Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Tumor microenvironment and...
Design, responsive modes, and...
Multi-stimuli responsive drug...
Conclusions and Prospects
Abbreviations
Acknowledgements
References
Introduction
Tumor microenvironment and...
Design, responsive modes, and...
Multi-stimuli responsive drug...
Conclusions and Prospects
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2017; 7(3):538-558. doi:10.7150/thno.16684 This issue Cite
Review
Current Multistage Drug Delivery Systems Based on the Tumor Microenvironment
Binlong Chen1, 2, Wenbing Dai1, ![]() , Bing He1,2, Hua Zhang1, Xueqing Wang1, Yiguang Wang1, 2, Qiang Zhang1, 2,
, Bing He1,2, Hua Zhang1, Xueqing Wang1, Yiguang Wang1, 2, Qiang Zhang1, 2, ![]()
1. Beijing Key Laboratory of Molecular Pharmaceutics and New Drug Delivery Systems, School of Pharmaceutical Sciences, Peking University, Beijing 100191, China
2. State Key Laboratory of Natural and Biomimetic Drugs, Beijing 100191, China
Received 2016-7-2; Accepted 2016-11-14; Published 2017-1-7
Citation:
Chen B, Dai W, He B, Zhang H, Wang X, Wang Y, Zhang Q. Current Multistage Drug Delivery Systems Based on the Tumor Microenvironment. Theranostics 2017; 7(3):538-558. doi:10.7150/thno.16684. https://www.thno.org/v07p0538.htm
Other stylesAbstract

The development of traditional tumor-targeted drug delivery systems based on EPR effect and receptor-mediated endocytosis is very challenging probably because of the biological complexity of tumors as well as the limitations in the design of the functional nano-sized delivery systems. Recently, multistage drug delivery systems (Ms-DDS) triggered by various specific tumor microenvironment stimuli have emerged for tumor therapy and imaging. In response to the differences in the physiological blood circulation, tumor microenvironment, and intracellular environment, Ms-DDS can change their physicochemical properties (such as size, hydrophobicity, or zeta potential) to achieve deeper tumor penetration, enhanced cellular uptake, timely drug release, as well as effective endosomal escape. Based on these mechanisms, Ms-DDS could deliver maximum quantity of drugs to the therapeutic targets including tumor tissues, cells, and subcellular organelles and eventually exhibit the highest therapeutic efficacy. In this review, we expatiate on various responsive modes triggered by the tumor microenvironment stimuli, introduce recent advances in multistage nanoparticle systems, especially the multi-stimuli responsive delivery systems, and discuss their functions, effects, and prospects.
Keywords: drug delivery systems, tumor microenvironment, multistage, stimuli-responsive, activatable nanoparticles
Introduction
Tumor-targeted nanoparticle delivery system is a landmark discovery and has been extensively studied in tumor therapy and diagnosis since the enhanced permeability and retention (EPR) effect was first described in 1986 [1] and the concept of nanomedicine emerged in the late 1990s [2]. To effectively deliver drugs to tumor tissues, nanoparticles need to remain stable during blood circulation and exhibit enhanced accumulation in the tumor. To maximize the therapeutic efficacy, nanoparticles should be able to penetrate into the deep tumor tissue, have high-affinity interaction with tumor cells and release the drugs outside or within the tumor cells. Enormous progress has been made in many aspects, such as PEGylation [3], size and surface property optimization, and targeting module (antibody and peptide ligand) modification [4, 5].
With regard to PEGylated passively targeted nanoparticles, many can successfully extravasate into the tumor tissue following intravenous injection. However, only few nanoparticles are capable of spreading throughout the entire tumor because of several biological barriers in the tumor microenvironment, such as the dense tumor stroma, abnormal angiogenesis, and elevated tumor interstitial fluid pressure (IFP). Furthermore, most nanoparticles are unable to deliver the drugs into tumor cells actively. Also, a balance needs to be found between the stability of the nanoparticles during the transport in blood circulation and the ideal tumor cellular uptake after extravasation into tumor tissue. For example, PEGylation increases nanocarrier escape from the blood opsonization processes and achieves perfect passive tumor targeting. Nevertheless, some studies have revealed that the outer PEG layer significantly prevents the uptake of nanoparticles into tumor cells. This is referred to as the “PEG dilemma” [6].
As for the ligand-modified actively targeted delivery systems, despite promising results in preclinical studies, most of the clinical trials did not yield the expected results. The likely reasons include not only the similar challenges as the passively targeted nanoparticles but also the heterogeneous expression of membrane receptors among individual patients and different tumors as well as during different tumor stages within a single tumor. Furthermore, there is a unique challenge for the actively targeted delivery systems. The “binding site barrier” phenomenon of actively targeted nanoparticles, resulting from the strong receptor-ligand interactions at the tumor periphery after preferential extravasation from the leaky tumor vasculature, can further obstruct the penetration of the extravasated nanoparticles into the tumor [7].
Therefore, more efficient drug delivery strategies are in great demand. Multistage drug delivery systems (Ms-DDS), also referred to as stimuli-responsive or activatable systems, show different behaviors during systemic circulation and in the tumor microenvironment. Ms-DDS have been developed in the 1970s when the first thermo-sensitive liposome for accelerated and quick release of drugs in the tumor was designed [8]. The stimuli of Ms-DDS can mainly be divided into two classes: 1) the extracorporeal physical stimuli, such as the magnetic-responsive superparamagnetic iron oxide-based nanoparticles [9, 10] and external light- [11], thermo- [12] or ultrasound-sensitive drug delivery systems [13]; 2) the endogenous tumor microenvironment and intracellular biological stimuli, including the acidic pH [14], over-expressed enzymes [15], hypoxia [16] and elevated reductive conditions within organelles [17]. Such stimuli can result in a change in nanoparticles size, zeta potential, hydrophobicity or carrier disassembly, which is favorable for nanoparticles penetration deep into the tumor as well as endocytosis by tumor cells, thus achieving much more efficient tumor cell targeting.
The progress made in Ms-DDS that are induced by extracorporeal physical stimuli in recent years has been exhaustively summarized and discussed [18]. In this review, we mainly describe Ms-DDS that are sensitive to the tumor microenvironment for tumor therapy and imaging. First, a brief introduction is provided to summarize the main biological responsive factors of the tumor microenvironment (acidic pH, enzymatic up-regulation, increased redox potential and hypoxia) followed by details about the responsive mechanisms. Next, we focus on the various responsive actions based on various stimuli, their functions for drug delivery, and design strategies for Ms-DDS in recent years. Finally, we introduce, in particular, the current multi-stimuli responsive DDS (Ms-DDS) for cancer therapy and discuss their advantages and limitations.
Tumor microenvironment and cellular biological stimuli
Compared with normal tissues or cells, several distinguishing biological/endogenous factors (e.g., pH, enzymes, redox, and hypoxia) in the tumor microenvironment or tumor cells have been used as stimuli to trigger specific changes in nanoparticles. Since these stimuli have been extensively studied and validated, we provide here a brief overview and summary.
The acidic pH
The acidic pH, including the pH of the extracellular tumor microenvironment (pHe 6-7) as well as the intracellular endosomes and lysosomes (pHi 4-6), is the most widely-used trigger for stimuli-responsive drug-delivery systems due to its universality in most solid tumors. The decreased pH is due to the Warburg effect, which was first proposed by Warburg in the 1930s [19]; it illustrates the phenomenon of increased glucose uptake and aerobic glycolysis in cancer cells leading to an increase in lactate production even when the oxygen levels are normal [20, 21]. The mechanisms of various pH-sensitive nanocarriers with different design basis (including ionizable chemical groups, acid-labile chemical bonds, pH-sensitive peptides, gas-generating systems and pH-responsive polymers) and specific responsive components with their respective functions are summarized in Table 1.
Despite extensive research and verification, exploiting the acidic pH in tumor microenvironment has its own shortcomings. First, the acidic pH is typically located far from the blood vessels which would lead to no response of pH-responsive nanoparticles in the perivascular regions. Furthermore, sometimes the pH differences between the normal tissue and tumor tissue are not significant enough to trigger the responsiveness.
Table 1
Mechanisms of pH-sensitive drug delivery systems.
| Mechanism | Species | Functions | Refs. |
|---|---|---|---|
| Ionizable chemical groups | Amines, phosphoric acids, carboxylic acids, acylsulfonamide | i. Molecular conformation change ii. Drug release iii. Positive charge | [75, 77, 95] |
| Acid-labile chemical bonds | Acetal, orthoester, hydrazone, imine, cis-aconyl | i. Hydrolytic cleavage ii. Size shrinking iii. Drug release iiii. Ligand re-emergence | [62, 177, 187] |
| pH-responsive polymer | Anionic polymers containing carboxylic groups (PAA, PMAA, PEAA, PPAA, PBAA, NIPAM, and PGA), and sulfonamide groups | i. Drug release | [189-190] |
| pH-responsive polymer | Cationic polymers containing tertiary amine groups, pyridine groups and imidazole groups | i Drug release ii. Positive charge iii. Proton sponge | [76, 85, 86, 118] |
| pH-sensitive peptide | GALA, pHLIP, histidine-containing acidic pH-activated cell-penetrating peptides | i. Molecular conformation change ii. Cell penetration | [155, 191] |
| Incorporate carbon dioxide-generating precursors | Sodium bicarbonate, ammonium bicarbonate | i. Carbon dioxide generation ii. Drug release | [132, 133] |
Table 2
Key hydrolases of enzyme-sensitive drug delivery systems.
| Class | Enzyme | Substrate | Position | Functions | |
|---|---|---|---|---|---|
| Proteases | CAPs (MMP-1/MMP-2/MMP-9) | GPLGVRGK, GPLGIAGQ, PVGLIP, PLGLAGr9 peptides, gelatin | Extracellular | Size-shrinking [116, 192] PEG deshielding [61,193] Drug release [194, 195] Morphology switch [196, 197] | |
| Proteases | Lysosomal enzyme (Cathepsin B) | GFLG peptide, Biotin-avidin system | Intracellular (mainly lysosome) | Drug release [198] Cap system [150, 199] | |
| Proteases | FAP-α | DRGETGPAC | Extracellular (mainly CAFs) | Fluorescence release [200] | |
| Peptidase | Amino peptidase | Unacylated amino acids | Extracellular | CPP activation [201] | |
| Peptidase | Dipeptidyl peptidase IV | Xaa-Pro dipeptides | Extracellular | CPP activation [201] | |
| Lipases | PLA2 | Phospholipids with a non-hydrolyzable ether bond in the 1-position, di(ethylene glycol) diacrylate hyaluronic acid | Extracellular | Drug release [202, 203] Cellular uptake [204] Size shrinking [205] | |
The upregulated enzyme
The upregulated enzymes by tumor cells and tumor stroma cells is another important trigger for the tumor microenvironment-responsive Ms-DDS [22, 23]. High concentrations of enzymes play a critical role in tumor cell growth, angiogenesis, invasion, and metastasis serving as a key stimulus for responsive DDS. Compared with nanoparticles modified with targeting molecules that recognize the receptors overexpressed by cancer cells, polymeric nanomaterials that are sensitive to enzymes in the tumor enable a tumor-confined, spatiotemporal drug release at the right time and in the right place thus achieving superior tumor targeting efficacy with reduced unwanted side effects in normal organs. There are several types of over-expressed enzymes in solid tumors, including proteases (e.g., matrix metalloproteinase and cathepsin B), peptidases (e.g., aminopeptidase), and lipases (e.g., phospholipase A2). The substrates and functions of the individual enzyme-responsive Ms-DDS are summarized in Table 2. The high level of enzymes in tumoral and perivascular regions make them favorable stimuli for different nano-sized Ms-DDS.
The hypoxia
The vascular network in the solid tumor is abnormal and incapable of delivering sufficient oxygen to the entire tumor tissue (especially the inner region with poor vascularization). Also, the over-proliferated tumor cells further accelerate the consumption of oxygen [24]. These two factors mainly account for the hypoxia in the tumor microenvironment. Evidently, the regions that are at a further distance from the blood vessels in the tumor microenvironment are typically hypoxic which is not favorable to the large nanoparticles with low penetration ability into deep tissue. A series of hypoxia-sensitive nanoparticle systems have been developed and can be divided into two main mechanisms. The first one is, nitroimidazole-based micelle systems that can undergo bioreduction to aminoimidazole, leading to a hydrophobic-to-hydrophilic transition and further triggering micelle disassembly [25]. The second mechanism entails the azobenzene group-employed siRNA nanocarriers which can be cleaved, leaving the PEI/siRNA complex with a positively charged surface for facilitating cellular uptake [26].
The reductive environment
Unlike the extracellular stimuli, redox potential is an intracellular stimulus that results from the different concentrations of glutathione (GSH) between the extracellular space (approximately 2-10 μM) and intracellular space (approximately 2-10 mM) [27]. The huge concentration gradient in GSH (about 100-1000 times) can efficiently ensure the responsiveness of the redox-sensitive Ms-DDS. Numerous intracellularly-triggered Ms-DDS are based on the reductive environment in the tumor cells. The disulfide bond that is prone to the rapid cleavage by GSH represents the most typical and efficient mechanism. By introducing the disulfide bond, Ms-DDS can achieve several functions including particle disassembly, nanocarrier degradation, and fast drug release [28-30]. Since the disulfide bond can be easily introduced into drugs and polymers, it is widely applied in the intracellularly-responsive Ms-DDS. Progress on redox-responsive Ms-DDS has been described previously [18]. Also, disulfide bond-based redox-responsive Ms-DDS have been utilized in several nanocarriers, such as zwitterionic dextran nanoparticles [31], single-walled carbon nanotubes [32], chitosan-based nanoparticles [33], PEGylation micelles [34], and mesoporous silica nanoparticles [35, 36]. These redox-sensitive Ms-DDS should be designed carefully to make them not only stable in the blood circulation but also in the tumor tissue before entering into tumor cells.
Other stimuli
In addition to the biological stimuli described above, the oxidative stress [37, 38], hyperthermia [39], and ATP [40] have also been applied in the Ms-DDS based on the tumor and cellular microenvironment. In the future, novel Ms-DDS responsive to other specific pathophysiological properties in the tumor microenvironment, such as high or unique levels of amino acids, functional proteins, and DNA fragments, will be designed and applied in tumor targeting [41].
Design, responsive modes, and functions of Ms-DDS
Taking advantage of the unique stimuli in the tumor microenvironment briefly mentioned above, conventional DDS can be redesigned or modified to become Ms-DDS for tumor targeted therapy or imaging. Upon reaching the tumor microenvironment, Ms-DDS receive the stimulation and then change the physicochemical features or structures, eventually realizing the desired functions. Different responsive modes of Ms-DDS and their functions are discussed in the following sections.
Various tumor microenvironment stimuli, especially the acidic pH and upregulated enzymes, can trigger approximately the same behavior of Ms-DDS in tumor tissues. Therefore, this section is arranged mainly according to the changes of nanoparticles (or responsive modes) instead of the stimuli as in the previous review [18]. Generally, the responsive modes in the tumor microenvironment include PEG detachment, targeting ligand exposure, surface charge reversal, and particle-size shrinkage. Ms-DDS with stimuli-responsive drug release and signal Turn-OFF/ON are the hotspots in the field in recent years and therefore are included as a separate sub-section.
3.1. PEG detachment
As mentioned above, the “PEG dilemma” phenomenon greatly hinders the interaction between nanoparticles and cell membrane, leading to poor cellular uptake and unsatisfactory tumor inhibition [42]. Therefore, several Ms-DDS have been developed to overcome this problem by tumor microenvironment-triggered PEG cleavage, which is referred to as sheddable nanoparticle systems. During blood circulation, the outer PEG layer can shelter the hydrophobicity and positive charge of the core, keeping the stability of nanoparticles and prolonging the blood circulation time. As shown in Fig. 1A, When the MS-DDS leaks into the tumor site, the linker between PEG and the particle core is cleaved by the stimuli in the tumor microenvironment and the PEG layer is detached, thus exposing the original nanoparticle with an enhanced surface-cell interaction and increasing the antitumor effect. Hypoxic-degradable nitroimidazole [26], redox-degradable disulfide bond [43], acid-degradable amide bond [44], and 2-propionic-3-methylmaleic anhydride (CDM) [45] have been applied in the PEG detachable Ms-DDS for improved chemotherapeutics and siRNA delivery. In the acid-degradable amide bond example, PEG-Dlinkm-PLGA was developed by introducing a pH-sensitive link (Dlinkm), which resulted in a significant increase in the cellular uptake of the pH sensitive dPEGNPPLGA when pretreated at pH 6.5 compared with pretreatment at pH 7.4. However, the intracellular amount of siRNA of the pH insensitive NPPLGA and NPPLA showed no difference when pretreated at different pH values [44].
Along with inner core exposure of the Ms-DDS, PEG detachment can also achieve several other effects, including ligand re-emergence, charge reversal, and particle disassembly, which will be further discussed in the following sections.
Targeting ligand re-emergence
Compared with the unmodified passive nanoparticles, the actively-targeted nanovehicles are a landmark discovery in tumor therapy. However, every coin has two sides. Although targeting ligand modification can promote cellular uptake, some problems such as the immunogenicity of antibodies, unfavorable circulation time, toxicity of targeting peptides, and non-specific uptake of nanocarriers by non-cancerous cells represent a significant problem. Take the cell-penetrating peptides (CPPs) as an example; it has previously been shown that the positively charged CPPs can target and penetrate into tumor cells in a receptor-independent manner which has been widely applied in tumor diagnosis and tumor therapy studies [46, 47]. However, the clinical in vivo application of CPPs is limited because of the two drawbacks. First, the positively charged amino acids can be recognized and scavenged by the reticuloendothelial system (RES), leading to instability in the physiological blood circulation. Second and more important, most CPPs exhibit nonspecific targeting to tumor cells and normal tissues resulting in severe toxicity and weakened therapy [48, 49]. It is desirable that the CPPS emerge and become effective only after the actively-targeted nanovehicles access the target-site. The emerging multistage nanocarriers with targeting ligands responsive to tumor microenvironment include the activatable cell-penetrating peptide (ACPP) systems, de-shielding systems, “pop-up” systems, and trojan-horse targeting systems.
Activatable cell-penetrating peptide systems
ACPP systems were first put forward in 2004 by Tsien's group [50] and have been developed and applied in a variety of nanoscale systems, including dendrimer [51], peptide-drug conjugation [52], polypeptides lipid nanoparticles [53], PEG-PLA micelles [54], and others. As shown in Fig. 1B (i), ACPP is a moiety that consists of three segments: CPP, enzyme-responsive peptide, and polyanionic sequences that can neutralize the positive charge of CPP. During circulation, the polyanionic sequences could shield the nonspecific adhesiveness of CPPs. Once MS-DDS permeate into tumors, the enzyme-sensitive peptide linkers are cleaved by proteases (main1y MPPs) and the CPPs appear on the surface to achieve perfect cellular uptake with reduced side effects. Recent studies have revealed that the cellular uptake of ACPP-conjugated cargo was significantly improved in almost all tissues, including tumors as well as normal tissues, which meant that the activation of ACPP was tumor-independent and quite possibly happened in the vasculature [55]. In 2007, Bae and co-authors constructed a pH-responsive ACPP system assembled by TAT-modified PLA-PEG micelles and ultra pH-sensitive copolymers of PEGylated poly (methacryloyl sulfadimethoxine) (PSD-b-PEG) [56]. In physiological condition, the anionic PSD can combine with the cationic TAT peptide through electrostatic interactions. In the acidic tumor microenvironment, on the other hand, the protonation of sulfonamide leads to separation from TAT allowing improved interaction with tumor cells. There are still deficiencies that remain to be overcome. As previously mentioned, it is too difficult for the short TAT peptide with limited positive charges to maintain a strong electrostatic interaction with PSD, thus leading to fast dissociation in the bloodstream with various serum proteins [57, 58].
To solve the problems mentioned above, some novel ACPP systems have been designed. Xiang's group developed a legumain protease-activated TAT-liposome for tumor targeting [59]. The substrate of endoprotease legumain, alanine-alanine-asparagine (AAN), was attached to the fourth lysine of TAT to create a branch in CPPs, decreasing the cell-penetrating capacity of TAT by 72.65%. While in the tumor microenvironment, AAN was directly cleaved by extracellular legumain overexpressed in a wide variety of tumors following which the transmembrane transport function of TAT was fully recovered. Therefore, much more doxorubicin loaded in AAN-TAT-modified liposomes (AAN-TAT-Lipo-Dox) accumulated in tumor tissues and entered into tumor cells, leading to a greatly increased tumoricidal effect (4-fold apoptotic rate of TAT-Lipo-Dox) significantly prolonging the survival duration and reducing the systemic toxicity. Compared with the former ACPP systems, the AAN-TAT system has the advantages of briefness with only two segments and directly blocking the core domain of penetration. Furthermore, since legumain is also upregulated in tumor-associated macrophages (TAMs), the AAN-TAT-modified nano cargos can target tumor cells as well as the tumor stroma.
The pH-sensitive and redox-sensitive strategies have also been applied in the novel ACPP systems. Radosz's group introduced succinyl amides to the lysine residues' amines of TAT (aTAT), masking the nonspecific interactions in the bloodstream. After entering the tumor microenvironment, the acidic pH quickly hydrolyzed the succinyl amides restoring the cell-penetration function of TAT. An aTAT-functionalized and DOX-loaded PEG-PCL micelle system was further constructed and exhibited good antitumor activity and low cardiotoxicity [57]. Furthermore, Leroux's group developed a redox-triggered azobenzene PEG chain self-immolative linkage conjugated to the lysine residues of CPPs for delivering nucleic acid drugs [60].
Shielded/de-shielded systems
Since PEG layers can shield the surface characteristics of nanoparticles (including zeta potential, hydrophobicity etc.), stimuli-sensitive PEG chains have also been applied to mask CPPs before entering into the tumor microenvironment. As shown in Fig. 1B (ii), Torchilin and colleagues modified the surface of the same nanoparticles with two PEG chains of different lengths as follows: the longer chain (PEG2000) was conjugated with an MMP2-sensitive peptide [61] or a pH-sensitive hydrazine bond [62], which acted as a hydrophilic corona to hide TAT peptide. This conjugated long chain TAT peptide was bonded to the terminal of the shorter chain (PEG1000), which was exposed in the tumor microenvironment. Therefore, the cellular uptake of the PTX-loaded nanoparticles with the shielded/de-shielded TAT modification was nearly 2-fold higher than that of the unresponsive nanoparticles and the tumor suppression efficacy was significantly improved.
Sofou et al. also designed an acid-triggered lipid vesicle system with tunable membrane surface topography [63]. Because some lipids, such as distearyl phosphatidyl serine, can be protonated by the acidic tumor microenvironment, heterogeneous membranes with phase-separated domains can be formed by the creation of hydrogen bonds among the newly protonated lipids. Thus, the PEGylated-titratable lipids were mixed with ligand-modified lipids to develop the acid-tunable heterogeneous lipid vesicles. Before protonation, the lipid vesicles exhibited homogeneity, and their outer PEG layer masked the ligands, leading to longer circulation in the blood stream. Once within the acid tumor microenvironment, the gathered PEGylated lipids exposed the shielded ligands to interact with tumor cells. In vitro and in vivo studies have further verified the superiority of the pH-triggered lipid-membrane systems [64]. However, the application of this Ms-DDS strategy is limited to lipid vesicles as most nanoparticles do not contain lipids. Additionally, the gathered PEG layers in the tumor microenvironment may also have some influence on blocking the interaction between targeting ligands and tumor cells.
“Pop-up” systems
In addition to the acid-triggered ACPP, Bae's group also developed a pH-induced targeting ligand pop-up system displayed in Fig. 1B (iii) [65, 66]. Acid-responsive poly (L-histidine) (polyHis) was synthesized and conjugated with PEG and poly (L-lactic acid) (pLLA) to prepare polyHis-b-PEG and pLLA-b-PEG-polyHis-ligand, respectively. Mixed micelles were designed with these two copolymers and performed differently in different pH environments. First, the ligands (biotin or TAT) were anchored to the core of micelles because of the hydrophobic interaction between the core and polyHis adjacent to the ligands and the targeting ability was shielded by the outer PEG shell during circulation (pH>7.0). Subsequently, after protonation by the acidic tumor microenvironment, the polyHis became hydrophilic bringing the ligands out of the PEG surface with recovered targeting effect. Contrary to the two above mentioned strategies, the hide and re-emergence of ligand in this system are reversible, which could further enhance the targeting efficiency.
Trojan-horse targeting strategy
The non-specific uptake of the conventional actively-targeted nanovehicles by non-cancerous cells could lead to side effects. Recently, Trojan-horse targeting strategy was reported by Wu and Tan's group to reduce this risk. Fe3O4 nanocrystals-loaded albumin nanoparticles modified by folic acid and pH-sensitive polymers (PP-FA-AN-FN) were prepared for tumor imaging [67]. PP-FA-AN-FN could hide or expose the targeting ligand (folic acid) of nanoparticles on demand. When nanoparticles enter into normal tissue (pH 7.4), folic acid molecules are hidden in hydrophilic pH-sensitive polymers which helps to avoid the recognition and capture of nanoparticles by non-cancerous cells. However, in tumor regions, pH-sensitive polymers shrink and the original hidden folic acid molecules are exposed and able to bind the receptors on the surface of cancer cells to execute the active targeting effect. Similar to the“Pop-up” systems, this strategy is also reversible. After passing through the normal tissue, the ligands of the unused actively-targeted nanoparticles are shielded and eventually are effectively exploited in the tumor region.
Charge reversal
The surface charge of nanoparticles is also an important factor that influences their stability, tumor targeting, and cellular uptake. Similar to the “PEG dilemma” mentioned above, there are several contradictions for the nanoparticle's charge. On one hand, negatively or neutrally charged surface is necessary for nanoparticles to remain stable in the bloodstream after intravenous administration, since positively charged nanoparticles tend to absorb and aggregate with plasma proteins and are easily recognized and eliminated by the RES [68, 69] causing severe cytotoxicity [70]. On the other hand, positively charged nanoparticles are superior due to the electrostatic interactions with tumor cell membranes that possess an overall negative charge, leading to significantly improved cellular uptake in tumor tissues [71]. Therefore, surface charge-switchable Ms-DDS, which have a negative or neutral charge in the bloodstream and convert to positive in the tumor microenvironment are desired for better tumor targeting (Fig. 1C). Hence, ionizable chemical groups (amine, imidazole, and acyl sulfonamide) as well as pH-cleavable chemical bonds (hydrazine and dimethyl maleic anhydride) are introduced to construct charge-convertible Ms-DDS.
DMA group-included, surface charge-reversal of Ms-DDS have been studied to enhance the cellular uptake and targeting of cancer stem cells (CSC) for the delivery of small molecular chemotherapeutics (such as DOX) and nucleic acid drugs (such as siRNA), and has been previously reviewed [72]. Therefore, we only give a brief introduction about some of the newly presented charge-alterable Ms-DDS in the following section.
Chitosan is a natural cationic polysaccharide composed of glucosamine with favorable, biodegradable, and nontoxic properties [73]. Additionally, the amino groups of chitosan can be protonated at a weak acidic pH (<6.5), which is approximately the pH of the tumor microenvironment. Thus, chitosan or glycol chitosan (GC) with improved water solubility, have been widely used as an outer shell to construct pH-triggered, charge-switching Ms-DDS, such as chitosan- or GC-coated silica nanospheres [74], superparamagnetic iron oxide [75], or liposomes [76]. In normal tissues and bloodstream, the zeta potential of these coated nanocarriers was approximately electrically neutral, and quickly increased to 10 mV after protonation within the acidic tumor microenvironment leading to a four-fold higher cellular uptake. Zhang's group designed a novel zwitterionic oligopeptide lipid (HHG2C18), which was synthesized using a small peptide containing glutamic acid and histidine, as well as two stearyl alkane chains [77]. The negatively charged liposomes prepared with HHG2C18 and PEGylated HHG2C18 (PEGHG2C18) exhibited a pH-responsive positively charge change within the tumor microenvironment because of the protonation of the imidazole group in histidine [78]. The zwitterionic oligopeptide lipid was further used to coat mesoporous silica nanoparticles to develop another pH-sensitive, charge-convertible nanovehicle (M-HHG2C18-L) for the co-delivery of erlotinib and doxorubicin for improved synergistic therapy of Lewis lung carcinoma [79].
In addition to oligopeptides, polyHis has also been conjugated with PEG [80, 81], PLGA [82], PEI [83], PEG-PLA [84], or together with PEG and PLGA [85]. These compounds have been used for the construction of various pH-induced positively charged tunable nanoparticles to improve tumor delivery and cellular uptake of several antitumor drugs, including oncolytic adenovirus (Ad), DOX, docetaxel, and paclitaxel. Protein drugs can also be delivered by an imidazole-based pH-sensitive positively charged PEGylated poly(β-amino ester) [86]. Zwitterionic chemical structures with pH-sensitive property have been used to design charge-switchable systems as well, such as carboxybetaine [87, 88] and phosphorylcholine [89, 90]. However, as mentioned previously, since carboxybetaine and phosphorylcholine can only be protonated at a pH<2 and pH<5, respectively, the pH-sensitivity and charge-switching abilities of these zwitterionic polymers are too weak to respond to the slightly acidic tumor microenvironment (6.8~6.3) [91]. Rotello and his colleagues developed a novel alkoxyphenyl acyl sulfonamide-modified zwitterionic gold nanoparticle system, which could achieve negative-positive charge switching at a pH of 6.5 with increased cellular uptake [91]. Therefore, the acyl sulfonamide group is more suitable for the acidic tumor microenvironment responsiveness than carboxy betaine and phophorylcholine.
A Layer-by-Layer (LBL) nanoparticle system with an acid-triggered positive charge that emerged in tumor hypoxia regions was developed by Hammond's group [92]. The LBL nanoparticles were assembled stepwise with iminobiotin-modified positively charged poly-L-lysine (PLL), neutravidin, and then biotin-functionalized PEG. The nanoparticles remained stable during circulation in vivo owning to the strong interaction of the biotin-avidin system and the shielding effect of the PEG layer [93]. Since the nonchemical bond between iminobiotin and neutravidin rapidly breaks down in acidic conditions, the outer PEG layer could be unshielded in hypoxic tumor areas with a low pH, exposing the positively charged PLL layer to interact with the negatively charged tumor cell membrane. Based on this technology, quantum dots [92], NIR-II probes [94], hyaluronan [95], anticancer drugs, and siRNA [96] have been successfully delivered into tumor tissues. Because the well-established platform can include a variety of nanoscale carriers and materials in the core, as well as co-loading two or more antitumor drugs with a synergistic effect, it will be promising for tumor theranostics, especially for combination therapies.
Another important function of charge reversal is, enhancing endosomal escape for efficient delivery of siRNA and protein drugs. Although RNA interference is a promising therapy for cancer, several challenges limit its application in the clinic [97, 98]. The major bottleneck for siRNA delivery that needs to be overcome is the endosomal escape, as the acidic pH as well as several enzymes within lysosomes can degrade the siRNA [99]. The charge-based strategies are the most common and efficient way for endosomal rupture and escape [100, 101]. The pH-sensitive polymers with buffering capacity have been utilized to achieve endosomal escape and intracellular delivery, such as HHG2C18-L [78, 79], poly (carboxybetaine)-modified lipoplexes [87], and DMA-modified PEG-PLL [102].
Size shrinkage
In addition to tumor accumulation, carrier-cell interaction, drug release behavior, and drug resistance, tumor penetration is critically important and hinders the in vivo tumor inhibition effect of most nanoparticles in solid tumors. Because of tumor heterogeneity [103, 104], proliferated tumor stroma cells, cross-linked extracellular matrix, aberrant tumor vessels [105], and abnormally elevated IFP [106], the distribution and penetration of traditional DDS in tumor tissues are greatly impeded leading to physical resistance. Two main strategies have been envisioned to resolve this problem. The more frequently applied approach is referred to as “tumor priming”, which modulates the tumor microenvironment. Decreasing cell density (tumor cells or stromal cells) by cytotoxic drugs [107-109], degrading the ECM by collagenase and hyaluronidase [110], improving the vascular perfusion via hyperthermia therapy [111], and reducing IFP through tyrosinase inhibitors [112] could exhibit a significant improvement in tumor penetration and inhibition of nanoparticles in solid tumors. However, these tumor priming methods are usually utilized with anotherdrug-loaded nanoparticles to achieve a combination therapy, which requires a great effort to balance and fine-tune the dosage, sequence, and inter dose interval of the two agents. More importantly, the individual administration of the tumor-priming system and tumor-therapy system contributes to poor compliance and increased toxicity. Another method for overcoming the physical barriers is optimizing the particle sizes to achieve stimuli-triggered size shrinkage. Previous studies have revealed that although nanoparticles with larger sizes (100 nm) achieved superior tumor accumulation because of the favorable EPR effect, improved tumor penetration and therapeutic efficacy were achieved using small nanoparticles with a 30 nm size due to the small interstitial space [113, 114]. To combine the advantages of the perfect EPR effect of large nanoparticles and superior tumor penetration of small nanoparticles, several ideal Ms-DDS with size-changeable abilities that respond to endogenous stimuli have been designed (Fig. 1D).
These systems are usually composed of a mothership nanocarrier with a large size (approximately 100 nm) and 10 nm-sized babyship nanocarriers encapsulated or conjugated on the surface. The mothership NPs are stable during systemic delivery and concentrate in tumor tissues via the EPR effect. Subsequently, the babyship NPs are released due to the acidic pH or overexpressed enzymes of the tumor, delivering drugs into the deep regions of the tumor. Fukumura and coworkers employed quantum dots (QDs) as a model for the babyship system and loaded them in an MMP-2-degradable 100-nm gelatin nanoparticle. This mothership delivery model penetrated up to 300 μm into HT-1080 tumors at 6 h post-injection [115]. However, drug-loaded nanoparticles with small size should be further explored to replace the QDs and the tumor inhibition efficacy also needs to be evaluated. Gao's laboratory synthesized DOX-coupled AuNPs with PEG layer modification which were also packed into MMPs-responsive gelatin NPs to achieve size-shrinkage [116]. Although these AuNPs exhibited the best distribution and growth suppression in 4T1 and B16F10 tumors, this platform may not be suitable for some solid tumors with heavy desmoplastic compositions. This is because the size of final AuNPs (59.3 nm) is still too large for deep tumor penetration and the PEGylation may hinder the cellular internalization due to the “PEG dilemma”. To circumvent these shortcomings, a novel size changeable polymeric clustered nanoparticles system (iCluster) has been fabricated by Wang and colleagues [117]. The iCluster was constructed with 100-nm PEG-PCL micelles and 5-nm dendrimers conjugated to the hydrophobic core via a pH-sensitive CDM group. In the acidic tumor microenvironment, platinum-linked dendrimers detached from the mothership, permeated into the deep tumor tissue, and easily entered tumor cells by charge interactions [118]. Hence, the novel iCluster/Pt achieved 95% tumor inhibition and 74.2% prolonged survival time and will be meaningful for developing novel Ms-DDS.
Triggered drug release
Drug release is one of the most concerning issues for most passively- and actively-targeted nano-sized DDS and plays a critical role in tumor therapy as the tumor inhibition effect of most chemotherapeutics is dose-dependent. Although many nanoparticles and biomaterials have been developed to achieve perfect tumor-targeted delivery, drug release is still a common problem that needs to be solved. Liposomes and self-assembled nanoparticles are two promising nanocarriers because of their perfect biocompatibility, long circulation, and tumor targeting efficacy. Nevertheless, both of these representative platforms have their problems with drug release. As for liposomes, although they exhibit excellent stability and rarely release the loaded drug in vitro and in blood circulation leading to satisfactory tumor accumulation [119], the loaded drug is still trapped within the liposomes and cannot reach the target sites in the tumor tissues and tumor cells. For example, in vitro cellular uptake of the widely used liposomal DOX nanoparticles reveals that unlike free DOX, liposomal DOX is mainly located in the cytoplasm [119, 120] instead of the nucleus, which is the therapeutic target of DOX. Therefore, the clinical therapeutic effect of Doxil® is compromised because of the low drug bioavailability in the tumor microenvironment [121]. As for self-assembled nanoparticles, the main drawback is drug leakage in the blood circulation, which is diametrically opposite to the liposome systems, leading to premature drug release [122]. Although augmenting the drug-carrier compatibility by improving drug's hydrophobicity or drug-carrier miscibility achieved improved in vivo stability and tumor-targeted delivery, the tumor inhibition effect was still unsatisfactory [123]. Hence, novel platforms, including triggered drug release systems based on the tumor microenvironment have been developed to liberate the loading cargo at the right time and in the right place. The microenvironment stimuli-triggered drug release can mainly be divided into three types based on the mechanism: nanoparticle disassembly systems, sensitive-bond cleavage systems, and cap systems.
Nanoparticle disassembly and destabilization
For amphiphilic polymer-based self-assembled nanoparticles, it is easy to achieve tumor microenvironment-triggered drug release by disassembling the nanoparticles. As the stability of assembled nanoparticles depends on the hydrophilic-hydrophobic structure, changes in either segment will lead to nanoparticle disassembly and loaded-cargo release, as shown in Fig. 1E (i). The protonation-induced hydrophobic-hydrophilic switch is the main mechanism of pH-triggered nanoparticles disassembly. Several polymers with acid-sensitive groups, such as poly His [124, 125] and poly (2-(diisopropylamino) ethylmethacrylate) (PDPA) [126, 127], have been used to deliver chemotherapeutics and siRNA into tumors, achieving cellular controlled release and escape from endosomes or lysosomes.
Hydrophobic 2-nitroimidazoles (NIs) can also be converted to hydrophilic 2-aminoimidazoles by tumor hypoxia. Thambi and co-authors modified the hydrophilic carboxymethyl dextran (CM-Dex) backbone with NIs to induce the self-assembly of NPs and encapsulate DOX. While penetrating into the hypoxia region of the tumor, NPs became destabilized due to the bioreduction of NIs, releasing DOX and achieving superior tumor inhibition [25]. However, it is difficult for NPs with particle size as large as 200 nm to penetrate into the deep regions of the tumor with poor oxygen concentration. Hence, hypoxia-triggered drug release systems may not be effective in most solid tumors.
In addition to the stimuli-triggered hydrophobic-hydrophilic switch, direct removal of either segment of the amphiphilic polymers can also lead to NP disassembly. For example, methoxy PEG and vitamin E were linked by disulfide bonds to construct a novel amphiphilic copolymer, which could further self-assemble into micelles loaded with atorvastatin. Upon entering tumor cells, the PEG segments can be cleaved by high glutathione concentration leading to Ms-DDS disassembly and fast drug release [128]. This same idea was also applied in PEG-SS-PCL micelles [34]. Also, PEG-phenyl acetamide hybrids with an enzymatic detachment of hydrophobic phenyl acetamide were designed by Amir et al. to achieve enzyme-triggered disassembly and cargo release [129].
Like nanoparticles disassembly, nanoparticles destabilization can also lead to fast drug release. This strategy is mainly combined with some stimuli-sensitive lipid vesicles or cross-linking structures as well as gas generation systems. GSH- and pH-sensitive biomaterials with a cystamine structure can cross-link to form a shell over the surface of the drug-loaded micelle core preventing drug release in the normal environment. After the cross-linked shell was degraded in the acidic and reductive tumor environment, the cumulative release of DOX was nearly seven-times higher [130]. Strategies have also been developed to improve drug release from liposomes in the tumor microenvironment and tumor cells. Wang's group inserted the pH-responsive malachite green carbinol base (MG) into liposomes and regulated the drug release through polarity transformation of the MG molecule and disordering of lipid bilayers [131]. Based on the acidic decomposition of bicarbonate ions, a novel pH-triggered CO2 gas generating liposome has been prepared using an NH4HCO3 gradient method to induce fast drug release [132]. This gas generation method was further introduced in hollow PLGA nanoparticles to quickly liberate DOX overcoming the multi-drug resistance (MDR) [133]. However, the triggered drug release efficacy of disassembled nanoparticles is superior when compared with stimuli-responsive destabilization. For example, a pH-sensitive disassembly micelle led to a higher than 80% cargo release in 4 h and could entirely liberate the encapsulated drugs, while most destabilization-based nanoparticles needed more than 12 h for drug release and a portion of drugs remained captive after 48 h.
Sensitive bond cleavage
Besides physical encapsulation, drugs can be conjugated to the surface or core of nanoparticles using stimuli-sensitive chemical bonds to achieve triggered release as shown in Fig. 1E (ii). Hydrazone bond, the most widely used acid-sensitive linkers, has been successfully applied to conjugate antitumor drugs with polymers since it was utilized to label PTX with PEG by Rodrigues [134]. Among various chemotherapeutics, anthracyclines such as DOX, are the most popular model drugs for hydrazone conjugation, as previously reviewed by Wu et al [135]. Gao et al used this method to modify DOX on the surface of Au nanoparticles to achieve intracellular drug release in acidic organelles promoting DOX accumulation in the nucleus [136]. However, because of the low acid sensitivity of hydrazone, DOX was only released below pH 5.0 taking more than two days for complete release. Therefore, the therapeutic window was not reached in time for tumor therapy. MMP-2 sensitive peptides, redox-responsive disulfide connected drug-conjugated nanoparticles [137-139], and a series of stimuli-sensitive prodrug systems [140-142] have been developed for tumor microenvironment-responsive cleavage and drug release. However, the same problems discussed above have also impaired the in vivo antitumor effects of these diverse nanoparticles. Shen and colleagues designed an injectable nanoparticle generator (iNPG) that partly solves the problems [143]. This iNPG is a 2.5 μm diameter nanoporous silicon particle that exhibits a natural tropism and accumulates in liver and lung. PLL-conjugated DOX with a hydrazone linker (pDOX) was prepared and loaded into nanopores of iNPG (iNPG-pDOX). When accumulated in liver and lung metastases, pDOX was liberated to assemble into nanoparticles for better cellular uptake and DOX was further released through the acidic tumor microenvironment-triggered cleavage. Because of the perfect liver and lung metastases capture, the released DOX concentration in tumor cells is capable of reaching the therapeutic window quickly, although the cleavage of hydrazone is still a slow process.
Hypoxia-responsive drug release in the tumor by cleavage of the linker has become a hotspot in recent years. The main linkers in the design of drug-polymer conjugate in response to hypoxia include indolequinone, benzoquinone, nitroaromatics and N-oxide [144]. It is of note that the efficiency of hypoxia-responsive drug release not only depends on the selection of the linker and structure of the conjugate but also on the cancer types as well as the grade and stage of the tumor. Hence, this strategy for drug release may only be applicable to specific circumstances.
Cap systems
Cap systems, also named the gatekeeper systems, can be used to achieve tumor microenvironment-triggered drug burst release (Fig. 1E iii)) and is most commonly applied in mesoporous silica nanoparticles (MSNs). Since their discovery in the 1990s, MSNs have exhibited promising applications in drug delivery and possess multiple advantages, including perfect biocompatibility, biodegradation and a highly porous interior structure with a distinctive drug loading capacity [145]. Several previous reviews have summarized the applications of MSNs in drug delivery and biomedicine [146-149]. Here, we will give a short introduction of the recent developments of tumor microenvironment-responsive MSNs with gatekeepers. Although capable of loading drugs at a level as high as 60%, MSNs without caps leak the loaded drugs in one hour which is an undesirable feature for the in vivo drug delivery. After modification with gatekeepers, the drug release from MSNs was significantly suppressed until stimuli were added or until their entry into the tumor microenvironment [150, 151]. Several molecules and nanoparticles can be used to cap the pores of MSNs including Fe3O4 [152], avidin [150, 153], peptide [154, 155], human serum albumin [151], β-cyclodextrin (β-CD) [156] and gelatin [157]. The removal of most of these caps is triggered by cancer-associated proteases (mainly the MMP family), because proteins and peptides have the advantage of better biocompatibility and lower toxicity and are easier to modify when compared with other caps triggered by acidic and redox conditions.
Signal Turn-OFF/ON system for tumor diagnosis
The main limitations for tumor imaging and diagnosis are low precision and signal-to-noise ratio (SNR). Most current imaging probes are always-on systems exhibiting the same signal in tumor and normal tissues. Although macromolecular conjugation and nanosized DDS can significantly promote tumor accumulation, a considerable amount of probes remains in nontarget tissues with slow clearance. This leads to a high background signal, which intensifies the difficulty for tumor visualization, especially for metastatic tumors with small volume. To address these problems, tumor microenvironment stimuli-based signal “Turn OFF/ON” nanoplatforms have been developed (Fig. 1F) [158-160]. A summary of the main signal turn off mechanisms as well as the related activatable nanocarrier systems is presented in Table 3. As shown, Föster resonance energy transfer (FRET, including homoFRET and heteroFRET) and the acidic pH environment of the tumor are the most widely used mechanisms to construct stimuli-triggered signal Turn OFF/ON nanocarriers. Photoinduced electron transfer (PeT) and H-type dimers are alternate photochemical mechanisms that are applied to induce probe fluorescence quenching and has been reviewed previously [161]. Hence, we will give several examples of signal Turn OFF/ON Ms-DDS based on the above mechanisms.
Table 3
Quench mechanism and platforms for tumor microenvironment-based Signal Turn Off/On.
| Quench Mechanism | Trigger | Systems | Advantages | |
|---|---|---|---|---|
| Intramolecular spirocyclic caging | γ-glutamyl transpeptidase | γ-glutamyl hydroxymethyl rhodamine green [206] | Activated within 10 min and detect tumors smaller than 1 mm diaeter. | |
| Intramolecular spirocyclic caging | β-galactosidase | hydroxymethyl rhodol derivatives bearing β-galactoside [207] | Fluorescence activation > 1400-fold and sensitively detect intracellular β-galactosidase activity | |
| Intramolecular photoinduced electron transfer (PeT) | β-galactosidase | 2-Me-4OMe TokyoGreen O-β-galactoside [163, 164] | Up to 440-fold fluorescence activation and visualization of intraperitoneal tumors as small as 200 μm | |
| H-dimer induced homoFRET | Antibody-receptor interaction and conformation change | photosensitizer-antibody conjugate [208] | Tumor cell-targeted photoimmunotherapy and fast cell death | |
| Conjugation induced homoFRET | Tumor-associated proteases | Fluorescence probe or photosensitizer conjugated PEG-PLL nanoparticles [168, 209] | 12-fold increase in near- infrared fluorescence signal in vivo, able to detect tumors with submillimeter-sized diameters; 6-fold On/Off of SOG with increased tumor inhibition | |
| heteroFRET with fluorescence quencher via direct conjugation | Matrix metalloproteinases | CuS-peptide-BHQ3 [210] | Tumor-activatable photoacoustic imaging with improved detection depth | |
| heteroFRET with fluorescence quencher via direct conjugation | Matrix metalloproteinases | Photosensitizer-peptide-BHQ3 [170] | 12-fold fluorescence increase and 18-fold 1O2 production after 3 h cleavage | |
| Restriction of intramolecular rotation (RIR) and prohibition of energy dissipation through nonradiative channels | Cathepsin B | Dual-targeted aggregation-induced emission fluorogens-peptide-target [211, 212] | 35-fold higher fluorescence as well as significant SOG in 1 h | |
| Energy/charge transfer and efficient exciton migration | Acidic extracellular tumor microenvironment | Cationic conjugated polyelectrolyte and gold nanoparticle hybrid [213] | 8.2-fold enhancement of fluorescence in acid within 1 h | |
| H-type homoaggregates via face-to-face stacking | Acidic pH-triggered fluorophore cleavage | PEGylated dendrimer with hydrazone bonds [214] | 6-fold fluorescence increase after 24 h | |
| assembly induced homoFRET | Acidic pH-triggered fluorophore cleavage | Dextran with acid sensitive bond [159] | Low background fluorescence in normal tissues with high tumor/normal tissue ratio | |
| assembly induced homoFRET | Acidic pH-triggered hydrophobicity change and particle disassembly | Ionizable block copolymers of poly(ethylene oxide) and tertiary amine containing poly-methacrylate [172-175] | Ultra pH-sensitivity of 0.25 pH unit, tunable pH transition (from 7.1 to 4.4), fast temporal response (<5 ms) and higher than 100-fold fluorescence On/Off | |
| assembly induced homoFRET | pH-triggered hydrophobicity change and particle disassembly | poly(β-benzyl-L-aspartate) based polymers [167] | 0.6 pH sensitivity with multifunctions (MRI and PDT) to overcome tumor heterogeneity and multidrug resistance | |
| assembly induced heteroFRET with fluorescence quencher | pH-triggered hydrophobicity change and particle disassembly | MPEG-PAEs with tertiary amine moiety [158] | Acid-induced fluorescence turn on with several pHt | |
| assembly induced heteroFRET with fluorescence quencher | pH-triggered hydrophobicity change and particle disassembly | Self-assembled oligopeptide nanoparticles [215] | More than 10-fold enhancement of fluorescence in acid within 10 min | |
| Intramolecular photoinduced electron transfer (PeT) | Acidic environment in lysosomes | Selenium-rubyrin-loaded nanoparticles functionalized with folate [166] | Tumor cell-targeting; 10-times SOG On/Off with complete tumor growth inhibition | |
| HomoFRET by absorption | High GSH level intracellular | Redox-sensitive MnO2 nanosheets [169] | High intracellular delivery efficiency and enhanced photodynamic therapy efficacy by reducing glutathione levels in tumor cells | |
Fig 1
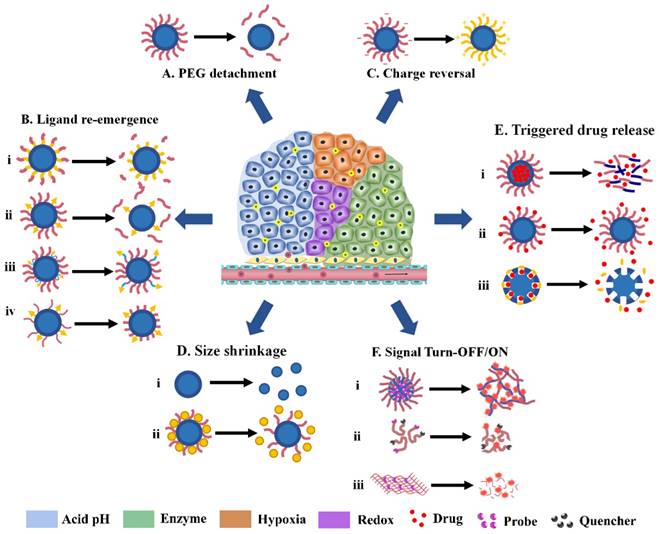
Various tumor microenvironment-activatable responsive modes. (A) PEG detachment; (B) Ligand re-emergence; (C) Charge reversal; (D) Size shrinkage; (E) Triggered drug release; (F) Signal Turn-OFF/ON.
Kobayashi and colleagues synthesized a novel acidic pH-responsive boron-dipyrromethene fluorescence probe (BODIPY), which was modified with N,N-dialkylated anilines at the C8 position that could form PeT with BODIPY fluorophore leading to fluorescence quenching in the neutral pH of normal tissues. After protonation of the aniline group, the disappearance of PeT led to 300-fold fluorescence emission enhancement in the acidic tumor microenvironment or organelles [162]. Substitution of the N-alkyl group of anilines with alkyl groups of different pKa further changed the pH of fluorescence turn on (pHa). Antibodies were then conjugated with the pHa-tunable fluorophore to achieve actively targeted imaging of the tumor microenvironment or acidic intracellular organelles. This pH-activatable, antibody-conjugated probe achieved 22-fold higher in vivo tumor imaging and visualization of metastatic tumors of 1 mm in diameter. However, it required nearly a 100-fold change of [H+] to completely turn on the signal, which may not be sensitive enough for detection in vivo. Kobayashi et al also designed a β-galactosidase-activated fluorescence probe based on PeT and achieved greater than 400-fold signal turn on [163, 164]. A pH-sensitive trifunctional photosensitizer based on the BODIPY structure was further developed and encapsulated in PEG-PLA nanomicelles. It achieved satisfactory tumor-triggered near-infrared imaging with a 10-fold tumor to normal tissue (T/N ratio) signal as well as improved photodynamic therapy [165]. However, due to the instability of physically encapsulated drugs in PEG-PLA micelles [123], most probes may be released during circulation resulting in high background signal and low bioavailability. These problems also need to be addressed in the PeT-based and pH-activatable rubyrin-loaded PEG-DSPE/PLGA nanoparticles [166].
pH-triggered nanocarrier disassembly [167], hydrazone bonds cleavage [159], enzyme-responsive peptide backbone breakage [168], and GSH-sensitive nanosheets disintegration [169] have been combined with the FRET effect to develop tumor microenvironment-responsive signal turn on nanoparticles systems to deliver photosensitizers. Because of the low quantum yield and weak fluorescence intensity, the homoFRET based “Turn OFF/ON” system of most photosensitizers exhibited low fluorescence and singlet oxygen generation (<10-fold) [167, 168]. Hence, fluorescence quenchers that can exhibit heteroFRET when close to fluorescence probes were introduced in triggered “Turn OFF/ON” systems, such as BHQ [158, 170]. Fan and colleagues utilized GSH-sensitive MnO2 nanosheet as a carrier and fluorescence quencher to deliver chlorin e6 (Ce6). On one hand, MnO2 could strongly absorb the emission fluorescence of Ce6, resulting in 60-fold quenching efficacy. On the other hand, when MnO2 nanosheets were disintegrated by intracellular GSH, the fluorescence and SOG ability of Ce6 recovered, leading to superior tumor inhibition and reduced side effects [169]. Although the On/OFF ratio of SOG was low (only 4-fold), which may be due to the photosensitivity of the SOSG probe [171], this nanoplatform maximized the functions of MnO2 nanosheets (high drug loading and quenching efficacy, and GSH-responsive degradation) and will be meaningful in biomedical applications.
As mentioned in section 3.5.1, protonation-induced micelle disassembly is also widely used to construct diagnostic signal-activatable Ms-DDS responding to the acidic tumor microenvironment. When encapsulated or aggregated in micelles, the fluorescence signal of a probe is greatly quenched because of FRET effect and is only turned on after pH-induced disassembly. Lee's group synthesized a series of poly (ethylene glycol) methyl ether-poly (β-amino esters) (MPEG-PAEs) with several pH transitions according to the tertiary moiety and the alkyl chain lengths [158]. Fluorescent dye and quencher were co-encapsulated and achieved fluorescence turn on within 0.5 pH unit. However, due to fast release, the quenched state of the encapsulated probe could be maintained no longer than 4 h greatly limiting the in vivo application of this pH-sensitive nanoparticles system. Another pH-sensitive, self-assembled nanocarrier for tumor theranostics was developed by Ling and coworkers [167]. They introduced the protonatable imidazole groups into copolymers and Ce6 was covalently conjugated achieving a 4-fold fluorescence turn on in 1 pH unit. Also, catechol groups were further modified to anchor iron oxide nanoparticles and exhibited pH-triggered T1 contrast turn on for the first time which will be promising for tumor acid-responsive MR imaging. Furthermore, Gao's group pushed the pH-sensitive Turn OFF/ON nanoparticles to a new height by using a copolymer with ionizable tertiary amine groups modified on hydrophobic blocks [172]. Based on the protonation of tertiary amine groups and FRET effect of conjugated fluorescence probes, the pH-activatable micellar nanoparticles achieved an ultra pH sensitivity of 0.25 pH unit with more than one hundred-fold fluorescence increase in 5 ms. Additionally, different alkane substitutions and copolymerization of two methacrylate monomers with different hydrophobicities could accurately control the pH transition of fluorescence leading to a broad pH tunability and making it possible to specifically recognize the tumor extracellular environment or different intracellular organelles with multicolored imaging [173, 174]. Finally, Gao et al combined the extracellular tumor microenvironment-sensitive nanoprobe (UPSe) with the intracellular sensitive nanoprobe (UPSi) to diagnose a broad range of tumors. This combination achieved higher than 300-fold signal amplification in tumor tissues with a 14-fold T/N tissue ratio and effectively detected small metastatic nodules smaller than 1 mm. To the best of our knowledge, these represent the most sensitive of all reported pH-sensitive fluorescence imaging nanoparticles [175]. This endeavor with the UPS nanosystem was highly praised in several journals and will be widely applied in drug delivery for tumor therapy.
Multi-stimuli responsive drug delivery systems for improved cancer therapy
Although most of the single-stimulus-triggered Ms-DDS mentioned above succeeded in overcoming one of the biological barriers, none of them could be “ever-victorious” for tumors with multi-barriers because of the complexity of the tumor microenvironment. For example, charge reversal and targeting ligand re-emergence systems may not be effective for solid tumors with dense stroma due to the poor penetration of nanoparticles larger than 50 nm. Additionally, although the size shrinkage platforms are capable of improving the tumor penetration, they have no advantage in the tumor cell internalization because of inefficient cellular uptake of a small size of babyship NPs [176]. In another case, one stimulus could not completely trigger a responsive behavior of drug delivery system. For instance, Mahmoud et al designed poly thioether ketal-based logic gate nanoparticles responsive to both the oxidative stress and reduced pH in tandem for the timed and accelerated release of encapsulated protein [37]. The results showed that only response to both stimuli could lead to degradation of polymeric nanoparticles and higher release of encapsulated protein within 24 hours.
An ideal Ms-DDS that is efficient for a majority of cancers should combine multi-stimuli-triggered strategies to further improve the drug delivery. As listed in Table 4, several activatable nanoplatforms with more than two responsive modes triggered by two or more biological stimuli have been developed in recent years. Because of the coexistence of several triggers in the tumor microenvironment, multiple combination schemes can be used in the design of multi-stimuli responsive drug delivery systems. Different types of responses to tumor microenvironment may occur one-by-one or simultaneously, depending on the design of multi-stimuli responsive drug delivery systems.
In the iCluster nanoparticles prepared by Wang's group, extracellular pH-unstable, CDM-triggered size shrinkage was combined with intracellular redox-sensitive drug release to overcome multiple barriers (Fig. 2). Therefore, after being discharged by the acidic tumor microenvironment, the tiny dendrimers were further reduced by intracellular GSH to liberate cisplatin (drug release), resulting in a fast cell-killing effect [117]. Furthermore, the protonation of tertiary amino groups in dendrimers could lead to a positively charged surface, which may further promote cellular uptake during size shrinkage. The stepwise size reduction and drug release combined strategy has also been applied in another nanovehicle system designed by Huang et al [177]. Two individual N-(2-hydroxypropyl) methacrylamide (HPMA) with opposite charges were developed to self-assemble the activatable nanovehicle through charge interactions. The anionic copolymers underwent charge reversal in the acidic extracellular microenvironment by breakage of DMA leading to nanovehicle disassembly and size reduction. The released linear conjugates with positive charge further penetrated into the tumor and were endocytosed by tumor cells. Finally, a second size shrinkage and nuclear-homing CPP-modified DOX detachment occurred in acidic intracellular organelles via hydrazone cleavage resulting in perfect nuclear targeting of chemotherapeutics (Fig. 3). Nevertheless, the multitude proteins and ionic compounds in the blood may influence the stability of charge-assembled nanoparticles resulting in a short circulation half-life and low bioavailability in vivo.
Different from the extra- and intracellular sequential multistage nanoplatforms described above, a novel sequential intra- and intercellular nanoparticles system with seven steps was developed by Zhang and colleagues [178]. As illustrated in Fig. 4, instead of size shrinkage, they designed a swelling-shrinking reversible mechanism to achieve intercellular delivery and tumor penetration. This strategy was realized by incorporating a cross-linked polyelectrolyte (N-lysinal-N'-succinyl chitosan and poly (N-isopropylacrylamide)) as the core with bovine serum albumin as an outer shell. In the neutral bloodstream environment, the slightly negatively charged surface and compact core successfully maintained its stability. After entering tumor cells, the amino groups of chitosan quickly protonated in endo-lysosomes and electrostatic repulsion was produced in the resulting positively charged core leading to simultaneous particle swelling, endosomal escape, and encapsulated drug release. Interestingly, the escaped nanoparticles could again shrink back to the initial state with small size in the neutral cytoplasm and could be further liberated from the dead cells infecting neighboring tumor cells similar to a virus. Therefore, these sequential intra-intercellular nanoparticles could kill tumor cells layer-by-layer and penetrate deep into the whole tumor tissue afterward. Zhang and colleagues also constructed another extracellular acidic microenvironment-triggered charge reversal system to achieve mitochondria targeting via three stages, including charge conversion, proton sponge effect, and mitochondrial binding [77].
Table 4
Multi-stimulus drug delivery systems
| Stimulus 1 | Stimulus 2 | Aim 1 | Aim 2 | Sequence | Ref. |
|---|---|---|---|---|---|
| pH-responsive targeting peptide (R8NLS) | Lysosomal enzyme-responsive sub-units | Positive charge for cellular uptake | Endosomal escape for nuclear targeting | One-by-one | [198] |
| pH-responsive zwitterionic oligopeptide lipids(pHe) | pH-responsive zwitterionic oligopeptide lipids(pHi) | Positive charge for cellular uptake | Endosomal escape for mitochondrial targeting | One-by-one | [78] |
| pH-sensitive MPEG-PLA-PAE | Size shrinkage and Positive charge for tumor penetration and cellular uptake | simultaneous | [187] | ||
| MMP2-sensitive | pH-triggered | Size shrinkage for tumor penetration | Dox release in lysosome | One-by-one | [116] |
| ROS-triggered | pH-triggered | Transformation and degradation of polymeric backbone for protein release | One-by-one | [37] | |
| pH-triggered | MMP2-sensitive | CPP re-emergence | simultaneous | [216] | |
| pH-sensitive anionic HPMA copolymer | pH-response hydrazine linkage | Size shrinkage, Positive charge for tumor penetration and cellular uptake | Nucleus entrance | Simultaneous then one-by-one | [177] |
| MMP-2 responsive peptide | GSH-responsive Au-S bond | Enhanced drug release | simultaneous | [217] | |
| pH-sensitive methylmaleic anhydride | GSH-responsive | Size shrinkage for tumor penetration | Enhanced drug release | one-by-one | [117] |
| pH-reversal protonation of chitosan | Positive charge for swelling and endosomal escape | Intercellular delivery | one-by-one | [178] | |
Fig 2
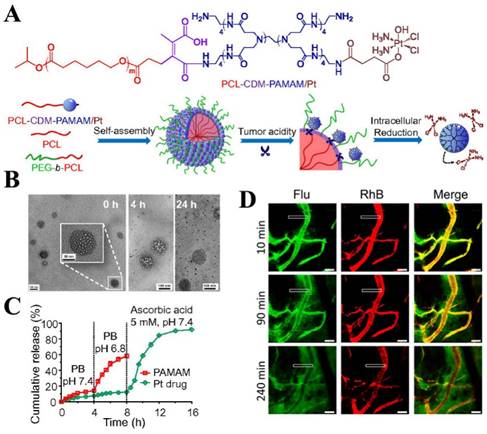
Clustered nanoparticles with extracellular pH-triggered size shrinkage and intracellular-activatable drug release. (A) Chemical structure and schematic of the iCluster/Pt. (B) TEM images of acid-triggered dendrimers released from iCluster. (C) pH- and redox-induced dendrimers liberation and Pt release. (D) Confocal images of liberated dendrimers penetration into tumors. Adapted from Ref [117] with permission from National Academy of Sciences.
Fig 3
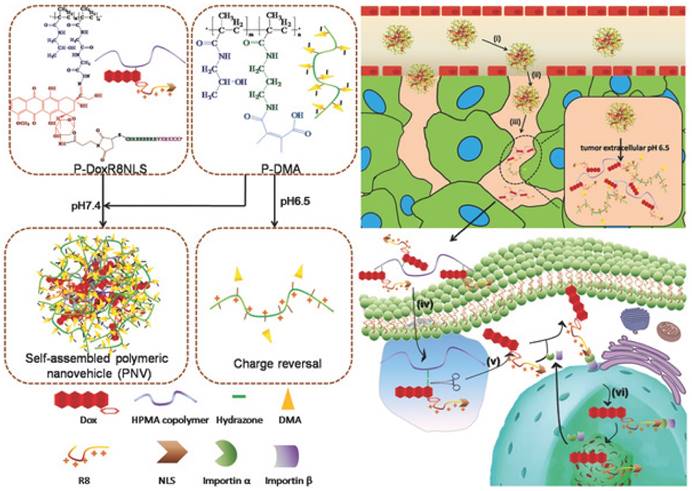
Extracellular- and intracellular-responsive stepwise size reduction and charge reversal for nuclear targeted delivery. Adapted from Ref [177] with permission from Wiley-VCH.
Fig 4
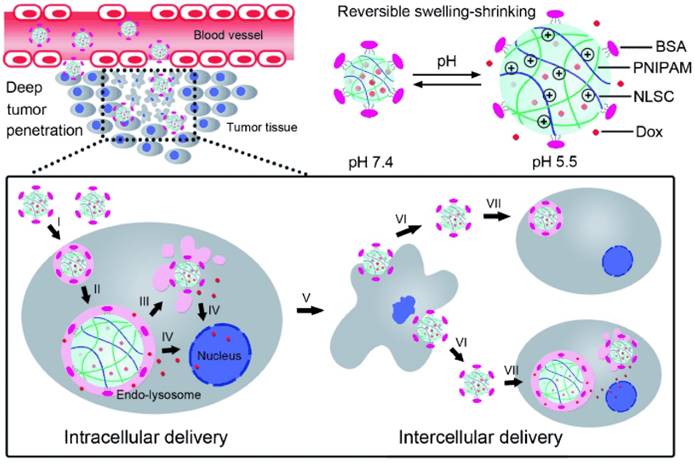
Cellular acid-induced, swelling-shrinking reversible nanoparticle for sequential intra-intercellular delivery. Adapted from Ref [178] with permission from Wiley-VCH.
Compared with single stimulus-triggered Ms-DDS, the multiple stimuli combined nanoplatform could integrate several activatable strategies into a single nanoparticles system with multiple functions to overcome several biological barriers during drug delivery. Hence, multi-stimuli based Ms-DDS will be feasible and effective in various types of cancers, as they will be less hindered by individual tumor differences. Thus, the advantages of different stimuli can be efficiently combined to maximize drug delivery and antitumor efficacy. However, some limitations still hinder the development of multi-stimuli-responsive Ms-DDS. To precisely control the spatio-temporal drug delivery, the main obstacle is the complexity of nanoparticles design, since a series of issues must be addressed; these include the selection of nanocarriers, modification of sequences of different sensitive groups by appropriate methods, and adjusting the time interval of different responses. Also, the timing and degree of the responses are more difficult to control in a real situation. Due to the heterogeneous expression and distribution of the stimuli in the tumor microenvironment, the multi-stimuli response may not work one-by-one or simultaneously as expected leading to the ineffectiveness of the systems. Therefore, a rational design of multi-stimuli triggered Ms-DDS would be much more difficult than a single-stimulus sensitive system.
Conclusions and Prospects
Since the discovery of liposomes in the 1960s, a variety of nanosized drug delivery systems, including polymer micelles, polymer nanoparticles, dendrimers, and polymer-drug conjugates, have been developed and applied to deliver chemotherapeutics, siRNA, antibodies, fluorescence probes, and contrast agents into tumors for cancer theranostics. A number of nanocarrier-based systems have been approved by the FDA for clinical use [179-182]. As estimated and statistically analyzed by the American Cancer Society, the cancer death rates during the last five years in the USA decreased by 1.8% and 1.4% per year in men and women, respectively [183], partly due to the application of drug delivery systems. However, as the second leading cause of death, cancer claimed millions of lives in the USA in 2015. Currently, the next generation of drug delivery systems with stimuli-responsiveness for better tumor therapy and diagnosis seems to enter the mainstream. In this article, we reviewed different types of stimuli in tumor microenvironment that can trigger changes in the properties of nanosize drug delivery systems. We have also outlined various strategies in developing tumor microenvironment-responsive Ms-DDS to enhance carrier-cell interactions (PEG detachment, targeting ligand re-emergence, and charge reversal), promote drug release and tumor penetration (size shrinkage), and amplify the signal in tumor imaging (signal Turn OFF/ON). We have also discussed some newly-emerging Ms-DDS with several combined stimuli-activatable models in recent years.
When different stimuli-activatable strategies are integrated into a single one delivery system, the sequence of different effects needs to be considered carefully. For instance, it is better for Ms-DDS to release the drug after cellular internalization and spatially next to the target sites such as nucleus or mitochondria. In spite of the fact that the extracellularly released drugs could diffuse deeply into tumor tissues with fewer barriers, a majority of drugs would be excreted by efflux proteins in most solid tumors with multidrug resistance resulting in low bioavailability and poor tumor suppression [184]. As shown in Fig. 5, there appear to be basic tenets for four sequential stages in using Ms-DDS for cancer theranostics: first, the designed Ms-DDS remain stable during their transport in blood stream and long circulation time; second, after accumulation in tumor tissues via EPR effect, the 100 nm NPs shrink to small ones with 10 nm size in diameter for better penetration into deep tumor tissues with poor vascularization; third, charge reversal or ligands re-emergence promotes cellular uptake; and fourth, after entering cells, cellular stimuli-triggered signal turn-on can further amplify diagnosis efficacy while endosomal escape and drug release achieve cellular targeting (mitochondria or nucleus) maximizing tumor suppression. Although this sequential tumor microenvironment-based Ms-DDS has not been developed yet, we believe that it would be a prospective activatable nanocarrier system for most tumors, especially for some resistant tumors with poor permeability.
While progress has been made in the design and evaluation of Ms-DDS, many difficult challenges remain, especially in their industrial production and clinical translation. About 77 products in clinical trials and 51 FDA-approved nanomedicines have been reported [182], none of which is Ms-DDS based on the tumor microenvironment. So far, only exogenous temperature- and magnetic-responsive DDS have reached the clinical stage [18]. There are two main reasons for the failure of Ms-DDS to reach the clinic: first one is the lower-than-expected in vivo outcome and the second is the sophisticated designs of the Ms-DDS which complicate the manufacturing process, reproducibility, and quality control. The molecular and cellular heterogeneity of tumor cells, the tumor stage, and individual variations will greatly determine the therapeutic efficacy in vivo [185]. Likewise, the low pH or the high concentration of reducing agents or the up-regulated enzymes in normal cells or non-tumoral tissues will, at times, lead to off-target effects [186]. So, the stimuli-sensitivity of the Ms-DDS should be well balanced. Thus, further optimization of the design of Ms-DDS and ideal choice of the indication are required for achieving the rapid clinical translation of Ms-DDS.
Fig 5
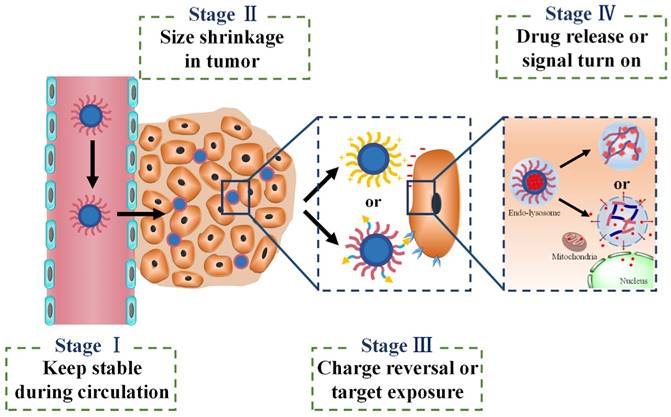
Four-stage drug delivery system applicable for various tumors.
Abbreviations
EPR effect: enhanced permeability and retention effect; IFP: interstitial fluid pressure; Ms-DDS: multistage drug delivery systems; PEI: polyethyleneimine; GSH: glutathione; CDM: 2-propionic-3-methylmaleic anhydride; PLGA: polylactic-glycolic acid; CPPs: cell-penetrating peptides; RES: reticuloendothelial system; ACPP: activable cell-penetrating peptide; PLA: poly(L-lactic acid); MPP: matrix metalloproteinase; AAN: alanine-alanine-asparagine; TAM: tumor-associated macrophage; PEG: polyethylene glycol; polyHis: poly(L-histidine); pLLA: poly(L-lactic acid); RES: reticuloendothelial system; CSC: cancer stem cell; DMA: 2,3-dimethylmaleic anhydride; GC: glycol chitosan; Ad: oncolytic adenovirus; LBL: Layer-by-Layer; PLL: poly-L-lysine; NIR: near-infrared; PDPA: poly(2-(diisopropylamino) ethylmethacrylate); NIs: nitroimidazoles; MDR: multi-drug resistance; iNPG: injectable nanoparticle generator; MSNs: mesoporous silica nanoparticles; β-CD: β-Cyclodextrin; ECM: extracellular matrix; QDs: quantum dots; iCluster: clustered nanoparticle system; SNR: signal to noise ratio; FRET: Föster resonance energy transfer; PeT: photoinduced electron transfer; BODIPY: boron-dipyrromethene; pHa: the pH of fluorescence turn on; T/N ratio: tumor to normal tissue; BHQ: black hole quencher; Ce6: chlorin e6; HPMA: N-(2-hydroxypropyl) methacrylamide.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81120059, 81573359) and the National Basic Research Program of China (2015CB932100).
The authors would like to thank Prof. Honggang Cui, Dr. Shawn Chen, and Professor Jun Wang for the manuscript invitation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yasuhiro Matsumura HM. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986;46:6387-92
2. Wagner V, Dullaart A, Bock AK. et al. The emerging nanomedicine landscape. Nat Biotechnol. 2006;24:1211-7
3. Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev. 2011;63(3):136-151
4. Durymanov MO, Rosenkranz AA, Sobolev AS. Current Approaches for Improving Intratumoral Accumulation and Distribution of Nanomedicines. Theranostics. 2015;5:1007-20
5. Chauhan VP, Jain RK. Strategies for advancing cancer nanomedicine. Nat Mater. 2013;12:958-62
6. Hatakeyama H, Akita H, Kogure K. et al. Development of a novel systemic gene delivery system for cancer therapy with a tumor-specific cleavable PEG-lipid. Gene Ther. 2007;14:68-77
7. Saga T, Neumann RD, Heya T. et al. Targeting cancer micrometastases with monoclonal antibodies: a binding-site barrier. Proc Natl Acad Sci U S A. 1995;92:8999-9003
8. Yatvin MB, Weinstein JN, Dennis WH. et al. Design of liposomes for enhanced local release of drugs by hyperthermia. Science. 1978;202:1290-3
9. Zhang F, Braun GB, Pallaoro A. et al. Mesoporous multifunctional upconversion luminescent and magnetic "nanorattle" materials for targeted chemotherapy. Nano Lett. 2012;12:61-7
10. Plassat V, Wilhelm C, Marsaud V. et al. Anti-Estrogen-Loaded Superparamagnetic Liposomes for Intracellular Magnetic Targeting and Treatment of Breast Cancer Tumors. Adv Funct Mater. 2011;21:83-92
11. Lu J, Choi E, Tamanoi F, Zink JI. Light-Activated Nanoimpeller-Controlled Drug Release in Cancer Cells. Small. 2008;4:421-6
12. Chen KJ, Liang HF, Chen HL. et al. A thermoresponsive bubble-generating liposomal system for triggering localized extracellular drug delivery. ACS Nano. 2013;7:438-46
13. Schroeder A, Honen R, Turjeman K. et al. Ultrasound triggered release of cisplatin from liposomes in murine tumors. J Control Release. 2009;137:63-8
14. Webb BA, Chimenti M, Jacobson MP. et al. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer. 2011;11:671-7
15. de la Rica R, Aili D, Stevens MM. Enzyme-responsive nanoparticles for drug release and diagnostics. Adv Drug Deliv Rev. 2012;64:967-78
16. Brown JM, Wilson WR. Exploiting tumour hypoxia in cancer treatment. Nat Rev Cancer. 2004;4:437-47
17. Go YM, Jones DP. Redox compartmentalization in eukaryotic cells. Biochim Biophys Acta. 2008;1780:1273-90
18. Mura S, Nicolas J, Couvreur P. Stimuli-responsive nanocarriers for drug delivery. Nat Mater. 2013;12:991-1003
19. Vander HM, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029-33
20. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891-9
21. He X, Li J, An S. et al. pH-sensitive drug-delivery systems for tumor targeting. Ther Deliv. 2013;4:1499-510
22. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161-74
23. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52-67
24. Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11:393-410
25. Thambi T, Deepagan VG, Yoon HY. et al. Hypoxia-responsive polymeric nanoparticles for tumor-targeted drug delivery. Biomaterials. 2014;35:1735-43
26. Perche F, Biswas S, Wang T. et al. Hypoxia-targeted siRNA delivery. Angew Chem Int Ed Engl. 2014;53:3362-6
27. Kuppusamy P, Li H, Ilangovan G. et al. Noninvasive imaging of tumor redox status and its modification by tissue glutathione levels. Cancer Res. 2002;62:307-12
28. Sun Y, Yan X, Yuan T. et al. Disassemblable micelles based on reduction-degradable amphiphilic graft copolymers for intracellular delivery of doxorubicin. Biomaterials. 2010;31:7124-31
29. Li J, Huo M, Wang J. et al. Redox-sensitive micelles self-assembled from amphiphilic hyaluronic acid-deoxycholic acid conjugates for targeted intracellular delivery of paclitaxel. Biomaterials. 2012;33:2310-20
30. Kim H, Kim S, Park C. et al. Glutathione-induced intracellular release of guests from mesoporous silica nanocontainers with cyclodextrin gatekeepers. Adv Mater. 2010;22:4280-3
31. Wu L, Zhang L, Shi G. et al. Zwitterionic pH/redox nanoparticles based on dextran as drug carriers for enhancing tumor intercellular uptake of doxorubicin. Mater Sci Eng C Mater Biol Appl. 2016;61:278-85
32. Zhang Z, Hou L, Yang X. et al. A novel redox-sensitive system based on single-walled carbon nanotubes for chemo-photothermal therapy and magnetic resonance imaging. Int J Nanomedicine. 2016;11:607-24
33. Lin C, Lu K, Wang S. et al. CD44-specific nanoparticles for redox-triggered reactive oxygen species production and doxorubicin release. Acta Biomater. 2016;35:280-92
34. Qu Q, Wang Y, Zhang L. et al. Nanoplatform with Precise Control over Release of Cargo for Enhanced Cancer Therapy. Small. 2016;12:1378-90
35. Maggini L, Cabrera I, Ruiz-Carretero A. et al. Breakable mesoporous silica nanoparticles for targeted drug delivery. Nanoscale. 2016;8:7240-7
36. Wu M, Meng Q, Chen Y. et al. Nanoparticles: Large Pore-Sized Hollow Mesoporous Organosilica for Redox-Responsive Gene Delivery and Synergistic Cancer Chemotherapy. Adv Mater. 2016;28:1963-9
37. Mahmoud EA, Sankaranarayanan J, Morachis JM. et al. Inflammation responsive logic gate nanoparticles for the delivery of proteins. Bioconjug Chem. 2011;22:1416-21
38. Kwon J, Kim J, Park S. et al. Inflammation-responsive antioxidant nanoparticles based on a polymeric prodrug of vanillin. Biomacromolecules. 2013;14:1618-26
39. Schwerdt A, Zintchenko A, Concia M. et al. Hyperthermia-induced targeting of thermosensitive gene carriers to tumors. Hum Gene Ther. 2008;19:1283-92
40. Naito M, Ishii T, Matsumoto A. et al. A phenylboronate-functionalized polyion complex micelle for ATP-triggered release of siRNA. Angew Chem Int Ed Engl. 2012;51:10751-5
41. Ge Z, Liu S. Functional block copolymer assemblies responsive to tumor and intracellular microenvironments for site-specific drug delivery and enhanced imaging performance. Chem Soc Rev. 2013;42:7289-325
42. Li SD, Huang L. Stealth nanoparticles: high density but sheddable PEG is a key for tumor targeting. J Control Release. 2010;145:178-81
43. Dong H, Tang M, Li Y. et al. Disulfide-bridged cleavable PEGylation in polymeric nanomedicine for controlled therapeutic delivery. Nanomedicine (lond). 2015;10:1941-58
44. Xu C, Zhang H, Sun C. et al. Tumor acidity-sensitive linkage-bridged block copolymer for therapeutic siRNA delivery. Biomaterials. 2016;88:48-59
45. Sun C, Shen S, Xu C. et al. Tumor Acidity-Sensitive Polymeric Vector for Active Targeted siRNA Delivery. J Am Chem Soc. 2015;137:15217-24
46. Zorko M, Langel U. Cell-penetrating peptides: mechanism and kinetics of cargo delivery. Adv Drug Deliv Rev. 2005;57:529-45
47. Fu J, Yu C, Li L. et al. Intracellular Delivery of Functional Proteins and Native Drugs by Cell-Penetrating Poly(disulfide)s. J Am Chem Soc. 2015;137:12153-60
48. Vives E. Present and future of cell-penetrating peptide mediated delivery systems: "is the Trojan horse too wild to go only to Troy?". J Control Release. 2005;109:77-85
49. Kondo E, Saito K, Tashiro Y. et al. Tumour lineage-homing cell-penetrating peptides as anticancer molecular delivery systems. Nat Commun. 2012;3:951
50. Jiang T, Olson ES, Nguyen QT. et al. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc Natl Acad Sci U S A. 2004;101:17867-72
51. Olson ES, Jiang T, Aguilera TA. et al. Activatable cell penetrating peptides linked to nanoparticles as dual probes for in vivo fluorescence and MR imaging of proteases. Proc Natl Acad Sci U S A. 2010;107:4311-6
52. Li SY, Cheng H, Qiu WX. et al. Protease-activable cell-penetrating peptide-protoporphyrin conjugate for targeted photodynamic therapy in vivo. ACS Appl Mater Interfaces. 2015;7:28319-29
53. Gao W, Xiang B, Meng TT. et al. Chemotherapeutic drug delivery to cancer cells using a combination of folate targeting and tumor microenvironment-sensitive polypeptides. Biomaterials. 2013;34:4137-49
54. Zhang B, Zhang Y, Liao Z. et al. UPA-sensitive ACPP-conjugated nanoparticles for multi-targeting therapy of brain glioma. Biomaterials. 2015;36:98-109
55. Duijnhoven SM, Robillard MS, Nicolay K. et al. Tumor targeting of MMP-2/9 activatable cell-penetrating imaging probes is caused by tumor-independent activation. J Nucl Med. 2011;52:279-86
56. Sethuraman VA, Bae YH. TAT peptide-based micelle system for potential active targeting of anti-cancer agents to acidic solid tumors. J Control Release. 2007;118:216-24
57. Jin E, Zhang B, Sun X. et al. Acid-Active Cell-Penetrating Peptides for in Vivo Tumor-Targeted Drug Delivery. J Am Chem Soc. 2013;135:933-40
58. Itaka K, Kataoka K. Progress and prospects of polyplex nanomicelles for plasmid DNA delivery. Curr Gene Ther. 2011;11:457-65
59. Liu Z, Xiong M, Gong J. et al. Legumain protease-activated TAT-liposome cargo for targeting tumours and their microenvironment. Nat Commun. 2014;5:4280
60. Lee SH, Moroz E, Castagner B. et al. Activatable Cell Penetrating Peptide-Peptide Nucleic Acid Conjugate via Reduction of Azobenzene PEG Chains. J Am Chem Soc. 2014;136:12868-71
61. Zhu L, Wang T, Perche F. et al. Enhanced anticancer activity of nanopreparation containing an MMP2-sensitive PEG-drug conjugate and cell-penetrating moiety. Proc Natl Acad Sci U S A. 2013;110:17047-52
62. Koren E, Apte A, Jani A. et al. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J Control Release. 2012;160:264-73
63. Karve S, Bandekar A, Ali MR. et al. The pH-dependent association with cancer cells of tunable functionalized lipid vesicles with encapsulated doxorubicin for high cell-kill selectivity. Biomaterials. 2010;31:4409-16
64. Bandekar A, Karve S, Chang MY. et al. Antitumor efficacy following the intracellular and interstitial release of liposomal doxorubicin. Biomaterials. 2012;33:4345-52
65. Lee ES, Na K, Bae YH. Super pH-Sensitive Multifunctional Polymeric Micelle. Nano Lett. 2005;5:325-9
66. Lee ES, Gao Z, Kim D. et al. Super pH-sensitive multifunctional polymeric micelle for tumor pHe specific TAT exposure and multidrug resistance. J Control Release. 2008;129:228-36
67. Shen Z, Wu H, Yang S. et al. A novel Trojan-horse targeting strategy to reduce the non-specific uptake of nanocarriers by non-cancerous cells. Biomaterials. 2015;70:1-11
68. He C, Hu Y, Yin L. et al. Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials. 2010;31:3657-66
69. Ma SF, Nishikawa M, Katsumi H. et al. Cationic charge-dependent hepatic delivery of amidated serum albumin. J Control Release. 2005;102:583-94
70. Fischer D, Li Y, Ahlemeyer B. et al. In vitro cytotoxicity testing of polycations: influence of polymer structure on cell viability and hemolysis. Biomaterials. 2003;24:1121-31
71. Bareford LM, Swaan PW. Endocytic mechanisms for targeted drug delivery. Adv Drug Deliv Rev. 2007;59:748-58
72. Du JZ, Mao CQ, Yuan YY. et al. Tumor extracellular acidity-activated nanoparticles as drug delivery systems for enhanced cancer therapy. Biotechnol Adv. 2014;32:789-803
73. Bhattarai N, Gunn J, Zhang M. Chitosan-based hydrogels for controlled, localized drug delivery. Adv Drug Deliv Rev. 2010;62:83-99
74. Deng Z, Zhen Z, Hu X. et al. Hollow chitosan-silica nanospheres as pH-sensitive targeted delivery carriers in breast cancer therapy. Biomaterials. 2011;32:4976-86
75. Crayton SH, Tsourkas A. pH-Titratable Superparamagnetic Iron Oxide for Improved Nanoparticle Accumulation in Acidic Tumor Microenvironments. ACS Nano. 2011;5:9592-601
76. Yan L, Crayton SH, Thawani JP. et al. A pH-Responsive Drug-Delivery Platform Based on Glycol Chitosan-Coated Liposomes. Small. 2015;11:4870-4
77. Mo R, Sun Q, Xue J. et al. Multistage pH-Responsive Liposomes for Mitochondrial-Targeted Anticancer Drug Delivery. Adv Mater. 2012;24:3659-65
78. Mo R, Sun Q, Li N. et al. Intracellular delivery and antitumor effects of pH-sensitive liposomes based on zwitterionic oligopeptide lipids. Biomaterials. 2013;34:2773-86
79. He Y, Su Z, Xue L. et al. Co-delivery of erlotinib and doxorubicin by pH-sensitive charge conversion nanocarrier for synergistic therapy. J Control Release. 2016;229:80-92
80. Choi J, Dayananda K, Jung S. et al. Enhanced anti-tumor efficacy and safety profile of tumor microenvironment-responsive oncolytic adenovirus nanocomplex by systemic administration. Acta Biomater. 2015;28:86-98
81. Choi J, Jung S, Kasala D. et al. pH-sensitive oncolytic adenovirus hybrid targeting acidic tumor microenvironment and angiogenesis. J Control Release. 2015;205:134-43
82. Hung CC, Huang WC, Lin YW. et al. Active Tumor Permeation and Uptake of Surface Charge-Switchable Theranostic Nanoparticles for Imaging-Guided Photothermal/Chemo Combinatorial Therapy. Theranostics. 2016;6:302-17
83. Hu J, Miura S, Na K. et al. pH-responsive and charge shielded cationic micelle of poly(L-histidine)-block-short branched PEI for acidic cancer treatment. J Control Release. 2013;172:69-76
84. Ouahab A, Cheraga N, Onoja V. et al. Novel pH-sensitive charge-reversal cell penetrating peptide conjugated PEG-PLA micelles for docetaxel delivery: in vitro study. Int J Pharm. 2014;466:233-45
85. Radovic-Moreno AF, Lu TK, Puscasu VA. et al. Surface Charge-Switching Polymeric Nanoparticles for Bacterial Cell Wall-Targeted Delivery of Antibiotics. ACS Nano. 2012;6:4279-87
86. Gao GH, Park MJ, Li Y. et al. The use of pH-sensitive positively charged polymeric micelles for protein delivery. Biomaterials. 2012;33:9157-64
87. Li Y, Liu R, Shi Y. et al. Zwitterionic poly(carboxybetaine)-based cationic liposomes for effective delivery of small interfering RNA therapeutics without accelerated blood clearance phenomenon. Theranostics. 2015;5:583-96
88. Li Y, Zhang X. Zwitterionic poly(carboxybetaine) modified liposomes enhancing tumor therapy without accelerated blood clearance phenomenon. J Control Release. 2015;213:e125
89. Jin Q, Xu JP, Ji J. et al. Zwitterionic phosphorylcholine as a better ligand for stabilizing large biocompatible gold nanoparticles. Chem Commun (Camb). 2008;26:3058-60
90. Zhao J, Chai YD, Zhang J. et al. Long circulating micelles of an amphiphilic random copolymer bearing cell outer membrane phosphorylcholine zwitterions. Acta Biomater. 2015;16:94-102
91. Mizuhara T, Saha K, Moyano DF. et al. Acylsulfonamide-Functionalized Zwitterionic Gold Nanoparticles for Enhanced Cellular Uptake at Tumor pH. Angew Chem Int Ed Engl. 2015;54:6567-70
92. Poon Z, Chang D, Zhao X. et al. Layer-by-Layer Nanoparticles with a pH-Sheddable Layer for in Vivo Targeting of Tumor Hypoxia. ACS Nano. 2011;5:4284-92
93. Poon Z, Lee JB, Morton SW. et al. Controlling in Vivo Stability and Biodistribution in Electrostatically Assembled Nanoparticles for Systemic Delivery. Nano Lett. 2011;11:2096-103
94. Dang X, Gu L, Qi J. et al. Layer-by-layer assembled fluorescent probes in the second near-infrared window for systemic delivery and detection of ovarian cancer. Proc Natl Acad Sci U S A. 2016;113:5179-84
95. Dreaden EC, Morton SW, Shopsowitz KE. et al. Bimodal Tumor-Targeting from Microenvironment Responsive Hyaluronan Layer-by-Layer (LbL) Nanoparticles. ACS Nano. 2014;8:8374-82
96. Deng ZJ, Morton SW, Ben-Akiva E. et al. Layer-by-Layer Nanoparticles for Systemic Codelivery of an Anticancer Drug and siRNA for Potential Triple-Negative Breast Cancer Treatment. ACS Nano. 2013;7:9571-84
97. Wang T, Shigdar S, Shamaileh HA. et al. Challenges and opportunities for siRNA-based cancer treatment. Cancer Lett. 2016 In press
98. Okholm AH, Kjems J. DNA nanovehicles and the biological barriers. Adv Drug Deliv Rev. 2016;106:183-91
99. Wittrup A, Ai A, Liu X. et al. Visualizing lipid-formulated siRNA release from endosomes and target gene knockdown. Nat Biotechnol. 2015;33:870-6
100. Nel AE, Madler L, Velegol D. et al. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8:543-57
101. Shen J, Kim HC, Su H. et al. Cyclodextrin and polyethylenimine functionalized mesoporous silica nanoparticles for delivery of siRNA cancer therapeutics. Theranostics. 2014;4:487-97
102. Chen S, Rong L, Lei Q. et al. A surface charge-switchable and folate modified system for co-delivery of proapoptosis peptide and p53 plasmid in cancer therapy. Biomaterials. 2016;77:149-63
103. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346-54
104. Denison TA, Bae YH. Tumor heterogeneity and its implication for drug delivery. J Control Release. 2012;164:187-91
105. Miao L, Lin CM, Huang L. Stromal barriers and strategies for the delivery of nanomedicine to desmoplastic tumors. J Control Release. 2015;219:192-204
106. Heldin CH, Rubin K, Pietras K. et al. High interstitial fluid pressure - an obstacle in cancer therapy. Nat Rev Cancer. 2004;4:806-13
107. Wang J, Lu Z, Wang J. et al. Paclitaxel tumor priming promotes delivery and transfection of intravenous lipid-siRNA in pancreatic tumors. J Control Release. 2015;216:103-10
108. Alvarez R, Musteanu M, Garcia-Garcia E. et al. Stromal disrupting effects of nab-paclitaxel in pancreatic cancer. Brit J Cancer. 2013;109:926-33
109. Chen B, Wang Z, Sun J. et al. A tenascin C targeted nanoliposome with navitoclax for specifically eradicating of cancer-associated fibroblasts. Nanomedicine. 2016;12:131-41
110. Kohli AG, Kivimäe S, Tiffany MR. et al. Improving the distribution of Doxil® in the tumor matrix by depletion of tumor hyaluronan. J Control Release. 2014;191:105-14
111. Frazier N, Ghandehari H. Hyperthermia approaches for enhanced delivery of nanomedicines to solid tumors. Biotechnol Bioeng. 2015;112:1967-83
112. Fan Y, Du W, He B. et al. The reduction of tumor interstitial fluid pressure by liposomal imatinib and its effect on combination therapy with liposomal doxorubicin. Biomaterials. 2013;34:2277-88
113. Cabral H, Matsumoto Y, Mizuno K. et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815-23
114. Wang J, Mao W, Lock LL. et al. The Role of Micelle Size in Tumor Accumulation, Penetration, and Treatment. ACS Nano. 2015;9:7195-206
115. Wong C, Stylianopoulos T, Cui J. et al. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc Natl Acad Sci U S A. 2011;108:2426-31
116. Ruan S, Cao X, Cun X. et al. Matrix metalloproteinase-sensitive size-shrinkable nanoparticles for deep tumor penetration and pH triggered doxorubicin release. Biomaterials. 2015;60:100-10
117. Li H, Du J, Du X. et al. Stimuli-responsive clustered nanoparticles for improved tumor penetration and therapeutic efficacy. Proc Natl Acad Sci U S A. 2016;113:4164-9
118. Sun Q, Sun X, Ma X. et al. Integration of Nanoassembly Functions for an Effective Delivery Cascade for Cancer Drugs. Adv Mater. 2014;26:7615-21
119. Dai W, Yang F, Ma L. Combined mTOR inhibitor rapamycin and doxorubicin-loaded cyclic octapeptide modified liposomes for targeting integrin α3 in triple-negative breast cancer. Biomaterials. 2014;35:5347-58
120. Qin C, He B, Dai W. et al. Inhibition of metastatic tumor growth and metastasis via targeting metastatic breast cancer by chlorotoxin-modified liposomes. Mol Pharm. 2014;11:3233-41
121. Slingerland M, Guchelaar HJ, Gelderblom H. Liposomal drug formulations in cancer therapy: 15 years along the road. Drug Discov Today. 2012;17:160-6
122. Chen H, Kim S, He W. et al. Fast release of lipophilic agents from circulating PEG-PDLLA micelles revealed by in vivo forster resonance energy transfer imaging. Langmuir. 2008;24:5213-7
123. Zhao Y, Fay F, Hak S. et al. Augmenting drug-carrier compatibility improves tumour nanotherapy efficacy. Nat Commun. 2016;7:11221
124. Lee ES, Na K, Bae YH. Doxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumor. J Control Release. 2005;103:405-18
125. Qiu L, Qiao M, Chen Q. et al. Enhanced effect of pH-sensitive mixed copolymer micelles for overcoming multidrug resistance of doxorubicin. Biomaterials. 2014;35:9877-87
126. Li S, Wu W, Xiu K. et al. Doxorubicin loaded pH-responsive micelles capable of rapid intracellular drug release for potential tumor therapy. J Biomed Nanotechnol. 2014;10:1480-9
127. Xu X, Wu J, Liu Y. et al. Ultra-pH-Responsive and Tumor-Penetrating Nanoplatform for Targeted siRNA Delivery with Robust Anti-Cancer Efficacy. Angew Chem Int Ed Engl. 2016;55:7091-4
128. Xu P, Yu H, Zhang Z. et al. Hydrogen-bonded and reduction-responsive micelles loading atorvastatin for therapy of breast cancer metastasis. Biomaterials. 2014;35:7574-7
129. Harnoy AJ, Rosenbaum I, Tirosh E. et al. Enzyme-Responsive Amphiphilic PEG-Dendron Hybrids and Their Assembly into Smart Micellar Nanocarriers. J Am Chem Soc. 2014;136:7531-4
130. Zhao X, Liu P. Reduction-Responsive Core-Shell-Corona Micelles Based on Triblock Copolymers: Novel Synthetic Strategy, Characterization, and Application As a Tumor Microenvironment-Responsive Drug Delivery System. ACS Appl Mater Interfaces. 2015;7:166-74
131. Liu Y, Gao F, Zhang D. et al. Molecular structural transformation regulated dynamic disordering of supramolecular vesicles as pH-responsive drug release systems. J Control Release. 2014;173:140-7
132. Liu J, Ma H, Wei T. et al. CO2 gas induced drug release from pH-sensitive liposome to circumvent doxorubicin resistant cells. Chem Commun (Camb). 2012;48:4869-71
133. Ke CJ, Chiang WL, Liao ZX. et al. Real-time visualization of pH-responsive PLGA hollow particles containing a gas-generating agent targeted for acidic organelles for overcoming multi-drug resistance. Biomaterials. 2013;34:1-10
134. Rodrigues P, Scheuermann K, Stockmar C. et al. Synthesis and in vitro efficacy of acid-sensitive poly(ethylene glycol) paclitaxel conjugates. Bioorg Med Chem Lett. 2003;13:355-60
135. Kanamala M, Wilson WR, Yang M. et al. Mechanisms and biomaterials in pH-responsive tumour targeted drug delivery: A review. Biomaterials. 2016;85:152-67
136. Ruan S, Yuan M, Zhang L. et al. Tumor microenvironment sensitive doxorubicin delivery and release to glioma using angiopep-2 decorated gold nanoparticles. Biomaterials. 2015;37:425-35
137. Chen WH, Luo GF, Lei Q. et al. MMP-2 responsive polymeric micelles for cancer-targeted intracellular drug delivery. Chem Commun (Camb). 2015;51:465-8
138. Chuan X, Song Q, Lin J. et al. Novel free-paclitaxel-loaded redox-responsive nanoparticles based on a disulfide-linked poly(ethylene glycol)-drug conjugate for intracellular drug delivery: synthesis, characterization, and antitumor activity in vitro and in vivo. Mol Pharm. 2014;11:3656-70
139. Song Q, Wang X, Wang Y. et al. Reduction Responsive Self-Assembled Nanoparticles Based on Disulfide-Linked Drug-Drug Conjugate with High Drug Loading and Antitumor Efficacy. Mol Pharm. 2016;13:190-201
140. Yin T, Wu Q, Wang L. et al. Well-Defined Redox-Sensitive Polyethene Glycol-Paclitaxel Prodrug Conjugate for Tumor-Specific Delivery of Paclitaxel Using Octreotide for Tumor Targeting. Mol Pharm. 2015;12:3020-31
141. Sun JD, Liu Q, Wang J. et al. Selective Tumor Hypoxia Targeting by Hypoxia-Activated Prodrug TH-302 Inhibits Tumor Growth in Preclinical Models of Cancer. Clin Cancer Res. 2012;18:758-70
142. Ueki N, Wang W, Swenson C. et al. Synthesis and Preclinical Evaluation of a Highly Improved Anticancer Prodrug Activated by Histone Deacetylases and Cathepsin L. Theranostics. 2016;6:808-16
143. Xu R, Zhang G, Mai J. et al. An injectable nanoparticle generator enhances delivery of cancer therapeutics. Nat Biotechnol. 2016;34:414-18
144. Wong PT, Choi SK. Mechanisms of drug release in nanotherapeutic delivery systems. Chem Rev. 2015;115:3388-432
145. He Q, Shi J. MSN anti-cancer nanomedicines: chemotherapy enhancement, overcoming of drug resistance, and metastasis inhibition. Adv Mater. 2014;26:391-411
146. Wang Y, Zhao Q, Han N. et al. Mesoporous silica nanoparticles in drug delivery and biomedical applications. Nanomedicine. 2015;11:313-27
147. Li Z, Barnes JC, Bosoy A. et al. Mesoporous silica nanoparticles in biomedical applications. Chem Soc Rev. 2012;41:2590-605
148. Ruehle B, Saint-Cricq P, Zink JI. Externally Controlled Nanomachines on Mesoporous Silica Nanoparticles for Biomedical Applications. Chemphyschem. 2016;17:1769-79
149. Shi S, Chen F, Cai W. Biomedical applications of functionalized hollow mesoporous silica nanoparticles: focusing on molecular imaging. Nanomedicine (Lond). 2013;8:2027-39
150. Schlossbauer A, Kecht J, Bein T. Biotin-Avidin as a Protease-Responsive Cap System for Controlled Guest Release from Colloidal Mesoporous Silica. Angew Chem Int Ed Engl. 2009;48:3092-5
151. Liu J, Zhang B, Luo Z. et al. Enzyme responsive mesoporous silica nanoparticles for targeted tumor therapy in vitro and in vivo. Nanoscale. 2015;7:3614-26
152. Gan Q, Lu X, Yuan Y. et al. A magnetic, reversible pH-responsive nanogated ensemble based on Fe3O4 nanoparticles-capped mesoporous silica. Biomaterials. 2011;32:1932-42
153. van Rijt SH, Bölükbas DA, Argyo C. et al. Protease-Mediated Release of Chemotherapeutics from Mesoporous Silica Nanoparticles to ex Vivo Human and Mouse Lung Tumors. ACS Nano. 2015;9:2377-89
154. Ashley CE, Carnes EC, Phillips GK. et al. The targeted delivery of multicomponent cargos to cancer cells by nanoporous particle-supported lipid bilayers. Nat Mater. 2011;10:389-97
155. Zhao Z, Meng H, Wang N. et al. A controlled-release nanocarrier with extracellular pH value driven tumor targeting and translocation for Drug Delivery. Angew Chem Int Ed Engl. 2013;52:7487-91
156. Zhang J, Yuan Z, Wang Y. et al. Multifunctional Envelope-Type Mesoporous Silica Nanoparticles for Tumor-Triggered Targeting Drug Delivery. J Am Chem Soc. 2013;135:5068-73
157. Zou Z, He X, He D. et al. Programmed packaging of mesoporous silica nanocarriers for matrix metalloprotease 2-triggered tumor targeting and release. Biomaterials. 2015;58:35-45
158. Ko JY, Park S, Lee H. et al. pH-Sensitive Nanoflash for Tumoral Acidic pH Imaging in Live Animals. Small. 2010;6:2539-44
159. Li C, Xia J, Wei X. et al. pH-Activated Near-Infrared Fluorescence Nanoprobe Imaging Tumors by Sensing the Acidic Microenvironment. Adv Funct Mater. 2010;20:2222-30
160. Lee S, Park K, Kim K. et al. Activatable imaging probes with amplified fluorescent signals. Chem Commun (Camb). 2008;36:4250-60
161. Kobayashi H, Ogawa M, Alford R. et al. New strategies for fluorescent probe design in medical diagnostic imaging. Chem Rev. 2010;110:2620-40
162. Urano Y, Asanuma D, Hama Y. et al. Selective molecular imaging of viable cancer cells with pH-activatable fluorescence probes. Nat Med. 2008;15:104-9
163. Kamiya M, Kobayashi H, Hama Y. et al. An enzymatically activated fluorescence probe for targeted tumor imaging. J Am Chem Soc. 2007;129:3918-3929
164. Urano Y, Kamiya M, Kanda K. et al. Evolution of fluorescein as a platform for finely tunable fluorescence probes. J Am Chem Soc. 2005;127:4888-94
165. Tian J, Zhou J, Shen Z. et al. A pH-activatable and aniline-substituted photosensitizer for near-infrared cancer theranostics. Chem Sci. 2015;6:5969-77
166. Tian J, Ding L, Xu HJ. et al. Cell-specific and pH-activatable rubyrin-loaded nanoparticles for highly selective near-infrared photodynamic therapy against cancer. J Am Chem Soc. 2013;135:18850-8
167. Ling D, Park W, Park SJ. et al. Multifunctional tumor pH-sensitive self-assembled nanoparticles for bimodal imaging and treatment of resistant heterogeneous tumors. J Am Chem Soc. 2014;136:5647-55
168. Choi Y, Weissleder R, Tung CH. Selective antitumor effect of novel protease-mediated photodynamic agent. Cancer Res. 2006;66:7225-9
169. Fan H, Yan G, Zhao Z. et al. A Smart Photosensitizer-Manganese Dioxide Nanosystem for Enhanced Photodynamic Therapy by Reducing Glutathione Levels in Cancer Cells. Angew Chem Int Ed Engl. 2016;55:5477-82
170. Zheng G, Chen J, Stefflova K. et al. Photodynamic molecular beacon as an activatable photosensitizer based on protease-controlled singlet oxygen quenching and activation. Proc Natl Acad Sci U S A. 2007;104:8989-94
171. Kim S, Fujitsuka M, Majima T. Photochemistry of singlet oxygen sensor green. J Phys Chem B. 2013;117:13985-92
172. Zhou K, Wang Y, Huang X. et al. Tunable, Ultrasensitive pH-Responsive Nanoparticles Targeting Specific Endocytic Organelles in Living Cells. Angew Chem Int Ed Engl. 2011;50:6109-14
173. Zhou K, Liu H, Zhang S. et al. Multicolored pH-Tunable and Activatable Fluorescence Nanoplatform Responsive to Physiologic pH Stimuli. J Am Chem Soc. 2012;134:7803-11
174. Ma X, Wang Y, Zhao T. et al. Ultra-pH-Sensitive Nanoprobe Library with Broad pH Tunability and Fluorescence Emissions. J Am Chem Soc. 2014;136:11085-92
175. Wang Y, Zhou K, Huang G. et al. A nanoparticle-based strategy for the imaging of a broad range of tumours by nonlinear amplification of microenvironment signals. Nat Mater. 2013;13:204-12
176. Kievit FM, Zhang M. Cancer nanotheranostics: improving imaging and therapy by targeted delivery across biological barriers. Adv Mater. 2011;23:H217-47
177. Li L, Sun W, Zhong J. et al. Multistage Nanovehicle Delivery System Based on Stepwise Size Reduction and Charge Reversal for Programmed Nuclear Targeting of Systemically Administered Anticancer Drugs. Adv Funct Mater. 2015;25:4101-13
178. Ju C, Mo R, Xue J. et al. Sequential intra-intercellular nanoparticle delivery system for deep tumor penetration. Angew Chem Int Ed Engl. 2014;53:6253-8
179. Peer D, Karp JM, Hong S. et al. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751-60
180. Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771-82
181. Shi J, Votruba AR, Farokhzad OC. et al. Nanotechnology in Drug Delivery and Tissue Engineering: From Discovery to Applications. Nano Lett. 2010;10:3223-30
182. Bobo D, Robinson KJ, Islam J. et al. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm Res. 2016;33:2373-87
183. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5-29
184. Ganoth A, Merimi KC, Peer D. Overcoming multidrug resistance with nanomedicines. Expert Opin Drug Deliv. 2015;12:223-8
185. Du J, Lane LA, Nie S. Stimuli-responsive nanoparticles for targeting the tumor microenvironment. J Control Release. 2015;219:205-14
186. Jhaveri A, Deshpande P, Torchilin V. Stimuli-sensitive nanopreparations for combination cancer therapy. J Control Release. 2014;190:352-70
187. Yu Y, Zhang X, Qiu L. The anti-tumor efficacy of curcumin when delivered by size/charge-changing multistage polymeric micelles based on amphiphilic poly (β-amino ester) derivates. Biomaterials. 2014;35:3467-79
188. Bajaj I, Singhal R. Poly (glutamic acid) - an emerging biopolymer of commercial interest. Bioresour Technol. 2011;102:5551-61
189. Lu T, Wang Z, Ma Y. et al. Influence of polymer size, liposomal composition, surface charge, and temperature on the permeability of pH-sensitive liposomes containing lipid-anchored poly(2-ethylacrylic acid). Int J Nanomedicine. 2012;7:4917-26
190. Chen H, Moore T, Qi B. et al. Monitoring pH-triggered drug release from radioluminescent nanocapsules with X-ray excited optical luminescence. ACS Nano. 2013;7:1178-87
191. Reshetnyak YK, Andreev OA, Segala M. et al. Energetics of peptide (pHLIP) binding to and folding across a lipid bilayer membrane. Proc Natl Acad Sci U S A. 2008;105:15340-5
192. Liu Y, Zhang D, Qiao Z. et al. A Peptide-Network Weaved Nanoplatform with Tumor Microenvironment Responsiveness and Deep Tissue Penetration Capability for Cancer Therapy. Adv Mater. 2015;27:5034-42
193. Yu H, Chen J, Liu S. et al. Enzyme sensitive, surface engineered nanoparticles for enhanced delivery of camptothecin. J Control Release. 2015;216:111-20
194. Basel MT, Shrestha TB, Troyer DL. et al. Protease-Sensitive, Polymer-Caged Liposomes: A Method for Making Highly Targeted Liposomes Using Triggered Release. ACS Nano. 2011;5:2162-75
195. Sandersjöö L, Jonsson A, Löfblom J. A new prodrug form of Affibody molecules (pro-Affibody) is selectively activated by cancer-associated proteases. Cell Mol Life Sci. 2015;72:1405-15
196. Kalafatovic D, Nobis M, Javid N. et al. MMP-9 triggered micelle-to-fibre transitions for slow release of doxorubicin. Biomater Sci. 2015;3:246-9
197. Ku T, Chien M, Thompson MP. et al. Controlling and Switching the Morphology of Micellar Nanoparticles with Enzymes. J Am Chem Soc. 2011;133:8392-5
198. Zhong J, Li L, Zhu X. et al. A smart polymeric platform for multistage nucleus-targeted anticancer drug delivery. Biomaterials. 2015;65:43-55
199. Cheng Y, Luo G, Zhu J. et al. Enzyme-Induced and Tumor-Targeted Drug Delivery System Based on Multifunctional Mesoporous Silica Nanoparticles. ACS Appl Mater Interfaces. 2015;7:9078-87
200. Ji T, Zhao Y, Wang J. et al. Tumor Fibroblast Specific Activation of a Hybrid Ferritin Nanocage-Based Optical Probe for Tumor Microenvironment Imaging. Small. 2013;9:2427-31
201. Bode SA, Hansen MB, Oerlemans RAJF. et al. Enzyme-Activatable Cell-Penetrating Peptides through a Minimal Side Chain Modification. Bioconjug Chem. 2015;26:850-6
202. Zhang H, Wang L, Xiang Q. et al. Specific lipase-responsive polymer-coated gadolinium nanoparticles for MR imaging of early acute pancreatitis. Biomaterials. 2014;35:356-67
203. Arouri A, Dathe M, Blume A. The helical propensity of KLA amphipathic peptides enhances their binding to gel-state lipid membranes. Biophys Chem. 2013;180-181:10-21
204. Arouri A, Mouritsen OG. Membrane-perturbing effect of fatty acids and lysolipids. Prog Lipid Res. 2013;52:130-40
205. Yang C, Wang X, Yao X. et al. Hyaluronic acid nanogels with enzyme-sensitive cross-linking group for drug delivery. J Control Release. 2015;205:206-17
206. Yasuteru U, Masayo S, Nobuyuki K. et al. Rapid Cancer Detection by Topically Spraying a γ-Glutamyltranspeptidase-Activated Fluorescent Probe. Sci Trans Med. 2011;3:110-9
207. Asanuma D, Sakabe M, Kamiya M. et al. Sensitive β-galactosidase-targeting fluorescence probe for visualizing small peritoneal metastatic tumours in vivo. Nat Commun. 2015;6:6463
208. Mitsunaga M, Ogawa M, Kosaka N. et al. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat Med. 2011;17:1685-91
209. Morimoto S. In-vivo imaging of tumors with protease activated near-infrared fluorescent probes. Tanpakushitsu Kakusan Koso. 2007;52:1774-5
210. Yang K, Zhu L, Nie L. et al. Visualization of Protease Activity In Vivo Using an Activatable Photo-Acoustic Imaging Probe Based on CuS Nanoparticles. Theranostics. 2014;4:134-41
211. Shi H, Liu J, Geng J. et al. Specific detection of integrin alphavbeta3 by light-up bioprobe with aggregation-induced emission characteristics. J Am Chem Soc. 2012;134:9569-72
212. Yuan Y, Zhang CJ, Gao M. et al. Specific light-up bioprobe with aggregation-induced emission and activatable photoactivity for the targeted and image-guided photodynamic ablation of cancer cells. Angew Chem Int Ed Engl. 2015;54:1780-6
213. Yuan Y, Ding D, Li K. et al. Tumor-Responsive Fluorescent Light-up Probe Based on a Gold Nanoparticle/Conjugated Polyelectrolyte Hybrid. Small. 2014;10:1967-75
214. Almutairi A, Guillaudeu SJ, Berezin MY. et al. Biodegradable pH-sensing dendritic nanoprobes for near-infrared fluorescence lifetime and intensity imaging. J Am Chem Soc. 2008;130:444-5
215. Zhao Y, Ji T, Wang H. et al. Self-assembled peptide nanoparticles as tumor microenvironment activatable probes for tumor targeting and imaging. J Control Release. 2014;177:11-9
216. Huang S, Shao K, Kuang Y. et al. Tumor targeting and microenvironment-responsive nanoparticles for gene delivery. Biomaterials. 2013;34:5294-302
217. Chen W, Xu X, Jia H. et al. Therapeutic nanomedicine based on dual-intelligent functionalized gold nanoparticles for cancer imaging and therapy in vivo. Biomaterials. 2013;34:8798-807
Author contact
![]() Corresponding authors: Wenbing Dai, E-mail: pawpaw009com; Qiang Zhang, E-mail: zqdodoedu.cn.
Corresponding authors: Wenbing Dai, E-mail: pawpaw009com; Qiang Zhang, E-mail: zqdodoedu.cn.
Citation styles
APA
Chen, B., Dai, W., He, B., Zhang, H., Wang, X., Wang, Y., Zhang, Q. (2017). Current Multistage Drug Delivery Systems Based on the Tumor Microenvironment. Theranostics, 7(3), 538-558. https://doi.org/10.7150/thno.16684.
ACS
Chen, B.; Dai, W.; He, B.; Zhang, H.; Wang, X.; Wang, Y.; Zhang, Q. Current Multistage Drug Delivery Systems Based on the Tumor Microenvironment. Theranostics 2017, 7 (3), 538-558. DOI: 10.7150/thno.16684.
NLM
Chen B, Dai W, He B, Zhang H, Wang X, Wang Y, Zhang Q. Current Multistage Drug Delivery Systems Based on the Tumor Microenvironment. Theranostics 2017; 7(3):538-558. doi:10.7150/thno.16684. https://www.thno.org/v07p0538.htm
CSE
Chen B, Dai W, He B, Zhang H, Wang X, Wang Y, Zhang Q. 2017. Current Multistage Drug Delivery Systems Based on the Tumor Microenvironment. Theranostics. 7(3):538-558.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.