Impact Factor
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
Introduction
Results
Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(6):609-617. doi:10.7150/thno.11222 This issue Cite
Research Paper
Charge and Hydrophobicity Effects of NIR Fluorophores on Bone-Specific Imaging
Kai Bao1,2*, Khaled A. Nasr1,3,*, Hoon Hyun1, Jeong Heon Lee1, Julien Gravier1, Summer L. Gibbs4 ![]() , Hak Soo Choi1
, Hak Soo Choi1 ![]()
1. Division of Hematology/Oncology, Department of Medicine, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA 02215;
2. Key Laboratory of Structure-based Drug Design and Discovery, Ministry of Education, Shenyang Pharmaceutical University, Shenyang, China, 110016;
3. Advanced Imaging Research Center, The University of Texas Southwestern Medical Center, Dallas, TX 75390;
4. Department of Biomedical Engineering, OHSU Center for Spatial Systems Biology, Knight Cancer Institute, Oregon Health & Science University, Portland, OR 97201.
*These authors contributed equally to this work.
Received 2014-12-2; Accepted 2015-1-23; Published 2015-3-1
Abstract

Recent advances in near-infrared (NIR) fluorescence imaging enabled real-time intraoperative detection of bone metastases, bone growth, and tissue microcalcification. Pamidronate (PAM) has been widely used for this purpose because of its high binding affinity toward bone and remarkable therapeutic effects. Herein we describe the development of a series of PAM-conjugated NIR fluorophores that varied in net charges and hydrophobicity, and compared their bone targeting efficiency, biodistribution, and blood clearance. Since the targeting moiety, PAM, is highly negatively charged but small, the overall in vivo bone targeting and biodistribution were mediated by the physicochemical properties of conjugated fluorophores.
Keywords: Near-infrared fluorophores, Bone targeting, Charge and hydrophobicity, Microcalcification, Zwitterionic fluorophores.
Introduction
Phosphonate derivatives have been widely used as targeting and therapeutic agents for bone imaging and bony disease treatments [1,2]. Bisphosphonates are known to exhibit high affinity for bone surfaces minerals and calcium salts [3]. Bisphosphonates have also been used as a bone-targeting vector in a variety of contrast agents for the detection and therapy of numerous bone diseases [4-6]. Clinically bone tissue imaging is based almost exclusively on x-ray or radiochemical methods. Recently the state-of-the-art imaging modalities such as computed tomography (CT), magnetic resonance imaging (MRI), single photon emission computed tomography (SPECT), positron emission tomography (PET), and their combinations have been used to detect changes in bone by using radioactive metal ions chelators conjugated to bone-binding bisphosphonates [7-10]. These methodologies have proven useful for diagnostic imaging; however, real-time imaging cannot be readily achieved using CT, MRI, SPECT or PET imaging. Real-time in vivo optical imaging using near-infrared (NIR) fluorescent light has received considerable interest in imaging of animal and human tissues including nerve [11], vasculature [12,13], tissue perfusion [14,15], lymph nodes [16-19], ureter [20-22], bile duct [23,24], and various cancers [25-28]. Additionally the NIR window (650-900 nm) has minimized tissue scattering, absorption, and autofluorescence [29]; therefore, it has the potential to provide highly sensitive, rapid, and non-radioactive bone imaging.
Previously we have reported the development of bone-specific NIR fluorophores by conjugating conventional dyes IRDye78 [30] and IRDye800-CW [31,32] with pamidronate (PAM). Although both Pam78 and Pam800 exhibit rapid and specific binding to hydroxyapatite (HA) in vitro and in vivo, the detailed molecular effect of conjugated fluorophores on bone targeting potency and in vivo performance has not been investigated.
Indeed, when conjugated with zwitterionic fluorophores, relatively large targeting ligands such as peptides and proteins were dominated by overall molecular charge and charge distribution, which played an important role to improve signal-to-background ratio (SBR) for fluorescence imaging [12,25]. We have also been systematically probing the relationship among the hydrodynamic diameter, shape, charge, and hydrophobicity of NIR fluorophores on in vivo targeting and biodistribution [33-35].
In this study, we hypothesize that zwitterionic or positively charged NIR fluorophores may improve or change the in vitro and in vivo performance of targeted small molecule ligands. Bone-specific bisphosphonate ligand PAM was used to conjugate with three spectrally similar heptamethine NIR fluorophores, and their targeting performance was explored on the basis of the final physicochemical properties including net charge, hydrophobicity, and polarity. The optical property and in vitro HA-binding of PAM-NIR fluorophore were also performed systematically.
Results
Synthesis of PAM-NIR fluorophores. PAM-ZW800-1, PAM-CW800, and PAM-ZW800-3a were synthesized by conjugating the protected PAM with ZW800-1 [12], IRDye800-CW [32], and ZW800-3a [12], respectively. First, as shown in Fig. 1a, the tetramethyl-protected pamidronate 6 was synthesized via a modified one-pot synthesis from our previously reported method [32]. The multi-step synthetic scheme initiated by reacting readily available Z-β-Alanine 1 with oxalyl chloride to produce an acyl chloride derivative 2 without isolation. In the following one-pot Arbuzov reaction, the acyl chloride 2 was reacted with trimethylphosphite to yield a phosphite derivative 3. A second crucial Arbuzov reaction with dimethyl(trimethylsilyl)phosphite led to the formation of the Cbz-protected tetramethylphosphonate 5. After catalytic hydrogenolysis, the tetramethyl protected pamidronate 6 was obtained without any isomerization, a limitation of the previously published method [31,32]. The conjugation of the NIR fluorophores ZW800-1, CW800, and ZW800-3a to compound 6 was completed by using dipyrrolidino(N-succinimidyloxy)carbenium hexafluorophosphate (HSPyU) and N,N-diisopropylethylamine (DIEA) in dry dimethyl sulfoxide (DMSO). In the fluorophore conjugation reaction, hydrolysis of the four contained methyl groups using trimethylsilyl bromide in dimethylformamide (DMF) yielded PAM-ZW800-1, PAM-CW800, and PAM-ZW800-3a (see details in Materials and Methods). The final compounds were purified by preparative HPLC and analyzed by mass spectrometry (Supplementary Material: Fig. S1). Prior to measurement of optical properties and in vivo performance, each PAM-NIR fluorophore was purified to ≥ 90% as measured using a photodiode array (PDA) at 210 nm absorbance.
Synthesis and characterization of PAM-ZW800-1, PAM-CW800 and PAM-ZW800-3a NIR fluorophores. (a) Synthetic scheme for compound 6 and PAM-NIR fluorophores. (b) Chemical structures and physicochemical properties of NIR fluorophores having systematically varying net charges. 3D molecular structure was depicted using MarvinSketch 6.2.2 (ChemAxon). Red: negative charge; blue: positive charge; and gray: hydrophobicity. LogD = partition coefficient; PSA = polar surface area; QY = quantum yield; ε = extinction coefficient.
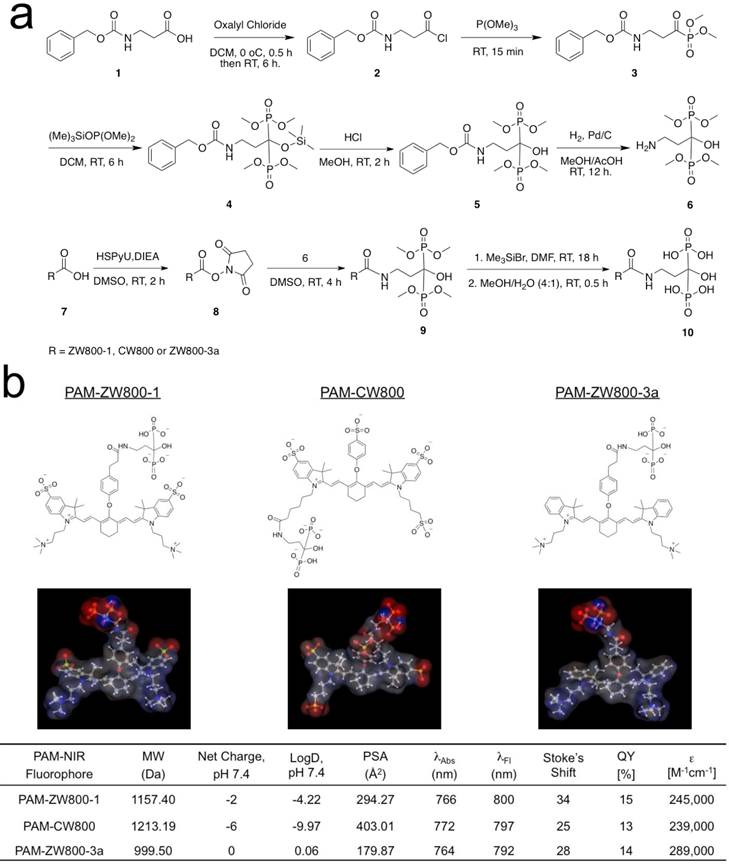
Characterizations of PAM-NIR fluorophores. Physicochemical and optical properties of the synthesized PAM-NIR fluorophores are summarized in Fig. 1b and Supplementary Material: Fig. S2. By varying the side chains of the polymethine core, it was possible to systematically modify the electron resonance, hydrophobicity and polarity. The 3D structures display differences in charge and hydrophobicity distributions over the molecular surface. Because of the flexible “O” atom on the meso carbon, there is a roughly 60° angle between the cyanine skeleton and the phenoxy propionic acid group, after conjugation with the PAM. The partition coefficient (logD values at pH 7.4) reflects the decreased molecular hydrophobicity after PAM conjugation in the order of PAM-ZW800-3a (0.06), PAM-ZW800-1 (-4.22), and PAM-CW800 (-9.97). The conjugation of PAM with the heptamethine fluorophores had minimal impact on the spectral properties of each NIR fluorophore. All PAM-NIR fluorophores exhibited maximum absorption around 770 nm, maximum fluorescence emission ≥ 790 nm, and relatively high extinction coefficients and quantum yields in 100% serum, which together minimize tissue autofluorescence and maximize fluorescence signal during intraoperative imaging.
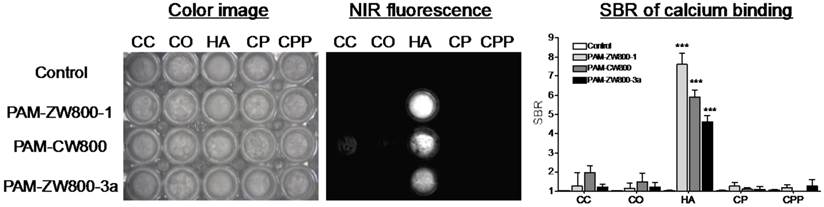
In vitro calcium salt binding specificity. The specificity of PAM-ZW800-1, PAM-CW800, and PAM-ZW800-3a for hydroxyapatite (HA) over other calcium salts was determined using NIR fluorescence imaging. 5 μM of each PAM-NIR fluorophore was incubated with HA and calcium carbonate (CC), calcium oxalate (CO), calcium phosphate (CP), and calcium pyrophosphate (CPP) for 30 min at room temperature with constant shaking, then washed 3 times with a 100-fold excess of phosphate-buffered saline (PBS). As shown in Fig. 2, all the fluorophores demonstrated strong affinity for HA compared to other calcium salts. In addition, PAM-ZW800-1 exhibited higher SBR when binding to HA than the other two PAM fluorophore derivatives (Fig. 2). This increase in SBR for PAM-ZW800-1 and PAM-CW800 over PAM-ZW800-3a can be attributed to the improved calcium binding via salt formation through the phenyl sulfonate moieties in the fluorophore backbone [36,37]. In addition, the hydroxyl group of HA contributes to the formation of strong hydrogen bonds with anions of fluorophores, which increased binding affinity significantly [36]. Of note, PAM-ZW800-1 had 8-fold higher specificity toward HA over other calcium salts, and permits NIR fluorescence detection of HA with high sensitivity.
Specificity of PAM-ZW800-1, PAM-CW800 and PAM-ZW800-3a for hydroxyapatite (HA) over other calcium salts. Each fluorophore was well mixed with calcium salts for 30 min and washed with excess of PBS prior to NIR fluorescence imaging. SBR was calculated by the fluorescence intensity of each fluorophore sample versus the signal intensity of each control sample (n = 3, mean ± s.d.). ***P < 0.001. Abbreviations: CC = calcium carbonate; CO = calcium oxalate; HA = hydroxyapatite; CP = calcium phosphate and CPP = calcium pyrophosphate.
In vivo NIR fluorescence imaging of bone tissue in mice. 10 nmol of each PAM-NIR fluorophore was administered intravenously to mice and allowed 4 h to distribute and clear prior to fluorescence imaging. As shown in Fig. 3a, PAM-CW800 provided high-sensitivity in the normal bones including jawbone, sternum, ribs, spinal cord, and tailbones. Interestingly PAM-ZW800-1 signals were found both in bone and cartilage tissues including knee joints, rib cage, and intervertebral discs. The increased cartilage signal (arrowheads) is likely due to the cartilage affinity of quaternary ammonium moiety in the ZW800-1 fluorophore [38]. One the other hand, PAM-ZW800-3a fluorescent signals were found not only in the bone and cartilage, but also in the liver with a high portion due to its high hydrophobicity. Control experiments were also performed with NIR fluorophores without PAM to show the inherent biodistribution and clearance of unconjugated fluorophores [12]. ZW800-1 showed high renal clearance, while CW800 and ZW800-3a represented elevated nonspecific background signals in the liver and skin (Supplementary Material: Fig. S3).
In vivo biodistribution and bone specific imaging using PAM-NIR fluorophores in mice. (a) 10 nmol (0.4 mg/kg) of each PAM-NIR conjugate was injected intravenously into 25 g nude mice 4 h prior to imaging. Arrowheads indicate cartilage tissues. Li = liver, scale bars = 1 cm. All NIR fluorescence images were collected with the same exposure time and are displayed with identical normalization. (b) Blood clearance (%ID/g) and blood half-life (mean ± 95% confidence intervals) in mice (n = 3, mean ± s.d.). (c) Organ biodistribution in mice. Animals were sacrificed at 30, 60, 90, 120, 180, and 240 min, and major tissues and organs were resected and imaged (n = 3, mean ± s.d.). *P < 0.05, **P < 0.01, and ***P < 0.001.
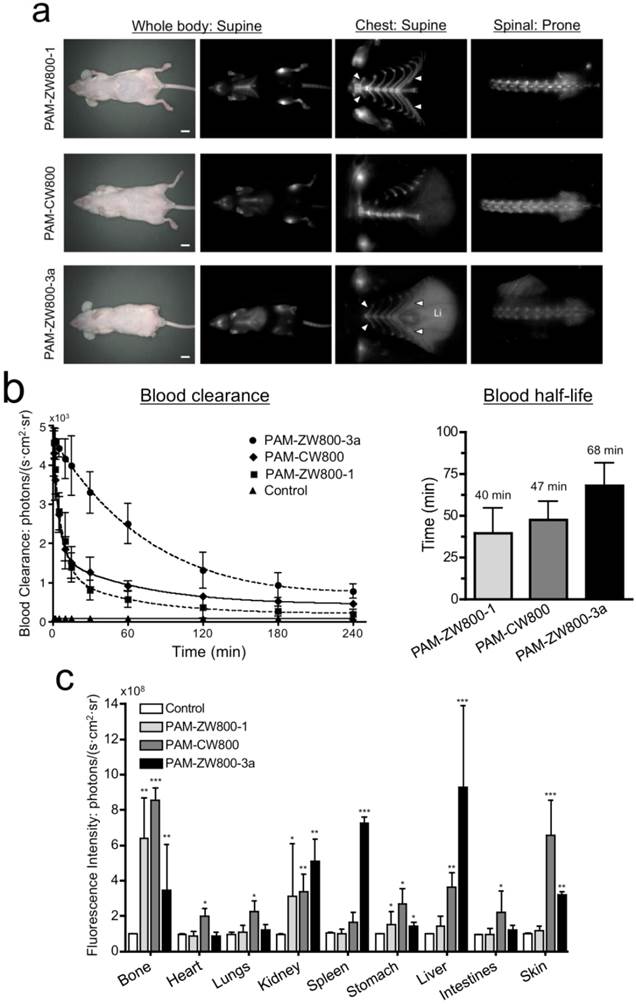
Blood half-life and organ biodistribution. Blood half-life of each PAM-NIR fluorophore was measured during the 4 h biodistribution and clearance time (Fig. 3b). The elimination half-life (t1/2β) of PAM-ZW800-1 and PAM-CW800 were similar and found to be 40 min and 47 min, respectively. PAM-ZW800-3a showed an extended t1/2 of 68 min, which was longer than that of fluorophore itself (28 min) due to the additional negative charges following conjugation to the PAM molecule [12]. As shown in Fig. 3c, PAM-ZW800-1 was eliminated from the body through renal clearance with minimal non-specific uptake in other organs and tissues, while specific fluorescence signal was seen in bone and cartilage. PAM-CW800 demonstrated bone specific targeting, however the uptake in skin (***P < 0.001) and intestine (*P < 0.05) was much higher than the other two agents. As demonstrated through fluorescence imaging in Fig. 3a, PAM-ZW800-3a showed bone targeting fluorescence signal, but also demonstrated significant uptake in the liver (***P < 0.001). These results establish the relationship between the surface charge and hydrophobicity of targeted small molecule fluorophores and the biodistribution, blood half-life, organ retention and clearance route following intravenous administration in vivo.
Discussion
Physicochemical properties of small molecules determine the fate of an intravenously injected fluorophore in the body [28,33-35]. Total surface charge and hydrophobicity are of significant importance to design targeted contrast agents for tissue-specific imaging and optimized in vivo performance [25]. PAM is a negatively charged small molecule with a molecular weight of 235.07 Da, Polar surface area (PSA) of 161.31 Å2, and logD of -6.92 at physiological pH. Comparison of the net charge at pH 7.4 between free fluorophores and PAM-NIR fluorophore conjugates were performed by using MarvinSketch 6.2.2 software, and PAM imparts an additional net charge of -3 to the total molecular charge and significantly increases the overall hydrophilicity and polarity. However, as demonstrated in the current work the optical properties of the conjugated NIR fluorophores were unchanged in the presence of PAM. When the in vitro specificity was examined, all PAM-NIR fluorophores demonstrated strong binding affinity to HA with significantly higher SBR by comparison to the other calcium salts. This indicates that PAM plays a dominant role in the binding and the changes in physicochemical properties have limited impact on the functionality of the ligand. Furthermore, the intraoperative mouse imaging clearly indicates all PAM conjugated NIR fluorophores had strong binding affinity for bone.
Although PAM maintained its ligand functionality in vitro and in vivo, the biodistribution and clearance patterns of the conjugates were strongly governed by the NIR fluorophores because the size of the targeting ligand is smaller than that of the effector domain. Since ZW800-1 has the balanced surface charge and relative hydrophilic property, PAM-ZW800-1 excreted to urine after active uptake into the skeleton. Additionally, the presence of 2 quaternary ammonium cations in the ZW800-1 molecule also led the molecule to accumulate in cartilage due to the strong affinity of these positively charge moiety for the negatively charged glycosaminoglycans [38]. The negatively charged PAM-CW800 (-6) accumulated in skin, liver, and intestines in addition to the strong uptake in bone tissue 4 h post-intravenous injection, which was enhanced by the presence of three phenyl sulfonate groups on the surface of the molecule [36,37]. In the case of PAM-ZW800-3a, the absence of phenyl sulfonate groups and presence of strong quaternary ammonium cations enhanced the affinity of ZW800-3a for cartilage. In addition, the logD value at pH 7.4 for PAM-ZW800-3a increased to 0.06, which resulted in high nonspecific uptake in the liver, spleen, and skin.
In conclusion, three bone-targeting nanoprobes that varied in net charges and hydrophobicity were developed systematically by conjugating PAM on the backbone of different NIR fluorophores. The in vivo data prove that overall molecular net charge and hydrophobicity of targeted fluorophores play a significant role in the biodistribution, blood half-life, organ accumulation and clearance route of intravenously administered contrast agents. Thus the overall molecular net charge and hydrophobicity will also have strong influence on the targetability, specificity and fluorophore selection for targeted contrast agents and is therefore an important consideration in the development of next generation organ-specific imaging probes and therapeutic agents. However, it should be noted that the introduction of multivalent ligands on the surface of small molecules or nanoparticles would change the pattern of biodistribution significantly because the conjugated ligands will be dominant over the effector domain [28,33].
Materials and Methods
Reagents: All chemicals were American Chemical Society grade or higher and commercially available unless noted otherwise. IRDye800CW-NHS was purchased from LI-COR (Lincoln, NE) and stored under nitrogen at -80°C. HPLC-grade solvents were purchased from Fisher Scientific (Pittsburgh, PA, USA) and Sigma-Aldrich (Saint Louis, MO, USA). Ultra dry DMSO was purchased from Acros Organics (Geel, Belgium).
NMR Spectroscopy: NMR spectra were recorded on a Varian 400 MHz NMR system. Chemical shifts, d, are represented in part per million (ppm) and coupling constants, J, in Hertz (Hz). Tetramethylsilane (TMS, 0 ppm) was used as the internal standard for proton NMR.
One-pot synthesis of protected bisphosphonate 5: 3-[(benzyloxycarbonyl)amino]-propionic acid (4.46 g, 20 mmol) was dissolved in 40 mL of dichloromethane and cooled down to 0° C under a nitrogen atmosphere. Oxalyl Chloride (8.57 mL, 100 mmol) was added and the reaction mixture was stirred at 0° C for 30 minutes, followed by stirring at room temperature for 6 h. The solvent was evaporated under vacuum and washed with additional dichloromethane to remove the excess oxalyl chloride. The product was dried under vacuum (0.001 Torr) for 2 h and trimethyl phosphite (2.73 g, 22 mmol) was added dropwise at 0° C over 5 minutes. The resulting mixture was allowed to reach room temperature after which it was stirred for an additional 15 minutes. Following removal of the volatile material under reduced pressure, dimethyl trimethylsilyl phosphite (4 g, 22 mmol) in dichloromethane (20 mL) was added at 0° C under a nitrogen atmosphere. The reaction mixture was stirred at room temperature for 6 h. The solvent was evaporated under vacuum and the resulting mixture was treated with 3 M hydrochloric acid (HCl, 40 mL) and methanol (20 mL) at room temperature for 2 h. The reaction mixture was extracted with ether (3 x 30 mL), washed with water and brine, and then dried over magnesium sulfate (MgSO4). The volatile organic solvent was evaporated under reduced pressure and washed with hexane (3 x 10 mL) to give dimethyl [3-(Benzyloxycarbonylamino)-1-(dimethoxyphosphoryl)-1-hydroxypropyl] phosphonate 5 as pale yellow oil (7.48 g, 88% yield). 1H NMR (400 MHz, CD3OD) δ 2.25 (m, 2H), 3.50 (t, J=6.0 Hz, 2H), 3.83 (s, 12H), 5.07 (s, 2H), 5.25 (brs, 1H), 5.61 (brs, 1H), 7.31-7.33 (m, 5H). Accurate mass TOF HRMS (m/z) [M+H]+ calculated for C15H26NO9P2, 426.1083, found 426.1085.
Synthesis of unprotected bisphosphonate 6: Dimethyl [3-(Benzyloxycarbonylamino)-1-(dimethoxyphosphoryl)-1-hydroxypropyl] phosphonate 5 (4.25 g, 10 mmol) was placed in a 500-mL Parr bottle under nitrogen and dissolved in 50 mL of cold denatured methanol. Palladium on activated carbon (0.27 g, 10 wt.%) was added carefully and the Parr shaker apparatus was assembled. The reaction was shaken at 50 psi hydrogen at room temperature until hydrogen uptake was complete (6-12 h). The palladium on activated carbon was filtered over a Celite pad and the solvent was evaporated under reduced pressure to give dimethyl [3-amino-1-(dimethoxyphosphoryl)-1-hydroxypropyl] phosphonate 6 as yellow solid (2.62 g, 90% yield). 1H NMR (400 MHz, CD3OD) δ 1.33 (t, J=7.4 Hz, 2H), 2.38 (brs, 2H), 2.49 (m, 1H), 3.21 (m, 2H), 3.30-3.90 (m, 12H). Accurate mass TOF HRMS (m/z) [M+H]+ calculated for C7H20NO7P2, 292.0715, found 292.0716.
Synthesis of PAM-ZW800-1, PAM-CW800, and PAM-ZW800-3a: To a solution of 10 mg of the NIR fluorophore in 0.5 mL of anhydrous DMSO, 3 equivalents (eq) of N,N-Diisopropylethylamine (DIEA) and 1.5 eq of Dipyrrolidino (N-succinimidyloxy) carbenium hexafluorophosphate (HSPyU) were added. The pH value of the mixture was maintained between 8-10 by adding additional DIEA when required. The reaction was stirred at room temperature in the dark for 2 h followed by the addition of 2 eq. of compound 6. After stirring at room temperature in the dark for 4 h, the reaction mixture was poured over 5 mL ice-cold water and purified by reverse-phase HPLC to give compound 9 as the green solid after concentration under vacuum. 5 eq. Trimethylsilyl bromide were added slowly to a solution of 5 mg compound 9 in 0.2 mL of dry DMF at 0 °C under nitrogen atmosphere. The reaction mixture was stirred at room temperature for 18 h in the dark and the solvent was evaporated off to dryness. Then 0.2 mL of CH3OH-H2O (4:1) was added to yield a bright green solution, which was stirred for 30 min at room temperature and purified by reverse-phase HPLC to give the PAM-NIR fluorophores (compound 10) as the green solid. PAM-ZW800-1: green solid (3.7 mg, 79% yield), accurate mass TOF HRMS (m/z) [M+2H]2+/2 calculated for C54H77N5O15P2S2 580.7161, found 580.7159. PAM-CW800, green solid (3.8 mg, 81% yield), accurate mass TOF HRMS (m/z) [M-2H]2-/2 calculated for C49H61N3O21P2S4 , 608.6084, found [M+H]+ 608.6087. PAM-ZW800-3a, green solid (3.4 mg, 74% yield), accurate mass TOF HRMS (m/z) [M+2H]2+/2 calculated for C54H77N5O9P2, 500.7593, found 500.7592.
Reverse phase HPLC: HPLC purification of compounds was performed on a Waters (Milford, MA, USA) prepHPLC 150 ml fluid handling unit equipped with a manual injector (Rheodyme 3725i) and a 2487 dual wavelength absorbance detector outfitted with a semi-preparative flow cell. A flow splitter (Upchurch Scientific, Oak Harbor, WA, USA) diverted a portion of the eluate into an evaporative light scatter detector (ELSD, Richards Scientific, Novato, CA, USA) with the nebulizer modified to reduce band broadening at low flow rates while the other part flowed into a fraction collector. The ELSD was set to 42 °C, with a nitrogen pressure at 3.5 bar and a gain of 8. HPLC purification was completed using a Symmetry Prep C18 column (19 x 150 mm, 7 µm particle size) and a dual-wavelength absorbance detector (254 and 700 nm). Solvent A was water with 10 mM TEAA and solvent B was absolute methanol. The purification utilized a linear gradient from 0% to 70% solvent B over 35 min. Mobile phase flow rate was 15 mL/min.
ES-TOF mass spectroscopic analysis: The purity of all compounds was measured using ultra-performance liquid chromatography (UPLC, Waters) combined with simultaneous ELSD, absorbance (photodiode array), fluorescence and electrospray time-of-flight (ES-TOF) mass spectrometry (MS). UPLC analyses were carried out using a Waters ACQUITY UPLC HSS C18 column (2.1x50 mm, 1.8 µm) and Xevo G2 QTof detector connected to a PC with a MassLynx 4.1 workstation. The mobile phase was solvent A = 0.1% formic acid in water, solvent B = 0.1% formic acid in acetonitrile with a linear gradient from 10% to 90% (from A to B for 5 minutes). The flow rate was 0.6 mL/min and all compounds were identified by mass to charge ratio.
Optical and physicochemical property analyses: All optical measurements were performed at 37°C in 100% fetal bovine serum (FBS), buffered with 50 mM HEPES, pH 7.4. The partition coefficient (logD at pH 7.4) of each fluorescent conjugate was calculated using JChem calculator plugins (ChemAxon, Budapest, Hungary). Fluorescence quantum yield (QY) was measured for each fluorescent conjugate using ICG in DMSO (QY 13%) as a calibration standard under conditions of matched absorbance at 765 nm. Absorbance and fluorescence spectra for all fluorescent conjugates were collected using online fiberoptic HR2000 absorbance (200-1100 nm) and USB2000FL fluorescence (350-1,000 nm) spectrometers (Ocean Optics, Dunedin, FL) with NIR excitation provided by a 770 nm NIR laser diode light source (Electro Optical Components, Santa Rosa, CA).
In vitro calcium salts specificity experiments: Calcium carbonate, calcium oxalate, hydroxyapatite, calcium phosphate and calcium pyrophosphate salts (25 mg/mL salts, 0.2-0.25 M calcium ions) were incubated separately with 5 µM of PAM-ZW800-1, PAM-CW800 and PAM-ZW800-3a in PBS. The calcium salts were vortexed continuously with the fluorophore conjugates at room temperature for 30 min. The crystals, calcium salts, were washed three times with a 100-fold excess of PBS, centrifuged and visualized in 96-well plate using our fluorescence imaging system [37]. All NIR fluorescence images were collected at identical exposure times and are displayed with equal normalization.
In vivo biodistribution and clearance: Animals were housed in an AAALAC-certified facility and were studied under the supervision of Beth Israel Deaconess Medical Center's IACUC in accordance with the approved institutional protocol (#101-2011). Male CD-1 mice weighing roughly 20 g were purchased from Charles River Laboratories (Wilmington, MA) and male NCr nu/nu mice 5 to 6 weeks of age weighing 22 g ± 3 g were purchased from Taconic Farms (Germantown, NY). Animals were anesthetized with 100 mg/kg ketamine and 10 mg/kg xylazine intraperitoneally (Webster Veterinary, Fort Devens, MA). 10 - 25 nmol (0.4 - 1.0 mg/kg) of the NIR fluorophore conjugate in saline were administered intravenously, and animals were imaged with our real-time intraoperative imaging system as described in detail previously [12,25]. For fluorescence excitation, 14 mWcm-2 of 745 to 779 nm-filtered light was used, and for emission, light was filtered using a 800 to 848 nm band pass filter. For each experiment, camera exposure time and image normalization was held constant. To quantify the blood clearance rate and urinary excretion, intermittent sampling from the tail vein was performed over the 4 h period following intravenous administration. Approximately 10-20 μL of blood were collected using glass capillary tubes at the following time points: 0, 1, 2, 5, 10, 15, 30, 60, 90, 120, 180, and 240 min. Custom intraoperative fluorescence imaging system was used to measure the fluorescence intensity of each sample, and the concentration was calculated based on the image SBR using a standard curve for each fluorophore. To measure total body excretion in mice, animals were sacrificed at 30, 60, 90, 120, 180, and 240 min, and major tissues and organs were resected and imaged. Using capillary tubes, the amount of NIR fluorophore in each organ/tissue was quantified by measuring fluorescent intensity (photons/s/cm2/sr).
Quantitative analysis: At each time point, the fluorescence and background intensity of a region of interest (ROI) over each tissue was quantified using custom imaging software and ImageJ v1.48 (NIH, Bethesda, MD). The signal-to-background ratio (SBR) was calculated as SBR = fluorescence/background, where background is the signal intensity of neighboring tissues such as muscle or skin obtained over the period of imaging time. All NIR fluorescence images for a particular fluorophore were normalized identically for all conditions of an experiment. At least three animals were analyzed at each time point. A one-way ANOVA followed by Tukey's multiple comparisons test were used to assess the statistical differences between multiple groups. A P value of less than 0.05 was considered significant: *P < 0.05, **P < 0.01, and ***P < 0.001. Results are presented as mean ± standard deviation (s.d.) where curve fitting was performed using Prism version 4.0a software (GraphPad, San Diego, CA).
Supplementary Material
Figures S1 - S3.
Acknowledgements
We thank Wolfgang Maison for many helpful discussions and experimental support. This study was supported by the NIH/NIBIB grant #R01-EB-011523; the contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Author Contributions
KB, KAN, HH, GP, YA, JHL, and SLG performed the experiments. KB, KAN, HH, and HSC reviewed, analyzed, and interpreted the data. KB, KAN, SLG, and HSC wrote the paper. All authors discussed the results and commented on the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Jung A, Bisaz S, Fleisch H. The binding of pyrophosphate and two diphosphonates by hydroxyapatite crystals. Calcified tissue research. 1973;11:269-80
2. Fleisch H. Bisphosphonates--history and experimental basis. Bone. 1987;8(Suppl 1):S23-8
3. van Beek ER, Lowik CW, Ebetino FH, Papapoulos SE. Binding and antiresorptive properties of heterocycle-containing bisphosphonate analogs: structure-activity relationships. Bone. 1998;23:437-42
4. Lipton A, Theriault RL, Hortobagyi GN, Simeone J, Knight RD, Mellars K. et al. Pamidronate prevents skeletal complications and is effective palliative treatment in women with breast carcinoma and osteolytic bone metastases: long term follow-up of two randomized, placebo-controlled trials. Cancer. 2000;88:1082-90
5. Ogawa K, Mukai T, Arano Y, Ono M, Hanaoka H, Ishino S. et al. Development of a rhenium-186-labeled MAG3-conjugated bisphosphonate for the palliation of metastatic bone pain based on the concept of bifunctional radiopharmaceuticals. Bioconjugate chemistry. 2005;16:751-7
6. Lam MG, de Klerk JM, van Rijk PP, Zonnenberg BA. Bone seeking radiopharmaceuticals for palliation of pain in cancer patients with osseous metastases. Anti-cancer agents in medicinal chemistry. 2007;7:381-97
7. Vitha T, Kubicek V, Hermann P, Kolar ZI, Wolterbeek HT, Peters JA. et al. Complexes of DOTA-bisphosphonate conjugates: probes for determination of adsorption capacity and affinity constants of hydroxyapatite. Langmuir: the ACS journal of surfaces and colloids. 2008;24:1952-8
8. Qiu L, Cheng W, Lin J, Luo S, Xue L, Pan J. Synthesis and biological evaluation of novel (99m)Tc-labelled bisphosphonates as superior bone imaging agents. Molecules. 2011;16:6165-78
9. Ogawa K, Washiyama K. Bone target radiotracers for palliative therapy of bone metastases. Current medicinal chemistry. 2012;19:3290-300
10. Rasulova N, Lyubshin V, Arybzhanov D, Sagdullaev S, Krylov V, Khodjibekov M. Optimal timing of bisphosphonate administration in combination with samarium-153 oxabifore in the treatment of painful metastatic bone disease. World journal of nuclear medicine. 2013;12:14-8
11. Park MH, Hyun H, Ashitate Y, Wada H, Park G, Lee JH. et al. Prototype nerve-specific near-infrared fluorophores. Theranostics. 2014;4:823-33
12. Choi HS, Nasr K, Alyabyev S, Feith D, Lee JH, Kim SH. et al. Synthesis and in vivo fate of zwitterionic near-infrared fluorophores. Angew Chem Int Ed Engl. 2011;50:6258-63
13. Hyun H, Bordo MW, Nasr K, Feith D, Lee JH, Kim SH. et al. cGMP-Compatible preparative scale synthesis of near-infrared fluorophores. Contrast Media Mol Imaging. 2012;7:516-24
14. Ashitate Y, Vooght CS, Hutteman M, Oketokoun R, Choi HS, Frangioni JV. Simultaneous assessment of luminal integrity and vascular perfusion of the gastrointestinal tract using dual-channel near-infrared fluorescence. Mol Imaging. 2012;11:301-8
15. Ashitate Y, Lee BT, Ngo LH, Laurence RG, Hutteman M, Oketokoun R. et al. Quantitative assessment of nipple perfusion with near-infrared fluorescence imaging. Ann Plast Surg. 2013;70:149-53
16. Tanaka E, Choi HS, Fujii H, Bawendi MG, Frangioni JV. Image-guided oncologic surgery using invisible light: completed pre-clinical development for sentinel lymph node mapping. Ann Surg Oncol. 2006;13:1671-81
17. Mieog JS, Troyan SL, Hutteman M, Donohoe KJ, van der Vorst JR, Stockdale A. et al. Toward optimization of imaging system and lymphatic tracer for near-infrared fluorescent sentinel lymph node mapping in breast cancer. Ann Surg Oncol. 2011;18:2483-91
18. Liu F, Lunsford EP, Tong J, Ashitate Y, Gibbs SL, Yu J. et al. Real-time monitoring of tumorigenesis, dissemination, & drug response in a preclinical model of lymphangioleiomyomatosis/tuberous sclerosis complex. PLoS One. 2012;7:e38589
19. Ashitate Y, Hyun H, Kim SH, Lee JH, Henary M, Frangioni JV. et al. Simultaneous mapping of pan and sentinel lymph nodes for real-time image-guided surgery. Theranostics. 2014;4:693-700
20. Tanaka E, Ohnishi S, Laurence RG, Choi HS, Humblet V, Frangioni JV. Real-time intraoperative ureteral guidance using invisible near-infrared fluorescence. J Urol. 2007;178:2197-202
21. Matsui A, Tanaka E, Choi HS, Kianzad V, Gioux S, Lomnes SJ. et al. Real-time, near-infrared, fluorescence-guided identification of the ureters using methylene blue. Surgery. 2010;148:78-86
22. Verbeek FP, van der Vorst JR, Tummers QR, Boonstra MC, de Rooij KE, Lowik CW. et al. Near-Infrared Fluorescence Imaging of Both Colorectal Cancer and Ureters Using a Low-Dose Integrin Targeted Probe. Ann Surg Oncol. 2014
23. Matsui A, Tanaka E, Choi HS, Winer JH, Kianzad V, Gioux S. et al. Real-time intra-operative near-infrared fluorescence identification of the extrahepatic bile ducts using clinically available contrast agents. Surgery. 2010;148:87-95
24. Ashitate Y, Stockdale A, Choi HS, Laurence RG, Frangioni JV. Real-time simultaneous near-infrared fluorescence imaging of bile duct and arterial anatomy. J Surg Res. 2012;176:7-13
25. Choi HS, Gibbs SL, Lee JH, Kim SH, Ashitate Y, Liu F. et al. Targeted zwitterionic near-infrared fluorophores for improved optical imaging. Nat Biotechnol. 2013;31:148-53
26. Hutteman M, Choi HS, Mieog JS, van der Vorst JR, Ashitate Y, Kuppen PJ. et al. Clinical translation of ex vivo sentinel lymph node mapping for colorectal cancer using invisible near-infrared fluorescence light. Ann Surg Oncol. 2011;18:1006-14
27. Lee JH, Park G, Hong GH, Choi J, Choi HS. Design considerations for targeted optical contrast agents. Quant Imaging Med Surg. 2012;2:266-73
28. Choi HS, Liu W, Liu F, Nasr K, Misra P, Bawendi MG. et al. Design considerations for tumour-targeted nanoparticles. Nat Nanotechnol. 2010;5:42-7
29. Frangioni JV. In vivo near-infrared fluorescence imaging. Curr Opin Chem Biol. 2003;7:626-34
30. Zaheer A, Lenkinski RE, Mahmood A, Jones AG, Cantley LC, Frangioni JV. In vivo near-infrared fluorescence imaging of osteoblastic activity. Nat Biotechnol. 2001;19:1148-54
31. Bhushan KR, Misra P, Liu F, Mathur S, Lenkinski RE, Frangioni JV. Detection of breast cancer microcalcifications using a dual-modality SPECT/NIR fluorescent probe. J Am Chem Soc. 2008;130:17648-9
32. Bhushan KR, Tanaka E, Frangioni JV. Synthesis of conjugatable bisphosphonates for molecular imaging of large animals. Angew Chem Int Ed Engl. 2007;46:7969-71
33. Choi HS, Frangioni JV. Nanoparticles for biomedical imaging: fundamentals of clinical translation. Mol Imaging. 2010;9:291-310
34. Choi HS, Ipe BI, Misra P, Lee JH, Bawendi MG, Frangioni JV. Tissue- and organ-selective biodistribution of NIR fluorescent quantum dots. Nano Lett. 2009;9:2354-9
35. Choi HS, Liu W, Misra P, Tanaka E, Zimmer JP, Itty Ipe B. et al. Renal clearance of quantum dots. Nat Biotechnol. 2007;25:1165-70
36. Moriguchi T, Yano K, Nakagawa S, Kaji F. Elucidation of adsorption mechanism of bone-staining agent alizarin red S on hydroxyapatite by FT-IR microspectroscopy. Journal of colloid and interface science. 2003;260:19-25
37. Hyun H, Wada H, Bao K, Gravier J, Yadav Y, Laramie M. et al. Phosphonated near-infrared fluorophores for biomedical imaging of bone. Angew Chem Int Ed Engl. 2014;53:10668-72
38. Freedman JD, Lusic H, Snyder BD, Grinstaff MW. Tantalum oxide nanoparticles for the imaging of articular cartilage using x-ray computed tomography: visualization of ex vivo/in vivo murine tibia and ex vivo human index finger cartilage. Angew Chem Int Ed Engl. 2014;53:8406-10
Author contact
![]() Corresponding authors: Summer L. Gibbs, Ph.D. Collaborative Life Sciences Building, 2730 SW Moody Ave, Mail Code: CL3SG, Portland, OR 97201. Office: 503-494-8940; Fax: 503-418-9311 Email: gibbssedu or Hak Soo Choi, Ph.D. 330 Brookline Ave. Room SL-436A, Boston, MA 02215. Office: 617-667-6024; Fax: 617-667-0214 Email: hchoiharvard.edu.
Corresponding authors: Summer L. Gibbs, Ph.D. Collaborative Life Sciences Building, 2730 SW Moody Ave, Mail Code: CL3SG, Portland, OR 97201. Office: 503-494-8940; Fax: 503-418-9311 Email: gibbssedu or Hak Soo Choi, Ph.D. 330 Brookline Ave. Room SL-436A, Boston, MA 02215. Office: 617-667-6024; Fax: 617-667-0214 Email: hchoiharvard.edu.