Impact Factor
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Light delivery strategies for...
Forward looking methodologies...
Targeted probes that confine...
Harnessing local and systemic...
Summary and future perspectives
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2016; 6(13):2458-2487. doi:10.7150/thno.16183 This issue Cite
Review
Beyond the Barriers of Light Penetration: Strategies, Perspectives and Possibilities for Photodynamic Therapy
Srivalleesha Mallidi1, Sriram Anbil1,2,3*, Anne-Laure Bulin1*, Girgis Obaid1*, Megumi Ichikawa1, Tayyaba Hasan1 ![]()
1. Wellman Center for Photomedicine, Massachusetts General Hospital, Boston, MA 02114;
2. Howard Hughes Medical Institute, Chevy Chase, MD, 20815;
3. The University of Texas School of Medicine at San Antonio, San Antonio, TX 78229.
*Authors contributed equally to the work.
Received 2016-5-16; Accepted 2016-9-1; Published 2016-10-23
Abstract

Photodynamic therapy (PDT) is a photochemistry based treatment modality that involves the generation of cytotoxic species through the interactions of a photosensitizer molecule with light irradiation of an appropriate wavelength. PDT is an approved therapeutic modality for several cancers globally and in several cases has proved to be effective where traditional treatments have failed. The key parameters that determine PDT efficacy are 1. the photosensitizer (nature of the molecules, selectivity, and macroscopic and microscopic localization etc.), 2. light application (wavelength, fluence, fluence rate, irradiation regimes etc.) and 3. the microenvironment (vascularity, hypoxic regions, stromal tissue density, molecular heterogeneity etc.). Over the years, several groups aimed to monitor and manipulate the components of these critical parameters to improve the effectiveness of PDT treatments. However, PDT is still misconstrued to be a surface treatment primarily due to the limited depths of light penetration. In this review, we present the recent advances, strategies and perspectives in PDT approaches, particularly in cancer treatment, that focus on increasing the 'damage zone' beyond the reach of light in the body. This is enabled by a spectrum of approaches that range from innovative photosensitizer excitation strategies, increased specificity of phototoxicity, and biomodulatory approaches that amplify the biotherapeutic effects induced by photodynamic action. Along with the increasing depth of understanding of the underlying physical, chemical and physiological mechanisms, it is anticipated that with the convergence of these strategies, the clinical utility of PDT will be expanded to a powerful modality in the armamentarium for the management of cancer.
Keywords: photodynamic therapy, light penetration
Introduction
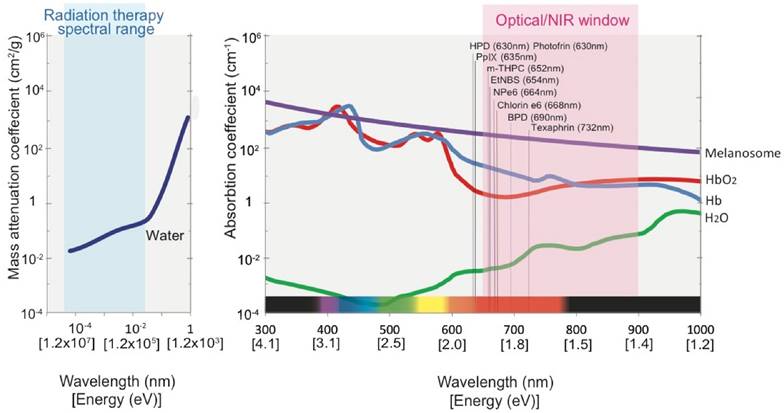
Visible and near infrared (NIR) radiation, although a miniscule part of the electromagnetic radiation spectrum, have provided us with a vast palette of applications in which we may not only “see” but also harness this energy for therapeutic purposes. The inquisitiveness that drove early pioneers to understand light-tissue interactions and to use electromagnetic radiation to peer at tissues residing deep within the body led to the identification and characterization of several physiological chromophores, including melanin, hemoglobin and water. As photonics technology advanced, thorough characterization of the wavelength dependent optical absorption and scattering coefficients of these common chromophores became possible, leading to the identification of the so called “optical window,” which exists between 600-900 nm light (Fig. 1). Absorption of light within the optical window by the common physiological chromophores is low, thereby allowing incident light between these wavelengths to penetrate more deeply into the tissue. For example, a ~70% reduction in optical absorption of melanin in the skin is observed (i.e., ~1.8-fold enhancement in penetration depth, ignoring the effects of tissue scattering) when the irradiation wavelength is changed from 500 nm to 700 nm. Despite the fact that the penetration depth of visible light does not exceed more than several millimeters, several diagnostic and therapeutic techniques utilizing visible and NIR radiation have significantly impacted the clinical standard of care over the past two decades, specifically in the treatment of age-related macular degeneration, dermatologic conditions, cancer, and various diagnostics and imaging applications [1]. Beyond these applications, visible and NIR light have been exploited to understand the physiology, microenvironment and treatment response of numerous pathologies in a multitude of preclinical studies [1].
Absorption spectrum of chromophores and water in the radiation therapy spectral range and visible to NIR spectral range. The optical window region where absorption of light due to physiological chromophores is low is shaded in pink. The absorption peaks of most commonly used photosensitizers for photodynamic therapy (PDT) are also depicted. Abbreviations: PpIX - Protoporphyrin IX, mTHPC - m-tetrahydroxyphenylchlorin, EtNBS - 5-ethylamino-9-diethylaminobenzo[α] phenothiazinium chloride, NPe6 - mono-L-aspartyl chlorin e6 and BPD - benzoporphyrin derivative monoacid A. HbO2 - Oxygenated hemoglobin, Hb - Deoxygenated hemoglobin, H2O - Water. Data adapted from Jacques et al [2] and National Institute of Standards and Technology database.
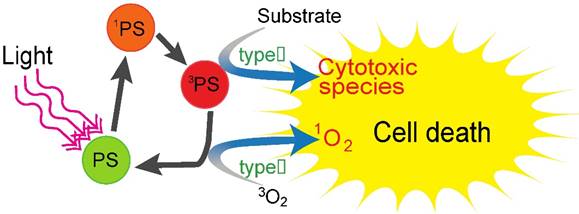
Photodynamic therapy (PDT), a light based cytotoxic therapy, has gained significant popularity as it offers temporal and spatial control of the treatment with minimal systemic toxicity [3]. PDT is a phototoxic therapy wherein the photosensitizer (PS, a photo-activatable molecule) is excited with light of a specific wavelength to generate reactive molecular species or free radicals that can react with the local microenvironment (Fig. 2). Spatial selectivity in PDT can be achieved by 1. Specifically targeting the PS to the tumor compartment by utilizing various methodologies such as immunoconjugates or nanoconstructs [4-8] and 2. Locally delivering light to the region of interest to cause damage to malignant tissue while sparing surrounding healthy tissues; both are critical requirements in treatment of diffuse tumors such as glioblastoma in the brain [3]. The translation of light based techniques such as PDT to pathologies that are deeply situated within the body is primarily restricted by the finite depth of light penetration into tissue. To date, the routine clinical use of PDT has been limited to superficial layers of tissues, such as the skin [9, 10], retina [11] and others, that are easily accessible. Delivering light to deeper tissues (e.g. large tumors) has been limited by a significant attenuation in potency as the light penetrates more deeply into tissue, thereby rendering it sub-cytotoxic as it reaches the target tissue and ultimately reducing the overall efficacy of PDT. In the context of cancer therapy, PDT has shown promise in its ability to treat superficial tumors resistant to standard therapies and also to eradicate residual disease in the surgical bed that may cause recurrence. Nevertheless, its applications for the treatment of tumors in deep tissue have been limited to date [3, 12-14].
Schematic representation of PDT mechanism of action. The photosensitizer (PS, a photo-activatable molecule) is excited from the ground state to the singlet excited state (1PS) with light of a specific wavelength. From this excited state, the PS undergoes intersystem crossing to an electronically different excited state lower in energy such as the triplet state (3PS). In its long-lived triplet state the PS reacts with local microenvironment to generate reactive molecular species or free radicals. These reactive species induce cell death. For example, energy from the PS triplet state is transferred to the ground-state triplet oxygen molecules (3O2) to generate reactive singlet oxygen (1O2) molecules.
PDT efficacy is determined by the interplay between light, the PS and the tissue microenvironment [15], and depends on several parameters such as the PS delivery-light-interval, overall light dose, the macroscopic and cellular PS localization, and the tumor oxygenation status, among others. Selective tissue damage can only be achieved when light and the PS are present in sufficient quantities at the desired location. Substantial efforts by several groups to enhance light delivery to deeper tissues are in progress; however, an upper limit exists on how far into the infrared region a PS can absorb light and still produce cytotoxic species. In photochemistry, the PS is typically electronically excited to the singlet excited state upon absorption of a photon. From this excited state, the PS molecule undergoes intersystem crossing to a longer lived triplet state, which can initiate photochemical reactions directly, giving rise to reactive free radicals, or transfer its energy to the ground-state triplet oxygen molecules (3O2) to generate reactive singlet oxygen (1O2) molecules. Specifically, the energy required to excite an oxygen molecule from its ground state to its singlet state is ~0.96 eV, creating an upper limit on the excitation wavelength to be around 850-900 nm depending on the energy level of the PSs' triplet state. Because most of the currently used PS's have absorption peaks in the 600 - 750 nm range (Fig. 1), the light irradiation window for PDT has been restricted to this range within the past few decades. Overall, the limitations stemming from the PS excitation wavelength and light delivery, coupled with the variability in clinical outcomes caused by inconsistencies due to inter- or intra- microenvironmental heterogeneity and the failure to customize the PDT dose in a patient-specific manner, historically has prevented PDT from gaining widespread acceptance as a first-line therapeutic modality.
PDT's therapeutic impact extends beyond the zone treated by light. Here, we review the current efforts and advances in the field of PDT to facilitate deep tissue therapy beyond the traditional barriers set by tissue optical properties. The first section of this review will discuss new developments in light delivery strategies that enable PS excitation in tissues deeper than previously possible. In the second section, we discuss new PS targeting strategies that enhance the selectivity and efficacy of PDT in deep tissue by reducing off-target toxicities. Throughout the review, the prospects for the clinical translation of PDT and the requirement for treatment monitoring techniques that enable accurate PDT dosimetry are discussed. Perspectives on combining PDT with current clinically-relevant treatments and other forward looking therapies such as mechanism-based combination regimens are discussed. We also discuss the impact of biomodulatory approaches that amplify the biotherapeutic effects induced by photodynamic action and the impact of harnessing local and systemic biological and immune responses to enhance deep-tissue PDT efficacy. We anticipate that the progress made in light delivery, dosimetry design, nanotechnology based combinations, biomodulating strategies, PS excitation and targeting strategies that have shown tremendous potential in preclinical studies will ultimately have significant clinical impact on customizing treatments and managing recalcitrant disease.
Light delivery strategies for deep tissue PDT
The term “photodynamic action” was coined by Prof. von Tappeiner in the early 1900's when he observed the toxic effect of an acridine dye on paramecia [16]. Since then, several PSs have been discovered or synthesized and their respective mechanisms of PDT have been unraveled. Together, the efforts to develop new PSs that can specifically localize to the target tissue, the development of Lasers, Light emitting diode (LEDs) and fiber optic technologies that can excite PSs at their optimal absorption peak, have enabled the translation of PDT to the clinic for a variety of disease applications. Various types of light sources ranging from inexpensive conventional arc lamps to expensive coherent, narrow bandwidth lasers have been utilized to excite PSs. It is well known that collimated laser beams scatter forward when interacting with tissues, and thereby have higher tissue penetration depth than non-coherent LED or arc lamps. These non-collimated light sources exhibit more divergent beam properties and therefore have reduced forward scattering of light making them unsuitable for treating deeper lesions [17].
Image-guided placement of fibers for deep-tissue PDT
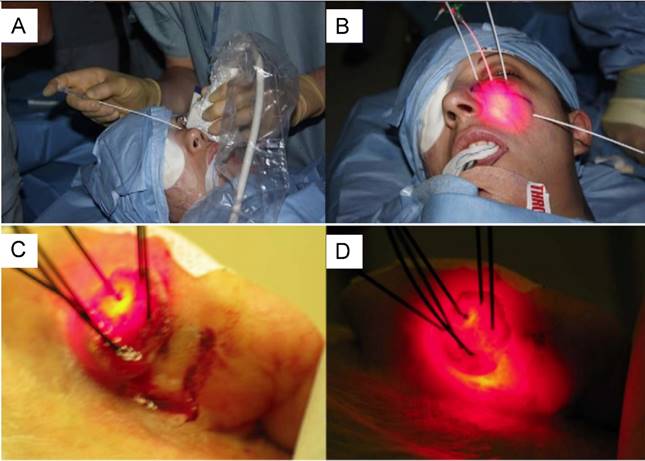
Historically, PDT has incorrectly been alleged only as a surface treatment because the application of external light may only treat superficial lesions. Availing advances in fiber optics and microendoscopic technology, PDT is now being extensively used in clinic with interstitial, endoscopic, intraoperative or laparoscopic light delivery systems. Laser light can be focused into thin optical fibers for delivery of light into deeper and difficult to access treatment sites. For example, in a recent clinical study by Jerjes et. al., [18] multiple fibers were placed under ultrasound guidance into various deep-seated pathologies such as head and neck tumors and vascular anomalies within the limbs (Fig. 3). More than one hundred patients were treated with PDT using the PS meso-tetrahydroxyphenyl chlorin (mTHPC). More than half the patients had a good response to the treatment while 5 patients became disease free. Of the patients harboring head and neck tumors, 80% reported improvements in breathing, swallowing and speech [18].
Examples of image-guided interstitial PDT for deeply situated tumors. A. Photograph of surgeon inserting needles under ultrasound-guidance for placing fibers in deep tissue. B. Light delivered to the heamangioma of the left infraorbital region through multiple fibers. Photographs of solid skin tumor in the ear with 6 fibers implanted under ultrasound guidance. C. One fiber is used for illumination while other 5 fibers are used for diagnostic purposes to evaluate light fluence, sensitizer concentration and tissue oxygenation. D. All the fibers are used in “transmit” mode to illuminate the whole tumor for PDT. Images adapted with permission from Jerjes et al [18] and Svanberg et al. [21]
Another notable case involved a pancreatic adenocarcinoma patient who underwent interstitial PDT after being initially considered unsuitable for surgical resection [19]. PDT was performed on this subject wherein the optical fiber was inserted into the tumor under CT guidance. PDT caused significant tumor damage and the patient subsequently was reclassified as a surgical candidate due to significant tumor shrinkage [19]. In another study by Huggett et al, all of the fifteen patients evaluated had PDT induced necrotic intratumoral lesions of up to 12 mm following a 40J light dose, and no noticeable side effects or toxicities [20]. Interstitial deep tissue PDT is also being evaluated in several other clinical trials as extensively reviewed by Svanberg et al [21]. These studies and other published trials have showcased the potential for PDT to treat deep-seated pathologies while inducing manageable to minimal toxicity. An attractive feature of interstitial PDT is that it also facilitates efficient dosimetric planning. Because fibers are placed in predetermined locations within the target site, they can not only be used to deliver light, but can simultaneously act as diagnostic sensors that can gauge important PDT parameters that critically impact the therapeutic response, such as the fluence rate, PS concentration, PS photobleaching, and the tissue oxygenation status [21, 22]. The low adverse event rates that have been reported in PDT treated patients, who were otherwise unsuitable for surgery or resistant to chemotherapy, point to the potentially important role that PDT can play in treating pathologies such as cancer. Furthermore, it should be noted that these studies were performed by coupling laser light into optical fibers. Indeed, coupling non-collimated light sources into fibers, though feasible, leads to a significant loss in the power at the fiber output, and has generally not been considered. Recent advances in LED light source technology have led to their ability to output hundreds of Watts. Along with enhanced portability stemming from battery powered sources and precision optical fiber coupling, these non-collimated and less expensive light sources will ease the translation of PDT to clinical procedures.
Pulsed or fractionated PDT regimes for achieving enhanced necrotic depth
Continuous wave (CW) lasers or light sources have traditionally been used for PDT. However, as the availability of pulsed lasers increased, several groups, including ours, have compared the effectiveness of pulsed lasers and CW irradiation for PDT since the late 1980s. Pulsed laser illumination was thought to enhance PDT efficacy primarily due to hypothesis that the downtime between light irradiation will: 1. Allow the tissue to re-oxygenate, making subsequent irradiations effective and 2. Allow re-accumulation of photosensitizer at the lesion [23]. While a few studies have shown that the necrotic depth induced by CW lasers is similar to thaFt seen with pulsed lasers, other studies have shown significant enhFancement in the necrotic depth resulting from pulsed irradiation [24-29]. For example, a study by the Bown group showed comparable outcomes between phthalocyanine (ALSPc) based PDT using an argon ion pumped CW dye laser with a copper vapour pumped dye pulsed laser (10KHz repetition rate) [28]. The same study also demonstrated that a low repetition rate with a high pulse energy source such as the flashlamp of a 5 Hz pumped dye laser is not an efficient irradiation source for PDT. Our group also demonstrated no statistically significant difference in the depth of necrosis 48 hrs post PDT with CW or the pulsed irradiation with the same average incident irradiance [30]. On the contrary, another study by Grecco et. al. demonstrated that a femtosecond laser irradiation produced twice as deep a necrotic zone compared to a CW laser at an equivalent dose (150 J/cm2) using the first-generation PDT sensitizer hemoatoporphyrin derivative (HpD) [31, 32]. Several differences, such as the type of PS and interval between irradiations etc, have made the comparison between pulsed and continuous PDT inconclusive. To determine factors that affect or increase the necrotic depth in a pulsed-PDT regime, Pogue et al simulated the deposited dose and reported that the pulsed laser irradiation can be beneficial for deep tissue PDT [33]; however, these results are modest and strongly depend on the PS, the laser pulse width, the pulse energy, and the repetition rate. In another study by Sterenborg et al [34], the simulations concluded that pulsed excitation in PDT is identical to CW for fluence rates below 4 × 108 Wm-2. At higher fluence rates, the effectiveness of pulse PDT drops significantly [34]. Despite promise for deep tissue PDT and the debate on the advantages of pulsed irradiation versus CW irradiation to produce optimal treatment outcomes, CW lasers appear to have gained more traction as clinically used light sources to date.
The type of light source used (CW or pulsed), the concentration of the PS at the treatment site also play an important role in determining the depth of necrosis induced by PDT. [35]. A lesion with a very high PS concentration may prevent light from penetrating to the deeper regions of the tumor due to a phenomenon known as PS self-shielding, in which saturated concentrations of the PS absorb a major portion of the incident light in the superficial layers. According to Pogue et al, a high intensity pulsed beam might have advantages over CW due to the transient change in absorption of the PS that allows the latter parts of the laser pulse to pass through the surface layers with less attenuation. In simple terms, the photobleaching or destruction of PS in the top layer will allow subsequent light to not be attenuated, and reach deeper tissues creating a “layer-by-layer” PDT effect [30]. Clinically, achieving high concentrations of PS may require either a localized intra-lesional PS injection or to limit PDT to nearly transparent tissues in which the PS absorption is much higher than that of tissue. A study by Rizvi et al also showed that high concentrations of PS may not translate to effective PDT therapy [36]. These observations point to the importance of “right” amount of PS and “right” light irradiance to obtain an effective treatment outcome. Another strategy utilized by our group and others to enhance PDT efficacy is to combine two or more PSs [37]. For example, Cincotta et al demonstrated that large RIF tumors were more effectively treated with a combination of Benzoporphyrin Derivative (BPD)-PDT and EtNBs-PDT compared to PDT with individual PS alone. This combination of PSs was chosen because each PS targets different compartments of the tumors (oxygenated vs hypoxic, vascular vs cellular) allowing for a better overall therapeutic outcome [38].
Another strategy to induce deep tissue phototoxicity is to perform repeated PDT or metronomic [39] PDT (slow infusion of PS and low dose light). In the realm of repeated PDT, studies have shown that fractionated PDT (i.e., PDT repeated with a prefixed time interval in one therapy session) induced necrosis to a depth 3 times greater than PDT alone [40]. In addition to affording a better treatment response profile, this PDT design also increases the feasibility of deep tissue PDT because it may allow for continuous accumulation of PSs at the treatment site, i.e., the first series of irradiation of PpIX in ALA-based PDT will lead to photobleaching of the PpIX and the time gap between irradiations will allow for resynthesis of PpIX to occur at the treatment site. The amount of PpIX reaccumulated at the treated site is demonstrated to be a function of the fluence rate of the first PDT dose [23, 41]. These studies indicate that clever PS delivery strategies together with appropriate light illumination strategies could lend themselves to more efficacious deep tissue PDT.
Image-guided dosimetry and treatment design for deep-tissue PDT
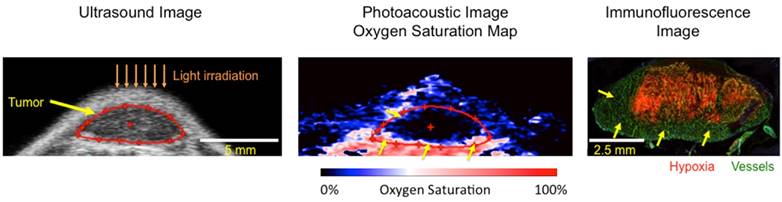
Tissue optical properties play a dominant role in determining the depth of the treatment zone during PDT [2, 42] and moreover, due to the variable vascular network and microenvironment in pathologies such as cancer, there is significant inter- and intra-lesion heterogeneity in treatment response. For example, the heterogeneous vascular network in tumors impacts PS uptake, thereby further altering the tissue optical properties. Understanding the spatial distribution of light in lesions and personalizing design strategies such as the placement of fiber optic probes or adjusting fluence rate based on real-time feedback on lesion properties (PS concentration, photobleaching, oxygenation content, etc.) is of the utmost importance to achieve predictable treatment outcomes from PDT. For example, Zhou et al demonstrated that personalizing the light dose based on pre-treatment measurements of the PS concentration within the lesion significantly reduced variability in treatment response [43]. Another important factor determining PDT efficacy is the PS-light-interval, wherein dosimetry and treatment planning can become complicated when considering damage to only the vascular compartment of the lesions and not to the surrounding tissue [44]. Fluorescence imaging has traditionally played a major role in PDT dosimetry by evaluating PS fluorescence and photobleaching [3, 15]; however, its penetration depth is limited and makes it difficult to gauge deeply-situated untreated regions. Other deep-tissue optical imaging techniques such as photoacoustic imaging [45] or diffuse optical imaging techniques [46] are currently being evaluated in several studies to understand the role of oxygen in PDT efficacy. In our recent studies, we showed that regions within the tumor that did not have complete vascular shutdown (i.e., no reduction in blood oxygen saturation) regrew post PDT [47]. Fig. 4 showcases an example of untreated regions within the subcutaneous tumor (xenograft with U87 glioblastoma cells) where there was no hypoxia due to vascular shutdown. Specifically, an ultrasound image (tumor structure), photoacoustic image (oxygen saturation), and immunofluorescence image (vasculature in green and hypoxic regions in red) of a tumor treated with BPD based PDT are shown in Fig. 4. Sufficient light dose (illumination at 690 nm) did not reach the bottom of the tumor (yellow arrows), thereby causing little to no damage to this region of the tumor. Given the heterogeneity in the tumor microenvironment, it is critical to incorporate imaging technologies that can sufficiently sample disease regions for markers such as vasculature, oxygen saturation, necrosis, blood flow changes etc. to assess potentially non-responsive areas and predict treatment response. Recently, techniques that directly monitor the singlet oxygen generated during PDT have also been employed to predict treatment response [45]. An extensive review of direct and indirect treatment response strategies in PDT have been provided elsewhere [15, 48] and are considered beyond the scope of this review. Overall, to achieve efficient therapeutic benefit from PDT, specifically also for deep tissue PDT, it is of paramount importance to monitor microenvironmental conditions and provide the “right or optimal” light dose (fluence rate and fluence) and illumination regime according to the photosensitizer concentration at the treatment site [49].
Utility of deep-tissue photoacoustic imaging to monitor PDT efficacy. The ultrasound image demarcates the location and size of the subcutaneous tumor (glioblastoma U87 cells). Spectroscopic photoacoustic imaging provides blood oxygen saturation map of the tumor at the same cross-section. The oxygen saturation maps are pseudo colored on a black (0%) to red (100%) scale. Immunofluorescence image at the same cross section of the tumor is obtained post-euthanasia. The vasculature is stained in green while red stain shows the hypoxic regions in the tumor. Hypoxic conditions are caused in PDT either due to consumption during the process or via vascular coagulation post-PDT. Here we observe that deeper tumor regions had no hypoxia stain (indicated by yellow arrows) or reduction in oxygen saturation indicating insufficient light dose reaching these deeper tissues. Incorporating therapy monitoring techniques to identify non-responsive or untreated areas is highly important and critical to prevent subsequent regrowth of these regions by designing appropriate therapy. Figure adapted from Mallidi et al. [47]
Forward looking methodologies for deep tissue PDT
To overcome the poor penetration depth of visible light into tissue, several alternatives involving penetrating radiation have been proposed and will be discussed in this section. Because the PS requires a threshold number of incident photons to initiate the cytotoxic photochemistry, the overarching goal of deep tissue PDT is to create an energy source that can locally activate the PS even at deeper depths. This source could be either self-activated, e.g. bioluminescent, or be comprised of other forms of electromagnetic radiation, e.g., near-infrared radiation (NIR), X-rays or γ-rays that are known to penetrate more deeply into tissues compared to visible light (Fig. 1). In situations where the PS cannot be directly excited by penetrating radiation, a transducer, usually consisting of a nanoparticle (NP), may be used to locally absorb the incoming radiation and transfer part of its energy to activate the PS [50]. In this section, we will review how bioluminescence, NIR light, and X-rays or γ -rays can be used to initiate PDT in deep tissues.
Chemi- and Bio-luminescent probes for PDT
Chemi- and bio-luminescent probes were the first self-emitters used to locally activate a PS in deep tissues. Both types of probes generate luminescent products, but contrary to chemiluminescence, the light emitted by bioluminescent probes is derived from enzymatic activity [51]. Phillip et al. were the first to report the use of chemiluminescent probes in the late 1980's [52]. They demonstrated in vivo that a peroxyoxalate chemiluminescent solution could activate the HpD Photofrin II, concluding that chemically activated luminescence could be a promising option for PDT in deep tissues. More recently, Huang et al. demonstrated that luminol activated by ferrous sulphate could excite the meso-tetraphenylporphyrin (TPP) PS inducing an effective decrease in the viability of Caco2 cells [53]. Yuan et al. confirmed these results by demonstrating a complete spectroscopic validation of the energy transfer between the oxidized luminol and the OPV, a cationic oligo (p-phenylene vinylene) PS [54]. Generation of ROS and cell death was confirmed in vitro in this chemi-luminescent based PDT study. The authors performed an in vivo study that demonstrated the combination of oxidized luminol and OPV could slow tumor growth with minimal systemic toxicity in HeLa tumor-bearing mice.
Despite their promise, chemiluminescent probes usually exhibit systemic toxicity that may limit their widespread adoption. A few years after the introduction of chemiluminescence based PDT, Carpenter et al. reported the first use of bioluminescence, a method with lower intrinsic toxicity than chemiluminescence, to excite a PS for PDT. The emission of oxyluciferin, a luminescent species produced by the oxidation of luciferin by the luciferase enzyme, was used to locally excite the PS hypericin. By demonstrating the ability of a bioluminescence molecule to transfer energy and excite the PS, this group opened up new possibilities to initiate PDT in deeper tissues than were previously possible. Later, Theodossiou et al. investigated the capacity of the oxyluciferin to activate the PS rose Bengal in vitro and induce cell death in murine fibroblasts [55]. Although Schipper et al. has more recently contested these results [56], the viability of cells transfected with the luciferase gene was reduced to (11±12)% when treated with 10 nM Rose Bengal. Schipper et al. strongly questioned the efficiency of the bioluminescence-activated PDT by showing that the light dose emitted by the bioluminescence probe (on the order of 10-9 mW.cm-2) was significantly lower than the doses typically employed in clinical trials for laser activated-PDT (~ 50 mW.cm-2) [56]. Besides this fundamental concern, several follow up studies demonstrated improved killing stemming from either bio- or chemi-luminescence activated PDT, highlighting our limited understanding of the mechanisms underlying these energy transfers since ostensibly the reduced energy densities emitted by the luminescent probes can still activate PS and impart cytotoxicity.
There is an intrinsic toxicity associated with the use of bioluminescence probes, although it is lower than that induced by chemiluminescent probes. To decrease this toxicity, Zhao et al. reported the synthesis of microcapsules containing the bioluminescent probe D-luciferin [57]. Once activated, D-luciferin emits a broad luminescence (520-680nm) that strongly overlaps with the absorption spectra of the PS's rose Bengal and hypericin. Microencapsulation decreased the direct toxicity of D-luciferin, in that MCF-7 cells treated directly with this formulation exhibited a viability of 93±1.8% compared to 55±3.7% following treatment with an equivalent concentration of free D-luciferin (9 μM). The microcapsules also enhanced the cytotoxicity of the bioluminescence/PS system while decreasing the cell viability from 96% to 88% via rose Bengal-PDT or to 71% with hypericin-PDT (both PSs were used at the equivalent concentrations of 0.03 mg.mL-1). To further reduce the toxicity, Hsu et al. proposed the use of a self-illuminated bioluminescent Rluc8-QD, which was previously synthesized by So et al., [58] to activate a PS [59]. Rluc8 is an enzyme that oxidizes a substrate, coelenterazine, to produce a characteristic fluorescence emission at 480nm. This energy can be transferred to quantum dots, (QD) by Forster Resonant Energy Transfer (FRET) or by Bioluminescent FRET (BRET), leading to their excitation. The QD energy relaxation is accompanied by an emission of luminescence centered in the red (655 nm) that can be used to activate a PS. Thus, BRET allows for shifting of the emission of the bioluminescent molecule, coelenterazine, to a wavelength that could excite a conjugated PS. In their study, Hsu et al. observed a 5-fold reduction in tumor volume compared to a control that used Foscan® loaded micelles as a PS self-activated by the conjugated coelenterazine/QD compound. In a more recent study, Kim et al. [60] used the same bioluminescent probe, Rluc8-QD, to activate a different PS, chlorin e6 (Ce6), to induce PDT. Besides reporting the efficacy of their conjugated system on cell viability and tumor growth suppression in vitro and in vivo respectively, the authors evaluated the equivalent light dose required to reach a similar killing efficiency as with laser activated PDT. This dose was estimated to be 2.2 mW.cm-2. In addition, a detailed in vivo study showed the impact of the RLuc8-QD mediated excitation of Ce6 on the reduction of cancer cells spread to sentinel and secondary lymph nodes, potentially indicating a promising new modality to decrease likelihood of distant metastases. Taken together, these results demonstrate a promising new method to induce PDT mediated cytotoxicity at depth. Even though the light dose produced by chemi- or bio-luminescent probes is lower than that usually required for PDT, this method still efficiently induces cytotoxicity, emphasizing the complexity and the limited understanding of the processes involved. Further investigations and thorough validations on the efficiency of these probes (either as free probes or in nanoparticle form) are required prior to clinical translation.
NIR radiation to induce PDT in deeper tissue
The most direct approach to excite PS for PDT in deeper tissue is to use radiation that lies within the NIR optical window (Fig. 1). Following absorption of a single photon, the excitation energy is below the production threshold of singlet oxygen molecules, allowing only type I PDT or oxygen independent-PDT [3]. Two photon (TP) processes use longer excitation wavelengths and can also be considered to excite the PS and induce photodynamic tissue damage. In the following sections we will discuss various options for PDT in deeper tissues using NIR radiation. These options are also illustrated in Fig 5A.
A. Three different ways to excite PSs for PDT using NIR radiation. Method 1 relies on direct two-photon excitation of the PS that, once excited, can undergo type I or type II photodynamic processes involving reactive molecular species and singlet oxygen. Methods 2 and 3 involve a nanotransducer. In the second scenario, the nanotransducer absorbs the NIR radiation through two-photon processes and transfers part of the energy to excite the PS either through a radiative or a non-radiative mechanism. The nanotransducers involved may be either organic chromophores that have a high TP-absorption cross-section, or optically active nanoparticulate entities such as Gold Nanorods (GNR), quantum dots (QD) or carbon quantum dots (CQD). Method 3 illustrates the use of up-converting nanoparticles that successively absorb two NIR photons through a metastable energy state and transfer part of this energy to the PS. B. This figure illustrates three ways to activate the PS using ionizing radiation such as X-rays. Because X-rays are used for radiation therapy, each of these activation routes could be combined with radiotherapy to enhance the overall efficiency of the tumor treatment. Method 1 involves direct excitation of the PS using ionizing radiation such as X-rays. In this case, the PSs are called radiosensitizers. Method 2 involves a local generation of light using the broad spectrum Cerenkov emission process, when a charged particle travels faster than light in a given matter and emits Cerenkov radiation. This emission presents a strong overlap with the absorption spectra of many PS and can subsequently be used to excite them. Method 3 shows the use of nanoscintillators that act as nanotransducers to locally convert ionizing radiation into visible light to excite PSs. The transfer from the nanoscintillator to the PS can either be radiative or non-radiative (FRET), and leads to activation of the PS. Abbreviations: NIR: Near Infrared, FL: Fluorescence, PH: Phosphorescence, ISC: Inter-System Crossing, MS: Metastable State, NRD: Non Radiative Decay, NER: Non-Elastic Relaxation and T: Thermalisation.
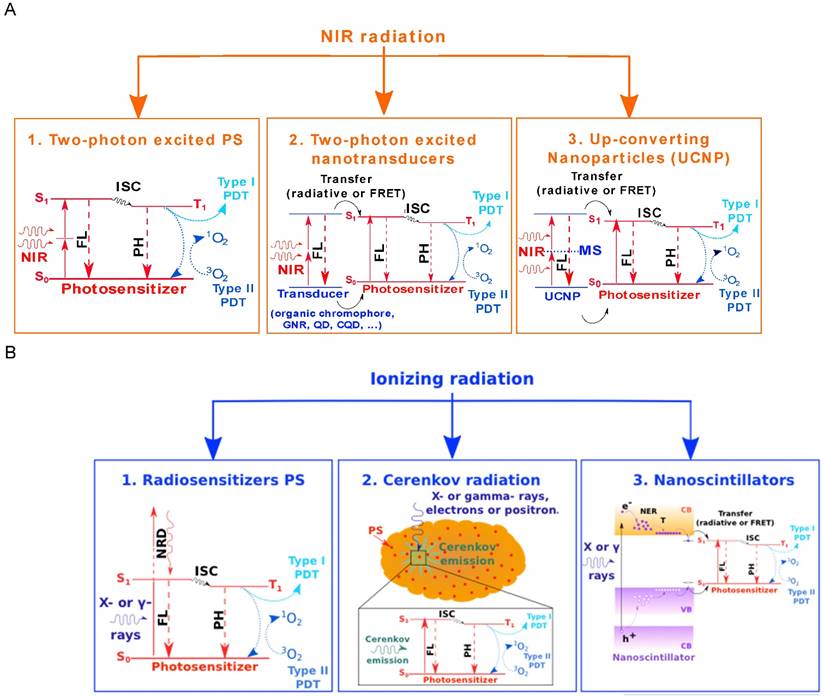
TP excited PS
Using TP processes to excite a PS would not only enhance the penetration depth but also localize the illumination area. Indeed, TP excitation is a non-linear process whose efficiency quadratically increases with the laser intensity [61]. Thus, the excitation is strongly confined to the focal plane. This confinement of light excitation is particularly relevant to avoid off-target toxicities. For example, in the treatment of age-related macular degeneration (AMD), TP excitation allows for the preservation of healthy tissues that have absorbed some PS and lie within the optical beam path, while still allowing for effective treatment of the target site [62]. Starkey et al. [63] demonstrated in murine xenograft models that TP-PDT could efficiently be used in vivo to induce tumor regression at depth. They showed that irradiating the mouse from the ventral side could treat a tumor located on the dorsal side.
The first studies of TP excited PS were reported in the 1980's, and were performed mainly using Nd:YAG lasers [64]. These results were controversial because ambient water molecules efficiently absorb the Nd:YAG laser emission (1064 nm) and induce hyperthermia in tissues, as was shown by Marchesini et al. [65]. In 1995, Lenz et al conducted a study of TP activated PDT in rat ears, and compared several PS including hematoporphyrin derivative (HPD) and phtalocyanine while controlling for the hyperthermia effect. Even though fluorescence intensity measurements demonstrated that TP processes could excite the PS, no cell killing was observed in these studies [66]. This pointed to the fact that TP excitation of a commonly used PS was not sufficient to induce PDT phototoxicity in vivo. Following this study, several commonly used PSs were investigated for TP-PDT, but the results mainly demonstrated that the efficiency was too low to induce cytotoxicity. Hence, commonly used PSs including PpIX, Photofrin or Visudyne, cannot be considered serious candidates for TP-PDT [67-69]. Several approaches have been investigated to improve the PS-TP absorption cross section [70] and during the last decade, several newly designed molecules have been synthesized as summarized in Table 1. The TP absorption cross-section as well as the singlet oxygen generation quantum yield have to be considered when evaluating the overall utility of the newly designed PSs for type II TP-PDT. An effective TP-PDT agent requires both the maximal TP absorption cross-section (gold nanorods for example) and a sufficient 1O2 quantum yield (porphyrin derivatives, for example). Despite a noteworthy increase in the TP absorption cross-section, the overall anti-tumor PDT efficacy usually remains low thereby limiting the use of TP-PDT.
Photosensitizers designed and synthesized for TP-PDT. For each compound, the excitation wavelength (in nanometers), the TP absorption cross section (in GM; 1 GM = 10-50 cm4s.Photon-1) and the singlet oxygen quantum yield are indicated.
| Compound | Excitation wavelength (nm) | TP absorption cross section (GM) | ΦΔ | Ref |
|---|---|---|---|---|
| Substituted difuranonaphtalene | 802 | 139 | 0.36 | [71] |
| Substituted distyryl benzene | / | 0.46 | ||
| Dendritic dimers around the PS | 887 | 7600 | / | [72] |
| Modified bare tetrapyrroles | ||||
| PyP | 780 | 8200 | 0.84 | [73] |
| yPyyPy | 9100 | 0.54 | ||
| Porphycenes | ||||
| PdTPPo | 770 | 1750 | 0.78 | [74] |
| TPPo | 2280 | 0.23 | ||
| Conjugated porphyrin dimers | 916 | 17000 | 0.60 | [75] |
| Symmetric squaraines derivatives | 806 | 17400 | 0.33 | [76] |
| Quadrupolar chromophores | 800 | 6000 | 0.51 | [77] |
| Gold Nanorods | 835 | 5.4.109 | 1.10-2 | [78] |
| Coumarin-based PS | 820 | 1556 | 0.49 | [79] |
| Porphyrin triphenylamine | 830 | 251 | 0.80 | [80] |
| Diketopyrrolopyrrole porphyrine conjugates | ||||
| DPP-ZnP | 910 | 2000 | 0.58 | [81] |
| DPP-ZnP-DPP | 4000 | 0.50 |
TP excited nano-transducer for PDT
In addition to developing new PSs with high TP absorption cross-sections, there has been an increased interest in using energy transducers to locally absorb incident NIR radiation to subsequently activate the PS. In most cases, NIR radiation is absorbed through TP processes by a nanotransducer (Fig. 5A2) that could have various origins. One option is to link the PS to chromophores that have strong TP absorption cross-sections. Under NIR radiation, the chromophores will be excited by multi-photon processes and will transfer part of the excitation energy to the PS by FRET. Bhawalkar et al. demonstrated that conjugating the PS to chromophores did not modify the photochemical properties of the PS, and demonstrated the ability for the linked PS to generate 1O2 [82]. Later, several studies were published that validated the concept of antenna chromophores, i.e. chromophores that activate the PS through FRET transfer following TP excitation [83-85]. Instead of chemically linking the PS to the chromophores, strategies that co-encapsulate them into silica nanoparticles (NPs) have also been proposed [86]. To improve the efficiency of the indirect activation of PDT with TP excitation nanoparticles, plasmonic gold nanorods (GNR) with higher TP absorption cross-sections can be used. Zhao et al. demonstrated enhanced 1O2 generation by using GNR combined with a porphyrin (T790) as PS [87]. Since TP excitation requires high incident laser intensity, the GNRs may undergo an irreversible deformation that leads to a loss of their emission properties. To prevent this type of photo-damage, Chen et al. synthesized mesoporous silica-encased GNRs that incorporate a PS for TP activated PDT and showed efficient generation of 1O2 and cell killing [88]. In addition to GNRs, QDs are potential candidates for TP mediated PS activation. In addition to exhibiting PS properties themselves [89, 90], QDs have demonstrated efficiency in acting as an energy transducer to activate PS bound to their surface via FRET [91]. These semi-conducting NPs are also characterized by a high TP absorption cross-section, making them uniquely suited for deep-tissue PDT. Because heavy metal containing QDs are quite toxic to cells, carbon QDs (CQD) appear as an attractive alternative. Fowley et al. reported the synthesis of high TP absorption cross-section CQDs combined with the PS PpIX. Under TP excitation, CQD absorb and transfer energy via FRET to the conjugated PS that then generates cytotoxic species. HeLa cells were exposed to different concentrations of CQD/PpIX conjugates and a viability reduction was demonstrated. In addition, the in vivo efficiency of this compound was shown in that it strongly reduced the size of fibrosarcoma tumors induced in mice [92]. Besides allowing for PDT activation at increased depth, TP-PDT also confines the excitation to the laser focal point. Though localized illumination of TP-PDT could be useful for certain applications, it can be a major limitation and unrealistic from a practical viewpoint for treating large and disseminated tumors. Broad therapeutic strategies, such as single photon PDT, could be used to treat larger tumor regions while TP-PDT could be reserved for subsequent precise tumor eradication at depth.
Upconverting Nanoparticles (UCNP)
Up-converting nanoparticles (UNCPs) are promising candidates for deep tissue PDT and have been extensively studied over the past few years [4, 93]. UCNPs are usually made of a ceramic lattice doped with rare earth ions that allow for sequential absorption of two photons through a metastable energy level. The lifetime of the metastable state is typically on the order of a microsecond, and is an order of magnitude longer than the lifetime of virtual states involved in TP processes. A consequence of the longer lifetime of the transitional state includes the possibility to use continuous wave lasers and, more importantly, lower power densities for UNCP excitation. For example, the power densities may be in the range of 1-103 W.cm-2 for UNCP excitation, whereas 106-109 W.cm2 are required for TP activation. Typically, after the absorption of two or more low energy NIR photons (usually around 980 nm) by UNCPs, a single higher energy photon is emitted in the visible range (Fig. 5A3). Since this process does not naturally occur in living systems, imaging rare earth materials through upconversion emission results in very low non-specific background for fluorescence and PDT, as is reviewed thoroughly by Moghe et al [94]. We will not go into details for this type of transducers because excellent and more exhaustive reviews have already been published on the subject [4, 93]. However, the large amount of in vitro and in vivo studies reporting an efficient UCNP induced PDT effect substantiate the promising role of UCNP to excite PS for PDT in deep tissue. More generally, rare earth materials used in upconversion or similar nanoparticles are also promising candidates for deep-tissue PDT based theranostics and medical imaging due to versatile synthesis, modification chemistries, photostability and relative safety. However, their in vivo bioclearance and toxicity still need thorough investigation to bolster their clinical applicability.
Ionizing radiation for PDT in deep tissues
Though better than visible radiation, NIR radiation still has a limited penetration depth of approximately 1 cm. Treatment of large superficial tumors may be possible with NIR light but tumors residing in deeper tissues remain unreachable without the secondary light delivery strategies discussed in section 2. To substantially improve the the penetration depth of excitation photons, X-rays that are already used for radiation therapy (RT) with little tissue penetration limitations, are interesting candidates even though they are known to cause intrinsic toxicities [95]. That being said, taking advantage of X-rays used during RT to simultaneously activate PSs, thereby potentiating the localized cytotoxic effect in deep tumors, may improve the overall treatment efficacy by affording PDT-RT combination effects and by decreasing the dose required for RT. This is because combining PDT and RT, which imparts cytotoxicity by mechanistically distinct avenues, may lead to treatment synergism. Although several in vitro studies have been published on the combination of PDT and RT, there is no consensus on the overall utility of this combination therapy. Berg et al. demonstrated that the time interval between RT and ALA-PDT is crucial and the treatment combination may lead either to an antagonistic or synergistic effect in adenocarcinoma cells (WiDr) [96]. Other studies report an additive effect between haematoporphyrin dimethyl ether-PDT [97] or indocyanine green-PDT [98] and RT on Eherlich ascites carcinoma cells and prostate carcinoma cell lines (PC3) respectively. More recently, Montazerabadi et al. demonstrated in vitro a synergistic effect on breast cancer cells (MCF-7) treated with both indocyanine green-PDT and RT [99]. Several methods of combining PDT and RT are illustrated in Figure 5B and will be discussed in the following sections.
PS that act as radiosensitizers
PSs that double as radiosensitizers (RSs) have been developed by several groups to combine the effects of PDT with RT and impart greater cytotoxicity in deep tissues. Luksiene et al. reported that Haematoporphyrin dimethyl ether (HPde) acts as a RS when injected at a concentration higher than 30 mg/kg of body weight in mice with transplanted tumors. The combination of PDT and RT for these mice led to a 4x reduction in relative tumor growth compared to radiation only [97]. In a subsequent study, the RS properties of three different PS (HPde, Photofrin II (PII) and hematopoprhyrin derivative (HPD)) were compared [100]. In this in vivo study, it was demonstrated that the HPde was the most efficient RS, followed by the PII and HPD. Furthermore, this study also showed that the RS effect of these PSs was cell line dependent. The interest in the development of dual PS and RS agents has diminished due to the low efficiency of PSs that act as RSs under direct excitation. Alternatively, various approaches were developed to locally generate visible light using X-ray irradiation that could excite the PS in deep tissue.
Cerenkov radiation to locally excite the PS
Cerenkov radiation has been proposed to generate light in deep tissues using ionizing radiation. Cerenkov emission is observed when charged particles, e.g. electrons or positrons, travel faster than the phase velocity of light in a given medium. Because there is a minimum velocity associated with this kind of radiation, there is also a minimum energy value required for these particles to be classified as Cerenkov. Thus, Cerenkov radiation can be generated either by β+ or β- emitter radioisotopes such as those used for positron emission tomography (PET) or X-ray based radiotherapy, which induce an electromagnetic cascade containing high-energy charged particles that interact with matter. The spectrum of Cerenkov emission is broad, is centered in the near ultra-violet range and can overlap with many PS excitation spectra (Fig. 5B2). In addition, the radionuclides and ionizing radiation used for radiotherapy can be used to generate Cerenkov emission making it particularly attractive for deep-tissue PDT. Although this approach is still relatively under-explored, two promising studies have been recently published. First, Axelsson et al. not only demonstrated that a measurable Cerenkov emission was produced in a water phantom following irradiation by X-rays (6-18MV) or electrons (6-18 MeV) delivered by a clinically used linear accelerator, but also that this Cerenkov emission was able to activate PpIX in solution [101]. This proof of concept demonstrated the potential role of Cerenkov radiation to induce PS excitation and PDT in deep tissues. More recently, Kotagiri et al. demonstrated that 64Cu radionuclide, usually used as a PET radiotracer and characterized by a high positron emission and fast decay, could induce Cerenkov radiation and excite TiO2 NPs that act as oxygen independent PS. In addition to demonstrating efficient cell killing in vitro, the authors presented in vivo experiments showing complete eradication of the tumor when NPs were combined with the 64Cu radionuclide, whereas tumors were unaffected in all the treatment control conditions [102]. Even though the number of published studies remains low, Cerenkov radiation seems to be a promising approach to activate the PS in deep tissues, either by using ionizing radiations utilized for RT (X-rays) or diagnostic purposes (radiotracers).
Nanoscintillators for X-ray conversion into visible light
A decade ago, Chen and Zhang [103] proposed a new approach that combined PSs with nanoscintillators. Nanoscintillators are nanoparticles (NP) that are able to convert ionizing radiation, such as X- or γ-rays into visible light. By locally converting the deep penetrating X-rays used for RT into visible light, nanoscintillators may act as a local excitation source for PS activation (Fig. 5B3). To enable the energy transfer (radiative or non-radiative) from the nanoscintillator to the PS, the PS excitation spectrum must overlap with the nanoscintillator's emission spectrum. Delivering nanoscintillator/PS constructs to tumors prior to radiation therapy (RT) may allow for excitation of the PS and induction of PDT, which, when combined with the cytotoxic effects of RT, could lead to synergistic treatment of tumors residing in deep tissue. The first experimental study of a conjugated nanoscintillator was published by Liu et al. who presented the synthesis of LaF3:Tb3+ nanoparticles conjugated to MTCP (meso-tetra(4-carboxyphenyl) porphine) as a PS and the generation of 1O2 following X-ray irradiation of the nanoscintillators [104]. Following this, a few other studies were published that reported the synthesis of new nanoscintillator conjugated PS compounds that showed similar energy transfer properties post excitation with X-ray irradiation. Most of these “proof of concept” studies are restricted to optical measurements (emission spectra, fluorescence decay, 1O2 chemical probes fluorescence properties) [105, 106] or in vitro experiments demonstrating reduction in viability due to nanoscintillator based PDT. For example, Abliz et al. reported a reduction in viability of human glioblastoma cells, from 80% to 10%, when micrometric gadolinium oxysulfide particles were combined with Photofrin II and irradiated with X-rays [107].
In order to help design useful nanoscintillator/PS conjugates with optimal size or composition that can induce cytotoxic effects in deep tissue following X-rays irradiation, it is necessary to better understand and characterize the underlying mechanisms. In this spirit, a study based on time-resolved spectroscopic measurements of terbium oxide nanoparticles (Tb2O3@SiO2 NPs) conjugated to a porphyrin PS revealed an energy transfer that occurs from the nanoscintillator to the PS, mainly as a non-radiative dipole-dipole transfer (FRET) [108]. This resulted in singlet oxygen generation following X-ray excitation. As FRET efficiency decreases in a manner inversely proportional to the distance between the emitter (luminescent ion in the nanoscintillator) and the absorber (PS) to the sixth power, this distance is a determining factor. By identifying FRET as a major contribution to the transfer mechanism, this study allows fixation of a condition on the distance between donor and acceptor centers in the nanoscintillator/PS systems. Another important parameter to estimate the overall efficiency of a PS-nanoscintillator conjugate is the 1O2 quantum yield upon X-ray irradiation. Clement et al deduced the quantum yield of a CeF3 nanoscintillator conjugated to verteporfin as a PS, by combining experimental measurements of the amount of 1O2 generated with the calculated amount of energy deposited in the scintillating NPs. To do so, they referred to a model firstly introduced by Morgan et al., that allows predicting the maximum amount of 1O2 generated under X-ray irradiation by estimating the amount of energy deposited in nanoscintillators during the irradiation [109]. Based on their model, Morgan et al came to the conclusion that only X-rays with energy below 300 keV, such as those used for brachytherapy, could induce sufficient cytotoxicity. Recently, these calculations were further refined using Monte Carlo simulations and a more accurate estimation of the energy deposited in nanoscintillator was provided by Bulin et al. [110]. Each of these simulation tools could efficiently be used to design the next nanoscintillators to use as local light source in combination with a PS to induce PDT in deep tissue.
With the advent of nanoscintillators, interest in combining PDT with RT appears to be growing. Chen et al. recently reported a study in murine subcutaneous tumor models where scintillating nanoparticles (SrAl2O4:Eu2+) were combined with merocyanine540 (MC540) as the PS. Following irradiation delivered by a X-ray tube (50kV, 70μA), the tumor size decreased approximately 8-fold in 12 days, and no effect was observed on the mice body weight [111]. Various nano-based approaches have been employed to incorporate scintillating materials and PSs. For example, Kascakova et al. reported the synthesis of lanthanide-based micelles integrating hypericin as a PS [112]. Although the authors reported a very low amount of 1O2 generated, the idea of incorporating lanthanides into the micelles could be easily extended to other types of nanocarriers, including liposomes. Due to their low density, the lanthanide complexes will very likely absorb only a small portion of the incoming X-rays, pointing to low PDT efficiency of such complexes and hence the need for further investigations. Another approach to initiate PDT via X-rays is using a nanoscintillator (LiYF4:Ce3+) combined with a semiconducting nanoparticle (ZnO). The strength of this approach lies in its ability to overcome the potential degradation of the PS under ionizing radiation while also diminishing the oxygen dependence of PDT, which is particularly limiting in hypoxic tissues for the majority of PSs that undergo oxygen-dependent type II reactions. Under irradiation, nanoscintillators convert X-rays into visible light that is able to excite the ZnO NPs. These NPs initiate type I photochemistry by generating cytotoxic hydroxyl radicals [113]. In vitro, this approach led to a reduction of HeLa cell viability to 70% after exposure to a 6 Gy dose applied either in normoxic (21% O2) or in hypoxic (2% O2) conditions. In vivo, when treated with NPs and exposed to a 8 Gy dose X-rays, the tumor size decreased by a factor 5 compared to the control group.
To induce a similar cytotoxic effect while decreasing the radiation dose, Ma et al. proposed to use NPs that exhibit an afterglow emission in addition to their scintillating properties [114]. By definition, afterglow is a delayed luminescence that persists after the irradiation ceases. This emission is due to the recombination of charges slowly released from shallow energy levels that have been trapped during irradiation. A significant reduction in the viability of PC3 cells was observed following incubation with the nanoconstruct (ZnS:Cu,Co conjugated with the PS tetrabromorhodamine-123) and exposure to X-ray radiation. More broadly, it is possible that a delayed emission may enable a reduction in the applied X-ray irradiation dose while still maintaining sufficient therapeutic efficacy. However, synthesizing biocompatible afterglow particles still remains a challenge and the efficiency of PS activation through this indirect phenomenon remains low. Overall, the development of nanoscintillator combined PSs is at a very nascent stage and more thorough in vitro and in vivo studies must be performed to realize the full clinical translational potential of this technique.
Targeted probes that confine phototoxicity
Theranostic approaches for imaging, diagnosis and monitoring PDT treatment of disease tissue often requires exogenous probes to act as selective contrast agents, which are specific to inherent aberrations and alterations in the target tissue. Advanced photoactive targeting probes have been deployed for a wide variety of preclinical and clinical applications, including the diagnosis of neoplastic tissue and the delineation of resectable tumor margins. [115-118] In relevance to this review, the array of optical theranostic targeting probes designed specifically for selective deep tissue PDT will be reviewed in the context of emerging technologies, their limitations and the essential steps required for successful future development. The design, synthesis and utility of such customizable targeting probes will also be discussed in this section.
Aside from light penetration, a specific limitation for deep tissue PDT is the inadequate tissue selectivity of phototoxicity. Although phototoxicity is confined to areas of irradiation, cellular discrimination of PDT within those areas is typically poor as a result of weak PS selectivity and strong tissue scattering of the excitation light. When irradiating neoplastic tissue in the vicinity of sensitive anatomical sites, such as the brain, off target phototoxicity could have drastic undesired effects, reducing the maximum tolerated PDT dose and limiting efficacy. This was exemplified by an in vivo canine brain tumor PDT study where high-dose therapy using 1800 J of 630 nm light and 2-4 mg/kg of the PS photofrin induced significant neurotoxicity and brain stem damage [119]. Reducing the administered PS dose to 0.75 mg/kg eliminated these adverse effects. Thus, selectivity of phototoxicity can be achieved by confining photosensitization to diseased tissue cells by targeting the PSs, regardless of the off-target incident light, thereby increasing the maximal tolerated PDT doses.
The critical parameters required for successful deep-tissue PDT are common for most bioconjugated diagnostic and therapeutic probes. These parameters revolve around the biological targeting probe's molecular weight, immunological effects dictating its circulation times, physiological clearance rates, physiological impact following PS conjugation, and the optical properties that ultimately govern their effectiveness as deep-tissue PDT agents. PS targeting can be achieved through three archetypal probe platforms as illustrated in Fig. 6, which will be discussed in this section. These platforms include probes for targeted delivery, probes for tumor-selective activation and probes that combine selective delivery and activation.
A schematic representation of the three archetypal targeted PDT probes that either selectively deliver PSs to tumor cells (A), are selectively activated within tumor tissue (B) or are activated within tumor cells following targeted delivery (C). A) Targeted probes conjugated to PS molecules can be delivered selectively to tumors cells through the blood or through direct contact on surfaces, whereby the probes actively bind to their targets and deliver their payload in a disease-specific manner. B) Microenvironmental characteristics of tumors, such as tumor-specific proteases that are upregulated during disease progression can be targeted with substrate-mimetic quenched PDT probes that become selectively activated within the tumor upon cleavage. C) Some activatable targeted probes combine the mechanisms of probe systems described in (B) and (C) to selectively deliver the PS payload to tumor cells, yet remain optically inactive until cancer-cell specific internalization, proteolytic degradation and PS dequenching.
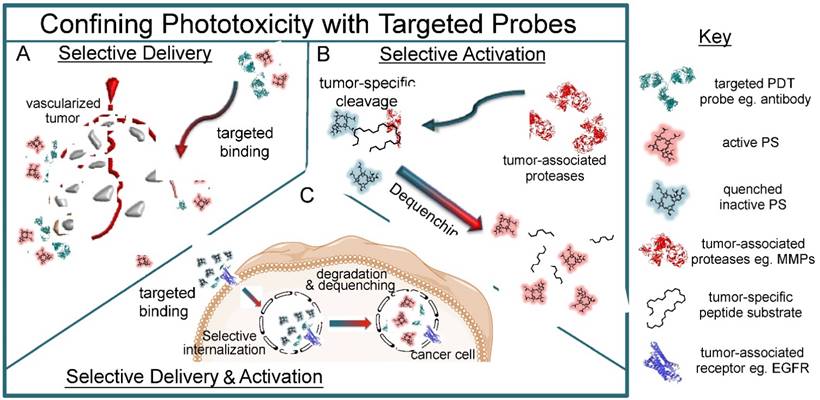
Probes for selective tumor cell delivery
The availability of FDA-approved therapeutic antibodies, such as cetuximab and trastuzumab have deemed them as fundamental platforms for selective PDT. Added advantages include their capacity for conjugating high PS payloads and their synergistic therapeutic effector function. The development of antibodies conjugated to PSs (photoimmunoconjugates, PICs), as shown in Fig. 6A, resulted in the innovation of a selective PDT treatment modality termed as photoimmunotherapy (PIT). PIT was pioneered in 1983 by the team of Julia Levy who conjugated the PS hematoporphyrin to anti-M-1 antibodies using l-ethyl-3-(3-dimethylaminopropy1)-carbodiimide HCL (EDC)-mediated amide coupling [120]. Increasing the PS-light intervals from 96 hrs to 192 hrs delayed tumor regrowth and extended median survival times of mice bearing subcutaneous M-1 tumors. Several adaptations of this PIC platform have since been developed, including multiple PS molecules stochastically conjugated to antibodies using similar covalent coupling techniques [121-127]. Both the glycosylation sites on the Fragment crystallizable (Fc) portion of full-length antibodies and the hinge region disulfides have been frequently utilized as handles for the site-specific conjugation of polymers carrying PS payloads so as to avoid PS interference with the antibody's binding capacity [128-132].
Since the seminal work by Levy et al., targeted imaging and fluorescence-guided surgical debulking assisted by an improved tumor margin delineation has attracted attention in the clinic. The clinical ambition of targeted theranostic probes modified with photosensitizing agents ultimately lies in the accurate photodiagnosis of malignant tissue followed by its selective PDT eradication. Towards this goal, fluorescent targeting probes have been clinically deployed for the surgical assistance of tumor resection. The earliest clinical use of targeted photoactive probes for the optical detection of cancer tissue in patients was reported in 1992 by Folli et al. who utilized a fluorescein conjugate of an anti-carcinoembryonic antigen antibody [115]. Although not used to intraoperatively assist surgical resection of the colonic carcinomas, in vivo selectivity of the probe was validated by ex vivo fluorescence imaging revealing heterogeneous labeling of cancer tissue with no fluorescence detected in the normal mucosa 24 hrs following intravenous administration. In 2011, van Dam et al. demonstrated for the first time an improvement in the sensitivity of intraoperative detection of ovarian cancer micrometastases in patients using a folate conjugate of fluorescein isothiocyanate (FITC) [116]. Through the selective binding of the folate-FITC probe to the tumors overexpressing folate receptor-α, median tumor nodule detection was improved 4.9-fold, as compared to conventional white light surgery. Inspired by these exciting advances, folate was conjugated to a porphyrin PS and investigated in a preclinical model of ovarian cancer micrometastases with the intent of selective in vivo PDT [133]. Although folate targeted PDT treatment was not attempted in vivo, folate-selective PS delivery resulted in the preferential accumulation of the PDT agent in tumor tissue with a mean tumor-to-normal ratio of 31:1 in the peritoneum. More recently, a conjugate of the FDA-approved anti-Epidermal Growth Factor Receptor (EGFR) antibody cetuximab and IRDye® 800CW has been deployed for first in-human trials of targeted antibody probes to mediate in vivo fluorescence tumor imaging and potentially guide surgical detection [117]. The clinical evolution of PICs is currently expanding to PDT applications in a Phase I trial for patients with head and neck cancer, where a cetuximab conjugate of the commercial photosensitizing fluorophore IRDye® 700DX is utilized [134]. However, a number of biophysical and biochemical parameters dictate the efficacy of targeted PDT probes, which will be further discussed in this section.
Molecular weight
The molecular weight of the targeted PDT probe plays a critical role in the pharmacokinetic behavior and biodistribution patterns of the conjugate; this has typically been investigated by using recombinant engineered biomolecules or through the fragmentation of full-length antibodies. Although oncogenic receptor antagonism and proapoptotic signaling is a fundamental therapeutic mechanism of targeted biologics, secondary immunological responses induced by the Fc fragments of intact full-length antibodies play a critical antitumor role [135]. Fc fragments are responsible for the antibody's effector function, which induces cytotoxicity through antibody-dependent cell cytotoxicity and complement-dependent cytolysis [135]. The effector function is in fact the predominant mechanism of toxicity for some targeted biologics, such as trastuzumab, and is abolished when utilizing antibody fragments devoid of Fc regions. In a thorough molecular imaging study, Sexton et al. compared the differential tumor distribution and biodistribution of an EGFR-specific Affibody® peptide (ca. 7 kDa) with the anti-EGFR antibody cetuximab (ca. 150 kDa) [118]. The authors found that the total tumoral concentrations of the IRDye800CW®-Affibody® peptide conjugates and IRDye680RD®-cetuximab antibody conjugates were equal, although intratumoral distributions varied. At the tumor margin, 72% of the probes present was the IRDye800CW®-Affibody®, whereas at the tumor interior, 55% of the probes present was the IRDye680RD®-cetuximab. Plasma clearance rates of the two probes were also size-dependent, where 66% of the IRDye800CW®-Affibody® probe was cleared within 1 min following intravenous injection, while only 37% the IRDye680RD®-cetuximab probe had been cleared at this time point. Thus, depending on the specific therapeutic need, photoactive targeted probes with varying pharmacokinetics and biodistribution properties may be selected to provide specific needs. For example, if fluorescence guidance of tumor resection is used and followed by photodynamic eradication of residual disease, then a targeted probe such as an Affibody® or an antibody fragment would be optimal for its superior ability to delineate and penetrate the tumor margin. Conversely, if whole tumor PDT is required to treat non-resectable neoplasia, a larger targeted probe such as a full length-antibody PIC with superior intratumoral distribution would be preferred. In addition, smaller molecular weight antibody fragments offer a certain advantage over full-length antibody conjugates, in that their faster physiological clearance rates shorten the time-frame necessary to achieve peak tumor levels [136]. For example, fragment antigen-binding (Fab) and F(ab)2 portions of antibodies have been shown to reach maximal tumor accumulation in 1-2 hrs, whereas a full-length IgG molecule requires 8 hrs to reach peak tumor concentrations following intravenous administration [136]. However, faster physiological clearance rates also limit the maximal tumor concentrations of the probe. Fab and F(ab)2 fragments have been shown to reach maximal tumor concentrations of 0.45% and 0.5% of the total injected dose, respectively, whereas full-length IgG antibodies reached 2.5% of the total injected dose, although requiring 6-7 hrs more to reach this maximal tumor accumulation [136]. Increased physiological exposure of targeted biologics by prolonging their serum half-lives is oftentimes desirable, as is the case for Etanercept, the therapeutic Tumor Necrosis Factor receptor fused to human IgG1 Fc fragments [137].
PICs composed of PSs conjugated to engineered antibody fragments devoid of Fc regions have also been thoroughly reported in the literature [138-141]. For example, Single-domain antibody fragments called nanobodies that are selective for EGFR have been conjugated to the PS IRDye700DX® and used for in vivo PDT of head and neck cancers [141]. The authors compared a monovalent nanobody PIC (15 kDa) with a biparatopic internalizing nanobody PIC (30 kDa) and found that the biparatopic variant exhibited a two-fold higher selectivity as compared to the monovalent equivalent. Furthermore, tumor-to-normal selectivity of both the monovalent and biaparatpic PIC was found to be approximately 7.5-fold higher at 24 hrs following administration than at 1 hr. Although the biparatopic nanobody PIC was more phototoxic in vitro than the monovalent equivalent, results of the in vivo PDT were the opposite. The authors attributed the improved in vivo PDT efficacy and apparent increase in tumor penetration of the monovalent nanobody PIC to its smaller size. However, this experimental evidence might allude to the binding site barrier hypothesis where the bivalent nanobody PIC (equilibrium dissociation constant (Kd) = 1.0 nM) exhibits a higher receptor affinity than the monovalent PIC (Kd = 1.9 nM) and is thus less able to penetrate the tumor. The binding site barrier hypothesis states that an inverse correlation exists between the binding affinity of targeted probes and their penetration within target tissue [142]. Thus, a lower affinity for the target may improve tumor penetration of the targeted probe and its homogenous distribution within the tumor, at the expense of reduced in vivo selectivity. In contrast, an increase in avidity has been shown to improve the biological activity of targeted therapeutics at the cellular level, where the affinity of target binding positively correlates with treatment response [136]. Watanbe et al. reported a comparative study of the biodistribution and selectivity of three different engineered Prostate-Specific Membrane Antigen (PSMA) PICs generated from IRDye700DX® PS conjugates of a full-length antibody (150 kDa), a minibody (80 kDa) and a diabody (50 kDa) [143]. Interestingly, the authors found that the antibody PIC and the minibody PIC had the highest selectivity towards a PSMA-positive tumor at 24 hrs following administration, with respect to a contralateral PSMA-negative tumor. However, the largest intertumoral difference of the nanobody PIC was observed at 6 hrs after injection. Although the selectivity of the minibody PIC was approximately 5.7-fold and 6.7-fold higher than the antibody PIC and the nanobody PIC, respectively, the antibody PIC was the most effective at delaying tumor regrowth following PDT. Furthermore, PDT using the antibody PIC prolonged mouse survival more than PDT using the minibody and nanobody PICs. The experimental evidence suggests that the impact of molecular weight on the PIC's overall efficacy must be viewed in conjunction with the multiple other influential properties. Therefore, the size of a PIC, in combination with other biochemical properties of PICs, may be selected to perform specific tasks.
Electrostatic charge
Although the molecular weight of the targeted probe is a critical aspect of its biological properties and efficacy, the electrostatic charge of the conjugate can be equally impactful. Hamblin et al. prepared anionic and cationic charged PICs from the IgG murine monoclonal antibody 17.1A that is specific for the Ep-CAM cell adhesion antigen found on HT-29 colorectal carcinoma cells and compared their biodistribution [132]. Following partial reduction of the hinge region disulfides, 17.1A was site-specifically conjugated with either a chlorin e6-bearing cationic polylsine carrier, or its succinylated anionic derivative. At 3 hrs following intravenous injection, the anionic 17.1A PIC delivered 12.58% of the total injected PS dose/g tissue to the tumor, whereas the cationic 17.1A PIC only delivered 2.27% of the injected dose/g tissue, which is in fact marginally lower than the tumor accumulation of the free PS (3.77% of the total injected dose/g tissue). Although the tumor-to-liver selectivity of the cationic and anionic 17.1A PIC was identical, the tumor-to-skin selectivity of the anionic PIC was approximately 1.68-fold higher than the cationic conjugate. These findings emphasized the drastic effects of electrostatics on PIC pharmacokinetics, where anionic PICs improve tumor delivery of the PS by 5.5-fold, as compared to the cationic counterpart; however, tumor selectivity was improved by only 1.68-fold [132]. Duksa et al. investigated the biodistribution of PICs formed of OC125 F(ab)2 fragments site specifically conjugated to cationic and anionic polylysine carriers of the PS chlorin e6 following intraperitoneal administration [139]. Contrary to previous findings from the aforementioned biodistribution study of cationic and anionic full-length antibody PICs [132] Duska et al. found that the cationic OC125 F(ab)2 PIC delivered 4.1-fold more PS to the ovarian cancer tissue than the anionic equivalent at 3 hrs following intraperitoneal injection. At 24 hrs following intraperitoneal administration, the cationic PIC delivered 4.5-fold higher amounts of PS that the anionic PIC. These findings underscore the impact of the electrostatic charge on the PIC's biodistribution and tumor selectivity, and how this impact is also dependent on other factors mentioned in this section such as route of administration and molecular weight of the probe. These findings emphasize that an accurate comparison of probe electrostatics can only be conclusive for the specific size of the probe used and for that specific route of administration.
Degree of probe conjugation
Importantly for PDT, the number of PSs conjugated per targeting probe has also been shown to exert a marked impact on its pharmacokinetics. Vrouenrats et al. investigated the pharmacokinetic behavior of a radiolabeled chimeric Ig anti-epican antibody, 125I-cMAb U36, conjugated to an amine-reactive tetrafluorophenyl ester of the porphyrin derivative TrisMPyPFCO2H [144]. The antibody-to-PS ratio was varied from 1:1.2, 1:2.1 and 1:3.0; the PIC was unstable and aggregated when 4 PS molecules were conjugated to each antibody. Tumor uptake was reduced when more PSs were conjugated to each antibody as HNX-OE head and neck mouse tumors were found to have 15.5, 8.6, 6.5, and 4.0 % injected dose (ID)/g tissue at 48 h following administration of the unconjugated, 1:1.2, 1:2.1 and 1:3.0 PICs, respectively. The reduced tumor delivery could be attributed to decreasing circulation half-lives as the degree of PS conjugation increased. At 48 h following injection, the 1:1.2, 1:2.1 and 1:3.0 PICs were found in the blood at 58%, 42%, and 26% of the unconjugated antibody in circulation, respectively. The expedited clearance rates with increasing degree of PS conjugation to the PICs did not correspond to higher tumor-to-normal ratios; on the contrary an inversely proportional relationship was observed. Interestingly, it has been shown that antibody-mediated agent delivery in tumors does not increase the total tumor uptake of the probes, as a sham non-selective IgG antibody accumulates in the tumor to the same degree as a tumor-specific IgG [136]. However, a stark difference in receptor binding and therapeutic efficacy is observed between the targeted antibody and the non-selective IgG. Activatable variants of the BPD-cetuximab PIC were developed by Savellano et al. who demonstrated that an increase in the degree of PS conjugation to the antibodies increased the static quenching of the BPD, reducing the photoactivity [145]. Spring et al., then utilized the quenched 7:1 BPD:cetuximab PICs, which exhibited a 7-fold quenching in photoactivity whilst intact, for the selective, tumor-specific activatable PDT of ovarian micrometastasis [146]. The activatable PIC leverages the specific cellular discrimination of antibody conjugates, regardless of total tumor concentration. The capacity for the tumor-activatable PIC to become activated following specific cancer cell binding, internalization and a proteolytic degradation adds further selectivity to PDT treatments that utilize targeted probes. In the presence of the micrometastatic ovarian tumors on the peritoneal wall, the intraperitoneally administered PIC selectively binds to the cancer cells, is internalized and activated (Fig. 7A). In the absence of disease, no fluorescence corresponding to the PIC is observed. Furthermore, the importance of selectivity is exemplified in this study by the absence of BPD in tumor-free colonic tissue when ovarian carcinoma-bearing mice are injected with the PIC (Fig. 7B). However, the non-selective free BPD accumulated in both the disease tissue, and the sensitive non-diseased colonic tissue. PDT using the tumor-activatable PIC in the peritoneal model of ovarian micrometastases allowed for a 17-fold increase in the tolerable PDT dose [146]. This allowed for a more complete PDT treatment procedure that minimizes off-target toxicity. Expanding this concept to solid tumors will improve cellular selectivity of deep tissue PDT, thus increasing the probability of complete tumor cell eradication, whilst retaining the viability of healthy tissue. Activatable antibody-based probes merge the concept of targeted PS delivery with tumor-selective activation, which is another mechanism for a class of targeted PDT probes that will be discussed in the next section.
A. In vivo microendoscopy of murine ovarian micrometastatic carcinoma showing selective binding and fluorescence activation of the activatable Cetuximab-BPD PIC upon cellular internalization and proteolytic digestion. Activation is highest at 8-24 hrs following intraperitoneal administration in disease-bearing mice, whereas activation of the PIC is not observed in the “no tumor” control mice (scale bars are 100 μm). B. Peritoneal distribution of BPD fluorescence in mice with disseminated ovarian tumors administered with free BPD (top) or the activatable Cetuximab-BPD PIC. The PIC (bottom) demonstrates selectivity and little activation at disease-free organs, such as the colon (white dotted line), as compared to the non-specific binding of free BPD (top). Figure adapted from Spring et al [146].
Probes for selective tumor activation
Photodynamic molecular beacons
Selectivity towards the target tissue that will be treated with PDT can be also imparted through the inherent biochemical lability of customized probes that are designed to be enzymatically or chemically cleaved, and thus activated, at the required site of action (Fig. 6B). Therefore, targeted PDT is no longer confined by selective tumor delivery and localized photo-irradiation, but is also regulated by tumor biochemistry and microenvironment. An archetypal example of such activatable technologies is referred to as 'molecular beacons', initially designed as sensors for biochemical binding and enzymatic processes. Matayoshi et al. were the first to report fluorogenic substrates for sensing HIV-1-encoded proteases that cleaved and activated the molecular beacon by recovering the fluorescence of an intramolecular FRET pair by up to 40-fold [147]. Such molecular beacons have evolved over the last two decades and have served as optical sensors of the tumor microenvironment and subsequently, tumor-selective photoactive therapeutics with the critical need to utilize NIR-absorbing PSs, as reviewed by Lovell et al [8]. Chen et al. were the first to report a photodynamic molecular beacon (PMB) consisting of a peptidic substrate of the apoptotic mediator caspase-3, flanked by the NIR PS pyropheophorbide a and a carotenoid singlet oxygen scavenger [148]. The team reported the solid-phase synthesis of a DEVD-containing peptide sequence, followed by the modular peripheral conjugation of the pyropheophorbide a and the distal liquid-phase conjugation of the carotenoid 1O2 scavenger. Caspase-3 hydrolysis of the peptide linker resulted in a 4-fold increase in 1O2 production, as measured by the phosphorescence emission at 1270 nm.
Matrix metalloprotease substrates
The technology was further expanded by developing a FRET-based PMB, which was activated by matrix metalloprotease-7 (MMP-7), an enzyme involved in tumor invasion and metastasis by the degradation of extracellular matrix and connective tissue [149]. The MMP-7 peptidic substrate was also prepared using solid phase synthesis and flanked by a pyropheophorbide PS and its respective FRET-based quenched, Black Hole Quencher 3 (BQH3). Within 3 hrs of incubation with MMP-7, the molecular beacon was reported to exhibit a 12-fold recovery of PS fluorescence, and more importantly for a PMB, a 19-fold increase of 1O2 generation following proteolysis. Extracellular cleavage by MMP-7 does not guarantee internalization of the PS cleavage product and phototoxicity. However, using confocal microscopy, the study confirmed that KB cells (MMP-7+) incubated with the PMB for 5 hrs, resulted in a significant intracellular presence of the pyropheophorbide cleavage product, with apparent mitochondrial localization. Conversely, BT20 cells (MMP-7-), minimal fluorescence corresponding to the PS cleavage product was observed. Furthermore, selective MMP-7 dependent activation of the PMB and the intracellular fate of the PS cleavage product directly translated into a selective photodynamic effect. The viability of KB cells (MMP-7+) incubated with 2.5 μM of the molecular beacon was reduced to almost 0% following irradiation at 7.5 J/cm2, whereas BT20 cell (MMP-7-) viability was essentially unaltered at the same irradiation conditions, even up to 4 μM of the probe. In vivo, there was some evidence of selective activation of the molecular beacon in the subcutaneous KB tumor, and following PDT the tumor volume regressed. However, the majority of the fluorescence signal originated from what appeared to be the liver, possibly through proteolytic digestion of the probe. The same team led by Gang Zheng very recently published a study presenting the compelling efficacy of a multi-MMP PMB with proven in vivo efficacy against vertebral metastases [150]. Treatment selectivity at such critical physiological sites is of the utmost importance, where preservation of the nervous system is a considerable challenge. The multi-MMP peptidic substrate GPLGLARK was flanked as before by the NIR PS pyropheophorbide and BQH3. PDT treatment using the MMP molecular beacon reduced tumor growth in vertebrate metastasis rat model by approximately 2-fold, significantly reduced numbers of tumor cells and osteoclasts, and increased the number of apoptotic cells, as compared to the respective controls.
Non-cleavable probes
Although not cleavable, an elegant alternative system is the more conventional oligonucleotide-based molecular beacon platform, typically utilized as genetic biosensors. More recently, the oligonucleotide molecular beacon technology was described in a photodynamic system, where complimentary nucleic acid binding to target-specific DNA sequences activates the photoactivity of the probe [151]. A 3' pyropheophorbide PS and 5' BlackBerry quencher phosphoramidite molecules, either as a monomer, duplex or triplex, flanked the nucleic acid sequence. Upon binding to the target DNA sequences, the molecular beacons were activated 30-fold, 40-fold and 100-fold when single, duplex or triplex quencher molecules were tethered. Likewise, 1O2 generation was recovered when the oligonucleotide beacon was exposed to the target DNA sequence. However, target DNA binding did not result in the maximal potential de-quenching of 300-fold when 3 quenchers molecules were tethered, underscoring a significant limitation in using hydrophobic FRET-pairs, as is typically the case for PMB. The in vitro and in vivo efficacy of this nucleic acid system was not explored further.
Non-oncological probes
Although not described for the treatment of cancer, enzymatic cleavage as a mediator for selective activation of PSs has also been deployed for antimicrobial PDT of antibiotic-resistant strains. We have previously reported the development of a photodynamic chemical probe susceptible to enzymatic cleavage by β-lactamase, the enzyme responsible for bacterial resistance to β-lactam antibiotics [152]. A cephalosporin, 7-amino-3-chloromethyl-3-cephem-4-carboxylic acid p-methoxybenzyl ester, containing a β-lactam ring was peripherally modified with two molecules of the PS 5-(4'-carboxybutylamino)-9-diethylaminobenzo[a]phenothiazinium chloride (EtNBS-COOH). Conjugation of two EtNBS molecules each to cephalosporin resulted in a 5-fold quenching of the PS. When cleaved by β-lactamase expressed by methicillin-resistant Staphylococcus Aureus (MRSA), the PSs were spatially separated and the fluorescence was dequenched. The EtNBS cleavage products were also capable of exerting a 76-96% photodynamic reduction in viability of multiple clinical MRSA strains following irradiation.
A major limitation of cleavable probes is their off-target non-specific cleavage or hydrolysis, which renders them ineffective and reduces the signal-to-noise ratio. Furthermore, in the context of deep tissue-PDT for cancer, sufficient intratumoral concentrations of small molecular weight probes can be limited by suboptimal pharmacokinetics, which may necessitate their improved delivery by multiple means, such as PEGylation [153]. Without altering the probe to promote cellular internalization, photodynamic molecular beacons are also restricted to extracellular activation, given that the cleavage product with PS is cell-permeable. Alternatively, intracellular cytosolic targets required for the activation of molecular probes can be reached through the conjugation of the probe to a cell penetrating peptide, or by internalization using nano-sized delivery vehicles, all of which are hindered their own limitations [154]. Stacking of hydrophobic macrocyclic photosensitizers with each other or with their respective FRET pairs can also limit the extent of dequenching following target-specific cleavage, and therefore requires the careful selection of PSs with appropriate opto-chemical properties.
Harnessing local and systemic biological responses to enhance deep-tissue PDT efficacy
The efficacy of PDT for disease sites at depth is limited by biophysical barriers such as hypoxia, poor vascularization, and necrotic tissue, features commonly observed in medium to large solid tumors. As a way of circumventing these limitations, several studies have been performed to exploit the secondary effects of PDT or PDT-based combination therapies to impact disease biology beyond the reach of light alone, thereby extending the therapeutic penetration to typically inaccessible disease sites. This section will discuss a number of approaches that impact disease at depth via PDT-induced secondary effects, such as immune stimulation or mechanistically cooperative PDT-based combination regimens.
PDT Induced Immune Stimulation
PDT impacts, and in some cases stimulates, the immune system through several mechanisms. When PDT damages cells through a combination of cytotoxic mechanisms, [155] endogenous intracellular molecules known as Damage Associated Molecular Patterns (DAMPs) are activated to repair treatment-induced damage within cells [156]. Intracellularly, DAMPs, such as heat-shock-protein 70 (HSP70), play an important damage control function in which they serve as chaperone proteins for impaired and misfolded proteins, thereby inhibiting stress induced apoptosis and aggregation of intracellular proteins. This pro-survival function is an important resistance pathway following PDT [157]. However, if cells are damaged beyond the point of repair, DAMPs, which are highly immunogenic, can be released into the extracellular space. HSP70 has been shown to bind to antigen presenting cells (APCs) and facilitate antigen cross presentation, leading to maturation of dendritic cells and activation of CD8+ cytotoxic T cells [158]. In addition, PDT-induced release of other heat shock family proteins, such as HSP47, 60, and 90, has been shown to be partially responsible for sensitization of APCs to tumor cells [159, 160]. Other DAMPs that have been implicated in sensitizing the immune system to tumor cells include calreticulin, phasphatidylserine, adenosine triphosphate, peroxiredoxin 1, HMGB1, BCL-2, and annexin A1, among others [157, 161-165]. Taken together, these results indicate a mechanism by which PDT-induced oxidative stress leads to cell death via necrosis or apoptosis, causing DAMP release and subsequent activation of the innate immune inflammatory cascade. Through additional mechanisms discussed below, mediators of the innate immune system are sensitized and primed to exhibit systemic anti-tumor activity in areas beyond the initial localized PDT target site [166, 167].
PDT based activation of the innate immune system is comprised of non-specific inflammation and secondary cytokine release leading to phagocyte recruitment and complement activation. Among the many cytokines that may be activated by PDT, interleukin (IL)-1 and IL-6 are thought to be of critical importance. They both serve as chemoattractants for various immune cells including neutrophils, phagocytes, and lymphocytes [168]. Neutrophil infiltration occurs within 5 minutes after PDT, and is mediated partially by IL-1 and IL-6 in addition to E-selectin expression by cells at the site of inflammation [169]. Several studies highlighted the importance of neutrophils in mediating PDT-induced cytotoxicity by demonstrating that PDT efficacy is reduced in neutrophil depleted tissues [170]. Furthermore, the application of granulocyte macrophage colony-stimulating factor (GM-CSF), which serves to increase neutrophil counts in tissues, potentiates overall PDT efficacy [171]. Cecic et al. also demonstrated the importance of the complement anaphylatoxin, C3a, in contributing to PDT-induced neutrophilia [172]. In this study, injecting a mouse model of fibrosarcoma with antibodies blocking the C3a receptor (C3aR) led to the inhibition of neutrophil accumulation in photofrin-PDT treated tissues.
The effects of PDT on the innate immune system suggest that PDT can lead to immune sensitization and prolonged anti-tumor immunity, even after the cessation of therapy [173]. Induction of anti-tumor immunity could significantly improve treatment outcomes, especially in deep tissue where the direct cytotoxic effects of PDT are oftentimes proportional to light penetration depths. Canti et al. published one of the first studies demonstrating the beneficial effects of PDT-induced anti-tumor immunity. Mice bearing MS-2 fibrosarcomas underwent a curative regimen comprising of PDT with the PS aluminum pthalocyanine disulfonate in addition to surgery. Upon tumor re-implantation following therapy, immunocompetent mice exhibited greater survival than immunosuppressed mice, suggesting a critical role of the immune system in yielding durable outcomes following treatment [174]. Another study in syngenic BALB/c mice demonstrated a 100% cure rate following photofrin-PDT of EMT6 mammary sarcomas. However, no long-term cures were observed in non-obese diabetic or severe combined immunodeficiency (SCID) nude mice. Transplantation of splenic T-lymphocytes from the immunocompetent mice to the SCID mice pre-PDT led to a delay in tumor recurrence. Furthermore, transplantation of a mixture of splenic T-lymphocytes and B-lymphocytes from the PDT treated immunocompetent mice to the SCID mice led to a 100% cure rate following PDT. The authors demonstrated the specificity of PDT-induced anti-tumor immunity by showing that transplantation of splenic T and B lymphocytes to SCID mice undergoing X-ray based radiation therapy did not lead to cures. Taken together, this study demonstrates that the efficacy of PDT-induced anti-tumor immunity relies both on innate and humoral immunity, and is a significant determinant of the overall outcome following photofrin-PDT [175]. Finally, BPD-PDT of poorly immunogenic mouse sarcomas (RIF1) in immunocompetent C3H/HeN mice led to the disappearance of tumors followed by an eventual recurrence. The authors then transfected the RIF-1 tumors with green fluorescent protein (GFP) of jellyfish origin, hypothesizing that tumors bearing a foreign protein would subsequently increase the tumors' visibility to the host's immune system. Indeed, BPD-PDT of the GFP transfected RIF1 tumors led to a 100% cure rate, suggesting that the GFP served as an immune sensitizing antigen that promoted sustained anti-tumor activity by the host immune system [176].
The ability of PDT to induce systemic anti-tumor immunity via tumor cell death and subsequent immune sensitization has led to several studies exploring the development of PDT generated anti-tumor vaccines. This technique is based on the generation and injection of tumor cell lysates following PDT ex vivo. Several studies demonstrated a robust anti-tumor immunity following immunization with PDT treated tumor cells, including EMT6 cells (human mammary sarcoma), P815 cells (mouse mastocytoma), and SCCVII cells (mouse squamous cell carcinoma) [157, 177, 178]. The mechanism underlying antitumor immunity likely relies heavily on DAMP release following PDT-induced cytotoxicity. Importantly, cells treated with Ultra-violet (UV) or ionizing radiation do not induce tumor immunization to the same extent as PDT, demonstrating the specificity of PDT in sensitizing immune cells to untreated tumor cells situated in deep tissue [157].
In addition to a PDT-induced systemic immune response, the impact of PDT on the microvasculature of pathologic tissues also has potential to induce therapeutic effects in deep tissue. PDT-induced endothelial damage leads to vessel contraction, vascular leakage, and basement membrane exposure [179]. In addition to serving as a potent signal for platelet aggregation via the coagulation cascade, basement membrane exposure leads to thromboxane release as well as complement cascade activation, thereby contributing to the acute inflammatory effects of PDT described above. The destructive effects of PDT on the vasculature, termed microvessel shutdown, have been exploited in therapeutic applications as a way to induce hypoxia in tumor or diseased tissue [180, 181]. The mechanism underlying PDT-induced microvessel shutdown may involve PDT based inhibition of nitric oxide (NO) release, a vasodilatory molecule, leading to local hypoxia. A study by Korebelik et al. suggested that PDT-induced inhibition of NO led to enhanced cure rates in endogenous NO producing tumors, indicating that the beneficial effects of PDT-induced vascular shutdown can be abrogated with vasodilation. In addition, tumors with low endogenous production of NO were more sensitive to Photofrin-PDT, and addition of NO inhibitors did not improve outcomes [182]. Local hypoxia at the target site coupled with PDT-induced oxidative stress to microvasculature via superoxide anion formation creates an area of tissue ischemia, which serves as a potent stimulator of complement activation and neutrophil infiltration, further enhancing the PDT-induced innate immune response in addition to creating areas of hypoxia at depth [176, 182]. The mechanistic benefits of PDT-induced vascular shutdown as part of a combination therapy are discussed further below.
PDT Based combination therapies
It is increasingly evident that the next-generation of cancer therapeutics will comprise of combinatorial approaches that overcome intrinsic resistance pathways via non-overlapping and interactive mechanisms that enhance overall survival and reduce recurrence rates. PDT forms the basis of promising anti-cancer regimens because the mechanisms underlying photodamage are distinct from most chemo-, radio-, or biological therapies. Therefore, PDT based combination regimens can improve outcomes due to the potential for mechanistic cooperativity, wherein the effects of PDT potentiate the effects of subsequent treatments and vice-versa, leading to synergistic reductions in tumor volume while maintaining a manageable toxicity profile. Due to these advantages, rationally designed PDT based combination therapies may help to overcome light-based limitations posed by deep tissue because the effects of secondary, non-light based, therapies can lead to increased cytotoxicity in target tissues that were either sub-optimally or minimally impacted by PDT. A selected series of studies demonstrating the potential for rationally designed PDT based combinations to improve outcomes in deep tissue are discussed below.
Numerous studies have demonstrated that the effectiveness of light-based combinations depends on mechanistic interactions between PDT, the subsequent therapy, and the disease being treated. In order to achieve synergism and impart an improved efficacy in deep tissue, PDT and the secondary therapy must induce interactive cytotoxic mechanisms in the target tissue whose consequences are greater than the additive effects of the individual modalities. Furthermore, the target tissue itself must also exhibit an amenable biological landscape to support any mechanistic cooperativity. This principle was highlighted as early as 1988, when Nahabendian et al. demonstrated disease and treatment specific interactions with HpD-PDT combined with either doxorubicin or cisplatin. It was found that treating BALB/c mice bearing EMT-6 tumor xenografts with HpD-PDT combined with doxorubicin led to a synergistic reduction in tumor burden and increased the depth of killing within the tumor. However, combining HpD-PDT with cisplatin led to an additive effect, and the authors noted that a significant proportion of the tumor was unaffected at depth for this regimen. In addition, RIF-1 tumors were not sensitive to either of the HpD-PDT based combinations, highlighting the importance of considering both the combination therapy as well as the disease being treated when designing comprehensive treatment regimens [183]. The sequence of component therapies in a larger combination also impacts the overall efficacy within deep tissue. For example, Zuluaga et. al. showed that mitomycin C treatment of mouse RIF-1 subcutaneous tumors prior to HpD-PDT was more effective at reducing tumor burden than vice-versa [184]. This finding has been echoed in a study published by our group demonstrating that BPD-PDT combined with carboplatin exhibited a sequence dependent synergism. Treating three-dimensional (3D) ovarian cancer nodules with carboplatin prior to BPD-PDT resulted in only an additive therapeutic effect, whereas a synergistic effect was observed when BPD-PDT preceded carboplatin [185]. This sequence dependent synergism was because BPD-PDT led to a disruption of the 3D nodules allowing a better penetration of carboplatin into the nodules (Fig. 8). These selected studies highlight the importance of considering both the biology of the target tissue to be treated as well as the sequence/composition of the regimen to be applied when designing PDT based combination therapies to yield efficacy in deep tissue.
Carboplatin poorly penetrates three-dimensional ovarian cancer nodules as revealed by a ring of dead cells (ethidium bromide - red) surrounded by a largely intact cluster of live cells (calcein - green) following carboplatin treatment (top image and graph). BPD-PDT disrupts the nodule structure, and may potentiate carboplatin therapy and enable increased therapeutic efficacy in deep tissue because of increased tumor cell surface area exposure to follow up chemotherapeutics (bottom image and graphs). Figure adapted from Rizvi et al [185].
An important advantage of PDT stems from its ability to sensitize tumors to follow up treatments that are individually ineffective due to intrinsic resistance pathways expressed by cancer cells. Because many of these secondary therapies can penetrate more deeply than light, there is significant opportunity to design combination therapies whereby initial cycles of PDT sensitize tumors to conventional chemo- or biologic therapies that then lead to enhanced eradication of tumor tissue at depth. Celli et. al. demonstrated that several pancreatic cancer cell lines (AsPC-1, BxPC-3, PANC-1, Capan-1, and Capan-2) exhibit baseline gemcitabine resistance that can be overcome via pre-treatment with BPD-PDT. The assumption made in that study is that BPD-PDT decreased expression of Bcl-XL, an anti-apoptotic protein, and shifted the Bax/Bcl-XL ratio towards a more pro-apoptotic balance, thereby creating an environment for enhanced gemcitabine efficacy [186]. In another study, platinum resistant ovarian cancer cells (OVCAR-5) pretreated with a PIC (anti-ovarian cancer antibody OC125 conjugated to chlorin e6-monoethylenediamine monamide as the PS) were resensitized to platinum therapy following PDT, suggesting that light based modalities have the ability to favorably modulate intrinsic tumor resistance pathways such that follow up therapies are more effective [187].
A significant barrier to deep tissue therapy stems from an inability to achieve sufficient drug delivery to these relatively inaccessible sites. PDT enhances vascular permeability leading to increased tumoral accumulation of macromolecular therapeutic agents [188]. Snyder et. al. showed that PDT treated tumors exhibited increased intratumoral, liposomally (0.1 μm diameter) encapsulated, doxorubicin compared to non-treated tumors [189]. This enhanced accumulation at tissue depths greater than those typically accessible by PDT significantly potentiated efficacy of both PDT and doxorubicin in combination, and little to no systemic toxicity was observed [189]. In addition, our recent studies have demonstrated that pretreatment with liposomal BPD (L-BPD)-PDT significantly increases the intracellular concentration of liposomal irinotecan (L-IRI) in vitro and improves localized tumor control in orthotopically implanted pancreatic cancer xenografts [180]. In addition to enhanced irinotecan accumulation, the study also showed other cooperative interactions with L-IRI to synergistically reduce tumor burden. L-IRI is a topoisomerase I inhibitor that binds topoisomerase-DNA complexes and prevents DNA religation, causing DNA strand breaks and apoptosis. Resistance to irinotecan is often mediated by ATP binding cassette sub-family G member 2 (ABCG2) efflux pumps expressed in the cell membrane and organelle membranes. Low dose L-BPD-PDT was shown to decrease expression of these efflux pumps, causing an increase in the intracellular L-IRI concentration, thereby leading to enhanced cell killing. Furthermore, L-IRI treatment led to the decreased expression of monocarboxylate transporter 4 (MCT-4), a prognostic indicator of pancreatic cancer progression that was shown to be upregulated in tumors treated with PDT alone. In a separate study, Gallagher-Colombo et. al. demonstrated that pretreating tumor xenografts with erlotinib, a small molecule inhibitor of EGFR, prior to BPD-PDT significantly extended the survival of mice compared with those treated with BPD-PDT or erlotinib alone [190]. The authors postulate that multiple mechanisms contributed to the potentiation of BPD-PDT efficacy. Not only was it observed that the intratumoral concentration of BPD was significantly greater following erlotinib treatment, but erlotinib also induced mild endothelial cell cytotoxicity that was likely potentiated by subsequent BPD-PDT, leading to significant vascular shutdown and enhanced tumor death. The results of these studies suggest a paradigm by which the individual components of a combination regimen mechanistically potentiate the effects of the combined therapy. Furthermore, these studies highlight the utility of PDT in improving intratumoral chemotherapy concentrations, in part by enhancing vascular permeability and improving chemotherapy diffusion into deep tissues, but also by interfering with the ability of target cells to efflux these drugs. As a result of these attributes, PDT-based combination must be carefully designed to simultaneously overcome modes of resistance to increase cytotoxicity at deep tissue while still maintaining a favorable systemic toxicity profile.
Significant efforts have been devoted towards applying PDT based combination therapies to target the tumor-associated vasculature as a way of inducing cancer cell cytotoxicity secondary to hypoxia at deeply residing tumors. In a selected example, Nowak-Sliwinska et. al. investigated the efficacy of a panel of angiostatic agents (sorafenib, erlotinib, sunitinib, and bevacizumab) in enhancing visudyne-PDT induced vascular shutdown [191]. Using a chorioallantoic membrane (CAM) model from a chicken embryo, it was found that PDT alone allowed significant regrowth of microvasculature two days after treatment, but PDT in combination with any of the anti-angiogenic agents led to sustained shutdown of microvasculature. Visudyne-PDT combined with sorafenib, which inhibits several tyrosine kinases including vascular endothelial growth factor receptor (VEGFR), platelet derived growth factor receptor, and raf family kinases, led to the most durable shutdown of microvasculature of all angiostatic agents tested in terms of reducing the number of vascular branching points and vessel density 48 hrs after treatment. Because several studies have demonstrated an important role for microvasculature shutdown in enhancing PDT-induced cytotoxicity and necrosis, this study along with several others demonstrate the importance of intelligently pairing angiostatic agents with PDT to impart greater tumor killing in deep tissue.
The benefits of multi-pathway inhibition to potentiate PDT efficacy have been well studied over the past two decades. In addition to limitations stemming from depth, a major hurdle in the treatment of solid malignancies stems from the ability of tumors to escape treatment through resistance pathways [192, 193]. To address this loophole, there has been recent interest in packaging synergistic multi-modality therapies with non-overlapping toxicities in nanoconstructs such that their release can be spatiotemporally controlled to synchronously inhibit these treatment escape pathways as they are expressed by tumors (Fig. 9). Spring et. al. have demonstrated the synthesis and characterization of a photoactivatable multi-inhibitor nanoliposome (PMIL) that contains the PS BPD in the lipid bilayer and encapsulates cabozantinib (XL184), an inhibitor of both the MET and VEGFR signaling pathways [194]. MET is the receptor tyrosine kinase for hepatocyte growth factor that promotes cancer cell motility, invasion, and metastatic escape in response to hypoxic conditions, which are characteristically seen in response to PDT-induced vascular shutdown and common VEGFR inhibition treatments. NIR excitation of BPD in the lipid bilayer of the PMIL leads to cabozantinib release such that target tissues experience simultaneous phototoxicity via BPD-PDT followed by a sustained inhibition of tumor survival pathways. It was found that a single cycle of the PMIL treatment led to a sustained reduction in both subcutaneous and orthotopic mouse models of pancreatic cancer. Notably, mouse tissues that did not undergo light irradiation, such as the liver and lymph nodes, also exhibited significant reductions in metastatic disease compared to completely untreated mice, suggesting that the secondary effects of BPD-PDT or systemic release of cabozantinib following light irradiation led to efficacy at distant sites. It also may be possible that localized anti-cMET therapy inhibits metastasis from the primary tumor. In another related study, photactivatable liposomes also containing BPD in the lipid bilayer but instead encapsulating bevacizumab significantly enhanced tumor reduction in a subcutaneous (AsPC-1 cells) mouse model of pancreatic cancer [195]. The strength of this approach lies in the mitigation of the pro-survival VEGF burst that is known to follow localized PDT [196]. Taken together, these findings have significant implications for treatment of deep tissue because multi-modal therapies formulated in a single package can impart cytotoxicity in deep tissue due to the incorporation of non-light dependent therapies that mechanistically and temporally cooperate with localized PDT.
Photoactivatable multi-inhibitor nanoliposomes (PMIL) enable synchronous suppression of survival pathways following PDT to enhance tumor cell death. Cartoon representation and cyro-electron microscopic (cryo-EM) image depicting a PMIL (A). These PMILs encapsulate cabozantinib (white arrow in the cryo-EM image), a multi-kinase inhibitor of the c-MET and VEGF signaling pathways in tumors (C), that is released upon light activation of BPD (B) in the PMIL bilayer (also denoted by a white arrow in the cryo-EM image in A). Light activation of PMILs leads to reduction of distant metastatic tumor burden in an orthotopic model of pancreatic cancer despite localized light irradiation (D). Figure adapted from Spring et al [197].
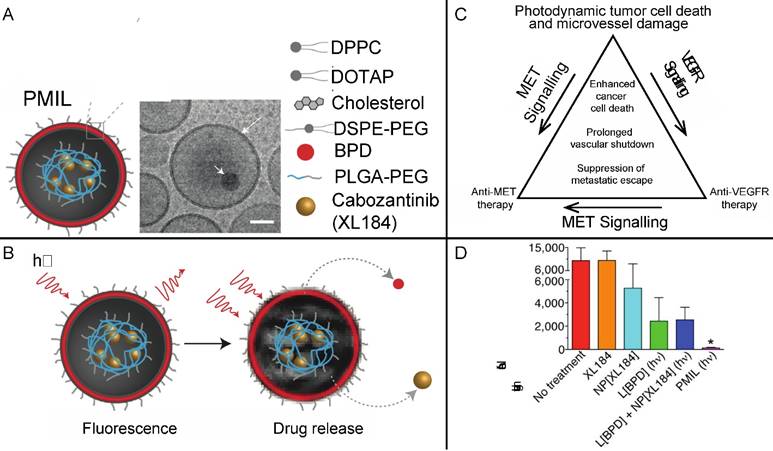
Summary and future perspectives
PDT is a light activated therapeutic modality that has demonstrated efficacy in many applications including, but not limited to, ophthalmology, oncology, infectious diseases, and cosmetic treatments [3, 13, 17, 197-201]. Particularly in oncologic applications, PDT efficacy in some cases is on par with and sometimes exceeds that of surgery, chemo or radiation therapy. Moreover, PDT lends itself to spatial and temporal control, thereby offering advantages such as reduced long-term morbidity and decreased systemic toxicities compared to standard chemo- or radiotherapy. Despite these advantages, PDT has largely been restricted to clinical trials or palliative adjunct treatments and its widespread acceptance as a mainstream clinical modality is hindered primarily due to following reasons 1. The need to coordinate consistent delivery of light to potentially heterogeneous treatment sites that can vary over many patients requires management and standardization of lasers, applicators, fiber optics, power meters etc. This is a particularly difficult task in context of deep tissue PDT because these already complicated technologies only increase in sophistication, creating a potentially expensive logistical barrier. 2. Heterogeneity in disease biology and topography necessitates customized, patient-specific, treatment planning that accounts for the size of the area to be treated, the light scattering or absorption properties of the target tissues, the depth to which the therapy must penetrate while sparring normal healthy tissue, 3. Intrinsic and extrinsic dosimetry parameters based on current clinically available imaging methods to gauge therapy insult in deep tissues need to be incorporated into treatment regimen and 4. Appropriate medical personnel such as radiation oncologist or medical physicist with some training in optical methods, image-guided fiber placement, dosimetry, and PDT and 5. Misconceptions centered on the limited penetration of PDT.
In addition to light penetration, several PS parameters such as selectivity, localization, quantum yield etc. are critical determinants of deep tissue PDT efficacy. Along with the challenges posed by optimizing these parameters, as discussed throughout this review, PS's also afford exciting opportunities to enhance the therapeutic efficacy of PDT because they are theranostic agents that induce photodynamic action and double as imaging contrast agents. Although changes in PS fluorescence have been correlated with PDT treatment efficacy in several studies, fluorescence imaging alone does not provide information in deep tissue due to limited light penetration. This highlights the need to exploit other imaging modalities, which still take advantage of the theranostic capabilities of PS at depth, that monitor relevant parameters such as photobleaching or oxygen saturation. Because no one imaging modality is likely to provide a sufficient amount of information to guide PDT at depth, there likely will be need to integrate several of these imaging methods into a single platform that can simultaneously monitor multiple parameters to guide therapy in deep tissues.
Parallel to the need for combined imaging platforms to monitor therapy at depth is the necessity to apply integrated multifunctional nanoparticles that can unify the biodistribution, pharmacokinetics and spatiotemporal synergy of both imaging and PDT based combination therapeutic modalities. There are two major criteria to facilitate translation of multifunctional nanoplatforms: therapeutic superiority and clinical safety. The introduction of any organic or inorganic nanomaterials into the body carries potential health risks, and thus substantial pre-clinical evidence showing an improvement in therapeutic efficacy versus the standard of care is required to justify their use. Nanoconstructs must demonstrate the capability to carry sufficient drug loads specifically to target tissue while retaining their simultaneous imaging capabilities. Furthermore, their pharmacokinetic profiles must ensure that the theranostic function has a tumor selectivity exceeding that of free agents. Some theranostic nanoconstructs that have been discussed in this review are based on radiologically or optically active inorganic nanomaterials, which have unknown long-term physiological effects. Of all nanomaterials explored to date, injectable Feraheme® (iron oxide nanoparticles) are the only ones currently approved by the FDA for iron deficiency. Even though it is approved, Feraheme® comes with warnings of extreme immune reactions and anaphylaxis. Similarly, inorganic nanomaterials with particularly attractive optical and photochemical properties, particularly for deep tissue PDT, may elicit unexpected reactions and tissue toxicities. Such examples include nanocrystals doped with heavy metal ions that are potentially hazardous. It is of paramount importance that physiological clearance of the nanomaterial to be used is definitively confirmed, their chemical and physical stability is maintained in the body, and that their theranostic utility does not negatively impact their systemic toxicity profile. Thorough safety analyses of the emerging theranostic nanotechnologies discussed in this review will inevitably precede any further clinical translation of multi-agent nanoplatforms for deep tissue PDT.
The traditional view that PDT is limited to superficial pathologies is being redefined with the emergence of new concepts and technologies in the realms of light delivery, PS delivery, and photodynamic activation as discussed in this review. As more and more PDT based modalities are moved into Phase II and III clinical trials, incorporation of personalized dosimetry (either implicit, explicit or surrogate) parameters integrated with clinically viable deep tissue imaging tools will become increasingly important to ensure durability of treatment response especially in deep tissues. Along these lines, newer platforms that deliver synergistic PDT based combination therapies and exploit the secondary biologic effects of PDT such as immune stimulation will likely gain traction in the coming years as they progress to the initial phases of the clinical translation. In the long term, the availability of biocompatible nanoparticles incorporating upconverting nanomaterials, radiosensitizers, and theranostic multi-functional agents, together with cleverly designed personalized light delivery strategies could substantially increase the footprint of PDT as a viable therapy that has significant impact in deep tissues and, more broadly, in enhancing overall outcomes.
Acknowledgements
This work was supported by NIH grants F32CA165881 (Mallidi), 5R01CA156177, R01 CA158415, R01-CA160998 (Hasan) and S10 ODO1232601 (Hasan) and funds from Canon USA Inc. Financial support by the Bullock-Wellman Postdoctoral Fellowship (Obaid) and Howard Hughes Medical Institute Medical Research Fellows Program (Anbil) is gratefully acknowledged. The authors would like to acknowledge Mr. Brian Battersby, Director of Computational Core, Wellman Center for Photomedicine for the help with cover art.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Parrish JA, Rosen CF, Gange RW. Therapeutic uses of light. Ann N Y Acad Sci. 1985;453:354-64
2. Jacques SL. Optical properties of biological tissues: a review. Phys Med Biol. 2013;58:R37-R61
3. Celli JP, Spring BQ, Rizvi I, Evans CL, Samkoe KS, Verma S. et al. Imaging and Photodynamic Therapy: Mechanisms, Monitoring, and Optimization. Chem Rev. 2010;110:2795-838
4. Lucky SS, Soo KC, Zhang Y. Nanoparticles in Photodynamic Therapy. Chem Rev. 2015;115:1990-2042
5. Rai P, Mallidi S, Zheng X, Rahmanzadeh R, Mir Y, Elrington S. et al. Development and applications of photo-triggered theranostic agents. Adv Drug Deliv Rev. 2010;62:1094-124
6. Chatterjee DK, Fong LS, Zhang Y. Nanoparticles in photodynamic therapy: an emerging paradigm. Adv Drug Deliv Rev. 2008;60:1627-37
7. Huang H-C, Hasan T. The “Nano” World in Photodynamic Therapy. Austin Journal of Biomedical Engineering. 2014;2:1-4
8. Lovell JF, Liu TWB, Chen J, Zheng G. Activatable Photosensitizers for Imaging and Therapy. Chem Rev. 2010;110:2839-57
9. De Rosa FS, Bentley MVLB. Photodynamic Therapy of Skin Cancers: Sensitizers, Clinical Studies and Future Directives. Pharm Res. 2000;17:1447-55
10. Zeitouni N. Photodynamic therapy for nonmelanoma skin cancers. Current review and update. Mol Immunol. 2003;39:1133-6
11. Mennel S, Barbazetto I, Meyer CH, Peter S, Stur M. Ocular Photodynamic Therapy - Standard Applications and New Indications (Part 1). Ophthalmologica. 2007;221:216-26
12. Agostinis P, Berg K, Cengel KA, Foster TH, Girotti AW, Gollnick SO. et al. Photodynamic therapy of cancer: An update. CA Cancer J Clin. 2011;61:250-81
13. Allison RR. Photodynamic therapy: oncologic horizons. Future Oncol. 2014;10:123-4
14. Brown SB, Brown EA, Walker I. The present and future role of photodynamic therapy in cancer treatment. Lancet Oncol. 2004;5:497-508
15. Wilson BC, Patterson MS, Lilge L. Implicit and explicit dosimetry in photodynamic therapy: a New paradigm. Laser Med Sci. 1997;12:182-99
16. Sharma SK, Mroz P, Dai T, Huang Y-Y, Denis TGS, Hamblin MR. Photodynamic Therapy for Cancer and for Infections: What Is the Difference? Isr J Chem. 2012;52:691-705
17. Weersink RA, Lilge L. Advances in Photodynamic Therapy. In: (ed.) Hamblin MR, Mroz P. Advances in Photodynamic Therapy: Basic, Translational and clinical. Boston: Artech House. 2008:93-110
18. Jerjes W, Upile T, Alexander Mosse C, Hamdoon Z, Morcos M, Morley S. et al. Prospective evaluation of 110 patients following ultrasound-guided photodynamic therapy for deep seated pathologies. Photodiagnosis and Photodynamic Therapy. 2011;8:297-306
19. Bown SG, Rogowska AZ, Whitelaw DE, Lees WR, Lovat LB, Ripley P. et al. Photodynamic therapy for cancer of the pancreas. Gut. 2002;50:549-57
20. Huggett MT, Jermyn M, Gillams A, Illing R, Mosse S, Novelli M. et al. Phase I/II study of verteporfin photodynamic therapy in locally advanced pancreatic cancer. British Journal of Cancer. 2014;110:1698-704
21. Svanberg K, Bendsoe N, Axelsson J, Andersson-Engels S, Svanberg S. Photodynamic therapy: superficial and interstitial illumination. J Biomed Opt. 2010;15:041502
22. Thompson MS, Johansson A, Johansson T, Andersson-Engels S, Svanberg S, Bendsoe N. et al. Clinical system for interstitial photodynamic therapy with combined on-line dosimetry measurements. Appl Opt. 2005;44:4023-31
23. de Bruijn HS, Kruijt B, van der Ploeg van den Heuvel A, Sterenborg HJCM, Robinson DJ. Increase in protoporphyrin IX after 5-aminolevulinic acid based photodynamic therapy is due to local re-synthesis. Photochem Photobiol Sci. 2007;6:857-64
24. Rausch PC, Rolfs F, Winkler MR, Kottysch A, Schauer A, Steiner W. Pulsed versus continuous wave excitation mechanisms in photodynamic therapy of differently graded squamous cell carcinomas in tumor-implanted nude mice. Eur Arch Otorhinolaryngol. 1993;250:82-7
25. Kawauchi S, Morimoto Y, Sato S, Arai T, Seguchi K, Asanuma H. et al. Differences between cytotoxicity in photodynamic therapy using a pulsed laser and a continuous wave laser: study of oxygen consumption and photobleaching. Lasers Med Sci. 2004;18:179-83
26. Miyamoto Y, Umebayashi Y, Nishisaka T. Comparison of phototoxicity mechanism between pulsed and continuous wave irradiation in photodynamic therapy. J Photochem Photobiol B. 1999;53:53-9
27. Shopova M, Stoichkov N, Milev A, Peev M, Georgiev K, Gizbreht A. et al. Photodynamic therapy of experimental tumours with Zn(II)-phthalocyanine and pulsed laser irradiation. Lasers Med Sci. 1995;10:43-6
28. Barr H, Boulos PB, MacRobert AJ, Tralau CJ, Phillips D, Bown SG. Comparison of lasers for photodynamic therapy with a phthalocyanine photosensitizer. Lasers Med Sci. 1989;4:7-12
29. Estevez JP, Ascencio M, Colin P, Farine MO, Collinet P, Mordon S. Continuous or fractionated photodynamic therapy? Comparison of three PDT schemes for ovarian peritoneal micrometastasis treatment in a rat model. Photodiagnosis Photodyn Ther. 2010;7:251-7
30. Pogue BW, Momma T, Wu HC, Hasan T. Transient absorption changes in vivo during photodynamic therapy with pulsed-laser light. Br J Cancer. 1999;80:344-51
31. Grecco C, Kurachi C, Bagnato VS. In vitro and in vivo evaluation of the induced PDT response using a femtosecond laser. Proc SPIE 7380, Photodynamic Therapy: Back to the Future. 2009:73804 Y-Y-6
32. Grecco Cv, Moriyama LT, Cosci A, Pratavieira So, Bagnato VS, Kurachi C. Necrosis response to photodynamic therapy using light pulses in the femtosecond regime. Lasers Med Sci. 2013;28:1177-82
33. Pogue BW, Lilge L, Patterson MS, Wilson BC, Hasan T. Absorbed photodynamic dose from pulsed versus continuous wave light examined with tissue-simulating dosimeters. Appl Opt. 1997;36:7257-69
34. Sterenborg HJ, van Gemert MJ. Photodynamic therapy with pulsed light sources: a theoretical analysis. Phys Med Biol. 1996;41:835-49
35. Wilson BC, Patterson MS. The physics of photodynamic therapy. Phys Med Biol. 1986;31:327
36. Rizvi I, Anbil S, Alagic N, Celli J, Celli JP, Zheng LZ. et al. PDT dose parameters impact tumoricidal durability and cell death pathways in a 3D ovarian cancer model. Photochem Photobiol. 2013;89:942-52
37. Kessel D, Reiners JJ. Enhanced efficacy of photodynamic therapy via a sequential targeting protocol. Photochem Photobiol. 2014;90:889-95
38. Cincotta LL, Szeto DD, Lampros EE, Hasan TT, Cincotta AHA. Benzophenothiazine and benzoporphyrin derivative combination phototherapy effectively eradicates large murine sarcomas. Photochem Photobiol. 1996;63:229-37
39. Bisland SK, Lilge L, Lin A, Rusnov R, Wilson BC. Metronomic Photodynamic Therapy as a New Paradigm for Photodynamic Therapy: Rationale and Preclinical Evaluation of Technical Feasibility for Treating Malignant Brain Tumors. Photochem Photobiol. 2004;80:22-30
40. Vijlder H, Sterenborg H, Neumann H, Robinson D, Haas E. Light Fractionation Significantly Improves the Response of Superficial Basal Cell Carcinoma to Aminolaevulinic Acid Photodynamic Therapy: Five-year Follow-up of a Randomized, Prospective Trial. Acta Derm Venereol. 2012;92:641-7
41. Mallidi S, Anbil S, Lee S, Manstein D, Elrington S, Kositratna G. et al. Photosensitizer fluorescence and singlet oxygen luminescence as dosimetric predictors of topical 5-aminolevulinic acid photodynamic therapy induced clinical erythema. J Biomed Opt. 2014;19:028001
42. Jacques SL. How tissue optics affect dosimetry of photodynamic therapy. J Biomed Opt. 2010;15:051608
43. Zhou X, Pogue BW, Chen B, Demidenko E, Joshi R, Hoopes J. et al. Pretreatment photosensitizer dosimetry reduces variation in tumor response. Radiation Oncology Biology. 2006;64:1211-20
44. Chen B, Pogue BW, Hoopes PJ. Vascular and cellular targeting for photodynamic therapy. Crit Rev Eukaryot Gene Expr. 2006;16:279-306
45. Woodhams JH, MacRobert AJ, Bown SG. The role of oxygen monitoring during photodynamic therapy and its potential for treatment dosimetry. Photochemical & Photobiological Sciences. 2007;6:1246-56
46. Sunar U, Rohrbach D, Rigual N, Tracy E, Keymel K, Cooper MT. et al. Monitoring photobleaching and hemodynamic responses to HPPH-mediated photodynamic therapy of head and neck cancer: a case report. Opt Express. 2010;18:14969-78
47. Mallidi S, Watanabe K, Timerman D, Schoenfeld D, Hasan T. Prediction of tumor recurrence and therapy monitoring using ultrasound-guided photoacoustic imaging. Theranostics. 2015;5:289-301
48. Pogue BW, Elliott JT, Kanick SC, Davis SC, Samkoe KS, Maytin EV. et al. Revisiting photodynamic therapy dosimetry: reductionist & surrogate approaches to facilitate clinical success. Phys Med Biol. 2016:57-89
49. Mallidi S, Spring BQ, Chang S, Vakoc B, Hasan T. Optical Imaging, Photodynamic Therapy and Optically Triggered Combination Treatments. Cancer J. 2015;21:194-205
50. Hu J, Tang Ya, Elmenoufy AH, Xu H, Cheng Z, Yang X. Nanocomposite-Based Photodynamic Therapy Strategies for Deep Tumor Treatment. SMALL. 2015;11:5860-87
51. Carpenter S, Fehr MJ, Kraus GA, Petrich JW. Chemiluminescent Activation of the Antiviral activity of Hypericin - A Molecular Flashlight. Proc Natl Acad Sci U S A. 1994;91:12273-7
52. Phillip MJ, Maximuke PP. Chemi-Luminescence and Hematoporphyrin Derivative - A Novel Therapy for Mammary Adenocarinomas in Mice. ONCOLOGY. 1989;46:266-72
53. Huang L, Chen T-C, Lin F-H. Luminol As In Situ Light Source in Meso-Tetraphenylporphyrin-Mediated Photodynamic Therapy. Curr Med Chem. 2013;20:1195-202
54. Yuan H, Chong H, Wang B, Zhu C, Liu L, Yang Q. et al. Chemical Molecule-Induced Light-Activated System for Anticancer and Antifungal Activities. J Am Chem Soc. 2012;134:13184-7
55. Theodossiou T, Hothersall JS, Woods EA, Okkenhaug K, Jacobson J, MacRobert AJ. Firefly luciferin-activated Rose Bengal: In vitro photodynamic therapy by intracellular chemiluminescence in transgenic NIH 3T3 cells. Cancer Res. 2003;63:1818-21
56. Schipper ML, Patel MR, Gambhir SS. Evaluation of firefly luciferase biolurninescence mediated photodynarnic toxicity in cancer cells. Mol Imaging Biol. 2006;8:218-25
57. Zhao J, Fei J, Gao L, Cui W, Yang Y, Wang A. et al. Bioluminescent Microcapsules: Applications in Activating a Photosensitizer. Chem Eur J. 2013;19:4548-55
58. So MK, Xu CJ, Loening AM, Gambhir SS, Rao JH. Self-illuminating quantum dot conjugates for in vivo imaging. Nat Biotechnol. 2006;24:339-43
59. Hsu C-Y, Chen C-W, Yu H-P, Lin Y-F, Lai P-S. Bioluminescence resonance energy transfer using luciferase-immobilized quantum dots for self-illuminated photodynamic therapy. Biomaterials. 2013;34:1204-12
60. Kim YR, Kim S, Choi JW, Choi SY, Lee S-H, Kim H. et al. Bioluminescence-activated deep-tissue photodynamic therapy of cancer. Theranostics. 2015;5:805-17
61. Denk W, Strickler JH, Webb WW. 2-Photon Laser Scanning Fluorescence Microscopy. Science. 1990;248:73-6
62. Samkoe KS, Clancy AA, Karotki A, Wilson BC, Cramb DT. Complete blood vessel occlusion in the chick chorioallantoic membrane using two-photon excitation photodynamic therapy: implications for treatment of wet age-related macular degeneration. J Biomed Opt. 2007:12
63. Spangler CW, Starkey J, Rebane A, Drobizhev M, Meng F, Gong A. Synthesis, characterization and two-photon PDT efficacy studies of triads incorporating tumor targeting and imaging components. Proc SPIE. 2008;6845:68450S
64. Patrice T, Lebodic MF, Lebodic L, Spreux T, Dabouis G, Hervouet L. Neodymium-Yttrium Aluminum Garnet Laser Destruction of Nonsensitized and Hematoporphyrin derivative-sensitized Tumors. Cancer Res. 1983;43:2876-9
65. Marchesini R, Melloni E, Pezzoni G, Savi G, Zunino F, Docchio F. et al. A Study on the Possible involvment of Nonlinear Mechanism of Light Absorption by HOD with Nd-YAG Laser. Lasers Surg Med. 1986;6:323-7
66. Lenz P. In-vivo Excitation of Photosensitizers by Infrared Light. Photochem Photobiol. 1995;62:333-8
67. Goyan RL, Cramb DT. Near-infrared two-photon excitation of protoporphyrin IX: Photodynamics and photoproduct generation. Photochem Photobiol. 2000;72:821-7
68. Karotki A, Khurana M, Lepock JR, Wilson BC. Simultaneous two-photon excitation of photofrin in relation to photodynamic therapy. Photochem Photobiol. 2006;82:443-52
69. Khurana M, Collins HA, Karotki A, Anderson HL, Cramb DT, Wilson BC. Quantitative in vitro demonstration of two-photon photodynamic therapy using Photofrin (R) and Visudyne (R). Photochem Photobiol. 2007;83:1441-8
70. Pawlicki M, Collins HA, Denning RG, Anderson HL. Two-Photon Absorption and the Design of Two-Photon Dyes. Angew Chem, Int Ed. 2009;48:3244-66
71. Frederiksen PK, Jorgensen M, Ogilby PR. Two-photon photosensitized production of singlet oxygen. J Am Chem Soc. 2001;123:1215-21
72. Ogawa K, Ohashi A, Kobuke Y, Kamada K, Ohta K. Strong two-photon absorption of self-assembled butadiyne-linked bisporphyrin. J Am Chem Soc. 2003;125:13356-7
73. Drobizhev M, Stepanenko Y, Dzenis Y, Karotki A, Rebane A, Taylor PN. et al. Extremely strong near-IR two-photon absorption in conjugated porphyrin dimers: Quantitative description with three-essential-states model. J Phys Chem B. 2005;109:7223-36
74. Arnbjerg J, Jimenez-Banzo A, Paterson MJ, Nonell S, Borrell JI, Christiansen O. et al. Two-photon absorption in tetraphenylporphycenes: Are porphycenes better candidates than porphyrins for providing optimal optical properties for two-photon photodynamic therapy? J Am Chem Soc. 2007;129:5188-99
75. Collins HA, Khurana M, Moriyama EH, Mariampillai A, Dahlstedt E, Balaz M. et al. Blood-vessel closure using photosensitizers engineered for two-photon excitation. Nat Photonics. 2008;2:420-4
76. Beverina L, Crippa M, Landenna M, Ruffo R, Salice P, Silvestri F. et al. Assessment of water-soluble pi-extended squaraines as one- and two-photon singlet oxygen photosensitizers: Design, synthesis, and characterization. J Am Chem Soc. 2008;130:1894-902
77. Velusamy M, Shen J-Y, Lin JT, Lin Y-C, Hsieh C-C, Lai C-H. et al. A New Series of Quadrupolar Type Two-Photon Absorption Chromophores Bearing 11,12-Dibutoxydibenzo[a,c]-phenazine Bridged Amines; Their Applications in Two-Photon Fluorescence Imaging and Two-Photon Photodynamic Therapy. Adv Funct Mater. 2009;19:2388-97
78. Zhao T, Shen X, Li L, Guan Z, Gao N, Yuan P. et al. Gold nanorods as dual photo-sensitizing and imaging agents for two-photon photodynamic therapy. Nanoscale. 2012;4:7712-9
79. Zou Q, Fang Y, Zhao Y, Zhao H, Wang Y, Gu Y. et al. Synthesis and in Vitro Photocytotoxicity of Coumarin Derivatives for One- and Two-Photon Excited Photodynamic Therapy. J Med Chem. 2013;56:5288-94
80. Hammerer F, Garcia G, Chen S, Poyer F, Achelle S, Fiorini-Debuisschert C. et al. Synthesis and Characterization of Glycoconjugated Porphyrin Triphenylamine Hybrids for Targeted Two-Photon Photodynamic Therapy. J Org Chem. 2014;79:1406-17
81. Schmitt J, Heitz V, Sour A, Bolze F, Ftouni H, Nicoud J-F. et al. Diketopyrrolopyrrole-Porphyrin Conjugates with High Two-Photon Absorption and Singlet Oxygen Generation for Two-Photon Photodynamic Therapy. Angew Chem, Int Ed. 2015;54:169-73
82. Bhawalkar JD, Kumar ND, Zhao CF, Prasad PN. Two-Photon Photodynamic Therapy. J Clin Laser Med Surg. 1997;15:201-4
83. Dichtel WR, Serin JM, Edder C, Frechet JMJ, Matuszewski M, Tan LS. et al. Singlet oxygen generation via two-photon excited FRET. J Am Chem Soc. 2004;126:5380-1
84. Oar MA, Serin JA, Dichtel WR, Frechet JMJ. Photosensitization of singlet oxygen via two-photon-excited fluorescence resonance energy transfer in a water-soluble dendrimer. Chem Mater. 2005;17:2267-75
85. Oar MA, Dichtel WR, Serin JM, Frechet JMJ, Rogers JE, Slagle JE. et al. Light-harvesting chromophores with metalated porphyrin cores for tuned photosensitization of singlet oxygen via two-photon excited FRET. Chem Mater. 2006;18:3682-92
86. Kim S, Ohulchanskyy TY, Pudavar HE, Pandey RK, Prasad PN. Organically modified silica nanoparticles co-encapsulating photosensitizing drug and aggregation-enhanced two-photon absorbing fluorescent dye aggregates for two-photon photodynamic therapy. J Am Chem Soc. 2007;129:2669-75
87. Zhao T, Yu K, Li L, Zhang T, Guan Z, Gao N. et al. Gold Nanorod Enhanced Two-Photon Excitation Fluorescence of Photosensitizers for Two-Photon Imaging and Photodynamic Therapy. ACS Appl Mater Interfaces. 2014;6:2700-8
88. Chen N-T, Tang K-C, Chung M-F, Cheng S-H, Huang C-M, Chu C-H. et al. Enhanced Plasmonic Resonance Energy Transfer in Mesoporous Silica-Encased Gold Nanorod for Two-Photon-Activated Photodynamic Therapy. Theranostics. 2014;4:798-807
89. Samia ACS, Dayal S, Burda C. Quantum dot-based energy transfer: Perspectives and potential for applications in photodynamic therapy. Photochemistry and photobiology. 2006;82:617-25
90. Juzenas P, Chen W, Sun Y-P, Neto Coelho MA, Generalov R, Generalova N. et al. Quantum dots and nanoparticles for photodynamic and radiation therapies of cancer. Adv Drug Deliv Rev. 2008;60:1600-14
91. Yaghini E, Seifalian AM, MacRobert AJ. Quantum dots and their potential biomedical applications in photosensitization for photodynamic therapy. Nanomedicine. 2009;4:353-63
92. Fowley C, Nomikou N, McHale AP, McCaughan B, Callan JF. Extending the tissue penetration capability of conventional photosensitisers: a carbon quantum dot-protoporphyrin IX conjugate for use in two-photon excited photodynamic therapy. Chem Commun. 2013;49:8934-6
93. Wang C, Cheng L, Liu Z. Upconversion Nanoparticles for Photodynamic Therapy and Other Cancer Therapeutics. Theranostics. 2013;3:317-30
94. Naczynski DJ, Tan M-C, Riman RE, Moghe PV. Rare earth nanoprobes for functional biomolecular imaging and theranostics. J Mater Chem B. 2014;2:2958-16
95. Morgan WF. Non-targeted and delayed effects of exposure to ionizing radiation: II. Radiation-induced genomic instability and bystander effects in vivo, clastogenic factors and transgenerational effects. Radiat Res. 2003;159:581-96
96. Berg K, Luksiene Z, Moan JH, Ma LW. Combined Treatment of Ionizing Radiation and Photosensitization by 5-Aminolevulinic Acid-incuded Protoporphyrin-IX. Radiat Res. 1995;142:340-6
97. Luksiene Z, Kalvelyte A, Supino R. On the combination of photodynamic therapy with ionizing radiation. J Photochem Photobiol B. 1999;52:35-42
98. Colasanti A, Kisslinger A, Quarto M, Riccio P. Combined effects of radiotherapy and photodynamic therapy on an in vitro human prostate model. Acta Biochim Pol. 2004;51:1039-46
99. Montazerabadi AR, Sazgarnia A, Bahreyni-Toosi MH, Ahmadi A, Aledavood A. The effects of combined treatment with ionizing radiation and indocyanine green-mediated photodynamic therapy on breast cancer cells. J Photochem Photobiol B. 2012;109:42-9
100. Luksiene Z, Juzenas P, Moan J. Radiosensitization of tumours by porphyrins. Cancer Lett. 2006;235:40-7
101. Axelsson J, Davis SC, Gladstone DJ, Pogue BW. Cerenkov emission induced by external beam radiation stimulates molecular fluorescence. Med Phys. 2011;38:4127-32
102. Kotagiri N, Sudlow GP, Akers WJ, Achilefu S. Breaking the depth dependency of phototherapy with Cerenkov radiation and low-radiance-responsive nanophotosensitizers. Nat Nanotechnol. 2015;10:370-9
103. Chen W, Zhang J. Using nanoparticles to enable simultaneous radiation and photodynamic therapies for cancer treatment. J Nanosci Nanotechnol. 2006;6:1159-66
104. Liu Y, Chen W, Wang S, Joly AG. Investigation of water-soluble x-ray luminescence nanoparticles for photodynamic activation. Appl Phys Lett. 2008:92
105. Cooper DR, Kudinov K, Tyagi P, Hill CK, Bradforth SE, Nadeau JL. Photoluminescence of cerium fluoride and cerium-doped lanthanum fluoride nanoparticles and investigation of energy transfer to photosensitizer molecules. Phys Chem Chem Phys. 2014;16:12441-53
106. Popovich K, Prochazkova L, Pelikanova IT, Vlk M, Palkovsky M, Jary V. et al. Preliminary study on singlet oxygen production using CeF3:Tb3+@SiO2-PpIX. Radiat Meas. 2016 -
107. Abliz E, Collins JE, Bell H, Tata DB. Novel applications of diagnostic X-rays in activating a clinical photodynamic drug: Photofrin II through X-ray induced visible luminescence from ``rare-earth'' formulated particles. J Xray Sci Technol. 2011;19:521-30
108. Bulin AL, Truillet C, Chouikrat R. et al. X-ray-Induced Singlet Oxygen Activation with Nanoscintillator-Coupled Porphyrins. J Phys Chem C. 2013;117:21583-9
109. Morgan NY, Kramer-Marek G, Smith PD, Camphausen K, Capala J. Nanoscintillator Conjugates as Photodynamic Therapy-Based Radiosensitizers: Calculation of Required Physical Parameters. Radiation research. 2009;171:236-44
110. Bulin A-L, Vasil'ev A, Belsky A. et al. Modelling energy deposition in nanoscintillators to predict the efficiency of the X-ray-induced photodynamic effect. Nanoscale. 2015;7:5744-51
111. Chen H, Wang GD, Chuang Y-J, Zhen Z, Chen X, Biddinger P. et al. Nanoscintillator-Mediated X-ray Inducible Photodynamic Therapy for In Vivo Cancer Treatment. Nano Lett. 2015;15:2249-56
112. Kascakova S, Giuliani A, Lacerda S, Pallier A, Mercere P, Toth E. et al. X-ray-induced radiophotodynamic therapy (RPDT) using lanthanide micelles: Beyond depth limitations. Nano Res. 2015;8:2373-9
113. Zhang C, Zhao K, Bu W, Ni D, Liu Y, Feng J. et al. Marriage of Scintillator and Semiconductor for Synchronous Radiotherapy and Deep Photodynamic Therapy with Diminished Oxygen Dependence. Angew Chem, Int Ed. 2015;54:1770-4
114. Ma L, Zou X, Bui B, Chen W, Song KH, Solberg T. X-ray excited ZnS:Cu,Co afterglow nanoparticles for photodynamic activation. Appl Phys Lett. 2014:105
115. Folli S, Wagnieres G, Pelegrin A, Calmes JM, Braichotte D, Buchegger F. et al. Immunophotodiagnosis of colon carcinomas in patients injected with fluoresceinated chimeric antibodies against carcinoembryonic antigen. Proc Natl Acad Sci U S A. 1992;89:7973-7
116. van Dam GM, Themelis G, Crane LM, Harlaar NJ, Pleijhuis RG, Kelder W. et al. Intraoperative tumor-specific fluorescence imaging in ovarian cancer by folate receptor-alpha targeting: first in-human results. Nat Med. 2011;17:1315-9
117. Rosenthal EL, Warram JM, de Boer E, Chung TK, Korb ML, Brandwein-Gensler M. et al. Safety and Tumor Specificity of Cetuximab-IRDye800 for Surgical Navigation in Head and Neck Cancer. Clin Cancer Res. 2015;21:3658-66
118. Sexton K, Tichauer K, Samkoe KS, Gunn J, Hoopes PJ, Pogue BW. Fluorescent affibody peptide penetration in glioma margin is superior to full antibody. PLoS One. 2013;8:e60390
119. Whelan HT, Schmidt MH, Segura AD, McAuliffe TL, Bajic DM, Murray KJ. et al. The role of photodynamic therapy in posterior fossa brain tumors. A preclinical study in a canine glioma model. J Neurosurg. 1993;79:562-8
120. Mew D, Wat CK, Towers GH, Levy JG. Photoimmunotherapy: treatment of animal tumors with tumor-specific monoclonal antibody-hematoporphyrin conjugates. J Immunol. 1983;130:1473-7
121. Hamblin MR, Bamberg MP, Miller JL, Hasan T. Cationic photoimmunoconjugates between monoclonal antibodies and hematoporphyrin: selective photodestruction of ovarian cancer cells. Appl Opt. 1998;37:7184-92
122. Soukos NS, Hamblin MR, Keel S, Fabian RL, Deutsch TF, Hasan T. Epidermal growth factor receptor-targeted immunophotodiagnosis and photoimmunotherapy of oral precancer in vivo. Cancer Res. 2001;61:4490-6
123. Mew D, Lum V, Wat CK, Towers GH, Sun CH, Walter RJ. et al. Ability of specific monoclonal antibodies and conventional antisera conjugated to hematoporphyrin to label and kill selected cell lines subsequent to light activation. Cancer Res. 1985;45:4380-6
124. Savellano MD, Hasan T. Targeting cells that overexpress the epidermal growth factor receptor with polyethylene glycolated BPD verteporfin photosensitizer immunoconjugates. Photochem Photobiol. 2003;77:431-9
125. Savellano MD, Hasan T. Photochemical targeting of epidermal growth factor receptor: a mechanistic study. Clin Cancer Res. 2005;11:1658-68
126. Abu-Yousif AO, Moor AC, Zheng X, Savellano MD, Yu W, Selbo PK. et al. Epidermal growth factor receptor-targeted photosensitizer selectively inhibits EGFR signaling and induces targeted phototoxicity in ovarian cancer cells. Cancer Lett. 2012;321:120-7
127. Mitsunaga M, Ogawa M, Kosaka N, Rosenblum LT, Choyke PL, Kobayashi H. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat Med. 2011;17:1685-91
128. Oseroff AR, Ohuoha D, Hasan T, Bommer JC, Yarmush ML. Antibody-targeted photolysis: selective photodestruction of human T-cell leukemia cells using monoclonal antibody-chlorin e6 conjugates. Proc Natl Acad Sci U S A. 1986;83:8744-8
129. Hamblin MR, Miller JL, Rizvi I, Loew HG, Hasan T. Pegylation of charged polymer-photosensitiser conjugates: effects on photodynamic efficacy. Br J Cancer. 2003;89:937-43
130. Goff BA, Hermanto U, Rumbaugh J, Blake J, Bamberg M, Hasan T. Photoimmunotherapy and biodistribution with an OC125-chlorin immunoconjugate in an in vivo murine ovarian cancer model. Br J Cancer. 1994;70:474-80
131. Del Governatore M, Hamblin MR, Shea CR, Rizvi I, Molpus KG, Tanabe KK. et al. Experimental photoimmunotherapy of hepatic metastases of colorectal cancer with a 17.1A chlorin(e6) immunoconjugate. Cancer Res. 2000;60:4200-5
132. Hamblin MR, Del Governatore M, Rizvi I, Hasan T. Biodistribution of charged 17.1A photoimmunoconjugates in a murine model of hepatic metastasis of colorectal cancer. Br J Cancer. 2000;83:1544-51
133. Azais H, Schmitt C, Tardivel M, Kerdraon O, Stallivieri A, Frochot C. et al. Assessment of the specificity of a new folate-targeted photosensitizer for peritoneal metastasis of epithelial ovarian cancer to enable intraperitoneal photodynamic therapy. A preclinical study. Photodiagn Photodyn Ther. 2016;13:130-8
134. https://clinicaltrials.gov/ct2/show/NCT02422979
135. Kimura H, Sakai K, Arao T, Shimoyama T, Tamura T, Nishio K. Antibody-dependent cellular cytotoxicity of cetuximab against tumor cells with wild-type or mutant epidermal growth factor receptor. Cancer Sci. 2007;98:1275-80
136. Muchekehu R, Liu D, Horn M, Campbell L, Del Rosario J, Bacica M. et al. The Effect of Molecular Weight, PK, and Valency on Tumor Biodistribution and Efficacy of Antibody-Based Drugs. Transl Oncol. 2013;6:562-72
137. Haraoui B, Bykerk V. Etanercept in the treatment of rheumatoid arthritis. Ther Clin Risk Manag. 2007;3:99-105
138. Molpus KL, Hamblin MR, Rizvi I, Hasan T. Intraperitoneal photoimmunotherapy of ovarian carcinoma xenografts in nude mice using charged photoimmunoconjugates. Gynecol Oncol. 2000;76:397-404
139. Duska LR, Hamblin MR, Bamberg MP, Hasan T. Biodistribution of charged F(ab')2 photoimmunoconjugates in a xenograft model of ovarian cancer. Br J Cancer. 1997;75:837-44
140. Heukers R, van Bergen en Henegouwen PM, Oliveira S. Nanobody-photosensitizer conjugates for targeted photodynamic therapy. Nanomedicine. 2014;10:1441-51
141. van Driel PB, Boonstra MC, Slooter MD, Heukers R, Stammes MA, Snoeks TJ. et al. EGFR targeted nanobody-photosensitizer conjugates for photodynamic therapy in a pre-clinical model of head and neck cancer. J Control Release. 2016;229:93-105
142. Juweid M, Neumann R, Paik C, Perez-Bacete MJ, Sato J, van Osdol W. et al. Micropharmacology of monoclonal antibodies in solid tumors: direct experimental evidence for a binding site barrier. Cancer Res. 1992;52:5144-53
143. Watanabe R, Hanaoka H, Sato K, Nagaya T, Harada T, Mitsunaga M. et al. Photoimmunotherapy targeting prostate-specific membrane antigen: are antibody fragments as effective as antibodies? J Nucl Med. 2015;56:140-4
144. Vrouenraets MB, Visser GW, Loup C, Meunier B, Stigter M, Oppelaar H. et al. Targeting of a hydrophilic photosensitizer by use of internalizing monoclonal antibodies: A new possibility for use in photodynamic therapy. Int J Cancer. 2000;88:108-14
145. Savellano MD, Hasan T. Targeting cells that overexpress the epidermal growth factor receptor with polyethylene glycolated BPD verteporfin photosensitizer immunoconjugates. Photochem Photobiol. 2003;77:431-9
146. Spring BQ, Abu-Yousif AO, Palanisami A, Rizvi I, Zheng X, Mai Z. et al. Selective treatment and monitoring of disseminated cancer micrometastases in vivo using dual-function, activatable immunoconjugates. Proc Natl Acad Sci U S A. 2014;111:E933-42
147. Matayoshi ED, Wang GT, Krafft GA, Erickson J. Novel fluorogenic substrates for assaying retroviral proteases by resonance energy transfer. Science. 1990;247:954-8
148. Chen J, Stefflova K, Niedre MJ, Wilson BC, Chance B, Glickson JD. et al. Protease-triggered photosensitizing beacon based on singlet oxygen quenching and activation. J Am Chem Soc. 2004;126:11450-1
149. Zheng G, Chen J, Stefflova K, Jarvi M, Li H, Wilson BC. Photodynamic molecular beacon as an activatable photosensitizer based on protease-controlled singlet oxygen quenching and activation. Proc Natl Acad Sci U S A. 2007;104:8989-94
150. Liu TW, Akens MK, Chen J, Wilson BC, Zheng G. Matrix metalloproteinase-based photodynamic molecular beacons for targeted destruction of bone metastases in vivo. Photochem Photobiol Sci. 2016;15:375-81
151. Lovell JF, Chen J, Huynh E, Jarvi MT, Wilson BC, Zheng G. Facile synthesis of advanced photodynamic molecular beacon architectures. Bioconjugate Chem. 2010;21:1023-5
152. Zheng X, Sallum UW, Verma S, Athar H, Evans CL, Hasan T. Exploiting a bacterial drug-resistance mechanism: a light-activated construct for the destruction of MRSA. Angew Chem, Int Ed. 2009;48:2148-51
153. Zhu L, Xie J, Swierczewska M, Zhang F, Quan Q, Ma Y. et al. Real-time video imaging of protease expression in vivo. Theranostics. 2011;1:18-27
154. Chen AK, Behlke MA, Tsourkas A. Efficient cytosolic delivery of molecular beacon conjugates and flow cytometric analysis of target RNA. Nucleic Acids Res. 2008;36:e69
155. Mroz P, Yaroslavsky A, Kharkwal GB, Hamblin MR. Cell Death Pathways in Photodynamic Therapy of Cancer. Cancers. 2011;3:2516-39
156. Tesniere A, Panaretakis T, Kepp O, Apetoh L, Ghiringhelli F, Zitvogel L. et al. Molecular characteristics of immunogenic cancer cell death. Cell Death Differ. 2008;15:3-12
157. Korbelik M, Sun J, Cecic I. Photodynamic therapy-induced cell surface expression and release of heat shock proteins: relevance for tumor response. Cancer Res. 2005;65:1018-26
158. Todryk S, Melcher AA, Hardwick N, Linardakis E, Bateman A, Colombo MP. et al. Heat shock protein 70 induced during tumor cell killing induces Th1 cytokines and targets immature dendritic cell precursors to enhance antigen uptake. J Immunol. 1999;163:1398-408
159. Verrico AK, Haylett AK, Moore JV. In vivo expression of the collagen-related heat shock protein HSP47, following hyperthermia or photodynamic therapy. Lasers Med Sci. 2001;16:192-8
160. Hanlon JG, Adams K, Rainbow AJ, Gupta RS, Singh G. Induction of Hsp60 by Photofrin-mediated photodynamic therapy. J Photochem Photobiol B. 2001;64:55-61
161. Hsieh YJ, Wu CC, Chang CJ, Yu JS. Subcellular localization of Photofrin determines the death phenotype of human epidermoid carcinoma A431 cells triggered by photodynamic therapy: when plasma membranes are the main targets. J Cell Physiol. 2003;194:363-75
162. Garg AD, Agostinis P. ER stress, autophagy and immunogenic cell death in photodynamic therapy-induced anti-cancer immune responses. Photochem Photobiol Sci. 2014;13:474-87
163. Zitvogel L, Kepp O, Kroemer G. Decoding cell death signals in inflammation and immunity. Cell. 2010;140:798-804
164. Riddell JR, Wang XY, Minderman H, Gollnick SO. Peroxiredoxin 1 stimulates secretion of proinflammatory cytokines by binding to TLR4. J Immunol. 2010;184:1022-30
165. Garg AD, Krysko DV, Vandenabeele P, Agostinis P. DAMPs and PDT-mediated photo-oxidative stress: exploring the unknown. Photochem Photobiol Sci. 2011;10:670-80
166. Castano AP, Mroz P, Hamblin MR. Photodynamic therapy and anti-tumour immunity. Nat Rev Cancer. 2006;6:535-45
167. Gollnick SO, Evans SS, Baumann H, Owczarczak B, Maier P, Vaughan L. et al. Role of cytokines in photodynamic therapy-induced local and systemic inflammation. Br J Cancer. 2003;88:1772-9
168. Kaplanski G, Marin V, Montero-Julian F, Mantovani A, Farnarier C. IL-6: a regulator of the transition from neutrophil to monocyte recruitment during inflammation. Trends Immunol. 2003;24:25-9
169. Fingar VH, Wieman TJ, Wiehle SA, Cerrito PB. The role of microvascular damage in photodynamic therapy: the effect of treatment on vessel constriction, permeability, and leukocyte adhesion. Cancer Res. 1992;52:4914-21
170. Cecic I, Stott B, Korbelik M. Acute phase response-associated systemic neutrophil mobilization in mice bearing tumors treated by photodynamic therapy. Int Immunopharmacol. 2006;6:1259-66
171. de Vree WJ, Essers MC, de Bruijn HS, Star WM, Koster JF, Sluiter W. Evidence for an important role of neutrophils in the efficacy of photodynamic therapy in vivo. Cancer Res. 1996;56:2908-11
172. Cecic I, Sun J, Korbelik M. Role of complement anaphylatoxin C3a in photodynamic therapy-elicited engagement of host neutrophils and other immune cells. Photochem Photobiol. 2006;82:558-62
173. Gollnick SO. Photodynamic Therapy and Antitumor Immunity. J Natl Compr Canc Netw. 2012;10:S40-S3
174. Canti G, Lattuada D, Nicolin A, Taroni P, Valentini G, Cubeddu R. Antitumor immunity induced by photodynamic therapy with aluminum disulfonated phthalocyanines and laser light. Anticancer Drugs. 1994;5:443-7
175. Krosl G, Korbelik M, Krosl J, Dougherty GJ. Potentiation of photodynamic therapy-elicited antitumor response by localized treatment with granulocyte-macrophage colony-stimulating factor. Cancer Res. 1996;56:3281-6
176. Castano AP, Liu Q, Hamblin MR. A green fluorescent protein-expressing murine tumour but not its wild-type counterpart is cured by photodynamic therapy. Br J Cancer. 2006;94:391-7
177. Gollnick SO, Vaughan L, Henderson BW. Generation of effective antitumor vaccines using photodynamic therapy. Cancer Res. 2002;62:1604-8
178. Zhang H, Ma W, Li Y. Generation of effective vaccines against liver cancer by using photodynamic therapy. Lasers Med Sci. 2009;24:549-52
179. Fingar VH. Vascular effects of photodynamic therapy. J Clin Laser Med Surg. 1996;14:323-8
180. Huang HC, Mallidi S, Liu J, Chiang CT, Mai Z, Goldschmidt R. et al. Photodynamic Therapy Synergizes with Irinotecan to Overcome Compensatory Mechanisms and Improve Treatment Outcomes in Pancreatic Cancer. Cancer Res. 2016;76:1066-77
181. Schmidt-Erfurth U, Hasan T. Mechanisms of action of photodynamic therapy with verteporfin for the treatment of age-related macular degeneration. Surv Ophthalmol. 2000;45:195-214
182. Korbelik M, Parkins CS, Shibuya H, Cecic I, Stratford MR, Chaplin DJ. Nitric oxide production by tumour tissue: impact on the response to photodynamic therapy. Br J Cancer. 2000;82:1835-43
183. Nahabedian MY, Cohen RA, Contino MF, Terem TM, Wright WH, Berns MW. et al. Combination cytotoxic chemotherapy with cisplatin or doxorubicin and photodynamic therapy in murine tumors. J Natl Cancer Inst. 1988;80:739-43
184. Zuluaga MF, Lange N. Combination of photodynamic therapy with anti-cancer agents. Curr Med Chem. 2008;15:1655-73
185. Rizvi I, Celli JP, Evans CL, Abu-Yousif AO, Muzikansky A, Pogue BW. et al. Synergistic enhancement of carboplatin efficacy with photodynamic therapy in a three-dimensional model for micrometastatic ovarian cancer. Cancer Res. 2010;70:9319-28
186. Celli JP, Solban N, Liang A, Pereira SP, Hasan T. Verteporfin-based photodynamic therapy overcomes gemcitabine insensitivity in a panel of pancreatic cancer cell lines. Lasers Surg Med. 2011;43:565-74
187. Goff BA, Bamberg M, Hasan T. Photoimmunotherapy of human ovarian carcinoma cells ex vivo. Cancer Res. 1991;51:4762-7
188. Luo D, Carter KA, Miranda D, Lovell JF. Chemophototherapy: An Emerging Treatment Option for Solid Tumors. Adv Sci. 2016:1600106
189. Snyder JW, Greco WR, Bellnier DA, Vaughan L, Henderson BW. Photodynamic Therapy A Means to Enhanced Drug Delivery to Tumors. Cancer Res. 2003;63:8126-31
190. Gallagher-Colombo SM, Miller J, Cengel KA, Putt ME, Vinogradov SA, Busch TM. Erlotinib Pretreatment Improves Photodynamic Therapy of Non-Small Cell Lung Carcinoma Xenografts via Multiple Mechanisms. Cancer Res. 2015;75:3118-26
191. Nowak-Sliwinska P, Weiss A, van Beijnum JR, Wong TJ, Ballini JP, Lovisa B. et al. Angiostatic kinase inhibitors to sustain photodynamic angio-occlusion. J Cell Mol Med. 2012;16:1553-62
192. Buytaert E, Dewaele M, Agostinis P. Molecular effectors of multiple cell death pathways initiated by photodynamic therapy. Biochim Biophys Acta. 2007;1776:86-107
193. Spring BQ, Rizvi I, Xu N, Hasan T. The role of photodynamic therapy in overcoming cancer drug resistance. Photochem Photobiol Sci. 2015;14:1476-91
194. Spring BQ, Bryan Sears R, Zheng LZ, Mai Z, Watanabe R, Sherwood ME. et al. A photoactivable multi-inhibitor nanoliposome for tumour control and simultaneous inhibition of treatment escape pathways. Nat Nanotechnol. 2016;11:378-87
195. Tangutoori S, Spring BQ, Mai Z, Palanisami A, Mensah LB, Hasan T. Simultaneous delivery of cytotoxic and biologic therapeutics using nanophotoactivatable liposomes enhances treatment efficacy in a mouse model of pancreatic cancer. Nanomedicine. 2016;12:223-34
196. Ferrario A, von Tiehl KF, Rucker N, Schwarz MA, Gill PS, Gomer CJ. Antiangiogenic treatment enhances photodynamic therapy responsiveness in a mouse mammary carcinoma. Cancer Res. 2000;60:4066-9
197. Saini R, Poh CF. Photodynamic therapy: a review and its prospective role in the management of oral potentially malignant disorders. Oral Dis. 2013;19:440-51
198. Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M. et al. Photodynamic therapy. J Natl Cancer Inst. 1998;90:889-905
199. Levy JG. Photodynamic therapy. Trends Biotechnol. 1995;13:14-8
200. Huang Z, Li L, Wang H, Wang X, Yuan K, Meyers A. et al. Photodynamic Therapy: An Update on Clinical Applications. J Innov Opt Health Sci. 2009;02:73-92
201. Triesscheijn M, Baas P, Schellens JH, Stewart FA. Photodynamic Therapy in Oncology. The Oncologist. 2006;11:1034-44
Author contact
![]() Corresponding author: Tayyaba Hasan, PhD, Wellman Center for Photomedicine, Harvard Medical School Massachusetts General Hospital 40 Blossom St, Boston, MA 02114. E-mail: thasanharvard.edu.
Corresponding author: Tayyaba Hasan, PhD, Wellman Center for Photomedicine, Harvard Medical School Massachusetts General Hospital 40 Blossom St, Boston, MA 02114. E-mail: thasanharvard.edu.