Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and methods
Result
Discussion
Conclusion
Supplementary Material
Acknowledgements
Abbreviations
References
Introduction
Materials and methods
Result
Discussion
Conclusion
Supplementary Material
Acknowledgements
Abbreviations
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(7):755-771. doi:10.7150/thno.10804 This issue Cite
Research Paper
Coating Solid Lipid Nanoparticles with Hyaluronic Acid Enhances Antitumor Activity against Melanoma Stem-like Cells
Hongxin Shen, Sanjun Shi, Zhirong Zhang, Tao Gong, Xun Sun ![]()
Key Laboratory of Drug Targeting and Drug Delivery Systems, Ministry of Education, West China School of Pharmacy, Sichuan University, Chengdu 610041, P. R. China.
Received 2014-10-14; Accepted 2015-2-17; Published 2015-4-5
Citation:
Shen H, Shi S, Zhang Z, Gong T, Sun X. Coating Solid Lipid Nanoparticles with Hyaluronic Acid Enhances Antitumor Activity against Melanoma Stem-like Cells. Theranostics 2015; 5(7):755-771. doi:10.7150/thno.10804. https://www.thno.org/v05p0755.htm
Other stylesAbstract

Successful anticancer chemotherapy requires targeting tumors efficiently and further potential to eliminate cancer stem cell (CSC) subpopulations. Since CD44 is present on many types of CSCs, and it binds specially to hyaluronic acid (HA), we tested whether coating solid lipid nanoparticles with hyaluronan (HA-SLNs)would allow targeted delivery of paclitaxel (PTX) to CD44-overexpressing B16F10 melanoma cells. First, we developed a model system based on melanoma stem-like cells for experiments in vitro and in mouse xenografts, and we showed that cells expressing high levels of CD44 (CD44+) displayed a strong CSC phenotype while cells expressing low levels of CD44 (CD44-) did not. This phenotype included sphere and colony formation, higher proportion of side population cells, expression of CSC-related markers (ALDH, CD133, Oct-4) and tumorigenicity in vivo. Next we showed that administering PTX-loaded HA-SLNs led to efficient intracellular delivery of PTX and induced substantial apoptosis in CD44+ cells in vitro. In the B16F10-CD44+ lung metastasis model, PTX-loaded HA-SLNs targeted the tumor-bearing lung tissues well and subsequently exhibited significant antitumor effects with a relative low dose of PTX, which provided significant survival benefit without evidence of adverse events. These findings suggest that the HA-SLNs targeting system shows promise for enhancing cancer therapy.
Keywords: Hyaluronic acid, Paclitaxel, CD44, Melanoma, Cancer stem-like cell
Introduction
Malignant cancer, associated with aggressive tumor growth, typically means poor prognosis. While surgery, chemotherapy and radiation therapy can slow or reduce malignancy, researchers are actively searching for more effective chemotherapeutic approaches that show not only strong antitumor activity but also excellent specificity to minimize adverse effects in healthy tissue. Since most antitumor drugs distribute nonspecifically throughout the body, packaging them into liposomes, nanoparticles, micelles and nanoemulsions can cause them to accumulate preferentially at target sites. One such drug delivery vehicle showing good therapeutic promise is surfactant-stabilized solid lipid nanoparticles (SLNs). SLNs are proven to display excellent physical stability, protect labile drugs from degradation, control drug release behavior, and allow for site-specific targeting [1].
Many cancers prove resistant to chemotherapy because the drugs do not adequately inhibit cancer stem cells (CSCs), low-abundance subpopulations of tumor cells that are capable of self-renewal and that differentiate into the bulk population of non-tumorigenic cancer cells [2]. CSCs are more resistant than non-CSCs populations to conventional cancer therapy, so eradicating CSCs is crucial for treating malignant cancers [3, 4]. Unfortunately, it is difficult to identify and isolate the CSCs. However, we could focus on demonstrating candidate CSC by virtue of surrogating biomarker expression which could be associated with behavioral bioassays (colony formation, SP cells and tumorigenesis in vivo). Taken together, therapeutic strategies with higher tumor targeting efficiency and the potential to eliminate cells displaying CSC characteristics may be more effective and harmless. Such a strategy may be achieved by preparations modified with molecules specifically binding with surface markers of certain tumor stem cells.
Among the reported tumor cell surface markers, CD44 targeting has received extensive attentions in the recent years [5]. CD44 is a ubiquitous multi-structural and multi-functional cell surface glycoprotein involved in cell migration, self-renewal, drug resistance and apoptosis resistance. CD44 may be an indicator of tumors and metastasis in malignant diseases. Furthermore, most solid cancer stem cells expressed a high level of CD44 such as melanoma [6], colon cancer [7], non-small-cell-lung carcinoma [8], prostate cancer [9], head and neck cancer [10], hepatocellular carcinoma, and human nasopharyngeal carcinoma [11]. CD44 also plays an essential role in the motility of CSCs [12]. More importantly, preparations targeting CD44 may kill the CSCs population and eliminate cancer because CD44 is related with the self-renewal of cancer stem cells [13]. In vitro up-regulation of CD44 can enhance breast cancer cell adherence to endothelium [14]. Knocking down CD44 by therapy increased apoptosis and decreased self-renewal ability of CSCs when using a mammosphere culture method [15]. Thus, CD44 may be a good anchor to targeting cells predicted to have high tumorigenic potential. CD44-targeted delivery strategy to realize its dual role in tumor targeting and elimination of CSC-rich subpopulations is a desirable approach for developing a more effective anticancer therapy. Efforts to target chemotherapeutics to CD44-overexpressing cells have so far relied on conjugating the drug delivery vehicles ether to anti-CD44 antibody or hyaluronic acid are widely applied to targeting CD44 for cancer therapy. Hyaluronic acid-conjugated nanoparticles have been extensively studied because of the following advantages over anti-CD44 antibody. Firstly, HA is on the out shell of particles which can protect nanoparticles and regulate the circulation time and bio-distribution. Secondly, HA as the main component of the extracellular matrix, has better biocompatibility than anti-CD44 antibody which induces rejection of heterologous antibodies in humans [16]. Furthermore, antibody is difficult to modify and it may set inherent limits to penetration [17]. Therefore, hyaluronic acid has been widely investigated for CD44-targeted cancer therapy.
Hyaluronic acid (HA) is a charged linear polysaccharide composed of repeating units of glucuronic acid and N-acetyl-D-glucosamine. It was reported that all CD44 isoforms have uniform affinity for HA [18, 19]. More importantly, contrary to HA oligomers, the native high molecular weight HA does not induce expression of genes involved in proliferation or inflammation [20] and counteracts proangiogenic effects of the oligomers [21]. Even if native HA can activate some signaling pathways, this occurs at levels far lower than with HA oligomers [20]. Taken these factors into consideration, it is preferred to use the high molecular weight HA as a “bioinert” component [22]. Many efforts have been made in delivering drug into tumor cells by HA-derived carriers [23, 24]. In our study, a delivery system based solid lipid nanoparticles was exploited for paclitaxel to inhibit B16F10 (melanoma) cancer stem-like cells and applied for treatment of lung cancer. This delivery system was specifically designed as cationic vectors because several studies reported that systemic delivery of cationic vectors mediated specific and efficient accumulation of the vectors within the lung [25, 26]. Serum-induced aggregation has been demonstrated to play an important role in the in vivo fate of cationic complexes and more importantly, provides a distinct alternative strategy for lung targeting [27, 28]. Therefore, lung tumor-specific delivery of drug for treating metastasis can be achieved by the choice of cationic material and hyaluronic acid coating which is applied to further target cancer stem-like cells.
Because of the high tumor specificity and biocompatibility unique properties of high molecular weight HA, we therefore designed an optimized hyaluronic acid solid lipid nanoparticles (HA-SLNs) which would mediate more efficient cellular uptake and notably facilitate the specific tumor cancer cells (CD44+) delivery of anticancer drugs such as paclitaxel (PTX). In addition, high expression of CD44 associated with the CSCs characteristic (sphere and colony formation assay, CSCs marker expression and CSC related transcription factor Oct-4 expression level, tumorigenic ability in vivo, SP cells) was testified through the comparison of the CD44 based-sorted B16F10-CD44+ (92.3%) and B16F10-CD44- (29.4%) cells. HA-SLNs/PTX treatment inhibited sphere formation of CD44+ cells and attenuated Oct-4 and SP cell level. Furthermore, tumor cell targeting ability facilitated by surface modification with HA and the tumor targeting mechanism of HA-SLNs/PTX in vivo were studied. The antitumor efficacy was evaluated on a B16F10-CD44+ lung metastasis model.
Materials and methods
Materials
Glyceryl monostearate (GMS) and soy phosphatidylcholine (SPC) were purchased from Taiwei Pharmaceutical Co., Ltd (Shanghai, China), and cholesterol (Chol) was obtained from Boao Biotech Co., Ltd (Shanghai, China). Sodium hyaluronate (molecular weight, 300kDa) was provided by Shandong Freda Biochem Co., Ltd (Shandong, China). 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), coumarin-6 and dimethyldioctadecylammonium bromide (DDAB) were commercially purchased from Sigma-Aldrich (St.Louis, MO, USA). Paclitaxel was provided by Haoxuan Biotech Co., Ltd (Xian, China). Commercial paclitaxel injection (30 mg, 5 mL) was obtained from Sichuan Taiji Pharmaceutical Co., Ltd (Sichuan, China). 1,1'-dioctadecyl-3,3,3',3'-tetramethylindodicarbocyanine, 4-chlorobenzenesulfonate salt (DID) was supplied by Biotium (USA).
Female C57BL/6 mice aged 6-8 weeks were obtained from Vital River Laboratories (Beijing, China). All animal experiments were approved by the Institutional Animal Care and Ethics Committee of Sichuan University.
Preparation of paclitaxel-loaded solid lipid nanoparticles (SLNs/PTX)
The SLNs/PTX was prepared by film-ultrasonic method. Briefly, GMS (7.5 mg), SPC (7.5 mg), Chol (5 mg), DDAB (5.5 mg) and PTX (1 mg) were dissolved in chloroform, and the solution was added to a round bottom flask. Solvent was removed by distillation under low pressure, and the resulting film was sonicated (80 W, 60 s) in the presence of 5 mL of ultrapure water. SLNs/PTX was obtained and storage at 4°C.
Determination of entrapment efficiency and drug loading
Entrapment efficiency (EE) and drug loading (DL) were measured using the sodium citrate ultracentrifugation method. The desired amount of SLNs/PTX was dispersed in 10% (w/v) sodium citrate solution and centrifuged for 40 min at 40,000 rpm (Beckman-Coulter, California, USA) to separate the free PTX from SLNs/PTX. The same amount of SLNs/PTX was dissolved in methanol and the amount of PTX was determined by HPLC (Agilent, USA) using a Kromasil ODS-1 C18 column (150 × 4.6 mm, 5 µm), and a mobile phase of acetonitrile-water (55:45, v/v) at a flow rate of 1.0 mL/min. The detection wave length was set at 227 nm.
Coating SLNs/PTX with hyaluronic acid (HA-SLNs/PTX)
The HA-SLNs/PTX were prepared by the electrostatic attraction method. Briefly, 1 mL of a 0.02% (w/v) solution of hyaluronic acid (300 kDa) was slowly added under vigorous stirring to a dispersion of SLNs/PTX in the weight ratio of 12:1. The preparations were stored at 4○C.
Characterization of HA-SLNs/PTX
The particles size of HA-SLNs/PTX were measured in triplicate using photon correlation spectroscopy (PCS; Malvern Zetasizer Nano ZS90, UK). Transmission electron microscopy (TEM) specimens were prepared by depositing the nanoparticles suspensions onto a copper grid followed by staining for 10 s with 1% phosphotungstic acid (pH 6.5). Morphology of HA-SLNs/PTX was analyzed using TEM instrument (H-600, Hitachi, Japan).
In vitro PTX release studies
PTX release in vitro from HA-SLNs was evaluated using the dialysis method. Briefly, 1 mL of free PTX solution (0.2 mg/mL PTX) and equal volumes of SLNs/PTX and HA-SLNs/PTX dispersions were loaded into separate dialysis bags, separately. Then the dialysis bags were immersed in 50 mL of release medium (0.2% Tween 80 in PBS, pH 7.4) at 37○C with stirring at 200 rpm. At the predetermined time points (0.5, 1, 2, 4, 6, 9, 12, 24, 36, 48, 72, 96 h), 1 mL of dialysis solution was removed, and 1 mL of fresh medium was added to the remaining release medium. The amount of PTX in the samples was then determined using the HPLC described above.
In vitro cellular uptake behavior of HA-SLNs/PTX
Murine melanoma B16F10 cells and A549 cells were obtained from the American Type Culture Collection (ATCC) and cultured in RPMI-1640 with 10% fetal bovine serum and 1% penicillin/streptomycin. Firstly, CD44-enriched cells (B16F10-CD44+) and CD44-low expressing cells (B16F10-CD44-) were sorted and the expression of CD44 determined by flow cytometry using the CytomicsTM FC500 flow cytometry (Beckman Coulter, Miami, FL, USA).
B16F10-CD44+ cells were seed into a 12-well plate and cultured overnight at 37°C and 5% CO2 in medium. Coumarin-6 (in the place of PTX) loaded in SLNs, heparin-SLNs and HA-SLNs (all with the same containing of coumarin-6) were added directly to the cells and cultured for 1 h. Then cells were harvested and subjected to flow cytometry analysis at the channel FL1 using an argon ion laser (CytomicsTM FC500, Beckman Coulter, Miami, FL). Furthermore, the analysis of interaction of HA-SLNs/PTX(coumarin-6) and SLNs/PTX(coumarin-6) with high CD44- and low CD44-expressing B16F10 cells, cells were treated as above.
For competitive inhibition studies to determine the cellular uptake of HA-SLNs/ PTX, the cells were preincubated with 1 mL of serum-free culture medium containing HA polymer (5 mg/mL) or anti-CD44 antibody (5 µg) for 30min and then were replaced by 1 mL HA-SLNs loading coumarin-6 (20 ng/well) followed by incubation for 1 h. Then, the uptake was determined by flow cytometry as described above. Furthermore, the uptake of treated cells was also quantified by confocal laser scanning microscope (CLSM).
In vitro cell proliferation and apoptosis assay
The in vitro cell viability of B16F10-CD44+ cells after treatment with different formulations was determined by MTT assay. All concentrations were expressed in PTX equivalents. Cells were seeded in 96-well culture plates (1 × 104 cells/well) and incubated for 24 h at 37°C prior to drug treatments. Cells were treated for 4 h with free PTX, cremophor/ethanol mixture (the same ratio in the commercial PTX injection, 1:1, v/v), SLNs, HA-SLNs, SLNs/PTX and HA-SLNs/PTX (2, 5, 10 or 20 µg/mL of PTX) and were further cultured for 48 h with RPMI 1640 supplemented with 10% fetal bovine serium (FBS). Then, MTT solution in PBS (5 mg/mL, 20 µL) was added to each well and samples were incubated for 4 h in the dark. Precipitates were resuspended by adding 150 µL DMSO. Cell viability was assessed by measuring the absorbance at 570 nm with a microplate reader (Thermo, Varioskan Flash).
B16F10-CD44+ cells were treated for 4 h with the formulations at a PTX concentration of 10 µg/mL, after which the medium was replaced with fresh medium. After 48 h culture, cells were rinsed with PBS, and cell nuclei were then stained for 5 min with 4'6-diamidino-2-phenylindole (DAPI). Nuclear morphology of cells was checked by a Zeiss fluorescence microscope (Ex 358 nm, Em 461 nm). In addition, annexin V-FITC/PI double staining assay was used to further examine B16F10-CD44+ cell apoptosis induced by the indicated preparations. In brief, the treated cells were collected and washed with PBS and then resuspended in 500 µL of binding buffer (HEPES buffered saline supplemented with 2.5 mM CaCl2). Cell suspension was treated for 25 min in the dark with FITC-conjugated annexin V (5 µL) and PI (5 µL). Then samples were analyzed by flow cytometry as described above.
Cancer stem-like cell characteristics of B16F10-CD44+ cells and ability of PTX preparations to suppress these characteristics
Sphere-formation assays
B16F10-CD44+ cells were seeded into 12-well plates (1 × 103 cells/well) and cultured for 2 weeks in DMEM/F12 medium containing 20 ng/mL of epidermal growth factor (EGF; Invitrogen) and 10 ng/mL of basic fibroblast growth factor -basic (bFGF; Invitrogen). Then each well was treated with PTX formulations (10 µg/mL) for 4 h, after which the medium was replaced with fresh medium and sphere colonies were imaged using light microscope (Zeiss Axiovert 40, Germany).
Soft agar colony formation assay
Agarose in PBS (1.2% or 0.7%, w/v) was melted and allowed to stabilize at 40○C in a waterbath. Agarose at 1.2% was mixed with an equal volume of 2 × RPMI 1640 medium and 1.5 mL of the mixture was added to 6-well plate and allowed to solidify. Agarose at 0.7% was mixed with equal volume of cells (1 × 103), and 1 mL of cell suspension was added to the plates and incubated for 2-3 weeks. After crystal violet staining, the cells were observed by light microscope.
Real-time PCR quantification of CD133, ALDH and Oct-4 mRNA expression
For the evaluation of mRNA expression, RNA was extracted using RNA isolation kits according to the manufacturer's instructions (Tian Gen, China). Total RNA (~ 50 ng) was subjected to reverse transcription using the following stem-loop primers (RiboBio, Guangzhou, China) to generate the first-strand cDNA: CD133, F: 5′-AAGACCCATTTCCCTTGAGTTT-3′, R:5′-TTCATAGCCCCAGGAGTGTTAT-3′; ALDH, F:5′-AAAATGTCTCCATCACTTGG-3′, R:5′-AAGTCTTTGCCAATGCAT AC-3′; Oct-4, F: 5'-CTCACCCTGGGCGTTCTCT-3', R: 5'-AGGCCTCGAAGCG ACAGA-3'. Then the cDNA products were used for Eva Green-quantitative real-time PCR reaction with the above primers on iQTM 5 Real Time PCR Detection System (Bio-Rad, USA). PCR reactions were conducted at 95°C for 20 s followed by 40 cycles of 95°C for 10 s, 60°C for 20 s and 70°C for 7 s. Quantitative expression data were analyzed using the iCycler iQTM 5 software and reported as normalized fold expression relative to a β-actin as internal control (n = 3).
Tumorigenicity of B16F10-CD44+ and B16F10-CD44- cells in mice
B16F10-CD44+ and B16F10-CD44- cells were injected subcutaneously into C57BL/6 mice (5 × 104 cells in 100 µL of saline) into the right and left abdomen, and tumor growth was monitored for 23 days after injection. Tumor length and tumor width were measured using Vernier gauges. Tumor volume was calculated using the following formula.
V = (tumor length) × (tumor width)2/2
Fluorescence-activated cell discrimination of SP cells
In triplicate, cultures of B16F10-CD44+ cells were left untreated or treated with either SLNs/PTX or HA-SLNs/PTX (equal PTX concentration 10 µg/mL). In parallel, triplicate cultures of B16F10-CD44- cells were also analyzed. Cells were washed twice with PBS and counted. Then, the cells were resuspended at a density of 1 × 106 cells/mL in DMEM containing 2% FBS and 10 mM HEPES and divided into two groups. One group of untreated B16F10-CD44+ cells, treated B16F10-CD44+ cells and B16F10-CD44- cells were incubated for 90 min at 37°C with the DNA binding dye Hoechst33342 (5 µg/mL final concentration; Sigma Aldrich, USA), with gentle agitation every 15 min. The second group of B16F10-CD44+ cells was pretreated with 100 µg/mL verapamil (Sigma Aldrich) for 20 min prior to and during Hoechst 33342 staining. All incubations with Hoechst dye were carried out in the dark. Cells were then washed twice with ice-cold PBS and resuspended in PBS containing 2% FBS and 10 mM HEPES. Cell suspensions were stored at 4°C in the dark, stained with PI (2 µg/mL final concentration) and subjected to fluorescence-activated cell sorting (BD Biosciences, USA).
Assessment of cellular pharmacology
Invasion assay
Tumor cell invasion was assessed using a Boyden system (Corning, USA) involving polycarbonate membranes (8 µm pore size) precoated with Matrigel (10 µg/mL, BD Biosciences). Lower chambers were filled with RPMI 1640 medium containing 10% FBS, and upper chambers were filled with RPMI 1640 medium containing 0.1% FBS. B16F10-CD44+ cells were treated or not with PTX formulations and added to upper chambers in triplicate (1×104 cells per well), then incubated 18 h at 37°C. Cells that had moved and adhered to the lower side of the membrane were fixed and stained with Crystal Violet, which was then dissolved with 33% acetic acid. Cell invasion was assessed by measuring the absorbance at 570 nm with a microplate reader (Thermo, Varioskan Flash).
Adhesion assay [29]
Adhesion ability was measured using the MTT assay. B16F10-CD44+ cells (5 × 104/well) previously treated with PTX formulations were seeded in triplicate into wells of a MatrigelTM (BD, 50 µg/mL)-coated 96-well plate. Non-adherent cells were removed 2 h later by PBS. After washing, and numbers of adherent cells and the total numbers of untreated cells were measured using the MTT assay at a wavelength of 570 nm. The optical density ratio due to adherent cells and total untreated cells reflects the proportion of cells that adhered to the MatrigelTM-coated 96-well plate.
Cell-cycle distribution
The effect of PTX on progression of B16F10-CD44+ cells through the cell cycle was assessed using a PI-based assay [30, 31]. Cells were seeded in to six-well plates (5 × 105 cells per well) and incubated for 24 h at 37°C, then, cultures were exposed to 2 mL of medium containing PTX (10 µg/mL) for 4 h, and finally in fresh medium for another 48 h. Cells were digested and merged with the floating fraction. After two washes in PBS, cells were fixed in cold 75% ethanol for 30 min at 4°C, washed twice again in PBS, and incubated for 15 min in 0.1 mL Triton X-100 (0.1%, v/v) to punch on the cytomembrane. Intracellular RNA was digested for 30 min at 37°C using ribonuclease A (20 µg/mL), then stained with PI (20 µg/mL) at 4°C for another 30 minutes in the dark. The proportions of cells in different stages of the cell cycle were measured using flow cytometry.
Wound healing assay
Migration and mobility of B16F10-CD44+ cells were assessed using a wound healing assay. Cultured cells were seeded into 12-well plates and grown to confluence, then the cell monolayer was scratched wound using a sterile pipette tip. Scratched cultures were exposed for 4 h to PTX formulations (equal to 10 µg/mL of PTX), then cultured in fresh medium for another 48 h. Digitized images of the wound area were captured using a Zeiss Axiovert 40 inverted microscope (Germany).
In vivo biodistribution and tumor targeting mechanism of HA-SLNs/PTX
B16F10-CD44+ cells were injected intravenously into C57BL/6 mice (3 ×105 cells in 100 µL of PBS) via the tail vein, and tumors were allowed to grow for 24 d. Tumor-bearing mice were randomly divided into four groups, each of which was injected intravenously in the tail vein with one of the following formulations at a dose of 1mg/kg: free PTX, SLNs/PTX, heparin-SLNs/PTX, HA-SLNs/PTX. After 1, 2 or 6 h, the heart, liver, spleen, lung, kidney, brain and blood were collected and stored. Tissues were weighed and mixed with physiological saline (1:2, w/v) and homogenized. The homogenate was mixed with methanol (1:3, v/v) and centrifuged and centrifuged (15000 rpm, 10 min), and the supernatant was collected. The concentration of PTX in the supernatant determined by LC-MS/MS (Agilent, USA) using a Diamonsil ODS column (50 × 4.6 mm, 1.8 µm) and methanol-0.1% formic acid (75:25, v/v) as mobile phase. The flow rate was 0.4 mL/min.
C57BL/6 mice created a murine lung metastasis model were received i.v. injections of SLNs/PTX and HA-SLNs/PTX (PTX represented by DID) corresponding to a dose of 50 µg/kg of DID. Three tumor-bearing lungs per group were then cryopreserved in tissue freezing medium (Leica) and cut into cross-sections using a microtome (Leica CM1950). Sections were mounted on glass slides and stained with DAPI and FITC-conjugated anti-CD44 antibody for fluorescence analysis. Sections were analyzed using confocal laser scanning microscopy (CLSM). Another three tumor-bearing lungs per group were ground up and cells were resuspended in PBS, incubated with FITC-conjugated anti-CD44 monoclonal antibody and analyzed by flow cytometry.
For the competitive inhibition study [32], HA-SLNs/PTX (1 mg/kg) was intravenously injected into tumor-bearing mice 3 min after intravenous administration of a high dose of HA (40 mg/kg). The PTX quantity of tissues were determined by LC-MS/MS described above.
In vivo therapeutic effects of HA-SLNs/PTX
B16F10-CD44+ cells (3 × 105) were injected into the tail vein of C57BL/6 mice to create a murine lung metastasis model [33], and starting at 9 d later, tumor-bearing mice received intravenous injections of the indicated samples (1mg of PTX per kg) via i.v. injection till day 14. On day 24, animals were sacrificed after six injections and the tumor-bearing lungs were collected and analyzed. The paraffin-embedded sections were stained with hematoxylin and eosin for histopathologic evaluation. Furthermore, for the survival analysis study, tumor-bearing mice were also treated from day 9 till 14, and the death date of mice was recorded.
Toxicity evaluation and Immunogenicity
To measure the liver toxicity difference for the reason of HA modification, the serum samples of C57BL/6 mice in the therapeutic experiment treated with PTX, SLNs/PTX and HA-SLNs/PTX were collected before sacrificed. Serum levels of aspartate aminotransferase (AST) and alanine transaminase (ALT) were then measured according to the manufacturer's protocol (Jiancheng, Nanjing, China).
C57BL/6 mice aged 6-8 weeks were injected intravenously with PTX (commercial PTX), PTX-loaded SLNs or PTX-loaded HA-SLNs (1 mg/kg PTX). Untreated mice were maintained in parallel. At 6 h after injection, serum was collected and assayed for the concentration of the proinflammatory cytokine interleukin-12 (IL-12) using a commercial ELISA kit according to the manufacturer's instructions (R&D Systems, Minneapolis, USA). Absorbance at 450 nm was measured using a microplate reader (Varioskan Flash).
Statistics
Results were expressed as mean ± standard deviation of triplicate measurements, unless otherwise noted. Inter-group differences were assessed for significance using one-way ANOVA followed by Student's t-test. The significance threshold was defined as p < 0.05, and such differences were marked with *, differences with an associated p < 0.01 were marked with **.
Result
Characteristics of HA-SLNs/PTX
Cationic SLNs/PTX were prepared using a film-ultrasonic method and found to have the following biophysical parameters: size, 160.6 ± 1.997 nm; poly dispersity index, 0.218 ± 0.012; and zeta potential, 46.3 ± 1.07 mV. They showed high EE and DL (3.58%). Simply mixing of HA and SLNs/PTX (1:12, w/w) led to the formation of nanoparticles through electrostatic interactions. The resulting HA-SLNs/PTX had an average size of 190.0 ± 1.474 nm, a polydispersity index (PDI) of 0.164 ± 0.011 and a zeta potential of 31.8 ± 0.306 mV. The reduced zeta potential of HA-SLNs/PTX was attributed to negatively charged hyaluronic acid bound to the surface of positively charged SLNs and the suitable zeta potential probably contributed to their good storage stability. The transmission electron photomicrographs (TEM) showed both SLNs/PTX and HA-SLNs/PTX to have a uniform, round shape and narrow size distribution (Fig. 1).
Fig 1
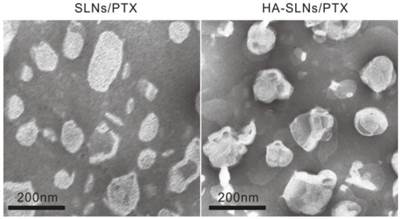
Transmission electron microscopy (TEM) of uncoated SLNs/PTX (left) and HA-SLNs/PTX (right), Scale bar, 200 nm.
In vitro release studies
The dynamic dialysis method was used to compare the drug release behavior of free PTX, SLNs/PTX and HA-SLNs/PTX. As shown in Fig. 2, PTX was released much more slowly from HA-SLNs/PTX than that from free PTX and SLNs/PTX. The release profiles could be divided into two phases: an initial burst-release phase, followed by sustained-release phase. During the first 12 h, 62.14% of drug was releases from the free PTX solution, compared to only 31.2% of PTX from HA-SLNs/PTX. The lower burst release was more beneficial, since the toxicity of PTX was normally associated with extremely high drug concentration.
Uptake of HA-SLNs/PTX by B16F10-CD44+ cells
B16F10-CD44+ cells took up HA-SLNs/PTX to a much greater extent than the nanoparticles lacking HA or the same nanoparticles coated with the similar mucopolysaccharide heparin, even though the heparin-coated vesicles showed a similar zeta potential (32.3 ± 1.56 mV) and particle size (183.3 ± 1.2192 nm). This suggested that uptake occurred through a specific, HA-mediated mechanism. Experiments in Fig. 3A and Supplementary Material: Fig. S1 suggested that the high affinity of HA-SLNs/PTX to the B16F10-CD44+ cells was not only attributed to nonspecific electrostatic attraction between the nanoparticles and the cell surface, but also because of specific HA binding to CD44.
Fig 2
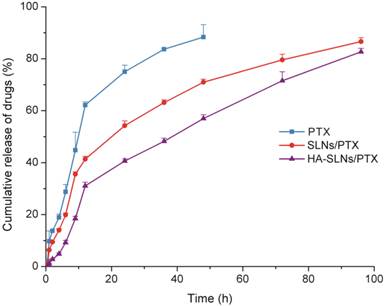
Release of PTX from free PTX (commercial PTX injection), SLNs/PTX and HA-SLNs/PTX in PBS containing 0.2% Tween 80. Results shown were mean ± SD (n = 3).
Fig 3
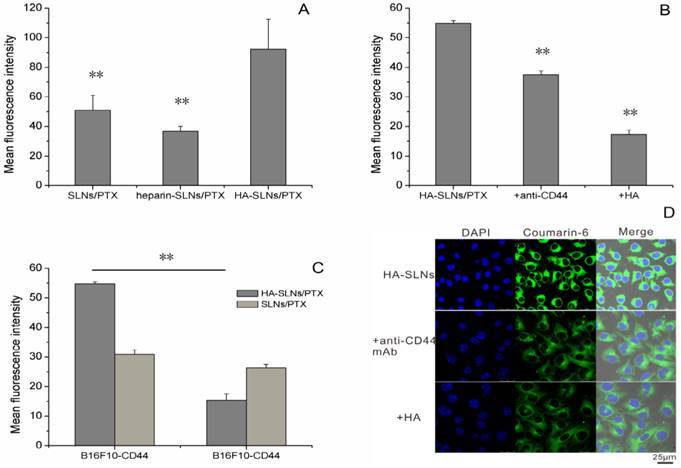
A) Flow cytometry analysis to measure the efficiency of cellular uptake of uncoated SLNs/PTX, heparin-coated SLNs/PTX and HA-SLNs/PTX (Coumarin-6 was incorporated in the place of PTX as a fluorescent marker). Each preparation was incubated with B16F10-CD44+ cells for 1 h. B) Efficiency of cell binding by HA-SLNs/PTX in the presence of excess free HA or anti-CD44 antibody (n = 3). C) Cell-binding ability of HA-SLNs/PTX (loading coumarin-6) in CD44 high expression- and low expression-cells. Data shown were mean ± SD (n = 3). D) Confocal laser scanning microscopy of uptake of HA-SLNs/PTX (loading coumarin-6) by B16F10-CD44+ cells at 37°C in the presence of excess free HA or anti-CD44 antibody. Scale bar, 25 μm. *p < 0.05, **p < 0.01, compared with cells treated with HA-SLNs/PTX.
To further examine the role of CD44 in the association of the HA-SLNs/PTX with the cells, the fluorescence loaded HA-SLNs/PTX were incubated with the cells to determine the cell-associated fluorescence in the presence and absence of monoclonal anti-CD44 mAb and hyaluronic acid. HA-SLNs/PTX binding efficiency was weaker in the presence of antibody and hyaluronic acid than that in its absence (Fig. 3B, Supplementary Material: Fig. S1), which suggested that anti-CD44 antibodies and hyaluronic acid inhibited the cell association of HA-SLNs/PTX. This is consistent with the idea that the cell surface marker CD44 helps mediate the efficient uptake of HA-SLNs/PTX.
This implies that our HA-coated nanoparticles may be taken up efficiently by CSCs, which express high levels of CD44, but not by non-CSCs. To test this possibility, we examined uptake efficiency of HA-SLNs and SLNs to our cell-sorted populations of B16F10-CD44+ cells (mean surface expression CD44 = 92.3%) and B16F10-CD44- cells (mean surface expression = 29.4%).While SLNs showed similar uptake to both cell populations, HA-SLNs bound to a much greater extent to the CD44+ cells than to CD44- cells (Fig. 3C, p < 0.01). This further supports the idea that coating SLNs with HA leads to CD44-mediated uptake, selectively targeting the nanoparticles to CSCs.
HA-SLNs/PTX induced improved cytotoxic effects and apoptosis
Dose-dependent cytotoxicity against B16F10-CD44+ and A549 cultures was compared for free PTX and for PTX delivered in SLNs or HA-SLNs in the dose range of 2-20 µg/mL using the MTT assay (Fig. 4A and Supplementary Material: Fig. S3). Greater concentrations of PTX led to greater growth inhibition. In B16F10-CD44+ cultures, HA-SLNs/PTX showed the lowest IC50 (11.13 ± 1.62 µg/mL), followed by SLNs/PTX (18.11 ± 3.79 µg/mL) and then free PTX (31.39 ± 4.81 µg/mL). Similar results were obtained in A549 cells, with the corresponding IC50 values of 23.99 ± 1.81, 28.90 ± 1.04 and 40.89 ± 2.72 µg/mL. In fact, cytotoxicity with PTX-loaded HA-SLNs at a dose of 10 µg/mL PTX was similar to that of PTX-loaded SLNs at a dose of 20 µg/mL PTX. These results suggest that the use of HA-SLNs enhances the ability of PTX to inhibit cell proliferation in vitro.
Fig 4
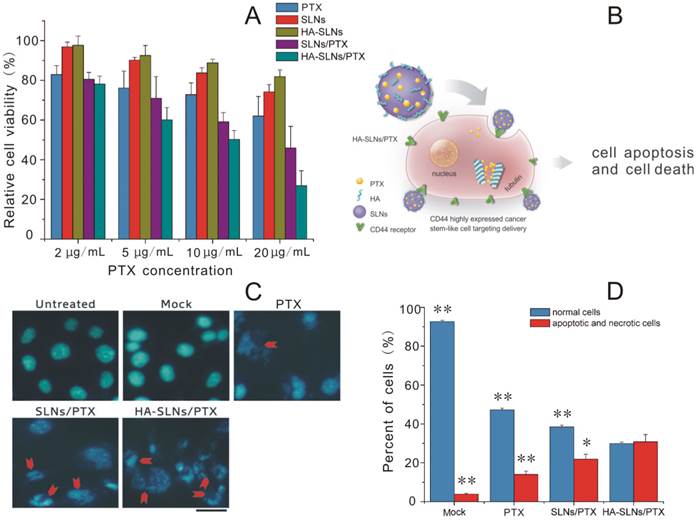
A) Viability of B16F10-CD44+ cells after treatment with PTX formulations based on the MTT assay. Cell viability was calculated as a percentage of the result with untreated cells and shown as mean ± SD (n = 3). B) Schematic illustration of B16F10-CD44+ cell killing by HA-SLNs/PTX based on the therapeutic effect of paclitaxel (PTX). C) Induction of apoptosis and morphological changes in B16F10-CD44+ cells by HA-SLNs/PTX. Mock conditions involved addition of all reagents except PTX. Arrows indicate apoptotic cells. Scale bar, 30 μm. D) Measurement of necrosis and apoptosis using flow cytometry to track FITC-labeled annexin V and propidium iodide. Shown were proportions of normal, apoptotic and necrotic cells after treatment with the indicated formulations. *p < 0.05, **p < 0.01, compared with the HA-SLNs/PTX group.
In addition to antiproliferative effects, we assessed the ability of PTX to induce apoptosis and morphological changes when delivered from SLNs or HA-SLNs. Double immunostaining with FITC-conjugated annexin V and PI was used to measure the numbers of apoptotic and necrotic cells, while morphology was assessed using DAPI staining. PTX-loaded HA-SLNs induced extreme chromatin condensation, nuclear fragmentation and apoptotic body formation in a substantially larger number of cells than the mock, PTX and SLNs/PTX did (Fig. 4C). Treating cells with PTX-loaded HA-SLNs led to a significantly higher fraction of cells in the annexin V+/PI+ quadrant (apoptotic and necrotic cells) than treating them with PTX-loaded SLNs (p = 0.0261) or PTX (p < 0.01) (Fig. 4D). Conversely, treating cultures with PTX-loaded HA-SLNs led to a significantly lower fraction of annexin V-/PI- (normal) cells than treating them with PTX-loaded SLNs or PTX (both p < 0.01; Fig. 4D).
Relationship of CD44 with CSC characteristics and preparations' suppressive effect
To identify whether surface CD44 expression correlates with CSC phenotype in our B16F10 experimental system, we used cell sorting to separate B16F10-CD44+ and B16F10-CD44- populations and we tested each population in the cell sphere-formation assay, widely used to identify stem cells based on their self-renewal ability at the single-cell level [34, 35]. We also tested both populations in the soft agar colony formation assay, which measures tumorigenicity by examining proliferation in a semisolid culture media [36]. Third, we compared levels of expression of the melanoma CSC markers ALDH [37, 38] and CD133 [39, 40] in both populations. Lastly, we compared the tumorigenicity of each cell type in a xenograft mouse model.
B16F10-CD44+ cells grew as spheroids and cell colonies under our experimental conditions, whereas B16F10-CD44- cells did not (Fig. 5A). Quantitative real-time PCR showed that B16F10-CD44+ cells expressed significantly higher levels of ALDH and CD133 (Fig. 5B). When mice were injected subcutaneously with 5 ×104 B16F10-CD44+ or CD44- cells, CD44+ cells formed tumors (average size, 219.55 mm3), whereas CD44- cells did not (Fig. 5D). Taken together, these findings suggest that B16F10-CD44+ cells, but not B16F10-CD44- cells, have substantial expression of CSC marker, self-renewal activity and tumorigenicity in vitro and in vivo, consistent with a CSC phenotype. This implies that PTX-loaded nanoparticles should target B16F10-CD44+ cells in order to provide effective antitumor therapy.
Side population (SP) cell was rarely part of the cell population which discharged nucleus-fluorescent dye to the extracellular. It was found in many types of cancer and usually enriched in therapy-resistant as well as tumorigenic cells. The identification of cancer stem-like cells had further raised the interest in the SP (side population) technique, and the SP phenotype might explain the resistance of a subpopulation of tumor cells to chemotherapy [41, 42]. The ''Side Population'' (SP) discrimination assay was a flow cytometry method used to detect stem cells based on the dye efflux properties of ABC transporters. Hoechst staining was applied to determine the SP cells. Importantly, the phenotyped SP subpopulations needed to be further confirmed by verapamil inhibition controls. In B16F10-CD44+ cells, we detected 56.2% SP cells while significant inhibition was observed in verapamil presented group and non-SP cells in B16F10-CD44- cells (Fig. 5E). When the cells preincubated with verapamil to block the ATP transporter, the SP proportion was reduced to 0.4%. This provides further evidence that the B16F10-CD44+ population contains substantial proportions of chemo-resistant and stem-like cells.
More importantly, Free PTX and PTX released from SLNs or HA-SLNs inhibited sphere formation by B16F10-CD44+ cells, with PTX-loaded HA-SLNs showing the greatest effect (Fig. 5C). In addition, treating B16F10-CD44+ cells with PTX-loaded SLNs reduced the proportion of SP cells from 56.2% to 14.5%, while treating them with HA-SLNs/PTX reduced the proportion to 6.7% (Fig. 5E).
Cellular pharmacology
Invasion assay
Using the Boyden chamber system, we compared the ability of different drug formulations to inhibit the invasion ability of B16F10-CD44+ cells in vitro. Invasion ability was measured as the number of cells that migrated to the bottom side of the membrane. HA-SLNs/PTX inhibited invasion to a significantly greater extent than other groups (Fig. 6A, p < 0.01).
Adhesion assay
We compared the ability of different drug formulations to inhibit the adhesion of cancer cells to the extracellular matrix and molecules on the cell surface, which we modeled using MatrigelTM, a mixture of components mimicking the extracellular matrix. Such adhesion is a key step during metastasis in vivo and a critical factor in extravasation and formation of new tumor foci [43]. Adhesion ability of B16F10-CD44+ cells was much lower after treatment with HA-SLNs/PTX (35.81%) than after treatment with SLNs/PTX (47.07%) or PTX (56.70%) (Fig. 6B).
Fig 5
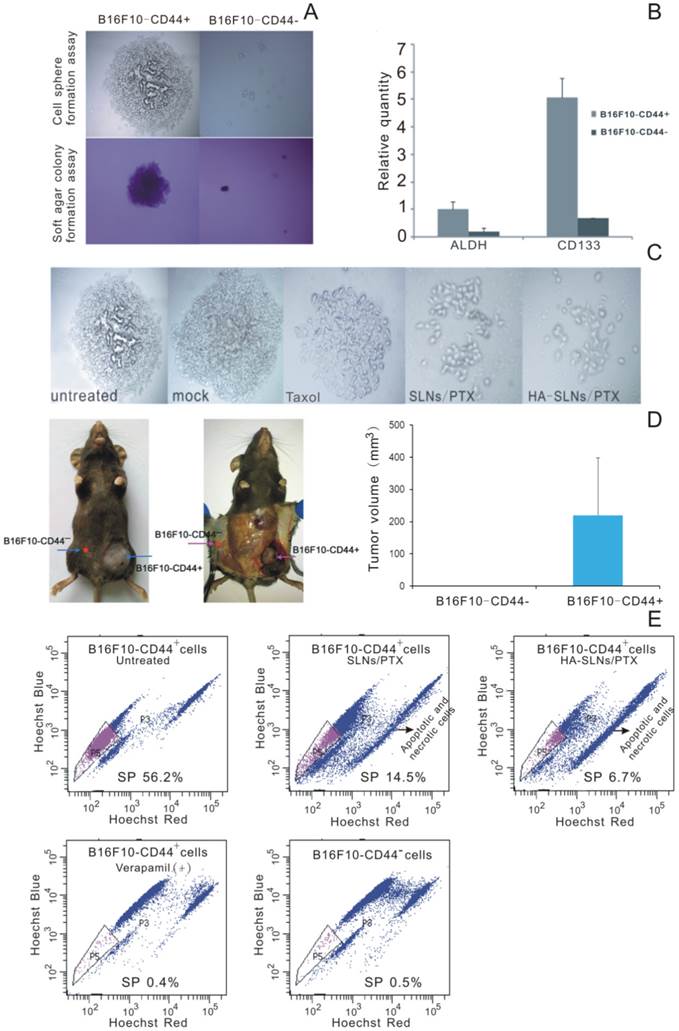
CSC phenotype of B16F10-CD44+ cells. A) Sphere formation and soft agar colony formation assays using B16F10-CD44+ and B16F10-CD44- cells. B) Quantitative RT-PCR determination of ALDH and CD133 expression. Data were shown as normalized fold expression relative to β-actin as an internal control (n = 3). C) Representative results of a spheres formation assay in which B16F10-CD44+ cell were cultured for 2-3 weeks, treated for 4 h with HA-SLNs/PTX and photographed 48 h later. D) Representative tumors and tumor volume in C57BL/6 mice injected subcutaneously with B16F10-CD44+ or B16F10-CD44- cells (5 × 104 cells/animal), and tumor growth was monitored for 23 days after injection. E) Identification of the side population (SP) and the proportion of SP cells in B16F10-CD44- cells and in B16F10-CD44+ cells (untreated or treated with SLNs/PTX or HA-SLNs/PTX). As a control for identify of SP cells, experiments were also performed in the presence of the ABC transporter inhibitor verapamil. Data were shown as mean ± SD (n = 3).
Fig 6
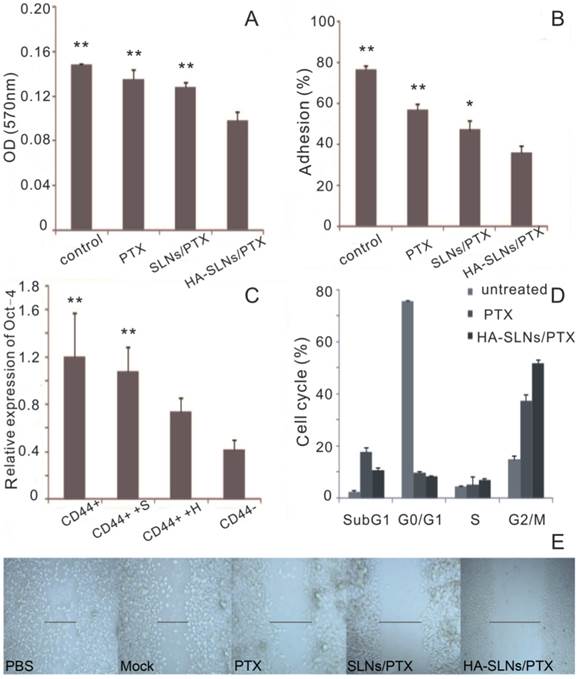
Effect of HA-SLNs /PTX on cellular pharmacology. A) Crystal violet staining assay to measure the effects on cell invasion at 570nm (n = 3). B) Effects on cell adsorption by MTT assays at 570 nm (n = 3). C) Quantitative RT-PCR analysis to measure the effects on Oct-4 expression. Cells were untreated (CD44+) or were pre-treated with SLNs/PTX (CD44++S) or HA-SLNs/PTX (CD44++H). In parallel, B16F10-CD44- cells (CD44-) were also analyzed. Data were shown as normalized fold expression relative to β-actin as internal control (n = 3). D) Flow cytometry analysis to measure the effects on the cell cycle distribution of B16F10-CD44+ cells. Cells were treated with PTX and HA-SLNs/PTX containing the same concentration (10 μg/mL). Data shown were mean ± standard deviation (n = 3). E) Wound healing assay to measure the effects on motility and migration. B16F10-CD44+ cell were treated with PTX formulations. *p < 0.05, **p < 0.01, compared with HA-SLNs/PTX.
Oct-4 expression
Oct-4, a transcription factor critical for CSC survival [44], plays an important role in drug resistance in several types of cancer [45]. Oct-4 is also essential for triggering an anti-apoptotic response to stress in embryonic stem cells [46]. Therefore, effective cancer therapy involves down-regulating Oct-4 expression, so we examined the ability of our drug formulations to do this.
After demonstrating that B16F10-CD44+ cells expressed higher levels of Oct-4 mRNA than B16F10-CD44- cells, consistent with our other results correlating CD44 expression with CSC phenotype, we compared Oct-4 expression in B16F10-CD44+ cells before and after treatment with PTX-loaded SLNs or PTX-loaded HA-SLNs. While both types of SLNs reduced Oct-4 expression, the reduction was significantly greater in the case of HA-SLNs/PTX (p < 0.01, Fig. 6C).
Cell cycle assay
PTX exerts anti-mitotic effects by inhibiting microtubule assembly and inducing tubulin self-association into coiled spiral aggregates, arresting cells in G2/M phase [47]. To estimate the in vitro antimitotic effect of PTX at the cellular level, the effect of PTX solution and HA-SLNs/PTX on the cell cycle of B16F10-CD44+ cells was evaluated, Treating B16F10-CD44+ cells with PTX-loaded HA-SLNs led to a much higher proportion of cells in G2/M (51.7%) than treating them with PBS (15.1%) or PTX (37.3%) (Fig. 6D).
Wound-healing assay
The in vitro wound-healing assay is a simple, inexpensive, and well-developed method to investigate the combined processes of cell migration, cell motility and proliferation [48]. B16F10-CD44+ cells migrated faster into the scratch, and the recovery of the wound area was nearly completed within one day in groups treated with PBS, mock (all reagents except PTX), while PTX, SLNs/PTX and HA-SLNs/PTX inhibited the wound closure of all scratches (Fig. 6E). These observations also illustrated that HA-SLNs/PTX-treated tumor cells were more significantly attenuated in cell motility and migration.
HA alters the distribution of PTX in mice and its tumor-targeting mechanism
Mice were injected intravenously with free PTX or with one of three types of PTX-loaded nanoparticles: SLNs, heparin-SLNs or HA-SLNs. Then the biodistribution of PTX was compared among the different conditions (Fig. 7A-C). PTX accumulated in the lungs to a greater extent when delivered in SLNs than when delivered as free drug, and the accumulation was significantly greater with PTX-loaded HA-SLNs than with any other drug formulation at all time points (p < 0.01). Our finding that accumulation was similar for SLNs and heparin-SLNs reinforces our in vitro evidence that coating nanoparticles with HA improves tumor targeting efficiency.
Fig 7
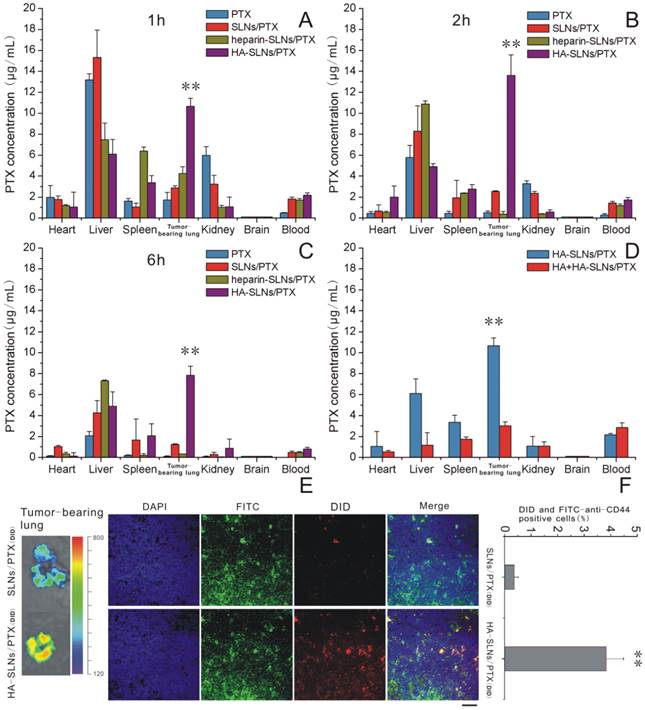
A-C) Biodistribution of PTX in various tissues at 1, 2 and 6 h after intravenous administration of different PTX formulations (n = 3). D) Biodistribution of PTX at 1 h after injection of HA-SLNs/PTX following pretreatment with a high dose of free HA. E) Representative ex vivo fluorescence imaging and confocal laser scanning microscopy of sections of tumor-bearing lung from mice treated with different drug formulations. Mice were injected intravenously SLNs/PTX and HA-SLNs/PTX (PTX represented by DID, red). After 2 h injection, mice were sacrificed and lungs removed. Tissue was frozen, fixed in 4% paraformaldehyde and stained with FITC-labeled anti-CD44 monoclonal antibody (green) and DAPI (blue). Scale bar, 30 μm. F) Flow cytometry detection of drug (DID) in B16F10-CD44+ cells in tumor-bearing lungs from C57BL/6 mice intravenously injected with DID (PTX) loaded SLNs/PTX and HA-SLNs/PTX. Animals were sacrificed at 2 h after i.v. injection and tumor-bearing lungs were lapped to single cell suspensions. The suspensions were incubated for 30 min at 4°C with FITC-labeled anti-CD44 monoclonal antibody, then analyzed by flow cytometry (n = 3). *p < 0.05, **p < 0.01, for comparisons of HA-SLNs/PTX with other groups (PTX, SLNs/PTX, heparin-SLNs/PTX).
The competitive inhibition study was performed to investigate the tumor-targeting mechanism of HA-SLNs/PTX. To analyze this question more directly, we analyzed biodistribution of PTX in mice first treated with a high dose of HA, followed by PTX-loaded HA-SLNs. HA pre-injection significantly reduced the amount of HA-SLNs/PTX in tumor-bearing lung tissue (p < 0.01, Fig. 7D), consistent with the idea that specific interaction between HA and CD44 mediates tumor targeting of the HA-SLNs/PTX.
Since studies of drug distribution in tumor-bearing lung tissue may not accurately reflect the real drug biodistribution in tumor cells, we also examined frozen tumor sections (Fig. 7E) and performed flow cytometry experiments with suspensions of tumor-bearing lung cells incubated with FITC-labeled anti-CD44 antibody (Fig. 7F). These experiments showed that PTX delivery to CD44+ cells was more efficient with HA-SLNs than with SLNs (p < 0.01).
Antitumor effect
The antitumor efficacy of PTX-loaded HA-SLNs was tested in a murine lung metastasis model; animal survival as well as tumor weight and histopathology were analyzed. Obvious tumors were detected in the lungs of animals treated with PTX (commercial PTX injection) or SLNs/PTX, but not in animals treated with HA-SLNs/PTX (Fig. 8A and 8C); similarly, average lung weight was significantly lower in mice treated with HA-SLNs/PTX than in the PBS and PTX groups (p < 0.01). While animals treated with PBS died relatively rapidly, animals treated with PTX, PTX-loaded SLNs or PTX-loaded HA-SLNs survived significantly longer (log-rank test, p< 0.01; Fig. 8D). Six consecutive injections of PTX-loaded HA-SLNs led to substantially longer lifespan than the equivalent doses of PTX (p = 0.032, log-rank test) or PTX-loaded SLNs (p = 0.049, log-rank test)
Fig 8
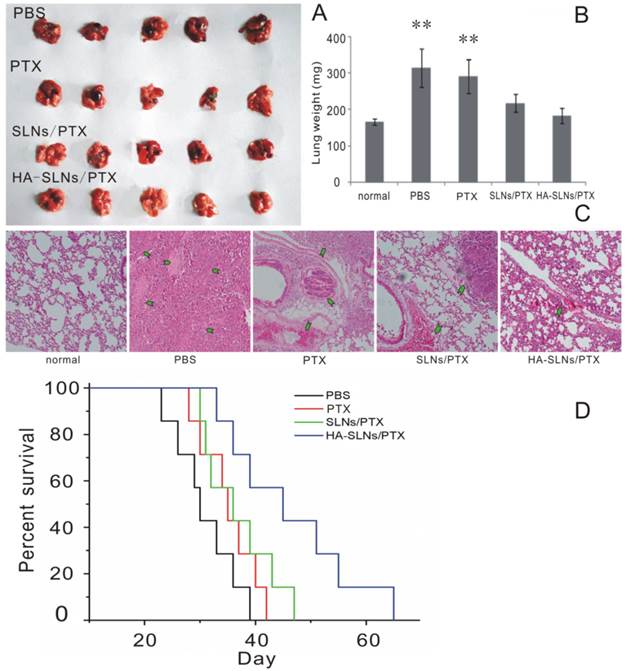
A) Photographs of B16F10-CD44+-bearing lungs on the day 24 after six consecutive intravenous injections of the indicated preparations. B) Enhanced antitumor effects of HA-SLNs/PTX based on tumor-bearing lung weight (n = 5). C) Histological staining of the tumor-bearing lungs tissue after treatment with HA-SLNs/PTX. Arrows, tumor nodules. D) Survival analysis of tumor-bearing mice (n = 7). *p < 0.05, **p < 0.01, compared with the group treated with HA-SLNs/PTX.
Fig 9
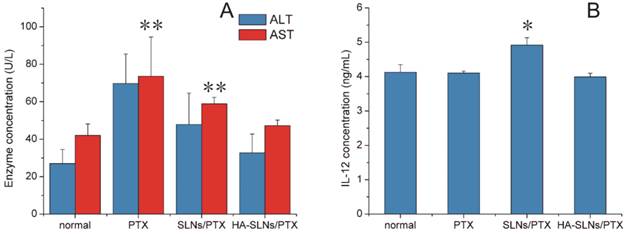
A) In vivo assessment of hepatotoxicity of HA-SLNs/PTX. Serum levels of ALT and AST were measured after the six dose of free PTX, SLNs/PTX or HA-SLNs/PTX. B) Serum concentrations of IL-12 at 6 h after intravenous injection of C57BL/6 mice with HA-SLNs/PTX (n = 5). *p < 0.05, **p < 0.01, compared with the group treated with HA-SLNs/PTX.
Toxicity and Immunogenicity
In addition to testing antitumor efficacy of our PTX formulations in vivo, we also examined its ability to induce inflammation and toxic effects. In particular, we investigated its hepatotoxicity, since a relatively large proportion of the drug accumulated in liver (Fig. 7A-C). C57BL/6mice treated with PTX-loaded SLNs showed significantly higher serum levels of the liver enzyme aspartate aminotransferase (AST) than animals treated with PTX-loaded HA/SLNs (p < 0.01, Fig. 9A). In fact, typical signs of hepatic injury were not detectable in mice treated with PTX-loaded HA-SLNs. These results suggest that adsorbing HA onto nanoparticles decreased liver toxicity.
We also investigated whether PTX-loaded HA-SLNs induced an immune response in C57BL/6 mice by evaluating serum concentrations of IL-12 after intravenous injection of different drug formulations. While PTX-loaded SLNs significantly increased IL-12 expression (p < 0.05), PTX-loaded HA-SLNs did not (Fig. 9B). Histopathology analysis failed to detect signs of inflammation or its consequences in animals treated with HA-SLNs/PTX (Fig. 8C).
Discussion
Standard oncology treatments such as chemotherapy, radiotherapy and surgical resection only shrink the bulk tumor which was companied with poor prognosis [49]. Therefore, a novel therapeutic strategy focuses on eradicating the candidate of CSCs which can render tumor resistant to chemotherapy resistance and can differentiate into tumor cells, leading to relapse and metastasis. Here we combine the traditional chemotherapeutic PTX with an HA-SLNs system that efficiently targets melanoma cancer stem-like cells. We show in vitro and in vivo that the HA-SLNs/PTX system effectively inhibits invasion, migration, and tumorigenicity of B16F10-CD44+ melanoma cancer stem-like cells. The primary factor in this system's success is an outer coating of HA, which interacts specifically with the CD44 expressed at high levels on the surface of CSCs. This interaction allows more efficient uptake, greater antitumor efficacy, and lower toxicity than we observed with SLNs lacking an HA coating.
Our drug delivery strategy exploits the strong binding affinity between HA and CD44, and reflects the fact that cancer treatments targeting CD44-expressing CSCs often show better therapeutic effect than other treatments [50]. Since this cell surface glycoprotein is highly expressed on CSCs in numerous types of malignancies, including melanoma, colon cancer, non-small cell lung carcinoma, prostate cancer, head and neck cancer, and breast cancer stem, our delivery system may prove useful for treating many of these cancers.
We created HA-SLNs through electrostatic attraction, a simple and controllable method that avoids the chemical reactions or organic solvents needed to attach HA covalently to the nanoparticles [24]. PTX-loaded HA-SLNs created using this approach showed good dispersity and remained stable at 4ºC for more than one week. We demonstrated in several experiments that the HA coating was essential for efficient uptake into B16F10-CD44+ cells, and that this uptake involved CD44-mediated processes. Uptake of HA-coated nanoparticles was greater than that of particles without a coating or with a coating of heparin, which shows glycosaminoglycan properties similar to those of HA but does not bind CD44. Uptake of HA-SLNs/PTX was blocked by addition of excess free HA or anti-CD44 antibody. Uptake of HA-SLNs/PTX was higher in B16F10-CD44+ cells than in B16F10-CD44- cells. The gap of uptake efficiency between SLNs/PTX and HA-SLNs/PTX group was greater in B16F10-CD44+ cells than in the A549 non-small cell lung cancer cell line, which expressed much less CD44 on the surface (66.7%) than B16F10-CD44+ (92.3%) (Fig.S2). PTX-loaded HA-SLNs arrested a greater proportion of cells in G2/M phase and induced apoptosis to a greater extent than PTX did.
As part of our study of the efficacy of PTX-loaded HA-SLNs, we compared in detail the CSC-like characteristics of B16F10-CD44+ and B16F10-CD44- cells. We showed that only CD44+ cells grew into spheres in serum-free medium, formed colonies in soft agar, and gave rise to tumors in mice. CD44+ cells also contained a higher proportion of SP cells. These results suggest, consistent with the literature linking CD44 expression and CSC phenotype, that B16F10-CD44+ cells exhibited more potent self-renewal ability, oncogenicity and resistance to chemotherapy than B16F10-CD44- cells. In addition, expression of Oct-4mRNA, which correlated positively with tumorigenicity and survival of CSCs [51], was 3-fold higher in B16F10-CD44+ cells. Treating B16F10-CD44+ cells with PTX-loaded HA-SLNs eliminated their ability to form spheres and down-regulated Oct-4 expression.
Coating our SLNs with HA led to much greater accumulation in tumor-bearing lung tissue than we observed with uncoated SLNs; this presumably reflects the enhanced permeability and retention (EPR) effect, the cationic nature of the delivery system and the ability of HA to bind CD44 specifically [52]. At the same time, coating the nanoparticles with HA reduced accumulation in the liver, translating to lower hepatoxicity. Consistent with these findings, experiments with tumor slices and flow cytometry showed that HA-SLNs delivered much more PTX to B16F10-CD44+ cells in tumors than uncoated SLNs did. This drug delivery was inhibited in the presence of excess free HA, consistent with the key interaction between HA and CD44 for nanoparticle uptake and drug delivery. The absence of this CD44 interaction may help explain why SLNs/PTX or heparin-SLNs/PTX showed much lower drug delivery efficacy and weaker antitumor effects.
The superior ability of HA-SLNs to deliver PTX to tumor cells in vitro and in vivo relative to free PTX and SLNs can be attributed to several factors. HA-SLNs/PTX bind specifically to CD44, allowing targeting of cancer stem-like cells, efficient receptor-mediated endocytosis and potent antitumor activity. In general, loading drugs into nanoparticles prolongs their half-life in the blood, whereas free PTX injected directly into the blood is rapidly eliminated [53]. The hydrophilic HA chains on the nanoparticle surface expand and swell in the body, which may create a cloud of chains at the particle surface and thereby reduce opsonization by blood proteins and uptake by macrophages. Such observations are also consistent with a higher accumulation of the drug in a more selective manner in cancer cells when animal were treated with HA-SLNs/PTX compared with free PTX and SLNs/PTX, allowing a higher efficacy and a reduction of side effect.
Nanoparticle-based drug delivery systems have the potential of targeting to reticuloendothelial system, resulting in greater drug accumulation in the liver, spleen and bone marrow [54]. This accumulation may induce toxicity. In particular, we investigated the liver toxicity of PTX preparations. Serum levels of AST and ALT were within the normal range in mice treated with PTX-loaded HA-SLNs. This may reflect the relatively low dose of PTX used (1 mg/kg) compared with previous similar antitumor efficacy with relatively high drug doses (3-10 mg per kg of body weight) [24, 55, 56], and the efficient accumulation of PTX in tumor tissue due to the presence of the HA coating.
High-molecular-weight HA is commonly regarded as an effective “bioinert” component of drug delivery systems. We verified this directly in our model system by measuring serum levels of IL-12 in mice treated with PTX-loaded HA-SLNs. No significant increase in IL-12 levels was observed, consistent with previous reports that high-molecular-weight HA does not induce expression of genes involved in proliferation or inflammation [20].
Conclusion
We have designed and tested a system for delivering PTX to lung tumors using HA-SLNs, which selectively targeted CD44 associated cancer stem-like cells. HA coated SLNs/PTX via electrostatic interactions, giving rise to well-dispersed nano-complexes that were taken up more efficiently by CD44+ cells and that induced apoptosis to a greater extent than uncoated SLNs or SLNs coated with heparin. As part of this work, we established that B16F10-CD44+ cells showed a strong CSC phenotype, whereas B16F10-CD44- cells did not. Treating B16F10-CD44+ cells with HA-SLNs/PTX inhibited sphere formation in vitro, down-regulated Oct-4 mRNA expression and reduced the proportion of SP cells in the population. Such treatment also inhibited invasion, migration and adhesion by B16F10-CD44+ cells. The specific interaction between CD44 and HA allowed HA-SLNs/PTX to accumulate in primary tumor tissue in lungs and to deliver the drug efficiently there. The result was a significant increase in survival rate. At the same time, HA-SLNs/PTX elicited minimal hepatotoxicity and inflammatory response. Taken together, these findings suggest that PTX-loaded HA-SLNs represent a compelling strategy for enhanced anticancer drug delivery.
Supplementary Material
Fig.S1 - S4.
Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (No. 81173011, 81422044) and the National Science & Technology Major Project of China (No. 2012ZX09304004).
Abbreviations
PTX: paclitaxel; SLN: solid lipid nanoparticles; HA: hyaluronic acid; CSCs: cancer stem cells; AST: aspartate amino-transferase; ALT: alanine amino-transferase; HPLC: high-performance liquid chromatography; LC-MS/MS: liquid chromatography tandem mass apectrometry; RT-PCR: real time-polymerase chain reaction; MTT: 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PBS: phosphate buffered saline; DAPI: 4,6-diamino-2-phenyl indole; PI: propidium iodide; DMSO: dimethyl sulfoxide; EE: entrapment efficiency; DL: drug loading; FBS: fetal bovine serum; PDI: polydispersity index; Chol: cholesterol; SPC: soy phosphatidylcholine; DDAB: dimethyldioctadecylammonium bromide; GMS: glyceryl monostearate; DID: 4-chlorobenzenesulfonate salt; CLSM: confocal laser scanning microscope; TEM: transmission electron microscopy; SP cell: side population cell; EPR: enhanced permeability and retention.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wissing SA, Kayser O, Muller RH. Solid lipid nanoparticles for parenteral drug delivery. Advanced drug delivery reviews. 2004;56:1257-72 doi:10.1016/j.addr.2003.12.002
2. Lee H, Kim JB, Park SY, Kim S-S, Kim H. Combination Effect of Paclitaxel and Hyaluronic Acid on Cancer Stem-Like Side Population Cells. Journal of Biomedical Nanotechnology. 2013;9:299-302 doi:10.1166/jbn.2013.1498
3. LaBarge MA. The difficulty of targeting cancer stem cell niches. Clinical cancer research. 2010;16:3121-9 doi:10.1158/1078-0432.CCR-09-2933
4. Wang Z, Li Y, Ahmad A, Azmi AS, Kong D, Banerjee S. et al. Targeting miRNAs involved in cancer stem cell and EMT regulation: An emerging concept in overcoming drug resistance. Drug resistance updates. 2010;13:109-18 doi:10.1016/j.drup.2010.07.001
5. Wu XD, Chen H, Wang XD. Can lung cancer stem cells be targeted for therapies? Cancer Treat Rev. 2012;38:580-8 doi: 10.1016/j.ctrv.2012.02.013
6. Dou J, Pan M, Wen P, Li Y, Tang Q, Chu L. et al. Isolation and identification of cancer stem-like cells from murine melanoma cell lines. Cellular & molecular immunology. 2007;4:467-72
7. Haraguchi N, Ohkuma M, Sakashita H, Matsuzaki S, Tanaka F, Mimori K. et al. CD133+CD44+ population efficiently enriches colon cancer initiating cells. Annals of surgical oncology. 2008;15:2927-33 doi:10.1245/s10434-008-0074-0
8. Leung ELH, Fiscus RR, Tung JW, Tin VPC, Cheng LC, Sihoe ADL. et al. Non-Small Cell Lung Cancer Cells Expressing CD44 Are Enriched for Stem Cell-Like Properties. Plos One. 2010 5. doi:10.1371/journal.pone.0014062
9. Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira SM, Garcia-Echeverria C. et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. P Natl Acad Sci USA. 2009;106:268-73 doi: 10.1073/pnas.0810956106
10. Han J, Kioi M, Chu WS, Kasperbauer JL, Strome SE, Puri RK. Identification of potential therapeutic targets in human head & neck squamous cell carcinoma. Head Neck Oncol. 2009 1. doi:10.1186/1758-3284-1-27
11. Su J, Xu XH, Huang QA, Lu MQ, Li DJ, Xue F. et al. Identification of Cancer Stem-like CD44(+) Cells in Human Nasopharyngeal Carcinoma Cell Line. Arch Med Res. 2011;42:15-21 doi:10.1016/j.arcmed.2011.01.007
12. Mine S, Fujisaki T, Kawahara C, Tabata T, Iida T, Yasuda M. et al. Hepatocyte growth factor enhances adhesion of breast cancer cells to endothelial cells in vitro through up-regulation of CD44. Exp Cell Res. 2003;288:189-97
13. Mimeault M, Batra SK. New advances on critical implications of tumor- and metastasis-initiating cells in cancer progression, treatment resistance and disease recurrence. Histol Histopathol. 2010;25:1057-73
14. Mine S, Fujisaki T, Kawahara C, Tabata T, Iida T, Yasuda M. et al. Hepatocyte growth factor enhances adhesion of breast cancer cells to endothelial cells in vitro through up-regulation of CD44. Exp Cell Res. 2003;288:189-97 doi:10.1016/S0014-4827(03)00184-8
15. Subramaniam V, Vincent IR, Gilakjan M, Jothy S. Suppression of human colon cancer tumors in nude mice by siRNA CD44 gene therapy. Exp Mol Pathol. 2007;83:332-40 doi: 10.1016/j.yexmp.2007.08.013
16. Dostalek M, Gardner I, Gurbaxani BM, Rose RH, Chetty M. Pharmacokinetics, pharmacodynamics and physiologically-based pharmacokinetic modelling of monoclonal antibodies. Clinical pharmacokinetics. 2013;52:83-124 doi:10.1007/s40262-012-0027-4
17. Ackerman ME, Pawlowski D, Wittrup KD. Effect of antigen turnover rate and expression level on antibody penetration into tumor spheroids. Molecular cancer therapeutics. 2008;7:2233-40 doi:10.1158/1535-7163.MCT-08-0067
18. Negi LM, Talegaonkar S, Jaggi M, Ahmad FJ, Iqbal Z, Khar RK. Role of CD44 in tumour progression and strategies for targeting. J Drug Target. 2012;20:561-73 doi: 10.3109/1061186x.2012.702767
19. De Stefano I, Battaglia A, Zannoni GF, Prisco MG, Fattorossi A, Travaglia D. et al. Hyaluronic acid-paclitaxel: effects of intraperitoneal administration against CD44(+) human ovarian cancer xenografts. Cancer chemotherapy and pharmacology. 2011;68:107-16 doi:10.1007/s00280-010-1462-2
20. Noble PW. Hyaluronan and its catabolic products in tissue injury and repair. Matrix Biol. 2002;21:25-9 doi: S0945-053x(01)00184-6Unsp 0945-053x/02/$Doi 10.1016/S0945-053x(01)00184-6
21. Deed R, Rooney P, Kumar P, Norton JD, Smith J, Freemont AJ. et al. Early-response gene signalling is induced by angiogenic oligosaccharides of hyaluronan in endothelial cells. Inhibition by non-angiogenic, high-molecular-weight hyaluronan. Int J Cancer. 1997;71:251-6 doi: 10.1002/(Sici)1097-0215(19970410)71:2<251::Aid-Ijc21>3.0.Co;2-J
22. Pardue EL, Ibrahim S, Ramamurthi A. Role of hyaluronan in angiogenesis and its utility to angiogenic tissue engineering. Organogenesis. 2008;4:203-14
23. Dufay Wojcicki A, Hillaireau H, Nascimento TL, Arpicco S, Taverna M, Ribes S. et al. Hyaluronic acid-bearing lipoplexes: physico-chemical characterization and in vitro targeting of the CD44 receptor. Journal of controlled release. 2012;162:545-52 doi:10.1016/j.jconrel.2012.07.015
24. Yang XY, Li YX, Li M, Zhang L, Feng LX, Zhang N. Hyaluronic acid-coated nanostructured lipid carriers for targeting paclitaxel to cancer. Cancer letters. 2013;334:338-45 doi:10.1016/j.canlet.2012.07.002
25. Bragonzi A, Boletta A, Biffi A, Muggia A, Sersale G, Cheng SH. et al. Comparison between cationic polymers and lipids in mediating systemic gene delivery to the lungs. Gene therapy. 1999;6:1995-2004 doi:10.1038/sj.gt.3301039
26. Zou SM, Erbacher P, Remy JS, Behr JP. Systemic linear polyethylenimine (L-PEI)-mediated gene delivery in the mouse. The journal of gene medicine. 2000;2:128-34 doi:10.1002/(SICI)1521-2254(200003/04)2:2<128::AID-JGM95>3.0.CO;2-W
27. Li S, Rizzo MA, Bhattacharya S, Huang L. Characterization of cationic lipid-protamine-DNA (LPD) complexes for intravenous gene delivery. Gene therapy. 1998;5:930-7 doi:10.1038/sj.gt.3300683
28. Li S, Tseng WC, Stolz DB, Wu SP, Watkins SC, Huang L. Dynamic changes in the characteristics of cationic lipidic vectors after exposure to mouse serum: implications for intravenous lipofection. Gene therapy. 1999;6:585-94 doi:10.1038/sj.gt.3300865
29. Qian H, Yu J, Li Y, Wang H, Song C, Zhang X. et al. RNA interference of metastasis-associated gene 1 inhibits metastasis of B16F10 melanoma cells in a C57BL/6 mouse model. Biology of the Cell. 2007;99:573-81 doi:10.1042/bc20060130
30. Galan-Malo P, Vela L, Gonzalo O, Calvo-Sanjuan R, Gracia-Fleta L, Naval J. et al. Cell fate after mitotic arrest in different tumor cells is determined by the balance between slippage and apoptotic threshold. Toxicol Appl Pharm. 2012;258:384-93 doi: 10.1016/j.taap.2011.11.021
31. Lu XY, Zhang FY, Qin L, Xiao FY, Liang W. Polymeric micelles as a drug delivery system enhance cytotoxicity of vinorelbine through more intercellular accumulation. Drug Delivery. 2010;17:255-62 doi: 10.3109/10717541003702769
32. Choi KY, Chung H, Min KH, Yoon HY, Kim K, Park JH. et al. Self-assembled hyaluronic acid nanoparticles for active tumor targeting. Biomaterials. 2010;31:106-14 doi:10.1016/j.biomaterials.2009.09.030
33. Chen Y, Zhu X, Zhang X, Liu B, Huang L. Nanoparticles modified with tumor-targeting scFv deliver siRNA and miRNA for cancer therapy. Molecular therapy. 2010;18:1650-6 doi:10.1038/mt.2010.136
34. Beier D, Hau P, Proescholdt M, Lohmeier A, Wischhusen J, Oefner PJ. et al. CD133(+) and CD133(-) glioblastoma-derived cancer stem cells show differential growth characteristics and molecular profiles. Cancer Res. 2007;67:4010-5 doi: 10.1158/0008-5472.Can-06-4180
35. Takaishi S, Okumura T, Tu SP, Wang SSW, Shibata W, Vigneshwaran R. et al. Identification of Gastric Cancer Stem Cells Using the Cell Surface Marker CD44. Stem Cells. 2009;27:1006-20 doi: 10.1002/Stem.30
36. Hwang-Verslues WW, Kuo WH, Chang PH, Pan CC, Wang HH, Tsai ST. et al. Multiple Lineages of Human Breast Cancer Stem/Progenitor Cells Identified by Profiling with Stem Cell Markers. Plos One. 2009 4. doi:10.1371/Journal.Pone.0008377
37. Luo Y, Dallaglio K, Chen Y, Robinson WA, Robinson SE, McCarter MD. et al. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cells. 2012;30:2100-13 doi:10.1002/stem.1193
38. Cortes-Dericks L, Froment L, Boesch R, Schmid R, Karoubi G. Cisplatin-resistant cells in malignant pleural mesothelioma cell lines show ALDH high CD44+ phenotype and sphere-forming capacity. BMC cancer. 2014;14:304. doi:10.1186/1471-2407-14-304
39. El-Khattouti A, Selimovic D, Haikel Y, Megahed M, Gomez CR, Hassan M. Identification and analysis of CD133(+) melanoma stem-like cells conferring resistance to taxol: An insight into the mechanisms of their resistance and response. Cancer letters. 2014;343:123-33 doi:10.1016/j.canlet.2013.09.024
40. Gazzaniga P, Cigna E, Panasiti V, Devirgiliis V, Bottoni U, Vincenzi B. et al. CD133 and ABCB5 as stem cell markers on sentinel lymph node from melanoma patients. European journal of surgical oncology. 2010;36:1211-4 doi:10.1016/j.ejso.2010.05.001
41. Chua C, Zaiden N, Chong KH, See SJ, Wong MC, Ang BT. et al. Characterization of a side population of astrocytoma cells in response to temozolomide. Journal of neurosurgery. 2008;109:856-66 doi:10.3171/JNS/2008/109/11/0856
42. Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nature reviews Cancer. 2005;5:275-84 doi:10.1038/nrc1590
43. Pan Y, Song QL, Lin YH, Lu N, Yu HM, Li XJ. GLB prevents tumor metastasis of Lewis lung carcinoma by inhibiting tumor adhesion actions. Acta pharmacologica Sinica. 2005;26:881-6 doi:10.1111/j.1745-7254.2005.00125.x
44. Hu TS, Liu SR, Breiter DR, Wang F, Tang Y, Sun SH. Octamer 4 small interfering RNA results in cancer stem cell-like cell apoptosis. Cancer Res. 2008;68:6533-40 doi: 10.1158/0008-5472.Can-07-6642
45. Linn DE, Yang X, Sun F, Xie Y, Chen H, Jiang R. et al. A Role for OCT4 in Tumor Initiation of Drug-Resistant Prostate Cancer Cells. Genes & cancer. 2010;1:908-16 doi:10.1177/1947601910388271
46. Guo Y, Mantel C, Hromas RA, Broxmeyer HE. Oct-4 is critical for survival/antiapoptosis of murine embryonic stem cells subjected to stress: Effects associated with Stat3/Survivin. Stem Cells. 2008;26:30-4 doi: 10.1634/stemcells.2007-0401
47. Bacus SS, Gudkov AV, Lowe M, Lyass L, Yung Y, Komarov AP. et al. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene. 2001;20:147-55 doi: 10.1038/sj.onc.1204062
48. Balekar N, Katkam NG, Nakpheng T, Jehtae K, Srichana T. Evaluation of the wound healing potential of Wedelia trilobata (L.) leaves. J Ethnopharmacol. 2012;141:817-24 doi: 10.1016/j.jep.2012.03.019
49. Wei X, Chen X, Ying M, Lu W. Brain tumor-targeted drug delivery strategies. Acta Pharmaceutica Sinica B. 2014;4:193-201 doi:10.1016/j.apsb.2014.03.001
50. Shi S, Han L, Gong T, Zhang Z, Sun X. Systemic delivery of microRNA-34a for cancer stem cell therapy. Angew Chem Int Ed Engl. 2013;52:3901-5 doi:10.1002/anie.201208077
51. Atlasi Y, Mowla SJ, Ziaee SAM, Bahrami AR. OCT-4, an embryonic stem cell marker, is highly expressed in bladder cancer. Int J Cancer. 2007;120:1598-602 doi: 10.1002/Ijc.22508
52. Audouy SAL, de Leij LFMH, Hoekstra D, Molema G. In vivo characteristics of cationic liposomes as delivery vectors for gene therapy. Pharmaceut Res. 2002;19:1599-605 doi: 10.1023/A:1020989709019
53. Williams J, Lansdown R, Sweitzer R, Romanowski M, LaBell R, Ramaswami R. et al. Nanoparticle drug delivery system for intravenous delivery of topoisomerase inhibitors. Journal of controlled release: official journal of the Controlled Release Society. 2003;91:167-72
54. Cuenca AG, Jiang H, Hochwald SN, Delano M, Cance WG, Grobmyer SR. Emerging implications of nanotechnology on cancer diagnostics and therapeutics. Cancer. 2006;107:459-66 doi:10.1002/cncr.22035
55. Zhang W, Shi Y, Chen Y, Hao J, Sha X, Fang X. The potential of Pluronic polymeric micelles encapsulated with paclitaxel for the treatment of melanoma using subcutaneous and pulmonary metastatic mice models. Biomaterials. 2011;32:5934-44 doi:10.1016/j.biomaterials.2011.04.075
56. Shen J, Yin Q, Chen L, Zhang Z, Li Y. Co-delivery of paclitaxel and survivin shRNA by pluronic P85-PEI/TPGS complex nanoparticles to overcome drug resistance in lung cancer. Biomaterials. 2012;33:8613-24 doi:10.1016/j.biomaterials.2012.08.007
Author contact
![]() Corresponding author: Xun Sun, West China School of Pharmacy, Sichuan University, Chengdu 610041, Sichuan, China. Tel: 86-28-85502307; fax: 86-28-85501615. E-mail address: sunxunedu.cn (X.Sun)
Corresponding author: Xun Sun, West China School of Pharmacy, Sichuan University, Chengdu 610041, Sichuan, China. Tel: 86-28-85502307; fax: 86-28-85501615. E-mail address: sunxunedu.cn (X.Sun)
Citation styles
APA
Shen, H., Shi, S., Zhang, Z., Gong, T., Sun, X. (2015). Coating Solid Lipid Nanoparticles with Hyaluronic Acid Enhances Antitumor Activity against Melanoma Stem-like Cells. Theranostics, 5(7), 755-771. https://doi.org/10.7150/thno.10804.
ACS
Shen, H.; Shi, S.; Zhang, Z.; Gong, T.; Sun, X. Coating Solid Lipid Nanoparticles with Hyaluronic Acid Enhances Antitumor Activity against Melanoma Stem-like Cells. Theranostics 2015, 5 (7), 755-771. DOI: 10.7150/thno.10804.
NLM
Shen H, Shi S, Zhang Z, Gong T, Sun X. Coating Solid Lipid Nanoparticles with Hyaluronic Acid Enhances Antitumor Activity against Melanoma Stem-like Cells. Theranostics 2015; 5(7):755-771. doi:10.7150/thno.10804. https://www.thno.org/v05p0755.htm
CSE
Shen H, Shi S, Zhang Z, Gong T, Sun X. 2015. Coating Solid Lipid Nanoparticles with Hyaluronic Acid Enhances Antitumor Activity against Melanoma Stem-like Cells. Theranostics. 5(7):755-771.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.