Theranostics
13.3
Impact Factor
 Global reach, higher impact
Global reach, higher impact
Theranostics 2016; 6(1):14-27. doi:10.7150/thno.13515 This issue Cite
Research Paper
Triple-Layered pH-Responsive Micelleplexes Loaded with siRNA and Cisplatin Prodrug for NF-Kappa B Targeted Treatment of Metastatic Breast Cancer
Haijun Yu1,#, ![]() , Chengyue Guo1,2,#, Bing Feng1, Jianping Liu1, Xianzhi Chen1, Dangge Wang1, Lesheng Teng2, Youxin Li2, Qi Yin1, Zhiwen Zhang1, Yaping Li1,
, Chengyue Guo1,2,#, Bing Feng1, Jianping Liu1, Xianzhi Chen1, Dangge Wang1, Lesheng Teng2, Youxin Li2, Qi Yin1, Zhiwen Zhang1, Yaping Li1, ![]()
1. State key Laboratory of Drug Research & Center of Pharmaceutics, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China;
2. College of Life Science, Jilin University, Changchun, 130012, China.
#: These two authors contributed equally to this study.
Received 2015-8-9; Accepted 2015-9-15; Published 2016-1-1
Citation:
Yu H, Guo C, Feng B, Liu J, Chen X, Wang D, Teng L, Li Y, Yin Q, Zhang Z, Li Y. Triple-Layered pH-Responsive Micelleplexes Loaded with siRNA and Cisplatin Prodrug for NF-Kappa B Targeted Treatment of Metastatic Breast Cancer. Theranostics 2016; 6(1):14-27. doi:10.7150/thno.13515. https://www.thno.org/v06p0014.htm
Other stylesAbstract

The combination of chemotherapy and RNA interference is a promising approach for efficient cancer therapy. However, the success of such a strategy is hampered by the lack of suitable vectors to coordinate small interfering RNA (siRNA) and chemotherapeutic drug into one single platform. We herein report a novel triple-layered pH-responsive micelleplex loading siRNA and alkylated cisplatin prodrug for NF-Kappa B targeted treatment of metastatic breast cancer. The micelles were self-assembled from poly(ethylene glycol)-block-poly(aminolated glycidyl methacrylate)-block-poly(2-(diisopropyl amino) ethyl methacrylate) (PEG-b-PAGA-b-PDPA) triblock copolymers. At pH 7.4, the cisplatin prodrug was encapsulated in the hydrophobic PDPA core and siRNA was loaded on the positively charged PAGA interlayer to form the micelleplexes. The PEG corona can prevent protein absorption during blood circulation, minimize non-specific interaction with the reticuloendothelial system, and prolong the systemic circulation of the micelleplexes. The positively charged PAGA interlayer can facilitate deep tumor penetration of the micelleplexes, which, upon cellular uptake, are dissociated in the early endosomes to release anticancer drug payload due to protonation of the PDPA core. Using a 4T1 breast cancer model, we demonstrate that this novel micelleplex co-loaded with cisplatin prodrug and siRNA-p65 is able to simultaneously inhibit tumor growth and suppress distant metastasis of the cancer cells by downregulating NF-kappa B expression. The results reported in this study suggest that siRNA and anticancer drug co-delivery using pH-responsive micelleplexes is a promising strategy for efficient treatment of metastatic cancer.
Keywords: Cancer Metastasis, Micelleplexes, pH-Responsive, RNAi, Chemotherapy.
Introduction
The dissemination of cancer cells from the primary tumor to nearby or distant organs, referred to as cancer metastasis, accounts for over 90% of cancer-caused human mortality.(1) Cancer metastasis is a complicated and multistep process, which is highly associated with angiogenesis and modulation of the microenvironment in target organs. Neoadjuvant chemotherapy is one mainstream option for clinical treatment of locally invasive or metastatic cancer.(2) However, evidence suggests that conventional chemotherapy using cytotoxic drugs tends to elicit the dissemination of cancer cells as a defense mechanism against the cytotoxic stress.(3) Antiangiogenic therapies were also extensively employed in the past to suppress tumor growth and metastasis by blocking the formation of new blood vessels.(4) Despite the promising potential, a combination of chemotherapy with antiangiogenic therapy, for instance, paclitaxel plus bevacizumab targeting vascular endothelia growth factor (VEGF) did not improve the overall survival rates compared to paclitaxel alone.(5) Similar to chemotherapy and radiotherapy, antiangiogenic therapy may also promote cancer metastasis by initiating more aggressive and invasive tumor phenotype.(6)
Recent advances in genomics and cancer biology have identified a positive correlation between cancer metastasis and over-activation of certain genes, for instance, the transcription factors Twist and nuclear factor kappa B (NF-κB).(7-9) Ectopic expression of Twist induces cell motility and invasion by promoting epithelial-mesenchymal transition (EMT) and reducing E-cadherin mediated cell-cell adhesion.(8) It has been reported that the distant metastasis of breast cancer could be significantly inhibited by blocking the Twist pathway.(12) On the other hand, NF-κB stimulates cell invasion and metastasis by upregulating matrix metalloproteinase (MMPs) and interleukin-8 (IL-8).(9,10) MMPs, more specifically, MMP-2 and MMP-9 are responsible for extracellular matrix (ECM) degradation and subsequent intravasation of the cancer cells from the primary tumor into either lymphatic or blood microvessels.(11) These findings imply a promising potential for a combination of chemotherapy with RNA interference (RNAi) therapy for treatment of metastatic breast cancer by inhibiting the proliferation and suppressing the motility of cancer cells.(13-15) To maximize the additive synergistic therapeutic efficacy of chemotherapy and RNAi, it is crucial to spatiotemporally co-localize anticancer drugs and siRNA in cancer cells. This can best be accomplished by using nanosized drug delivery systems (NDDS).(16,17) The NDDS can not only elongate the time of small molecular drugs in blood circulation but also passively accumulate inside the tumor via the enhanced penetration and retention (EPR) effect. Unfortunately, most of the currently used NDDS displayed unsatisfactory or only marginally improved therapeutic benefits due to the presence of several well-defined biological barriers.(17) First, NDDS administered systemically need to remain stable in the plasma during blood circulation. Second, to interact with tumor physiology, NDDS should efficiently evade reticuloendothelial system (RES) of the liver and renal clearance by the kidneys. Third, NDDS should be able to unload the therapeutics inside the cancer cells in a timely manner.(18,19) Furthermore, an additional prerequisite for siRNA and anticancer drug co-delivery is to integrate hydrophilic siRNA and hydrophobic anticancer drugs into one single platform. This represents a significant challenge for NDDS design since most of the clinically approved chemotherapeutic drugs display poor water solubility.
To address the aforementioned limitations of current NDDS and improve their therapeutic efficacy against metastatic breast cancer, we herein designed and developed a novel triple-layered micelleplex platform for the co-delivery of a hydrophobic cisplatin prodrug and siRNA. The micelles are self-assembled from a pH-responsive triblock copolymer poly(ethylene glycol)-block-poly(aminolated glycidyl methacrylate)-block-poly(2-(diisopropylamino) ethyl methacrylate) (PEG-b-PAGA-b-PDPA) and composed of three separated functional domains, which are a hydrophilic poly(ethylene glycol) (PEG) corona, a cationic intermediate layer, and a hydrophobic PDPA core, respectively. The PEG shell is integrated to shield protein absorption, prevent non-specific interaction with the RES, and elongate the systemic circulation of the siRNA-loaded micelleplexes. The aminolated interlayer possesses positive charge at physiological neutral pH due to protonation of the primary amino groups. Thus the interlayer can protect siRNA from serum degradation during blood circulation, and facilitate tumor penetration of the micelleplexes once they reach the tumor site.(20) In the PDPA segment can stabilize the micelleplexes during blood circulation and encapsulate the anticancer drug by forming a hydrophobic core. The PDPA core can be specifically dissociated in the early endosomes (pH = 5.9~6.2)(21,22) and trigger payload release via protonation of the PDPA tertiary amine at pH below 6.3,(23,24) thus avoiding siRNA degradation in lysosomes and drug elimination through the endocytic recycling pathway (Fig. 1).(25) It is expected that this novel micelleplex co-loading cisplatin prodrug and siRNA could simultaneously inhibit the growth and distant metastasis of highly invasive breast tumors by suppressing NF-κB expression.
Results and discussion
Synthesis and characterization of PEG-b-PAGA-b-PDPA triblock copolymer and micelles
To prepare the triple-layered pH-responsive micelleplexes, a parental triblock copolymer poly(ethylene glycol)-block-poly(glycidyl methacrylate)-block-poly(2- (diisopropylamino) ethyl methacrylate) (PEG-b-PGA-b-PDPA) was first synthesized by sequential atom transfer radical polymerization (ATRP) of glycidyl methacrylate (GA) and 2-(diisopropylamino) ethyl methacrylate (DPA) using mPEG225-Br as a macroinitiator (Fig. S1). The polymerization degree of PGA and PDPA segments was determined to be 45 and 50 respectively by 1H-nuclear magnetic resonance (1H-NMR) spectra examination (Fig. S2). The PGA block of PEG-b-PGA-b-PDPA copolymer was then reacted with a set of oligoamines (i.e. ethylenediamine EDA, diethylenetriamine DETA, triethylenetetramine TETA and tetraethylenepentamine TEPA) to obtain aminolated PEG-b-PEDAGA-b-PDPA, PEG-b-PDETAGA-b-PDPA, PEG-b-PTETAGA-b-PDPA or PEG-b-PTEPAGA-b-PDPA triblock copolymer. The resulting copolymers were denoted as PEDA, PDETA, PTETA or PTEPA respectively, according to the abbreviation of oligoamine used for aminolation. The successful aminolation of PGA segment was verified by 1H-NMR spectra examination (Fig. S2& Fig. S3). Two control diblock copolymers of mPEG225-b-PDPA50 and EDA-aminolated mPEG225-b-PGA45 (namely, PEG-b-PDPA and PEG-b-PEDA, respectively) were also synthesized by ATRP method (Fig. S4). Cationic micelles from PEG-b-PAGA-b-PDPA triblock copolymer were then prepared using a solvent precipitation method as described previously.(23) The cationic micelles prepared from different copolymers displayed similar surface charge around 16.0 mV, confirming the presence of aminolated interlayer in the micelles. The number-averaged hydrodynamic diameter of all cationic micelles was around 40 nm as determined by using dynamic light scattering measurement.
The pH-responsive property of the triblock copolymer was characterized by acid-base titration study. Diblock copolymers of PEG-b-PEDA and PEG-b-PDPA were shown as control (Fig. 2a). The pH titration curve of PEDA showed two pH transitions: the initial titration displayed narrow pH-responsive behavior due to deprotonation of PDPA tertiary amino groups (pH 6.2~6.4), and later titration (pH 6.4~9.0) was similar to that of PEG-b-PEDA (Fig. 2b), suggesting a composite pH-responsive behavior of PEDA triblock copolymer. PEG-b-PEDA diblock copolymer displayed response to a broad pH range from 6.0 to 9.0. This could be attributed to the stepwise protonation of the primary and secondary amino groups of EDA, since these two types of amino groups possess distinct pKa value of 9.1 and 6.3, respectively.(26) To visualize acid-induced micelle dissociation, PEDA micelles were loaded with a polarity-responsive and pH-insensitive fluorescent dye nile red (NR). As expected, at pH ≥ 6.4, strong fluorescence signal assigned to NR was detected due to encapsulation of NR in the hydrophobic PDPA core. At pH ≤ 6.2, the fluorescence emission of NR was completely quenched due to micelle dissociation and NR release into the aqueous phase (Fig. 2c).(27) The acid-induced dissociation of the cationic micelles was further examined by transmission electron microscopic (TEM) measurement. At pH 7.4, all PEG-b-PAGA-b-PDPA micelles displayed spherical morphology with particle size around 30~50 nm. They were dissociated to form amorphous aggregates due to protonation of PDPA core at acidic pH 6.0 (Fig. 2d). A minor fraction of the micelles intended to form aggregate at pH 7.4 when investigated by TEM examination that might be caused by the formation of hydrogen bonds between amino groups of different micelles during the dehydration process.
Figure 1
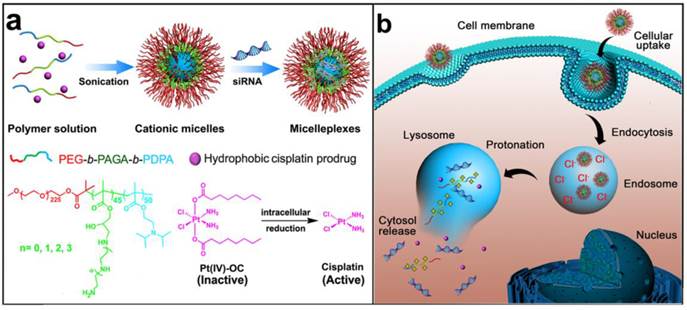
Schematic illustration for the preparation and intracellular activation of siRNA and cisplatin prodrug co-loaded pH-responsive micelleplexes: (a) Pt(IV)-OC was encapsulated in the hydrophobic core and siRNA was loaded on the intermediate layer of the micelleplexes; (b) Intracellular acidic pH-induced activation of micelleplexes. The micelles were dissociated inside acidic late endosome/lysosome due to protonation of the PDPA core. Pt(IV)-OC prodrug and siRNA were then simultaneously released into cytosol due to the strong hemolytic activity of the cationic triblock copolymers.
Figure 2
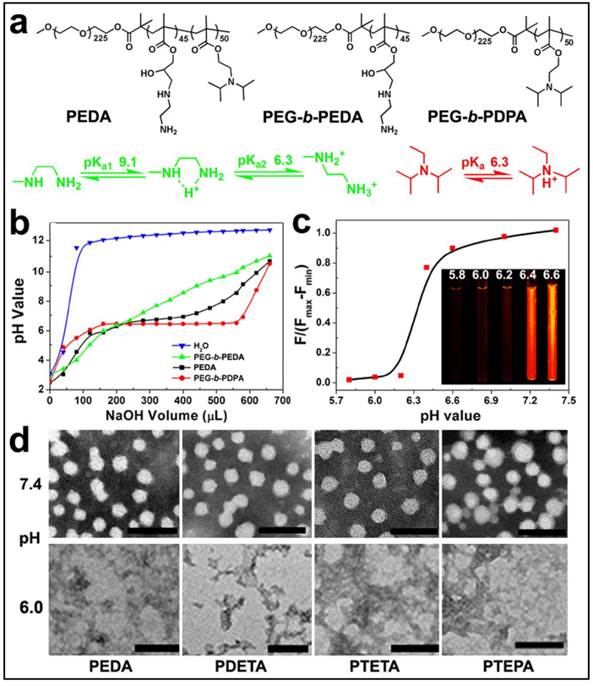
Characterization of PEG-b-PAGA-b-PDPA triblock copolymer and micelles: (a) Chemical structure of PEDA triblock copolymer, PEG-b-PEDA and PEG-b-PDPA diblock copolymers, respectively; (b) pH titration curves of PEDA, PEG-b-PEDA and PEG-b-PDPA copolymers; (c) Fluorescence spectra and fluorescent images of NR-loaded PEDA micelles at different pH; (d) Representative TEM images of PEDA, PDETA, PTETA and PTEPA micelles examined at neutral (pH 7.4) or acidic (pH 6.0) (scale bar = 100 nm).
To demonstrate intracellular micelle dissociation in endosome/lysosome vesicles, PEDA micelles was labeled with a pH-insensitive fluorescence dye tetramethyl rhodamine (TMR) through co-assembling 85 wt.% of aminolated triblock copolymer with 15 wt.% of TMR-conjugated PEG-b-PDPA diblock copolymer. Three molecules of TMR was conjugated on each PEG-b-PDPA diblock copolymer via ester bonds (Fig. S5).(27) Around 1.0 wt.% TMR was loaded into each micelle nanoparticle. At pHs above 6.3 (i.e. pH = 6.4 or 7.4), the as-prepared micelles displayed a quenched fluorophore signal owing to Förster resonance energy transfer (FRET) effect between the TMR molecules.(28) At weakly acidic pHs (pH ≤ 6.2), the fluorescent emission of TMR recovered dramatically due to PDPA protonation and micelle disassemble (Fig. 3a). Next, 4T1 murine breast cancer cells were pre-treated with 200 nM bafilomycin-A1 (Baf-A1), and then incubated with TMR-labeled PEDA micelles. Baf-A1 is a potent inhibitor for vacuolar-type H+-ATPase (V-ATPase) proton pump. The V-ATPase is responsible for the acidification of the endocytic vesicles (i.e., endosomes or lysosomes).(29,30) Fig. 3b demonstrated that 4T1 cells displayed “silenced” TMR signals after 12 h co-incubation with Baf-A1 and TMR-labeled micelles when examined using confocal laser scanning microscopy (CLSM). This result is caused by the inhibition of endosome acidification and corresponding lack of dissociation of the PDPA core. In contrast, strong fluorescence signal assigned to TMR was observed in Baf-A1 untreated cell control due to micelle dissociation in the early endosomes.(16) Taken together, the pH titration curves, fluorescence changing pattern of both NR-loaded and TMR-labeled micelles consistently suggested a sharp responsiveness of PEG-b-PAGA-b-PDPA micelles to weakly acidic environment, implying good potential of these micelles for drug release in early endosomes.
Given the strong buffer capacity of PEG-b-PAGA-b-PDPA micelles in the pH range of 6.2 to 7.4, we next examined their membrane destabilization ability by a hemolytic assay. Fig. 3c illustrated that no hemoglobin was released from the red blood cells (RBC) when treated with the micelles at pH 7.4, implying good blood compatibility of the micelles at the neutral pH condition. In significant contrast, at acidic pH = 6.2 simulating the weakly acidic environment of early endosome, dramatic hemoglobin release was induced by all four kinds of micelles in a concentration-dependent manner. More specifically, over 30%, 50% and 70% of hemoglobin was released at a PEDA concentration of 25, 50 and 100 μg/mL, respectively, implying an acidic pH-dependent membrane destabilization capacity of the PEG-b-PAGA-b-PDPA micelles.
Good serum stability is a prerequisite for systemic administration of NDDS. The influence of serum concentrations on the colloidal stability of micellar nanoparticles was examined using dynamic light scattering (DLS) measurement. As shown in Fig. 3d, consistent hydrodynamic particle sizes were found for all PEG-b-PAGA-b-PDPA micelles when the serum contents were increased from 0 to 20% (V/V), along with a slight increase of the polydispersity index (PDI) of the particle size distribution. The DLS data suggested good serum stability of the cationic micelles that could most likely be attributed to the shielding effect of PEG corona.
Figure 3
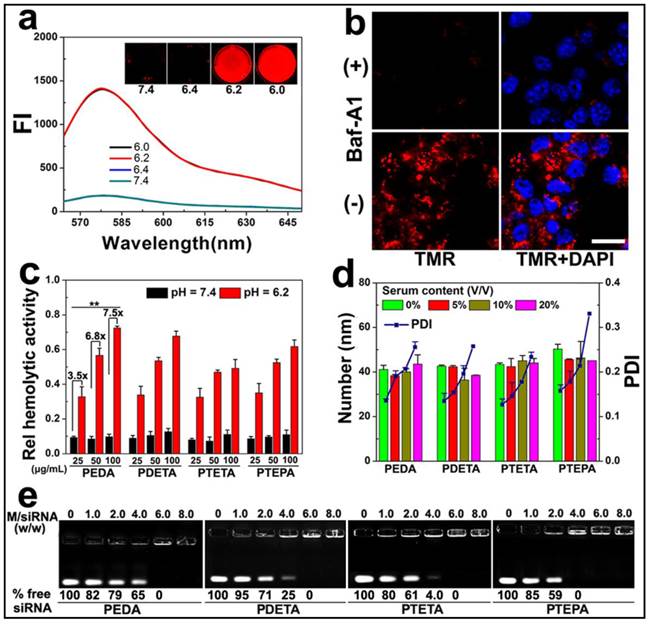
Intracellular dissociation, serum stability, blood compatibility and siRNA binding affinity of PEG-b-PAGA-b-PDPA micelles with different oligoamine side chain: (a) Fluorescence curves of TMR-labeled PEDA micelles versus pH (the insert shows the fluorescence images of the micelles at different pH values); (b) CLSM images of 4T1 cells treated with TMR-labeled PEDA micelles, Baf-A1 inhibited the intracellular dissociation of PEDA micelles, as indicated by the lack of TMR fluorescence in Baf-A1 pre-treated cells (scale bar = 100 μm); (c) Hemolytic activity of the pH-responsive micelles (**, p < 0.01); (d) Change of hydrodynamic particle size and polydispersity (PDI) versus serum concentrations; (e) siRNA binding ability of PEDA, PDETA, PTETA or PTEPA micelles as determined by gel retardation assay.
The siRNA binding ability of the cationic PEG-b-PAGA-b-PDPA micelles was examined by gel shift assay. As shown in Fig. 3e, a positive correlation was found between the chain length of oligoamines and the siRNA binding ability of the micelles. For instance, 65%, 25%, 4.0% and no free siRNA was detected when it was mixed with PEDA, PDETA, PTETA or PTEPA micelles respectively, at a micelle to siRNA weight ratio of 4.0. This result suggested the strongest siRNA binding affinity of PTEPA micelles which can most likely be attributed to the highest secondary amino density of TEPA among all the oligoamines.(30)
Intracellular distribution and GFP knockdown efficiency of micelleplexes
To examine the intracellular uptake and distribution of siRNA-loaded micelleplexes, PEDA micelles were covalently labeled with TMR dye as described above. The micelles were then loaded with fluorescein-modified siRNA (siRNA-FAM) to form the micelleplexes. A time course study was first performed to examine the internalization efficacy of the PEDA micelleplexes. The intracellular fluorescence intensity of both FAM and TMR was increased over time when examined by flow cytometric measurement, indicating that the cellular uptake of the micelleplexes was dependent on incubation time (Fig. S6). The intracellular distribution of PEDA micelleplexes was then investigated using CLSM examination. As shown in Fig. 4a, after 12 h incubation, considerable red fluorescence signal assigned to TMR was observed in A549 cells, implying successful internalization and dissociation of the PEDA micelles. The red spots of TMR were not completely colocalized with the green ones of FAM, indicating successful cytosol release of siRNA payload due to the strong hemolytic activity of the triple-layered micelles (Fig. S7). The majority of the red fluorescence spots were localized surrounding the cellular nuclei and colocalized with the lysosome vesicles as demonstrated by lysosome staining assay, indicating the majority of the micellar nanoparticles were entrapped inside the lysosome vesicles (Fig. S8).
To screen the optimized micelle composition for siRNA delivery, the siRNA transfection efficacy of four kinds of micelleplexes (i.e. PEDA, PDETA, PTETA and PTEPA) was examined in A549 cells with stable green fluorescence protein expression (A549-GFP). Commercially available transfection reagent Lipofectamine-2000 (Lipo-2000) was used as a parallel control. Strong GFP expression was observed in siRNA-NC treated A549-GFP cells. On the contrary, significant decrease of GFP expression was found in the cells transfected with siRNA-GFP micelleplexes, implying successful RNAi-specific GFP knockdown in A549-GFP cells (Fig. 4b&c and Fig. S9). A quantitative analysis of the GFP knockdown efficacy using flow cytometry revealed that PEDA micelleplexes possessed the highest RNAi-binding efficacy among four kinds of micelleplexes. For instance, PEDA silenced over 80% of GFP expression in A549-GFP cells at a micelle to siRNA weight ratio of 20, much more efficient than other kinds of micelles and Lipo-2000 (Fig. 4d). The GFP knockdown study suggests a negative correlation between the siRNA binding ability and target gene silencing efficacy of our pH-responsive micelleplexes. This could most likely be caused by the low efficiency of PTEPA micelles to release siRNA payload intracellularly since PTEPA micelles displayed the strongest siRNA binding ability among all micelles. The micelles bearing different ologoamine side chains were further screend by a cytotoxocity assay. It was found that PEDA micelles with or without siRNA complexation caused the lowest cytotoxicity among all the micelles as determined by a sulforhodamine B (SRB) staining assay (Fig. 4e & Fig. S10). Thus we selected PEDA micelleplexes prepared at micelle to siRNA weight ratio of 20 for RNAi studies in vitro and in vivo.
Inhibition of tumor metastasis in vitro
The ability of PEDA micelleplexes to silence NF-κB subunit p65 was tested in 4T1 cancer cells, a murine breast cancer cell line with high potential for lung metastasis. As shown in Fig. 5a&b, siRNA-p65-loaded PEDA (PEDA/p65) micelleplexes significantly suppressed p65 expression in 4T1 cells in a siRNA concentration dependent manner. Approximately 50% of p65 expression was silenced at a siRNA-p65 concentration of 150 nM. Knockdown of p65 protein notably inhibited metal matrixproteinase-9 (MMP-9) expression in 4T1 cells (Fig. 5c&d) abolishing 40% of MMP-9 expression in 4T1 cells at a siRNA-p65 concentration of 150 nM. Furthermore, silencing of p65 in 4T1 cells reduced over 70% of cell cycle regulator Cyclin D1 (Fig. 5e). The influence of p65 knockdown on the proliferation rate of 4T1 cells was examined using flow cytometric assay. Notable increase of G0/G1 ratio (from 41.91% to 63.83%) was found in siRNA-p65-transfected cells, implying significant inhibition of cell proliferation induced by Cyclin D1 downregulation (Fig. S11).
The motility of siRNA-p65 transfected 4T1 cells was then examined by wound healing assay. Compared to the untreated cell control that healed thoroughly, p65-silenced 4T1 cells displayed negligible wound reparation ability (Fig. 5f). A quantitative wound healing analysis showed that the wound reparation ability of 4T1 cells was significantly reduced by over 95% following transfection with siRNA-p65-loaded PEDA micelleplexes. This translates into 20-fold lower motility than that of the cell control treated with siRNA-NC (Fig. 5g). A transwell assay was also performed to evaluate the ability of cancer cells to migrate to adjacent tissue or traffic through the extracellular matrix. The photographs presented in Fig. 5f indicate that following transfection with siRNA p65, the number of 4T1 cells passing through the membrane was notably less than that of the cells treated with siRNA-NC. The migration and invasion capability of 4T1 cells were reduced by 4.4 and 1.5-fold, respectively, when treated with siRNA-p65-loaded micelleplexes (Fig. 5g). This could most likely be attributed to p65 knockdown-induced MMP-9 downregulation.
Figure 4
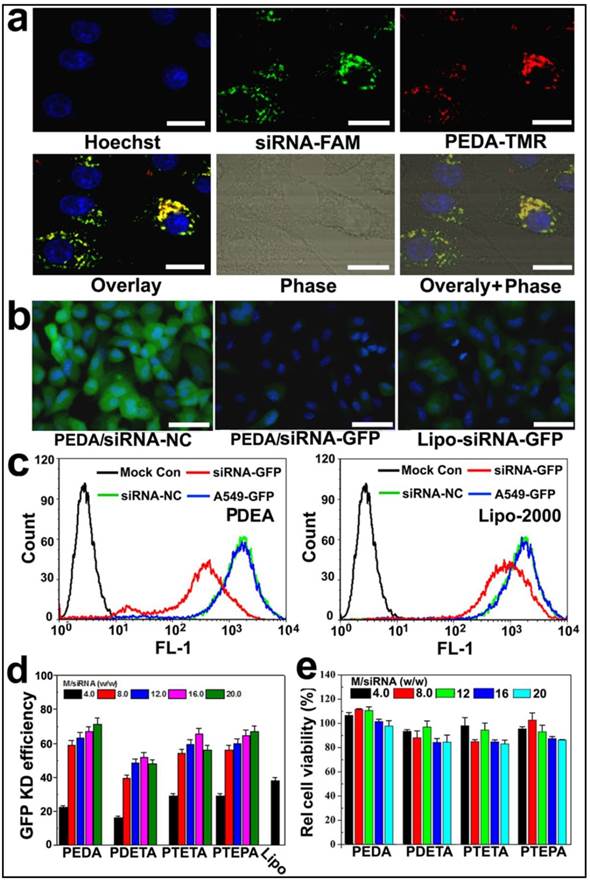
Cellular uptake and target gene knockdown of siRNA-loaded pH-responsive micelleplexes: (a) Intracellular distribution of siRNA-FAM loaded PEDA micelleplexes determined by CLSM examination (scale bar = 50 μm); (b) Fluorescence images of A549-GFP cells transfected by siRNA-GFP loaded PEDA micelleplexes or lipoplexes (scale bar = 150 μm); (c) Flow cytometry determined GFP expression in A549-GFP cells transfected with siRNA-GFP loaded PEDA micelleplexes or lipoplexes; (d) Influence of micelle to siRNA weight ratio on GFP knockdown (KD) efficiency of siRNA-GFP loaded micelleplexes; (e) Relative viability of A549-GFP cells after 24 h incubation with siRNA-loaded micelleplexes.
Figure 5
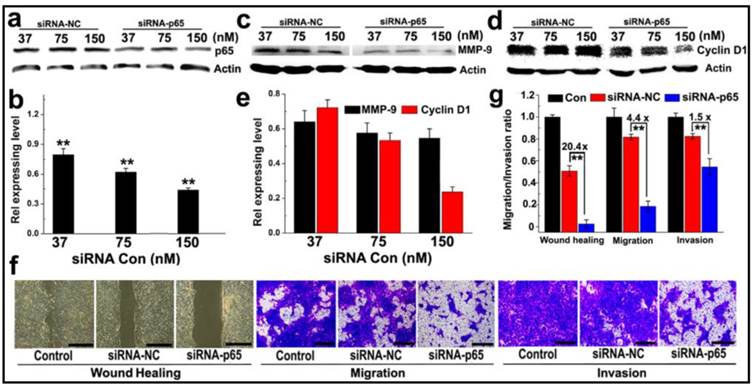
Anti-metastasis in murine 4T1 breast cancer cells in vitro: (a-c) Western-blotting assay determined (a&b) p65; (c-e) MMP-9 and Cyclin D1 expression in 4T1 cells transfected with PEDA/siRNA-p65 micelleplexes in vitro; (f) Representative images determined by wound healing, migration and invasion assay of siRNA-p65 transfected 4T1 cells (scale bar = 200 μm); (g) Quantitative analysis of the wound healing, migration and invasion potential of siRNA-p65 transfected 4T1 cells. The wound healing, migration and invasion ratios of siRNA transfected cells were normalized with that of untreated cells control (**, p < 0.01).
Biodistribution and pharmacokinetics of siRNA and cisplatin prodrug co-loaded micelleplexes
To test the potential of PEDA micelles for co-delivery of siRNA and anti-cancer drug, we first synthesized a hydrophobic cisplatin prodrug Pt(IV)-OC by esterification of Pt(IV)-OH with octanoic anhydride. This was performed by following the procedure reported by Farokhzad et al (see Fig. S12 & Fig. S13 for synthesis and MS spectrum, respectively).(17) Pt(IV)-OC was then encapsulated into the core of PEDA micelles through hydrophobic interaction with PDPA moiety. After loading 1.0%(wt) Pt(IV)-OC prodrug, the number averaged hydrodynamic diameters of PEDA micelles changed from 41.6±1.8 to 54.9±4.3 nm, which increased further to 67.2±4.7 nm following siRNA complexation. The Pt(IV)-OC-loaded PEDA micelles displayed positive surface charge of 16.0±1.1 mV, which decreased to 11.8±2.4 mV after siRNA loading (Fig. 6a&b). The acidic pH-induced dissociation of the micelleplexes was not impaired by Pt(IV)-OC-loading and siRNA complexation as confirmed by TEM examination (Fig. 6c). The cytotoxicity of cisplatin, Pt(IV)-OC and PEDA micelleplexes loading siRNA-NC/Pt(IV)-OC or siRNA-p65/Pt(IV)-OC (denoted as M/Pt(IV)-OC/NC and M/Pt(IV)-OC/p65, respectively) was evaluated by SRB staining assay. Cisplatin and Pt(IV)-OC prodrug showed comparable cytotoxicity, suggesting effective intracellular reduction of the prodrug into activate status. A comparison of the dose-response curves showed significantly lower IC50 values of the prodrug-loaded micelleplex than that of cisplatin or prodrug (Fig. 6d). This could most likely be explained by nanoparticle-enhanced internalization of the alkylated cisplatin prodrug.(17) Moreover, M/Pt(IV)-OC/p65 displayed higher cytotoxicity than M/Pt(IV)-OC/NC in all selected drug concentrations. The ability of siRNA and Pt(IV)-OC co-loaded micelleplexes to induce cellular apoptosis was also examined in 4T1 cells. When incubated with siRNA-NC loaded PEDA micelleplexes (M/NC) or Pt(IV)-OC, 14.8% and 35.4% of 4T1 cells underwent apoptosis, respectively. The apoptotic cell percentage increased notably to 95.1% after incubation with M/Pt(IV)-OC/p65 micelleplexes (Fig. S14). To elucidate the possible underlying mechanism, caspase-3 activation was examined by immunostaining analysis. Significant caspase-3 elevation was detected in 4T1 cells treated with M/Pt(IV)-OC/p65, suggesting caspase-3 activation might be the dominant mechanism to induce cellular apoptosis (Fig. S15). Taken together, all these results verified that the simultaneous delivery of siRNA-p65 and Pt(IV)-OC is able to concurrently sensitize 4T1 breast cancer cells to platinum treatment by suppressing p65 expression.
Figure 6
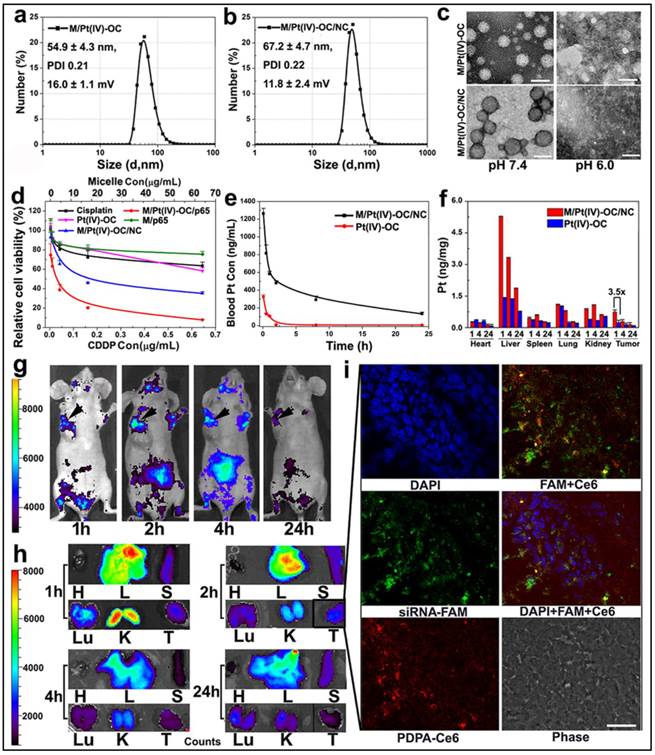
Physical characterization and in vivo behavior of siRNA and Pt(IV)-OC co-loaded PEDA micelleplexes: (a&b) Size distribution of (a) PEDA/Pt(IV)-OC micelles and (b) PEDA/Pt(IV)-OC/siRNA micelleplexes; (c) TEM image of PEDA/Pt(IV)-OC micelles, and PEDA/Pt(IV)-OC/siRNA micelleplexes examined at pH = 7.4 or 6.0 (scale bar = 100 nm); (d) In vitro cytotoxicity, and (e) pharmacokinetics of PEDA/Pt(IV)-OC/siRNA micelleplexes; (f) Tissue distribution of Pt(IV)-OC prodrug when delivered with PEDA micelleplexes; (g) In vivo and (h) ex-vivo distribution of siRNA-loaded micelleplex in 4T1 orthotopic tumor-bearing mice (H: heart; L: liver; S: spleen; Lu: lung; T: tumor; the black arrows in Figure g indicated the location of 4T1 tumors); (i) CLSM examination determined intratumoral distribution of siRNA-loaded micelleplexes 2 h post tail vein injection (scale bar = 50 μm). The PEDA micelles and siRNA were labeled with Ce6 and FAM, respectively.
The pharmacokinetic behavior and biodistribution of Pt(IV)-OC, systemically administered either in free drug form or micelleplex formulation, were quantitatively examined by ICP-MS measurement. The maximal drug concentration and area under the curve of Pt(IV)-OC were significantly increased by 3.9 and 17.7-fold over those of free Pt(IV)-OC respectively, when delivered with PEDA micelles (Fig. 6e and Table S1). The tissue distribution of Pt(IV)-OC was examined in nude mice bearing orthotopic 4T1 tumor at 1, 4 or 24 h time point post intravenous (i.v.) injection. Fig. 6f showed that free Pt(IV)-OC was mainly found in the lung, liver and kidney, with negligible tumor distribution, which could be explained by the rapid clearance of small molecular drug by the kidneys and Kupffer cells. When delivered with PEDA micelles, the tumoural concentration of Pt(IV)-OC was notably increased by 3.2-fold when examined at 1 h. This could be mainly attributed to the passive tumor targeting ability of PEDA micelles through the EPR effect.
The in vivo and ex-vivo distribution of siRNA and Pt(IV)-OC co-loaded PEDA micelleplexes was examined by whole body imaging. For this purpose, PEG-b-PDPA diblock copolymer was conjugated with a fluorescence dye chlorine e6 (Ce6) (see Fig. S5 for synthesis). The PEG-b-PDPA-Ce6 copolymer was thoroughly purified by dialyzing against DMSO and Milli-Q water to remove free Ce6. The complete removal of any trace free Ce6 was verified by high performance liquid chromatography (HPLC) examination (Fig. S16). Each PEG-b-PDPA molecule was conjugated with three Ce6 molecules as determined by UV-Vis spectroscopic measurement. PEG-b-PDPA-Ce6 copolymer was then mixed with PEDA triblock copolymer at a weight ratio of 15 to 85 to obtain Ce6-labled PEDA micelles. The Ce6-labeled micelles were then loaded with siRNA-FAM for tracking siRNA distribution in vivo. As shown in Fig. 6g&h, tumor accumulation of PEDA micelleplexes was found 1 h post i.v. injection. The fluorescence intensity increased slightly at 2 h, and then dropped slowly over time due to blood washing of the micelleplexes. It is worth noting that strong fluorescence signal was found in the kidney 1 h post nanoparticle injection, this could be most likely caused by fast disassembly and retention of the cationic micelleplexes at the kidney glomerular basement membrane as reported by Davis et al.(31)
The intratumoral distribution of siRNA-loaded PEDA micelleplexes was further investigated by CLSM examination of the tumor sections. As shown in Fig. 6i, green fluorescence assigned to FAM and red fluorescence of Ce6 were both found throughout the tumor tissue when examined at the 2 h time point. The fluorescence dots of FAM and Ce6 were distributed mainly surrounding the nucleus and were partially co-localized, indicating successful internalization of the micelleplexes and cytosol release of siRNA payload in vivo. We had recently reported that the micellar nanoparticles with neutral surface charge tended to distribute in the perivascular areas of solid tumor without notable tumor penetration,(32) even though they can accumulate in the tumor site via EPR effect. On the contrary, the tumor distribution images clearly indicated good tumor penetration and cellular entry of PEDA micelleplexes. Our observation is also consistent with the literature reports that cationic nanoparticles can penetrate deeper inside the solid tumors than their counterparts bearing neutral or anionic surface charge.(20,33) This could be most likely attributed to the positive surface charge of the micelleplexes facilitating their interaction with the ECM and cell membrane post extravasation from the blood vessel at the tumor site.
Inhibition of tumor growth and metastasis in vivo
Encouraged by the improved cytotoxicity in 4T1 cells and the significant accumulation in the orthotopic 4T1 tumor of siRNA and Pt(IV)-OC co-loaded PEDA micelleplexes, their ability to inhibit tumor growth was also evaluated in vivo. The 4T1 tumor bearing nude mice were randomly divided into six groups when the tumor volume reached 50 mm3. The mice were then systemically injected with siRNA and prodrug co-loaded PEDA micelleplexes or other formulations at an equal siRNA and prodrug dose of 3.0 and 0.60 mg/kg, respectively. The micelle dose was 60 mg/kg accordingly. The injection was repeated for 6 times at an interval of 2 days. As shown in Fig. 7a, the tumors of M/Pt(IV)-OC/p65 group grew much slower than those of M/Pt(IV)-OC/NC or M/p65 group. A hematoxylin and eosin (H&E) staining assay revealed significant DNA degradation in the tumor sections of the M/Pt(IV)-OC/p65 group (Fig. 7b). The tumor slices of each mouse group were further examined by the tunnel staining assay. Significant cellular apoptosis was detected in M/p65/Pt(IV)-OC group in comparison with that of M/Pt(IV)-OC/NC or cisplatin group (Fig. 7c).
The ability of M/Pt(IV)-OC/p65 to suppress lung metastasis of the orthotopicallly implanted 4T1 tumors was studied in the six groups of mice. At the 24th day post first injection, the mice were sacrificed. The lungs were collected and photographed. Metastatic nodules of 4T1 cells from the primary tumor were found in the lungs of the PBS or M/NC group as confirmed by bioluminescence imaging (BLI) (Fig. 7d&e). Although M/p65 and cisplatin displayed comparable tumor growth inhibition effect, significantly less metastasis nodules and weaker BLI signal were found in the lungs of M/p65 group. This indicated a much better tumor metastasis inhibition ability of siRNA-p65 than that of cisplatin that could be attributed to the p65-knockdown-induced metastasis suspension as demonstrated in the cell culture studies in vitro. Most notably, M/p65/Pt(IV)-OC treatment significantly inhibited both the growth of the primary tumors and the dissemination of 4T1 cells to lung, suggesting a cumulative therapeutic outcome of Pt(IV)-OC-mediated chemotherapy and siRNA-p65-directed RNAi therapy.
A notable liver and kidney distribution of the micelleplexes was detected in the biodistribution study. To evaluate the systemic toxicity of the micelleplexes, the liver and kidney of the tumor bearing ice were examined using a histological analysis at the end of the anti-tumor study. Cisplatin-treatment induced obvious tubular atrophy and necrosis due to its severe nephrotoxicity. On the contrary, neither siRNA-loaded nor siRNA and Pt(IV)-OC co-loaded PEDA micelleplexes caused notable damage in the liver and kidney (Fig. S17). Biochemical analysis of blood was further performed to evaluate the influence of micelleplex-injection on the liver function. There was no significant difference between the liver function of PBS or micelleplex-injected mouse groups when tested for several factors including the serum levels of albumin, globulin, alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, total bilirubin level and total protein level (Table S2). Both the H&E staining and results from liver function analysis implied satisfactory biosafety of the micelleplex nanoparticles co-loaded with siRNA and cisplatin-prodrug.
Metastasis is one of the leading challenges for breast cancer therapy. The dissemination of cancer cells to a secondary location is a multistep and complicated process. One significant driving force for cancer metastasis is the uncontrolled proliferation of cancer cells causing hypoxia and acidic tumor microenvironment due to insufficient oxygen supply and medium exchange in deep tumor.(34) This is believed to stimulate local invasion of cancer cells by initiating the EMT of cancer cells.(35) The second underlying reason for tumor metastasis is the genetic instability of cancer cells. The ectopic activation of NF-κB in cancer cells could promote EMT and matrix degradation by eliciting MMP-9 expression.(36) In addition, over-activation of NF-κB is also closely related to the chemoresistance of cancer cells by up-regulating anti-apoptotic gene Bcl-2.(37,38) Considering this complex, multi-factorial nature of metastasis, it is essential to treat metastatic cancer by simultaneously targeting several different mechanisms.
Figure 7
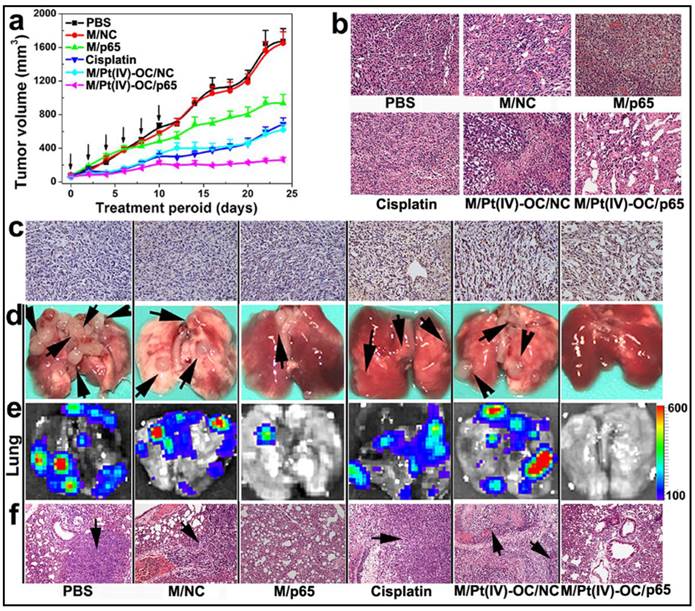
PEDA micelleplex mediated siRNA-p65 and Pt(IV)-OC prodrug co-delivery significantly inhibited the growth of primary tumor and suppressed lung metastasis: (a) Tumor growth curves in a mouse model bearing 4T1 tumors after treatment with various formulations (the black arrows indicated the injection time points); (b) H&E and (c) tunnel staining of tumor sections; (d-f) Lung metastasis of 4T1 breast tumor determined by (d) photograph, (e) BLI imaging, and (f) H&E staining (the black arrows indicated the location of metastasis nodules in the lung) (200 × for H&E and tunnel staining images). The black arrows in d and f indicate the presence of metastasis nodules in the lungs.
We hypothesized that a combination of cytotoxic chemotherapy with RNAi therapy could address the current challenge for treatment of metastatic breast cancer. To verify our hypothesis, we firstly selected cisplatin as the therapeutic reagent. Cisplatin is a first-line anticancer drug widely used for chemotherapy of a large variety of carcinomatous diseases, including lung, ovarian and blood cancers.[40] However, the clinical application of cisplatin is severely hindered by its poor pharmacokinetics, low bioavailability, undesired side effect and tendency to induce resistance.(41) Alkylation of cisplatin has recently been demonstrated as a practical strategy to convert cisplatin into a hydrophobic prodrug form.(17,42) In this study, we employed octanoic anhydride as an alkylation reagent to prepare hydrophobic cisplatin prodrug Pt(IV)-OC, which can be specifically activated with intracellular GSH in cancer cells. In parallel, we selected siRNA-p65 targeting the NF-κB pathway for RNAi therapy to inhibit both the growth of the primary tumor and distant metastasis of the cancer cells.
We previously reported a set of PDPA-based pH-responsive micellar nanoparticles for delivery of siRNA(22) and anti-cancer drugs.(23,24) These nanoparticles displayed a sharp pH transition around 6.3 in response to the weakly acidic microenvironment of early endosome via protonation of the PDPA core. In this study, we developed triple-layered micelleplexes by successfully integrating the hydrophobic cisplatin prodrug and therapeutic siRNA into one single micelleplex nanoplatform. These unique micelleplexes were responsive to the weakly acidic microenvironment of early endosomes and therefore could be specifically dissociated in the early endosomes. The intracellular dissociation of the micelleplexes could be blocked by Baf-A1 treatment verifying the endosome pH-responsive property of this novel micelleplex. In cell culture studies, we demonstrated that siRNA-p65-loaded micelleplexes could completely inhibit migration and invasion of 4T1 cells by suppressing gelatinase MMP-9 expression. Also, a combination of siRNA-p65 and cisplatin prodrug caused significant apoptosis of 4T1 cells by downregulating Cyclin D1 and the anti-apoptotic gene Bcl-2.(42) In the anti-tumor study in vivo, we found that siRNA-p65 and cisplatin prodrug co-loaded micelleplexes simultaneously inhibited the growth and metastasis of orthotopic 4T1 breast tumor. We propose a synergistic therapeutic mechanism of the siRNA and cisplatin prodrug co-loaded micelleplexes in vivo. Pt(IV)-OC primarily inhibited growth of the primary tumor by inducing cellular apoptosis. Simultaneously, siRNA-p65 suppressed both the growth of the primary tumor and distant metastasis of cancer cells.
Compared to the NDDS reported so far, the present micelleplex platform possesses several obvious advantages. Firstly, the PEG-b-PAGA-b-PDPA micelles display well-defined and triple-layered topological structure, thus allowing integration of very different functions into one micellar nanoplatform. The micelles can encapsulate both the hydrophobic anti-cancer drug inside the hydrophobic PDPA core, and compact hydrophilic siRNA on the positively charged interlayer. The PDPA core can stabilize the micelleplexes at physiological pH and ensure high serum stability during blood circulation. Secondly, the hydrophobic to hydrophilic transition of PDPA core uses proton transfer and non-covalent disassembly rendering a fast response (< 5 ms) to endosomal acidic pH upon protonation of the tertiary amino groups, allows timely and burst release of the chemotherapeutic payload in the early endosome vesicles.(44,16) Although conventional NDDS prepared with poly(ε-caprolactone) (PCL) or poly(lactide-co-glycolide) (PLGA) had been extensively exploited for cancer therapy, these nanoparticles displayed low efficiency of drug release since it was highly dependent on the degradation of polymer backbone.(53,54) This might induce acquired drug resistance due to continued expose of cancer cells to a suboptimal drug concentration.(33) Nanoparticles with pH-responsive properties were also extensively exploited for drug and gene delivery application recently.(45-47) Most of these nanovehicles were based on the breaking of pH-labile covalent bonds to induce nanoparticle degradation. This strategy is time consuming (generally several hours to days), and their pH-responsive property is non-specific to the tumoural or intracellular acid microenvironment.(48-52) Furthermore, the PAGA and PDPA segments display a composite pH-responsive property and strong proton sponge capacity in the weak acid condition (i.e. pH 6.3 ~ 7.0) (Fig. 2a&b). Consequently, the micelleplexes could synergistically induce endosome/lysosome disruption and facilitate cytosol release of siRNA and hydrophobic drugs.(24) Moreover, the micelleplex platform co-loaded with siRNA and anti-cancer drugs could simultaneously inhibit the growth of primary tumor as well as the distant metastasis of the cancer cells.
Conclusion
In summary, we reported a unique micelleplex platform with sharp pH-responsive property for delivery of siRNA and the cisplatin prodrug. The multifunctional micelleplexes exhibited sharp response to weakly acidic microenvironment of early endosomes. In cell culture studies, PEDA micelleplexes loaded with siRNA-p65 could completely abolish the migration and invasion ability of 4T1 breast cancer cells by blocking the NF-κB pathway. Furthermore, the micelleplexes co-loaded with cisplatin prodrug and siRNA-p65 could passively accumulate into the orthotopic 4T1 breast tumor. Most significantly, the micelleplexes inhibited both the growth of primary tumor and the lung metastasis of cancer cells. Given the defined chemical structure, as well as the superior anti-tumor property of our triple-layered micelleplexes, the results presented in this study imply a promising potential of this novel platform co-loading siRNA and anti-cancer drugs not only for treatment of metastatic breast cancer, but also other kinds of invasive solid tumors.
Supplementary Material
Supplementary materials and methods, Supplementary tables and figures.
Acknowledgements
Financial support from the National Basic Research Program of China (2013CB932704), the National Natural Science Foundation of China (81373359 and 81270047) and the Youth Innovation Promotion Association CAS is gratefully acknowledged.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer. 2006;6:449-58
2. Voduc KD, Cheang MC, Tyldesley S, Gelmon K, Nielsen TO, Kennecke H. Breast cancer subtypes and the risk of local and regional relapse. J Clin Oncol. 2010;28:1684-91
3. Daenen LG, Roodhart JM, van Amersfoort M, Dehnad M, Roessingh W, Ulfman LH, Derksen PW, Voest EE. Chemotherapy enhances metastasis formation via VEGFR-1-expressing endothelial cells. Cancer Res. 2011;71:6976-85
4. Eckhardt BL, Francis PA, Parker BS, Anderson RL. Strategies for the discovery and development of therapies for metastatic breast cancer. Nat Rev Drug Discov. 2012;11:479-97
5. Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, Shenkier T, Cella D, Davidson NE. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666-76
6. De Bock K, Mazzone M, Carmeliet P. Antiangiogenic therapy, hypoxia, and metastasis: risky liaisons, or not? Nat Rev Clin Oncol. 2011;8:393-404
7. Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927-39
8. Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22:725-36
9. Huber MA, Azoitei N, Baumann B, Grünert S, Sommer A, Pehamberger H, Kraut N, Beug H, Wirth T. NF-kappa B is essential for epithelial-mesenchymal transition and metastasis in a model of breast cancer progression. J Clin Invest. 2004;114:569-81
10. Wu Y, Deng J, Rychahou PG, Qiu S, Evers BM, Zhou BP. Stabilization of snail by NF-kappa B is required for inflammation-induced cell migration and invasion. Cancer Cell. 2009;15:416-28
11. Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25:9-34
12. Ma L, Reinhardt F, Pan E, Soutschek J, Bhat B, Marcusson EG, Teruya-Feldstein J, Bell GW, Weinberg RA. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat Biotechnol. 2010;28:341-7
13. Schramek D, Sendoel A, Segal JP, Beronja S, Heller E, Oristian D, Reva B, Fuchs E. Direct in vivo RNAi screen unveils myosin IIa as a tumor suppressor of squamous cell carcinomas. Science. 2014;343:309-13
14. M. Saraswathy, S. Q. Gong, Recent developments in the co-delivery of siRNA and small molecule anticancer drugs for cancer treatment. Mater. Today. 2014;17:298-306
15. Schroeder A, Heller DA, Winslow MM, Dahlman JE, Pratt GW, Langer R, Jacks T, Anderson DG. Treating metastatic cancer with nanotechnology. Nat Rev Cancer. 2011;12:39-50
16. Yu H, Zou Y, Wang Y, Huang X, Huang G, Sumer BD, Boothman DA, Gao J. Overcoming endosomal barrier by amphotericin B-loaded dual pH-responsive PDMA-b-PDPA micelleplexes for siRNA delivery. ACS Nano. 2011;5:9246-55
17. Xu X, Xie K, Zhang XQ, Pridgen EM, Park GY, Cui DS, Shi J, Wu J, Kantoff PW, Lippard SJ, Langer R, Walker GC, Farokhzad OC. Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc Natl Acad Sci USA. 2013;110:18638-43
18. Sun Q, Sun X, Ma X, Zhou Z, Jin E, Zhang B, Shen Y, Van Kirk EA, Murdoch WJ, Lott JR, Lodge TP, Radosz M, Zhao Y. Integration of nanoassembly functions for an effective delivery cascade for cancer drugs. Adv Mater. 2014;26:7615-21
19. Ernsting MJ, Murakami M, Roy A, Li SD. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J Control Release. 2013;172:782-94
20. Yim H, Park SJ, Bae YH, Na K. Biodegradable cationic nanoparticles loaded with an anticancer drug for deep penetration of heterogeneous tumours. Biomaterials. 2013;34:7674-82
21. Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nat Rev Mol Cell Biol. 2010;11:50-61
22. Wang Y, Zhou K, Huang G, Hensley C, Huang X, Ma X. et al. A nanoparticle-based strategy for the imaging of a broad range of tumours by nonlinear amplification of microenvironment signals. Nat. Mater. 2014;13:204-12
23. Yu P, Yu H, Guo C, Cui Z, Chen X, Yin Q, Zhang P, Yang X, Cui H, Li Y. Reversal of doxorubicin resistance in breast cancer by mitochondria-targeted pH-responsive micelles. Acta Biomater. 2015;14:115-24
24. Yu H, Xu Z, Chen X, Xu L, Yin Q, Zhang Z, Li Y. Reversal of lung cancer multidrug resistance by pH-responsive micelleplexes mediating co-delivery of siRNA and paclitaxel. Macromol Biosci. 2014;14:100-9
25. Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5:121-32
26. Miyata K, Oba M, Nakanishi M, Fukushima S, Yamasaki Y, Koyama H, Nishiyama N, Kataoka K. Polyplexes from poly(aspartamide) bearing 1,2-diaminoethane side chains induce pH-selective, endosomal membrane destabilization with amplified transfection and negligible cytotoxicity. J Am Chem Soc. 2008;130:16287-94
27. Yu H, Xu Z, Wang D, Chen X, Zhang Z, Li Y. Intracellular pH-activated PEG-b-PDPA wormlike micelles for hydrophobic drug delivery. Polymer Chem. 2013;4:5052-55
28. Zhou K, Wang Y, Huang X, Luby-Phelps K, Sumer BD, Gao J. Tunable, ultrasensitive pH-responsive nanoparticles targeting specific endocytic organelles in living cells. Angew Chem Int Ed. 2011;50:6109-14
29. Bowman EJ, Siebers A, Altendorf K. Bafilomycins: a class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc Natl Acad Sci USA. 1988;85:7972-6
30. Johnson LS, Dunn KW, Pytowski B, McGraw TE. Endosome acidification and receptor trafficking: bafilomycin A1 slows receptor externalization by a mechanism involving the receptor's internalization motif. Mol Biol Cell. 1993;4:1251-66
31. Salcher EE1, Kos P, Fröhlich T, Badgujar N, Scheible M, Wagner E. Sequence-defined four-arm oligo(ethanamino)amides for pDNA and siRNA delivery: Impact of building blocks on efficacy. J Control Release. 2012;164:380-6
32. Zuckerman JE, Choi CH, Han H, Davis ME. Polycation-siRNA nanoparticles can disassemble at the kidney glomerular basement membrane. Proc Natl Acad Sci USA. 2012;109:3137-42
33. Yu H, Cui Z, Yu P, Guo C, Feng B, Jiang T, Wang S, Yin Q, Zhong D, Yang X, Zhang Z, Li Y. pH and near-infrared light-responsive micelles with hyperthermia-triggered tumor penetration and cytoplasm drug release reverse doxorubicin resistance in breast cancer. Adv Funct. Mater. 2015;25:2489-2500
34. Ojea-Jiménez I1, García-Fernández L, Lorenzo J, Puntes VF. Facile preparation of cationic gold nanoparticle-bioconjugates for cell penetration and nuclear targeting. ACS Nano. 2012;6:7692-702
35. Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12:895-904
36. Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265-73
37. Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420-8
38. Wang CY, Cusack JC Jr, Liu R, Baldwin AS Jr. Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kappaB. Nat Med. 1999;5:412-7
39. Tong L, Yuan Y, Wu S. Therapeutic microRNAs targeting the NF-kappa B signaling circuits of cancers. Adv Drug Deliv Rev. 2015;81:1-15
40. Nishiyama N, Okazaki S, Cabral H, Miyamoto M, Kato Y, Sugiyama Y, Nishio K, Matsumura Y, Kataoka K. Novel cisplatin-incorporated polymeric micelles can eradicate solid tumors in mice. Cancer Res. 2003;63:8977-83
41. Cohen SM, Lippard SJ. Cisplatin: from DNA damage to cancer chemotherapy. Prog Nucleic Acid Res Mol Biol. 2001;67:93-130
42. Zheng YR, Suntharalingam K, Johnstone TC, Yoo H, Lin W, Brooks JG, Lippard SJ. Pt(IV) prodrugs designed to bind non-covalently to human serum albumin for drug delivery. J Am Chem Soc. 2014;136:8790-8
43. Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559-64
44. Wang Y, Zhou K, Huang G, Hensley C, Huang X, Ma X, Zhao T, Sumer BD, DeBerardinis RJ, Gao JM. nanoparticle-based strategy for the imaging of a broad range of tumours by nonlinear amplification of microenvironment signals. Nat Mater. 2014;13:204-12
45. Li L, Gu W, Chen J, Chen W, Xu ZP. Co-delivery of siRNAs and anti-cancer drugs using layered double hydroxide nanoparticles. Biomaterials. 2014;35:3331-9
46. Creixell M, Peppas NA. Co-delivery of siRNA and therapeutic agents using nanocarriers to overcome cancer resistance. Nano Today. 2012;7:367-79
47. Xiong XB, Lavasanifar A. Traceable multifunctional micellar nanocarriers for cancer-targeted co-delivery of MDR-1 siRNA and doxorubicin. ACS Nano. 2011;5:5202-13
48. Bachelder EM, Beaudette TT, Broaders KE, Dashe J, Fréchet JM. Acetal-derivatized dextran: an acid-responsive biodegradable material for therapeutic applications. J Am Chem Soc. 2008;130:10494-5
49. Bae Y, Fukushima S, Harada A, Kataoka K. Design of environment-sensitive supramolecular assemblies for intracellular drug delivery: polymeric micelles that are responsive to intracellular pH change. Angew Chem Int Ed. 2003;42:4640-3
50. Griset AP, Walpole J, Liu R, Gaffey A, Colson YL, Grinstaff MW. Expansile nanoparticles: synthesis, characterization, and in vivo efficacy of an acid-responsive polymeric drug delivery system. J Am Chem Soc. 2009;131:2469-71
51. Potineni A, Lynn DM, Langer R, Amiji MM. Poly(ethylene oxide)-modified poly(beta-amino ester) nanoparticles as a pH-sensitive biodegradable system for paclitaxel delivery. J Control Release. 2003;86:223-34
52. Wagner E. Polymers for siRNA delivery: inspired by viruses to be targeted, dynamic, and precise. Acc Chem Res. 2012;45:1005-13
53. Shuai X, Ai H, Nasongkla N, Kim S, Gao J. Micellar carriers based on block copolymers of poly(ε-caprolactone) and poly(ethylene glycol) for doxorubicin delivery. J Control Release. 2004;98:415-26
54. Avgoustakisa K, Beletsia A, Panagia Z, Klepetsanisa P, Karydasb A G, Ithakissiosa D.S. PLGA-mPEG nanoparticles of cisplatin: in vitro nanoparticle degradation, in vitro drug release and in vivo drug residence in blood properties. J Control Release. 2002;79:123-35
Author contact
![]() Corresponding authors: Dr. Haijun Yu, Prof. Dr. Yaping Li, State key Laboratory of Drug Research & Center of Pharmaceutics, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: hjyuac.cn, ypliac.cn; Tel/Fax: +86-21-2023-1979
Corresponding authors: Dr. Haijun Yu, Prof. Dr. Yaping Li, State key Laboratory of Drug Research & Center of Pharmaceutics, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China. E-mail: hjyuac.cn, ypliac.cn; Tel/Fax: +86-21-2023-1979
Citation styles
APA
Yu, H., Guo, C., Feng, B., Liu, J., Chen, X., Wang, D., Teng, L., Li, Y., Yin, Q., Zhang, Z., Li, Y. (2016). Triple-Layered pH-Responsive Micelleplexes Loaded with siRNA and Cisplatin Prodrug for NF-Kappa B Targeted Treatment of Metastatic Breast Cancer. Theranostics, 6(1), 14-27. https://doi.org/10.7150/thno.13515.
ACS
Yu, H.; Guo, C.; Feng, B.; Liu, J.; Chen, X.; Wang, D.; Teng, L.; Li, Y.; Yin, Q.; Zhang, Z.; Li, Y. Triple-Layered pH-Responsive Micelleplexes Loaded with siRNA and Cisplatin Prodrug for NF-Kappa B Targeted Treatment of Metastatic Breast Cancer. Theranostics 2016, 6 (1), 14-27. DOI: 10.7150/thno.13515.
NLM
Yu H, Guo C, Feng B, Liu J, Chen X, Wang D, Teng L, Li Y, Yin Q, Zhang Z, Li Y. Triple-Layered pH-Responsive Micelleplexes Loaded with siRNA and Cisplatin Prodrug for NF-Kappa B Targeted Treatment of Metastatic Breast Cancer. Theranostics 2016; 6(1):14-27. doi:10.7150/thno.13515. https://www.thno.org/v06p0014.htm
CSE
Yu H, Guo C, Feng B, Liu J, Chen X, Wang D, Teng L, Li Y, Yin Q, Zhang Z, Li Y. 2016. Triple-Layered pH-Responsive Micelleplexes Loaded with siRNA and Cisplatin Prodrug for NF-Kappa B Targeted Treatment of Metastatic Breast Cancer. Theranostics. 6(1):14-27.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.