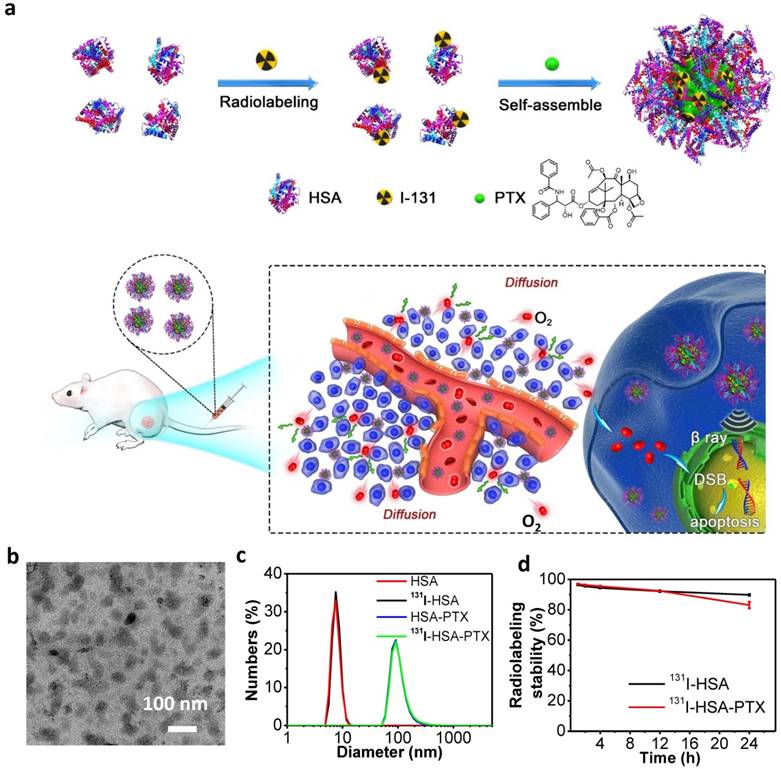
Preparation and characterization of 131I-HSA-PTX nanoparticles. (a) A schematic illustration to show the preparation of 131I-HSA-PTX nanoparticles for in vivo combined chemo-RIT. (b) A TEM image of HSA-PTX nanoparticles. (c) Dynamic light scattering (DLS) data of HSA, 131I-HSA, HSA-PTX and 131I-HSA-PTX. 131I-labeling induced no obvious size changes of HSA-PTX and HSA. (d) Radiolabeling stability of 131I-HSA-PTX and 131I-HSA incubated with serum at 37 oC for 24 h. Great radiolabeling stabilities were observed for both formulations.
Figure 2
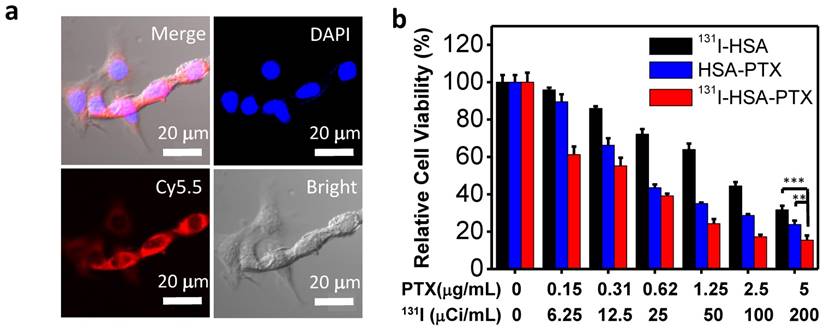
In vitro cell experiments. (a) Confocal fluorescence images of 4T1 cells incubated with Cy5.5-labeled HSA-PTX for 6 h. (b) The relative viabilities of 4T1 cells incubated with different concentrations of 131I-HSA, HSA-PTX and 131I-HSA-PTX for 72 h. P values were calculated by ANOVA (***p < 0.001, **p < 0.01, and *p < 0.05).
Before studying the therapeutic efficacy of 131I-HSA-PTX, we firstly investigated the cellular uptake of HSA-PTX. 4T1 murine breast cancer cells were incubated with Cy5.5-labeled HSA-PTX. After 6 h of incubation, the cells were washed with phosphate buffer solution (PBS) for three times and imaged by a confocal fluorescence microscope. Strong red fluorescence signals were observed in the cytoplasm of cells, suggesting efficient cellular uptake of those nanoparticles (Figure 2a). The cytotoxicity of different formulations of nanoparticles was then quantitatively measured by the cell counting kit-8 (CCK-8) assay. Murine breast cancer 4T1 cells were incubated with 131I-HSA, HSA-PTX or 131I-HSA-PTX at different concentrations for 72 h. 131I-HSA-PTX nanoparticles appeared to be more toxic than 131I-HSA or HSA-PTX alone at the same respective concentrations (Figure 2b). However, based on cell viability data, the synergistic cancer cell killing effect for in vitro combined radio-RIT with 131I-HSA-PTX was not that significant.
Radionuclide I-131 with both β-emission and γ-emission could not only be used for radiotherapy, but also act as a contrast agent for gamma imaging. In this work, nude mice bearing 4T1 tumors with thyroids blocked with gavage of cold KI were intravenously (i.v.) injected with free 131I (200 μCi of 131I), 131I-HSA (200 μCi of 131I) or 131I-HSA-PTX (200 μCi of 131I corresponding to 200 μg of PTX), and imaged by a small animal gamma imaging system at different time points post injection (p.i.). It was found that free 131I after i.v. injection could be rapidly excreted from the mouse body without noticeable tumor retention, while both 131I-HSA and 131I-HSA-PTX after i.v. injection exhibited obvious tumor accumulation (Figure 3a). Note that the tumor signals for 131I-HSA-PTX injected mice appeared to be much stronger than that for 131I-HSA injected mice, indicating that 131I-HSA-PTX nanoparticles possessed greater tumor homing ability owning to the enhanced permeability and retention (EPR) effect.
In order to further understand the in vivo behaviors of different formulations of 131I, we next systematically investigated the blood circulation and biodistribution of 131I-HSA and 131I-HSA-PTX nanoparticles. Mice bearing 4T1 tumors were i.v. injected with 131I-HSA (200 μCi of 131I) or 131I-HSA-PTX nanoparticles (200 μCi of 131I corresponding to 200 μg of PTX). The blood was drawn from the right side of orbital venous plexus of mice at different time points p.i. and measured by a gamma counter to determine the radioactivity of blood samples. The blood circulation half-lives of 131I-HSA-PTX (t1/2α=1.53 h, t1/2β=31.9 h) were significantly prolonged compared with free 131I and 131I-HSA (t1/2α=0.51 h, t1/2β=11.7 h) (Figure 3b), owing to the enlarged nanoparticle sizes in the 131I-HSA-PTX formulation to prevent rapid metabolism [32-37]. For the biodistribution study, those mice were sacrificed at 24 h p.i. The major organs and tissues were collected and weighted for radioactivity measurement by the gamma counter. Consistent to gamma imaging results, 131I-HSA-PTX exhibited higher tumor uptake than that of 131I-HSA (Figure 3c). Notably, the measured uptake of 131I-HSA-PTX in reticuloendothelial system (RES) such as liver and spleen appeared to be relatively low compared to many other types of nanoparticles (Figure 3c), likely owing to the dissociation of those nanoparticles after macrophage uptake and the subsequent metabolism of albumin. At 72 h post injection (Supporting Information Figure S1), the majority of radioactivity from 131I-HSA-PTX nanoparticles could be excreted from the body normal organs, while its clearance from the tumor was found to be relatively slower owing to the EPR effect. Such a feature is favorable for reducing the systemic toxicity of such radio-nanomedicine.
Figure 3
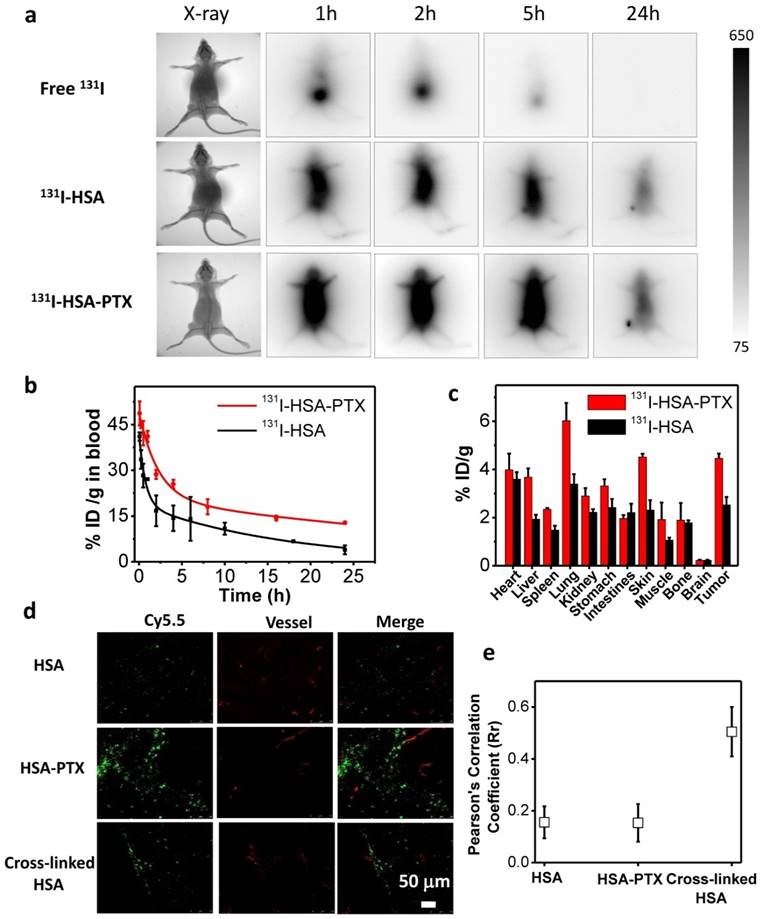
In vivo behaviors of 131I-HSA-PTX nanoparticles. (a) Gamma imaging of mice after i.v. injection of free 131I (up), 131I-HSA (middle) and 131I-HSA-PTX (down). Free 131I was rapidly excreted from the body via urine. Obvious tumor uptake of 131I-HSA and 131I-HSA-PTX showed up at 24 h post injection. (b) The blood circulation profiles of 131I-HSA and 131I-HSA-PTX. (c) The biodistribution of 131I-HSA-PTX and 131I-HSA measured at 24 h post injection. (d) Confocal fluorescence micrographs of tumor slices collected from mice injected with Cy5.5-labeled HSA, HSA-PTX or cross-linked HSA. The red signals were from the fluorescence of anti-CD31-stained blood vessels. (e) The statistics of Pearson's correlation coefficient (Rr) from (d). A higher Rr value indicates a higher level of co-localization between nanoparticles and blood vessels.
It is known that the therapeutic efficacy of energetic β-particles (β-ray) is closely associated with their positions and distribution inside the tumor [38]. However, the dense tumor extracellular matrix structure and aberrant tumor vasculatures often would lead to high interstitial fluid pressure and further block intra-tumor penetration of nanoparticles, and finally resulted in ineffective destruction of tumor cells locating far from blood vasculatures by nanomedicine, particularly if nanoparticles with relatively large sizes are used [39, 40]. To examine the intra-tumoral distribution of nanoparticles upon i.v. injection, HSA was labeled with Cy5.5, a fluorescent dye, and used to construct self-assembled Cy5.5-HSA-PTX nanoparticles by mixing with PTX, or cross-linked Cy5.5-HSA nanoparticles with the addition of glutaraldehyde. Nude mice bearing 4T1 tumors (~200 mm3) were i.v. injected with Cy5.5-HSA, Cy5.5-HSA-PTX, or cross-linked Cy5.5-HSA nanoparticles, and sacrificed at 24 h p.i. to collect tumors for frozen section (Figure 3d). For tumors from mice injected with cross-linked Cy5.5-HSA, which had the same size with Cy5.5-HSA-PTX nanoparticles, Cy5.5 fluorescence signals (green) were found to be largely co-localized or nearby tumor blood vessels stained by anti-CD31 (red) (Figure 3d&e), indicating the limited intra-tumoral penetration of cross-linked albumin nanoparticles with relatively large sizes (about 100 nm). On the other hand, for tumors from mice i.v. injected with free Cy5.5-HSA, scattered but rather weak Cy5.5 signals were found in the tumor, consistent to the relatively low tumor uptake of free albumin as observed by radiolabeling data. Interestingly, strong Cy5.5 fluorescence signals located far from blood vessels were found in tumors on mice i.v. injected with Cy5.5-HSA-PTX (Figure 3d&e), indicating both high tumor accumulation and efficient intra-tumoral diffusion of Cy5.5-HSA in this formulation. It has been reported that the gradual dissociation of self-assembled HSA-PTX nanoparticles in the body could result in improved size-dependent diffusion ability [41-43]. Moreover, the reduced tumor interstitial fluid pressure induced by PTX may be another reason of the excellent intra-tumoral diffusion ability [44, 45], which would help to kill tumor cells locating far from blood vasculatures.
According to literature, PTX, a classic chemotherapy drug, could also enhance the tumor blood flow and oxygenation through normalization of the tumor vasculature and decrease of interstitial fluid pressure [21, 22]. In order to prove that HSA-PTX nanoparticles could improve the tumor oxygenation, photoacoustic (PA) imaging, an optoacoustic and noninvasive imaging tool, which provides high-definition volumetric images of tissues by measuring light-induced sound waves from optically absorbing structures [46, 47], was conducted. The tumor oxygenation was measured by vascular saturated O2 (sO2), which was assessed using the standard multispectral PA imaging in the tumor in vivo with two excitation wavelengths at 750 nm and 850 nm for deoxygenated and oxygenated hemoglobin, respectively [48, 49]. PA imaging results showed that sO2 in tumors of mice treated with HSA-PTX (200 μg of PTX) was dramatically increased compared with the control group (Figure 4a and 4d). More importantly, the sO2 induced by PTX in the tumor remained at a high level 48 h after i.v. injection HSA-PTX. Considering that the radionuclide-labeled nanomaterials could accumulate in the tumor and continuously emit ionization rays for cancer therapy, the increased tumor oxygen content over a long period of time may be able to persistently enhance RIT efficacy.
To further prove that PTX delivered by HSA-PTX nanoparticles could relieve tumor hypoxia, immunofluorescence staining assay was conducted. Mice bearing 4T1 tumors were i.v. injected with HSA or HSA-PTX. At 24 h post injection, mice were then i.v. injected with pimonidazole hydrochloride. Tumors were surgically excised 90 min later, and then sliced for staining. Compared to tumors on mice treated with HSA, those on mice treated with HSA-PTX showed obviously weakened pimonidazole-stained (green) hypoxic area, indicating that PTX could efficiently improve the tumor oxygenation (Figure 4b and 4e). In addition, hypoxia situation in the area far from blood vessels was also greatly reversed, likely owing to the decreased interstitial fluid pressure and increased oxygen diffusion induced by PTX [21, 22]. Besides pimonidazole-based hypoxia staining, HIF-1α antibody staining was also used for evaluation of the tumor hypoxia state [50, 51]. HIF-1α, one of the two protein subunits composing HIF-1 complex, is continuously synthesized and degraded under normoxic conditions, and would accumulate rapidly following exposure to low oxygen tensions [52]. The over-expression of HIF-1α in solid tumors could mediate both DNA damage repair and angiogenesis, and finally lead to the radio-resistance [25, 53]. In our work, we also found that the HIF-1α expression level in tumors was sharply suppressed after HSA-PTX treatment (Figure 4c and 4f), favorable for effective RIT cancer treatment.
Finally, encouraged by the abilities of those PTX-bound albumin nanoparticles to passively target tumors and relieve tumor hypoxia, we then used 131I-HSA-PTX as a therapeutic agent for in vivo combined chemo-RIT. Nude mice bearing subcutaneous 4T1 tumors were randomly divided into four groups including: 1) PBS, 2) 131I-HSA (200 μCi of 131I), 3) HSA-PTX (200 μg of PTX), 4) 131I-HSA-PTX (200 μCi of 131I corresponding to 200 μg of PTX). Those nanoparticles were administrated via tail vein. This process was repeated every 4 days for a total of four treatment sessions (give at day 0, 4, 8 and 12). The tumor volumes were monitored by a caliper every other day (Figure 5a). Compared to the PBS group, RIT (131I-HSA) or chemotherapy (HSA-PTX) alone group could only slightly reduce the tumor growth speed (Figure 5a). Remarkably, the combination therapy by 131I-HSA-PTX was found to be much more effective, and appeared to be obviously stronger than the predicted additive effect of RIT and chemotherapy (Figure 5b), indicating the significant synergistic effect during combination therapy (combination index, CI=0.55). Microscopy images of hematoxylin and eosin (H&E) stained and TUNEL stained tumor slices also uncovered that the combined chemo-RIT by 131I-HSA-PTX exerted the most significant damages to tumor cells in vivo (Figure. 5c).
Though the synergistic effect of combined chemotherapy and RIT by 131I-HSA-PTX nanoparticles was not so obvious at the in vitro cellular level, excellent synergistic therapeutic result was observed in vivo. Such a difference may be attributed to the unique in vivo behaviors of 131I-HSA-PTX nanoparticles. Compared to 131I-HSA, 131I-HSA-PTX nanoparticles with prolonged blood half-life were found to show high tumor specific uptake and great intra-tumoral diffusion ability, both favorable for improved cancer therapy. In addition, the PTX-induced enhancement of tumor local oxygenation and suppression of HIF-1α expression in the 131I-HSA-PTX treatment group could help to overcome the hypoxia-associated radio-resistance and increase the radio-sensitivity of solid tumors during RIT, contributing to the synergistic therapeutic outcome in the combined chemo-RIT with 131I-HSA-PTX.
Figure 4
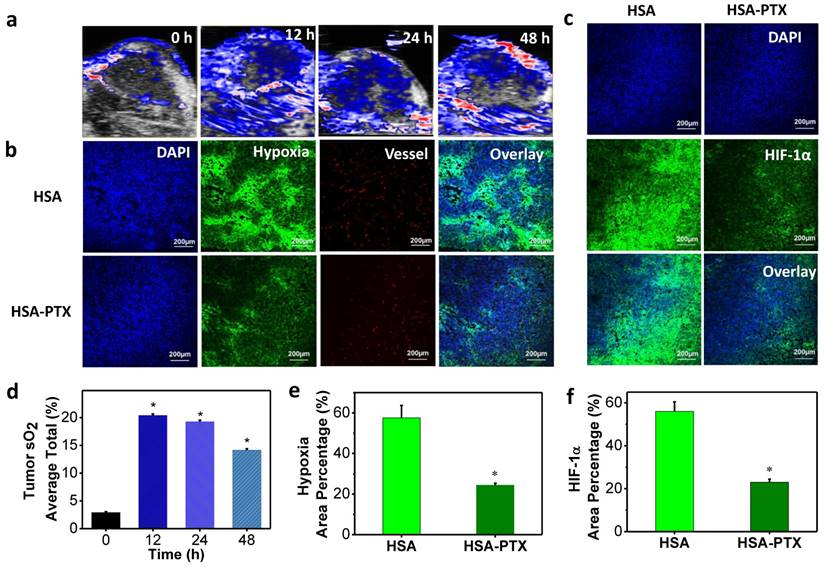
Modulation of tumor microenvironment after treatment with HSA-PTX. (a) Photoacoustic images of tumors after i.v. injection of HSA-PTX taken at different time points. (b) Representative immunofluorescence images of tumor slices collected from mice 24 h post i.v. injection of HSA or HSA-PTX. The cell nuclei, blood vessels, and hypoxia areas were stained with DAPI (blue), anti-CD31 antibody (red), and anti-pimonidazole antibody (green), respectively. (c) Anti-hypoxia inducible factor 1-alpha (anti-HIF-1α) antibody (green) stained tumor slices collected from mice 24 h post i.v. injection of HSA or HSA-PTX. (d) Tumor average total sO2 after HSA-PTX treatment from (a). Dramatic increase of sO2 after i.v. injection of HSA-PTX revealed the relived hypoxia state in the tumor. (e&f) Percentages of hypoxia-positive areas (e) and HIF-1α-positive areas (f) based on immunofluorescence staining results. More than 10 images were analyzed for each sample to obtain the statistic information. P values were calculated by ANOVA (*p < 0.001).
Figure 5
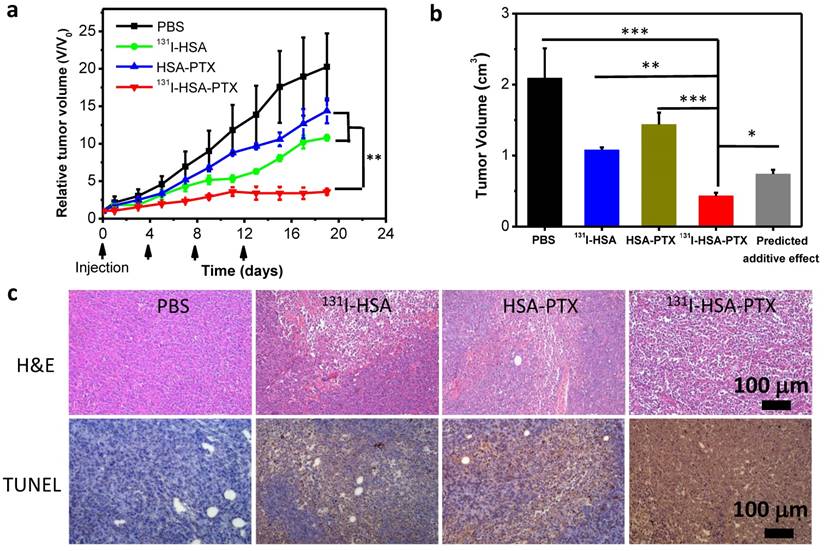
In vivo combination therapy. (a) Tumor growth curves of mice with different treatments given at day 0, 4, 8 and 12. Doses for each injection per mouse: 200 μg of PTX, 200 μCi of 131I. The tumor volumes were normalized to their initial sizes. (b) Average tumor volumes from mice 19 d after various treatments were initiated. The predicted additive effect was calculated by multiplying the tumor growth inhibition ratios of RIT alone and chemotherapy alone. (c) Images of H&E stained and TUNEL stained tumor slices from mice after different treatments. The most severe tumor damage and the highest level of tumor cell apoptosis were observed in the group after combined chemo-RIT with 131I-HSA-PTX. P values in (a&b) were calculated by ANOVA (***p < 0.001, **p < 0.01, and *p < 0.05).
Conclusion
In conclusion, a biocompatible / biodegradable nano-agent combining chemotherapy and RIT was constructed by simply mixing 131I-labeled HSA with PTX. In this formulation of nanoparticles, radionuclide 131I emitting strong beta and gamma rays could serve as a theranostic agent for nuclear imaging guided RIT, while PTX as a typical anticancer drug could induce the self-assembly of HSA, so as to improve the blood circulation half-life, tumor specific uptake and intra-tumoral diffusion ability. Notably, PTX bound to HSA nanoparticles delivered into the tumor could also remarkably enhance the tumor local oxygen level to overcome hypoxia-associated radio-resistance. As the results, the combined chemo-radioisotope therapy with 131I-HSA-PTX nanoparticles achieved excellent synergistic in vivo therapeutic effect. Therefore, our work presents a rather simple method to develop an albumin-based theranostic agent for in vivo synergistic chemo-RIT, based on a formulation similar to the clinical used nanoparticle drug, Abraxane. Considering the inherent biocompatibility of HSA, as well as the facile and reliable fabrication process, our agent may indeed have significant potential for clinical translation as a new nano/nuclear-medicine drug in cancer combination therapy.
Supplementary Material
Figure S1.
Acknowledgements
This work was partially supported by the National Research Programs from Ministry of Science and Technology (MOST) of China (2016YFA0201200), the National Natural Science Foundation of China (51525203), a Jiangsu Natural Science Fund for Distinguished Young Scholars (BK20130005), Collaborative Innovation Center of Suzhou Nano Science and Technology, and a Project Funded by the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Corsini MM, Miller RC, Haddock MG. Adjuvant radiotherapy and chemotherapy for pancreatic carcinoma: the Mayo Clinic experience (1975-2005). J. Clin. Oncol. 2008;26:3511-6
2. Bartelink H, Roelofsen F, Bosset JF. Concomitant radiotherapy and chemotherapy is superior to radiotherapy alone in the treatment of locally advanced anal cancer: results of a phase III randomized trial of the European Organization for Research and Treatment of Cancer Radiotherapy and Gastro. J. Clin. Oncol. 1997;15:2040-9
3. Lee YL, Lee YJ, Ahn SJ, Choi TH, Moon BS, Cheon GJ. et al. Combined radionuclide-chemotherapy and in vivo imaging of hepatocellular carcinoma cells after transfection of a triple-gene construct, NIS, HSV1-sr39tk, and EGFP. Cancer Lett. 2010;290:129-38
4. Steel GG, Peckham MJ. Exploitable mechanisms in combined radiotherapy-chemotherapy: the concept of additivity. Int. J. Radiat. Oncol. 1979;5:85-91
5. Nelson DF. Radiotherapy in the treatment of primary central nervous system lymphoma (PCNSL). J. Neuro-Oncol. 1999;43:241-7
6. Lavik E, Von RH. The role of nanomaterials in translational medicine. Acs Nano. 2011;5:3419-24
7. Ma X, Zhao Y, Liang XJ. Theranostic nanoparticles engineered for clinic and pharmaceutics. Accounts Chem. Res. 2011;44:1114-22
8. Li X, Liu W, Sun L, Aifantis KE, Yu B, Fan Y. et al. Effects of physicochemical properties of nanomaterials on their toxicity. J. Biomed. Mater. Res. A. 2015;103:755-63
9. Zhong X, Yang K, Dong Z, Yi X, Wang Y, Ge C. et al. Polydopamine as a biocompatible multifunctional nanocarrier for combined radioisotope therapy and chemotherapy of cancer. Adv. Funct. Mater. 2015;25:7327-36
10. Yi X, Yang K, Liang C, Zhong X, Ning P, Song G. et al. Imaging-guided combined photothermal and radiotherapy to treat subcutaneous and metastatic tumors using iodine-131-doped copper sulfide nanoparticles. Adv. Funct. Mater. 2015;25:4689-99
11. Absar S, Nahar K, Choi S, Ahsan F, Yang VC, Kwon YM. Serum albumin-protamine conjugate for biocompatible platform for targeted delivery of therapeutic macromolecules. J. Biomed. Mater. Res. A. 2014;102:2481-90
12. Chen Q, Wang C, Zhan Z, He W, Cheng Z, Li Y. et al. Near-infrared dye bound albumin with separated imaging and therapy wavelength channels for imaging-guided photothermal therapy. Biomaterials. 2014;35:8206-14
13. Green MR, Manikhas GS, Afanasyev B, Makhson AM, Bhar P, Hawkins MJ. Abraxane®, a novel Cremophor®-free, albumin-bound particle form of paclitaxel for the treatment of advanced non-small-cell lung cancer. Ann. Oncol. 2006;17:1263-8
14. Miele E, Spinelli GP, Miele E, Tomao F, Tomao S. Albumin-bound formulation of paclitaxel (Abraxane ABI-007) in the treatment of breast cancer. Int. J. Nanomed. 2009;4:99-105
15. Chen Q, Liang C, Wang C, Liu Z. An imagable and photothermal “Abraxane-Like” nanodrug for combination cancer Therapy to treat subcutaneous and metastatic breast tumors. Advanced Materials. 2015;27:903-10
16. Chen Q, Wang X, Wang C, Feng L, Li Y, Liu Z. Drug-induced self-assembly of modified albumins as nano-theranostics for tumor-targeted cmbination therapy. Acs Nano. 2015;9:5223-33
17. Chen Q, Liu X, Chen J, Zeng J, Cheng Z, Liu Z. A self-assembled albumin-based nanoprobe for in vivo ratiometric photoacoustic pH imaging. Adv. Mater. 2015;27:6820-27
18. Chen Q, Liu Z. Albumin carriers for cancer theranostics: A conventional platform with new promise. Adv. Mater. 2016 DOI: 10.1002/adma.201600038
19. Baruzzi A, Contin M, Perucca E, Albani F, Riva R. DOTA conjugate with an albumin-binding entity enables the first folic acid-targeted 177Lu-radionuclide tumor therapy in mice. J. Nucl. Med. 2012;36:1360-1
20. Mei L, Junxing H, Dongsheng Z, Xingmao J, Jia Z, Hong Y. et al. Hepatoma-targeted radionuclide immune albumin nanospheres:131I-antiAFPMcAb-GCV-BSA-NPs. Anal. Cell. Pathol. 2016;2016:1-8
21. Taghian AG, Abiraad R, Assaad SI, Casty A, Ancukiewicz M, Yeh E. et al. Paclitaxel decreases the interstitial fluid pressure and improves oxygenation in breast cancers in patients treated with neoadjuvant chemotherapy: clinical implications. Am. J. Physiol. 2005;23:1951-61
22. Danhier F, Danhier P, Saedeleer CJD, Fruytier AC, Schleich N, Rieux AD. et al. Paclitaxel-loaded micelles enhance transvascular permeability and retention of nanomedicines in tumors. Int. J. Pharm. 2015;479:399-407
23. Moeller BJ, Richardson RA, Dewhirst MW. Hypoxia and radiotherapy: opportunities for improved outcomes in cancer treatment. Cancer Metast. Rev. 2007;26:241-8
24. Huang LE, Bindra RS, Glazer PM, Harris AL. Hypoxia-induced genetic instability— Calculated mechanism underlying tumor progression. J. Mol. Med. 2007;85:139-48
25. Bristow RG, Hill RP. Hypoxia and metabolism. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer. 2008;8:180-92
26. Giaccia A, Siim BG, Johnson RS. HIF-1 as a target for drug development. Nat. Rev. Drug Discov. 2003;2:803-11
27. Liu J, Zhang L, Yang Z, Zhao X. Controlled release of paclitaxel from a self-assembling peptide hydrogel formed in situ and antitumor study in vitro. Int. J. Nanomed. 2011;6:2143-53
28. Li Y, Wu Z, Wei H, Chao Q, Jing Y, Zhou J. et al. Globular Protein-Coated Paclitaxel Nanosuspensions: Interaction Mechanism, Direct Cytosolic Delivery, and Significant Improvement in Pharmacokinetics. Mol. Pharm. 2015;12:1485-1500
29. De BT, Taourel P, Tubiana JM, Kuoch V, Ducreux M, Lumbroso J. et al. Hepatic intraarterial 131I iodized oil for treatment of hepatocellular carcinoma in patients with impeded portal venous flow. Radiology. 1999;212:665-8
30. Worth AJ, Zuber RM, Hocking M. Radioiodide 131I therapy for the treatment of canine thyroid carcinoma. Aust. Vet. J. 2005;83:208-14
31. Gong G, Xu Y, Zhou Y, Meng Z, Ren G, Zhao Y. et al. Molecular switch for the assembly of lipophilic drug incorporated plasma protein nanoparticles and in vivo image. Biomacromolecules. 2012;13:23-8
32. Giu M, VasilEv AV. Degradation of endo- and exogenous albumin in normal and starved rats. Voprosy Medit͡sinskoĭ Khimii. 1992;38(5):10-2
33. Eppel GA, Pratt LM, Greive KA, Comper WD. Exogenous albumin peptides influence the processing of albumin during renal passage. Nephron. 2002;92(1):156-64
34. Simpson LO, Shand BI. The implications of the changes in the nature of the urinary proteins which occur in albumin overload-induced proteinuria in normal mice. Int. J Eep. Pathol. 1983;64:6-14
35. Trieu V, Cordia J, Yang A, Beals B, Ci S, Louie L. et al. Pharmacokinetic and ADME study of nanoparticle albumin-bound 17AAG (nab-17AAG) in mice. Cancer Res. 2008;68:5747-5747
36. Chen Q, Wang C, Zhan Z, He WW, Cheng ZQ, Li YY. et al. Near-infrared dye bound albumin with separated imaging and therapy wavelength channels for imaging-guided photothermal therapy. Biomaterials. 2014;35:8206-14
37. Li SD, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol. Pharm. 2008;5:496-504
38. Kassis AI, Adelstein SJ. Radiobiologic principles in radionuclide therapy. J. Nucl. Med. 2005;46(Suppl 1):4S-12S
39. Ozin GA. Nanotechnology-based precision tools for the detection and treatment of cancer. Cancer Res. Treat. 2015;35:481-501
40. Lei M, Huang L. Exploring the tumor microenvironment with nanoparticles. Cancer Res. Treat. 2015;166:193-226
41. Sabroso CM, Torres-Suárez AI. Objective: tumor strategies of drug targeting at the tumor mass level. Clin. Transl. Oncol. 2014;16:1-10
42. Ruttala HB, Ko YT. Liposome encapsulated albumin-paclitaxel nanoparticle for enhanced antitumor efficacy. Pharm. Res. 2015;32:1002-16
43. Zhang H, Verkman AS. Microfiberoptic measurement of extracellular space volume in brain and tumor slices based on fluorescent dye partitioning. Biophys. J. 2010;99:1284-91
44. Torosean S, Flynn B, Axelsson J, Gunn J, Samkoe KS, Hasan T. et al. Nanoparticle uptake in tumors is mediated by the interplay of vascular and collagen density with interstitial pressure. Nanomed. Nanotechnol. 2013;9:151-8
45. Ng CP, Pun SH. A perfusable 3D cell-matrix tissue culture chamber for in situ evaluation of nanoparticle vehicle penetration and transport. Biotechnol. Bioeng. 2008;99:1490-501
46. Li L. Photoacoustic Imaging. Pathobiology of Human Disease. 2014;2:3912-24
47. Zhang Y, Hong H, Cai W. Photoacoustic imaging. Cold Spring Harb. Protoc. 2011;9:3912-24
48. Jöbsis FF. Noninvasive, infrared monitoring of cerebral and myocardial oxygen sufficiency and circulatory parameters. Science. 1977;198:1264-7
49. Prasad P, Gordijo CR, Abbasi AZ, Maeda A, Ip A, Rauth AM. et al. Multifunctional albumin-MnO₂ nanoparticles modulate solid tumor microenvironment by attenuating hypoxia, acidosis, vascular endothelial growth factor and enhance radiation response. Acs Nano. 2014;8:3202-12
50. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat. Med. 2006;12:895-904
51. Gong H, Chao Y, Xiang J, Han X, Song G, Feng L. et al. Hyaluronidase To enhance nanoparticle-based photodynamic tumor therapy. Nano Lett. 2016;16:2512-21
52. Salceda S, Caro J. Hypoxia-inducible factor 1α (HIF-1α) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions its steablization by hypoxia depends on redox-induced changes. J. Biol. Chem. 1997;272:22642-7
53. Dewhirst MW, Cao Y, Li CY, Moeller B. Exploring the role of HIF-1 in early angiogenesis and response to radiotherapy. Radiother. Oncol. 2007;83:249-55
Author contact
![]() Corresponding author: kyangedu.cn, zliuedu.cn
Corresponding author: kyangedu.cn, zliuedu.cn
Citation styles
APA
Tian, L., Chen, Q., Yi, X., Wang, G., Chen, J., Ning, P., Yang, K., Liu, Z. (2017). Radionuclide I-131 Labeled Albumin-Paclitaxel Nanoparticles for Synergistic Combined Chemo-radioisotope Therapy of Cancer. Theranostics, 7(3), 614-623. https://doi.org/10.7150/thno.17381.
ACS
Tian, L.; Chen, Q.; Yi, X.; Wang, G.; Chen, J.; Ning, P.; Yang, K.; Liu, Z. Radionuclide I-131 Labeled Albumin-Paclitaxel Nanoparticles for Synergistic Combined Chemo-radioisotope Therapy of Cancer. Theranostics 2017, 7 (3), 614-623. DOI: 10.7150/thno.17381.
NLM
Tian L, Chen Q, Yi X, Wang G, Chen J, Ning P, Yang K, Liu Z. Radionuclide I-131 Labeled Albumin-Paclitaxel Nanoparticles for Synergistic Combined Chemo-radioisotope Therapy of Cancer. Theranostics 2017; 7(3):614-623. doi:10.7150/thno.17381. https://www.thno.org/v07p0614.htm
CSE
Tian L, Chen Q, Yi X, Wang G, Chen J, Ning P, Yang K, Liu Z. 2017. Radionuclide I-131 Labeled Albumin-Paclitaxel Nanoparticles for Synergistic Combined Chemo-radioisotope Therapy of Cancer. Theranostics. 7(3):614-623.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.