Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2016; 6(4):545-557. doi:10.7150/thno.13352 This issue Cite
Research Paper
EMMPRIN-Targeted Magnetic Nanoparticles for In Vivo Visualization and Regression of Acute Myocardial Infarction
Irene Cuadrado1, Maria Jose Garcia Miguel Piedras1, Irene Herruzo1, Maria del Carmen Turpin1, Borja Castejón1, Paula Reventun2, Ana Martin1, Marta Saura2, Jose Luis Zamorano1, Carlos Zaragoza1 ![]()
1. Cardiology Department. University Francisco de Vitoria/Hospital Ramón y Cajal. Ctra. Colmenar Viejo, km. 9100. 28034 Madrid, Spain.
2. Department of Physiology, University of Alcalá, School of Medicine. Ctra. Madrid Barcelona, Km 3,300. 28875 Alcalá de Henares, Madrid, Spain.
Received 2015-7-27; Accepted 2015-12-9; Published 2016-2-15
Abstract

Inhibition of extracellular matrix (ECM) degradation may represent a mechanism for cardiac protection against ischemia. Extracellular matrix metalloproteinase inducer (EMMPRIN) is highly expressed in response to acute myocardial infarction (AMI), and induces activation of several matrix metalloproteinases (MMPs), including gelatinases MMP-2 and MMP-9. We targeted EMMPRIN with paramagnetic/fluorescent micellar nanoparticles conjugated with the EMMPRIN binding peptide AP-9 (NAP9), or an AP-9 scrambled peptide as a negative control (NAPSC). We found that NAP9 binds to endogenous EMMPRIN in cultured HL1 myocytes and in mouse hearts subjected to ischemia/reperfusion (IR). Injection of NAP9 at the time of or one day after IR, was enough to reduce progression of myocardial cell death when compared to Control and NAPSC injected mice (infarct size in NAP9 injected mice: 32%±6.59 vs Control: 46%±9.04 or NAPSC injected mice: 48%±7.64). In the same way, cardiac parameters were recovered to almost healthy levels (LVEF NAP9 63% ± 7.24 vs Control 42% ± 4.74 or NAPSC 39% ± 6.44), whereas ECM degradation was also reduced as shown by inhibition of MMP-2 and MMP-9 activation. Cardiac magnetic resonance (CMR) scans have shown a signal enhancement in the left ventricle of NAP9 injected mice with respect to non-injected, and to mice injected with NAPSC. A positive correlation between CMR enhancement and Evans-Blue/TTC staining of infarct size was calculated (R:0.65). Taken together, these results point to EMMPRIN targeted nanoparticles as a new approach to the mitigation of ischemic/reperfusion injury.
Keywords: Ischemia/Reperfusion, Matrix metalloproteinases, nanoparticles, EMMPRIN, AP9.
Introduction
Early coronary artery reperfusion is the most effective procedure for the treatment of acute myocardial infarction (AMI)1. However, reperfusion associated damage often worsens cardiac integrity, in part by eliciting an extensive inflammatory response in which proinflammatory macrophages play a pivotal role through the expression of extensive amounts of proinflammatory cytokines and extracellular matrix metalloproteinases (MMPs), thus contributing to ventricular injury2-3.
MMPs are proteolytic degrading enzymes that cleave several proteins, including extracellular matrix components. In the heart, MMP activation induces cardiac myocyte necrosis and heart failure, and elicits abnormal ventricular remodeling 4. In this regard, the extracellular matrix metalloproteinase inducer EMMPRIN (CD147) regulates the expression of several MMPs and plays a role in the inflammatory response to ischemia in monocytes and cardiac cells 5.
We and others have shown that EMMPRIN is highly expressed in monocytes and cardiac myocytes in response to AMI 6, and promotes the expression and activity of several MMPs, including MMP-2, MMP-9, MT1-MMP, and MMP-137.
We hypothesized that targeted inhibition of EMMPRIN may represent a new mechanism of cardiac protection, by preventing ECM degradation and myocyte cell death, thereby preserving heart contractility following ischemic reperfusion.
Materials and Methods
Reagents
General cell culture supplies were purchased from BD Biosciences (Spain); calf serum was from BioWhittaker (Verviers, Belgium). Cell culture-grade gelatin, trypsin, antibiotics, hematoxylin-eosin, Trichrome Masson staining reagents, Evans blue, and TTC dyes were from Sigma (Spain). HRP-conjugated anti-mouse secondary antibody and liquid DAB substrate were from Dako (Carpinteria, CA). Anti-MMP-2, MMP-9, CD68, and FITC-conjugated secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-heavy chain cardiac myosin antibody was from Abcam (Abcam, UK). HRP-conjugated anti-rabbit secondary antibody was from Sigma-Aldrich. Amersham ECL detection kit was from GE (GE Healthcare Life Sciences, Spain). Centrifugation concentrators were from Sartorius (Fischer Scientific, Spain).
Cells
The HL1 cardiomyocyte cell line was kindly donated by Dr. Antonio Bernad.
Animals
Wild type C57BL/6 mice were purchased from Charles River, and housed in our animal facilities in isolated rooms. All animal procedures were approved by the National Research Ethics Committee and conformed to EU Directive 86/609/EEC and recommendation 2007/526/EC regarding the protection of animals used for experimental and other scientific purposes (enacted under Spanish law 1201/2005).
Animal model of Ischemia/Reperfusion
Ischemia was induced by coronary artery ligation. Twelve-week-old mice were intraperitoneally anesthetized with ketamine/xylazine (100 and 10 mg/kg, respectively), intubated with a 1-mm steel tube, and ventilated (2ml, 80 strokes/min). The thorax was opened between the second and the third ribs and widened with the aid of a mouse retractor. The pericardium was opened and LAD was occluded for 30 min close to its bifurcation by using a 6-0 silk suture. Blanching of the ventricle provided direct evidence of coronary occlusion. Reperfusion was performed by ligation release. After the procedure, the chest was closed, negative pressure restored, and the skin sutured. Control mice (sham) were included in the assays, in which the same procedure was performed except for occlusion of the left coronary artery.
Determination of infarct size
Myocardial infarct size was determined by double staining with Evans Blue/TTC dyes. Animals were anesthetized and a 0.2% Evans Blue solution was injected into the aorta, allowing for uniform distribution of the dye. Animals were sacrificed, the hearts frozen, sliced in 1mm ring sections perpendicular to the LV long axis, and incubated with 0.5% TTC solution for 5 minutes. Healthy tissue (blue), area at risk (red), and infarct tissue (pale white) areas were subjected to morphometric analysis by using the Motic Images Plus software.
Echocardiography
Mouse hearts were visualized by echocardiography over time, using a high-frequency micro-ultrasound system (Vevo 2100; VisualSonics, Toronto, ON, Canada). Mice were anesthetized with 1.5% isoflurane gas, resulting in a heart rate of approximately 400 beats/min. Anesthetized animals were placed on the Integrated Rail System and Mouse Handling Table (provided by the manufacturer), which allows simultaneous acquisition of temperature data (37°C, by using the integrated heating pad) and electrocardiography (lead II) throughout the experiment. The chest of the mouse was carefully shaved, and warm ultrasound transmission gel was applied to ensure optimal image quality. Parasternal short-axis-view images of the heart were recorded using a 30-MHz RMV scan head in a B-mode to allow M-mode recordings by positioning the cursor in the parasternal short-axis view perpendicular to the interventricular septum and posterior wall of the left ventricle. From these recordings, the following parameters were determined using the on-site software cardiac package (Visual Sonics): left-ventricle end-diastolic diameter, left-ventricle end-diastolic volume, ejection fraction, and shortening fraction.
Histology and immunohistochemistry
Histological and immunohistochemical procedures were performed as previously described 7. In brief, mouse hearts were embedded in paraffin and 10, 4-μm serial sections (300 μm apart) were obtained from the apex to the mid LAD coronary artery. Immunohistochemical detection of EMMPRIN was performed by incubating sections with the corresponding primary and secondary antibodies. Red fluorescence emitted by NAP9 and NAPSC was detected at 600 nm wave-length in a Leica TCS SP5 II confocal microscope. Red and green intensities from each serial section (8-10 slides/section) were analyzed with the “co-localization finder” plugin NIH Image J64 software.
Flow cytometry
The effect of NAP9 and NAPSC on cell viability was determined by flow cytometry (FACS scanner, Becton Dickinson) using propidium iodide (RD systems) and annexin V conjugates (Life Technologies), as provided by the manufacturers.
Immunoblot analysis
Isolation of protein lysates and immunoblots were performed as described 7.
Peptide and Nanoprobe composition
Peptide AP9: YKLPGHHHHYRP.
Peptide AP9-scrambled: HYJLPGHRHPYHK.
The paramagnetic nanoparticles were prepared by the lipid film hydration method, with modifications as described 15. In brief, a film was prepared by rotary evaporation of Gd-DTPA-bis (GdDTPA-BSA), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyethylene glycol)-2000] (DSPE-PEG2000), DSPE-PEG2000-maleimide, and Rhodamine-PE, in a molar ratio 50:39:1:10, dissolved in a mixture of chloroform/methanol (4:1 v/v). The lipid film was hydrated with HEPES buffer, pH 6.7, and 150 mM NaCl, and the solution was rotated at 65°C for 1 hour. AP9 and the corresponding scrambled peptides were modified by adding a terminal cysteine residue (Fig. 1) to bind to the maleimide moiety at a molar ratio 1:40 (micelle:peptide). Uncoupled peptides were separated with centrifuge concentrators of 100 kDa. The number of peptide molecules bound per micelle was calculated by HLPC, estimating 4 ± 2 and 5 ± 1 the number of peptides present in NAP9 and NAPSC respectively.
Structure and chemical properties of NAP9 and NAPSC. A. Aminoacid composition of AP9 and AP9 scrambled peptides. B. Composition of NAP9 and NAPSC micelles containing AP9 and AP9 scrambled peptides respectively. C. Structure of micelle components and conjugation of Cys-peptides to maleimide moiety. D. Physical and chemical properties of NAP9, NAPSC, and empty micelles.
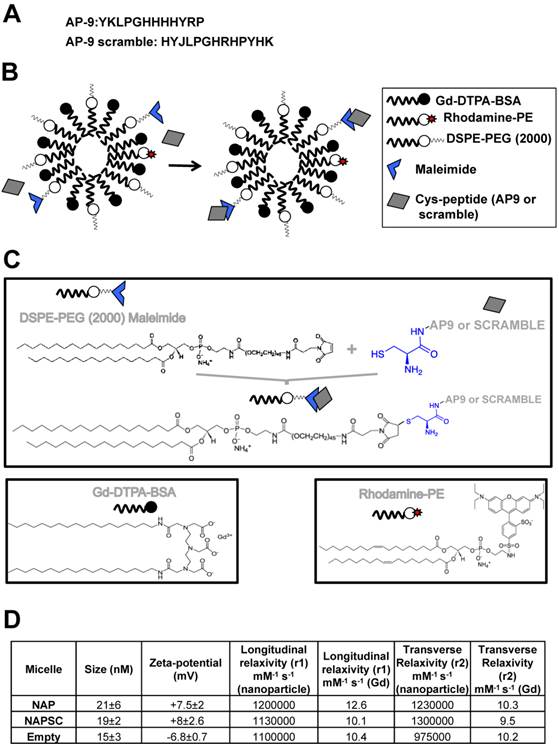
Physical and chemical properties, including Zeta-potential, nanoparticle size (calculated as hydrated diameter with dynamic laser light scattering (DSL, Malvern Zetasizer)), nanoparticle morphology (visualized with transmission electron microscopy (TEM)), longitudinal and transverse relaxivities, were calculated. Longitudinal relaxivities were evaluated by using inversion recovery sequences with 15 inversion times. Transverse relaxivities were calculated from spin-echo images and different echo times. In both cases, a series of images with different T1 and T2 weighting were generated. The r1 and r2 values were estimated from the slope of longitudinal and transverse relaxation rates vs Gd-DTPA or nanoparticle 8,9,10.
Affinity measurements
AP-9 peptide was labeled with Fluorescein isothiocyanate (FITC) (FluoroTAG-Conjugated KIT (Sigma-Aldrich)), according to the manufacturer´s instructions. FITC-labeled AP-9 (FAP-9) peptide and NAP9 micelles were used to examine affinity to endogenous EMMPRIN in cardiac myocytes. 105 adherent HL1 cells were plated into microwell plates and incubated with different concentrations ((10-7 M- 10-12 M) of FAP-9 or NAP9 for 5 hours at 37 °C. Binding experiments were performed in the absence or presence of a 100-fold excess AP-9 non-labelled peptide for competition assays. Specificity was tested by incubation FAP-9 or NAP-9 with 100 μM specific anti-EMMPRIN antibody. At the end of incubation, the cells were resuspended, then pelleted down, and fluorescence was measured in the supernatant (free peptide or micelle), and in the pellet (peptide or micelle bound to endogenous EMMPRIN) fractions. Kd values were calculated with GraphPad Prism software.
Cardiac Magnetic Resonance (CMR)
All magnetic resonance imaging (MRI) experiments were performed on a Bruker Biospec 47/40 (Bruker Biospin, Ettlingen, Germany) operating at 4.7 Tesla equipped with a 6 cm gradient system capable of reaching a maximal gradient strength of 450 mT/m.
Animals were anesthetized with a mixture of isoflurane and oxygen and then placed in prone position inside a 3.5 cm birdcage coil (Bruker Biospin, Ettlingen, Germany). Image acquisition was gated to the ECG and respiration signals using an external monitoring and gating system (SAII, New York, USA).
To localize the short axis image planes, two and four chambers images were acquired along the entire heart of the mouse. For these images an ECG and respiration-gated gradient echo images were used. The parameter for these experiments were: Repetition Time (TR) = 15 ms; Echo Time (TE) = 2.5 ms; Number of averaged experiments (NEX) = 2; Field of view (FOV) = 3 x 3 cm2; Slice Thickness (SLTH) = 1.0 mm; Acquired Matrix = 128 x 128.
Cardiac and respiratory triggered gradient echo images on the selected planes were acquired before and after the contrast agent injection. The measurements after injection were repeated every 15, 30, 60, and 120 minutes and in some cases 24 hours after the injection. The images on each plane were acquired separately. The parameters for these experiments were: TR = 200 ms; TE = 2.3 ms; flip angle = 80o; NEX = 8; matrix = 128 x 128; FOV = 2.5 x 2.5 cm2; SLTH = 1.0 mm.
Image analysis: For in vivo MR images, the signal-to-noise ratio (SNR) of the region of interest (ROI) is defined by SNRROI= IROI/Inoise where IROI is the intensity of either left ventricle of interest (for SNRlv) or surrounding muscle (for SNRm); Inoise is the standard deviation (S.D.) outside the animal. The normalized enhancement ratio of left ventricle (NERlv) to muscle is defined as:
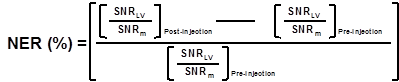
SNR=Signal to noise ratio= Mean signal intensity/SD.
Eight MR images of the matched (pre- and post-contrast) slices for each mouse were used for analysis.
Statistical analysis
Unless otherwise specified, data are expressed as means ± SD. Cell culture experiments were performed in triplicate, and conditions were assayed in duplicate on each replicate. Animal experiments were performed in triplicate, and the numbers of animals and replicates are specified in the text. Whenever comparisons were made with a common control, significance of differences was tested by analysis of variance followed by Dunnett's modification of the T test. Differences were considered significant at p<0.05. Error bars represent ± SD.
Results
NAP9 nanoparticle binds to EMMPRIN in vitro and in vivo
Targeted micelles were generated by adding a cysteine residue at the N-terminus of EMMPRIN binding peptide AP-9 (NAP9), and linked to the maleimide moiety of the micelles. Control micelles were generated by adding a cysteine residue to an AP-9 scrambled peptide and conjugated as before (NAPSC) (Fig. 1A, B, and C). Physical and chemical properties of the micelles are shown (Fig 1D).
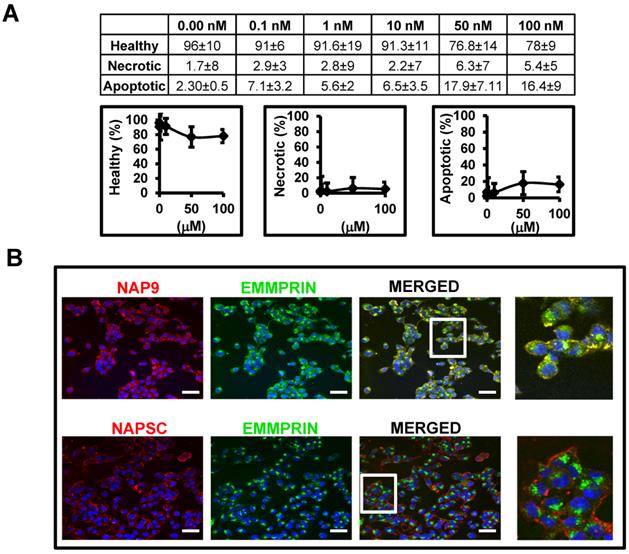
Affinity of AP-9 peptide and NAP9 micelles to endogenous EMMPRIN were calculated in HL1 myocytes (see methods for details) showing predicted binding affinities Kd=8 ± 0.6 nM, and Kd=6 ± 0.9 nM respectively. In the same way, nanoprobe cytotoxicity was tested in HL1 myocyte cell cultures 48 hours after incubation with increasing amounts of NAP9, and the percentages of living, necrotic, and apoptotic cells were determined by flow cytometry (Fig. 2A). We selected a concentration of 10 nM NAP9 as the maximal nanoparticle concentration with no significant effect on cell viability (Fig. 2A), and visualized by confocal microscopy (Cy3, red, Fig 2B). Co-localization of micelles with endogenous EMMPRIN (FYCT, green, Fig. 2B) was restricted to NAP9 positive cells (Merged, co-localization in yellow. Fig. 2B).
NAP9 binds to EMMPRIN in HL1 myocyte cells. A. Percentage of healthy, necrotic, and apoptotic HL1 myocytes incubated with NAP9 at the dosages shown. B. Confocal microscopy visualization of NAP9, NAPSC, and EMMPRIN in HL1 myocytes. Cells were incubated 1 hour with 10nM NAP9 or NAPSC. Nanoprobe uptake was evaluated by detection of Rhodamine (NAP9, and NAPSC, red). Endogenous EMMPRIN was detected in green (anti-EMMPRIN primary antibodies and FITC-conjugated secondary antibodies (green)), Co-localization was detected by merging both fluorescing signals (Merged panels, yellow). Scale bars, 50 μm (n= 3 by triplicate).
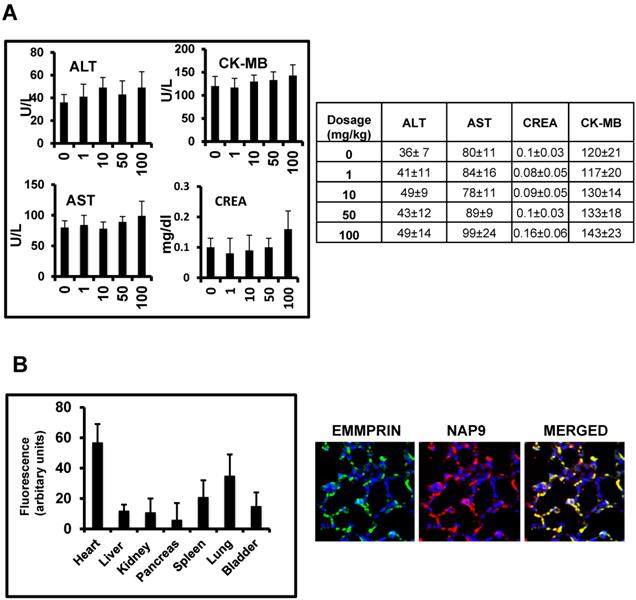
In vivo toxicity of NAP9 was assayed in healthy mice 10 days after IV administration of 1, 10, 50 and 100 mg/kg NAP9, by measuring serum levels of aspartate transaminase (AST) and alanine transaminase (ALT), as markers of liver performance, creatinine, an indicator of kidney functionality, and creatinine kinase-MB, indicative of cardiac necrosis, showing no significant signs of liver, kidney or heart muscle toxicity (Fig. 3A). In addition, NAP9 biodistribution was tested by confocal microscopy in the heart, liver, kidney, pancreas, spleen, lungs and bladder of 50 mg/Kg NAP9 injected mice 24 hours after IR, showing the higher uptake of NAP9 in the hearts and lungs of these mice (Fig. 3B, left). It may be that the levels of NAP9 present in the lungs respond to a significant accumulation of micelles trapped in the pulmonary microvasculature. Nevertheless, no aggregates were visualized; rather a strong co-localization of NAP9 with endogenous EMMPRIN in the pulmonary alveoli was detected by confocal microscopy (Fig. 3B, right).
In vivo toxicity of NAP9. A. Mice were injected with NAP9 at the dosages shown. After 10 days, plasma levels of hepatic alanine transaminase (ALT), aspartate transaminase (AST), creatinine (Crea) and creatinine kinase (CK-MB) were measured. (n=9 mice). B. Tissue distribution of 50 mg/Kg NAP9 in the same mice as before, as detected by confocal microscopy. Right. Confocal microscopy visualization of endogenous EMMPRIN (green) and NAP9 (red) in crossed sections of pulmonary tissue. Merged plane shows co-localization of both signals in yellow.
NAP9 uptake into mouse hearts is increased in response to IR
Mice were subjected to IR and injected with 1, 10, 50, or 100 mg/kg NAP9, NAPSC or saline (Control) and 24 hours after IR, cardiac parameters were evaluated by cardiac ultrasound; the animals were then sacrificed for further analysis (Fig. 4A).
NAP9 binds to EMMPRIN in mouse hearts subjected to IR. A. Study design. B. Confocal microscopy visualization of NAP9, or NAPSC (rhodamine, red), and endogenous EMMPRIN (FITC, green) in healthy hearts (upper panels) and in hearts subjected to IR (lower panels). Scale bars: 50 μm (n=9 mice/condition). C. Confocal microscopy visualization of NAP9 (rhodamine, red) and the macrophage marker CD68 (FlTC, green) or the muscle cell myosin heavy chain (MHC, FITC, green) in hearts subjected to IR (n=9 mice).
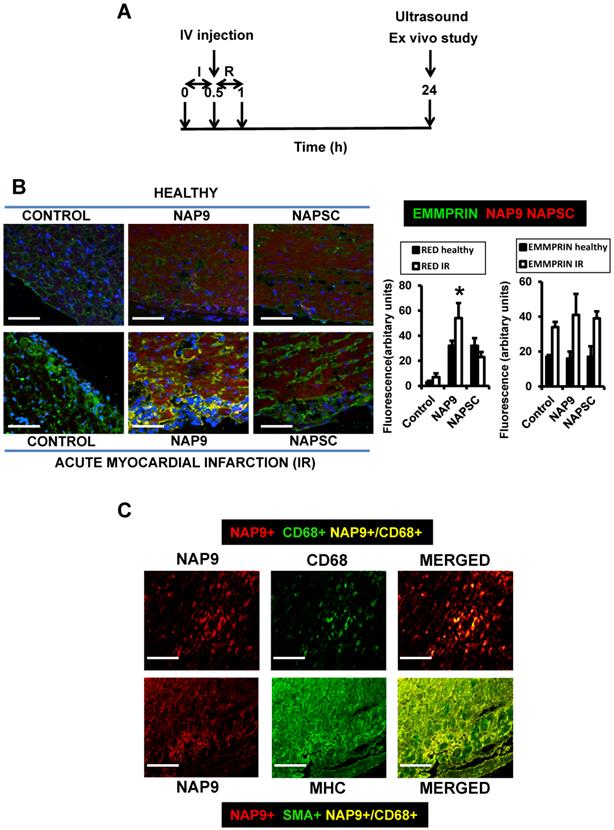
NAP9 uptake was detected by confocal microscopy in the hearts of healthy mice, and in mice subjected to IR. A dosage of 50mg/kg NAP9 was selected as the amount of NAP9 visualized by confocal microscopy with lower toxicity (Figs. 3A, and 4B).
The expression of endogenous EMMPRIN was stimulated in cardiac cells as a result of IR, as expected (Fig. 4B, Control lower panel), and NAP9 but not NAPSC co-localized with endogenous EMMPRIN in the heart (NAP9 and NAPSC lower panels, yellow).
The uptake of NAP9 was maximal in the heart sections of mice subjected to IR, when compared to the hearts of healthy mice, or with mice subjected to IR and injected with 50 mg/Kg NAPSC (Fig. 4B lower panel, NAP9).
Double immunostaining of heart sections of mice subjected to IR with Rhodamine and anti-MHC antibody, or with rhodamine and anti-CD68 antibody shows specificity of NAP9 uptake by cardiac myocytes and infiltrated macrophages (Fig. 4C).
NAP9 induces cardiac protection in mice subjected to IR
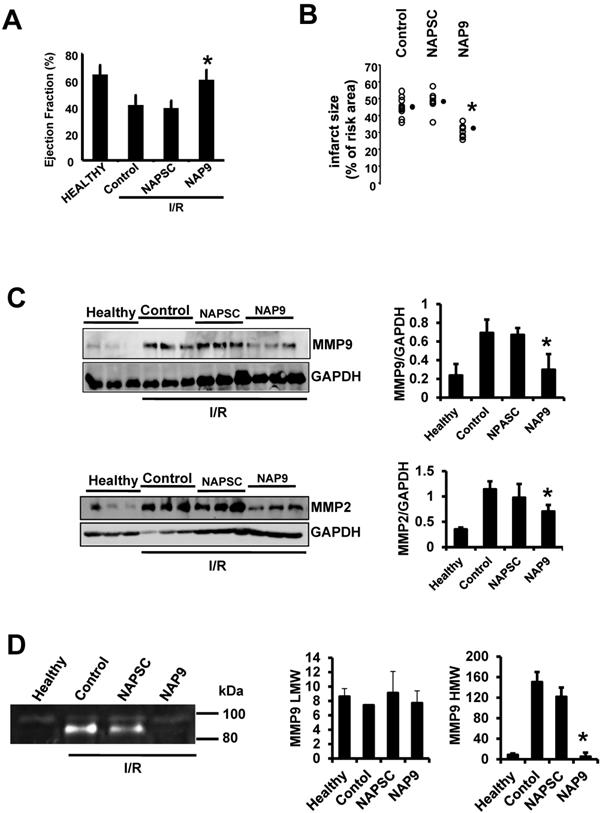
IV injection of 50 mg/kg NAP9 into mice subjected to IR improved heart contractility (Fig. 5A. LVEF: NAP9 63% ± 7.24 vs Control 42% ± 4.74 or NAPSC 39% ± 6.44 respectively), and reduced cardiac necrosis (Fig. 5B, Infarct size: NAP9 32%±6.59 vs Control 46%±9.04 or NAPSC 48%±7.64), when compared to mice injected with NAPSC or saline (Control) under the same conditions. Table 1 lists the cardiac parameters collected after M-mode ultrasound evaluation of mice.
NAP9 induces cardiac protection in mice subjected to IR. A. LVEF values from non ischemic (healthy) mice and from mice who underwent IR for 24 hours and were injected with 50 mg/Kg NAP9, NAPSC, or saline (Control) right after ischemia (n=9 mice/group. Mean ± SD; *p <0.05, NAP9 vs Control). B. Measurement of LV necrotic area as percentage with respect to the area at risk, detected by double Evans Blue/TTC staining (n = 18 mice/group; mean ± SD; *p <0.05, NAP9 vs Control). C. Immunoblot detection of MMP9 (upper panel), and MMP-2 (lower panel) in non ischemic (Healthy) mice, and in mice injected with 50 mg/Kg NAP9, NAPSC, or saline (Control) right after reperfusion. GAPDH was used as loading control (n=9 mice/group; mean ± SD; *p <0.05, NAP9 vs Control and NAPSC). D. Detection of MMP-9 by Gelatin zymography in the same hearts as in C. The two bands of 92 kDa and 87kDa correspond with the sizes of proMMP-9 and active-MMP-9 respectively. LMW: low molecular weight. HMG: high molecular weight (n=9 mice/group; mean ± SD; *p <0.05, NAP9 vs Control and NAPSC).
Cardiac function.
| Healthy | Control (Saline) | NAP9 | NAPSC | |
|---|---|---|---|---|
| LVDS (mm) | 2.752±0.49 | 3.593±0.65 | 3.012±0.53 | 3.415±0.69 |
| LVDD (mm) | 4.201±0.21 | 4.551±0.51 | 4.302±0.37 | 4.617±0.49 |
| VS (ml) | 25.271±2.88 | 52.953±7.03 * | 32.217±4.11 | 50.216±7.06 § |
| VD (ml) | 66.408±10.46 | 79.355±11.52 | 72.396±6.14 | 81.046±9.32 |
| SV (ml) | 41.137±3.2 | 26.402±1.25 | 40.179±5.61 | 30.83±5.22 |
| EF (%) | 61.946±4.38 | 33.271±5.92 * | 55.499±4.25 | 38.04±4.32 § |
| FS (%) | 34.497±2.95 | 21.05±2.45 | 29.986±4.74 | 26.034±1.89 |
| Heart rate (bpm) | 389±12 | 388±12 | 402±11 | 395±17 |
LVDS, left-ventricle end-systolic diameter; LVDD, left-ventricle end-diastolic diameter; VS, systolic volume; VD, diastolic volume; SV, systole-diastole volume difference; EF, left-ventricle ejection fraction; FS, shortening fraction.
*p<0.05 Healthy vs Control. §p<0.05 Healthy vs NAPSC.
We previously found that IR induces the expression of EMMPRIN in mouse hearts7, leading to the expression and activity of different MMPs, including gelatinases A and B (MMP-2, and 9 respectively), two crucial ECM-degrading enzymes of heart tissue in response to ischemic conditions. To further illustrate the contribution of NAP9 in the survival of cardiac myocytes, we analyzed the levels of MMP-2, and MMP-9, indicative of EMMPRIN activation in the heart. We found that in mice injected with 50 mg/Kg NAP9, the levels of MMP-2 and MMP-9 were almost reduced to those found in healthy animals (Fig 5B, and C). Taken together, these results show that NAP9 improves heart contractility at least in part by targeting EMMPRIN thorough the AP9-conjugated peptide, since empty micelles (Fig. 5B, Control micelles) or micelles containing AP-9 scrambled peptides (Fig. 5B, NAPSC micelles) did not reduce the levels of MMP-2 and MMP-9, nor did they improve heart contractility.
Non invasive Magnetic Resonance Imaging of NAP9 correlates with ex vivo LV infarction size
To test whether NAP9 could have a theranostic effect, we performed CMR scans in healthy mice or in mice subjected to IR, and injected with 50 mg/Kg NAP9, NAPSC, or saline (Control) (Fig. 6A). In healthy animals, neither NAP9 nor NAPSC caused enhancement of the hearts at 24 hours post injection (Fig. 6B upper panels). By contrast, in mice under IR, injection of NAP9 caused a significant enhancement of the heart when compared to Control and NAPSC injected animals (Fig. 6B. Right. Graphical representation corresponding to 3 independent experiments showing normalized enhancement ratios: Control IR 2.24 ± 4.86, NAPSC IR 6.96 ± 2.93%, NAP9 IR 24.39 ± 7.22%).
Non invasive CMR visualization of NAP9 in mice subjected to IR, correlates with ex vivo Evans Blue/TTC measures taken in the same mice. A. CMR study design. B. Parasternal short axis view of hearts from healthy mice (upper), or mice who underwent IR for 24 hours (IR, lower), and were injected with 50 mg/Kg NAP9, NAPSC, or saline (Control). Bottom. Detailed magnification of NAP9-injected healthy mice, or mice under IR. Green lines show Gadolinium enhanced areas corresponding to nanoprobe uptake. Blue and red dotted lines are marking external and internal LV walls respectively. Right graph shows the result of 3 independent assays (n=9 mice/condition; mean ± SD; *p <0.05, NAP9 vs Control and NAPSC in AMI). C. Scattered plot representing the extension of NAP9 uptake as detected by CMR (Y axis) with respect to the extension of infarct size, estimated by Evans Blue/TTC staining (X-axis) (R=0.69; p<0.05).
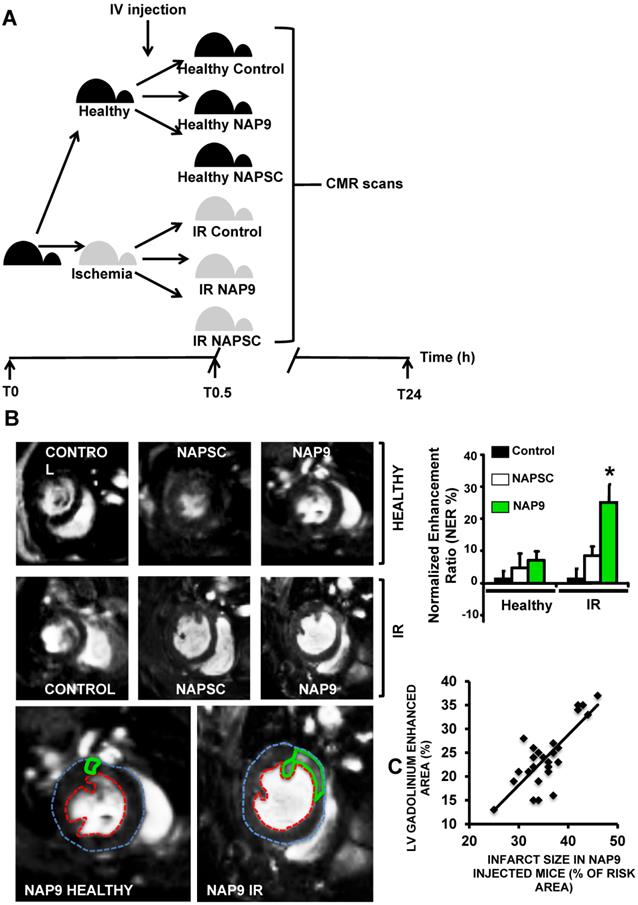
The same mice were used to evaluate ex vivo cardiac damage by Evans Blue/TTC staining, detecting a positive correlation between NAP9 uptake, as detected by CMR, with ex vivo measurement of infarct size (Fig. 6C; R=0.65, p<0.05).
The cardioprotective effect of NAP9 is maximal within the first 24 hours after injection
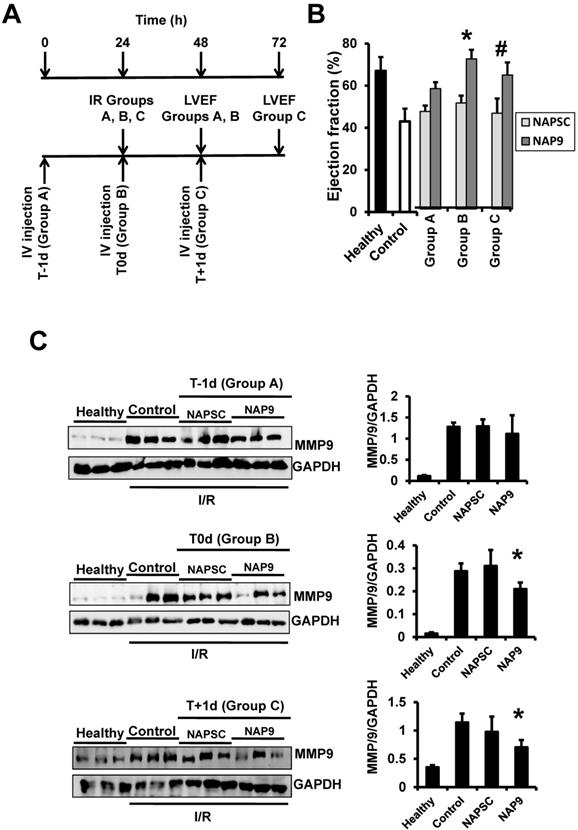
We found that NAP9 induces cardiac protection in mouse hearts subjected to IR. To determine the best time for nanoprobe administration, mice were injected with 50mg/kg NAP9, or NAPSC, 1 day before IR (t-1d, group A), at the time of ischemia (t0d, group B), and 1 day after IR (t+1d, group C) (Fig. 7A), and ventricular function (Fig. 7B), and MMP-9 expression (Fig 7C) were analyzed 24 hours after the procedures. The cardioprotective effect of NAP9 was maximal in mice injected with NAP9 at the time of ischemia (t0) and one day after (t1) IR, with respect to control mice and to mice injected with 50 mg/Kg NAPSC and subjected to IR (Fig. 7B,C). By contrast, administration of NAP9 before ischemia (t-1) had a non significant effect on LVEF (Fig. 7B), and MMP-9 expression (Fig. 7C).
NAP9 has no effect before IR. A. Study design. B. LVEF of healthy hearts or hearts from mice subjected to IR and injected one day before (Group A: T-1d) at the same time (Group B: T0d) or one day after (Group C: T+1d) with 50 mg/Kg NAP9, NAPSC, or saline (Control), and measured 24 hours after injection (see schematic representation of the assay) (n=10 mice/condition; mean ± SD; *p <0.05, NAP9 t0 vs NAP9 t-1d; p <0.05, NAP9 t+1d vs NAP9 t-1d). B Immunoblot detection of MMP-9 in the same hearts as before. GADP was used as loading control (n=9 mice/group *p <0.05, NAP9 vs Control).
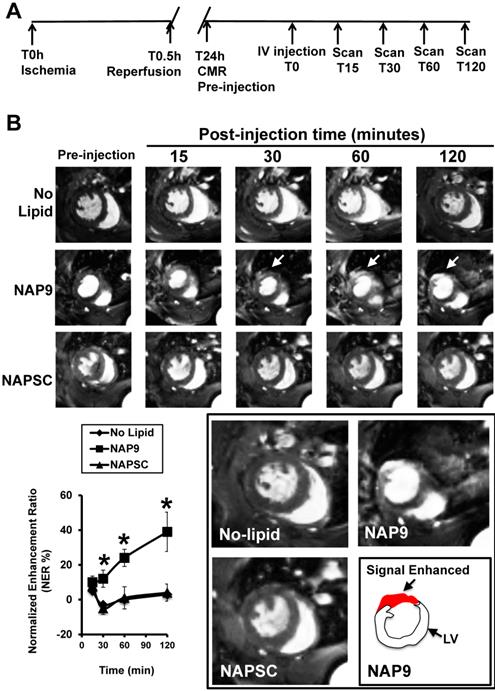
Mice were subjected to IR and injected 24 hours later with 50 mg/Kg NAP9 or NAPSC, and CMR scans were performed at different times after injection (Fig. 8A). NAP9 was visualized within the first 30 minutes and the signal was enhanced within the next two hours post injection (Fig. 8B). Taken together, these results suggest that NAP9 might be used to inhibit and to quantify the extension of myocardial cell death during the onset and progression of AMI by targeting EMMPIN in vivo.
NAP9 is detected in mouse hearts within the first 30 minutes of injection. A. Study design and timeline of imaging assessments. B. Parasternal shot axis view of mouse hearts at the times indicated after injection of 50mg/Kg NAP9 or NAPSC. White arrows point to gadolinium areas of enhancement. Bottom left: Normalized enhancement ratios. (n=9 mice/condition. mean ± SD; *p <0.05, NAP9 vs No lipid and NAPSC). Bottom right, magnification of CMR images recorded 120 minutes after injection. The area of signal enhancement is represented in red.
Discussion
Extracellular matrix degradation is a key step that precedes cardiac remodeling as a result of coronary artery occlusion, and subsequent ischemia and reperfusion12. In the current study, we found that targeting extracellular matrix metalloproteinase inducer (EMMPRIN) with paramagnetic fluorescent micellar nanoparticles successfully reduced reperfusion damage in hearts subjected to AMI. Likewise, extracellular matrix degradation as detected by expression of matrix metalloproteinase MMP-9 was notably reduced in mice injected with NAP9, when compared with NAPSC, containing an AP scrambled peptide. CMR scans in mice subjected to IR revealed a signal enhancement after injection with NAP9, whereas scans in mice injected with nanoparticles conjugated with AP9 scrambled peptide (NAPSC) did not show differences with respect to Control mice. Confocal microscopy detection of NAP9 in the same hearts confirmed the result, and in addition, NAP9 co-localized with endogenous EMMPRIN. A positive correlation between infarct size and CMR signal enhancement was also identified, which may suggest that future consideration should be given to this technology for fast, non-invasive detection and heart performance improvement in humans subjected to AMI.
Targeting of EMMPRIN was investigated as a therapeutic mechanism against numerous pathologies including several types of cancer, and kidney, liver, lung, and pancreatic diseases 13-19. The involvement of EMMPRIN in cardiovascular diseases was also evidenced 20. In atherosclerosis, induction of EMMPRIN by monocyte and proinflammatory macrophages plays a pivotal role in foam cell and plaque formation 21, smooth muscle cell proliferation and migration 22, and platelet activation; it significantly contributes to plaque burden, as well 23. In the heart, infiltrated monocytes and cardiac myocytes produce large amounts of EMMPRIN in response to ischemia, promoting MMP-mediated extracellular matrix degradation and cardiac cell death 24-26. In this regard, we found that antibody-mediated EMMPRIN inhibition reduced infarct size in murine models of IR 7.
Paramagnetic micelles have been used to target cells 27 and molecules 28 in the onset and progression of several diseases, by coupling different molecules to target the desired cell type, enzyme, binding protein or, very recently, pathophysiological conditions like apoptosis, angiogenesis, or even ischemia29-31. We targeted EMMPRIN by using gadolinium based-paramagnetic micelles containing specifically EMMPRIN binding peptides for non-invasive visualization of EMMPRIN by molecular MRI.
The use of antibody-mediated targeting of proteins has been successfully evaluated in animal models of disease 32. However, such an immunological approach was only effective in very few studies in humans, and therefore the translation to the clinic is limited for reasons that include immune cross reactivity, antibody-mediated side effects, or lack of desired therapeutic improvement. To avoid undesired immune reactivity, an efficient alternative is the use of smaller binding molecules to the target of interest, including specific binding peptides 15, 33. To our knowledge, the use of specific EMMPRIN-binding peptides has only been tested in vitro to inhibit EMMPRIN downstream MMP activation in 293 cells 34, in monocyte THP-1 cells 35, and in peripheral blood monocytes isolated from patients with rheumatoid arthritis 36. We found, for the first time, that paramagnetic nanoparticles conjugated with AP9 peptides bind to EMMPRIN as detected by confocal microscopy, and in vivo by molecular CMR. In addition, administration of NAP9 reduces downstream expression of MMP-2 and MMP-9 in the heart, and improves cardiac contraction when compared to AP9-scrambled conjugated nanoparticles. Nevertheless, a future translation to the bed side will require additional nanoparticle formulation to avoid potential nanoparticle-derived side effects.
A close correlation between the CMR signal enhancement and myocardial infarction was calculated in NAP9 injected mice subjected to IR. However, NAP9 had no effect when administered 1 day before IR, whereas upon administration at the time of and one day after IR, NAP9 improved LVEF and reduced MMP-9 expression, which may be explained due to reduced expression of EMMPRIN in healthy hearts.
In conclusion, NAP9 nanotherapy was shown to be effective to improve performance of hearts subjected to IR, restoring functional parameters and preventing MMP-mediated ECM degradation. Consequently, further studies in other species will be crucial for future testing in humans.
Abbreviations
EMMPRIN: Extracellular matrix metalloproteinase inducer. AMI: Acute myocardial infarction. MMP-9: Matrix metalloproteinase-9. MMP-13: Matrix metalloproteinase-13. MRI: Magnetic resonance imaging. LVEF: Left ventricle ejection fraction. LAD: Left anterior descending coronary artery.
Acknowledgements
Sources of funding
This work was supported by research grants from CZ: “Ministerio de Economía y Competitividad” SAF 2011-28375, “Instituto de Salud Carlos III” PI14/02022 (Plan Estatal de I+D+i 2013-2016) and co-financed by the European Development Regional Fund ''A way to achieve Europe'' (ERDF), and “Universidad Francisco de Vitoria 2014”. MS: “Ministerio de Economía y Competitividad” SAF 2012-35141 “Junta Castilla la Mancha” PPII2014-023-P co-financed by the European Development Regional Fund ''A way to achieve Europe'' (ERDF).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hausenloy DJ. Conditioning the heart to prevent myocardial reperfusion injury during ppci. Eur Heart J. 2012;1:13-32
2. Fernandez-Velasco M, Gonzalez-Ramos S, Bosca L. Involvement of monocytes/macrophages as key factors in the development and progression of cardiovascular diseases. Biochem J. 2014;458:187-193
3. Newby AC. Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol. 2008;28:2108-2114
4. Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, Schoen FJ, Kelly RA, Werb Z, Libby P, Lee RT. Targeted deletion of Matrix Metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55-62
5. Schmidt R, Bultmann A, Ungerer M, Joghetaei N, Bulbul O, Thieme S, Chavakis T, Toole BP, Gawaz M, Schomig A, May AE. Extracellular Matrix Metalloproteinase Inducer regulates matrix metalloproteinase activity in cardiovascular cells: Implications in acute myocardial infarction. Circulation. 2006;113:834-841
6. Seizer P, Ochmann C, Schonberger T, Zach S, Rose M, Borst O, Klingel K, Kandolf R, MacDonald HR, Nowak RA, Engelhardt S, Lang F, Gawaz M, May AE. Disrupting the EMMPRIN (CD147)-Cyclophilin A interaction reduces infarct size and preserves systolic function after myocardial ischemia and reperfusion. Arterioscler Thromb Vasc Biol. 2011;31:1377-1386
7. Tarin C, Lavin B, Gomez M, Saura M, Diez-Juan A, Zaragoza C. The extracellular matrix metalloproteinase inducer EMMPRIN is a target of nitric oxide in myocardial ischemia/reperfusion. Free Radic Biol Med. 2011;51:387-395
8. Meiboom S, Gill D. Modified spin-echo method for measuring nuclear relaxation times. Rev. Sci. Instrum. 1958;29:688-691
9. Carr HY, Purcell EM. Effects of diffusion on free precession in nuclear magnetic resonance experiments. Phys. Rev. 1954;94:630-638
10. Messroghli DR, Radjenovic A, Kozerke S, Higgins DM, Sivananthan MU, Ridgway JP. Modified Look-Locker inversion recovery (MOLLI) for high-resolution T1 mapping of the heart. Magnetic Resonance in Medicine. 2004;52:141-146
11. Chen W, Cormode DP, Vengrenyuk Y, Herranz B, Feig JE, Klink A, Mulder WJ, Fisher EA, Fayad ZA. Collagen-specific peptide conjugated HDL nanoparticles as MRI contrast agent to evaluate compositional changes in atherosclerotic plaque regression. JACC. 2013;6:373-384
12. Zavadzkas JA, Stroud RE, Bouges S, Mukherjee R, Jones JR, Patel RK, McDermott PJ, Spinale FG. Targeted overexpression of tissue inhibitor of matrix metalloproteinase-4 modifies post-myocardial infarction remodeling in mice. Circ Res. 2014;114:1435-1445
13. Hao J, Madigan MC, Khatri A, Power CA, Hung TT, Beretov J, Chang L, Xiao W, Cozzi PJ, Graham PH, Kearsley JH, Li Y. In vitro and in vivo prostate cancer metastasis and chemoresistance can be modulated by expression of either cd44 or cd147. PloS one. 2012;7:e40716
14. Kong LM, Liao CG, Zhang Y, Xu J, Li Y, Huang W, Bian H, Chen ZN. A regulatory loop involving miR-22, Sp1, and c-Myc modulates CD147 expression in breast cancer invasion and metastasis. Cancer Res. 2014;74:3764-3778
15. Le Floch R, Chiche J, Marchiq I, Naiken T, Ilc K, Murray CM, Critchlow SE, Roux D, Simon MP, Pouyssegur J. CD147 subunit of lactate/H+ symporters MCT1 and hypoxia-inducible MCT4 is critical for energetics and growth of glycolytic tumors. Proc Natl Acad Sci U S A. 2014;108:16663-16668
16. Kosugi T, Maeda K, Sato W, Maruyama S, Kadomatsu K. CD147 (EMMPRIN/Basigin) in kidney diseases: from an inflammation and immune system viewpoint. Nephrol Dial Transplant. 2015;30:1097-103
17. Zhang DW, Zhao YX, Wei D, Li YL, Zhang Y, Wu J, Xu J, Chen C, Tang H, Zhang W, Gong L, Han Y, Chen ZN, Bian H. HAb18G/CD147 promotes activation of hepatic stellate cells and is a target for antibody therapy of liver fibrosis. J Hepatol. 2012;57:1283-1291
18. Gwinn WM, Damsker JM, Falahati R, Okwumabua I, Kelly-Welch A, Keegan AD, Vanpouille C, Lee JJ, Dent LA, Leitenberg D, Bukrinsky MI, Constant SL. Novel approach to inhibit asthma-mediated lung inflammation using anti-CD147 intervention. J Immunol. 2006;177:4870-4879
19. Li L, Tang W, Wu X, Karnak D, Meng X, Thompson R, Hao X, Li Y, Qiao XT, Lin J, Fuchs J, Simeone DM, Chen ZN, Lawrence TS, Xu L. HAb18G/CD147 promotes pSTAT3-mediated pancreatic cancer development via CD44s. Clin Cancer Res. 2013;19:6703-6715
20. Seizer P, Gawaz M, May AE. Cyclophilin A and EMMPRIN (CD147) in cardiovascular diseases. Cardiovasc Res. 2014;102:17-23
21. Seizer P, Geisler T, Bigalke B, Schneider M, Klingel K, Kandolf R, Stellos K, Schreieck J, Gawaz M, May AE. EMMPRIN and its ligand cyclophilin A as novel diagnostic markers in inflammatory cardiomyopathy. Int J cardiol. 2010;163:299-304
22. Venkatesan B, Valente AJ, Reddy VS, Siwik DA, Chandrasekar B. Resveratrol blocks interleukin-18-EMMPRIN cross-regulation and smooth muscle cell migration. Am J Physiol Heart Circ Physiol. 2009;297:H874-886
23. Schmidt R, Bultmann A, Fischel S, Gillitzer A, Cullen P, Walch A, Jost P, Ungerer M, Tolley ND, Lindemann S, Gawaz M, Schomig A, May AE. Extracellular matrix metalloproteinase inducer (CD147) is a novel receptor on platelets, activates platelets, and augments nuclear factor kappaB-dependent inflammation in monocytes. Circ Res. 2008;102:302-309
24. Reddy VS, Prabhu SD, Mummidi S, Valente AJ, Venkatesan B, Shanmugam P, Delafontaine P, Chandrasekar B. Interleukin-18 induces EMMPRIN expression in primary cardiomyocytes via JNK/Sp1 signaling and MMP-9 in part via EMMPRIN and through AP-1 and NF-kappaB activation. Am J Physiol Heart Circ Physiol. 2010;299:H1242-1254
25. Nie R, Xie S, Du B, Liu X, Deng B, Wang J. Extracellular matrix metalloproteinase inducer (EMMPRIN) is increased in human left ventricle after acute myocardial infarction. Arch Med Res. 2009;40:605-611
26. Xie SL, Chen YY, Zhang HF, Deng BQ, Shu XR, Su ZZ, Lin YQ, Nie RQ, Wang JF. Interleukin 18 and extracellular matrix metalloproteinase inducer cross-regulation: Implications in acute myocardial infarction. Transl Res. 2015;165:387-95
27. Mulder WJ, Strijkers GJ, Briley-Saboe KC, Frias JC, Aguinaldo JG, Vucic E, Amirbekian V, Tang C, Chin PT, Nicolay K, Fayad ZA. Molecular imaging of macrophages in atherosclerotic plaques using bimodal PEG-micelles. Magn Reson Med. 2007;58:1164-1170
28. Chen W, Jarzyna PA, van Tilborg GA, Nguyen VA, Cormode DP, Klink A, Griffioen AW, Randolph GJ, Fisher EA, Mulder WJ, Fayad ZA. RGD peptide functionalized and reconstituted high-density lipoprotein nanoparticles as a versatile and multimodal tumor targeting molecular imaging probe. The FASEB J. 2010;24:1689-1699
29. Hwang H, Kwon J, Oh PS, Lee TK, Na KS, Lee CM, Jeong HS, Lim ST, Sohn MH, Jeong HJ. Peptide-loaded nanoparticles and radionuclide imaging for individualized treatment of myocardial ischemia. Radiology. 2014;273:160-167
30. Yu Q, Liu Y, Cao C, Le F, Qin X, Sun D, Liu J. The use of pH-sensitive functional selenium nanoparticles shows enhanced in vivo VEGF-siRNA silencing and fluorescence imaging. Nanoscale. 2014;6:9279-9292
31. Liu J, Liu Y, Bu W, Bu J, Sun Y, Du J, Shi J. Ultrasensitive nanosensors based on upconversion nanoparticles for selective hypoxia imaging in vivo upon near-infrared excitation. J Am Chem Soc. 2014;136:9701-9709
32. Marsden CJ, Eckersley S, Hebditch M, Kvist AJ, Milner R, Mitchell D, Warwicker J, Marley AE. The use of antibodies in small-molecule drug discovery. J Biomol Screen. 2014;19:829-838
33. Liu Z, Wang F. Development of rgd-based radiotracers for tumor imaging and therapy: Translating from bench to bedside. Curr Mol Med. 2013;13:1487-1505
34. Chen Z, Mi L, Xu J, Yu J, Wang X, Jiang J, Xing J, Shang P, Qian A, Li Y, Shaw PX, Wang J, Duan S, Ding J, Fan C, Zhang Y, Yang Y, Yu X, Feng Q, Li B, Yao X, Zhang Z, Li L, Xue X, Zhu P. of HAb18G/CD147 in invasion of host cells by severe acute respiratory syndrome coronavirus. J Infect Dis. 2005;191:755-760
35. Zhou J, Zhu P, Jiang JL, Zhang Q, Wu ZB, Yao XY, Tang H, Lu N, Yang Y, Chen ZN. Involvement of CD147 in overexpression of MMP-2 and MMP-9 and enhancement of invasive potential of PMA-differentiated THP-1. BMC Cell Biol. 2005;6:25
36. Yang Y, Lu N, Zhou J, Chen ZN, Zhu P. Cyclophilin A up-regulates MMP-9 expression and adhesion of monocytes/macrophages via CD147 signalling pathway in rheumatoid arthritis. Rheumatology (Oxford). 2008;47:1299-1310
Author contact
![]() Corresponding author: c.zaragoza.profes.
Corresponding author: c.zaragoza.profes.