Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(8):905-918. doi:10.7150/thno.11304 This issue Cite
Research Paper
Erythropoietin Improves the Accumulation and Therapeutic Effects of Carboplatin by Enhancing Tumor Vascularization and Perfusion
Dennis Doleschel1, Anne Rix1, Susanne Arns1, Karin Palmowski1,2, Felix Gremse1, Ruth Merkle3, Florian Salopiata3,4, Ursula Klingmüller3,4, Michael Jarsch5, Fabian Kiessling1 ![]() , Wiltrud Lederle1
, Wiltrud Lederle1 ![]()
1. Experimental Molecular Imaging, Medical Faculty, RWTH Aachen University, Aachen, Germany.
2. Department of Pneumology and Critical Care Medicine, Thoraxklinik, University of Heidelberg, Heidelberg, Germany.
3. Division of Systems Biology of Signal Transduction, DKFZ-ZMBH Alliance, German Cancer Research Center (DKFZ), Heidelberg, Germany.
4. Translational Lung Research Center (TLRC), Member of the German Center for Lung Research (DZL), Heidelberg, Germany.
5. Roche Pharmaceutical Research and Early Development, Pharmaceutical Sciences, Roche Innovation Center, Penzberg, Germany.
Received 2014-12-10; Accepted 2015-2-27; Published 2015-5-1
Abstract

Recombinant human erythropoietin (rhuEpo) is currently under debate for the treatment of chemotherapy-induced anemia due to clinical trials showing adverse effects in Epo-treated patients and the discovery of the erythropoietin-receptor (EpoR) in tumor and endothelial cells. Here, using Epo-Cy5.5 as theranostic near-infrared fluorescent probe we analyzed the effects of rhuEpo as co-medication to carboplatin in non-small-cell-lung-cancer (NSCLC)-xenografts with different tumor cell EpoR-expression (H838 ~8-fold higher than A549). Nude mice bearing subcutaneous A549 and H838 NSCLC-xenografts received either only carboplatin or carboplatin and co-medication of rhuEpo in two different doses. Tumor sizes and relative blood volumes (rBV) were longitudinally measured by 3D-contrast-enhanced ultrasound (3D-US). Tumoral EpoR-levels were determined by combined fluorescence molecular tomography (FMT)/ micro computed tomography (µCT) hybrid imaging. We found that rhuEpo predominantly acted on the tumor endothelium. In both xenografts, rhuEpo co-medication significantly increased vessel densities, diameters and the amount of perfused vessels. Accordingly, rhuEpo induced EpoR-phoshorylation and stimulated proliferation of endothelial cells. However, compared with solely carboplatin-treated tumors, tumor growth was significantly slower in the groups co-medicated with rhuEpo. This is explained by the Epo-mediated vascular remodeling leading to improved drug delivery as obvious by a more than 2-fold higher carboplatin accumulation and significantly enhanced tumor apoptosis. In addition, co-medication of rhuEpo reduced tumor hypoxia and diminished intratumoral EpoR-levels which continuously increased during carboplatin (Cp) -treatment. These findings suggest that co-medication of rhuEpo in well balanced doses can be used to improve the accumulation of anticancer drugs. Doses and indications may be personalized and refined using theranostic EpoR-probes.
Keywords: Angiogenesis, Lung cancer, Chemotherapy, Non-Invasive Imaging, Theranostic Agent, Fluorescence Molecular Tomography
Introduction
Lung cancer is the worldwide leading cause of cancer related deaths [1], the predominant histologic subtype being non-small-cell-lung-cancer (NSCLC) [2,3]. Despite improvements in therapy e.g. combination chemotherapy and VEGF or EGFR-targeting approaches, NSCLCs are still associated with a poor clinical prognosis [1,4]. Platinum-based combination chemotherapy, recommended as first line treatment in NSCLC [5,6], damages the hematopoietic cells in the bone marrow and is additionally nephrotoxic, thus affecting the Epo-producing cells in the renal cortex [7-9]. In consequence, a high number of patients treated with platinum-derivatives suffer from chemotherapy-induced anemia (CIA) which is associated with a decreased quality of life and an enhanced risk of hypoxia-induced treatment resistance [10]. Alternatively to blood transfusions, severe CIA is treated in the clinics with erythropoiesis stimulating agents (ESAs) such as recombinant human Epo (rhuEpo) [10].
The use of ESAs is currently under controversial debate since several clinical trials and meta-analyses, including studies on NSCLCs, reported a negative clinical outcome with ESA-treatment, e.g. a lower survival rate for these patients [11-15]. The additional discovery of EpoR-transcripts and EpoR-protein on tumor and endothelial cells [16] has further intensified the debate, as Epo-treatment of tumors with functional EpoR and EpoR-mediated signaling might promote cancer growth by either supporting tumor cell survival/ proliferation or by stimulating the endothelium and thus promoting angiogenesis. Preclinical studies revealed controversial results [17], showing on the one hand that Epo had no effect on tumor growth [18] and in line with this, no functional EpoR could be detected on tumor cells [19-21]. On the other hand, tumor growth-promoting effects [22] and even enhanced metastasis [23] were observed in response to Epo-treatment which could in some studies be attributed to stimulation of angiogenesis [24,25].
In order to investigate the effects of Epo longitudinally in vivo, we have recently developed a near infrared (NIR)-labeled theranostic EpoR-probe (Epo-Cy5.5). This probe is therapeutically fully bioactive [26] and could sensitively assess the EpoR-levels of A549 and H838 NSCLC-xenografts in vivo that differ in tumor cell EpoR-expression [26]. Binding of the EpoR-probe was also observed at the tumor endothelium, suggesting that Epo might have an effect on endothelial cells in the NSCLC-xenografts. In the present study, the theranostic EpoR-probe was used to longitudinally analyze the effects of Epo on EpoR-expression in these NSCLC-xenografts during carboplatin-treatment. In parallel, we investigated the effects of Epo on chemotherapy accumulation and therapy outcome.
Materials and Methods
Cell lines and cell culture
The NSCLC cell lines A549 and H838 (both ATCC) were cultivated in Dulbecco's Modified Eagle's Medium (DMEM), 1% penicillin-streptomycin, and 10% fetal calf serum (all Invitrogen). Human umbilical vein endothelial cells (HUVEC, c-pooled, PromoCell) were cultured in VascuLife VEGF Cell Culture Medium (Lifeline Cell Technology). Both cell lines express the EpoR, but the expression level of A549 is approximately 8 times lower than of H838 [26].
Effect of Epo on HUVEC proliferation in vitro
HUVECs (4 x 104 cells / well) were seeded in 6 well plates (Becton Dickinson). One day after seeding, the cells were treated daily with 300 ng/ml or 75 ng/ml of rhuEpo-β (epoetin β, Roche) for four days. These doses correspond to the amount expected per ml blood in the mice in vivo. Untreated cells were used as controls. The cell medium was renewed all 24 h and the cell number was determined using a cell counter (Cedex XS, Innovatis). The analysis was done in triplicates for each condition.
Quantitative immunoblotting
HUVECs were seeded three days in advance and washed three times with DMEM without additives and then starved for 3 hours in DMEM with 1% penicillin/streptomycin, 2 mM L-glutamine (Gibco) and 1 mg/mL BSA. The cells were stimulated for 10 min with 150 ng/ml rhuEpo-β, washed with ice-cold PBS and directly lysed with 1.25 x NP-40 lysis buffer (1.25% NP-40, 187.5 mM NaCl, 25 mM Tris pH 7.4, 12.5 mM NaF, 1.25 mM EDTA pH 8.0, 1.25 mM ZnCl2 pH 4.0, 1.25 mM MgCl2, 1.25 mM Na3VO4, 12.5% glycerol) supplemented with aprotinin and AEBSF (Sigma). After removal of the cell debris, the lysate was used for determination of protein concentration (Pre-diluted Protein Assay Standards (BSA), Thermo Scientific; BCA Protein Assay Reagent A&B, Pierse®). Immunoprecipitations were performed with antibodies against EpoR (R&D, MAB 307) using protein A sepharose beads. Immunoprecipitates were eluted the following day by boiling in 2 x sample buffer for 3 min at 95°C, separated by 10% SDS-PAGE and transferred to a nitrocellulose membrane (Schleicher & Schuell). Proteins were immobilized with Ponceau S solution, blocked with 5 % BSA. The membranes were incubated with mouse antibodies to phosphotyrosine (pTyr) (Upstate, clone 4G10) and horseradish peroxidase-coupled anti-mouse antibodies (Dianova) containing 5 % BSA. To remove antibodies, membranes were treated with β-mercaptoethanol and SDS [27] and subsequently incubated with rabbit antibodies to EpoR (Santa Cruz, C-20) and horseradish peroxidase-coupled anti-rabbit antibodies (Dianova). Detection was performed using ECL substrate (GE Healthcare) and acquired with the CCD camera-based ImageQuant LAS 4000 (GE Healthcare). For quantification, the ImageQuant TL version 7.0 software (GE Healthcare) was used.
Effect of Epo on EpoR-expression of HUVECs under normoxic and hypoxic conditions
HUVECs were seeded in a number of 5 x 105 per flask (T75, Greiner Bio-One GmbH). After 24h, the cells were washed with 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer [28], and treated for 48h with 300 ng/ml of rhuEpo-β under normoxic (95% air, 5% CO2) and hypoxic (2% O2, 5% CO2) conditions. Untreated cells in both culture conditions were used as controls (n = 3 per culture and treatment condition). Thereafter, the cells were washed in cold phosphate buffered saline (PBS) and removed from the flask by scraping. Cell pellets were frozen at -80°C.
Expression of EpoR mRNA in HUVECs was analyzed by quantitative real time PCR (RT-qPCR) as described earlier [26]. In short, total RNA from 3 x 106 HUVECs was isolated using the RNeasy Mini Plus Kit (Qiagen) according to the manufacturer's instructions. For quantitative RT-PCR, cDNA was generated with the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and analyzed using the LightCycler 480 with the Universal ProbeLibrary platform (UPL, Roche Applied Science). Crossing point values were calculated using the Second Derivative Maximum method of the LightCycler 480 Basic Software (Roche Applied Science). PCR efficiency was assumed to be 100 %. Fold-changed EpoR-expression levels relative to untreated HUVECs at normoxia were calculated. UPL probes and primer sequences were: EpoR_for 5'-ttggaggacttggtgtgtttc-3', EpoR_rev 5'-agcttccatggctcatcct-3', Probe#69.
Animal study design and therapy
All experiments were approved by the governmental review committee on animal care. NSCLC-xenografts were induced by s.c. injection of either 5 × 106 H838 cells in 100 µl Matrigel (Phenol free, Growth Factor Reduced, BD Biosciences) or 4 x 106 A549 cells in 100 µl DMEM into the right flank of immunodeficient female CD-1 nude mice (Charles River).
A total number of 15 mice per NSCLC-model were included into the therapy study. At a tumor size of ~ 50 mm3, the mice were randomly divided into three treatment groups, including five animals each. All animals were treated once weekly i.p. with 60 mg/kg body weight carboplatin (Actavis) for three weeks, corresponding to a dose and treatment scheme that showed good efficacy [29]. Carboplatin was chosen as chemotherapeutic drug due to lower digestive, neuro- and nephrotoxicity compared to cisplatin [6]. Two animal groups were additionally treated with two different doses of rhuEpo-β, 5 µg/ kg body weight or 20 µg/ kg body weight, respectively. These correspond to dosages of 75 and 300 ng/ml in a 30 g mouse with a mean blood volume of 2 ml. Epo was applied trice per week for a period of three weeks and injected s.c. because of its short blood half-life [30]. Dosing and treatment schedule are equivalent to common treatment guidelines for humans [31]. Epo was not administered alone since in cancer patients, Epo is mainly applied for counterbalancing the side-effects of cytotoxic therapy. Additionally, the sole application of Epo increases the hemoglobin and hematocrit levels, thus increasing the risk of thromboembolisms and changing the physiology of the animals which hampers the comparability of the treatment groups. The Hb levels of the mice were controlled once weekly during Epo- and carboplatin-treatment by blood withdrawal (maximally 10 µl). Animals were sacrificed at day 21 of therapy. To assess the therapeutic efficacy of the used carboplatin dose, tumor sizes of carboplatin-treated and untreated mice were compared (n=5 per group).
Imaging Protocols
Ultrasound measurements
For both NSCLC-models, the longitudinal (week 1, 2 and 3) ultrasound scans were performed on anesthetized NSCLC-model mice (isoflurane, 2% v/v, AbbVie) at a small animal high frequency ultrasound system (Vevo2100; VisualSonics) equipped with a MS-250 transducer using the linear contrast mode (B-mode) [32]. For determining the tumor size and the relative blood volume, 3D contrast-enhanced ultrasound was performed as described previously [33]. In brief, before contrast agent injection, a first 3D scan was done to assess the tumor volume and to record the baseline values before contrast agent injection. For scanning, the ultrasound transducer was moved computer-operated across the tumor, acquiring a 3D data set. Thereafter, hard-shelled, gas filled poly-n-butylcyanoacrylate (PBCA) microbubbles [34] (5 x 106 per injection in 50 µl PBS) were injected via the tail vein, and a second 3D ultrasound scan was performed. The relative blood volume was determined by subtracting the voxel intensities of the baseline from the second 3D ultrasound scan as described [33] using the Imalytics Preclinical software (Philips). Due to the high inter-individual tumor heterogeneity, the tumor sizes and relative blood volumes measured during the therapy were normalized to the initial values before treatment start.
Fluorescence molecular tomography (FMT) and micro computed tomography (µCT)
The longitudinal (week 1, 2 and 3) quantitative determination of the EpoR-levels in the two NSCLC-models in vivo was done as described previously using the near infrared florescent (NIRF) theranostic probe Epo-Cy5.5 and an FMT-scanner which reconstructs the fluorescence based on diffuse trans-illumination and emission measurements using a CCD-camera (FMT2500 LX, PerkinElmer). Epo-Cy5.5 is fully bioactive as shown by induction of a similar increase in proliferation of bone marrow cells as rhuEpo [26]. 24 h before scanning, Epo-Cy5.5 was injected intravenously via a tail vein catheter (1 ng, diluted in 0.9 % NaCl, 100 µl injection volume). For scanning, the mice were anesthetized using isoflurane (2% v/v). In order to precisely localize the organs, a µCT scan was performed directly before the FMT measurement at a dual energy µCT system (Tomoscope Duo, CT-Imaging GmbH, Germany) as described [26]. The mice were kept anesthetized and held in a fixed position in a multimodal animal bed for the duration of both µCT and FMT scans. Quantitative 3D FMT-and 3D µCT-data were co-registered. The organs were segmented based on the anatomical µCT-data and the Epo-Cy5.5 concentrations were determined using the Imalytics Preclinical software [35].
Since Epo-Cy5.5 is completely cleared from the body after 5 days [26], no background levels occured in the repetitive measurements (week 1, 2, 3) with intervals of six days between each injection.
Indirect immunofluorescence
For both NSCLC-models, tumors were resected after sacrifice of the mice at the day 21 of therapy, frozen in liquid nitrogen vapor and cut in 7-10 μm slices. Fixation of the frozen sections and the staining procedure were performed as described [36]. Vessel density, vessel maturation, angiogenesis and tumor hypoxia were assessed using primary antibodies against CD31 (rat anti-mouse, BD Biosciences), α-smooth muscle actin (biotinylated anti α-SMA, Progen) and hypoxia-inducible factor (HIF) -1α (rabbit anti-human and anti-mouse, Novus Biologicals) as well as corresponding secondary antibodies (donkey anti-rat-FITC, streptavidin-Cy-3, and donkey anti-rabbit-Cy-3, all Dianova). Cell nuclei were counterstained by 4′, 6-diamidino-2-phenylindole (DAPI; Invitrogen). Apoptosis was detected using the 'In Situ Cell Death Detection Kit, TMR red' (Roche Diagnostics GmbH, Mannheim, Germany). Stained sections were examined and photographed at a fluorescence microscope equipped with appropriate detection filters (Axio Imager M2, Carl Zeiss, MicroImaging GmbH) and a digital camera (AxioCam MRm Rev.3, Carl Zeiss, MicroImaging GmbH). Each tumor was photographed by taking consecutive adjacent images in 50 x magnification in order to cover the whole tumor area. All images were fused to illustrate the whole tumor section. Quantification was done using the AxioVision Rel 4.8 software (Carl Zeiss, MicroImaging GmbH) for determining tumor vascularization (CD31+ area fraction), vessel maturation (α-SMA+ vessel/ total vessel number), hypoxia (HIF-1α+ area fraction) and apoptosis (TUNEL+ area fraction). Vessel sizes were determined by measuring the filled area of each vessel.
Determination of the tumor platinum concentration (ICP-MS)
Platinum concentrations in the tumors were measured by inductively coupled plasma mass spectrometry (ICP-MS) as described [37]. In brief, the tumor tissue was completely digested in a closed vessel microwave reaction system (MLS ethos plus, MPV-100/HAT) after addition of 1.5 ml nitric acid (65%) to 1.5 ml hydrogen peroxide and 1 mL of rhodium as internal standard). The amount of platinum was determined using a high-resolution sector field inductively coupled plasma mass spectrometer [ICP-MS (Elan-DRCII, PerkineElmer)] equipped with an injector (2.0 mm i.d. Quartz, Part No.: WE02-3916). A quartz cyclonic spray chamber (Part No.: WE02-52222) equipped with a Meinhard Type A quartz nebulizer was used for sample introduction. Data were acquired at medium resolution using rhodium as an internal standard. The platinum content was expressed in micrograms per kg tumor tissue.
Statistics
A one -way ANOVA combined with a Bonferroni post-test was applied using GraphPadPrism 5.0 (Graph-Pad) in order to assess differences between the groups. p < 0.05 was considered as statistically significant (*), p < 0.01 was considered as highly significant (**).
Results
Co-medication of Epo during carboplatin-treatment inhibits the growth but increases the vascularization in NSCLC-xenografts
The effects of Epo co-medication were analyzed in two NSCLC-xenograft models that markedly differ in tumor cell EpoR-expression, H838 showing approximately 8 fold higher levels than A549 [26]. Both NSCLC-models responded to chemotherapy with carboplatin leading to tumor size reductions to 65% in A549 and to 73% in H838 and to an increase in apoptosis to 209% and 217%, respectively after three weeks of treatment as compared with untreated tumors (Supplementary Figure 1). In order to investigate the effects of Epo co-medication on growth and vascularization of these NSCLCs, one animal group was treated solely with carboplatin, and two animal groups additionally received different doses of rhuEpo-β (5 µg/ kg or 20 µg/ kg, respectively). Epo was not administered alone in order to avoid thromboembolisms in the animals during repetitive administration over three weeks.
For both NSCLC-models, Epo had a normalizing effect on the Hb-levels of the carboplatin-treated mice. While the levels decreased from week two to three in mice that received only carboplatin, the decrease was lower in mice co-treated with 5 µg/ kg of Epo and physiological Hb-levels were measured in the groups co-treated with 20 µg/ kg of Epo at week three of therapy (Supplementary Figure 2).
The tumor volumes were determined longitudinally by 3D ultrasound. In both NSCLC-models irrespective of the tumor cell EpoR-expression, tumor growth was inhibited in animals co-treated with Epo while an increase was observed in the groups that received only carboplatin (Fig. 1 A). Differences in mean size between Epo co-medicated and only carboplatin-treated tumors were significant at all timepoints for A549 and H838 tumors (*p < 0.05, **p < 0.01). Both Epo-doses (5 and 20 µg/ kg, respectively) exerted similar effects.
Epo co-medication reduces tumor growth but increases the relative blood volume in carboplatin-treated NSCLC-xenografts. A: The tumor volumes, as measured by 3D ultrasound, were significantly lower in Epo co-medicated (Cp + Epo 5 and Cp + Epo 20) A549 (left) and H838 (right) compared with only carboplatin-treated (Cp) tumors starting from week one of therapy (*p < 0.05, **p < 0.001, n = 5 per group). B: In contrast, the relative blood volume (rBV), as determined by 3D contrast-enhanced ultrasound, was higher in Epo co-medicated A549 (left) and H838 (right) compared with the only carboplatin-treated tumors (*p < 0.05, n = 5 per group). C: Representative maximum intensity over time (MIOT) projections show higher ultrasound contrast in Epo co-medicated A549 (left) and H838 (right) tumors compared with only carboplatin-treated tumors, demonstrating an increased amount of perfused tumor vessels. Cp: carboplatin, Epo 5: rhuEpo 5 µg/kg, Epo 20: rhuEpo 20 µg/kg.
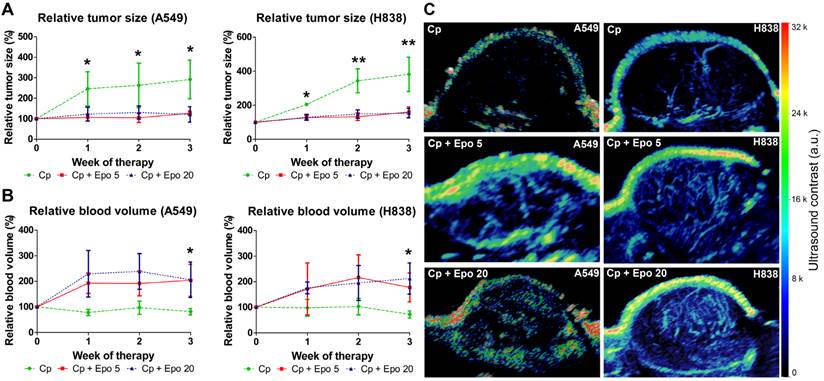
In contrast to the reduced tumor growth, 3D contrast-enhanced ultrasound revealed that in both NSCLC-models, the relative blood volume (rBV), representing the perfused vasculature, was increased in tumors receiving Epo co-medication in comparison with solely carboplatin-treated groups. Again, the trend was similar for both Epo-concentrations (Fig. 1 B, C). Differences in mean rBV between Epo- and only carboplatin-treated tumors were significant at week three of therapy in both NSCLC-models (*p < 0.05).
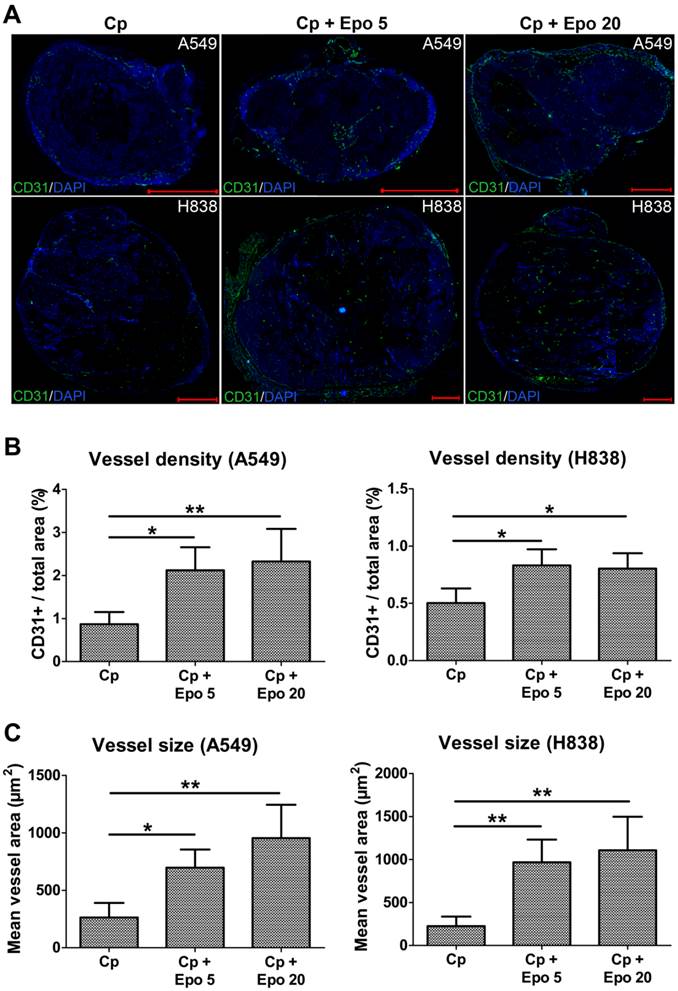
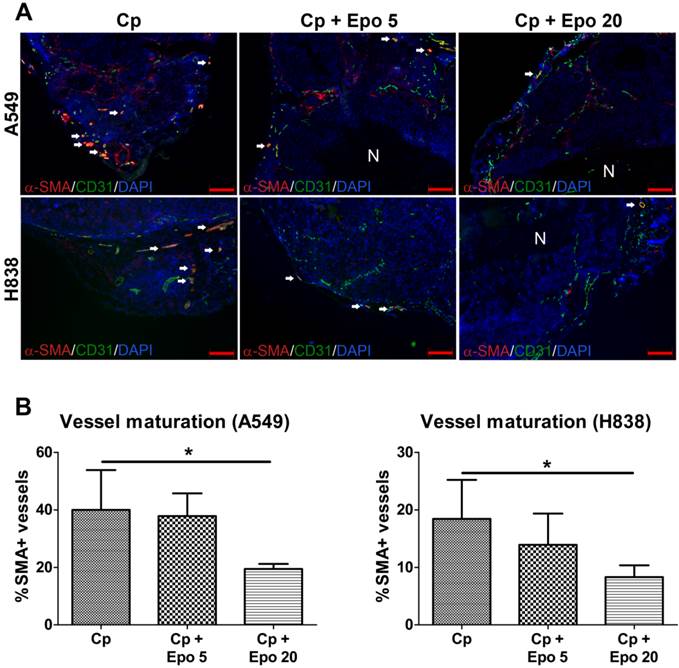
Immunofluorescent staining of tumor sections at day 21 and quantification confirmed the significantly higher vessel density and mean vessel size in tumors of Epo co-medicated compared with only carboplatin-treated animals (Fig. 2, *p < 0.05, **p < 0.01). In addition, vessel maturation was significantly lower in A549 and H838 tumors co-treated with the higher Epo dose (Fig. 3 A and B, n = 5, *p < 0.05). These findings suggested vessel-remodeling effects of Epo.
Epo co-medication leads to higher vessel density and size. Representative immunostainings for endothelial cells (CD31, green) and cell nuclei (DAPI, blue) of A549 and H838 tumor sections at day 21 (A) and quantification of the CD31+ area fraction (B) demonstrates the significantly higher vessel density in Epo co-medicated (Cp + Epo 5 and Cp + Epo 20) A549 and H838 compared with the only carboplatin-treated (Cp) tumors (n = 5 per group, *p < 0.05, **p < 0.001, scale bars: 1000 µm). C: Quantification of the mean area of a vessel in A549 (left) and H838 (right) demonstrates a significantly increased vessel size in additionally Epo-treated tumors (n = 5 per group, *p < 0.05, **p < 0.001). Cp: carboplatin, Epo 5: rhuEpo 5 µg/kg, Epo 20: rhuEpo 20 µg/kg.
Epo reduces vessel maturation. A: Representative immunostainings for α-SMA (red), endothelial cells (CD31, green) and cell nuclei (DAPI, blue) are shown for A549 and H838 (mature vessels indicated by arrows, scale bars: 200 µm). B: Quantification of vessel maturation shows significantly lower percentages of α-SMA-positive, mature tumor vessels in the groups co-medicated with 20 µg/kg Epo (Cp + Epo 20) in A549 and H838 compared to the only carboplatin-treated (Cp) tumors (*p < 0.05, n = 5). Cp: carboplatin, Epo 5: rhuEpo 5 µg/kg, Epo 20: rhuEpo 20 µg/kg, N: necrosis.
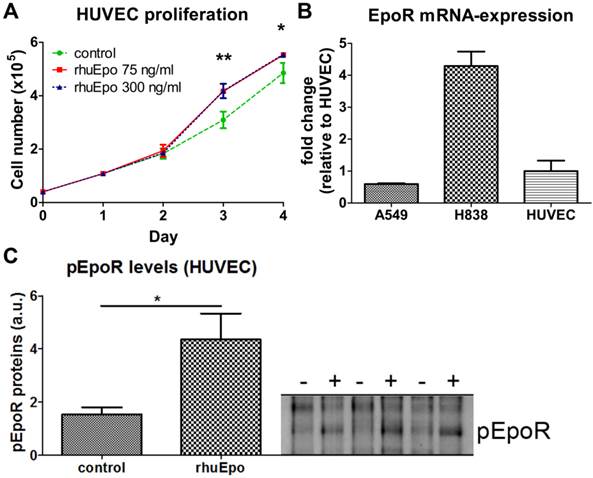
In order to analyze whether Epo exerts direct effects on endothelial cells, HUVECs were stimulated in vitro with Epo-doses corresponding to the amount expected per ml blood in vivo. When assuming a 30 g mouse with a mean blood volume of 2 ml, 5 µg/kg and 20 µg/kg of Epo correspond to 70 ng/ml and 300 ng/ml, respectively. At day three and four of Epo-stimulation, significantly higher cell numbers were measured for both Epo-concentrations, demonstrating that Epo directly enhances endothelial cell proliferation (Fig. 4 A, *p < 0.05, **p < 0.01). Analysis of EpoR mRNA-expression demonstrated that the transcript levels in HUVECs were 1.7-fold higher than in A549 but 4.3-fold lower than in H838 (Fig. 4 B). At the protein level, a 2.8 fold increased amount of phosphorylated EpoR was detected in upon Epo-stimulation indicating Epo-mediated signaling (Fig. 4 C).
Epo stimulates endothelial cell proliferation and EpoR-expression in vitro. A: Stimulation of HUVECs with Epo in vitro significantly increased the cell numbers starting at day three compared with non-stimulated controls (n = 3 per condition, *p < 0.05, **p < 0.001). B: Comparison of EpoR mRNA expression in HUVECs with A549 and H838 reveals higher transcript levels than in A549 (0.6 fold) and lower levels compared with H838 (4.3 fold). C: Phosphorylated EpoR was detected on HUVECs by immunoblotting. Significant pEpoR up-regulation was measured in HUVECs after stimulation with rhuEpo-β (300 ng/ml) (+) in comparison to untreated control cells (-) (n = 3, *p < 0.05).
Epo enhances the accumulation of carboplatin in the NSCLCs leading to increased apoptosis
One possible explanation for the significantly reduced tumor growth observed in the combined carboplatin- and Epo-treated tumors is an improved delivery and thus stronger accumulation of the chemotherapeutic drug due to the increased vascularization and perfusion. In order to test this hypothesis, the platinum concentration was measured in A549 and H838 tumors of day 21 by inductively coupled plasma mass spectrometry (ICP-MS). Indeed, significantly higher platinum concentrations (1.5-fold increase for A549 and 2.2-fold increase for H838) were found in Epo co-treated compared with only carboplatin-treated tumors (Fig. 5 A, A549: rhuEpo-β: 1161.25 µg/ kg ± 118.34; only carboplatin: 803.00 µg/ kg ± 210.08, *p < 0.05; H838: rhuEpo-β: 1471.75 µg/ kg ± 749.87; only carboplatin: 673.25 µg/ kg ± 274.81, *p < 0.05).
Epo co-medication improves the accumulation of carboplatin in the tumors leading to enhanced apoptosis. A: Significantly higher platinum concentrations were measured in the Epo co-medicated (Cp + Epo) A549 (left) and H838 (right) tumors of day 21 by ICP-MS compared to the only carboplatin-treated (Cp) groups (controls: n = 5, rhuEpo-β: n = 10; *p < 0.05). B: Representative stainings (TUNEL in red, cell nuclei in blue) of whole tumor sections showing a lower degree of apoptosis in the only carboplatin-treated (Cp) compared with additionally Epo-treated tumors (Cp + Epo 5, Cp + Epo 20, scale bars: 1000 µm). C: Quantification of the TUNEL-+ area fraction demonstrates an enhanced apoptosis in additionally Epo-treated A549 (left) and H838 (right) tumors at day 21 (n = 5 per group, *p < 0.05, **p < 0.001). Cp: carboplatin, Epo 5: rhuEpo 5 µg/kg, Epo 20: rhuEpo 20 µg/kg.
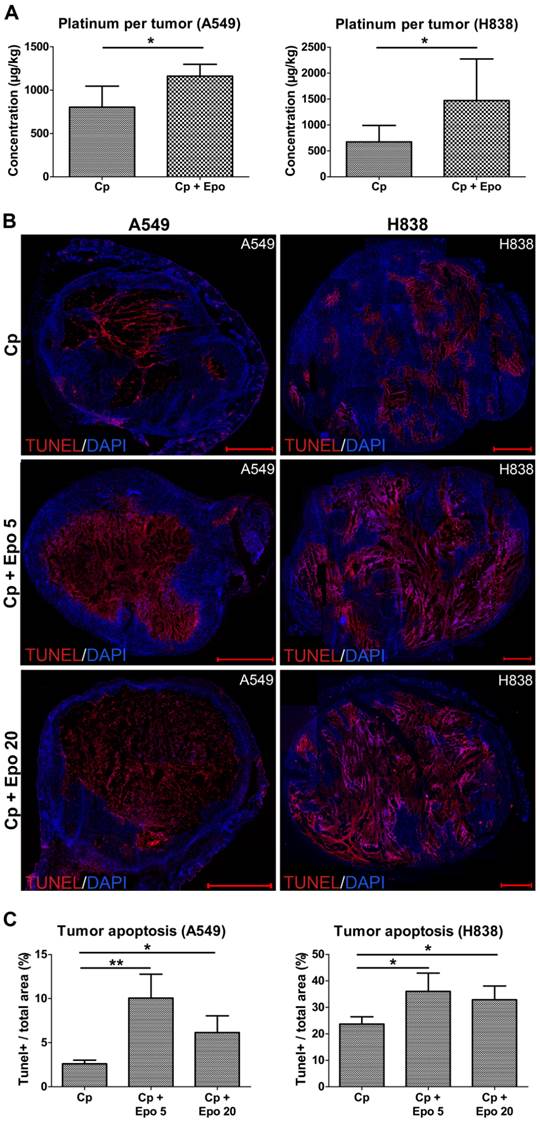
Next, we analyzed whether the higher carboplatin accumulation had an effect on apoptosis in the tumor tissue. For both NSCLC-models, TUNEL-staining and quantification of tumor sections at day 21 revealed significantly higher apoptotic rates in the groups that had additionally received Epo as compared with solely carboplatin-treated groups (Fig.5, representative stainings of whole tumor sections shown in B, quantification in C, *p < 0.05, **p < 0.01). The lower TUNEL-positive area in the A549 tumors treated with 20 µg/kg of Epo can be explained by a massive tumor cell loss which was validated by quantification of the DAPI+ area (Supplementary Figure 3). The apoptotic rate of untreated tumors in comparison was 48% (for A549) and 46% (for H838) lower than the solely carboplatin-treated groups (Supplementary Figure 1 C).
The EpoR-levels in the NSCLC-xenografts change during therapy
In order to analyze whether Epo co-medication had an effect on EpoR-expression, the EpoR-levels were measured longitudinally by µCT/FMT hybrid imaging using the NIR-labeled EpoR-probe Epo-Cy5.5 [26]. In both NSCLC-models, the Epo-Cy5.5 levels continuously increased during sole carboplatin-treatment, pointing to an up-regulation of EpoR in the tumor tissue (Fig. 6 C, Supplementary Fig. 4). However, in the H838 model, Epo-Cy5.5 accumulation was markedly lower in tumors co-medicated with Epo, reaching significant differences to solely carboplatin-treated tumors in week three of therapy (Fig. 6 A - C, *p < 0.05). A marginal decrease in EpoR-expression was also observed in the Epo co-medicated A549 tumors (Fig. 6 C, Supplementary Fig. 4). Since the tumor vascularization was significantly higher in the Epo co-medicated groups, which could have facilitated passive probe accumulation, the reduced Epo-Cy5.5 levels cannot be explained by a pharmacological effect but rather by down-regulation of EpoR.
Epo co-medication has differential effects on EpoR-expression but reduces tumor hypoxia. The EpoR-status in the tumors was longitudinally assessed by µCT/FMT hybrid imaging using Epo-Cy5.5. A: µCT/FMT fusion image demonstrating higher Epo-Cy5.5 accumulation in only carboplatin-treated (Cp) H838 (left panel) versus tumors additionally treated with 20 µg/kg of Epo (Cp + Epo 20, right panel) after three weeks of treatment. Note the different signal intensities in the tumors (lined in red), whereas they are comparable in the bone marrow of pelvis and tibia (arrowheads). B: 3D rendering of reconstructed µCT/FMT-data of the abdominal part of a H838 tumor-bearing mouse at day 21. Specific accumulation of Epo-Cy5.5 (blue) is visible in the EpoR-positive organs, the bone marrow of pelvis, spinal cord and hind legs (white), the tumor (green) and the kidneys (yellow). C: Epo-Cy5.5 levels show a similar increase in carboplatin- and additionally Epo-treated A549 tumors (upper graph), whereas in H838, significantly lower levels are recorded for combinatorial Epo-treated compared with only carboplatin-treated tumors in the third week of therapy (lower graph) (n = 5, *p < 0.05). D: Immunostainings for HIF-1α (red), endothelial cells (CD31, green) and cell nuclei (DAPI, blue) of representative A549 tumor sections at day 21. Note the reduced hypoxia in the additionally Epo-treated groups (scale bars: 200 µm). E: Quantification of HIF-1α staining reveals a significantly lower hypoxia in additionally Epo-treated A549 and H838 tumors at day 21 (n = 5, *p < 0.05). Cp: carboplatin, Epo 5: rhuEpo 5 µg/kg, Epo 20: rhuEpo 20 µg/kg.
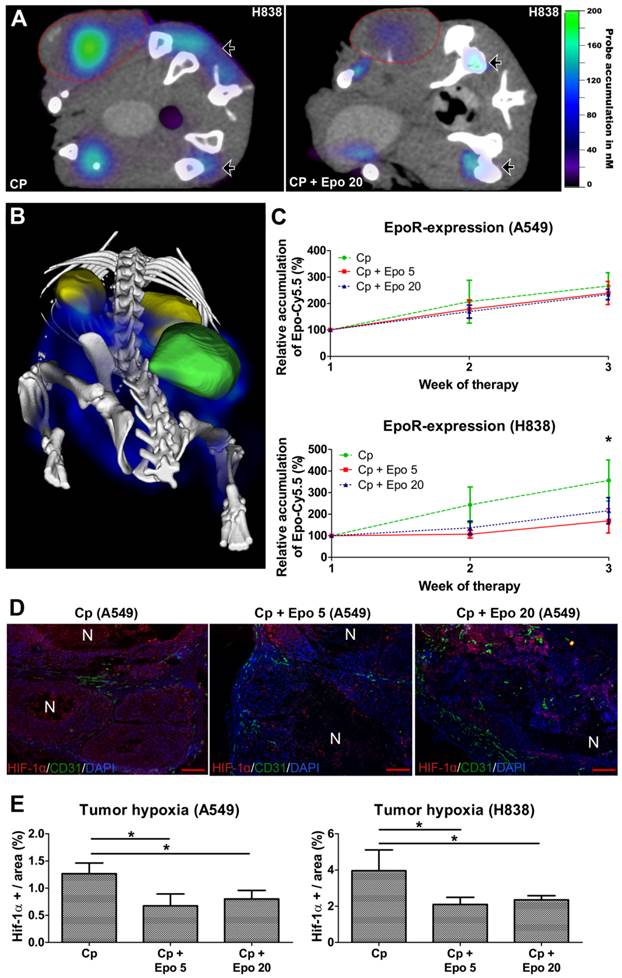
Since EpoR-expression is up-regulated by hypoxia in tumor cells [38] and HUVECs (Supplementary Fig. 5), the hypoxic state was analyzed in A549 and H838 tumors of day 21 by HIF-1α staining (Fig. 6 D). The degree of hypoxia was lower in carboplatin-treated A549 than H838 tumors (A549: 1.27 % ± 0.2; H838: 3.96 % ± 1.15). However, in both NSCLC-models, Epo co-medication resulted in significantly lower hypoxia compared with solely carboplatin-treated tumors (Fig. 6 E, *p < 0.05).
Discussion
Due to clinical studies with worse outcome in several cancer patient groups treated with Epo and the putative discovery of the EpoR in tumor and endothelial cells, erythropoiesis stimulating agents (ESAs) like rhuEpo-β have become subject to intense debates with respect to their use in the treatment of CIA. Pre-clinical studies that were undertaken to elucidate the role of Epo revealed controversial results with regard to tumor growth [22,24]. Since EpoR expression in tumors is currently discussed to play a crucial role in the effects of Epo in tumor growth and progression, we have recently developed a theranostic NIRF-probe (EpoCy5.5) that could highly sensitively differentiate EpoR-expression in two NSCLC-xenograft models in vivo and that maintained its therapeutic activity [26]. Using this probe we here longitudinally investigated the effects of rhuEpo-β on tumor growth during three weeks of carboplatin-treatment in two NSCLC-xenografts with different EpoR-levels of the tumor cells, A549 with very low and H838 with approximately 8 fold higher expression [26]. In both NSCLC-models, tumor growth was significantly reduced in the groups co-medicated with Epo (5 and 20 µg/kg). However, Epo exerted stimulatory effects on the tumor endothelium, leading to a significant increase in relative blood volume, vessel density and size, as well as lower vessel maturation. In line with the in vivo-results, Epo stimulated the proliferation of HUVECs with functional EpoR. This strongly suggests that Epo promotes vessel remodeling by enhancing endothelial cell proliferation and angiogenesis. Our findings are in accordance with results of other groups that describe increased endothelial cell proliferation, survival and angiogenesis in response to Epo-application [39,40]. In different pre-clinical studies, even enhanced tumor growth and progression were observed upon Epo administration [24,25]. However, in these studies Epo was applied as sole therapeutic agent [22,24,25]. Here it may directly lead to tumor progression by stimulating endothelial and tumor cells. In contrast to these findings, we observed a significantly reduced tumor growth in the Epo-treated groups which can be explained by the combined administration of Epo and the chemotherapeutic drug carboplatin. In co-medication with carboplatin, the Epo-mediated vascular remodeling leading to enhanced vascularization and perfusion improved the delivery and treatment efficacy of carboplatin in the xenografts as obvious by the higher intratumoral platinum concentrations measured by ICP-MS and the enhanced apoptosis. Similar observations of reduced tumor growth due to increased drug accumulation were obtained in squamous cell and colon carcinoma xenografts co-treated with 5-Fluoruracil and Epo [41]. Compared to angiogenic factors like VEGF-A that induce the formation of tortuous, leaky, highly permeable and instable vessels, which are only partially perfused and tend to collapse [42,43], Epo co-medication resulted in enhanced vessel perfusion and function in the NSCLC-xenografts, as obvious by the significantly increased rBV measured by ultrasound and the increased vessel size. This suggests that in combination with chemotherapy, Epo can have beneficial effects on the therapeutic outcome by improving the vascularization and thus drug delivery [44]. This hypothesis is supported by a recent clinical study on esophagogastric cancer patients, demonstrating that additional Epo-administration increased chemotherapeutic efficacy, resulting in a clear trend to improved survival [45]. By improving the tumor vascularization, Epo co-medication may not only increase the accumulation of low molecular weight anticancer agents but is suggested to also enhance the delivery of larger drug-loaded carrier systems such as liposomes, polymers, micelles or nanoparticles including theranostic nanomedicine formulations [46,47]. However, tumor accumulation of nanoparticles depends on both, the size of the nanoparticles and the enhanced permeability and retention (EPR) -effect of the tumor. The latter is strongly influenced by the vessel density, the vascular leakiness, the distribution space, and the composition of the extracellular matrix. In this context, the Epo-mediated decrease in vessel maturation that we observed in our NSCLC-models is suggested to enhance vessel permeability. Thus, in particular for long circulating nanomedicine formulations the enhancement of EPR induced by Epo may lead to an improved tumor accumulation. On the other hand, the effect of Epo on the tumor vasculature might vary dependent on the degree of angiogenesis and vessel maturity. Thus, subsequent studies including various nanomedicine formulations and tumor models with different aggressiveness and angiogenesis are planned in order to carefully evaluate the effect of Epo co-medication on tumor vascularization, drug delivery and therapeutic outcome.
The significantly increased tumor apoptosis that we observed in the Epo co-medication groups is in clear contrast to its described cyto-protective effects against chemotherapeutic drugs. rhuEpo-α in concentrations of 30 U/ ml corresponding approximately to 150 ng/ ml enhanced the survival of glioblastoma cells in response to cisplatin-treatment in vitro [48]. The observation that Epo did not exert tumor cell protective effects in our in vivo study but rather had stimulatory effects on the tumor endothelium can be explained by the different accessibilities of the EpoR-positive compartments to the hormone and thus varying local doses. Whereas the tumor endothelium is directly accessible to Epo circulating in the bloodstream, the hormone has to extravasate and penetrate the interstitium in order to reach the tumor cells, resulting in a considerably lower exposure. Based on the FMT-measurements with the fully bioactive NIRF-probe Epo-Cy5.5, we approximate an accumulation rate of 0.03% of the injected dose in the xenografts, thus the Epo-concentrations in the tumor cell compartment are estimated to be substantially below the described cyto-protective doses. Therefore, we assume that while the Epo-doses attaining the vessel compartment in the tumor were sufficient to stimulate the tumor endothelium due to the better accessibility, the amount of Epo that had reached the tumor cell compartment was too low to counterbalance the cytotoxic effects of carboplatin. This suggests that there is an optimal Epo-dose for improving drug delivery by vascular remodeling without promoting tumor cell survival, which strongly motivates the application of a companion diagnostic or theranostic EpoR-probe.
Using Epo-Cy5.5, we investigated the EpoR-status in the NSCLC-xenografts by µCT/FMT hybrid imaging [26]. In both tumor models, the EpoR-levels continuously increased with ongoing carboplatin-treatment. In view of the cyto-protective effects of Epo [48], the observed EpoR-upregulation can be explained by a rescue response in the tumor due to the cytotoxic effects of carboplatin and by induction of hypoxia as demonstrated for HUVECs in vitro. Epo co-medication reduced EpoR-expression in the tumors which was predominantly evident in the H838-model with higher tumor cell EpoR-expression and stronger tumor hypoxia. The histological analysis confirmed that additional Epo treatment significantly decreased hypoxia in both NSCLC-models, most probably due to the improved vascularization leading to enhanced oxygen supply. We therefore conclude that the improved oxygenation induces down-regulation of the survival receptor EpoR, thus EpoR-down-regulation may be a marker for vascular priming towards improved drug delivery. Since the changes in EpoR expression can be monitored using EpoR-probes such as Epo-Cy5.5, these may be valuable for personalizing Epo-medication in different tumor types and making its administration safer. With regard to clinical translation, the utility of the optical EpoR-probe is limited due to restrictions in tissue penetration and sensitivity. Therefore, we have recently developed 68Ga-DOTA-rhuEpo as sensitive EpoR PET probe that may be applicable in human studies as companion diagnostic agent [49].
Furthermore, Epo may also support radiotherapy by counteracting tumor hypoxia, since the sensitivity of tumors towards radiation rapidly decreases when the partial oxygen pressure declines [50]. Accordingly, a recent study revealed that combination of Epo with radiation therapy of human squamous cell carcinoma xenografts resulted in significantly reduced tumor growth compared to untreated controls [51].
In summary, co-administration of Epo in well balanced doses led to significantly improved carboplatin accumulation by increasing tumor vascularization and perfusion in two lung cancer xenografts. Thus, the administration of Epo may be a promising concept to modulate EPR and improve drug accumulation in tumors. However, it is not yet clear to which extent our findings can be translated to other tumor models and therapies. In this context, diagnostic or theranostic EpoR-probes are suggested to be valuable in order to identify responsive patient populations and to define the optimal Epo-doses for vascular remodeling towards improved drug delivery and tumor oxygenation.
Supplementary Material
Supplementary Figure 1-5.
Abbreviations
CIA: chemotherapy-induced anemia
Cp: Carboplatin
Epo: erythropoietin
EpoR: erythropoietin receptor
EPR: enhanced permeability and retention
ESA: erythropoiesis stimulating agent
FMT: fluorescence molecular tomography
Hb: hemoglobin
HIF: hypoxia-inducible factor
HUVEC: human umbilical vein endothelial cell
ICP-MS: inductively coupled plasma mass spectrometry
NIR: near infrared
NSCLC: non small cell lung cancer
PBCA: poly-n-butylcyanoacrylate
PBS: phosphate buffered saline
PET: positron emission tomography
rBV: relative blood volume
rhuEpo: recombinant human erythropoietin
s.c.: subcutaneous
µCT: micro computed tomography
VEGF: vascular endothelial growth factor
Acknowledgements
This work was supported by the German Ministry for Education and Research (BMBF)/ Bundesministerium für Bildung und Forschung (BMBF) project numbers 0315415C and 0316042F, and by the German Center for Lung Research (DZL).
Author contributions
D.D. Performed animal experiments, mainly analyzed the data, wrote and edited the manuscript, participated in study design and coordination. A.R. Participated in animal and cell experiments and in data analyses. S.A. Performed in vitro experiments, participated in data analyses. K.P. Participated in study design and edited the manuscript. F.G. Participated in data analyses and edited the manuscript. R.M. and F.S. Contributed in vitro experiments participated in data analyses and edited the manuscript. U.K. Gave conceptional advice and edited the manuscript. M.J. Participated in study design and edited the manuscript. F.K. Designed and coordinated the experiments, and edited the manuscript. W.L. Designed and coordinated the experiments, analyzed the data, wrote and edited the manuscript.
Competing Interests
M.J. is employee of Roche Pharmaceutical Research and Early Development, Pharmaceutical Sciences, Roche Innovation Center, Penzberg, Germany.
All other authors have no competing interests.
References
1. Keith RL, Miller YE. Lung cancer chemoprevention: current status and future prospects. Nat Rev Clin Oncol. 2013;10(6):334-43
2. Rinaldi M, Cauchi C, Gridelli C. First line chemotherapy in advanced or metastatic NSCLC. Ann Oncol. 2006;17(suppl 5):v64-v67
3. Minna JD, Gazdar AF, Sprang SR, Herz J. A Bull's Eye for Targeted Lung Cancer Therapy. Science. 2004;304(5676):1458-61
4. Yu Y, He J. Molecular classification of non-small-cell lung cancer: diagnosis, individualized treatment, and prognosis. Front Med. 2013;7(2):157-71
5. Vansteenkiste J, De Ruysscher D, Eberhardt WEE, Lim E, Senan S, Felip E. et al. Early and locally advanced non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(Suppl 6):vi89-vi98
6. Peters S, Adjei AA, Gridelli C, Reck M, Kerr K, Felip E. et al. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(Suppl 7):vii56-vii64
7. Jelkmann W. Molecular biology of erythropoietin. Intern Med. 2004;43(8):649-59
8. Del Mastro L, Gennari A, Donati S. Chemotherapy of non-small-cell lung cancer: role of erythropoietin in the management of anemia. Ann Oncol. 1999;10(Suppl 5):S91-S94
9. Spivak JL. The anaemia of cancer: death by a thousand cuts. Nat Rev Cancer. 2005;5(7):543-55
10. Kiss Z, Elliott S, Jedynasty K, Tesar V, Szegedi J. Discovery and basic pharmacology of erythropoiesis-stimulating agents (ESAs), including the hyperglycosylated ESA, darbepoetin alfa: an update of the rationale and clinical impact. Eur J Clin Pharmacol. 2010;66(4):331-40
11. Henke M, Mattern D, Pepe M, Bézay C, Weissenberger C, Werner M. et al. Do erythropoietin receptors on cancer cells explain unexpected clinical findings? J Clin Oncol. 2006;24(29):4708-13
12. Wright JR, Ung YC, Julian JA, Pritchard KI, Whelan TJ, Smith C. et al. Randomized, double-blind, placebo-controlled trial of erythropoietin in non-small-cell lung cancer with disease-related anemia. J Clin Oncol. 2007;25(9):1027-32
13. Bohlius J, Langensiepen S, Schwarzer G, Seidenfeld J, Piper M, Bennett C. et al. Recombinant human erythropoietin and overall survival in cancer patients: results of a comprehensive meta-analysis. J Natl Cancer Inst. 2005;97(7):489-98
14. Bohlius J, Schmidlin K, Brillant C, Schwarzer G, Trelle S, Seidenfeld J. et al. Recombinant human erythropoiesis-stimulating agents and mortality in patients with cancer: a meta-analysis of randomised trials. Lancet. 2009;373(9674):1532-42
15. Tonelli M, Hemmelgarn B, Reiman T, Manns B, Reaume MN, Lloyd A. et al. Benefits and harms of erythropoiesis-stimulating agents for anemia related to cancer: a meta-analysis. CMAJ. 2009;180(11):E62-E71
16. Dagnon K, Pacary E, Commo Fdr, Antoine M, Bernaudin M, Bernaudin JF. et al. Expression of erythropoietin and erythropoietin receptor in non-small cell lung carcinomas. Clin Cancer Res. 2005;11(3):993-9
17. Hardee ME, Arcasoy MO, Blackwell KL, Kirkpatrick JP, Dewhirst MW. Erythropoietin biology in cancer. Clin Cancer Res. 2006;12(2):332-9
18. Hardee ME, Kirkpatrick JP, Shan S, Snyder SA, Vujaskovic Z, Rabbani ZN. et al. Human recombinant erythropoietin (rEpo) has no effect on tumour growth or angiogenesis. Br J Cancer. 2005;93(12):1350-5
19. Elliott S, Sinclair A, Collins H, Rice L, Jelkmann W. Progress in detecting cell-surface protein receptors: the erythropoietin receptor example. Ann Hematol. 2014;93(2):181-92
20. Laugsch M, Metzen E, Svensson T, Depping R, Jelkmann W. Lack of functional erythropoietin receptors of cancer cell lines. Int J Cancer. 2008;122(5):1005-11
21. Swift S, Ellison AR, Kassner P, McCaffery I, Rossi J, Sinclair AM. et al. Absence of functional EpoR expression in human tumor cell lines. Blood. 2010;115(21):4254-63
22. Kumar SM, Zhang G, Bastian BC, Arcasoy MO, Karande P, Pushparajan A. et al. Erythropoietin receptor contributes to melanoma cell survival in vivo. Oncogene. 2012;31(13):1649-60
23. Hedley BD, Chu JE, Ormond DG, Beausoleil MS, Boasie A, Allan AL. et al. Recombinant human erythropoietin in combination with chemotherapy increases breast cancer metastasis in preclinical mouse models. Clin Cancer Res. 2011;17(19):6151-62
24. Okazaki T, Ebihara S, Asada M, Yamanda S, Niu K, Arai H. Erythropoietin promotes the growth of tumors lacking its receptor and decreases survival of tumor-bearing mice by enhancing angiogenesis. Neoplasia. 2008;10(9):932-9
25. Yang J, Xiao Z, Li T, Gu X, Fan B. Erythropoietin promotes the growth of pituitary adenomas by enhancing angiogenesis. Int J Oncol. 2012;40(4):1230-7
26. Doleschel D, Mundigl O, Wessner A, Gremse F, Bachmann J, Rodriguez A. et al. Targeted near-infrared imaging of the erythropoietin receptor in human lung cancer xenografts. J Nucl Med. 2012;53(2):304-11
27. Klingmüller U, Lorenz U, Cantley LC, Neel BG, Lodish HF. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell. 1995;80(5):729-38
28. Beleslin-Cokic BB, Cokic VP, Yu X, Weksler BB, Schechter AN, Noguchi CT. Erythropoietin and hypoxia stimulate erythropoietin receptor and nitric oxide production by endothelial cells. Blood. 2004;104(7):2073-80
29. Aharinejad S, Fink M, Abri H, Nedwed S, Schlag MG, Macfelda K. et al. Efficient carboplatin single therapy in a mouse model of human testicular nonseminomatous germ cell tumor. J Urol. 2002;167(1):368-74
30. Macdougall IC, Eckardt KU. Novel strategies for stimulating erythropoiesis and potential new treatments for anaemia. Lancet. 2006;368(9539):947-53
31. Blackwell KL, Kirkpatrick JP, Snyder SA, Broadwater G, Farrell F, Jolliffe L. et al. Human Recombinant Erythropoietin Significantly Improves Tumor Oxygenation Independent of Its Effects on Hemoglobin. Cancer Res. 2003;63(19):6162-5
32. Bzyl J, Palmowski M, Rix A, Arns S, Hyvelin JM, Pochon S. et al. The high angiogenic activity in very early breast cancer enables reliable imaging with VEGFR2-targeted microbubbles (BR55). Eur Radiol. 2013;23(2):468-75
33. Rix A, Lederle W, Siepmann M, Fokong S, Behrendt FF, Bzyl J. et al. Evaluation of high frequency ultrasound methods and contrast agents for characterising tumor response to anti-angiogenic treatment. Eur J Radiol. 2012;81(10):2710-6
34. Fokong S, Siepmann M, Liu Z, Schmitz G, Kiessling F, Gätjens J. Advanced characterization and refinement of poly N-butyl cyanoacrylate microbubbles for ultrasound imaging. Ultrasound Med Biol. 2011;37(10):1622-34
35. Gremse F, Theek B, Kunjachan S, Lederle W, Pardo A, Barth S. et al. Absorption Reconstruction Improves Biodistribution Assessment of Fluorescent Nanoprobes Using Hybrid Fluorescence-mediated Tomography. Theranostics. 2014;4(10):960-71
36. Lederle W, Linde N, Heusel J, Bzyl J, Woenne EC, Zwick S. et al. Platelet-derived growth factor-B normalizes micromorphology and vessel function in vascular endothelial growth factor-A-induced squamous cell carcinomas. Am J Pathol. 2010;176(2):981-94
37. Jayapaul J, Hodenius M, Arns S, Lederle W, Lammers T, Comba P. et al. FMN-coated fluorescent iron oxide nanoparticles for RCP-mediated targeting and labeling of metabolically active cancer and endothelial cells. Biomaterials. 2011;32(25):5863-71
38. Dioum EM, Chen R, Alexander MS, Zhang Q, Hogg RT, Gerard RD. et al. Regulation of Hypoxia-Inducible Factor 2α Signaling by the Stress-Responsive Deacetylase Sirtuin 1. Science. 2009;324(5932):1289-93
39. Warren JS, Zhao Y, Yung R, Desai A. Recombinant human erythropoietin suppresses endothelial cell apoptosis and reduces the ratio of Bax to Bcl-2 proteins in the aortas of apolipoprotein E-deficient mice. J Cardiovasc Pharmacol. 2011;57(4):424-33
40. Adelibieke Y, Shimizu H, Saito S, Mironova R, Niwa T. Indoxyl Sulfate Counteracts Endothelial Effects of Erythropoietin Through Suppression of Akt Phosphorylation. Circ J. 2013;77(5):1326-36
41. Tóvári J, Gilly R, Rásó E, Paku S, Bereczky B, Varga N. et al. Recombinant human erythropoietin alpha targets intratumoral blood vessels, improving chemotherapy in human xenograft models. Cancer Res. 2005;65(16):7186-93
42. Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology. 2005;69(Suppl 3):4-10
43. Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D. et al. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91(3):1071-121
44. Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10(6):417-27
45. Thomaidis T, Weinmann A, Sprinzl M, Kanzler S, Raedle J, Ebert M. et al. Erythropoietin treatment in chemotherapy-induced anemia in previously untreated advanced esophagogastric cancer patients. Int J Clin Oncol. 2013;19(2):288-96
46. Kobayashi H, Watanabe R, Choyke PL. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics. 2013;4(1):81-9
47. Yu MK, Park J, Jon S. Targeting Strategies for Multifunctional Nanoparticles in Cancer Imaging and Therapy. Theranostics. 2012;2(1):3-44
48. Belenkov AI, Shenouda G, Rizhevskaya E, Cournoyer D, Belzile JP, Souhami L. et al. Erythropoietin induces cancer cell resistance to ionizing radiation and to cisplatin. Mol Cancer Ther. 2004;3(12):1525
49. Fuge F, Doleschel D, Rix A, Gremse F, Wessner A, Winz O. et al. In-vivo detection of the erythropoietin receptor in tumours using positron emission tomography. Eur Radiol. 2014;25(2):472-9
50. Vaupel P, Thews O, Hoeckel M. Treatment resistance of solid tumors: role of hypoxia and anemia. Med Oncol. 2001;18(4):243-59
51. Lövey J, Bereczky B, Gilly R, Kenessey I, Rásó E, Simon E. et al. Recombinant Human Erythropoietin alpha Improves the Efficacy of Radiotherapy of a Human Tumor Xenograft, Affecting Tumor Cells and Microvessels. Strahlenther Onkol. 2008;184(1):1-7
Author contact
![]() Corresponding authors: Wiltrud Lederle, Experimental Molecular Imaging; Medical Faculty, RWTH Aachen University, Pauwelsstraße 30, 52074 Aachen, Germany, Phone: +49-241-80-80116, Fax: +49-241-80-82006, E-mail: wlederlede. Fabian Kiessling, Experimental Molecular Imaging; Medical Faculty, RWTH Aachen University, Pauwelsstraße 30, 52074 Aachen, Germany, Phone: +49-241-80-80117, Fax: +49-241-80-82006, E-mail: fkiesslingde
Corresponding authors: Wiltrud Lederle, Experimental Molecular Imaging; Medical Faculty, RWTH Aachen University, Pauwelsstraße 30, 52074 Aachen, Germany, Phone: +49-241-80-80116, Fax: +49-241-80-82006, E-mail: wlederlede. Fabian Kiessling, Experimental Molecular Imaging; Medical Faculty, RWTH Aachen University, Pauwelsstraße 30, 52074 Aachen, Germany, Phone: +49-241-80-80117, Fax: +49-241-80-82006, E-mail: fkiesslingde