Theranostics
13.3
Impact Factor
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results and discussion
Conclusions
Supplementary Materials
Acknowledgements
References
Introduction
Materials and Methods
Results and discussion
Conclusions
Supplementary Materials
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(9):985-994. doi:10.7150/thno.11938 This issue Cite
Research Paper
DNA Aptamer Selected against Pancreatic Ductal Adenocarcinoma for in vivo Imaging and Clinical Tissue Recognition
Xiaoqiu Wu1*, Zilong Zhao1*, Huarong Bai1, Ting Fu1, 2, Chao Yang1, 3, Xiaoxiao Hu1, Qiaoling Liu1, Carole Champanhac2, I-Ting Teng2, Mao Ye1 ![]() , Weihong Tan1, 2
, Weihong Tan1, 2 ![]()
1. Molecular Science and Biomedicine Laboratory, State Key Laboratory for Chemo/Bio Sensing and Chemometrics, College of Biology, College of Chemistry and Chemical Engineering and Collaborative Research Center of Molecular Engineering for Theranostics, Hunan University, Changsha 410082, China
2. Departments of Chemistry, Department of Physiology and Functional Genomics, Center for Research at Bio/Nano Interface, Shands Cancer Center, University of Florida Genetics Institute and McKnight Brain Institute, University of Florida, Gainesville, FL 32611-7200, USA
3. College of Life and Environmental Science, National Navel Orange Engineering Research Center, Gannan Normal University, Economic & Technological Development Zone, Ganzhou 341000, China
* Xiaoqiu Wu and Zilong Zhao contributed equally.
Received 2015-2-19; Accepted 2015-4-9; Published 2015-6-6
Citation:
Wu X, Zhao Z, Bai H, Fu T, Yang C, Hu X, Liu Q, Champanhac C, Teng IT, Ye M, Tan W. DNA Aptamer Selected against Pancreatic Ductal Adenocarcinoma for in vivo Imaging and Clinical Tissue Recognition. Theranostics 2015; 5(9):985-994. doi:10.7150/thno.11938. https://www.thno.org/v05p0985.htm
Other stylesAbstract

In this work, we have developed a truncated DNA aptamer, termed XQ-2d, with high affinity and specificity for pancreatic ductal adenocarcinoma (PDAC). Aptamer XQ-2d selectively binds to PL45 cells with a dissociation constant in the nanomolar range, as determined by its recognition of PL45 tumor cells in mice. Moreover, XQ-2d shows better recognition ratio for 40 tissue sections of clinical PDAC samples (82.5%) compared to the initial cell-SELEX selection library (5%). Therefore, XQ-2d can be considered a promising candidate as a tool for PDAC diagnosis and treatment.
Keywords: aptamer, cell-SELEX, pancreatic ductal adenocarcinoma, molecular probe
Introduction
Pancreatic adenocarcinoma (PAC) is one of the most lethal cancers. Pancreatic ductal adenocarcinomas (PDAC), as the main class of PAC, account for around 95% cases of PAC [1]. Despite development in detection and management of PAC in the past 50 years, prognosis of PAC remains very frustrating, and the 5-year survival rate is less than 5% [2]. Surgery is the only curative option for patients with malignant disease localized to the pancreas [3]. Unfortunately, 80-85% of patients are diagnosed at the advanced unresectable stage by the absence of early detection tests and recognizable signs or symptoms for most patients with localized disease [4, 5]. These circumstances call for the development of novel and effective molecular probes specifically recognizing PAC to facilitate early diagnosis and effective therapeutics.
Early work to develop better diagnostic and therapeutic molecular probes focused on the use of antibodies for tumor recognition and targeted therapy [6, 7]. However, the use of antibodies as molecular probes often lacks ideal pharmacokinetics, and such probes easily lose their activity during chemical modification and storage [8]. As an alternative molecular probe, aptamers, often termed chemical antibodies, have attracted extensive attention in bioanalysis and biomedicine. Aptamers are single-stranded oligonucleotides generated through an in vitro iterative selection process known as Systematic Evolution of Ligands by EXponential enrichment (SELEX) [9, 10]. Aptamers recognize their targets, such as small organic molecules, proteins, and intact cells, by folding into distinct tertiary structures [11]. Besides high binding affinity and specificity, aptamers also present many advantages, including convenient synthesis, easy chemical modification, no immunogenicity, rapid tissue penetration, low variability between different batches and long-term stability [8].
SELEX against live cells (cell-SELEX) has enabled the generation of aptamers that specifically bind target molecules in their native conformations on the cell surface without prior knowledge of the molecular signatures of target cells [12-14]. An improved specificity can be also achieved by performing an additional subtractive selection to produce aptamers against a given cell phenotype, even differentiating tumor cells from normal cells [15, 16]. Up to now, many aptamers have been selected by cell-SELEX against various types of cancer, such as leukemia [12], glioblastoma [13], liver cancer [17] and non-small cell lung cancer [18], and these aptamers have played important roles in molecular imaging [19, 20], capture of circulating tumor cells [21-23], targeted therapy [24] and biomarker discovery [25].
Herein, cell-SELEX was directed against the pancreatic ductal adenocarcinoma PL45 cell line with the human telomerase reverse transcriptase (hTERT) immortalized pancreatic ductal epithelial cell line (hTERT-HPNE) used for subtractive selection. After 15 cycles of selection, aptamer XQ-2 was able to differentiate PL45 cells from hTERT-HPNE cells, and it was, therefore, screened out. Aptamer XQ-2 bound to PL45 cells with a Kd in the nanomolar range. Truncation and chemical modification of aptamer XQ-2 were studied in detail by flow cytometry. In vivo and ex vivo fluorescence imaging demonstrated that the selected aptamer could target the tumor site after intravenous injection into nude mice grafted with PL45 cells. Moreover, laser scanning confocal microscopy imaging displayed that aptamer XQ-2d, a truncated version of aptamer XQ-2, could specifically recognize PDAC tissue with a detection ratio of 82.5%.
Materials and Methods
Cell lines and cell culture
Human pancreatic ductal adenocarcinoma cell line PL45, hTERT-HPNE cell line, CCRF-CEM cell line and Ramos cell line were obtained from the American Type Culture Collection (ATCC). Gastric cancer cell line (MGC-803), breast cancer cell lines (MCF-7 and MDA-MB-231), lung cancer cell lines (H1299, 95-C and 95-D), colon carcinoma cell line (HCT116), melanoma cell line (SK23), human embryonic kidney epithelial cell line (HEK293) and human embryonic lung fibroblast cell line (MRC-5) were obtained from the cell bank of Xiangya Hospital (Changsha, China). All cell lines were cultured at 37 °C in a humid atmosphere with 5% CO2. PL45 cells, MDA-MB-231 cells, MCF-7 cells, MRC-5 cells, SK23 cells, HEK293 cells, DU145 cells and 95-C cells were cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS, Hyclone) and 100 U/mL penicillin-streptomycin. MGC-803 cells, HCT116 cells, H1299 cells, 95-D cells, CCRF-CEM cells, Ramos cells and HL60 cells were cultured in RPMI-1640 medium supplemented with 10% FBS and 100 U/mL penicillin-streptomycin. 75% glucose-free DMEM medium (with additional 2 mM L-glutamine and 1.5 g/L sodium bicarbonate) and 25% M3:Base™ were mixed and then supplemented with 5% FBS, 10 ng/mL human recombinant EGF, 5.5 mM D-glucose and 750 ng/mL puromycin to make the complete growth medium for culturing hTERT-HPNE cells.
DNA library, primers and buffers
The library used in the cell-SELEX was a 42-nucleotide randomized region flanked by 19-nucleotide sequences for primer annealing (5'-ACC GAC CGT GCT GGA CTC A (N)42 A CTA TGA GCG AGC CTG GCG-3'). For PCR amplification, FAM-labeled forward primer (5'-FAM-ACC GAC CGT GCT GGA CTC A-3') and biotin-labeled reverse primer (5'-biotin-CGC CAG GCT CGC TCA TAG T-3') were used. All DNA sequences used in the cell-SELEX were purchased from Sangon Biotech Co. Ltd. (Shanghai, China). Washing buffer was prepared with Dulbecco`s phosphate buffered saline (D-PBS) supplemented with 4.5 g/L of glucose and 5 mM of MgCl2. Binding buffer was prepared with D-PBS supplemented with 4.5 g/L of glucose, 5 mM of MgCl2, 0.1 mg/mL of yeast tRNA, and 1 mg/mL of BSA.
Cell-SELEX procedures
Cell-SELEX was carried out according to previous reports with minimal modification [12, 14]. The initial DNA library was dissolved in binding buffer. After denaturing at 95 °C for 5 min and immediate cooling on ice for 10 min, the initial library was incubated with PL45 cells cultured in a 100-mm culture dish with 90% confluence for 2 h at 4 °C. After incubation, the supernatant solution was removed, and the cells were washed with washing buffer. Then PL45 cells were harvested and transferred to 500 µL of water, and the cell-binding DNAs were collected by heating at 95 °C for 10 min. The cell-binding DNA was used as a template to prepare the evolved DNA pool with FAM-labeled forward primer and biotin-labeled reverse primer by PCR (6-14 cycles of 30 s at 95 °C, 30 s at 58 °C, and 30 s at 72 °C, followed by 5 min at 72 °C). The double-stranded DNA (dsDNA) product was then separated from the PCR solution by streptavidin-coated sepharose beads (GE Healthcare). After treating with 0.2 M of NaOH, FAM-labeled ssDNA was separated from dsDNA, desalted and lyophilized for the next round of selection.
Starting from the third round, the evolved ssDNA pool was first incubated with hTERT-HPNE cells in a 60-mm culture dish with 90% confluence at 4 °C for 30 min for subtractive selection. The unbound ssDNAs were then removed and applied on PL45 cells. To increase the stringency of the selection, the positive incubation time was shortened from 2 h to 1 h as the number of selection rounds increased, and the washing times were also extended gradually from 2 to 3 times. At the same time, negative incubation time was gradually increased from 1 h to 2 h. From the 9th round of selection, two successive subtractive selections were performed to maximize the removal of ssDNA binding to normal pancreas cells. The evolved ssDNA pool generated from the 15th round was PCR-amplified and then high-throughput sequenced using Illumina MiSeq (Sangon Biotech Co., Ltd. Shanghai, China).
Flow cytometric analysis
To monitor the enrichment, 3 x 105 PL45 cells or hTERT-HPNE cells were incubated with 250 nM of FAM-labeled evolved ssDNA pools and library in 200 µL of binding buffer at 4 °C for 1 h. To analyze the binding ability of aptamer, each cell line (3 x 105 cells) was incubated with 250 nM of aptamer or library in 200 µL of binding buffer at 4 °C for 1 h. To measure the dissociation constant (Kd) of aptamers, target PL45 cells (3 x 105) were incubated with different concentrations of aptamer in 200 µL of binding buffer at 4 °C for 1 h.
After incubation, each cell sample was washed three times with 500 µL of washing buffer and then resuspended in 500 µL of binding buffer for flow cytometry analysis (BD FACSVerse™ flow cytometer). All of the experiments for the binding assay were repeated three times. The Kd of aptamer on PL45 cells was determined by fitting the dependence of fluorescence intensity of cell/aptamer complex on aptamer concentration with the equation Y =B max X/( Kd + X) using SigmaPlot software (Jandel Scientific).
Target type analysis and internalization analysis
For target type analysis, PL45 cells were detached with 200 µL 0.25% trypsin or 0.1 mg/ml Proteinase K for 10 min, followed by the addition of FBS to inhibit proteinase activity. After washing with D-PBS, the detached cells were incubated with 250 nM of aptamer in 200 µL of binding buffer at 4 °C for 1 h. After washing, the cell samples were analyzed by flow cytometry.
For internalization analysis, PL45 cells were incubated with 250 nM of FAM-labeled aptamer or library in 200 µL of binding buffer at 37 °C for 2 h. After incubation, cell samples were treated with 200 µL of 0.25% trypsin or 0.1 mg/ml Proteinase K at 37 °C for 10 min before FBS was added to inhibit proteinase function. After washing, the cell samples were analyzed by flow cytometry.
Confocal microscopy imaging
1.5 x 105 cells were seeded in a 35-mm glass bottom dish and cultured for 24 h. After washing with cold washing buffer, the cells were incubated with Cy5-labeled aptamer (250 nM) in 1 mL of binding buffer at 4 °C for 1 h. After washing twice, the cells were imaged by a FV1000-X81 confocal microscope (Olympus, Japan). The images were analyzed by FV10-ASW Version 3.1.
Stability of aptamer in serum
3 µM of FAM-labeled aptamer or 2'-O-methyl-modified aptamer were incubated in DMEM with 10% FBS for different times at 37 °C. At the assigned time, samples were flash frozen in a dry ice/ethanol bath and then stored at -80 °C until all samples were harvested. Samples were then thawed on ice and run on 3% agarose gels. Band density was assayed by a molecular imager (Bio-Rad).
In vivo and ex vivo fluorescence imaging
Male athymic BALB/c (BALB/c-nude) mice were purchased from the Shanghai SLAC Laboratory Animal Co., Ltd. Four- to six-week-old nude mice received a subcutaneous injection of 5 x 106 PL45 cells into the backside. Tumors were then allowed to grow over a period of 15 to 20 days until reaching 0.5-1.5 cm in diameter.
Tumor-bearing BALB/c nude mice were anesthetized with both tranquilizer and anesthetic. Once the mice were anesthetized to be motionless, 4.5 nmol of Cy5-labeled aptamer or library was injected intravenously via the tail vein. At certain time points, fluorescence images of live mice were collected by an IVIS Lumina II in vivo imaging system (Caliper LifeSicence, USA). For the ex vivo fluorescence experiment, tumor-bearing mice intravenously injected with Cy5-labeled aptamers or library were sacrificed by cervical dislocation under narcosis 1 h after injection. After anatomization, the dissected organs, including liver, kidney, spleen, lung, heart and tumor tissue, were imaged with the IVIS Lumina II in vivo imaging system, as described above.
Staining of human tumor tissue sections using selected aptamer
The tissue microarray was purchased from US Biomax (Xi'an AiLiNa Biotechnology Co., Ltd. China). The pretreatment of Formalin fixed, paraffin embedded (FFPE) tissue sections was described previously [18]. Briefly, all the tissue sections were deparaffinized in xylene to remove paraffin (15 min x 2) and then immersed in a degraded ethanol series (100%, 95%, 90%, 80%, and 70%) at 5 min intervals to rehydrate. After washing with D-PBS buffer, the hydrated tissue sections were heated in citrate buffer (0.01 M, pH 6.0) at 95 °C for 20 min to retrieve antigens. The prepared tissue sections blocked with binding buffer, 20% FBS and 0.1mg/ml Salmon Sperm DNA for 60 min were incubated with 250 nM Cy5-labeled aptamer or library in 200 µL of binding buffer for 30 min on ice in the dark. A FV1000-X81 confocal microscope was used for imaging. The images were analyzed by FV10-ASW Version 3.1 (Olympus) and displayed with the intensity scale ranging from 670 to 2940.
Results and discussion
Cell-SELEX against PDAC PL45 cells
To generate aptamers against PDAC cancer cells, the human PDAC PL45 cell line was used as the target, and the normal hTERT-HPNE cell line was used for counter selection. The cell-SELEX process is schematically shown in Figure 1. For the first two rounds of selection, ssDNA library was only applied on PL45 cell monolayer for positive selection. From the third round of selection, the DNA library was first incubated with normal hTERT-HPNE cells to remove nonspecific sequences. The unbound DNA was collected and then incubated with target PL45 cells for positive selection. After washing, the bound DNA was eluted and amplified for next-round selection. During selection, the target cell-binding ability of evolved ssDNA library was monitored by flow cytometry. When PL45 cells were incubated with FAM-labeled ssDNA pools from an increasing number of selection rounds, steady increases in fluorescence intensity from the target cells were observed (Figure 2A). However, almost no increase in fluorescence signal was observed for hTERT-HPNE cells after incubation with FAM-labeled ssDNA pool from the 15th selection round (Figure 2B). Therefore, the ssDNA pool evolved from the 15th round of selection was high-throughput sequenced with Illumina MiSeq.
The binding ability of DNA aptamer
After sequencing, the aptamer candidates were grouped based on homogeneity, and the first six groups were synthesized for further research. The results from flow cytometry and confocal microscopy imaging demonstrated that aptamer XQ-2 had good binding to the target PL45 cell line, but not to the negative cell line, hTERT-HPNE (Figure 3). These results suggested that aptamer XQ-2 might be capable of distinguishing PL45 cells from normal cells. Therefore, aptamer XQ-2 was further tested for its binding affinity to the PL45 cell line. To accomplish this, PL45 cells were incubated with different concentrations of FAM-labeled aptamers or initial library at 4 °C, followed by monitoring the fluorescence intensity of cell sample by flow cytometry. After the geometric mean fluorescence (GMF) intensity of cell samples treated with library was subtracted from that of cell samples treated with XQ-2, the equilibrium dissociation constants (Kd) of XQ-2 for PL45 cells were obtained by fitting the dependence of fluorescence intensity of samples on the concentration of the aptamers to the equation Y =B max X/( Kd +X). As shown in Supplementary Figure S1, the Kd of aptamer XQ-2 for PL45 cells was about 82.5 nM, demonstrating that the selected aptamer XQ-2 could bind with high affinity to target PL45 cells.
Figure 1
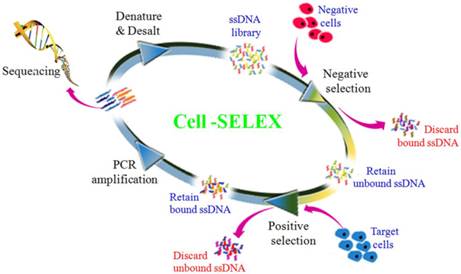
Scheme of systematic enrichment of aptamers against PDAC PL45 cell line. The DNA library was incubated with hTERT-HPNE cells (negative cells) to remove negative cell-binding sequences. The unbound DNAs were collected and then incubated with target PL45 cell line for positive selection. After washing, the bound DNAs were eluted and amplified by PCR for next-round selection. After enrichment, DNA sequencing was performed to identify individual aptamer sequences.
Figure 2
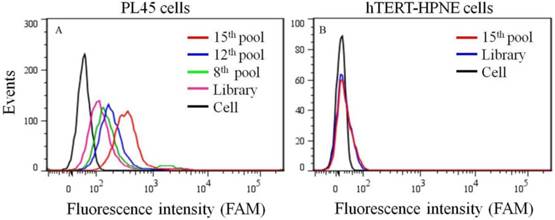
Binding ability of enriched ssDNA library and unselected library to PL45 cells (a) and hTERT-HPNE cells (b). The concentration of ssDNA pool was 250 nM.
Figure 3
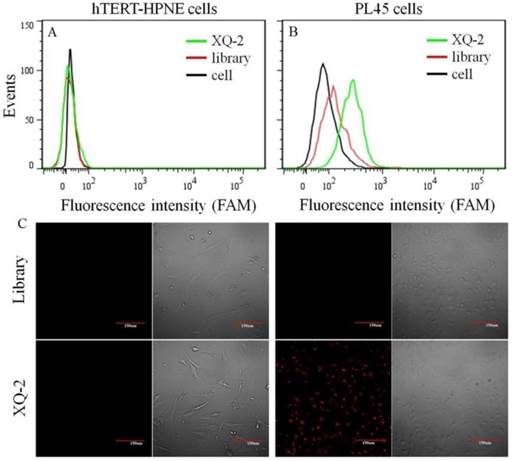
Flow cytometric assay (a, b) and confocal microscopy assay (c) of the binding of aptamer XQ-2 (250 nM) to hTERT-HPNE cells and PL45 cells. The unselected initial library (250 nM) was used as control. Scale bar = 100 µm.
To test the binding ability of aptamer XQ-2 for other cell lines, 14 cell lines, including 12 cancer cell lines and 2 normal cell lines, were incubated with 250 nM of FAM-labeled aptamers at 4 °C for 1 h, respectively. As shown in Figure 4, aptamer XQ-2 strongly bound three leukemia cell lines (HL60, CCRF-CEM, and Ramos), prostate cancer DU145 cell line, gastric cancer MGC-803 cells, breast cancer MDA-MB-231 cell line and two lung cancer cell lines (95-C and H1299). However, it showed little binding affinity for four other cancer cell lines, including breast cancer MCF-7, lung cancer 95-D, colon carcinoma HCT116 and human melanoma SK23 cells, when compared with FAM-labeled library. Moreover, aptamer XQ-2 showed no binding ability to two other normal cell lines: primary fibroblast MRC5 and human embryonic kidney epithelial HEK293. These results indicate that the target of aptamer XQ-2 is highly expressed in HL60, CCRF-CEM, Ramos, DU145, MGC-803, MDA-MB-231, 95-C and H1299 cancer cell lines, but little in MCF-7, 95-D, HCT116 and SK23 cancer cell lines. It could therefore be expected that the selected aptamer XQ-2 could be used as molecular probe for recognizing some kinds of cancer cells, as well as differentiating their subtypes.
Determination of target type for aptamer XQ-2
Because of its overexpression in certain kinds of cancer cell lines, the target of aptamer XQ-2 was further characterized. To determine if the binding target of XQ-2 is an extracellular membrane protein, the binding affinity of XQ-2 against PL45 cells treated with trypsin or proteinase K was investigated. For PL45 cells treated with trypsin or proteinase K for 10 min, as shown in Figure 5A, XQ-2 lost its binding ability. The significant loss of binding ability of XQ-2 to cells treated with proteinase suggests that the binding targets of XQ-2 are most likely extracellular proteins. To investigate whether XQ-2 could be internalized into PL45 cells, the intracellular fluorescence signals of the targeted cells were analyzed by flow cytometry after removing the fluorescence signals on the cell surface by trypsin or proteinase K. As shown in Figure 5B, the removal of the fluorescence signals on the cell surface caused the disappearance of fluorescence signals of target cells, indicating there were little intracellular fluorescence signals. Thus, it might be deduced from these data that aptamer XQ-2 mainly localized on the target PL45 cell surface after incubation at 37 °C for 2 h.
Figure 4
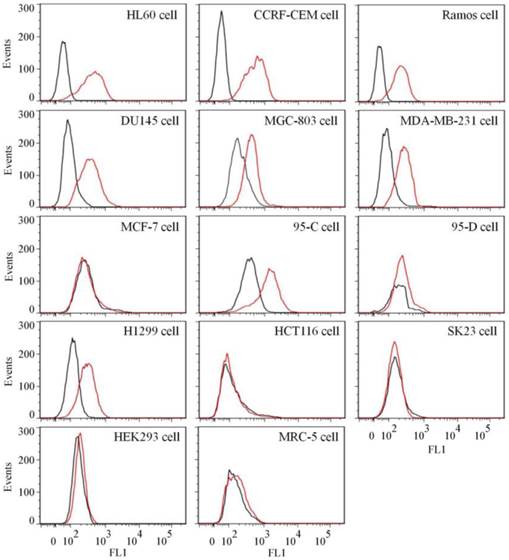
Binding ability of aptamer XQ-2 (250 nM, red line) on different cell lines. The unselected initial library (250 nM, black line) was used as control.
Figure 5
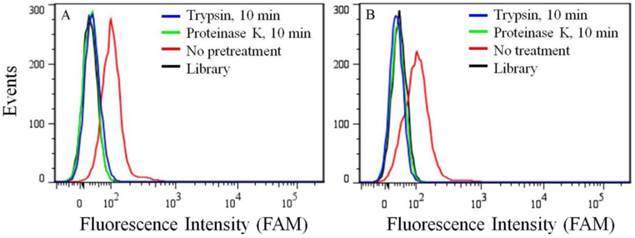
(a) Binding ability of aptamer XQ-2 (250 nM) to PL45 cells with no pretreatment and pretreatment with proteinase K or trypsin for 10 min. (b) Internalized assay of aptamer XQ-2 (250 nM) in PL45 cells after incubation at 37 °C for 2 h.
Figure 6
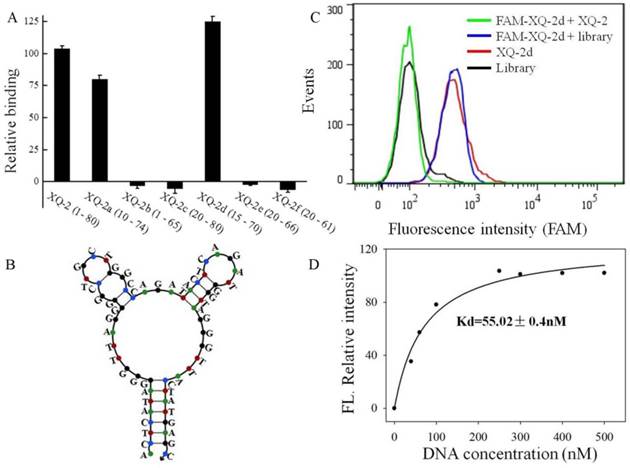
Truncation of aptamer XQ-2 and characterization of XQ-2d. (a) Binding ability of different truncated versions by removing nucleotides at 5' and 3'-terminus (250 nM) on target PL45 cells. (b) Secondary structure of XQ-2d predicted by Nupack. (c) Binding assay of XQ-2d (250 nM) to PL45 cells in the presence of XQ-2 (2.5 µM) or unselected library (2.5 µM). (d) Dissociation constant of truncated XQ-2d for target PL45 cells.
Sequence truncation and chemical modification of aptamer XQ-2
To reduce the cost of DNA synthesis, the full-length aptamer XQ-2 (80nt, 1-80) is required truncation, but with minimal loss in binding ability. Six kinds of truncated versions of the XQ-2 aptamer were synthesized by gradually removing the nucleotides at the 5' and 3' termini (Supplementary Table S1) and then testing the binding ability to PL45 cells by flow cytometry. It could be seen that XQ-2a (65nt, 10-74) and XQ-2d (56nt, 15-70) kept strong binding affinity (Figure 6A). However, further truncation resulted in reduction of the binding ability shown by full-length XQ-2. The structure of XQ-2d was predicted by Nupack as a stem-loop structure with two hairpins on the loop (Figure 6B). When the stem of XQ-2d was replaced by other base pairs or random bases, or the loop of XQ-2d was replaced by random bases, respectively (Supplementary Table S2), these substitutions caused the loss of binding ability of XQ-2d (Supplementary Figure S2), indicating that the bases from stem and loop are responsible for the binding function of XQ-2d. When PL45 cells were incubated with XQ-2d in the presence of 15-fold XQ-2 or initial library, it was found that XQ-2, but not library, could inhibit the binding of XQ-2d to PL45 cells (Figure 6C). Since the binding inhibition might be derived from by the fact that the binding site of XQ-2d on the targeted PL45 cell surface were occupied by 10-fold molar excess of XQ-2, this competition assay proved that XQ-2 and XQ-2d bound to the same protein target on the surface of PL45 cells. Thus, XQ-2d was suggested as the most suitable truncated form of XQ-2 still able to maintain strong binding affinity based on their respective Kds (55.02 nM, Figure 6D vs. 82.5 nM, Supplementary Figure S1), while still retaining the selectivity of full-length XQ-2, which had little, to no, binding to hTERT-HPNE cells (Supplementary Figure S3).
To increase the stability in serum for further application, the four bases in the 5' and 3' termini of XQ-2d were replaced by either phosphorothioate oligonucleotides or 2'-O-methyl oligonucleotides (Supplementary Table S3). Flow cytometry showed that the substitution with phosphorothioate oligonucleotides weakened the binding ability of XQ-2d to the target PL45 cells; however, the 2'-O-methyl substitution showed binding ability similar to that of XQ-2d (Supplementary Figure S4). The stability of XQ-2d in DMEM cell medium 10% FBS, with or without 2'-methyl oligonucleotides, was further tested by agar electrophoresis. It could be seen that nearly all XQ-2d was degraded 24 h after incubation in 10% FBS; however, the 2'-O-methyl substitution could last 36 h in 10% FBS (Supplementary Figure S5). These results suggested that the modification of XQ-2d with 2'-O-methyl oligonucleotides could provide a nuclease-resistant aptamer for further applications.
In vivo and ex vivo fluorescence imaging
To test whether aptamer XQ-2d retained its recognition ability in vivo, a systematic comparative investigation was performed to validate the selectivity of Cy5-labeled XQ-2d in illuminating PDAC in vivo, using Cy5-labeled library as the control. A weak fluorescence signal could be seen in the tumor site only 5 minutes after injecting XQ-2d into PDAC tumor-bearing mouse through tail vein, followed by a gradual increase of fluorescence signal at the tumor site 1 h post-injection (Figure 7, lower). However, the fluorescence signal significantly decreased at 2 h, and almost disappeared at 3 h post-injection. After injection of Cy5-labeled library into BALB/c-nude mice grafted with PDAC through tail vein, no fluorescence signal was seen at the tumor site during the entire procedure (Figure 7, upper). The biodistribution of Cy5-labeled aptamer in mice was also examined at 1 h post-injection. It could be seen that the tumor site from mice injected with Cy5-labeled aptamer presented higher fluorescence signal than that from mice injected with Cy5-labeled library (Supplementary Figure S6). Aptamer-injected tumor-bearing mice also presented more accumulation in tumor than heart, lung or spleen (Supplementary Figure S6), although liver and kidney tissue both presented a fluorescence signal after injection of XQ-2d (data not shown). These results suggested that aptamer XQ-2d possessed in vivo PDAC-targeting ability, providing a potential molecular probe for diagnosis and therapy of PDAC.
Imaging PDAC clinical tissues with aptamer
To test the recognition ability of XQ-2d in clinical samples, laser confocal fluorescence microscopy was used to image 48-core tissue arrays with 40 PDAC cases and 8 normal cases stained with Cy5-labeled aptamer or library, respectively. It could be seen that 33 of the 40 PDAC tissues and 2 of the 8 normal tissues displayed a fluorescence signal after incubation with Cy5-labeled XQ-2d (Figure 8 and Supplementary Figure S7). When another tissue array from the same patients was incubated with Cy5-labeled library, 2 of the 40 PDAC tissues and 2 of the 8 normal tissues presented binding to XQ-2d. Therefore, the binding ratio of XQ-2d to PDAC tissues was 82.5%, which was higher than the binding ratio of library to PDAC tissues (5%) from same patients (Table 1). The binding ratio of XQ-2d to normal tissues was 25%. However, it was noted XQ-2d bound to normal tissues as well as library, indicating that the binding of XQ-2d to normal tissues was mainly nonspecific. These results indicate that the target of aptamer XQ-2d was also expressed in patients with PDAC. Therefore, aptamer XQ-2d holds potential as an effective molecular diagnostic reagent for recognizing PDAC tumor tissue.
Table 1
Tissue imaging results with XQ-2d
| ssDNA | Tissue section | Number of sample | Positive rate |
|---|---|---|---|
| Cy5-labeled XQ-2d | PDAC tissue | 40 | 82.5% |
| Normal pancreatic tissue | 8 | 25% | |
| Cy5-labeled library | PDAC tissue | 40 | 5% |
| Normal pancreatic tissue | 8 | 25% |
Figure 7
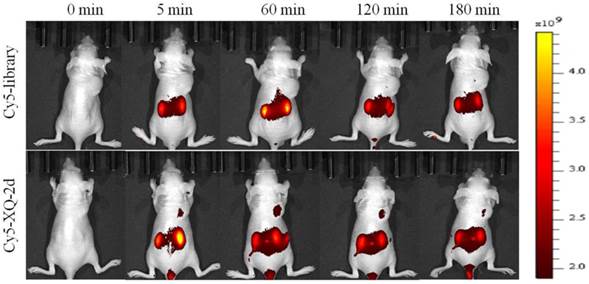
PDAC tumor-bearing mice were intravenously injected with Cy5-labeled library or Cy5-labeled aptamer XQ-2d and then imaged. Upper: timelapse in vivo fluorescence imaging after injection of Cy-5 labeled library. Lower: timelapse in vivo fluorescence imaging after injection of Cy-5 labeled library.
Figure 8
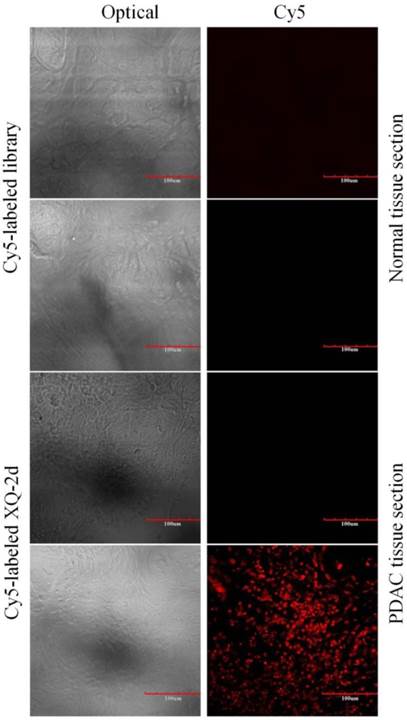
Representative florescence images of PDAC tissue sections and normal tissue sections stained with Cy5-labeled aptamer XQ-2 (250 nM) or Cy5-labeled library (250 nM). Scale bar = 100 µm.
Conclusions
In summary, a DNA aptamer against the PDAC PL45 cell line has been successfully selected by cell-SELEX through 15 rounds of evolved enrichment. The Kd value of aptamer on PL45 cells is in the nanomolar range. The target of aptamer XQ-2 has been preliminarily determined as a membrane protein on the cell surface. Structural studies resulted in a truncated XQ-2d with recognition ability to target cell PL45 cells equal to that of XQ-2. In vivo imaging and recognition of clinical samples indicate that XQ-2/XQ-2d has potential for the diagnosis and molecular imaging of PDAC. Moreover, since XQ-2/XQ-2d has high binding affinity in differentiating cancer cells and normal cells, or subtypes of tumor cell lines, aptamers targeting PDAC with subsequent imaging capability might facilitate understanding the molecular mechanisms underlying oncogenesis.
Supplementary Materials
Figures S1-S7 and Tables S1-S3.
Acknowledgements
This work is supported by the National Key Scientific Program of China (2011CB911000), NSFC grants (NSFC 2013CB932702, NSFC 21405041, NSFC 21221003, NSFC 21327009), and China National Instrumentation Program 2011YQ03012412. It is also supported by grants awarded by the National Institutes of Health (GM079359 and CA133086).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bardeesy N, DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002;2:897-909
2. Wolfgang CL, Herman JM, Laheru DA. et al. Recent progress in pancreatic cancer. CA Cancer J Clin. 2013;63:318-48
3. Vincent A, Herman J, Schulick R. et al. Pancreatic cancer. Lancet. 2011;378:607-20
4. Lee MX, Saif MW. Screening for early pancreatic ductal adenocarcinoma: an urgent call. JOP. 2009;10:104-8
5. Fong ZV, Winter JM. Biomarkers in pancreatic cancer: diagnostic, prognostic, and predictive. Cancer J. 2012;18:530-8
6. Folli S, Westermann P, Braichotte D. et al. Antibody-indocyanin conjugates for immunophotodetection of human squamous cell carcinoma in nude mice. Cancer Res. 1994;54:2643-9
7. Neri D, Carnemolla B, Nissim A. et al. Targeting by affinity-matured recombinant antibody fragments of an angiogenesis associated fibronectin isoform. Nat Biotechnol. 1997;15:1271-5
8. Jayasena SD. Aptamers: an emerging class of molecules that rival antibodies in diagnostics. Clin Chem. 1999;45:1628-50
9. Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505-10
10. Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818-22
11. Stoltenburg R, Reinemann C, Strehlitz B. SELEX-a (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol Eng. 2007;24:381-403
12. Shangguan DH, Li Y, Tang ZW. et al. Aptamers evolved from live cells as effective molecular probes for cancer study. Proc Natl Acad Sci USA. 2006;103:11838-43
13. Daniels DA, Chen H, Hicke BJ. et al. A tenascin-C aptamer identified by tumor cell SELEX: systematic evolution of ligands by exponential enrichment. Proc Natl Acad Sci USA. 2003;100:15416-21
14. Sefah K, Shangguan DH, Xiong XL. et al. Development of DNA aptamers using Cell-SELEX. Nat Protoc. 2010;5:1169-85
15. Tan WH, Donovan MJ, Jiang JH. Aptamers from cell-based selection for bioanalytical applications. Chem Rev. 2013;113:2842-62
16. Fang XH, Tan WH, Aptamers generated from cell-SELEX for molecular medicine. a chemical biology approach. Acc Chem Res. 2010;43:48-57
17. Shangguan DH, Cao ZH, Meng L. et al. Cell-specific aptamer probes for membrane protein elucidation in cancer cells. J Proteome Res. 2008;7:2133-9
18. Zhao ZL, Xu L, Shi XL. et al. Recognition of subtype non-small cell lung cancer by DNA aptamers selected from living cells. Analyst. 2009;134:1808-14
19. Hicke BJ, Stephens AW, Gould T. et al. Tumor targeting by an aptamer. J Nucl Med. 2006;47:668-78
20. Shi H, He XX, Wang KM. et al. Activatable aptamer probe for contrast-enhanced in vivo cancer imaging based on cell membrane protein-triggered conformation alteration. Proc Natl Acad Sci USA. 2011;108:3900-5
21. Sheng W, Chen T, Kamath R. et al. Aptamer-enabled efficient isolation of cancer cells from whole blood using a microfluidic device. Anal Chem. 2012;84:4199-206
22. Shen Q, Xu L, Zhao LB. et al. Specific capture and release of circulating tumor cells using aptamer-modified nanosubstrates. Adv Mater. 2013;25:2368-73
23. Zhao W, Cui CH, Bose S. et al. Bioinspired multivalent DNA network for capture and release of cells. Proc Natl Acad Sci USA. 2012;109:19626-31
24. Zhu GZ, Zheng J, Song EQ. et al. Self-assembled, aptamer-tethered DNA nanotrains for targeted transport of molecular drugs in cancer theranostics. Proc Natl Acad Sci USA. 2013;110:7998-8003
25. Shangguan DH, Meng L, Cao ZH. et al. Identification of liver cancer-specific aptamers using whole live cells. Anal Chem. 2008;80:721-8
Author contact
![]() Corresponding authors: Prof. Weihong Tan, E-mail: tanufl.edu; Prof. Mao Ye, E-mail: yemaocsucom
Corresponding authors: Prof. Weihong Tan, E-mail: tanufl.edu; Prof. Mao Ye, E-mail: yemaocsucom
Citation styles
APA
Wu, X., Zhao, Z., Bai, H., Fu, T., Yang, C., Hu, X., Liu, Q., Champanhac, C., Teng, I.T., Ye, M., Tan, W. (2015). DNA Aptamer Selected against Pancreatic Ductal Adenocarcinoma for in vivo Imaging and Clinical Tissue Recognition. Theranostics, 5(9), 985-994. https://doi.org/10.7150/thno.11938.
ACS
Wu, X.; Zhao, Z.; Bai, H.; Fu, T.; Yang, C.; Hu, X.; Liu, Q.; Champanhac, C.; Teng, I.T.; Ye, M.; Tan, W. DNA Aptamer Selected against Pancreatic Ductal Adenocarcinoma for in vivo Imaging and Clinical Tissue Recognition. Theranostics 2015, 5 (9), 985-994. DOI: 10.7150/thno.11938.
NLM
Wu X, Zhao Z, Bai H, Fu T, Yang C, Hu X, Liu Q, Champanhac C, Teng IT, Ye M, Tan W. DNA Aptamer Selected against Pancreatic Ductal Adenocarcinoma for in vivo Imaging and Clinical Tissue Recognition. Theranostics 2015; 5(9):985-994. doi:10.7150/thno.11938. https://www.thno.org/v05p0985.htm
CSE
Wu X, Zhao Z, Bai H, Fu T, Yang C, Hu X, Liu Q, Champanhac C, Teng IT, Ye M, Tan W. 2015. DNA Aptamer Selected against Pancreatic Ductal Adenocarcinoma for in vivo Imaging and Clinical Tissue Recognition. Theranostics. 5(9):985-994.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.