Impact Factor
- Issue 14; 2026
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Volume 16; 2026
- Advance Articles
- Past Issues
- Cover Images
- Cover Suggestion
- Index & Coverage
- Special Issues
1. Introduction
2. Theranostics for macrophage...
3. Limitations and Future...
4. Conclusions
Abbreviations
Acknowledgements
References
Author Biography
International Journal of Biological Sciences
International Journal of Medical Sciences
 Global reach, higher impact
Global reach, higher impact

Theranostics 2015; 5(2):150-172. doi:10.7150/thno.9476 This issue Cite
Review
Macrophage Targeted Theranostics as Personalized Nanomedicine Strategies for Inflammatory Diseases
Sravan Kumar Patel, Jelena M. Janjic ![]()
Graduate School of Pharmaceutical Sciences, Duquesne University, 600 Forbes Ave, Pittsburgh, PA, 15282, USA.
Received 2014-4-23; Accepted 2014-6-28; Published 2015-1-1
Abstract
Inflammatory disease management poses challenges due to the complexity of inflammation and inherent patient variability, thereby necessitating patient-specific therapeutic interventions. Theranostics, which integrate therapeutic and imaging functionalities, can be used for simultaneous imaging and treatment of inflammatory diseases. Theranostics could facilitate assessment of safety, toxicity and real-time therapeutic efficacy leading to personalized treatment strategies. Macrophages are an important cellular component of inflammatory diseases, participating in varied roles of disease exacerbation and resolution. The inherent phagocytic nature, abundance and disease homing properties of macrophages can be targeted for imaging and therapeutic purposes. This review discusses the utility of theranostics in macrophage ablation, phenotype modulation and inhibition of their inflammatory activity leading to resolution of inflammation in several diseases.
Keywords: macrophages, theranostics, inflammation, phenotype, photodynamic therapy and photothermal therapy.
1. Introduction
Recognizing the heterogeneity of diseases and inter-patient variability, there has been a shift in disease management from a generalized approach towards personalized treatment strategies. Development of dedicated therapeutic interventions in collaboration with specific disease markers (companion diagnostics) was the first step towards achieving this goal. This strategy was originally referred to as 'theranostics' i.e. specific therapy based on molecular diagnosis.[1] An appropriate example is Herceptin® for the treatment of breast cancer, which expresses a specific biomarker, human epidermal growth factor receptor 2. Theranostics are also defined as an integration of therapeutic and diagnostic or imaging functionalities on a single platform that can diagnose, deliver drugs and monitor the therapy leading to individualized treatment.[2, 3] By fusing imaging and therapeutic functionalities together, it is possible not only to visualize and track the biodistribution of the theranostic in real-time but also to predict efficacy and toxicities based on the tissue accumulation.[3] This information can be used to adjust or modify the treatment strategy.
Nanotechnology has taken center stage in the development of theranostics.[4] Nanoparticles possess promising features to be utilized as theranostics, namely high surface-area-to-volume ratio that yield high therapeutic and imaging agent loading, surface functionalization with targeting ligands and small size for extravasation to leaky vasculature.[5] They can be functionalized to modulate the release based on environmental stimuli such as pH, temperature, enzymes, and redox potential [6]. Blood circulation times of nanoparticles can be enhanced by surface functionalization with a hydrophilic polymer, polyethylene glycol (PEG). [5] High therapeutic and imaging payload, combined with targeted delivery, can increase the therapeutic and imaging efficacy while reducing off-site toxicity. Nanoconstructs such as gold nanoparticles [7] and carbon nanotubes [8, 9] also possess inherent theranostic functionalities due to their photothermal and optical properties.
Due to the highly variable treatment efficacies and the heterogeneity of cancer, much theranostic research is focused on oncology. Similar to cancer, inflammatory disease management poses a challenge due to the complexity of inflammation and inherent patient variability.[10-13] This necessitates the use of patient-specific therapeutic interventions. Cancer, cardiovascular, neurodegenerative and autoimmune diseases have an active inflammatory component.[14-17] Inflammatory diseases are a major contributor to global health costs estimated at $57.8 billion in 2010.[18] As a result, theranostics are now being investigated for inflammatory diseases. Additionally, theranostic technology transfer to inflammation research has become feasible, as the principles governing the theranostic design and the biological players involved coincide with cancer.[19]
Inflammation can be broadly defined as a host's response to infection, injury or metabolic imbalance to restore homeostasis.[20] Aberrant or prolonged inflammation can produce significant endogenous tissue injury leading to several diseases.[21, 22] Macrophages are a major cellular component present in the inflammatory milieu.[23] The inflammatory mediators produced by macrophages cause significant tissue injury leading to initiation, promotion and progression of many diseases.[24] Due to their abundance and pathogenic roles, macrophages have been targeted for therapeutic and imaging purposes. For example, visualization of macrophage infiltration by targeted imaging agents has been used to assess disease severity and treatment efficacy.[25-27] Anti-inflammatory therapies targeting macrophages by specific ablation [28], inhibition of their infiltration [29] and reduction of pro-inflammatory mediator release [30] has shown efficacy in rheumatoid arthritis (RA), atherosclerosis, vascular injury and cancer. However, anti-inflammatory therapies have shown variable efficacy results across the patient population.[31, 32] In some instances, significant depletion of macrophages has been associated with immunosuppression, infection [33, 34] and reduced wound healing.[35] This combination of therapeutic and harmful effects can be attributed to the different activated states of macrophages in disease environments.[36]
To delineate the protective and detrimental effects of targeting macrophages, there is a need to bring therapy and diagnosis together. In this regard, theranostics could provide essential information about the delivery of drug carriers to macrophages as well as their biodistribution, treatment efficacy and toxicity profile in real-time, leading to better therapeutic intervention. This review focuses on the exploration of macrophages as targets for theranostics with the intention of simultaneous imaging and therapy. A brief introduction on the role of macrophages in diseases and an overview of molecular imaging techniques used in imaging macrophages is presented. Studies utilizing nano- and micron-sized particles as theranostics for macrophages in different disease models are described. Ultimately, critical analysis of the current theranostic approaches in macrophage modulation and the potential opportunities and limitations for clinical translation are discussed.
1.1. Role of macrophages in inflammation and pathogenesis
Macrophages are a type of leukocyte belonging to the mononuclear phagocytic system. They are derived from progenitor cells of hematopoietic origin in bone marrow.[37] They are replenished on a regular basis from circulating blood monocytes.[37] Macrophages are constitutively present in all the tissues where they participate in tissue survival, regulation and modeling processes.[36] Their primary function is to clear by phagocytosis apoptotic cells and cellular debris generated during tissue remodeling and necrosis. In addition to these homeostatic roles, macrophages are essential immune cells participating in both innate and adaptive immunity.[23] During inflammation, macrophages are recruited to remove the injurious stimuli and aid in the wound healing process.[38]
Inflammation represents a highly complex network of cellular and sub-cellular components, which work in a regulated fashion to defend the host against deleterious stimuli. [21] In response to injurious stimuli, local endothelial and immune cells release inflammatory mediators, which increase blood flow and vascular permeability. Leukocytes such as neutrophils and macrophages migrate to the site of injury based on released chemotactic factors (cytokines and chemokines) by a process known as diapedesis. The inflammatory endothelium and local immune cells also upregulate cell adhesion molecules (CAMs) such as selectins, integrins and cadherins, which further aid in the adhesion and transmigration of leukocytes.[39] Amongst leukocytes, neutrophils are the first responders to the site of injury and act to remove the stimuli by phagocytosis and release of inflammatory mediators. Neutrophils are present in the first few hours to days, while macrophages are recruited by further downstream signals to aid in resolution and repair.[39] This cascade is known as acute inflammation which is self-limiting and results in the restoration of homeostasis. Failure to cease the injurious stimuli leads to chronic inflammation.
During the chronic phase, macrophages are continuously recruited and release inflammatory mediators such as chemokines, cytokines, lipid mediators, proteases and reactive oxygen species (ROS).[23] These mediators cause detrimental effects to the host leading to initiation, exacerbation and progression of several infectious and non-infectious diseases. This review majorly focuses on pathologies involving non-infectious chronic inflammatory conditions. One example of these is rheumatoid arthritis (RA), in which macrophages have a prominent role through their production of inflammatory mediators that cause considerable inflammation and joint destruction.[40] Another example is Type 2 diabetes, in which accumulation of macrophages is associated with several complications such as neuropathy, nephropathy and atherosclerosis.[41] A third example is atherosclerosis, in which macrophages uptake low density lipoprotein and transform into lipid-laden cells called foam cells that contribute to initiation and progression of atherosclerotic lesions.[42] Additionally, proteases produced by macrophages degrade the extracellular matrix (ECM), which leads to plaque rupture and myocardial infarction (MI). Also, tumor-associated macrophages (TAMs) are involved in several tumorigenic activities leading to angiogenesis and metastasis.[43] Similarly, macrophages are involved in several stages of pathogenesis in neurological, auto-immune, cardiovascular and pulmonary diseases.[40, 44, 45]
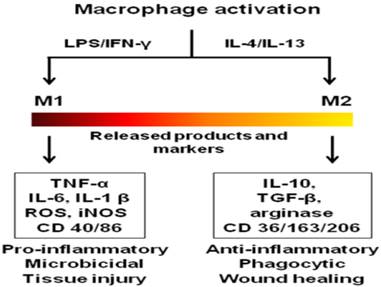
In addition to the discussed pathogenic roles, macrophages also display protective functions in chronic inflammatory diseases. Depending on the pathological environment, macrophages adapt to different phenotypes which exacerbate or resolve the disease.[36] They are broadly classified as classically activated (M1) pro-inflammatory and alternatively activated anti-inflammatory (M2) phenotypes mirroring Th1 and Th2 states of T cells.[46] Due to the vast biochemical and physiological differences within M1 and M2, it was suggested that macrophages form part of a continuum and possess overlapping functions of different macrophage subsets.[47] For the purpose of this review, the two extreme states of macrophages, M1 and M2 are considered. Figure 1 shows the activation states and the biomarkers of macrophage phenotypes. The diverse roles of macrophages are attributed to these phenotypes. For example, TAMs exhibit M1 and M2 phenotypes depending on the local cytokine milieu. M2 macrophages are involved in the release of growth factors, angiogenesis and degradation of the ECM leading to metastasis, while M1 macrophages exhibit tumoricidal activity.[48] In atherosclerosis, macrophages participate in the initiation, progression and rupturing of atheroma.[15] In contrast, a subset of macrophages (M2) is involved in the resolution and tissue remodeling of the disease.[49] Likewise, macrophages play a diverse role in autoimmune [50], pulmonary [44] and neurological diseases.[45] Therefore, macrophages represent promising targets for the treatment and diagnosis of inflammatory diseases.
Activation states, released products and functions of macrophage phenotypes. The colored bar indicates macrophage phenotype continuum with M1 and M2 at the extreme ends. LPS-lipopolysaccharide (bacterial toxin), IL-interleukin; IFN-γ-interferon gamma; TNF-tumor necrosis factor; ROS-reactive oxygen species; iNOS-inducible nitric oxide synthase; TGF-transforming growth factor; CD-cluster of differentiation.
1.2. Imaging modalities
Molecular imaging techniques are indispensible for understanding the role of macrophage dynamics in a pathophysiological setting. Molecular imaging has been applied to study macrophage infiltration, modulation and spatio-temporal distribution in neurological diseases [25, 26], atherosclerosis [51, 52] and RA.[53] In these cases, imaging macrophages provided opportunities to assess the severity of the disease [54] and therapy efficacy [27] which could help in strategizing the treatments. Several imaging agents can be incorporated for the purpose of detection, quantification and in vivo localization of theranostics or labeled macrophages. Imaging modalities and imaging agents used in macrophage theranostic applications are briefly reviewed here. Imaging modalities differ in their spatio-temporal resolution, depth of penetration, acquisition time, safety, cost and clinical applicability. Most imaging modalities utilize electromagnetic radiation of different energies to obtain anatomical and functional information. They can be broadly divided into those utilizing ionizing (high energy) and non-ionizing (low energy) radiation with the exception of ultrasound imaging, which uses high frequency sound waves.
1.2.1. Nuclear imaging
Nuclear imaging techniques such as positron emission tomography (PET) and single-photon emission computed tomography (SPECT) detect β or γ-radiation from radionuclides localized within the body. PET detects two coincident high energy photons that result from annihilation of β+ and surrounding tissue electrons.[55] SPECT detects the decay of γ-rays emitting radioisotopes (99mTc, 123I). PET and SPECT are 3D-tomographic techniques with nanomolar to picomolar detection sensitivity [56], but with low spatial (1-4 mm) resolution.[57] They are widely used non-invasive clinical imaging tools. However, the use of ionizing radiation is a concern with nuclear imaging techniques. The requirement for cyclotrons to produce radioisotopes and their short half lives (typically a few hours), may reduce the time available to perform the required quality controls. The development of long-lived radioisotopes such as 124I (t1/2 = 4.2 days) has increased their clinical and preclinical research applications. To obtain anatomical context, PET and SPECT are usually coupled with computed tomography (CT), magnetic resonance imaging (MRI) and ultrasound imaging. CT utilizes ionizing radiation (X-rays) whereas the latter use non-ionizing radiation and sound waves respectively. Combined nuclear and ionizing radiation-based imaging modalities, although providing excellent sensitivity and resolution, are limited due to potential radiation exposure, especially for multiple imaging sessions.
1.2.2. Magnetic resonance imaging
MRI is a commonly used non-invasive clinical diagnostic tool utilizing non-ionizing radio waves. MRI has high spatial resolution (~100 µm) and unlimited tissue penetration.[56, 58] Clinically, MRI is used for detection, staging, image guided surgery and assessment of therapy responses in many diseases. In MRI, a strong magnetic field aligns magnetically susceptible nuclei such as 1H and 19F. Radio frequency pulses are then used to excite the nuclei and their transverse and longitudinal relaxation properties (T1 and T2) are detected to construct an image.[58] Based on the tissue environment, the density and relaxation properties of 1H nuclei vary leading to contrast. Tissues with low proton density such as lungs are not readily visible in MRI. In addition, it has low sensitivity (10-3 to 10-9 M) and requires long acquisition time. MRI can evidently delineate soft tissues and can be used for repetitive imaging sessions. Signal to noise ratio (SNR) in MRI can be increased by targeted contrast agents.
1H MRI contrast agents
Most contrast agents are paramagnetic gadolinium or superparamagnetic iron oxide (SPIO) constructs. They function by altering the relaxation time of the surrounding protons, thereby producing contrast. 1H contrast agents are not detected directly, but their effect on relaxation of surrounding water molecules produces contrast.[58, 59] Contrast agent-based MRI has been used clinically for gastrointestinal and blood pool contrast as well as to visualize many pathological conditions. To obtain high target concentrations, contrast agents are incorporated in nanoparticles and can be targeted to a specific tissue. The sensitivity of MRI contrast agents is usually in 10-3 M range.[60] Nanoparticles incorporating iron oxide and gadolinium agents have been used to image macrophages in pathologies such as atherosclerosis [52], RA [61] and multiple sclerosis.[62] Although contrast can enhance the local signal, the inherent inhomogeneities of tissues limit the unambiguous detection of a pathological condition.[63]
19F MRI
19F MRI detects organic fluorine, which is introduced exogenously into the host. Similar to 1H, the fluorine (19F) nucleus has a half spin, comparable MRI sensitivity (83%) and resonance (differs by 6%) to 1H.[64] Therefore, 1H MRI machines can detect 19F nuclei by tuning to an appropriate frequency. 19F MRI is not a contrast imaging as there is virtually no imageable endogenous fluorine present in the body. To obtain 19F images, non-toxic perfluorocarbons (PFCs) with a large number of fluorine atoms are introduced into the body in biocompatible formulations.[58, 65] 1H MRI can be registered during the same session to obtain anatomical context for the 19F signal. Similar to 1H MRI, 19F MRI has 10-3 M detection sensitivity.[66] To increase SNR, gradient echos, ultrafast radial sequences and paramagnetic ions in close proximity to PFCs can be used.[66] Nanoparticles, liposomes and nanoemulsions incorporating PFCs for 19F MRI are extensively reported.[64, 67] PFC nanoemulsions have been utilized to study cell-cell interactions in a whole animal [64], label macrophages for in vivo inflammation quantification [68] and track the biodistribution of drug loaded nanoparticles.[69-71] Successful in vivo macrophage visualization using 19F MRI has also been reported in cancer [67, 72], diabetes [68], RA [73], pulmonary inflammation [74], and transplantation models.[75] Unlike 1H contrast agents, 19F nuclei can be quantified in vivo due to near zero background [76] and imaging prior to PFC administration is not required. Clinical use of 19F MRI is still in its infancy, however recently clinical trials have been initiated to use 19F MRI in cell based immunotherapy.[77]
1.2.3. Optical imaging
Optical imaging is a widely employed tool for cellular, sub-cellular and whole animal imaging in preclinical research, due to its low cost and high detection sensitivity (picomolar to femtomolar).[82] Optical imaging utilizes electromagnetic radiation in the ultraviolet, visible and near-infrared (NIR) region to produce images based on the emitted light. A source of detected light can be from bioluminescence or fluorescence. Bioluminescence imaging utilizes the chemical reaction between luciferase and a substrate to produce detectable light. Bioluminescence imaging is primarily restricted to preclinical studies as genetically modified luciferase expressing cells are required. Fluorescence imaging is one of the most commonly used techniques where light of an appropriate wavelength is used to excite an endogenous or externally introduced fluorescent moiety and the light emitted at a longer wavelength is detected.[82] Light scattering and tissue attenuation effects with visible light based fluorophores limit their use for deep tissue in vivo imaging. Fluorophores active in the NIR (700-1000) region can reduce the tissue effects and increase penetration depth up to few centimeters.[83] Fluorescence imaging is regularly used in preclinical setting to visualize bioaccumulation of drug carrying nanoparticles to macrophages. Many organic and inorganic fluorescent dyes are used for this purpose such as cyanine derivatives (indocyanine green) and quantum dots. [84] With the availability of different wavelength lasers and emission filters, a combination of fluorescent reporters can be used to study the complex inflammatory mechanisms. Fluorescence reporters can also be equipped with pH, enzyme, temperature, and redox sensitivity.[85] For example, in vivo fluorescence molecular tomography (FMT) was used to visualize protease enzyme activity in macrophages labeled with a protease active fluorescent reporter in a mouse atherosclerosis model.[80] Clinically, fluorescence imaging is used for surface tissue imaging (breast), intravital microscopy and real-time image guided surgery.[86] However, it is limited by quenching and photobleaching effects of fluorophores.
1.2.4. Multimodal imaging
Since no imaging modality ideally serves all research and medical needs, multiple imaging agents are integrated into a single platform. Multimodal nanoparticles with complementary imaging agents offer high sensitivity detection, anatomical localization and data validation from different imaging modalities.[56] Nanoparticles incorporating combinations of optical, MR and PET imaging agents have been reported.[65, 72] Imaging macrophages labeled with PET and MRI multimodal nanoparticles has been demonstrated.[81] Availability of multimodal imaging instruments combining MRI, CT, optical, PET and ultrasound have helped to obtain anatomical and molecular imaging simultaneously. Functional PET imaging has been combined with CT to precisely locate the radio isotope labeled macrophages.[78] Similarly, combination of functional 19F MRI and fluorescence with anatomical MRI and CT respectively has been reported. [74, 80]
2. Theranostics for macrophage detection and therapy
The pathogenic roles of macrophages coupled with their phagocytic nature and abundance are the key features that make them feasible targets for theranostics. Although macrophages can readily phagocytose nanoparticles, several attributes, namely size, shape, surface charge, targeting ligands, and surface functionalization with PEG influence their in vivo targeting efficiency and performance. The physicochemical characteristics of macrophage-targeted nanosystems have been reviewed elsewhere.[87] Macrophages express several receptors that can be used for active targeting with ligands such as dextran [7, 88], tuftsin [89], mannose [90], and hyaluronate.[87] The disrupted vasculature in the inflammatory environment provides an additional opportunity to passively target theranostic nanosystems.[19] As blood monocytes are continuously recruited to sites of inflammation, monocytes and macrophages have an intrinsic disease-homing property which can be utilized for targeting theranostics.[19] Through these properties, macrophages have been used as Trojan horses to deliver drugs and imaging agents to the disease site.[91, 92]
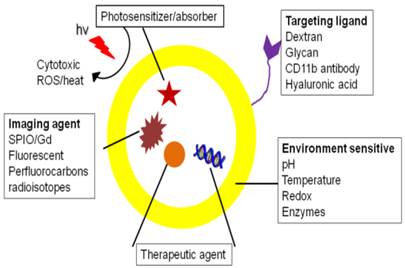
Theranostics that target macrophages and associated pro-inflammatory substances have been investigated to visualize and treat the underlying pathology. In this section, theranostic applications are categorized based on therapeutic approaches, divided into ablation and non-ablation sections. Macrophage ablation uses photo and chemotherapy, while non-ablation approaches include phenotype change, and inhibition of macrophage infiltration and pro-inflammatory mediators. A schematic of multifunctional theranostics for macrophages is shown in figure 2.
Schematic showing a theranostic with targeting, imaging and therapeutic functionalities.
2.1. Theranostics for macrophage ablation
Reduction in macrophages deprives the pathological environment of macrophage-mediated pro-inflammatory mechanisms, and thus reduces inflammation.[7] Selective macrophage ablation was the major strategy used in experimental studies to investigate their role in pathogenesis of several diseases. Macrophage depletion has been applied in the treatment of atherosclerosis [93], restenosis [94], RA [28] and cancer.[95] However, the lack of selectivity for disease specific macrophages could lead to immunosuppression and infections as observed previously.[33-35] In this section, the potential utility of theranostics to enhance selective macrophage depletion using photo and chemotherapy based approaches is presented.
2.1.1. Photodynamic therapy
Photodynamic therapy (PDT) is an externally activatable treatment modality utilizing photosensitizers that produce cytotoxic singlet oxygen upon illumination.[96] Photosensitizers are chemicals that absorb light and react with oxygen to produce ROS, singlet oxygen and radicals, which are cytotoxic and damage the tissue.[96] Their inherent fluorescence and the ability to produce ROS makes photosensitizers theranostic agents. Since photosensitizer accumulation and photo-irradiation are required to release the cytotoxic products, PDT is a selective technique. PDT is clinically used to treat cancer, skin conditions and other surface accessible pathologies.[96] Typical therapy includes photosensitizer administration, time delay allowing accumulation of photosensitizers in the diseased region (while washing out from other tissues) and finally photo-irradiation of the tissue. Accumulation in the diseased tissue is driven by increased vascular permeability. Ideal photosensitizers are non-fluorescent and non-toxic in the absence of light, and provide selectivity to the target cells. Photosensitizers that are active in the NIR region are particularly attractive because background absorption, autofluorescence and non-specific tissue toxicity can be minimized.[2] Clinically used photosensitizers are derivatives of porphyrins, chlorophylls and cyanine dyes.[97] Theranostics have promising applications in PDT, since the imaging component can be used for light dosimetry and guidance, real-time therapy modulation and therapeutic efficacy assessment.[2]
Small molecule photosensitizers such as 5-aminolevulinic acid (ALA) have been used for macrophage PDT in RA [98] and atherosclerosis.[99] ALA preferentially accumulates in inflamed tissues and converts to a fluorescent photosensitizer, protoporphyrin IX [100]. Due to its low molecular weight and non-selectivity, ALA has been shown to accumulate not only in macrophages, but also in endothelial cells.[98, 99] Endothelial cell destruction could be detrimental in diseases such as atherosclerosis because they are involved in plaque stabilization. Low molecular weight photosensitizers have also showed short half lives, thereby requiring multiple administrations that could lead to skin sensitivities.[2]
To overcome rapid elimination from the target tissue, non-specific accumulation, high dose requirements, and low water solubility, nanoparticles incorporating photosensitizers have been developed. In order to increase tissue retention, Schmitt et al. developed chitosan-hyaluronate nanogels incorporating different anionic porphyrin-based photosensitizers and evaluated them using a mouse arthritis model.[101] The fluorescence of the photosensitizer aided in the assessment of cellular uptake and joint residence time, the latter of which was higher for nanogels when compared to free photosensitizer. Since the nanogels were functionalized with a targeting ligand, hyaluronate, selective uptake in macrophages (but not in fibroblasts) was observed due to the increased phagocytosis. In addition to selective uptake, in vitro macrophage depletion was observed only with the combination of nanogels and light demonstrating the selectivity of the theranostic. Imaging also helped to select the delay time required before PDT, which was obtained from the time showing the highest joint fluorescence. Based on these results, PDT (diode laser at 652 nm) was performed 2.5 h after injection of nanogels in the synovial cavity of an arthritic mouse. PDT and a standard corticosteroid treatment showed comparable serum amyloid A content. Serum amyloid A is an acute phase protein used in clinical diagnosis of RA. In this study, the theranostic helped increase the retention and assess the uptake, joint residence and delay time. However, invasive local injection could reduce the patient acceptance of this theranostic. Further exploration of nanogel retention and PDT efficacy after intravenous (i.v.) administration is warranted. Since one of the photosensitizers used in the study (chlorin e6) has previously shown skin toxicity [102], photosensitivity tests are required. Finally, the mechanism of cell death needs to be explored as PDT can cause necrosis and apoptosis [2] and necrotic cell death can lead to tissue inflammation.[103]
Another major area where macrophage targeted theranostics for PDT has been studied is atherosclerosis. In atherosclerosis, macrophages contribute to plaque instability leading to stroke and MI. McCarthy et al. developed cross-linked dextran-coated iron oxide (CLIO) nanoparticles for depletion of atherosclerotic macrophages.[104] CLIO nanoparticles are widely used in drug delivery and imaging because macrophages can selectively uptake these nanoparticles.[105] A fluorescence and MR visible (Fig. 3A) multimodal theranostic was constructed by conjugating CLIO to a NIR fluorescent dye, Alexa Fluor 750 (AF750) and a photosensitizer, 5-(4-carboxyphenyl)-10,15,20-triphenyl-2,3-dihydroxychlorin (TPC). In vitro studies in mouse macrophages showed cytotoxicity of the theranostic only upon illumination (Fig. 3B); however, this preparation had stability problems associated with flocculation and low conjugation efficiency of the hydrophobic TPC.
PDT using dextran cross-linked iron oxide (CLIO) theranostic nanoparticles (TNP). A) 1H MRI and fluorescence images of TNPs. B) Percent cell viability, as determined by the MTS assay, of human macrophages after incubation (1 h) with the respective nanoparticles (0.2 mg Fe per mL) and light treatment (42 mWcm-2, 7.5 J). The TNP dark experiment consisted of TNP exposed cells that did not receive PDT treatment. Control cells were incubated with saline. MNP-magnetic nanoparticles, MFNP-magnetofluorescent nanoparticles (fluorescent labeled iron oxide), TNP-theranostic nanoparticles (fluorescent/iron oxide/photosensitizer). C) Phototoxicity (650 nm, 50 mWcm-2) of second generation CLIO-THPC (photosensitizer) theranostics compared with a conventional photosensitizer, chlorin e6. D) Intravital fluorescence microscopy image (top) of CLIO-THPC localized to carotid atheroma. Fluorescence image obtained in the AF750 channel demonstrates particle uptake. Fluorescence angiogram utilizing fluorescein-labeled dextran outlines the vasculature (middle). Merged image of the two fluorescence channels (bottom). E) Fluorescence microscopy (excitation: 750 nm) of an aortic root plaque section, 24 hours after fluorescent CLIO-THPC injection shows subendothelial deposition of CLIO-THPC in atheroma of atherosclerotic mouse (ApoE-/-). F) Skin photosensitivity of chlorin e6 versus CLIO-THPC based on the change in thickness in the treated edema paw 24 hours after laser irradiation (** p=0.009, * p=0.02). Figures adapted and reprinted with permission from references [102, 104].
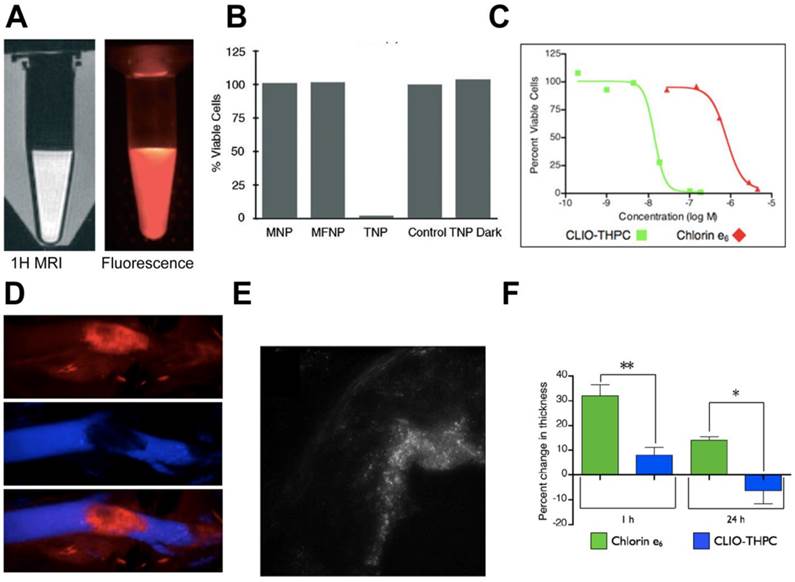
In a subsequent study, second generation CLIO nanoparticles with a hydrophilic photosensitizer, meso-tetra(m-hydroxyphenyl)chlorin (THPC) were reported.[102] Compared to TPC, a 3-fold increased loading was observed with THPC. As shown in figure 3C, fluorescently labeled nanoparticles (CLIO-THPC-AF750) exhibited cytotoxicity upon illumination with significantly lower LD50 of 14 nM compared to a conventional photosensitizer, chlorin e6 (800 nM). PDT (650 nm laser) was performed on exposed carotid artery of an atherosclerotic mouse after i.v. administration of nanoparticles. Intravital fluorescence microscopy, fluorescence imaging and histology showed nanoparticle accumulation in the apoptotic macrophages of the atherosclerotic plaque (Fig. 3D-E). THPC-free nanoparticles were PDT inactive showing the inertness of the delivery vehicle. Compared to chlorin e6, CLIO-THPC showed minimum skin phototoxicity (Fig. 3F) validating the benefit of using targeted nanoparticles. The construct was rationally designed to impart therapeutic and dual mode imaging capabilities, but the multistep synthesis and purification involved in preparation warrants further optimization. Also, the presence of residual organic impurities and solvents needs to be carefully evaluated. From a clinical standpoint, MRI can precisely visualize the in vivo localization of CLIO nanoparticles prior to PDT, while the NIR dye can aid in microendoscopic fluorescence imaging for image guided therapy. Meanwhile, although the scaffold showed efficient macrophage delivery and potent phototoxicity, the effect of macrophage ablation on plaque reduction was not shown. Even though non-invasive i.v. administration was utilized, surgical exposure of carotid artery reduces the potential applicability in a clinical setting. The nanoconstruct could be modified with a longer wavelength (>800 nm) photosensitizer to enable deep tissue imaging and therapy without the need for surgical exposure.
To further improve selectivity towards plaque rupturing macrophages, Shon et al. reported an enzyme sensitive theranostic.[106] A protease activated photodynamic agent (L-SR15) was utilized to selectively deplete cathepsin-B (Cat B) producing macrophages.[107] Cathepsin, a protease enzyme involved in matrix degradation, can cause plaque rupture.[108] In its native state, L-SR15 is non-toxic and non-fluorescent, but proteolytic cleavage by Cat B releases the fluorescent photosensitizer, chlorin e6. Fluorescence imaging after multiple i.v. administrations showed accumulation of L-SR15 in a mouse atherosclerotic model. Cat B activity, in response to PDT, was assessed in vivo and ex vivo using a Cy7-Cat B sensing fluorescent probe. L-SR15 showed enhanced fluorescence and reduced Cat B activity compared to D-SR16 (a Cat B inactive chlorin e6 conjugate) and saline. Cat B reduction was exhibited only by carotid arteries subjected to PDT, thus showing the selectivity of L-SR15. Skin phototoxicity assessments showed significantly larger damage in areas treated with free chlorin e6 compared to L-SR15. The study clearly demonstrated the multilevel selectivity of the construct, which means that theranostic accumulation, Cat B specific activation and external photo illumination are required for macrophage ablation. However, the efficiency of this strategy is compromised by the requirements for multiple injections and PDT with invasive surgery. Also, further studies are required to investigate the effect of Cat B producing macrophage depletion on plaque stability and size. Finally, translating this theranostic to other diseases is not feasible as the role of Cat B is primarily implicated in atherosclerosis. Nevertheless, such stimuli-sensitive theranostics promise increased selectivity while sparing benign cells. Similarly, thrombin-sensitive photosensitizer theranostics have been applied for increased selectivity in thrombin-rich synovial sites of RA.[109, 110]
The major aims of photodynamic theranostics in macrophage ablation are to increase retention, targeting and selectivity to reduce off-target toxicities. It is important to note that in certain diseases, a subset of macrophages show beneficial effects (e.g. M2 macrophages in atherosclerosis) and their depletion could hamper wound healing. A more selective targeting of macrophage subsets, such as enzyme activated PDT, could overcome this problem. There is also a need for imaging and treatment of deep tissues to widen the spectrum of applications for these theranostics. To some extent, NIR activatable photosensitizers can increase the penetration depth of PDT. Furthermore, MRI and PET contrast agents can help to visualize the theranostic accumulation in deep tissues, while microendoscopic techniques could be used to deliver light and guide the therapy. These multimodal constructs have already been utilized in vivo for cancer [111] and the technology could be transferred to inflammatory diseases.
2.1.2. Photothermal therapy
Similar to PDT, photothermal therapy (PTT) is an externally activatable light therapy. PTT utilizes photoabsorbers such as organic dyes to generate localized hyperthermia upon illumination by visible-NIR light. [2] Hyperthermia can induce cell death by protein denaturation and disruption of the cytoskeleton. [7] Unlike photosensitizers, photoabsorbers do not produce cytotoxic oxygen products. Preferably, NIR-absorbing (650-900 nm) material is used to enable treatment of deep tissues (up to few centimeters). To overcome the limitations of photobleaching and reduced absorbance by small molecule organic dyes, several nanomaterials such as gold nanorods (GNRs), nanoshells and carbon nanotubes have been studied as NIR absorbing materials.[9, 88, 112] Additionally, these materials have optical properties that can be used for in vitro and in vivo imaging. Since external irradiation is required to generate heat, PTT is a targeted therapy similar to PDT.
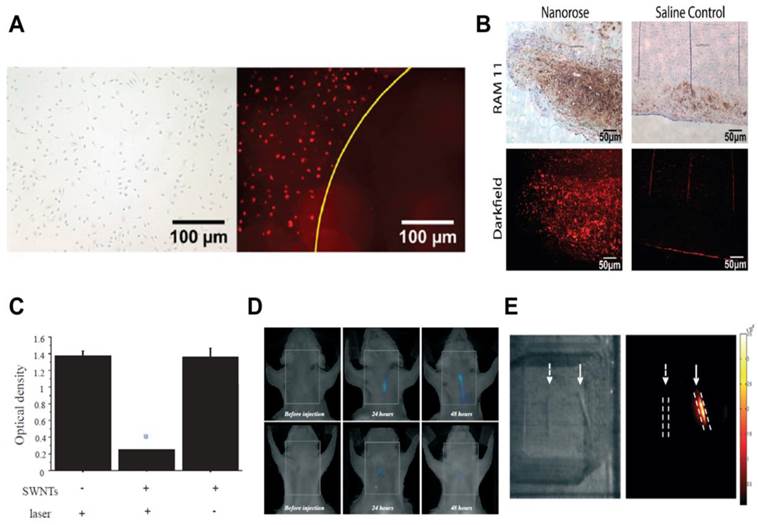
Nanoparticles containing gold are promising for PTT as they are non-photobleaching, generate rapid heat, and possess optical properties for imaging.[2] Ma et al. reported the use of dextranated iron oxide nanoparticles with surface gold coating (nanoroses) for thermal ablation of atherosclerotic macrophages.[112] Dextran coating provided colloidal stability during preparation and storage (for 8 months stability), apart from acting as a ligand for macrophages. NIR reflectance imaging showed selective uptake of nanoroses by macrophages compared to endothelial and smooth muscle cells, while NIR irradiation exhibited macrophage death by apoptosis in vitro (Fig. 4A). In an in vivo rabbit atherosclerosis model, i.v. administration of nanoroses and ex vivo analysis showed their presence in plaque macrophages (Fig. 4B). Since these nanoroses incorporate iron oxide, they provide an additional imaging capability, MRI. Furthermore, the authors prepared multimodal nanoroses by a one-step self-assembly process, which shows potential for large scale production. Although the nanoroses showed uptake in plaque macrophages, further studies are needed to ascertain their in vivo efficacy.
PTT of macrophages using gold nanoroses (A-B) and carbon nanotubes (C-E) in atherosclerosis. A) PTT was performed on macrophages in vitro with a single 50 ns pulse (755 nm, 18 J/cm2). After irradiation without nanoroses, a bright field image indicates that the macrophages were intact (left). After irradiation with nanoroses, a dark field image shows a zone of macrophage ablation (right). B) Histological sections of atherosclerotic rabbit aorta injected (i.v.) with nanoroses or saline control. Macrophages (brown color RAM 11 stain) are co-present with nanoroses (bright red reflections). C) Significant decrease in macrophage viability (with SWNTs), assessed by MTT assay, 24 hours after thermal treatment (*p<0.05). D) Representative serial in vivo FMT images of a carotid-ligated mouse (top) and a sham-operated mouse (bottom) before and after injection of Cy5.5-conjugated SWNTs. The low intensity signal in sham mice represents some inflammation from the surgical procedure. E) White light image (left) and intrinsic SWNT NIR fluorescence image (right) of ex vivo ligated carotid arteries. Solid arrows point to ligated carotid artery from a mouse injected with SWNTs showing high NIR signal, but the ligated carotid artery from a mouse without SWNTs show no signal (dashed arrows). Images reproduced with permission from references [9, 112].
Compared to spherical gold nanoparticles, GNRs produce intense absorption with tunable absorption properties, and can be readily synthesized.[113] GNRs also possess optical imaging properties providing contrast for dark field imaging in the cellular environment, and two-photon luminescence.[113] Pissuwan et al. synthesized GNRs displaying longitudinal plasmon resonance around 710 nm conjugated to a macrophage targeting monoclonal antibody (CD11b).[114] Approximately 80 % cell death was observed upon NIR irradiation (650 nm) compared to 1 % for unexposed macrophages. Targeted GNRs exhibited cytotoxicity in macrophages, but not in Chinese hamster ovary cells, thus showing their selectivity. However, only a moderate increase in cell death compared to naked GNRs was observed, which is probably due to the similar cellular uptake of targeted and naked GNRs. Modification of nanorods for absorption in the far NIR (>800 nm) could increase the penetration depth and selectivity for their in vivo utility. Choi et al. reported dextran coated GNRs absorbing at a slightly longer wavelength (760 nm).[7] In comparison with the PEGylated GNRs, dextranated nanorods showed almost complete cell death upon NIR illumination (808 nm) which can be attributed to the higher uptake (2.5 fold) in macrophages. Though GNRs showed efficient macrophage ablation, further studies are required to characterize the mechanism of cell death before pursuing in vivo applications.
In addition to gold-based nanoconstructs, single walled carbon nanotubes (SWNTs) have also been used for PTT. Compared to GNRs, SWNTs can achieve comparable thermal destruction at a 10-fold lower dose and 3-fold lower power.[115] SWNTs have intrinsic luminescence above 800 nm, which can be used to image their uptake.[115] A recent study by Kosuge et al. showed the feasibility of using SWNTs as theranostics for PTT and imaging of atherosclerotic macrophages.[9] In NIR light (808 nm) exposed culture, more than 90% ablation of macrophages was observed at nanomolar concentrations of SWNTs, while SWNTs or light alone showed no toxicity (Fig. 4C). In vivo and ex vivo fluorescence imaging showed significant (p<0.05) signal accumulation in ligated carotids compared to mice receiving sham surgery (Fig. 4D-E). PTT (808 nm) of excised carotids showed apoptotic macrophages in ligated carotid arteries with SWNTs, but not in control groups without SWNTs or without light exposure. Ex vivo PTT is uncomplicated where focused illumination and plaque access is relatively easy compared to in vivo whole animal PTT. Therefore, further studies can attempt to perform PTT in live mice. Furthermore, the selectivity of SWNTs could be increased by surface coating with targeting ligands such as dextran. Since carbon nanotubes have shown toxicities previously [116], a detailed investigation for biocompatibility is needed to use this nanoconstruct for selective in vivo macrophage eradication in vascular tissue without causing detrimental effects to the vasculature.
Based on the studies presented, PTT for macrophage ablation could be promising in inflamed tissues that are superficial and accessible by NIR light, such as RA and atherosclerosis. Several challenges need to be addressed, such as focused irradiation of target tissues, release of cellular products post cell death, light penetration depth in clinical subjects and dissipation of heat by blood flow. Toxicities due to long term accumulation of photoabsorbers and inflammatory responses caused by damaged tissues during light therapy [117, 118] mandates in-depth investigation of this approach.
2.1.3. Cytotoxic chemotherapy
Limited light penetration makes photo-based therapies impractical for inflammatory conditions in deeper tissues where surgical or invasive endoscopic interventions become inevitable. In these conditions, target-specific accumulation of cytotoxic chemotherapeutic drugs could be an alternative. Several cytotoxic drugs such as clodronate, glucocorticoids, doxorubicin and methotrexate are utilized for this purpose.[119-121] Systemic release of a cytotoxic drug can reduce the global macrophage population, thereby affecting homeostasis and making the host prone to infections.[33, 34] In this regard, theranostics could be employed to target cytotoxic drugs to inflamed tissue actively (using ligands) or passively (using size) while the imaging functionality could help visualize biodistribution and thus predict toxicities.
For this purpose, the effect of prednisolone phosphate (PLP) encapsulated in MRI-fluorescence active liposomes was studied on TAMs.[120] PLP is an anti-inflammatory glucocorticoid with high anticancer activity, but its effect on tumor reduction comes at the expense of severe side effects. Therefore, PLP has been previously incorporated in liposomes to increase tumor targeting and reduce the side effects. Liposomal PLP (PLP-L) has shown enhanced tumor reduction compared to PLP alone, though the mechanism was not fully understood.[122] This very detailed study demonstrated the utility of combined imaging and therapy to understand the mechanism of PLP action at the tumor site. Fluorescence was used for histological analysis and microscopy, while contrast enhanced MRI with gadolinium (Gd) liposomes was used to evaluate drug delivery to the tumor site in a mouse melanoma model. T1-weighted imaging showed intra-tumoral distribution of Gd-PLP-L based on pixel contrast enhancement (Fig. 5A). Longitudinal relaxivity changes (ΔR1, enhanced relaxivity compared to pre-contrast administration) indicated accumulation of liposomes at the tumor site after single dose i.v. administration (Fig. 5B). ΔR1 after Gd-PLP-L injection showed that the accumulation increased with tumor volume. Treatment outcome was assessed by measuring excised tumor volume using MRI. Irrespective of the tumor accumulation assessed by MRI, drastic tumor reduction was observed. Gd-PLP-L and PLP-L showed similar therapeutic efficacy while drug free Gd-L showed no effect. However, only 5% of Gd-PLP-L was accumulated in TAMs and a 90% drop in systemic leukocyte count was observed. The authors concluded that the systemic drop in leukocyte count as opposed to TAM depletion could be the reason for therapeutic efficacy. While systemic reduction could reduce the tumor-infiltrating monocytes, a high drop in their count can make the host susceptible to infections. This theranostic could be modified with a targeting agent for increased accumulation in TAMs. Future studies should evaluate the effect of PLP-L on macrophage phenotype and their contribution to tumor reduction, because glucocorticoids have previously shown to change macrophage phenotype.[123] The relaxivity changes in the tumor environment could be a function of local vascular changes during treatment and the clearance of Gd, which could lead to errors in quantifying PLP-L tissue accumulation. Alternatively, background free 19F MRI using PFC formulations could be used. While the use of FDA approved liposomes and Gd makes this platform promising for clinical translation, detailed in vitro and in vivo characterizations for toxic effects are needed.
Studies with cytotoxic chemotherapeutic theranostics. A) T1-weighted images of a slice through a tumor-bearing mouse pre- and post- (5 min and 85 min) Gd-PLP-L theranostic administration. The red line indicates the tumor boundary and the yellow color represents contrast-enhanced pixels that show enhanced tumor accumulation at 85 min. B) T1 distribution maps of a slice through a tumor-bearing mouse before and after (2 h and 24 h) Gd-PLP-L theranostic administration, “T” represents the tumor. C) Schematic of SPIO-PLGA nanoparticles with surface Avidin for conjugation to antibodies. D) Extended release of methotrexate (MTX) and clodronate (Clod) from PLGA nanoparticles with and without SPIO. MTX and Clod shows 100% and 35% release respectively after 8 days, while iron release was negligible. Figures adapted and reproduced with permission from references [120, 121].
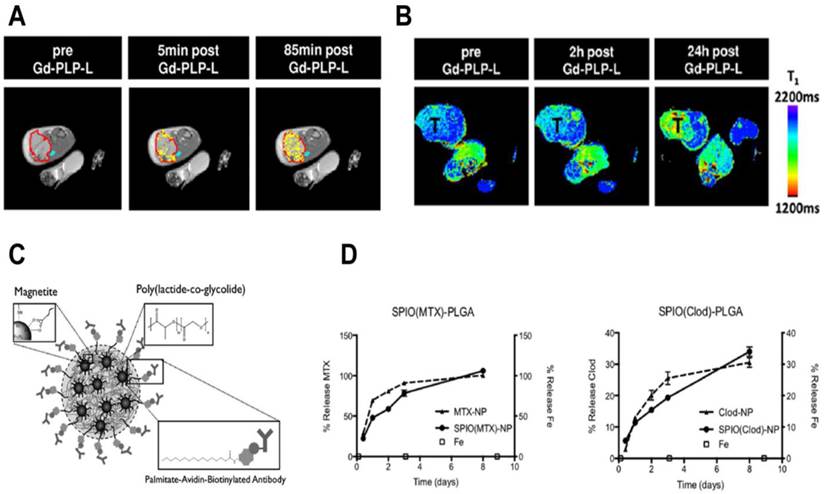
In order to retain imaging capabilities during controlled drug release, SPIO-PLGA (poly(lactic-co-glycolic acid)) nanoparticle theranostics were reported.[121] The MR contrast based theranostic platform has incorporated cytotoxic methotrexate or clodronate to target macrophages. Fatty acids were used to load SPIO clusters in the PLGA matrix and surface functionalize with macrophage specific antibody (anti-F4/F80). A schematic of the nanoparticle is shown in figure 5C. A fluorescent dye (coumarin-6) was incorporated to enable confocal imaging, which showed enhanced uptake in RAW 264.7 and bone marrow derived macrophages (BMDMs). Most importantly, the nanoparticles did not alter the metabolic functions of macrophages. Methotrexate or clodronate encapsulated nanoparticles showed controlled drug release over several days and a negligible iron oxide release (Fig. 5D). Drug-loaded nanoparticles exposed to macrophages showed cytotoxicity while free drugs or naked nanoparticles did not induce cytotoxicity. The versatility of the formulation was shown by effective internalization of nanoparticles functionalized with a tumor targeting agent in B16 melanoma cells, which are not phagocytic. In vivo biodistribution studies in a xenograft tumor model displayed enhanced blood pool kinetics and tumor retention. These results suggest that the antibody conjugated SPIO-PLGA theranostic is expected to localize at inflammation sites and reduce the macrophage burden, while the clustered SPIO provides enhanced contrast for MRI. Since the iron release from nanoparticles was minimal, the target tissue could be imaged with high contrast during drug release, thus avoiding multiple administrations of contrast agent for therapeutic monitoring. Besides prolonged release, target site retention of nanoparticles is crucial for longitudinal imaging studies. Since the theranostic was constructed with clinically approved materials, it could have high utility, provided future studies attempt to increase the retention. Finally, the long term effects of SPIO accumulation in the body due to high retention and slow release should be investigated.
2.1.4. Summary
Macrophage ablation therapies are inherently aggressive; therefore, selectivity is very crucial. Although light-mediated theranostics are selective, they were shown to cause significant tissue damage and induce inflammatory reactions, which can lead to recurrence of the disease.[124] From a technological perspective, guiding light to the anatomically complex locations without causing collateral damage is a challenge. Chemotherapeutic ablation of macrophages is useful to treat deep-tissue pathologies which are inaccessible by PDT and PTT. Conferring the theranostic with tissue selectivity by targeting ligands, stimuli sensitivity, or external activation, is vital in order to confine the activity to the target tissue. These aggressive approaches are valuable in life-threatening diseases such as cancer and less invasive alternatives need to be explored.
2.2. Theranostics utilizing non-ablation approaches
Reducing the pathogenic activity of macrophages could be promising since the detrimental effects of macrophage ablation and the released toxic products can be avoided. Macrophages produce a plethora of pro-inflammatory mediators that damage the host tissue and these can be targeted by theranostics. This section discusses non-ablation based applications of macrophage theranostics.
2.2.1. Macrophage phenotype modulation
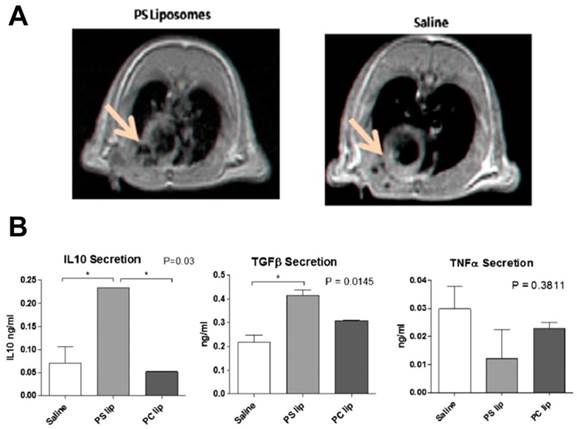
As mentioned earlier in this review, macrophages in inflammation present with different phenotypes that participate in pro- and anti-inflammatory processes. In post-MI, M2 macrophages participate in wound healing process. During the acute phase, pro-inflammatory M1 macrophages dominate the infarct site and the duration of this phase affects the infarct size and remodeling process. Harel-Adar et al. reported the utility of phosphatidylserine (PS)-presenting liposomes to modulate macrophage phenotype in MI.[125] PS is expressed on apoptotic cells, which are recognized by macrophages and cleared. Apoptotic cells produce anti-inflammatory effects by inhibiting production of pro-inflammatory cytokines from macrophages concomitant with the secretion of anti-inflammatory cytokines. In this comprehensive study, micrometer size liposomes encapsulating iron oxide were surface modified with PS. The presence of iron oxide enabled visualization of PS-liposome accumulation at the infarct site after femoral vein injection in a rat model of acute MI (Fig. 6A). Following uptake of PS-liposomes by macrophages, an increase in anti-inflammatory cytokines (TGF-β and IL-10) and a reduction in pro-inflammatory tumor necrosis factor- α (TNF-α) were seen in both in vitro and in vivo (Fig. 6B). The mannose receptor CD206 was upregulated while the CD86 surface marker was downregulated showing the shift of macrophage phenotypes from M1 to M2. Histological studies revealed the co-localization of PS-liposomes and macrophages. PS-liposome treated rats showed increased angiogenesis, preservation of scars and prevention of heart dilation and left ventricle remodeling, indicating effective wound healing. Though not evaluated in this study, MRI can be potentially used for dynamic visualization of PS-liposome infarct accumulation and monitoring the response. The contribution of iron oxide to the phenotype change requires further investigation because iron oxide has been previously known to produce anti-inflammatory cytokine release consistent with the M2 phenotype.[126] It should also be noted that excessive scar tissue formation by M2 macrophages could lead to arrhythmias. Nevertheless, this construct has promising potential for clinical translation as FDA approved liposomes and iron oxides were used for constructing the theranostic and clinically viable i.v. administration was employed.
Macrophage phenotype modulation using iron oxide loaded PS-liposomes. A) 1H MRI showing contrast changes with the accumulation of PS liposomes (arrow). B) Cytokine analysis of supernatants from macrophage cultures exposed to PS liposomes. PS-liposomes show an increase in IL-10 and TGF-β and a reduction in TNF-α compared to saline and phosphatidyl choline (PC) liposomes. Figures adapted and reproduced with permission from reference [125].
Another approach to increase M2-activity is by increasing the infiltration of M2 macrophages to the MI site. The mineralocorticoid receptor antagonist, Eplerenone, has previously shown to increase macrophage infiltration and M2 polarization leading to infarct healing effects.[127] Theranostics for macrophage phenotype changes could also be effective in other diseases such as cancer. Celecoxib, a selective COX-2 inhibitior, has been shown to change the TAM phenotype from pro-tumor (M2) to tumoricidal M1-like.[128] Specific receptors are expressed by macrophages displaying different phenotype. Therefore, these can be used for selective targeting.[129, 130] The advantage of this approach is that, compared to aggressive macrophage depletion, modulation of macrophage phenotype is non-destructive and the likelihood of benign tissue damage is minimal since no cytotoxic products are released.
2.2.2. Inhibition of macrophage infiltration
Recruitment of macrophages to the inflammation site increases the pro-inflammatory activities which can exacerbate disease. Besides selective depletion strategies, macrophage infiltration can be reduced by inhibiting the mediators involved in their recruitment. Bindarit, an inhibitor of monocyte chemoattractant protein-1 (MCP-1), and Natalizumab, a leukocyte adhesion molecule inhibitor, have shown anti-inflammatory effects.[131] However, clinically CAM inhibitors have been associated with serious infections due to reduced global leukocyte activity. Alternatively, theranostics could be promising tools to study the effects due to the inhibition of these mediators by imaging macrophage infiltration.
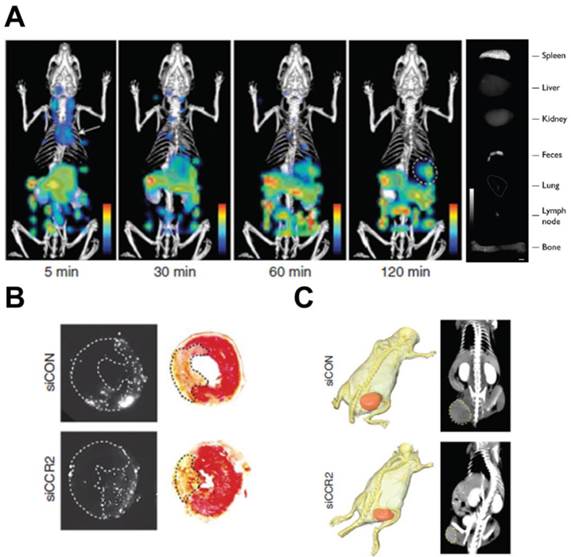
Inflammatory monocyte/macrophage recruitment to pathological site is mediated by MCP-1 and its receptor, C-C chemokine receptor type 2 (CCR2). The non-inflammatory monocyte subset does not depend on this chemokine/chemokine-receptor. [132] MCP-1 is upregulated in inflammatory monocytes/macrophages leading to enhanced movement to the disease site. Leuschner et al. developed a fluorescently-tagged siRNA (small interfering RNA)-loaded theranostic lipid nanoparticle for CCR2 silencing activity in inflammatory monocytes.[133] A detailed study outlining the utility of this delivery vehicle in MI, atherosclerosis, diabetes and cancer showed compelling results. Dynamic FMT-CT and ex vivo fluorescence reflectance imaging was used to visualize nanoparticle/siRNA biodistribution (Fig. 7A), which were predominantly present in splenic, bone marrow and blood inflammatory monocytes. MI is characterized by a strong inflammatory response, which negatively affects the infarct size and heart function. Compared to a control siRNA (siCON), CCR2 siRNA (siCCR2) showed a 71% reduction in inflammatory monocytes while the infarct/area-at-risk ratio was reduced by 34% in a mouse MI model (Fig. 7B). Consistent with previous reports [134], in vivo imaging studies showed the spleen as the major organ of theranostic accumulation. Splenectomy performed at the time of MI showed splenic monocytes as a major target for siCCR2. This theranostic was also applied in a pancreatic islet transplantation model, which shows high infiltration of inflammatory monocytes participating in graft rejection. In a streptozotocin-induced murine diabetes model, siCCR2 nanoparticles showed increased islet survival and graft function compared to untreated and siCON groups. In atherosclerosis, a 3 week siCCR2 therapy showed 82% reduction in inflammatory monocytes and a 38% reduction in lesion size. TAMs are involved in pro-tumorigenic functions and their numbers show negative prognosis. In a xenograft tumor model, siCCR2 reduced the tumor size compared to siCON (Fig. 7C) concomitant with a significant reduction in TAMs (54%). The fluorescence imaging component of the theranostic is used for in vivo biodistribution studies and histological analysis. As a theranostic, this construct has limited clinical applicability due to the imaging depth limitation associated with fluorescent agents. Since splenic monocytes were the major targets, there is a potential for side effects due to the reduction in systemic monocyte activity.
MCP-1 inhibition by a fluorescent theranostic. A) FMT-CT longitudinal imaging of siCCR2 nanoparticles show spleen accumulation at 120 min (circled region). Ex vivo imaging of the organs show high fluorescence in the spleen. B) Reduced infarct risk area in siCCR2 compared to siCON, imaged by fluorescence reflectance imaging. C) X-ray CT image showing tumor reduction in an siCCR2 treated mouse compared to siCON (circled region). Figures reproduced with permission from reference [133].
Majmudar et al. [135] evaluated the therapeutic efficacy of this theranostic by imaging macrophages in an atherosclerosis model employing clinically used dextran coated nanoparticles (DNPs). Zirconium-89 radiolabeled (89Zr) DNPs were further conjugated to a NIR dye for cellular visualization. Therapeutic and imaging agents were incorporated in distinct nanoparticles. Multimodal PET and MR imaging showed reduced macrophage-labeled DNPs in siCCR2 compared to siCON treated mice, concomitant with plaque reduction. A potential future expansion of this concept would be to combine 89Zr-DNPs and siCCR2 for simultaneous imaging and therapy. DNPs have been previously used to incorporate siRNA.[136] Whether the combination of siCCR2 and DNPs will increase target site accumulation can be answered by further studies. These findings show that silencing inflammatory-monocyte CCR2 has great therapeutic potential using nanoparticles across the disease models. Furthermore, only inflammatory monocyte/macrophages depend on MCP-1/CCR2, silencing CCR2 should not affect the function of wound healing macrophages.
In addition to MCP-1, CAMs can also be targeted to reduce macrophage infiltration. CAMs are over expressed in the inflammatory environment and aid the adherence and transmigration of leukocytes and lymphocytes to the target tissue. CAMs have been used for targeting nanoparticles to the inflamed tissue.[137] Theranostics incorporating CAM inhibitors such as natalizumab could be functionalized to reduce side effects and increase target site accumulation. Macrophage theranostics incorporating CAM inhibitors have not yet been investigated, but could hold potential for future studies.
2.2.3. Inhibition of pro-inflammatory mediators
Macrophages secrete several pro-inflammatory mediators in response to injurious stimuli, which if unchecked can lead to significant tissue injury. Inflammatory mediators such as prostaglandins, TNF-α and cytotoxic oxygen products are upregulated in macrophages during inflammation. Thesecould be targeted for imaging and therapeutic purposes.
Tumor necrosis factor-α
TNF-α is a potent pro-inflammatory cytokine produced primarily by activated M1 macrophages. Dysregulated production of TNF-α has been implicated in cancer, RA, inflammatory bowel disease (IBD), and Alzheimer's disease.[138, 139] Clinically used anti-TNF-α therapeutics, infliximab and etanercept are associated with severe infections and malignancies due to their non-selectivity.[140] Nanoparticles inhibiting TNF-α expressed by inflammatory macrophages have been widely investigated. Nanoparticles facilitate targeted delivery of TNF-α inhibitors such as siRNA to inflammatory macrophages and increase their oral absorption.[141, 142] The following studies present theranostics that incorporate TNF-α siRNA to target macrophages, thereby increasing oral absorption and site specific delivery.
Microparticles encapsulating Map4k4 siRNA were prepared for oral delivery to inhibit systemic TNF-α.[141] Map4k4 is an enzyme involved in the TNF-α signaling pathway. It was proposed that gut associated macrophages can take up microparticles and infiltrate other organs leading to TNF-α reduction. Microparticles were prepared by a series of alkaline, acid and solvent extractions from baker's yeast and fluorescently tagged (GeRPs). The outer shell of GeRPs contained 1,3-β-glucan to facilitate receptor-mediated uptake by macrophages through β-glucan receptors. Fluorescent labeling of GeRPs and siRNA with distinct fluorescent reporters enabled in vitro and in vivo macrophage uptake assessment using confocal microscopy. Fluorescence cellular imaging of tissue macrophages showed that orally administered siRNA-GeRPs migrated to liver, spleen and lungs thereby reducing systemic TNF-α. The formulation protected mice from LPS-induced lethality. However, multiple preparation and purification steps, along with a dosing frequency of 8 days, present challenges in the large scale production and clinical utility of this theranostic.
A subsequent study by Yin et al. reported supramolecular self-assembled nanoparticles (SSNPs) involving a simpler formulation method and with multiple functionalities.[142] In SSNPs, chitosan was used for permeation enhancement, mannose for macrophage uptake, cysteamine for mucoadhesion, and a polypeptide for cell internalization and endosomal escape. Similar to the previous study, fluorescent dye (Cy3 or Dy800) labeled siRNA-SSNPs showed uptake in intestine, lung, liver and spleen. In LPS-induced mouse hepatic injury, a single oral dose of SSNPs produced an 80% reduction in systemic TNF-α, which is comparable to multiple administrations of GeRPs.[141] The increased efficacy could be attributed to the multivalent targeting functionality of the nanoparticles and also the use of TNF-α specific siRNA. Clearly, this system is an improvement over GeRPs in terms of potency and dosing regimen. Furthermore, a widely accepted oral route of administration and high potency are promising features of this platform. However, a large number of components can be a concern for scalability and a systemic reduction in TNF-α could lead to opportunistic infections similar to current antibody therapies, necessitating careful monitoring.
Komano et al. reported the use of anti-TNF-α siRNA-loaded liposomes called wraposomes (WS) targeting synovial macrophages in a mouse model of arthritis.[143] First, biodistribution studies were performed using a fluorescent labeled non-therapeutic siRNA liposome (Cy5-siRNA/WS). Fluorescence imaging showed specific accumulation in arthritic joints after i.v. administration and increased fluorescence compared to naked siRNA. Synovial macrophages (70%) were the major cellular targets of the siRNA/WS. To evaluate therapeutic efficacy, Cy5-siRNA was replaced with a non-fluorescent anti-TNF-α siRNA which showed reduced arthritic score, paw thickness and TNF-α compared to controls. As the construct showed negligible accumulation in liver, spleen and blood, side effects related to systemic TNF-α inhibition are likely to be minimal. Despite the absence of targeting ligands, WS showed high selectivity for synovial macrophages. Although the same construct is used for both imaging and therapy, the inter-subject variability could lead to differences in bioaccumulation. For future studies, co-incorporation of imaging agents and TNF-α siRNA in the same construct can enable individualized assessment of bioaccumulation, which could vary due to the differences in arthritic inflammation among subjects. [13]
Cyclooxygenase-2
Cyclooxygenase (COX) enzymes, COX-1 and COX-2 are involved in several homeostatic functions. COX enzymes convert arachidonic acid into potent inflammatory mediators called prostaglandins. COX-1 is constitutively present and involved in 'housekeeping' functions whereas COX-2 is mainly implicated in inflammation.[144] COX-2 plays an important role in the pathogenesis of inflammatory diseases, where it is over-expressed.[145, 146] A subclass of non-steroidal anti-inflammatory drugs (NSAIDs) that selectively inhibit COX-2 has proven effective against cancer, atherosclerosis, RA, IBD, and neurodegenerative diseases.[146-149] Development of COX-2 specific PET and fluorescence imaging agents to detect cancer and inflammation have demonstrated the role COX-2 plays in inflammatory diseases.[150, 151] COX-2 is highly induced in M1 (pro-inflammatory) macrophages.[36] Due to its ubiquitous presence in inflammatory diseases, theranostics inhibiting macrophage COX-2 activity could find wide applicability. In cancer, COX-2 silencing has delayed tumor formation and reduced metastasis. Lisposomal COX-2 siRNA co-incorporated with iron or gadolinium-based MRI contrast agents were used to visualize COX-2 siRNA delivery to tumors.[152] The presence of MRI contrast agent allowed for tracking biodistribution and cancer tissue accumulation. Though the study did not evaluate the effect of the COX-2 siRNA theranostic in macrophages, it could be applied to inflammatory diseases involving macrophages.
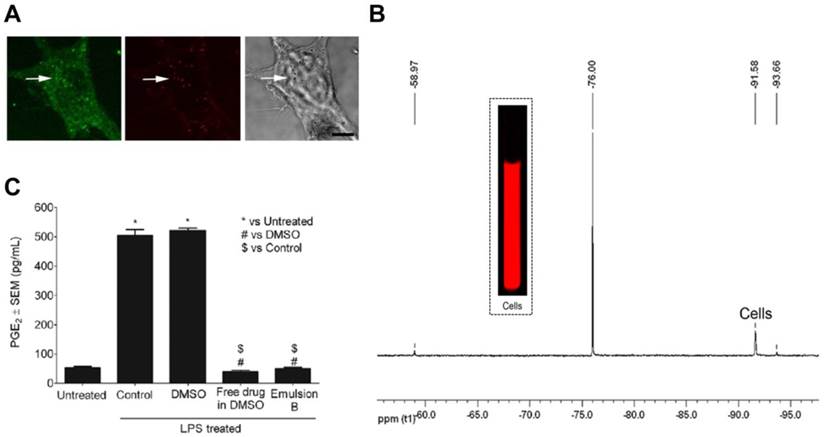
We have recently developed a PFC theranostic incorporating celecoxib for COX-2 inhibition in activated macrophages.[153] NIR fluorescent reporter and celecoxib incorporated non-toxic PFC nanoemulsions were prepared using microfluidization. Dual mode imaging capabilities helped us to assess nanoemulsion uptake in macrophages by confocal microscopy, NIR fluorescence imaging and 19F NMR (Fig. 8A-B). A high correlation between 19F NMR and fluorescence signal intensities at different doses indicated that either of the imaging modalities can be utilized. In LPS activated macrophages, celecoxib-loaded nanoemulsion showed complete shutdown of COX-2 activity assessed by PGE2 release compared to controls (Fig. 8C). The presence of PFC aided in quantitative uptake measurements by 19F NMR (1011 19F per cell) and could help visualize macrophages in inflammatory diseases with 19F MRI. In addition to COX-2 inhibition, celecoxib has shown to reduce MCP-1 [146] and CAMs [148] in other studies, which could further reduce the inflammatory activity. Taken together these effects, theranostics utilizing COX-2 inhibitors have promising potential against inflammatory diseases. Further in vivo studies to evaluate the effect of our celecoxib theranostic on macrophage phenotype in inflammatory disease models are currently underway. Compared to using biologics such as siRNA and oligonucleotides, well studied small molecule COX-2 inhibitors have greater potential for clinical translation.
A multimodal theranostic for COX-2 in macrophages. A) Confocal microscopy of macrophages showing cell membrane in green (FITC) and nanodroplets in red (Cellvue® burgundy dye). B) Representative 19F NMR and NIRF imaging of cells labeled with PFC theranostic. C) PGE2 estimation in supernatants of cells exposed to different treatments followed by 4 h LPS activation (*, #, $ = p<0.0001). Figures reproduced from reference [153].
Reactive oxygen species
Reactive oxygen species (ROS) is a collective term for highly reactive small chemical molecules possessing oxygen, such as hydrogen peroxide (H2O2), superoxide and hydroxyl radicals, which are byproducts of cellular metabolism.[154, 155] ROS are involved in cell signaling pathways in several biological and pathophysiological settings. Excessive ROS causes oxidative stress and is implicated in several inflammatory diseases. H2O2 is a precursor of several ROS and is overproduced in an inflammatory environment making it a selective marker of inflammation. Activated macrophages and neutrophils are the major source of ROS in the inflammatory environment.[154]
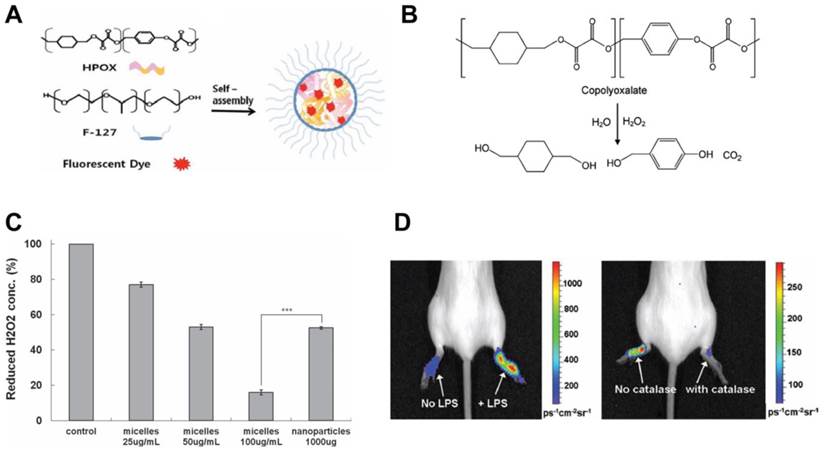
Cho et al. showed in vitro and in vivo the anti-inflammatory activity of chemiluminescent antioxidant micelles incorporating the antioxidant, hydroxyl benzyl alcohol (HBA) and a fluorescent dye (Fig. 9A).[156] Pluronic® F127 micelles were prepared with biodegradable copolyoxalate polymers conjugated to HBA. Fluorescent dye was incorporated in the micelle for close proximity. The presence of H2O2 resulted in a POCL (peroxylate chemiluminescence) reaction leading to the transfer of energy to the nearby fluorescent dye (Fig. 9B). In LPS activated macrophages, the theranostic micelles reduced H2O2 production compared to control and HBA-nanoparticles (Fig. 9C). Micelles were injected at the site of inflammation induced by LPS in a mouse model. Chemiluminescence was observed at the inflammation site and reduced when H2O2 degrading catalase was present due to inhibition of the POCL reaction (Fig. 9D). Micelles were specific to H2O2 but no other ROS metabolites, showing the specificity of the theranostic. Since luminescence as well as release of HBA occurs in the presence of H2O2, this theranostic shows high inflammation sensing potential. Intravenous administration of these micelles could therefore provide selectivity for diseased sites, thereby avoiding non-selective systemic reduction in H2O2, which could shutdown cell signaling pathways leading to metabolic imbalance. The study shows the promising features of this theranostic as an in vitro, ex vivo and preclinical diagnostic and therapeutic tool. Clinically, such constructs could find potential applications in surface accessible inflammatory conditions similar to PDT and PTT, where fluorescence intensity can be used to sense ROS activity and hence the treatment efficacy. Redox sensitive MR contrast agents [158] and ROS sensing PET imaging agents were reported before, which could find clinical applicability as a H2O2-responsive theranostic in macrophage-mediated diseases.
A hydrogen peroxide sensitive anti-oxidant theranostic. A) A schematic showing formation of chemiluminescent micelles where antioxidant HPOX (HBA-incorporated copolyoxalate) and fluorescent dyes are co-incorporated in the hydrophobic core. B) Degradation of copolyoxalate in the presence of H2O2 and water releases energy which activates nearby fluorescent dye. C) Graph showing reduction of H2O2 by HPOX micelles and nanoparticles in solution, P < 0.001. D) Chemiluminescent image of H2O2 generated by locally injected HPOX micelles during LPS-induced inflammation (left). Image showing inhibited POCL reaction of HPOX micelles by H2O2 degrading catalase (right). Figures adapted and reproduced with permission from references [156, 157].
Myeloid-related protein-8/14
In addition to the above discussed targets, macrophages express many enzymatic, protein and lipid based targets for therapeutic intervention. Myeloid-related protein-8/14 (Mrp-8/14) is a member of the S100-family of Ca2+-modulated proteins.[159] It participates in vascular inflammation, macrophage recruitment and pro-inflammatory cytokine release. Mrp-8/14 is found abundantly in atherosclerotic plaques making it a target for anti-inflammatory theranostic. Mrp-8/14 is detected in the cytoplasm of neutrophils and macrophages. Maiseyeu et al. targeted Mrp-8/14 in a mouse atherosclerotic model using a multivalent theranostic containing PS, ω-carboxynonanoyl-cholesteryl ester and an anti-Mrp antibody coupled with gadolinium nanoparticle (aMrp-NP).[159] The ω-carboxynonanoyl-cholesteryl ester (expressed on foam cells) and PS were utilized for uptake by macrophages, while the anti-Mrp antibody was used as an anchor for retention in atherosclerotic plaques. These functionalities gave the nanoparticle a multivalent targeting capability. In vitro studies in BMDMs of the atherosclerotic mouse showed neutralization of Mrp 8/14 pro-inflammatory effects with aMrp-NP possibly by antigen-antibody interactions. In vivo studies in the murine atherosclerotic model confirmed the presence of aMrp-NP around the plaque by MRI. Nanoparticles were accumulated mostly in neutrophils, monocytes and endothelial cells. Although, in vivo theranostic utility was not shown, multivalent targeting has the potential to be effective in plaque reduction. Also, the presence of PS might enhance the therapeutic effect by changing the macrophage phenotype and targeting a subset of macrophages as observed previously.[125] Further in vivo studies to outline the therapeutic effects of these functionalities are warranted.
2.3. Summary
Inhibition of macrophage infiltration and pro-inflammatory mediator production has been the main strategy for current clinical anti-inflammatory therapies. Nanoparticles and more specifically, theranostics are promising platforms to assess and limit the toxicities of these therapies. Based on the presented studies, theranostics can facilitate evaluation of macrophage uptake, biodistribution of the drug carrier, targeted delivery, increased oral absorption and environment sensitive drug release.
3. Limitations and Future Perspective
A significant growth in the field of theranostics has occurred in the last decade, primarily for cancer and inflammatory diseases. However, clinical translation of theranostics still remains a distant dream. One of the greatest challenges for the translation is the feasibility of large-scale economic production. Theranostics contain multiple active functionalities (Fig. 2) and usually require several preparation, purification and processing steps, which present scale-up challenges. As a result, most of the developed theranostics are confined to the laboratory and preclinical research. Regulatory approval of theranostics could hit many roadblocks due to the complexity of their design and the materials used with minimum characterization. Utilizing FDA approved well-characterized excipients, imaging and therapeutic agents could speed up the regulatory approval process. Yet, safety of individual components may not translate into safety of the theranostic system as a whole. Therefore, rigorous toxicological characterization of theranostics needs to be performed. Macrophage theranostics could require additional characterization for phenotype modulation effects because components of the reported theranostics such as glucocorticoids, celecoxib, iron oxide and liposomes have been shown to change the macrophage phenotype. [123, 125, 126, 128]
As detailed in this review, macrophage targeted theranostics have shown promising results in preclinical models for simultaneous imaging and therapy. However, several limitations need to be addressed. For example, the majority of the presented theranostics utilized optical imaging, whose clinical applicability is limited by light penetration. While, PET and SPECT can be used for evaluation studies in preclinical and clinical subjects, regular use of these modalities for diagnosis and therapy monitoring are limited by radiation exposure. Theranostics that incorporate paramagnetic MRI contrast agents have potential, but the inherent tissue background limits unambiguous detection of macrophages. Although 19F MRI is in the developmental stages, this imaging technique holds great potential due to near-zero background leading to quantitative detection. The low sensitivity of MRI, which requires a large amount of imaging agents necessitates careful evaluation for their biocompatibility, elimination and toxicity.
Since macrophages exhibit varied phenotypes, development of theranostics that selectively target specific phenotypes could overcome the toxicities associated with the non-selective macrophage ablation by PDT, PTT and cytotoxic drugs. Current anti-inflammatory therapies are majorly concentrated in targeting inflammatory mediators such as TNF-α, prostaglandins (COX-2) and inhibition of macrophage migration. In this regard, biologics such as siRNA showed potent anti-inflammatory properties, but were usually associated with severe toxicities. While theranostics could limit these toxicities by site-specific targeting, challenges associated with large scale production of biologics [160] could add to the existing limitations of theranostics for clinical translation. Alternately, small molecule anti-inflammatory drugs with well characterized efficacy, toxicity and safety profiles could be applied for theranostic research. Amongst these drugs, NSAIDs have shown efficacy across inflammatory disease models, but their effect on macrophage modulation is not well understood. For example, celecoxib reduces inflammation by inhibiting COX-2 production, whereas in cancer, it has shown to increase the pro-inflammatory activity of TAMs by changing their phenotype to M1-like. An interesting area for future research could be the application of theranostics to elucidate the mechanism of action of such drugs on macrophage activity. In this regard, theranostics could help visualize macrophages in the inflammatory environment, while the incorporated drug modulates their activity.
4. Conclusions
Macrophages are widely present and generally accessible cellular components in inflammatory diseases. In this review, a number of molecular targets of macrophages that are indicated in several diseases were discussed and the potential of theranostics in simultaneous imaging and therapy were demonstrated. The presented theranostics showed selectivity for disease-specific macrophages by light-based activation, the use of targeting ligands, enzyme and redox sensitive drug release. Studies also showed the utility of imaging for the assessment of macrophage uptake, biodistribution, visualization of target accumulation and therapy monitoring. Macrophage targeted theranostics have demonstrated efficacy in several inflammatory diseases either by reduction of macrophage numbers or inhibition of inflammatory activity. Theranostics have also been shown to increase the oral absorption of biologics through macrophage-mediated uptake. Based on these results, it can be concluded that the theranostics targeting of macrophages holds great promise for diagnosis and treatment of the inflammatory diseases, leading to to effective disease management.
Abbreviations
PEG: polyethylene glycol; RA: rheumatoid arthritis; ROS: reactive oxygen species; CAM: cell adhesion molecule; ECM: extracellular matrix; TAM: tumor-associated macrophages; MI: myocardial infarction; PET: positron emission tomography; SPECT: single-photon emission computed tomography; CT: computed tomography; MRI: magnetic resonance imaging; SNR: signal to noise ratio; PFC: perfluorocarbon; NIR: near-infrared; FMT: fluorescence molecular tomography; PDT: photodynamic therapy; ALA: 5-aminolevulinic acid; CLIO: cross-linked iron oxide; i.v.: intravenous; Cat B: cathepsin B; PTT: photothermal therapy; GNRs: gold nanorods; SWNT: single walled carbon nanotubes; PLP: prednisolone phosphate; PLGA: poly(lactic-co-glycolic acid); SPIO: superparamagnetic iron oxide; PS: phosphatidylserine; MCP-1: monocyte chemoattractant protein-1; CCR2: C-C chemokine receptor 2; DNP: dextran nanoparticles; TGF-β: transforming growth factor-β; TNF-α: tumor necrosis factor-α; SSNPs: supramolecular self-assembled nanoparticles; COX: cyclooxygenase; NSAID: non-steroidal anti-inflammatory drug; PGE2: prostaglandin E2; HBA: hydroxyl benzyl alcohol; POCL: peroxylate chemiluminescence; BMDMs: bone marrow derived macrophages.
Acknowledgements
The authors thank Michael J. Patrick from Carnegie Mellon University, Christina Bagia from Duquesne University and Natasha Baker from University of Pittsburgh for their critical reading and editing of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Pene F, Courtine E, Cariou A, Mira JP. Toward theragnostics. Critical care medicine. 2009;37:S50-8
2. Rai P, Mallidi S, Zheng X, Rahmanzadeh R, Mir Y, Elrington S. et al. Development and applications of photo-triggered theranostic agents. Advanced drug delivery reviews. 2010;62:1094-124
3. Mura S, Couvreur P. Nanotheranostics for personalized medicine. Advanced drug delivery reviews. 2012;64:1394-416
4. Janib SM, Moses AS, MacKay JA. Imaging and drug delivery using theranostic nanoparticles. Advanced drug delivery reviews. 2010;62:1052-63
5. Couvreur P. Nanoparticles in drug delivery: past, present and future. Advanced drug delivery reviews. 2013;65:21-3
6. Ganta S, Devalapally H, Shahiwala A, Amiji M. A review of stimuli-responsive nanocarriers for drug and gene delivery. Journal of Controlled Release. 2008;126:187-204
7. Choi R, Yang J, Choi J, Lim EK, Kim E, Suh JS. et al. Thiolated dextran-coated gold nanorods for photothermal ablation of inflammatory macrophages. Langmuir: the ACS journal of surfaces and colloids. 2010;26:17520-7
8. Boncel S, Muller KH, Skepper JN, Walczak KZ, Koziol KK. Tunable chemistry and morphology of multi-wall carbon nanotubes as a route to non-toxic, theranostic systems. Biomaterials. 2011;32:7677-86
9. Kosuge H, Sherlock SP, Kitagawa T, Dash R, Robinson JT, Dai H. et al. Near infrared imaging and photothermal ablation of vascular inflammation using single-walled carbon nanotubes. Journal of the American Heart Association. 2012;1:e002568
10. Swindell WR, Xing X, Stuart PE, Chen CS, Aphale A, Nair RP. et al. Heterogeneity of inflammatory and cytokine networks in chronic plaque psoriasis. PloS one. 2012;7:e34594
11. Drazen JM, Silverman EK, Lee TH. Heterogeneity of therapeutic responses in asthma. British medical bulletin. 2000;56:1054-70
12. Kuliczkowski W, Witkowski A, Polonski L, Watala C, Filipiak K, Budaj A. et al. Interindividual variability in the response to oral antiplatelet drugs: a position paper of the Working Group on antiplatelet drugs resistance appointed by the Section of Cardiovascular Interventions of the Polish Cardiac Society, endorsed by the Working Group on Thrombosis of the European Society of Cardiology. European heart journal. 2009;30:426-35
13. Lindberg J, af Klint E, Ulfgren AK, Stark A, Andersson T, Nilsson P. et al. Variability in synovial inflammation in rheumatoid arthritis investigated by microarray technology. Arthritis research & therapy. 2006;8:R47
14. Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? The Lancet. 2001;357:539-45
15. Libby P. Inflammation in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:2045-51
16. Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM. et al. Inflammation and Alzheimer's disease. Neurobiology of aging. 2000;21:383-421
17. Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. 2010;140:784-90
18. GBI Research, 2011. Anti-Inflammatory Therapeutics Market to 2017 - Respiratory Diseases and Arthritis Continue to Dominate: PR Newswire. http://www.prnewswire.com/news-releases/anti-inflammatory-therapeutics-market-to-2017---respiratory-diseases-and-arthritis-continue-to-dominate-132437538.html
19. Fang J, Nakamura H, Maeda H. The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Advanced drug delivery reviews. 2011;63:136-51
20. Medzhitov R. Inflammation 2010: new adventures of an old flame. Cell. 2010;140:771-6
21. Nathan C. Points of control in inflammation. Nature. 2002;420:846-52
22. Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O'Neill LA. et al. Resolution of inflammation: state of the art, definitions and terms. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2007;21:325-32
23. Fujiwara N, Kobayashi K. Macrophages in inflammation. Current Drug Targets-Inflammation & Allergy. 2005;4:281-6
24. Pollard JW. Trophic macrophages in development and disease. Nature Reviews Immunology. 2009;9:259-70
25. Hasan DM, Mahaney KB, Magnotta VA, Kung DK, Lawton MT, Hashimoto T. et al. Macrophage imaging within human cerebral aneurysms wall using ferumoxytol-enhanced MRI: a pilot study. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:1032-8
26. Petry KG, Boiziau C, Dousset V, Brochet B. Magnetic resonance imaging of human brain macrophage infiltration. Neurotherapeutics: the journal of the American Society for Experimental NeuroTherapeutics. 2007;4:434-42
27. Tang TY, Howarth SP, Miller SR, Graves MJ, Patterson AJ, JM UK-I. et al. The ATHEROMA (Atorvastatin Therapy: Effects on Reduction of Macrophage Activity) Study. Evaluation using ultrasmall superparamagnetic iron oxide-enhanced magnetic resonance imaging in carotid disease. Journal of the American College of Cardiology. 2009;53:2039-50
28. Barrera P, Blom A, Van Lent PL, Van Bloois L, Beijnen JH, Van Rooijen N. et al. Synovial macrophage depletion with clodronate-containing liposomes in rheumatoid arthritis. Arthritis & Rheumatism. 2000;43:1951-9
29. Young L, Katrib A, Cuello C, Vollmer-Conna U, Bertouch JV, Roberts-Thomson PJ. et al. Effects of intraarticular glucocorticoids on macrophage infiltration and mediators of joint damage in osteoarthritis synovial membranes: Findings in a double-blind, placebo-controlled study. Arthritis & Rheumatism. 2001;44:343-50
30. Catrina AI, Trollmo C, af Klint E, Engstrom M, Lampa J, Hermansson Y. et al. Evidence that anti-tumor necrosis factor therapy with both etanercept and infliximab induces apoptosis in macrophages, but not lymphocytes, in rheumatoid arthritis joints: extended report. Arthritis and rheumatism. 2005;52:61-72
31. Buch MH, Conaghan PG, Quinn MA, Bingham SJ, Veale D, Emery P. True infliximab resistance in rheumatoid arthritis: a role for lymphotoxin alpha? Annals of the rheumatic diseases. 2004;63:1344-6
32. Szefler S, Leung D. Glucocorticoid-resistant asthma: pathogenesis and clinical implications for management. European Respiratory Journal. 1997;10:1640-7
33. Burnett SH, Kershen EJ, Zhang J, Zeng L, Straley SC, Kaplan AM. et al. Conditional macrophage ablation in transgenic mice expressing a Fas-based suicide gene. Journal of leukocyte biology. 2004;75:612-23
34. Cailhier JF, Partolina M, Vuthoori S, Wu S, Ko K, Watson S. et al. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. The Journal of Immunology. 2005;174:2336-42
35. Mirza R, DiPietro LA, Koh TJ. Selective and specific macrophage ablation is detrimental to wound healing in mice. The American journal of pathology. 2009;175:2454-62
36. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nature reviews Immunology. 2011;11:723-37
37. Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656-61
38. van Ravenswaay Claasen H, Kluin PM, Fleuren G. Tumor infiltrating cells in human cancer. On the possible role of CD16+ macrophages in antitumor cytotoxicity. Laboratory investigation. 1992;67:166-74
39. Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428-35
40. Kinne RW, Stuhlmuller B, Burmester GR. Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis research & therapy. 2007;9:224
41. Tesch G. Role of macrophages in complications of type 2 diabetes. Clinical and Experimental Pharmacology and Physiology. 2007;34:1016-9
42. Glass C. The macrophage foam cell as a target for therapeutic intervention. Nat Med. 2002;8:1235-42
43. Baay M, Brouwer A, Pauwels P, Peeters M, Lardon F. Tumor cells and tumor-associated macrophages: secreted proteins as potential targets for therapy. Clinical & developmental immunology. 2011;2011:565187
44. Melgert BN. Macrophage Heterogeneity in Respiratory Diseases. Mediators of Inflammation. 2013;2013:769214
45. Gate D, Rezai-Zadeh K, Jodry D, Rentsendorj A, Town T. Macrophages in Alzheimer's disease: the blood-borne identity. Journal of neural transmission. 2010;117:961-70
46. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. The Journal of clinical investigation. 2012;122:787-95
47. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature Reviews Immunology. 2008;8:958-69
48. Mantovani A, Bottazzi B, Colotta F, Sozzani S, Ruco L. The origin and function of tumor-associated macrophages. Immunology today. 1992;13:265-70
49. Gui T, Shimokado A, Sun Y, Akasaka T, Muragaki Y. Diverse roles of macrophages in atherosclerosis: from inflammatory biology to biomarker discovery. Mediators of inflammation. 2012;2012:1-14
50. Kinne RW, Brauer R, Stuhlmuller B, Palombo-Kinne E, Burmester G-R. Macrophages in rheumatoid arthritis. Arthritis research. 2000;2:189-202
51. Trivedi RA, JM UK-I, Graves MJ, Cross JJ, Horsley J, Goddard MJ. et al. In vivo detection of macrophages in human carotid atheroma: temporal dependence of ultrasmall superparamagnetic particles of iron oxide-enhanced MRI. Stroke; a journal of cerebral circulation. 2004;35:1631-5
52. Trivedi RA, Mallawarachi C, Jean-Marie U, Graves MJ, Horsley J, Goddard MJ. et al. Identifying inflamed carotid plaques using in vivo USPIO-enhanced MR imaging to label plaque macrophages. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:1601-6
53. Gent YY, Voskuyl AE, Kloet RW, van Schaardenburg D, Hoekstra OS, Dijkmans BA. et al. Macrophage positron emission tomography imaging as a biomarker for preclinical rheumatoid arthritis: findings of a prospective pilot study. Arthritis and rheumatism. 2012;64:62-6
54. Brochet B, Deloire M, Touil T, Anne O, Caille J, Dousset V. et al. Early macrophage MRI of inflammatory lesions predicts lesion severity and disease development in relapsing EAE. Neuroimage. 2006;32:266-74
55. Dobrucki LW, Sinusas AJ. PET and SPECT in cardiovascular molecular imaging. Nature Reviews Cardiology. 2009;7:38-47
56. Chacko AM, Hood ED, Zern BJ, Muzykantov VR. Targeted nanocarriers for imaging and therapy of vascular inflammation. Current opinion in colloid & interface science. 2011;16:215-27
57. Moses WW. Fundamental limits of spatial resolution in PET. Nuclear Instruments and Methods in Physics Research Section A: Accelerators, Spectrometers, Detectors and Associated Equipment. 2011;648:S236-S40
58. Srinivas M, Heerschap A, Ahrens ET, Figdor CG, Vries IJM. 19F MRI for quantitative in vivo cell tracking. Trends in biotechnology. 2010;28:363-70
59. Geraldes CF, Laurent S. Classification and basic properties of contrast agents for magnetic resonance imaging. Contrast media & molecular imaging. 2009;4:1-23
60. Fass L. Imaging and cancer: a review. Molecular oncology. 2008;2:115-52
61. Kim J, An H, Rieter W, Esserman D, Taylor-Pashow K, Sartor R. et al. Multimodal optical and Gd-based nanoparticles for imaging in inflammatory arthritis. Clinical and experimental rheumatology. 2009;27:580-6
62. Dousset V, Brochet B, Deloire M, Lagoarde L, Barroso B, Caille J-M. et al. MR imaging of relapsing multiple sclerosis patients using ultra-small-particle iron oxide and compared with gadolinium. American journal of neuroradiology. 2006;27:1000-5
63. Wolters M, Mohades SG, Hackeng TM, Post MJ, Kooi ME, Backes WH. Clinical Perspectives of Hybrid Proton-Fluorine Magnetic Resonance Imaging and Spectroscopy. Investigative radiology. 2013;48:341-50
64. Srinivas M, Boehm-Sturm P, Figdor CG, de Vries IJ, Hoehn M. Labeling cells for in vivo tracking using 19F MRI. Biomaterials. 2012;33:8830-40
65. Janjic JM, Ahrens ET. Fluorine-containing nanoemulsions for MRI cell tracking. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2009;1:492-501
66. Schmid F, Höltke C, Parker D, Faber C. Boosting 19F MRI—SNR efficient detection of paramagnetic contrast agents using ultrafast sequences. Magnetic Resonance in Medicine. 2013;69:1056-62
67. Weibel S, Basse-Luesebrink TC, Hess M, Hofmann E, Seubert C, Langbein-Laugwitz J. et al. Imaging of Intratumoral Inflammation during Oncolytic Virotherapy of Tumors by (19)F-Magnetic Resonance Imaging (MRI). PloS one. 2013;8:e56317
68. Srinivas M, Morel PA, Ernst LA, Laidlaw DH, Ahrens ET. Fluorine-19 MRI for visualization and quantification of cell migration in a diabetes model. Magnetic Resonance in Medicine. 2007;58:725-34
69. Caruthers SD, Cyrus T, Winter PM, Wickline SA, Lanza GM. Anti-angiogenic perfluorocarbon nanoparticles for diagnosis and treatment of atherosclerosis. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2009;1:311-23
70. Lanza GM, Winter PM, Caruthers SD, Hughes MS, Hu G, Schmieder AH. et al. Theragnostics for tumor and plaque angiogenesis with perfluorocarbon nanoemulsions. Angiogenesis. 2010;13:189-202
71. Winter PM, Neubauer AM, Caruthers SD, Harris TD, Robertson JD, Williams TA. et al. Endothelial alpha(v)beta3 integrin-targeted fumagillin nanoparticles inhibit angiogenesis in atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2006;26:2103-9
72. Balducci A, Wen Y, Zhang Y, Helfer BM, Hitchens TK, Meng WS. et al. A novel probe for the non-invasive detection of tumor-associated inflammation. OncoImmunology. 2013;2:e23034
73. Balducci A, Helfer BM, Ahrens ET, O'Hanlon CF, Wesa AK. Visualizing arthritic inflammation and therapeutic response by fluorine-19 magnetic resonance imaging (19F MRI). Journal of Inflammation. 2012;9:1-10
74. Ebner B, Behm P, Jacoby C, Burghoff S, French BA, Schrader J. et al. Early Assessment of Pulmonary Inflammation by 19F MRI In VivoCLINICAL PERSPECTIVE. Circulation: Cardiovascular Imaging. 2010;3:202-10
75. Hitchens TK, Ye Q, Eytan DF, Janjic JM, Ahrens ET, Ho C. 19F MRI detection of acute allograft rejection with in vivo perfluorocarbon labeling of immune cells. Magnetic Resonance in Medicine. 2011;65:1144-53
76. Kadayakkara DK, Ranganathan S, Young W-B, Ahrens ET. Assaying macrophage activity in a murine model of inflammatory bowel disease using fluorine-19 MRI. Laboratory Investigation. 2012;92:636-45
77. Celsense New Release: The First Patient Treated Using Celsense MRI Cell Tracking Technology; 2013. http://www.celsense.com/april11_first_patient.html
78. Nahrendorf M, Zhang H, Hembrador S, Panizzi P, Sosnovik DE, Aikawa E. et al. Nanoparticle PET-CT imaging of macrophages in inflammatory atherosclerosis. Circulation. 2008;117:379-87
79. Morishige K, Kacher DF, Libby P, Josephson L, Ganz P, Weissleder R. et al. High-resolution magnetic resonance imaging enhanced with superparamagnetic nanoparticles measures macrophage burden in atherosclerosis. Circulation. 2010;122:1707-15
80. Nahrendorf M, Waterman P, Thurber G, Groves K, Rajopadhye M, Panizzi P. et al. Hybrid in vivo FMT-CT imaging of protease activity in atherosclerosis with customized nanosensors. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:1444-51
81. Jarrett BR, Correa C, Ma KL, Louie AY. In vivo mapping of vascular inflammation using multimodal imaging. PloS one. 2010;5:e13254
82. Weissleder R, Ntziachristos V. Shedding light onto live molecular targets. Nature medicine. 2003;9:123-8
83. Frangioni JV. In vivo near-infrared fluorescence imaging. Current opinion in chemical biology. 2003;7:626-34
84. Luo S, Zhang E, Su Y, Cheng T, Shi C. A review of NIR dyes in cancer targeting and imaging. Biomaterials. 2011;32:7127-38
85. Demchenko AP, Mély Y, Duportail G, Klymchenko AS. Monitoring biophysical properties of lipid membranes by environment-sensitive fluorescent probes. Biophysical journal. 2009;96:3461-70
86. Kosaka N, Ogawa M, Choyke PL, Kobayashi H. Clinical implications of near-infrared fluorescence imaging in cancer. Future Oncology. 2009;5:1501-11
87. Chellat F, Merhi Y, Moreau A, Yahia L. Therapeutic potential of nanoparticulate systems for macrophage targeting. Biomaterials. 2005;26:7260-75
88. Lim YT, Cho MY, Choi BS, Noh Y-W, Chung BH. Diagnosis and therapy of macrophage cells using dextran-coated near-infrared responsive hollow-type gold nanoparticles. Nanotechnology. 2008;19:375105
89. Jain S, Amiji M. Tuftsin-Modified Alginate Nanoparticles as a Noncondensing Macrophage-Targeted DNA Delivery System. Biomacromolecules. 2012;13:1074-85
90. Kelly C, Jefferies C, Cryan SA. Targeted liposomal drug delivery to monocytes and macrophages. Journal of drug delivery. 2011;2011:727241
91. Choi J, Kim HY, Ju EJ, Jung J, Park J, Chung HK. et al. Use of macrophages to deliver therapeutic and imaging contrast agents to tumors. Biomaterials. 2012;33:4195-203
92. Choi M-R, Stanton-Maxey KJ, Stanley JK, Levin CS, Bardhan R, Akin D. et al. A cellular Trojan Horse for delivery of therapeutic nanoparticles into tumors. Nano letters. 2007;7:3759-65
93. Verheye S, Martinet W, Kockx MM, Knaapen MW, Salu K, Timmermans J-P. et al. Selective clearance of macrophages in atherosclerotic plaques by autophagy. Journal of the American College of Cardiology. 2007;49:706-15
94. Danenberg HD, Fishbein I, Gao J, Mönkkönen J, Reich R, Gati I. et al. Macrophage depletion by clodronate-containing liposomes reduces neointimal formation after balloon injury in rats and rabbits. Circulation. 2002;106:599-605
95. Zeisberger S, Odermatt B, Marty C, Zehnder-Fjällman A, Ballmer-Hofer K, Schwendener R. Clodronate-liposome-mediated depletion of tumour-associated macrophages: a new and highly effective antiangiogenic therapy approach. British journal of cancer. 2006;95:272-81
96. Celli JP, Spring BQ, Rizvi I, Evans CL, Samkoe KS, Verma S. et al. Imaging and photodynamic therapy: mechanisms, monitoring and optimization. Chemical reviews. 2010;110:2795-838
97. Allison RR, Downie GH, Cuenca R, Hu X-H, Childs CJH, Sibata CH. Photosensitizers in clinical PDT. Photodiagnosis and Photodynamic Therapy. 2004;1:27-42
98. Kirdaite G, Lange N, Busso N, Van Den Bergh H, Kucera P, So A. Protoporphyrin IX photodynamic therapy for synovitis. Arthritis & Rheumatism. 2002;46:1371-8
99. Peng C, Li Y, Liang H, Cheng J, Li Q, Sun X. et al. Detection and photodynamic therapy of inflamed atherosclerotic plaques in the carotid artery of rabbits. Journal of Photochemistry and Photobiology B: Biology. 2011;102:26-31
100. Fotinos N, Campo MA, Popowycz F, Gurny R, Lange N. 5-Aminolevulinic Acid Derivatives in Photomedicine: Characteristics, Application and Perspectives. Photochemistry and photobiology. 2006;82:994-1015
101. Schmitt F, Lagopoulos L, Käuper P, Rossi N, Busso N, Barge J. et al. Chitosan-based nanogels for selective delivery of photosensitizers to macrophages and improved retention in and therapy of articular joints. Journal of Controlled Release. 2010;144:242-50
102. McCarthy JR, Korngold E, Weissleder R, Jaffer FA. A Light-Activated Theranostic Nanoagent for Targeted Macrophage Ablation in Inflammatory Atherosclerosis. Small. 2010;6:2041-9
103. Tong L, Cheng J-X. Gold nanorod-mediated photothermolysis induces apoptosis of macrophages via damage of mitochondria. Nanomedicine. 2009;4:265-76
104. McCarthy JR, Jaffer FA, Weissleder R. A macrophage-targeted theranostic nanoparticle for biomedical applications. Small. 2006;2:983-7
105. Tassa C, Shaw SY, Weissleder R. Dextran-coated iron oxide nanoparticles: a versatile platform for targeted molecular imaging, molecular diagnostics, and therapy. Accounts of chemical research. 2011;44:842-52
106. Shon S-M, Choi Y, Kim J-Y, Lee DK, Park J-Y, Schellingerhout D. et al. Photodynamic Therapy Using a Protease-Mediated Theranostic Agent Reduces Cathepsin-B Activity in Mouse Atheromata In Vivo. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1360-5
107. Choi Y, Weissleder R, Tung C-H. Selective antitumor effect of novel protease-mediated photodynamic agent. Cancer research. 2006;66:7225-9
108. Lutgens SP, Cleutjens KB, Daemen MJ, Heeneman S. Cathepsin cysteine proteases in cardiovascular disease. The FASEB Journal. 2007;21:3029-41
109. Gabriel D, Busso N, So A, van den Bergh H, Gurny R, Lange N. Thrombin-sensitive photodynamic agents: a novel strategy for selective synovectomy in rheumatoid arthritis. Journal of controlled release: official journal of the Controlled Release Society. 2009;138:225-34
110. Gabriel D, Lange N, Chobaz-Peclat V, Zuluaga MF, Gurny R, van den Bergh H. et al. Thrombin-sensitive dual fluorescence imaging and therapeutic agent for detection and treatment of synovial inflammation in murine rheumatoid arthritis. Journal of controlled release: official journal of the Controlled Release Society. 2012;163:178-86
111. Spernyak JA, White III WH, Ethirajan M, Patel NJ, Goswami L, Chen Y. et al. Hexylether derivative of pyropheophorbide-a (HPPH) on conjugating with 3gadolinium (III) aminobenzyldiethylenetriaminepentaacetic acid shows potential for in vivo tumor imaging (MR, fluorescence) and photodynamic therapy. Bioconjugate chemistry. 2010;21:828-35
112. Ma LL, Feldman MD, Tam JM, Paranjape AS, Cheruku KK, Larson TA. et al. Small multifunctional nanoclusters (nanoroses) for targeted cellular imaging and therapy. ACS nano. 2009;3:2686-96
113. Tong L, Wei Q, Wei A, Cheng JX. Gold Nanorods as Contrast Agents for Biological Imaging: Optical Properties, Surface Conjugation and Photothermal Effects†. Photochemistry and photobiology. 2009;85:21-32
114. Pissuwan D, Valenzuela SM, Killingsworth MC, Xu X, Cortie MB. Targeted destruction of murine macrophage cells with bioconjugated gold nanorods. Journal of Nanoparticle Research. 2007;9:1109-24
115. Robinson JT, Welsher K, Tabakman SM, Sherlock SP, Wang H, Luong R. et al. High performance in vivo near-IR (> 1 μm) imaging and photothermal cancer therapy with carbon nanotubes. Nano research. 2010;3:779-93
116. Shvedova AA, Kisin ER, Mercer R, Murray AR, Johnson VJ, Potapovich AI. et al. Unusual inflammatory and fibrogenic pulmonary responses to single-walled carbon nanotubes in mice. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2005;289:L698-L708
117. Tatar O, Adam A, Shinoda K, Yoeruek E, Szurman P, Bopp S. et al. Influence of verteporfin photodynamic therapy on inflammation in human choroidal neovascular membranes secondary to age-related macular degeneration. Retina. 2007;27:713-23
118. F irczuk M, Nowis D, Gołąb J. PDT-induced inflammatory and host responses. Photochemical & Photobiological Sciences. 2011;10:653-63
119. Chakravarthy KV, Davidson BA, Helinski JD, Ding H, Law WC, Yong KT. et al. Doxorubicin-conjugated quantum dots to target alveolar macrophages and inflammation. Nanomedicine. 2011;7:88-96
120. Kluza E, Yeo SY, Schmid S, van der Schaft DW, Boekhoven RW, Schiffelers RM. et al. Anti-tumor activity of liposomal glucocorticoids: The relevance of liposome-mediated drug delivery, intratumoral localization and systemic activity. Journal of controlled release: official journal of the Controlled Release Society. 2011;151:10-7
121. Ragheb RR, Kim D, Bandyopadhyay A, Chahboune H, Bulutoglu B, Ezeldein H. et al. Induced clustered nanoconfinement of superparamagnetic iron oxide in biodegradable nanoparticles enhances transverse relaxivity for targeted theranostics. Magnetic resonance in medicine. Magnetic Resonance in Medicine. 2013;70:1748-60
122. Kluza E, Heisen M, Schmid S, van der Schaft DW, Schiffelers RM, Storm G. et al. Multi-parametric assessment of the anti-angiogenic effects of liposomal glucocorticoids. Angiogenesis. 2011;14:143-53
123. Hofkens W, Storm G, van den Berg W, van Lent P. Inhibition of M1 macrophage activation in favour of M2 differentiation by liposomal targeting of glucocorticoids to the synovial lining during experimental arthritis. Annals of the rheumatic diseases. 2011;70:A40-A
124. Tracy EC, Bowman MJ, Henderson BW, Baumann H. Interleukin-1alpha is the major alarmin of lung epithelial cells released during photodynamic therapy to induce inflammatory mediators in fibroblasts. British journal of cancer. 2012;107:1534-46
125. Harel-Adar T, Mordechai TB, Amsalem Y, Feinberg MS, Leor J, Cohen S. Modulation of cardiac macrophages by phosphatidylserine-presenting liposomes improves infarct repair. Proceedings of the National Academy of Sciences. 2011;108:1827-32
126. Siglienti I, Bendszus M, Kleinschnitz C, Stoll G. Cytokine profile of iron-laden macrophages: implications for cellular magnetic resonance imaging. Journal of neuroimmunology. 2006;173:166-73
127. Fraccarollo D, Galuppo P, Bauersachs J. Novel therapeutic approaches to post-infarction remodelling. Cardiovascular research. 2012;94:293-303
128. Nakanishi Y, Nakatsuji M, Seno H, Ishizu S, Akitake-Kawano R, Kanda K. et al. COX-2 inhibition alters the phenotype of tumor-associated macrophages from M2 to M1 in ApcMin/+ mouse polyps. Carcinogenesis. 2011;32:1333-9
129. Shen J, Chelvam V, Cresswell G, Low PS. Use of Folate-Conjugated Imaging Agents To Target Alternatively Activated Macrophages in a Murine Model of Asthma. Molecular pharmaceutics. 2013;10:1918-27
130. Melancon MP, Lu W, Huang Q, Thapa P, Zhou D, Ng C. et al. Targeted imaging of tumor-associated M2 macrophages using a macromolecular contrast agent PG-Gd-NIR813. Biomaterials. 2010;31:6567-73
131. Stüve O, Bennett JL. Pharmacological properties, toxicology and scientific rationale for the use of natalizumab (Tysabri®) in inflammatory diseases. CNS drug reviews. 2007;13:79-95
132. Melgarejo E, Medina MÁ, Sánchez-Jiménez F, Urdiales JL. Monocyte chemoattractant protein-1: a key mediator in inflammatory processes. The international journal of biochemistry & cell biology. 2009;41:998-1001
133. Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G. et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nature biotechnology. 2011;29:1005-10
134. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P. et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science. 2009;325:612-6
135. Majmudar MD, Yoo J, Keliher EJ, Truelove JJ, Iwamoto Y, Sena B. et al. Polymeric Nanoparticle PET/MR Imaging Allows Macrophage Detection in Atherosclerotic PlaquesNovelty and Significance. Circulation research. 2013;112:755-61
136. Abedini F, Hosseinkhani H, Ismail M, Domb AJ, Omar AR, Chong PP. et al. Cationized dextran nanoparticle-encapsulated CXCR4-siRNA enhanced correlation between CXCR4 expression and serum alkaline phosphatase in a mouse model of colorectal cancer. International journal of nanomedicine. 2012;7:4159-68
137. Tlaxca JL, Rychak JJ, Ernst PB, Konkalmatt PR, Shevchenko TI, Pizzaro TT. et al. Ultrasound-based molecular imaging and specific gene delivery to mesenteric vasculature by endothelial adhesion molecule targeted microbubbles in a mouse model of Crohn's disease. Journal of controlled release: official journal of the Controlled Release Society. 2013;165:216-25
138. Esposito E, Cuzzocrea S. TNF-alpha as a therapeutic target in inflammatory diseases, ischemia-reperfusion injury and trauma. Current medicinal chemistry. 2009;16:3152-67
139. Tobinick E, Gross H, Weinberger A, Cohen H. TNF-alpha modulation for treatment of Alzheimer's disease: a 6-month pilot study. Medscape General Medicine. 2006;8:25
140. Scheinfeld N. A comprehensive review and evaluation of the side effects of the tumor necrosis factor alpha blockers etanercept, infliximab and adalimumab. Journal of dermatological treatment. 2004;15:280-94
141. Aouadi M, Tesz GJ, Nicoloro SM, Wang M, Chouinard M, Soto E. et al. Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature. 2009;458:1180-4
142. Yin L, Song Z, Qu Q, Kim KH, Zheng N, Yao C. et al. Supramolecular Self-Assembled Nanoparticles Mediate Oral Delivery of Therapeutic TNF-alpha siRNA against Systemic Inflammation. Angewandte Chemie International English. 2013;52:5757-61
143. Komano Y, Yagi N, Onoue I, Kaneko K, Miyasaka N, Nanki T. Arthritic joint-targeting small interfering RNA-encapsulated liposome: implication for treatment strategy for rheumatoid arthritis. The Journal of pharmacology and experimental therapeutics. 2012;340:109-13
144. Koki A, Khan NK, Woerner BM, Dannenberg A, Olson L, Seibert K. et al. Cyclooxygenase-2 in human pathological disease. Advances in Experimental Medicine and Biology. 2002;507:177-84
145. Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S. et al. Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:5473-8
146. Wang K, Tarakji K, Zhou Z, Zhang M, Forudi F, Zhou X. et al. Celecoxib, a selective cyclooxygenase-2 inhibitor, decreases monocyte chemoattractant protein-1 expression and neointimal hyperplasia in the rabbit atherosclerotic balloon injury model. Journal of cardiovascular pharmacology. 2005;45:61-7
147. Deeks JJ, Smith LA, Bradley MD. Efficacy, tolerability, and upper gastrointestinal safety of celecoxib for treatment of osteoarthritis and rheumatoid arthritis: systematic review of randomised controlled trials. British Medical Journal. 2002;325:619
148. Gallicchio M, Rosa A, Dianzani C, Brucato L, Benetti E, Collino M. et al. Celecoxib decreases expression of the adhesion molecules ICAM-1 and VCAM-1 in a colon cancer cell line (HT29). British journal of pharmacology. 2008;153:870-8
149. Venkatesan P, Puvvada N, Dash R, Prashanth Kumar B, Sarkar D, Azab B. et al. The potential of celecoxib-loaded hydroxyapatite-chitosan nanocomposite for the treatment of colon cancer. Biomaterials. 2011;32:3794-806
150. Uddin MJ, Crews BC, Blobaum AL, Kingsley PJ, Gorden DL, McIntyre JO. et al. Selective visualization of cyclooxygenase-2 in inflammation and cancer by targeted fluorescent imaging agents. Cancer research. 2010;70:3618-27
151. Uddin MJ, Crews BC, Ghebreselasie K, Huda I, Kingsley PJ, Ansari MS. et al. Fluorinated COX-2 inhibitors as agents in PET imaging of inflammation and cancer. Cancer Prevention Research. 2011;4:1536-45
152. Mikhaylova M, Stasinopoulos I, Kato Y, Artemov D, Bhujwalla Z. Imaging of cationic multifunctional liposome-mediated delivery of COX-2 siRNA. Cancer gene therapy. 2008;16:217-26
153. Patel SK, Zhang Y, Pollock JA, Janjic JM. Cyclooxgenase-2 Inhibiting Perfluoropoly (Ethylene Glycol) Ether Theranostic Nanoemulsions—In Vitro Study. PloS one. 2013;8:e55802
154. Lee D, Khaja S, Velasquez-Castano JC, Dasari M, Sun C, Petros J. et al. In vivo imaging of hydrogen peroxide with chemiluminescent nanoparticles. Nature materials. 2007;6:765-9
155. Lee I-J, Hwang O, Yoo D-H, Khang G-S, Lee D-W. Detection of Hydrogen Peroxide in vitro and in vivo Using Peroxalate Chemiluminescent Micelles. Bulletin of the Korean Chemical Society. 2011;32:2187-92
156. Cho S, Hwang O, Lee I, Lee G, Yoo D, Khang G. et al. Chemiluminescent and Antioxidant Micelles as Theranostic Agents for Hydrogen Peroxide Associated-Inflammatory Diseases. Advanced Functional Materials. 2012;22:4038-43
157. Park H, Kim S, Kim S, Song Y, Seung K, Hong D. et al. Antioxidant and anti-inflammatory activities of hydroxybenzyl alcohol releasing biodegradable polyoxalate nanoparticles. Biomacromolecules. 2010;11:2103-8
158. Tu C, Osborne EA, Louie AY. Activatable T(1) and T(2) magnetic resonance imaging contrast agents. Annals of biomedical engineering. 2011;39:1335-48
159. Maiseyeu A, Badgeley MA, Kampfrath T, Mihai G, Deiuliis JA, Liu C. et al. In vivo targeting of inflammation-associated myeloid-related protein 8/14 via gadolinium immunonanoparticles. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:962-70
160. Schellekens H. Follow-on biologics: challenges of the 'next generation'. Nephrology Dialysis Transplantation. 2005;20:iv31-iv6
Author Biography
Sravan Kumar Patel obtained his Bachelors in Pharmacy (Hons.) from Birla Institute of Technology & Science, Pilani, India and MS in Pharmacy from University of Georgia, Athens, USA. He is currently a Ph. D. student at Graduate School of Pharmaceutical Sciences in Mylan School of Pharmacy, Duquesne University. His research work focuses on the development and application of theranostic nanoemulsions for drug delivery to macrophages in varied disease models.
Dr. Jelena M. Janjic is an Assistant Professor in Pharmaceutical Sciences of Duquesne University, Pittsburgh, USA. She is also a faculty member of McGowan Research Institute for Regenerative Medicine. She has co-authored publications in reputed journals such as Journal of American Chemical Society, Biomaterials, PLoS One. Dr. Janjic is the founder and co-director of the Chronic Pain Research Consortium at Duquesne University. Her research focuses on the development and application of theranostics as new therapies for cancer, chronic pain and inflammatory diseases.
![]() Corresponding author: Jelena M. Janjic, Email- janjicjedu, Tel-(412) 396-6369 Fax-(412) 396-4660.
Corresponding author: Jelena M. Janjic, Email- janjicjedu, Tel-(412) 396-6369 Fax-(412) 396-4660.