Theranostics
13.3
Impact Factor
- Current Issue
- Advance Articles
- Volume 16; 2026
- Volume 15; 2025
- Volume 14; 2024
- Volume 13; 2023
- Volume 12; 2022
- Archive
- Cover Images
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results and discussion
Conclusions
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results and discussion
Conclusions
Supplementary Material
Acknowledgements
References
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(4):431-442. doi:10.7150/thno.10891 This issue Cite
Research Paper
Cancer Cell-Specific Oligopeptides Selected by an Integrated Microfluidic System from a Phage Display Library for Ovarian Cancer Diagnosis
Chih-Hung Wang1, Chen-Hsun Weng1, Yu-Jui Che1, Kuan Wang4, Gwo-Bin Lee1, 2, 3 ![]()
1. Department of Power Mechanical Engineering, National Tsing Hua University, Hsinchu, Taiwan
2. Institute of Biomedical Engineering, National Tsing Hua University, Hsinchu, Taiwan
3. Institute of NanoEngineering and Microsystems, National Tsing Hua University, Hsinchu, Taiwan
4. Nanomedicine Program and Institute of Biological Chemistry, Academia Sinica, Taipei, Taiwan
Received 2014-10-23; Accepted 2015-1-7; Published 2015-2-5
Citation:
Wang CH, Weng CH, Che YJ, Wang K, Lee GB. Cancer Cell-Specific Oligopeptides Selected by an Integrated Microfluidic System from a Phage Display Library for Ovarian Cancer Diagnosis. Theranostics 2015; 5(4):431-442. doi:10.7150/thno.10891. https://www.thno.org/v05p0431.htm
Other stylesAbstract

Ovarian cancer is one of the leading causes of female mortality worldwide. Unfortunately, there are currently few high-specificity candidate oligopeptide targeting agents that can be used for early diagnosis of this cancer. It has been suggested that cancer-specific oligopeptides could be screened from a phage display library. However, conventional methods are tedious, labor-intensive, and time consuming. Therefore, a novel, integrated microfluidic system was developed to automate the entire screening process for ovarian cancer cell-specific oligopeptides. An oligopeptide screened with microfluidic chip-based technique was demonstrated to have high affinity to ovarian cancer cells and demonstrated relatively low binding to other cancer cells, indicating a high specificity. Furthermore, the developed method consumed relatively low volumes of samples and reagents; only 70 μL of reactant was used within the whole experimental process. Each panning process was also significantly shortened to only 7.5 hours. Therefore, the screened oligopeptide could be used to isolate ovarian cancer cells in a rapid manner, thus greatly expediting the diagnosis and its application as oligopeptide targeting agent for theranostics of this cancer.
Keywords: microfluidic chip, oligopeptide targeting agent, ovarian cancer, phage display library
Introduction
Ovarian cancer is one of the most common cancers among women worldwide. According to epidemiological surveys, the incidence of this cancer is higher in industrialized countries than developing countries [1]. Other studies have found that the ovarian cancer rate is gradually increasing in Taiwan, from 2.08 per 100,000 women in 1979 to 7.87 per 100,000 women in 2008 [2]. Moreover, ovarian cancer has the highest mortality rate among gynecological cancers [3]. Although the 5-year survival rate is approximately 92% for early-stage ovarian cancer, it is difficult to diagnose due to its clinically negligible symptoms. Therefore, it may progress undetected to later stages, and the 5-year survival rate for those that were diagnosed with the advanced stage was only 30% [4].
Ovarian cancers are divided into ovarian epithelial carcinoma, sex cord stromal tumors, and malignant germ cell tumors, depending on their original location in the ovary. Epithelial ovarian cancer is further classified into five histological types, including clear cell, endometrioid, mucinous, serous, and undifferentiated [5]. In industrialized countries, approximately 80 to 85% of ovarian carcinomas belong to the serous type [6]. Physical examinations, the CA-125 blood test, computed tomography (CT) scan, transvaginal ultrasound (US), magnetic resonance imaging (MRI), positron emission tomographic scanning (PET), and histological examinations have been commonly used for ovarian cancer diagnosis. However, these diagnostic methods either require expensive equipment and well-trained personnel or suffer from low specificity. Therefore, a new method for early diagnosis of ovarian cancer is of great need.
Recently, biomarkers have been extensively explored in cancer diagnosis and therapy. A cancer biomarker is a substance, including nucleic acids, peptides, and polysaccharides, whose concentration is significantly elevated in the bodies of those with cancer. Therefore, a cancer biomarker could be used as an indicator for determining the existence, stage, and/or subtype of cancer [7]. Many biomarkers have been developed for cancer diagnostics, such as alpha-fetoprotein (AFP) for liver cancer [8], breast cancer 1/2 (BRCA1/2)/human epidermal growth factor receptor 2 (HER-2) for breast cancer [9], carcinoembryonic antigen (CEA) for colorectal cancer, and prostate-specific antigen (PSA) for prostate cancer [10-11].
Five genes, including cancer antigen 125 (CA-125) or carbohydrate antigen 125, human epididymis protein 4 (HE4), mucin 1 (MUC1), vascular endothelial growth factor (VEGF), and claudin 3 were selected by recursive descent partition (CART) to serve as potential targeting agents for diagnosis of epithelial ovarian cancer [12], and CA-125 and HE4 have seen widespread usage. The levels of CA-125 in blood is currently the most often marker for ovarian cancer diagnosis [13]. Overexpression of CA-125 was observed in 0.2-5.9% of healthy women and 2.2-27.8% of those with ovarian cancer [15]. Levels of CA-125 increased by 80% in those with the serous type of ovarian cancer [14]. However, the CA-125 blood test is not considered a good screening test for ovarian cancer since it can also be detected in blood of individuals with other cancers, such as pancreatic, lung, breast, and gastrointestinal cancers [15]. Moreover, some other conditions, such as pregnancy and inflammation, can lead to elevated levels of CA125. Although HE4 has higher specificity than CA-125 for ovarian cancer, its expression is lower [16-17]. Therefore, biomarkers with high affinity and specificity to ovarian cancer are of great need such that early diagnosis of this cancer is feasible and the 5-year survival rate of those afflicted can be significantly improved.
To address this issue, several novel approaches have emerged to improve candidate targeting agent discovery for ovarian cancer diagnosis. Some genomic, proteomic, metabolomics, and lipidomic approaches have been employed [18-21]. Among these methods, the phage display library for oligopeptide screening and the single-strain DNA library for aptamer screening are the most common and powerful tools; both methods involve the binding of high-affinity molecules (i.e., probes) against specific targets in vitro or in vivo [22-24]. The phage display library is a powerful peptide ligand discovery platform that could be applied to study protein-protein, protein-peptide, and protein-DNA interactions through use of bacteriophages that encode a suite of proteins to be potentially hybridized to the test targets (e.g., a cancer cell) [25].
Typically, oligopeptide targeting agent screening with the phage display library requires iterative rounds of target recognition, washing, and amplification of screened phages. Then, respective gene for the screened peptide can be isolated by DNA cloning and transformation and then sequenced. Moreover, these sequences of screened peptides can be simply determined and predicted by DNA sequencing. For instance, the Ph.DTM-12 phage display library was commonly used in cancer candidate oligopeptide targeting agent screening [26]. With this method randomly synthesized (with 4.1 × 105 possible combination for 12-mers of amino acid) are individually randomly combined that was fused with to a short linker tetrapeptide sequence (Gly-Gly-Gly-Ser) of the coat protein (pIII) of the M13 phage, which constitutes as the display library [27]. These fused oligopeptides were expressed on the surface of the phage particle that was applied to an anchor with a corresponding target during the phage display screening process. The affinity binding (panning) steps of the phage display method involves three processes, including a target binding, a washing process to remove excessive and or non-specific phages and a multiplication process in E. coli. However, the entire process is time-consuming, sometimes requiring weeks to months for displayed peptides to be confirmed.
In recent years, microfluidic technology has been utilized for various biomedical applications since it may greatly reduce sample and reagent consumption and can be performed in an automated and compact fashion [28]. Some microfluidic devices and systems have been used for phage-target binding studies or for phage amplification in phage display screening systems [29-34]. For instance, Liu et al. (2009) developed a microfluidic device that could control magnetic and fluidic forces to increase selection stringency and specificity for selected targets [31]. Alternatively, Wang et al. (2011) used a live cell-adherent based microfluidic channel to rapidly select phage-displayed peptides. The simple micro-device could alleviate relatively complicated protocol which was commonly performed in conventional selection methods [32]. Furthermore, Cung et al. (2012) developed multiplexed microfluidic phage display device for high-throughput and automated screening [33]. These developed devices or systems exhibited rapid target recognition when compared with conventional methods. However, the whole panning procedures, including target binding, wash, and phage amplification could not simultaneously integrate into one microfluidic system in an automatic format.
In this study, a new microfluidic system was therefore developed such that all phage display screening steps could be automated and hands-on time could be further reduced. Moreover, target cells were conjugated onto the surface of magnetic beads to increase specific binding capacity for phage screening. In order to increase the affinity and specificity of ovarian cancer candidates screened in the developed microfluidic system, other cancer cells were used as negative controls to identify oligopeptides with low specificity for ovarian cancer cells. The high specificity of the selected oligopeptide was further confirmed with cell binding assays. Finally, binding affinity and fluorescent staining were also performed to evaluate the efficacy of the screened oligopeptide to serve as a suitable candidate oligopeptide targeting agent for early diagnosis of ovarian cancer.
Materials and Methods
Experimental cell lines
The ovarian cancer cell lines, BG1 (serous type) [35], A2780 (serous type, Sigma No. 93112517), TOV21G (clear cell type, ATCC® CRL-11730TM) were obtained from Dr. Keng-Fu Hsu (Department of Obsterics and Gynecology, National Cheng Kung University, Taiwan). TOV112D (endometrioid type, ATCC® CRL-11731TM) and IGROV1 (endometrioid type) [36] were provided by Dr. Ming-Shiou Jan (Institute of Microbiology and Immunology, College of Medicine, Chung Shan Medical University, Taiwan). The other tested cell lines included BxPC3 (human pancreatic cancer cell, ATCC® CRL-1687TM), HepG2 (human liver cancer cell, ATCC® HB-8065TM), MCF7 (human mammary gland/breast cancer cell, ATCC® HTB-22TM), and HeLa (human cervix cancer cell, ATCC® CCL-2TM) were provided by Dr. Kuang-Hung Cheng (Institute of Biomedical Sciences, National Sun Yat-Sen University, Kaohsiung, Taiwan). HCT8 cells (human colon cancer cell, ATCC® CCL-244TM) were provided by Dr. Hwan-You Chang (Institute of Molecular Medicine, National Tsing Hua University, Hsinchu, Taiwan). M2-10B4 cells (mouse bone marrow stromal cell, ATCC® CRL-1972TM) were provided by Dr. Shiaw-Min Hwang (Food Industry Research Development Institute, Hsinchu, Taiwan). All cell culture media and conditions were in accordance with product sheets of American type culture collection. E. coli TOP10 (tetracycline resistant, Invitrogen, USA) and ER2738 (tetracycline resistant, New England BioLabs, USA) were used for DNA transformation and phage amplification, respectively.
Phage display panning procedures on the microfluidic system
A phage display library was used in microfluidic format for candidate oligopeptide targeting agent screening. The working principle of the phage display-based oligopeptide targeting agent screening process is schematically shown in Figure 1. The chamber contained 5 μL of 1011 pfu (plaque forming units) of Ph.DTM-12 phage display library, 5 μL of 4×104 of Dynabeads® epithelial-enriched magnetic beads (Invitrogen, USA), and 10 μL of 105 trypsinized BG1 ovarian cancer cells. All samples and reagents were transported to the reaction chamber and incubated at room temperature for 30 min (Fig. 1A). Next, the phage-cell-bead complexes were collected by applying an external magnet. The unbound cells, free phages, and cellular debris were removed with three washes with 1× phosphate buffered saline (PBS, Invitrogen, USA) from the washing buffer chamber (Fig. 1B). Then, the washed complexes were mixed with 20 μL of freshly prepared E. coli ER2738 and 50 μL of lysogeny broth (LB, Sigma, USA) containing 25 µg mL-1 of tetracycline (Sigma, USA) and transported into the bacterial culture chamber for 5 hr culture in order to multiply the screened phages (Fig. 1C). The optimal culture time was determined by the relationship between the BG1 cell mortality and the growth rate of E. coli ER2738 in LB broth (Supplementary Material: Fig. S1). The trypan blue viability test was used to discriminate between viable and non-viable cells. In a 1:1 dilution of the tested cell suspension with 0.4% trypan blue solution (BioRad, USA), non-viable cells are stained blue, while viable cells are left unstained. Note that the survival rate of BG1 cells decreased significantly to 25% after one hour when they were grown under non-serum and growth factor contained bacterial growth medium. The BG1-captured phages were then released and attached to F pili on the outer membranes of the E. coli. Next, the phage DNA was multiplied into E. coli (Fig. 1D). Moreover, ten-fold serial dilutions of tested phages collected from different infection periods in LB broth were further incubated with E. coli until reaching the latter's mid-log phase. All of the reactants were mixed with melt top agar and immediately poured onto an IPTG (isopropyl β-D-1-thiogalactopyranoside, Sigma, USA)/Xgal (5-bromo-4-chloro-3-indolyl-beta-D-galacto-pyranoside, Sigma, USA)/LB plate. After overnight incubation at 37℃, approximately 100 plaques were counted for a relative diluted factor to get an original phage titer as plaque forming unit (PFU) (Supplementary Material: Fig. S1B). The PFU values reflected the number of the infected phages within E. coli. For these reasons, the PFU/CFU ratio at the 3rd, 5th, and 7th hour were measured to confirm the optimal E. coli culture period for maximum phage amplification in E. coli on the microfluidic system.
Figure 1
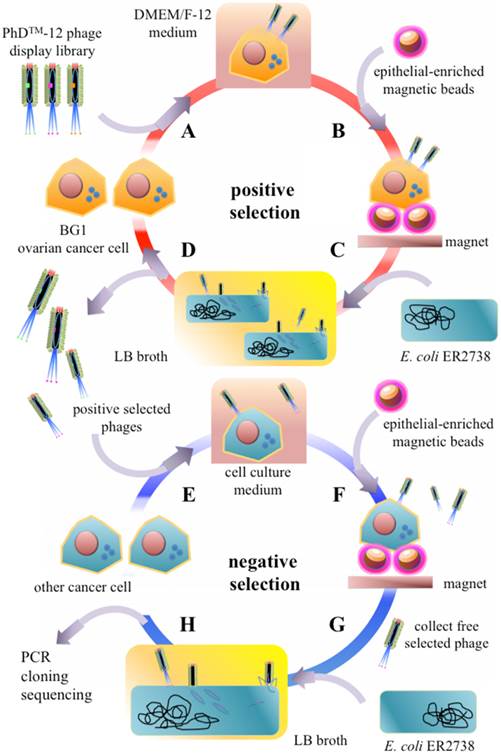
The working principle for screening of ovarian cancer cell-specific oligopeptides from a phage display library on an integrated microfluidic system. The screening system combined sequential positive (A-D) and negative selection (E-H) steps. (A) A Ph.DTM-12 phage display library was incubated with ovarian cancer cells (BG1). (B) Epithelial-enriched magnetic beads were used to capture BG1 cells, and cell-bead complexes were collected with an external magnet. (C) Free, un-captured phages were removed by the washing buffer, and the phage-cell-bead complexes were cultured with E. coli in LB broth. (D) Screened phages were cloned in E. coli and used in the next round of panning. (E) After five positive selection panning rounds, the positively selected phages were collected and incubated with other cancer cells. (F) Epithelial-enriched magnetic beads were used to capture other cancer cells. Then, these complexes were collected with an external magnet. (G) Free, unbound phages were collected and cloned into E. coli cells. (H) A portion of the gene encoding the selected, displayed oligopeptide was amplified by PCR, cloned, and sequenced.
Five continuous panning, included incubate, washing and multiplication steps were applied as in the “positive selection” experiments, which aimed to screen the candidate oligopeptide targeting agents for ovarian cancer cells (Fig. 1E). Finally, 1×105 trypsinized BxPC3, MCF7, HeLa, and HCT8 human cancer cells were used to further incubate with the positive-selected phages. This negative selection process could remove the peptides with less specificity and the remaining phages in supernatant were demonstrated to have high specificity with ovarian cancer cells and low affinity with other cancer cells. All of selected phages were further referenced with the manual guideline that and were cloned with TOPO® TA cloning (PCR®II-TOPO, ampicillin resistant, Invitrogen, USA) and transformed into E. coli TOP10 strain. Next, the DNA clones representing the phage library-selected oligopeptide were amplified by polymerase chain reaction (PCR) and sequenced [37].
Microfluidic chip design and fabrication
In order to perform the phage display-based screening on the developed microfluidic chip system, several microfluidic devices were integrated within a single chip, including suction-type micropumps, micromixers, normally-closed microvalves, microchannels, and microchambers (Figure 2A). The chip contained several chambers for samples (i.e., ovarian cancer or other cancer cells) and the following reagents: Dynabeads® epithelial-enriched magnetic beads, oligopeptide pool of the phage display library, E. coli ER2738, LB culture media, and washing buffer (1× PBS). Furthermore, reaction chambers and bacterial culture chambers were also integrated into the chip. This microfluidic chip consisted of two polydimethylsiloxane (PDMS) layers, one thick air chamber layer, and one thin liquid channel layer for fluid transportation. A glass substrate (0.7 mm, G-Tech Optoelectronics, Taiwan) was used as the substrate for the microfluidic chip that was bonded with the two PDMS layers via oxygen plasma treatment [38]. The master molds of the microfluidic chip were formed on polymethymethacrylate (PMMA) substrates with a computer-numerical-control (CNC) machine (EGX-400, Roland, Japan) for casting of the thick and thin PDMS layers [39]. All the microfluidic structures were then fabricated with a PDMS casting technique from molds on the PMMA substrate. An elastomer (Sylgard 184A silicon elastomer, Dow Corning, USA) and a curing agent (Sylgard 184B elastomer curing agent, Dow Corning, USA) were mixed well in a 10:1 ratio by weight and poured onto the PMMA master mold at 80°C over 2 hr to form the cured PDMS microfluidic structures.
The two PDMS layers were bonded to the glass substrate by oxygen plasma treatment to form the complete microfluidic chip [39]. The suction-type micropumps/micromixers were activated by applying a negative gauge pressure that produced a suction force to generate the deflection of the PDMS memebranes for fluid transportation and sample/reagent incubation [40]. Different air pressures were applied to optimize the pumping rate for the developed device by transporting 20 μL of double-distilled water (ddH2O) through the micropump to another microchamber. The maximum pumping rate could be then determined and used to operate the microfluidic chip. Furthermore, the suction-type micropumps could be used to mix the samples with the reagents [41]. Moreover, normally-closed microvalves were used in the developed microfluidic system to prevent fluid backflow during the sample/reagent transportation processes [40].
Phage display library and synthesized peptides
The Ph.DTM-12 phage display library kit (New England Biolabs, USA) was used to screen specific oligopeptides binding to BG1 ovarian cancer cells in this study. The library contains 12-mer random oligopeptide sequences that have been inserted into the N terminal region of the minor coat protein (cpIII) of the M13 phage. The Ph.DTM-12 display library contains 1010 pfu µL-1 of the phage mixtures. The E. coli strain ER2738 was used for M13 phage multiplying. After five rounds of positive and one round of negative panning, the BG1-anchored phages were collected, and the inserted DNA sequences were amplified with a forward cloning primer (5'-CCTTTAGTGGTACCTTTCTA-3') and a reverse cloning primer (5'-CTTTCAACAGTTTCGGCCGA-3'). All of the amplified, 95-base-pair (bp) products were further cloned within a pCR®II-TOPO® vector and transformed into E. coli TOP10 strain and selected from an LB-agar plate containing 100 µg mL-1 of ampicillin (Sigma). These selected DNAs were then purified and sequenced in one direction with the M13 reverse primer (synthesized by Mission Biotech. Co., Taiwan). The DNA sequences were compared to others on the NCBI database (http://www.ncbi.nlm.nih.gov) with the Basic Local Alignment Search Tool (BLAST), and Clustal W (ExPAsy, SIB Bioinformatics Resource Portal, http://www.expasy.org) was used to align sequences.
Two frequently-appearing oligopeptides, named oligopeptide I and II were then synthesized (>98% purity) by Protech Technology Enterprise Co. (Taiwan); the former was conjugated to fluorescein isothiocyanate (FITC; λexcitation=490 nm, λemission=525 nm), and the latter to tetramethylrhodamine (TMR; λexcitation=550 nm, λemission=573 nm) at carboxyl termini. These fluorescently-labeled oligopeptides were then used for flow cytometric assays and fluorescence microscopy analyses, both described below.
Binding capability of selected peptides with tested cancer cells
The binding capacity of the screened oligopeptides with target cancer cells was evaluated. The protocol for the conjugation of the oligopeptides to the magnetic beads can be found in a previous work [42]. Dilutions of 100, 10, and 1 nM of the oligopeptide I were conjugated to Dynabeads® MyOneTM carboxylic acid beads (Cat No. 65012, Invitrogen, USA) by covalent binding. Different concentrations of the oligopeptide I conjugated magnetic beads were further incubated with 105 CFU of BG1 ovarian cancer cells (target cells) at room temperature for 30 min. These magnetic beads were then bound with the BG1 cells and collected under an external magnetic field (2000 Gauss) after 1× PBS washing [43]. The binding capability was then estimated by quantifying the number of captured cells under optical microscope. For comparison, the purified, 100-nM oligopeptide I- or II-conjugated magnetic beads were also incubated with 105 CFU of either other ovarian cancer cell types or cells from non-ovarian cancers at room temperature for 30 min. The unbound or low-affinity cells were discarded during seven washes with 1× PBS. The captured cell number was calculated with a hemocytometer (Paul Marienfeld, Germany), and the binding capacity of the screened oligopeptides for different cell types was calculated as follows:
Binding capability (%) = the total number of captured cells/105 CFU of BG1 cells × 100
Binding affinity of screened peptides to ovarian cancer cells
Binding affinity of the two synthesized oligopeptides to BG1 ovarian cancer cells was assessed with flow cytometry. The intensity of the fluorescence signals was measured by an Accuri® C6 flow cytometer (BD Biosciences, USA), and the flow cytometric data were analyzed by the manufacturer's software. First, the BG1 cells were trypsinized with 1× Trypsin-EDTA (ethylenediaminetetraacetic acid, Sigma, USA) and washed three times with 1× PBS. The washed 105 BG1 cells were incubated with serially-diluted concentrations of fluorescent-labeled oligopeptides at room temperature for 30 min on a microtube Intelli-mixer (C2 program 20 rpm, ELMI, Latvia). The mixtures were then centrifuged at 1200 rpm for 5 min at room temperature, and the supernatant was removed and resuspended in 100 µL of 1× PBS buffer. The mean fluorescence intensity of the oligopeptide-BG1 complexes was determined and analyzed by GraphPad Prism 6 (GraphPad Software, USA) to calculate the dissociation constant (Kd) of the two oligopeptides with the tested cancer cells.
In vitro binding analysis by fluorescence microscopy
The screened oligopeptides were initially selected from suspended BG1 cells from a phage display library on an integrated microfluidic system. Fluorescence staining was further used to verify whether the screened, synthesized oligopeptides could bind suspended BG1 cells. Either 10 nM of FITC-labeled oligopeptide I (10 nM) along with a 1:1000 dilution of CellMask™ Deep Red Plasma membrane stain (Invitrogen, USA) or 10 nM of TMR-labeled oligopeptide II with CellMask™ Green Plasma Membrane Stain (Invitrogen, USA) were mixed with magnetic beads and 105 CFU of BG1 ovarian cancer cells at room temperature for 30 min. The magnetic bead-bound cells were collected after six washes with 1×PBS (5 min each wash). The fluorescent-labeled cells were observed under a fluorescence microscope (BX43, Olympus, Japan).
The epithelial-enriched magnetic system was used to combine with phage display library for novel ovarian cancer oligopeptide targeting agents screening in previously study. The BG1 cells required pretreatment with 1× trypsin-EDTA at 37℃ for 5 min. It was necessary to confirm that the selected oligopeptides could bind to BG1 cells in vitro, as well as in vivo. For adherent cell observations, fluorescent-labeled oligopeptides and plasma membrane stains were incubated with target cells as described above. After six 1× PBS washes, treated adherent BG1 cells were fixed with 4% paraformaldehyde (PFA, Sigma, USA) in PBS at 37℃, and 5% CO2 for 10 min. Next, fixed cells were washed thrice with 1× PBS (5 min each wash). Then, the BG1 cells were immersed in 0.01% Triton X-100 (Sigma, USA) in PBS at 37℃ and 5% CO2 for 5 min. After three additional washes with 1× PBS, a 1:5000 dilution of DAPI (4',6'-diamidino-2-phenyllndole, Invitrogen, USA) was added to the treated cells at 37℃ and 5% CO2 for 10 min to stain the nuclei. The labeled cells were observed by a BX43 inverted fluorescence microscope and fluorescent images were captured by NIS Elements BR (ver. 4.20) software (Nikon, Japan).
Results and discussion
Optimization of E. coli culture period
As schematically shown in Figure 1, a new strategy to screen cell-specific oligopeptides that could recognize ovarian cancer cells (BG1) from the Ph.DTM-12 display library was implemented on the developed microfluidic system. The optimal bacterial culturing time was found to be 5 hours. The epithelial-enriched magnetic beads were used to capture the BG1 cells or other cancer cells and collected by using an external magnet. Afterwards, E. coli ER2738 was incubated with the phage-BG1 cell-magnetic beads complex in LB broth. The eukaryotic cells swelled and were lysed in the culture process with bacterial culture medium. Then, phage DNA was cloned into E. coli bacterial cytoplasm, multiplied, re-assembled and secreted as mature phages from E. coli. The bacterial growth curve was first determined to calculate the total colony number of E. coli ER2738 cells in the LB broth during each hour of cell culture, and it was found that the log growth phase was initiated by the 3rd hour (Supplementary Material: Fig. S1A). Also, the survival rate of BG1 cells in bacterial LB broth was inferred from 0.4% trypan blue staining. Fig S1 shows that the survival rate dramatically decreased to 15% by the 3rd hour and was lower than 5% at the 7th hour. The PFU/CFU ratio was measured to be 4.50±0.28, 4.93±0.52, and 1.72±0.77 at the 3rd, 5th, and 7th hour, respectively. Thus, a 5-hr incubation was determined to be the optimal E. coli culture period for ovarian cancer oligopeptide targeting agents screening in one round of panning by the developed microfluidic system. With a 5-hr cloning period, the entire panning protocol required 7.5 hours, significantly less time than the 3-4 days required conducting conventional protocols.
Characterization of the micropumps
In the microfluidic chip, all fluid transfer was performed pneumatically. As shown in Fig. 2A, two PDMS layers were equipped with microchambers, micropumps, microvalves, microchannels, and air inlets. The dimensions of the microfluidic chip were measured to be 6.5 cm in length and 6.0 cm in width (Fig. 2B). Note that the red color indicates fluid chambers connected to the microchannels. The blue color indicates the air channels necessary to control fluid transportation. Among these microfluidic devices, the suction-type micropump was the most important for transporting of samples and reagents. The relationship between the pumping rate of the micropump and the applied gauge pressure is shown in Fig. 2C. The maximum pumping rate was measured to 2.75 µL s-1 at an applied gauge pressure of -80 kPa under 1 KHz. Note that this same device could also be used as a micromixer [44].
Figure 2
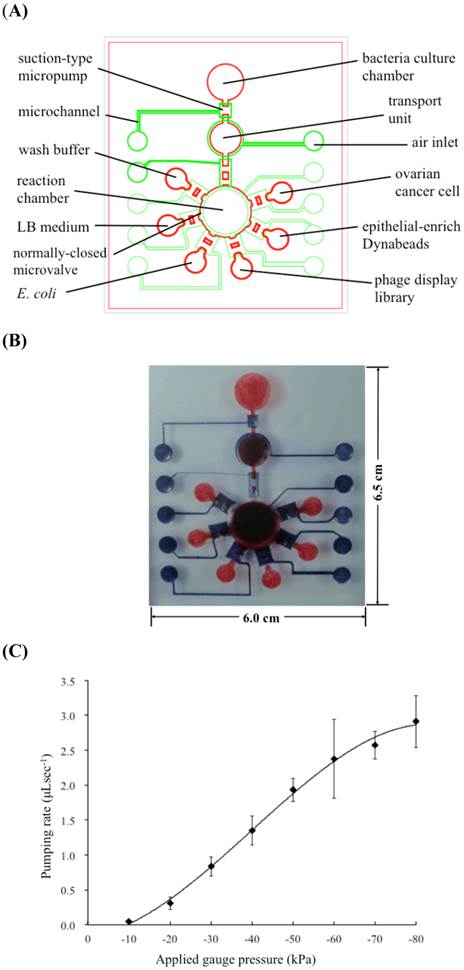
(A) Schematic layout of the integrated microfluidic chip. The suction-type micropumps, normally-closed microvalves, microchannels, reaction chambers and air inlets were integrated on a single chip. (B) A photograph of the microfluidic chip. (C) Relationship between the pumping rate and the applied gauge pressure. The maximum pumping rate was measured to be 2.75 µL s-1 at an applied gauge pressure of -80 kPa. The error bars of standard deviation were determined by triple replicates.
Characterization of the screened oligopeptides
The candidates for ovarian cancer cell-specific oligopeptide targeting agents were collected after five continuous positive selection rounds and one negative selection rounds, all of which were automated on the developed microfluidic chip. The respective nucleic acid sequences of the collected phage oligopeptides were amplified by PCR and ligated into a pCR®II-TOPO® vector. The pool of collected phages from every round of panning was also confirmed by PCR followed by electrophoresis (Supplementary Material: Fig. S2). As shown in Fig. S2, 95-bp PCR products corresponding to the putative oligopeptides of the collected phages were successfully amplified in every round, and 243 clones were collected and sequenced. These DNA sequences could be arranged into four groups and 19 independent sequences. In this study, members of groups I and II represented 15.63% and 9.38% of all clones, respectively, and the peptides encoded by two particular clones were considered to show potential as ovarian cancer oligopeptide targeting agents and were hence used to evaluate the capacity of the developed microfluidic system.
The amino acid sequences of oligopeptides I (N'-MPHPTKNFDLYV-C') and II (N'-ALWPPNLHAWVP-C') were conceptually translated from the selected DNA sequences, and these peptides were predicted to be hydrophilic and hydrophobic by ExPasy, respectively. The former appears to be an antiporter/transporter, peptidase, or ATPase. Furthermore, the group II was proposed to have high homology to a fatty acid-CoA ligase or a NADH dehydrogenase based on BLAST analysis of the peptide sequence. Such proteins have found to be overexpressed in cancer cells in other studies, as well [45-48]. This suggests that the phage display-based microfluidic chip developed herein is effective at selecting oligopeptide targeting agents for cancer diagnosis.
Binding capacity of screened oligopeptides
Three different concentrations of oligopeptide I (100, 10, and 1 nM) were surface-coated onto magnetic beads to determine the optimal quantity required to result in maximum binding to suspended BG1 ovarian cancer cells (Fig. 3). Note that the trypsinized BG1 cells were used as an internal control (Fig. 3A). The magnetic beads lacking an oligopeptide I coat are shown in Fig. 3B and indicate that uncoated beads cannot capture BG1 cells. Experimental data showed that when higher concentrations of oligopeptide-coated magnetic beads were used, more BG1 cells could be captured (Figs. 3C-3E). However, a 100 nM concentration of oligopeptide I-conjugated beads resulted in few captured liver cancer cells (Fig. 3F), suggesting that the selected oligopeptide had higher specificity to ovarian cancer cells than other cancer cells.
To further demonstrate this specificity, cells of different cancers were also incubated with magnetic beads surface-coated with oligopeptide I or II. As shown in Table 1, the experimental results demonstrated that the screened oligopeptide I had higher cell binding rates, 71.24±2.69 and 48.27± 3.03% with the serous cell types BG1 and A2780, respectively. Conversely, the oligopeptide I had relatively low binding rates (2-3-fold lower than with BG1 and A2780) with other cancer cell types (Table 1), including non-serous ovarian cancer cells. However, cellular binding capacity ranged from 10 to 30% for cancer cells incubated with magnetic beads coated with oligopeptide II, with the exception of lung cancer cells (A2780; 46.14±0.51%) and mouse bone marrow stromal cancer cells (M2-10B4; 45.61 ±3.84%).
Table 1
Binding capacity of the screened oligopeptides for different cell types.
| species | organ | cell type | cell binding rate (%) | |
|---|---|---|---|---|
| oligopeptide I | oligopeptide II | |||
| human | ovary | BG1/serous | 71.24 ± 2.69 | 22.37 ± 4.58 |
| A2780/serous | 48.27 ± 3.03 | 46.14 ± 0.51 | ||
| TOV112D/endometrioid | 22.89 ± 4.15 | 35.33 ± 10.09 | ||
| TOV21G/clear cell | 16.88 ± 2.36 | 26.76 ± 2.09 | ||
| IRGOV1/endometrioid | 15.57 ± 3.75 | 29.24 ± 1.92 | ||
| cervix | HeLa | 26.02 ± 2.01 | 8.91 ± 1.57 | |
| breast | MCF7 | 17.58 ± 1.51 | 21.70 ± 1.61 | |
| colon | HCT8 | 22.37 ± 1.89 | 11.49 ± 0.80 | |
| liver | HepG2 | 15.77 ± 5.25 | 5.38 ± 0.16 | |
| pancreas | BxPC3 | 12.89 ± 0.21 | 16.28 ± 0.47 | |
| mouse | bone marrow | M2-10B4 | 6.64 ± 2.27 | 45.61 ± 3.84 |
Note that 100 nM FITC-labeled oligopeptide I was used in this study. Three consecutive experiments were performed.
Figure 3
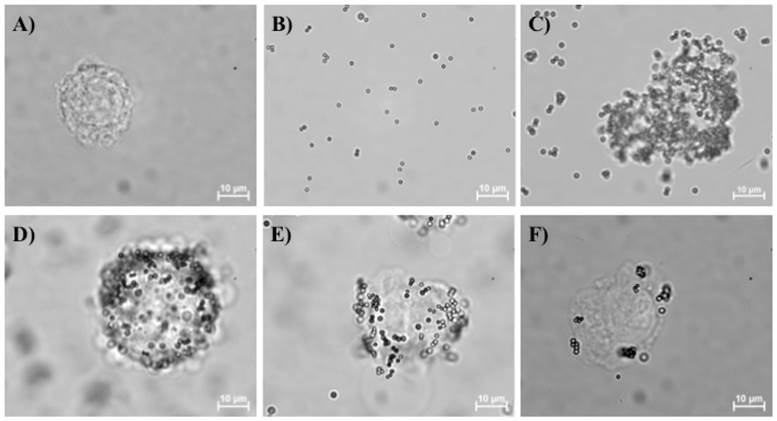
BG1 cells captured with magnetic beads coated with different concentrations of the screened oligopeptides. The BG1 cells alone (A) and the unlabeled magnetic beads (B) have also been shown. In panels C, D, and E, oligopeptide I has been coated onto the surfaces of magnetic beads at 100, 10, and 1 nM, respectively, and the beads were incubated with 105 CFU of suspended BG1 ovarian cancer cells, collected with an external magnetic field and washed with 1x PBS. In (F) 105 CFU of suspended HepG2 cells (liver cancer cells) were incubated with magnetic beads coated with oligopeptide I to demonstrate the lack of binding between this peptide and cells of other cancers.
Figure 4
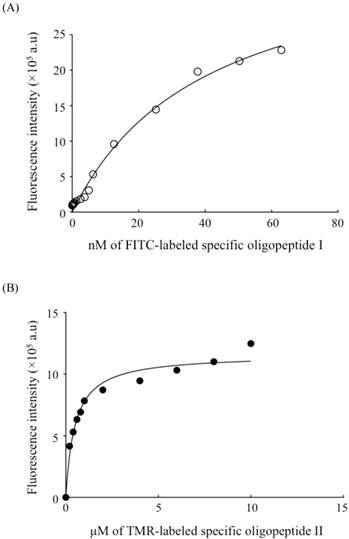
The binding affinity analysis of oligopeptide I (A) and oligopeptide II (B) with BG1 ovarian cancer cells by flow cytometry. Serial dilutions of FITC-labeled oligopeptide I (○) and TMR-labeled oligopeptide II (●) were used to measure the gated fluorescence intensity. The fitted curve of the dissociation constant (Kd) was determined with GraphPad Prism 6 software.
In general, oligopeptide I showed a higher binding affinity to serous types of ovarian cancer cells while oligopeptide II showed similar binding capacity to three major ovarian cancer cell types: serous (BG1), endometrioid (TOV112D and IRGOV1), and clear cells (TOV21G). Additionally, with the exception of A2780 and M2-10B4 cells, there were no significant difference in the binding capacity of oligopeptide II between ovarian cancer cells and other cancer cells; this would preclude its ability to be an oligopeptide targeting agent for ovarian cancer cells. The specificity of the peptide may be strongly dependent on the concentration. Although the high concentration (100 nM) of oligopeptide I could get higher binding capacity for BG1 ovarian cancer cells (target cells), the binding capacity of the other cancer cells were also enhanced by using this high concentration of oligopeptide I. Therefore, the specificity at this high concentration (100 nM) may not be satisfactory. However, at low concentrations (from 0 to 1.25 nM) of FITC-labeled oligopeptide I, we further performed a cytometry assay to determine the ratio between the fluorescence labeled cells and the total tested cancer cells (as shown in Supplementary Material: Table S1). It indicated the binding capacity for various tested cancer cells with oligopeptide I. In this table, 1 nM oligopeptide I was used to measure the fluorescence intensity of FITC labeled cancer cells. The ratio between the fluorescence labeled cells and the total tested cancer cells was then indicated as the binding capacity of the tested cells with oligopeptide I. The binding capacity at this low concentration (1 nM) of oligopeptide I was measured to be 25.42 ± 3.11% and 13.52 ± 1.47% , respectively, for serous types of ovarian cancer cells, BG1 (target cells) and A2780. Although the binding capacity of 1 nM oligopeptide I was lower than the one for 100 nM oligopeptide I, the binding capacity of other tested cancer cells for oligopeptide I at this low concentration was significantly decreased (Table 1 and S1).
In addition, Epithelial-enriched magnetic beads coated with BerEP4 antibodies are commonly used to capture various epithelial cellular membranes [43]. However, such epithelial-enriched beads exhibited a high binding affinity with lung cancer cells (A549) and breast cancer cells (MCF7) and a relatively low binding affinity for ovarian (<50%) and other cancer cells [49]. Hence, the oligopeptide I identified herein shows a higher binding affinity for serous-type ovarian cancer cells than previously identified oligopeptide targeting agents.
Binding affinity of screened oligopeptides
The dissociation constant (Kd) has been commonly used to determine the affinity between a ligand and a protein. In this study, Kd was defined as the binding strength between the screened oligopeptide and the tested cellular membrane receptors. The Kd values of the two selected oligopeptides were measured with a flow cytometric assay [40] and found to be 40.8±6.6 and 490.3±16.3 nM, respectively (Fig. 4A-B). Furthermore, a saturation specificity binding assay was used by using a one-site-specificity binding equation to determine the Kd in this study. In addition, a competition test for FITC-labeled oligopeptide I was performed with unlabeled oligopeptide I by using a flow cytometry assay. Serial dilutions of FITC-labeled oligopeptide I (○) with 1 nM (□) and 10 nM (△) of unlabeled oligopeptide I were added to test FITC-labeled oligopeptide I which competed with FITC-labeled oligopeptide I for cell binding (Supplementary Material: Figure S4). The fitted curve of the dissociation constant (Kd) was measured by using the gated fluorescence intensity and then determined with GraphPad Prism 6 software (Figure S4). The result showed that the unlabeled oligopeptide I could compete with FITC-labeled oligopeptide I to recognize the targeting binding site of ovarian cancer cells BG1. The Kd value of an aptamer specific for lung cancer stem cells was reported to be 15.32 nM by microfluidic SELEX based system [50], and the binding affinity of tumor-targeting peptides for non-small cell lung cancer cells was measured to be 0.0071-40 nM, as screened with a similar phage display library in a conventional method [51]. According to previous works reported previously [51], the Kd value of the tumor biomarker was about in the order of 102 nM either using SELEX based microfluidic systems or conventional phage display library methods. Therefore, the selected oligopeptide I by using phage display based microfluidic device was also similar to this Kd range. The result demonstrated that oligopeptide I had a comparable Kd value as a promising targeting binding agent for ovarian cancer.
In vitro immunofluorescence analysis
Immunofluorescence analysis was performed to evaluate the binding capacity of the screened oligopeptides. FITC-labeled oligopeptide I and TMR-labeled oligopeptide II were first used to stain the suspended BG1 cells. Figure 5 shows that the screened oligopeptides could be bound to suspended BG1 cells by immunofluorescence microscopy. The epithelial-enriched magnetic beads were first bound to BG1 cells (Fig. 5A). After incubation and collection, the bead-bound BG1 cells incubated with FITC-labeled oligopeptide I presented green fluorescent signals (Fig. 5B), and those incubated with the CellMask Deep Red Plasma Membrane Stain Kit expressed red fluorescent signal (Fig. 5C). The result of the merged photograph shows that the FITC-labeled oligopeptide I could bind to the plasma membrane of BG1 cells (Fig. 5D). Similarly, the TMR-labeled oligopeptide II and CellMask™ Green Plasma Membrane Stain kit were used to observe the binding capacity of oligopeptide II with the suspended BG1 cells (Figs. 5E-5H). The immunofluorescence results were consistent with the previous results in terms of appearing frequency, binding capacity, and affinity. Collectively, it appears that oligopeptide I had higher specificity and affinity for BG1 ovarian cancer cells than oligopeptide II. Then, these screened oligopeptides are then suitable candidates for in vitro or in vivo ovarian cancer diagnosis.
The same staining protocols were used to stain adherent BG1 cells with FITC-labeled oligopeptide I and TMR-labeled oligopeptide II (Figs. 6B-6C and 6F-6G, respectively). The CellMaskTM plasma membrane staining kit was used as a cell membrane indicator and DAPI was used as a marker of the cell boundary. DAPI was further used to stain nuclei to confirm the location of the adherent BG1 cells (Figs. 6A and 6E). Similar to the fluorescence staining results for the suspended BG1 cells, the screened oligopeptide I demonstrated stronger immunofluorescence intensity with adherent BG1 cells (Figs. 6B and 6D). Conversely, the TMR-labeled oligopeptide II exhibited a faint red fluorescence signal in the cytoplasm of adherent BG1 cells (Figs. 6E and 6H). The adherent BG1 cell was mimicked as in vitro status of ovarian cancer for specific oligopeptide recognized in this study. The oligopeptide I therefore demonstrated a promising targeting agent potential for clinical ovarian cancer diagnosis.
Figure 5
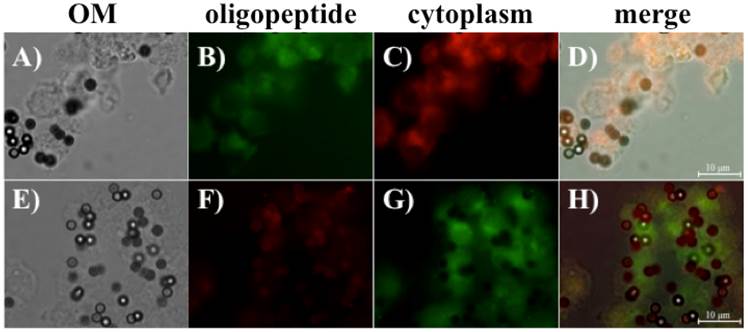
Fluorescent microscope images revealing that oligopeptides I and II can bind to suspended ovarian cancer cells BG1, as well as those captured by epithelial-enriched magnetic beads. The oligopeptide-BG1-captured magnetic beads complex was collected with an external magnet and observed under optical microscopy (OM) (A and E). The FITC-labeled oligopeptide I (B) and the CellMask™ Deep Red Plasma Membrane Stain Kit (C) were used to stain BG1 cells. The merged photographs (D) showed that the FITC-labeled oligopeptide I bound to BG1 ovarian cells (D). Similarly, the TMR-labeled oligopeptide II (F) and the CellMask™ Green Plasma Membrane Stain Kit (G) were used to stain BG1 cells, and the merged image is shown in (H).
Figure 6
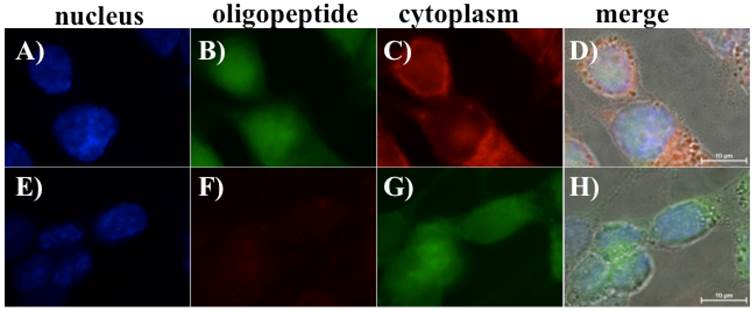
Fluorescent microscope images revealing that oligopeptides I and II can bind to adherent BG1 ovarian cancer cells. A monolayer of BG1 cells was treated with fluorescent-labeled oligopeptides and cellular membrane dyes. DAPI-stained nuclei are shown in (A) and (E). FITC-labeled oligopeptide I (B) and the CellMask™ Deep Red Plasma Membrane Stain Kit were used to stain BG1 cells (C). The merged image indicates that the FITC-labeled oligopeptide I can bind to BG1 ovarian cancer cells (D). Similarly, the TMR-labeled oligopeptide II (F) and the CellMask™ Green Plasma Membrane Stain kit were used to stain BG1 cells (G), and the corresponding merged image is shown in (H).
Conclusions
The novelty of this study is the development of a new microfluidic system to automate the entire phage display screening process. In addition to micropumps, different microfluidic devices such as microvalves, micromixers and an E. coli culture chamber were integrated on a single chip. Furthermore, in order to increase the affinity and specificity of ovarian cancer candidates screened in the developed microfluidic system, other cancer cells were used as negative selection. It has been demonstrated that an integrated microfluidic system could be used to automate the screening process for oligopeptide targeting agents specific to ovarian cancer cells. One of the screened oligopeptides from the phage display library demonstrated clinical potential for rapid diagnosis of cancer; the microfluidic system could automatically finish one round of panning within 7.5 hours, which is much faster than conventional methods. Two oligopeptides were chosen to evaluate the affinity and specificity of the screened oligopeptide targeting agents. Experimental data showed that oligopeptide I had a high binding rate (71.24±2.69%) and a high binding affinity (Kd= 40.8 ± 6.6 nM) with ovarian cancer cells. Compared to the binding capability with other types of ovarian cancer cells and other cancer cells, oligopeptide I had higher specificity for the serous type of ovarian cancer cells. In addition, according to the putative amino acid alignment, sequence of oligopeptide I was demonstrated that similar with some candidates of cancer oligopeptide targeting agents. Moreover, the fluorescent staining results indicated that oligopeptide I could bind to the cellular membrane of ovarian cancer cells in vitro. The merged fluorescence photograph confirmed that oligopeptide I could bind with ovarian cancer cell BG1 in this study. Summarized these results, the selected oligopeptide I was predicated that used to in vitro/or in vivo ovarian cancer diagnosis. Importantly, the developed phage display-based microfluidic system could become a powerful tool for future cancer or novel emergency disease oligopeptide targeting agent screening.
Supplementary Material
Figures S1-S4 and Table S1.
Acknowledgements
The authors thank the Ministry of Science and Technology of Taiwan (NSC 102-2221-E-007-054-MY3) for funding this work. Partial financial supports from the “Towards A World-class Class University Project” and the Nanomedicine Program, Academia Sinica, are also greatly appreciated. The authors also thank Dr. Keng-Fu Hsu (Department of Obsterics and Gynecology, National Cheng Kung University, Taiwan), Dr. Ming-Shiou Jan (Institute of Microbiology and Immunology, College of Medicine, Chung Shan Medical University, Taiwan), Dr. Kuang-Hung Cheng (Institute of Biomedical Sciences, National Sun Yat-Sen University, Kaohsiung, Taiwan), Dr. Hwan-You Chang (Institute of Molecular Medicine, National Tsing Hua University, Hsinchu, Taiwan) and Dr. Shiaw-Min Hwang (Food Industry Research Development Institute, Hsinchu, Taiwan) for providing tested cell strains in this study.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Parazzini F, Franceschi S, La Vecchia C. et al. Review: the epidemiology of ovarian cancer. Gynecol Oncol. 1990;43:9-23
2. Chiang YC, Chen CA, Chiang CJ. et al. Trends in incidence and survival outcome of epithelial ovarian cancer: 30-year national population-based registry in Taiwan. J Gynecol Oncol. 2013;4:342-51
3. Huang YW, Kuo CT, Stoner K. et al. An overview of epigenetics and chemoprevention. FEBS Lett. 2011;582:2129-36
4. Yurkovetsky Z, Skates S, Lomakin A. et al. Development of a multimarker assay for early detection of ovarian cancer. J Clin Oncol. 2010;28:2159-66
5. Soslow RA, Histologic subtypes of ovarian carcinoma. an overview. Int J Gynecol Pathol. 2008;27:161-74
6. Jeffrey S, Iren HS, Moutaz H. et al. The histologic type and stage distribution of ovarian carcinomas of surface epithelial origin. Int J Gynecol Pathol. 2004;23:41-4
7. Chen VW, Ruiz B, Killeen JL. et al. Pathology and classification of ovarian tumors. Cancer. 2003;97:2631-42
8. Masuzaki R, Karp SJ, Omata M. New serum markers of hepatocellular carcinoma. Semin Oncol. 2012;39:434-39
9. Marić P, Ozretić P, Levanat S. et al. Tumor markers in breast cancer-evalutation of their clinical usefulness. Coll Antropol. 2011;35:241-7
10. Yamashita K, Watanabe M. Clinical significance of tumor markers and an emerging perspective on colorectal cancer. Cancer Sci. 2009;100:195-9
11. Stephan C, Ralla B, Jung K. Prostate-specific antigen and other serum and urine markers in prostate cancer. Biochim Biophys Acta. 2014;1846:99-112
12. Lu KH, Patterson AP, Wang L. et al. Selection of potential markers for epithelial ovarian cancer with gene expression arrays and recursive descent partition analysis. Clin Cancer Res. 2004;10:3291-300
13. Suh KS, Park SW, Castro A. et al. Ovarian cancer biomarkers for molecular biosensors and translational medicine. Expert Rev Mol Diagn. 2010;10:1069-83
14. Markowska J, Manys G, Kubaszewska M. Value of CA-125 as a marker of ovarian cancer. Eur J Gynaecol Oncol. 1992;13:360-5
15. Carlson KJ, Skates SJ, Singer DE. Screening for ovarian cancer. Ann Intern Med. 1994;121:124-32
16. Moore RG, Miller MC, Steinhoff MM. et al. Serum HE4 levels are less frequently elevated than CA125 in women with benign gynecologic disorders. Am J Obstet Gynecol. 2012;206:351.e1-8
17. Shah CA, Lowe KA, Paley P. et al. Influence of ovarian cancer risk status on the diagnostic performance of the serum biomarkers mesothelin, HE4, and CA125. Cancer Epidemiol Biomarkers Prev. 2009;18:1365-72
18. Li TH, Kung HJ, Mack PC. et al. Genotype and genomic profiling of non-small-cell lung cancer: Implication for current and future therapies. J Clin Oncol. 2013;31:1039-49
19. Kume H, Muraoka S, Kuga T. et al. Discovery of colorectal cancer biomarker candidates by membrane proteomic analysis and subsequent verification using selected reaction monitoring (SRM) and tissue microarray (TMA) analysis. Mol Cell Proteomics. 2014;13:1471-1484
20. Millard RW, Rosevear PR. Metabolomics seeking a unique biomarker signature for coronary artery syndromes. J Am Coll Cardiol. 2012;59:1642-4
21. Lu N, Wei D, Chen F. et al. Lipidomic profiling and discovery of lipid biomarkers in snow alga Chlamydomonas nivalis under salt stress. Eur J Lipid Sci Tech. 2012;114:253-65
22. Xiao N, Cheng D, Wang Y. et al. Identification of a high affinity TAG-72 binding peptide by phage display selection. Cancer Biol Ther. 2011;11:22-31
23. Li J, Zhang Q, Pang Z. et al. Identification of peptide sequences that target to the brain using in vivo phage display. Amino Acids. 2011;42:2373-81
24. Van Simaeys D, López-Colón D, Sefah K. et al. Study of the molecular recognition of aptamers selected through ovarian cancer cell-SELEX. PLoS One. 2010;5:e13770
25. Smith GP, Filamentous fusion phage. novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315-7
26. Wu J, Park JP, Dooley K. et al. Rapid development of new protein biosensors utilizing peptides obtained via phage display. PLoS One. 2011;6:e24948
27. Tesar M1, Beckmann C, Röttgen P. et al. Monoclonal antibody against pIII of filamentous phage: an immunological tool to study pIII fusion protein expression in phage display systems. Immunotechnology. 1995;1:53-64
28. Mark D, Haeberie S, Roth G. et al. Microfluidic lab-on-a-chip platforms: requirements, characteristics and applications. Chem Soc Rev. 2010;39:1153-82
29. Persson J, Augustsson P, Laurell T. et al. Acoustic microfluidic chip technology to facilitate automation of phage display selection. FEBS J. 2008;275:5657-66
30. Bessette PH, Hu X, Soh HT. et al. Microfluidic library screening for mapping antibody epitopes. Anal Chem. 2007;79:2174-8
31. Liu Y, Adams JD, Turner K. et al. Controlling the selection stringency of phage display using a microfluidic device. Lab Chip. 2009;9:1033-6
32. Wang J, Liu Y, Teesalu T. et al. Selection of phage-displayed peptides on live adherent cells in microfluidic channels. Proc Natl Acad Sci USA. 2011;108:6909-14
33. Cung K, Slater RL, Cui Y. et al. Rapid, multiplexed microfluidic phage display. Lab Chip. 2012;12:562-5
34. Matochko WL, Ng S, Jafari MR. et al. Uniform amplification of phage display libraries in monodisperse emulsions. Methods. 2012;58:18-27
35. Noseda A, Berens ME, Piantadosi C. et al. Neoplastic cell inhibition with new ether lipid analogs. Lipids. 1987;22:878-83
36. Bénard J, Silva JD, Blois MCD. et al. Characterization of a human ovarian adenocarcinoma line, IGROV1, in tissue culture and in nude mice. Cancer Res. 1985;45:4970-9
37. Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol. 2005;23:1105-16
38. Wang CH, Lee GB. Automatic bio-sampling chips integrated with micro-pumps and micro-valves for disease detection. Biosen Bioelectron. 2005;12:419-25
39. Weng CH, Lien KY, Yang SY. et al. A suction-type, pneumatic microfluidic device for liquid transport and mixing. Microfluid Nanofluid. 2011;10:301-10
40. Weng CH, Hsieh IS, Hung LY. et al. An automatic microfluidic system for rapid screening of cancer stem-like cell-specific aptamers. Microfluid Nanofluid. 2013;14:753-65
41. Weng CH, Yeh WM, Ho KC. et al. A microfluidic system utilizing molecularly imprinted polymer films for amperometric detection of morphine. Sens Actuators B Chem. 2007;121:576-82
42. Wang CH, Chang CJ, Wu JJ. et al. An integrated microfluidic device utilizing vancomycin conjugated magnetic beads and nanogold-labeled nucleotide probes for rapid pathogen diagnosis. Nanomed Nanoech Biol Med. 2014;10:809-18
43. Chang CM, Chiu LF, Wang PW. et al. A microfluidic system for fast detection of mitochondrial DNA deletion. Lab Chip. 2011;11:2693-700
44. Wang CH, Lai HC, Liou TM. et al. A DNA methylation assay for detection of ovarian cancer cells using a HpaII/MspI digestion-based PCR assay in an integrated microfluidic system. Microfluid Nanofluid. 2013;15:575-85
45. Nanus DM. Of peptides and peptidase: the role of cell surface peptidase in cancer. Clin Cancer Res. 2003;9:6307-9
46. Fais S, De Milito A, You H. et al. Targeting vacuolar H+-ATPase as a new strategy against cancer. Cancer Res. 2007;22:10627-30
47. Monaco ME, Creighton CJ, Lee P. et al. Expression of long chain fatty acyl-CoA synthetase 4 in breast and prostate cancer is associated with sex steroid hormone receptor negativity. Trans Oncol. 2010;3:91-8
48. Hua YQ, Jia XF, S MX. et al. Plasma membrane proteomic analysis of human osteosarcoma and osteoblastic cells: revealing NDRG1 as a marker for osteosarcoma. Tumor Biol. 2011;32:1013-21
49. Hung LY, Wang CH, Hsu KF. et al. An on-chip Cell-SELEX process for automatic selection of high-affinity aptamers specific to different histologically classified ovarian cancer cells. Lab Chip. 2014;14:4017-28
50. Yang L, Nolan JP. High-throughput screening and characterization of clones selected from phage display libraries. Cytometry Part A. 2007;71A:625-31
51. McGuire MJ, Gray BP, Li S. et al. Identification and characterization of a suite of tumor targeting peptides for non-small cell lung cancer. Sci Rep. 2014;4:4480
Author contact
![]() Corresponding author: Dr. Gwo-Bin Lee. E-mail: gwobinnthu.edu.tw; Tel: +886-3-5715131 Ext. 33765; Fax: +886-3-5742495
Corresponding author: Dr. Gwo-Bin Lee. E-mail: gwobinnthu.edu.tw; Tel: +886-3-5715131 Ext. 33765; Fax: +886-3-5742495
Citation styles
APA
Wang, C.H., Weng, C.H., Che, Y.J., Wang, K., Lee, G.B. (2015). Cancer Cell-Specific Oligopeptides Selected by an Integrated Microfluidic System from a Phage Display Library for Ovarian Cancer Diagnosis. Theranostics, 5(4), 431-442. https://doi.org/10.7150/thno.10891.
ACS
Wang, C.H.; Weng, C.H.; Che, Y.J.; Wang, K.; Lee, G.B. Cancer Cell-Specific Oligopeptides Selected by an Integrated Microfluidic System from a Phage Display Library for Ovarian Cancer Diagnosis. Theranostics 2015, 5 (4), 431-442. DOI: 10.7150/thno.10891.
NLM
Wang CH, Weng CH, Che YJ, Wang K, Lee GB. Cancer Cell-Specific Oligopeptides Selected by an Integrated Microfluidic System from a Phage Display Library for Ovarian Cancer Diagnosis. Theranostics 2015; 5(4):431-442. doi:10.7150/thno.10891. https://www.thno.org/v05p0431.htm
CSE
Wang CH, Weng CH, Che YJ, Wang K, Lee GB. 2015. Cancer Cell-Specific Oligopeptides Selected by an Integrated Microfluidic System from a Phage Display Library for Ovarian Cancer Diagnosis. Theranostics. 5(4):431-442.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.